

Abstract
The molecular etiology linking the pathogenic mutations in the Huntingtin (Htt) gene with Huntington’s disease (HD) is unknown. Prior work suggests a role for Htt in neuronal autophagic function and mutant HTT protein disrupts autophagic cargo loading. Reductions in the bioavailability of the essential metal manganese (Mn) are seen in models of HD. Excess cellular Mn impacts autophagic function, but the target and molecular basis of these changes are unknown. Thus, we sought to determine if changes in cellular Mn status impact autophagic processes in a wild-type or mutant Htt-dependent manner. We report that the HD genotype is associated with reduced Mn-induced autophagy and that acute Mn exposure increases autophagosome induction/formation. To determine if a deficit in bioavailable Mn is mechanistically linked to the autophagy-related HD cellular phenotypes, we examined autophagosomes by electron microscopy. We observed that a 24 h 100 uM Mn restoration treatment protocol attenuated an established HD ‘cargo-recognition failure’ in the STHdh HD model cells by increasing the percentage of filled autophagosomes. Mn restoration had no effect on HTT aggregate number, but a 72 h co-treatment with chloroquine (CQ) in GFP-72Q-expressing HEK293 cells increased the number of visible aggregates in a dose-dependent manner. As CQ prevents autophagic degradation this indicates that Mn restoration in HD cell models facilitates incorporation of aggregates into autophagosomes. Together, these findings suggest that defective Mn homeostasis in HD models is upstream of the impaired autophagic flux and provide proof-of-principle support for increasing bioavailable Mn in HD to restore autophagic function and promote aggregate clearance.
Introduction
Huntington’s disease (HD) is an age-progressive neurodegenerative disease characterized by the primary symptom, chorea, uncontrolled motor behavior. However, symptoms and progression are highly variable between patients. There is no cure and few symptomatic treatments for this fatal disease. An expanded CAG repeat within exon 1 of the htt gene produces the mutated protein; however, it is still unclear how mutant HTT protein (mutHTT) causes specific cell death in the medium spiny neurons of the striatum (1–3). Though debated, it has been shown that mutHTT aggregates (or at least some forms of mutHTT aggregates) contribute to eventual cell death (4–7). Autophagy is the primary process by which mutHTT aggregates can be degraded. However, it is thought that loss-of-function of wild-type (WT) htt, via the HD mutation, disrupts autophagy-mediated aggregate clearance. In fact, WT htt has binding motifs for both p62 and ULK-1/LC3, critical proteins for labeling and incorporation of autophagic cargo, respectively. Acting as an autophagy scaffold, WT HTT protein promotes proximal localization of autophagic cargo and the autophagosome itself (8–10). Recent evidence shows that HD cells exhibit autophagy ‘cargo recognition failure’: autophagosomes are ineffectively loaded with cargo, resulting in reduced rates of macroautophagy and an accumulation of lipids and mitochondria within the cells. This deficit is suspected to impede aggregate clearance and thought to be caused by an abnormal association between mutHTT and the ubiquitin-binding protein, p62, a proposed ‘linker’ protein between autophagosomes and their respective cargo (11). Upregulation of autophagy via both mTOR-dependent and independent mechanisms can facilitate mutHTT aggregate clearance and rescue HD phenotypes. Genetic and pharmacological manipulation of autophagy via mTOR-dependent and independent mechanisms have been shown to promote mutHTT aggregate clearance, rescue medium spiny neuron health and normalize motor behavior in flies and mice (4, 12–20). Given this evidence, there has been a large impetus in the field to (1) understand how mutHTT dysregulates autophagy and (2) how to manipulate autophagy pathways to promote autophagic function in HD and potentially target this process pharmacologically.
Manganese (Mn) is an essential metal, a co-factor for a variety of enzymatic processes and a potent modulator of cell signaling, but in excess is neurotoxic. The striatum, the region of the brain most vulnerable in HD, and other basal ganglia nuclei, typically have the highest levels of Mn in the brain, suggesting Mn plays a particularly important, yet poorly understood, role in this brain region (21, 22). HD models exhibit striatal-specific reduced Mn uptake suggestive of a HD-dependent, brain-specific Mn deficiency (23–26). This deficit in net Mn uptake confers increased resistance to Mn cytotoxicity in HD cells. This also manifests as dysregulation in the Mn-dependent arginase–citrulline pathway and ATM, p53 and AKT signaling in these models, though we posit that many other Mn-responsive pathways and processes are also affected (27, 28).
Recent studies have shown that Mn exposure causes rapid, yet temporally bi-phasic, increases in common autophagy markers (p62, LC3, Beclin) in a variety of cell types and mouse models. However, increased expression of autophagy proteins has been associated with both activation and inhibition of autophagy by Mn (29–36). Mn also is known to activate ERK, AKT, ATM, AMPK, mTOR and various transcription factors (TFEB, CREB, p53, FOXOs, etc.), all of which regulate autophagy (25, 37–46). Evidence supports a neuroprotective role for autophagy in the context of Mn neurotoxicity. However, given the intricacies of interpreting autophagy data, the mechanism by which Mn impinges on autophagic function, particularly in the context of HD, is unknown. Given the role of Mn-responsive cell signaling proteins in the regulation of autophagy and a potentially striatal-specific Mn deficiency in HD models, we hypothesized that elevating extracellular Mn levels to restore subcellular Mn homeostasis would improve autophagic functions in HD cells via normalization of ‘Mn-starved’ cell signaling processes. In this study, we set out to (1) employ a battery of autophagy methodologies to rigorously assess the effects of Mn on autophagy and (2) evaluate the effects of Mn on autophagic flux and cargo sequestration in HD cells.
Results
Mn increases p62 and LC3-II/I expression after 24 h in WT cells, presence of HD mutation suppresses this effect
Previous reports have demonstrated that Mn exposure increases expression of common autophagy proteins—namely Beclin, LC3-II and p62 (29–31, 33, 35). However, elevated expression of these proteins can be a result of either increased autophagic flux or decreased autophagic clearance. Thus, it is still unclear whether upregulation of these proteins by Mn is indicative of inhibition or activation of autophagy. Here we defined how Mn impinges on autophagy, particularly in the STHdh striatal neuroprogenitor cell model (Q7 and Q111, and HD models, respectively), which is a widely used mouse striatal cell line model of HD. LC3 and p62 expression was measured, as markers of autophagy. To assess LC3 turnover, we calculated an LC3-II/I ratio, as LC3II is the lipidated form of LC3I and runs faster on sodium dodecyl sulfate (SDS)-PAGE (52). A 24 h Mn exposure caused significant increases in both p62 and the LC3-II/I ratio in WT cells, the increase in p62 being greater in magnitude (Fig. 1a, b and d). The increase in LC3-II/I ratio was driven primarily by a decrease in LC3I rather than an increase in LC3II, consistent with the lipidation of LC3I to LC3II during the synthesis/incorporation of autophagy (Fig. 1a). Mn-induced p62 and LC3-II/I was significantly blunted in HD cells. This finding is consistent with the reduced net Mn uptake phenotype observed in these cells (24, 27). To assess whether Mn-induced p62/LC3 were dependent on Mn-dependent kinases (PI3K and mTOR) (48, 49) we exposed STHdh cells to a series of small molecule inhibitors to see if they modulated Mn-induced autophagy (Supplementary Material, Fig. S1b). NU7441, a DNApk inhibitor known to activate autophagy (50), induced p62 expression, similar to Mn, and also exhibited synergistic, additive effects when co-exposed with Mn (Fig. 1a and b; Supplementary Material, Fig. S1a). Co-exposure with NU7441 did not ameliorate impaired Mn-induced p62 or LC3II/I in Q111 cells (Supplementary Material, Fig. S1a and b) (NU7441 inhibits mTOR and PI3K at 5 uM, thus only NU7441 (1 uM) was quantified and more specific inhibitors of PI3K and mTOR were used) (45, 89). Interestingly, Mn-induced p62 was completely abrogated by PI3K inhibition (LY294002) but not affected by mTOR inhibition (Rapamycin), suggesting Mn-induced p62 is PI3K, but not mTOR, dependent (Fig. 1a and e). LY294002 and rapamycin did not have a significant effect on Mn-induced LC3-II/I, though both inhibitors were trending to block Mn-induced LC3-II/I (Fig. 1a and c). LY294002 and Rapamycin were validated to inhibit basal and Mn-induced p-AKT and p-S6, respectively, in these cells, at the same concentrations, in a previous study in our laboratory (45).
Figure 1.
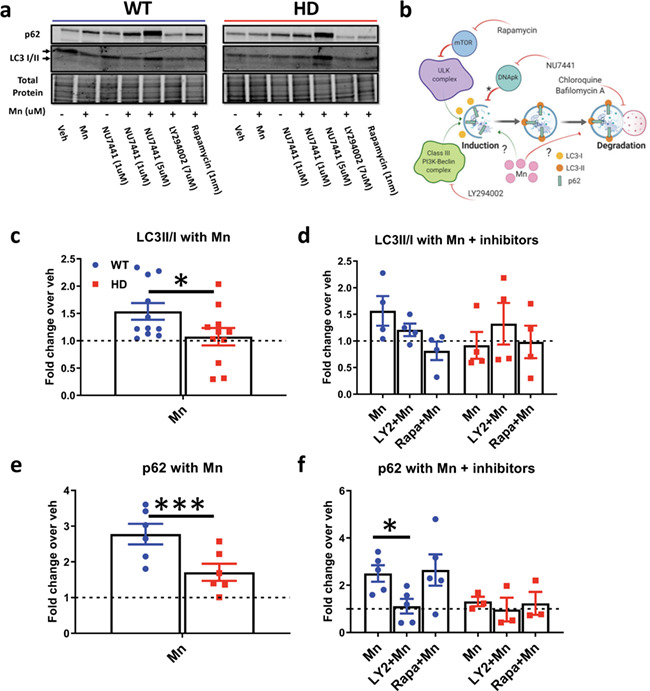
Mn increases expression of LC3-II/I and p62 which is reduced in Q111 cells. (a) Representative western blot of STHdh Q7/Q7 and Q111/Q111 after 24 h exposure with Mn (50 uM), NU7441 (1/5 uM), LY294002 (7 uM) or Rapamycin (1 nM). LC3-I = top arrow, LC3-II = bottom arrow. (b) Diagram of how different proteins and small molecules (NU7441, rapamycin and LY294002) impinge on autophagy. Asterisks, unclear mechanism by which DNApk inhibition leads to autophagy. (c) Quantification of LC3-II/I in cells treated with Mn only. Asterisks, significant by paired t-test. N = 11. (d) Quantification of LC3-II/I in cells treated with Mn and LY294002 or Rapamycin. Two-way analysis of variance (ANOVA); treatment = F(2, 6) = 2.665; P = 0.1485. N = 4. (e) Quantification of p62 in cells treated with Mn only. Asterisks, significant by paired t-test. N = 6. (f) Quantification of p62 in cells treated with Mn and LY294002 or Rapamycin. Two-way ANOVA; treatment = F(2, 6) = 5.579; P = 0.0428. N = 3. For c–f, each cell line’s vehicle is normalized to 1. NU7441 was used as a control for Figure 1a blot and quantified in Supplementary Material, Figure S1A and B. Blue = WT; red = HD. Error bars = SEM. *Significant by Dunnet’s post-hoc test was used to compare back to Mn treated in c and e. *P < 0.05, **P < 0.01, ***P < 0.001.
Mn induces autophagy by increasing autophagosome induction and formation
To determine whether Mn activates autophagic synthesis/incorporation or inhibits autophagic degradation we assessed autophagic flux via lysosomal autophagy inhibitors (chloroquine, CQ; bafilomycin A, BafA) to block autophagosome degradation, thus allowing for assessment of autophagosome induction and formation only (i.e. autophagy activators, but not inhibitors, will induce autophagic protein expression if the degradation of autophagosome is blocked; Supplementary Material, Fig. S1c–f) (53). We first determined concentrations of inhibitors that completely inhibit autophagosome degradation. To determine the effective saturating concentrations for CQ and BafA, we utilized stable, dsRed-LC3-II expressing WT and HD STHdh cells. We found that 10 uM CQ and 10 nM BafA caused maximal accumulation of LC3II puncta without incurring cell death or morphological changes, while super-saturating 20 μM CQ caused extensive, ‘swiss-cheese-like’ vacuolization in WT STHdh, and 20 nM BafA reduced cell size and caused cell death (Supplementary Material, Figs S2 and 3). Thus, we exposed STHdh cells to Mn and 10 uM CQ or 10 nM BafA for 24 h and monitored p62 and LC3II/I expression. Mn increased p62 and LC3-II/I in WT and HD cells and LC3-II/I in WT cells, even in the presence of saturating concentrations of CQ (Fig. 2a–c) or BafA (Fig. 2a, d and e). This supports our hypothesis that Mn increases autophagic induction/formation. We observed similar increases in magnitude of p62 and LC3-II/I in HD cells compared to WT cells with BafA and slightly blunted responses with CQ. However, Mn did not significantly increase LC3II/I in HD cells. The observed p62 doublet in BafA-treated conditions is consistent with reports in mouse brain and liver; however, we only quantified the bottom band as this was not observed in every condition (51).
Figure 2.
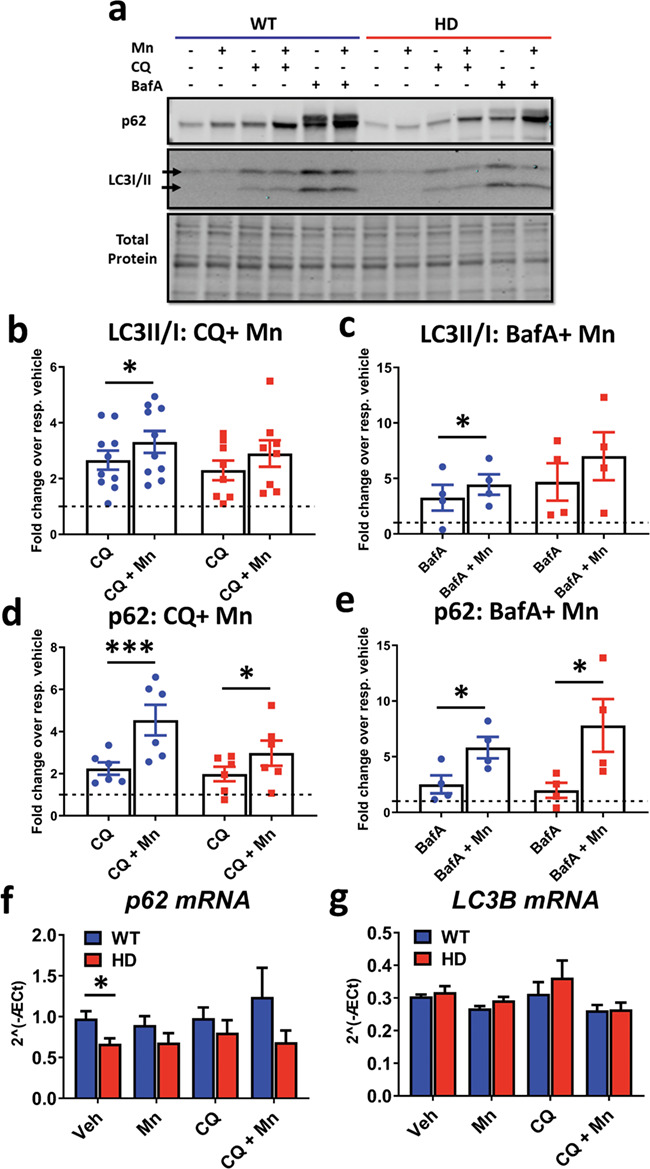
Mn increases LC3-II/I and p62 expression in the presence of CQ and BafA. (a) Representative western blot of p62 and LC3-II/I in SThdh Q7/Q7 and Q111/Q111 after 24 h exposure with Mn (50 uM), CQ (10 uM) and BafA (10 nM). LC3-I = top arrow, LC3-II = bottom arrow. (b) Quantification of LC3-II/I in cells treated with CQ+Mn. Two-way ANOVA; treatment = F(1, 10) = 18.08; P = 0.0017. N = 8. (c) Quantification of LC3-II/I in cells after treatment with BafA+Mn. Two-way ANOVA; treatment = F(1, 3) = 2.374; P = 0.2211. N = 6. (d) Quantification of p62 in cells after treatment with Mn+CQ. Two-way ANOVA; treatment = F(1, 3) = 229.2; P = 0.006. N = 4. (e) Quantification of p62 in cells after treatment with BafA+Mn. N = 4. Two-way ANOVA; treatment = F(1, 3) = 10.13; P = 0.0500. In BafA-treated conditions, only the bottom band of p62 was quantified in order to compare to CQ-treated conditions where only one band was present. (f and g) qRT-PCR analysis for p62 (f) and LC3B (g) in STHdh Q7/Q7 and Q111/Q111 after 24 h treatment with Mn (50 uM) and/or CQ (10 uM). N = 4 for qRT-PCR results. Two-way ANOVA for p62; treatment = F(1.296,3.887) = 0.7953; P = 0.4590. Two-way ANOVA; genotype = F(1, 3) = 17.63; P = 0.0247; two-way ANOVA for LC3B; treatment = F(1.296,3.887) = 0.7953; P = 0.4590. Two-way ANOVA; genotype = F (1, 3) = 17.63; P = 0.0247. Blue = WT; red = HD. Error bars = SEM. Asterisks, significant difference from vehicle by post-hoc comparisons. *P < 0.05, **P < 0.01, ***P < 0.001. For b–e, Sidak’s multiple comparison test compared CQ/BafA and Mn+CQ/BafA for each genotype. For f and g, Dunnet’s multiple comparison test compared all treatments back to vehicle for each genotype.
Mn-induced LC3 and p62 protein expression is not due to an increase in mRNA expression
We tested the hypothesis that Mn upregulates p62 and LC3B expression by increasing their mRNA expression. Mn and/or CQ exposure did not significantly change p62 or LC3B mRNA expression in either genotype (Fig. 2f and g). We did not observe a difference in basal LC3B mRNA between Q7 and Q111 cells, but observed a genotype difference in basal p62 mRNA. However, as no large increases in mRNA expression were observed, the Mn-induced modulation of protein markers of autophagy is not consistent with a transcriptional mechanism in these cells.
Exposure with CQ/BafA increases net Mn uptake in Q111 cells, attenuating defective Mn-induced autophagy
To gain mechanistic insight into why Mn exposure, under conditions where autophagy degradation is inhibited, induces LC3-II/I and p62 in HD cells to the same magnitude as WT cells (despite established reductions in net Mn accumulation in HD cells; Fig. 2a–e), we tested the hypothesis that CQ and BafA are capable of normalizing Mn uptake in HD cells. Using the cellular fura-2 Mn uptake assay (CFMEA) developed in our laboratory (26), we assessed net Mn uptake after 24 h exposure with Mn and CQ/BafA. Interestingly, CQ and BafA increased net Mn uptake in both Q7 and Q111 cell lines exposed to 25 μM Mn (Fig. 3a and b). The lower 25 μM Mn concentration was used to test for the possibility of an Mn uptake ceiling effect in Q7 cells at the higher concentration. After exposure to 50 μM Mn, (the same concentration used in previous experiments) we found CQ and BafA normalized net Mn uptake between the control and HD cell lines (Fig. 3a and b). Furthermore, these increases were completely blocked with addition of 5 mM 3-methyladenine (3MA), an autophagy inhibitor that blocks activation of autophagy by inhibiting class III PI3K (Fig. 3d). Additionally, 3MA alone was able to reduce net Mn uptake in these cells. 3MA reduction of Mn uptake could be due to inhibition of class III PI3K, or possibly through off target inhibition of class I PI3K (Fig. 3c), consistent with our recent report that inhibition of class I PI3K in these cells reduces net Mn uptake (45). We found that CQ and BafA increased net Mn uptake in other cell lines (3T3, HEK293, SH-SY5Y), though to a lesser extent than seen for STHdh cells (Fig. 3e). These data (1) explain why Mn-induced autophagy is similarly affected in WT and HD STHdh cells that are co-exposed with Mn+CQ, despite the reduce Mn accumulation usually seen in the HD STHdh cells and (2) suggest that autophagy plays a novel role in Mn homeostasis which contributes to the impaired Mn uptake in HD cells.
Figure 3.
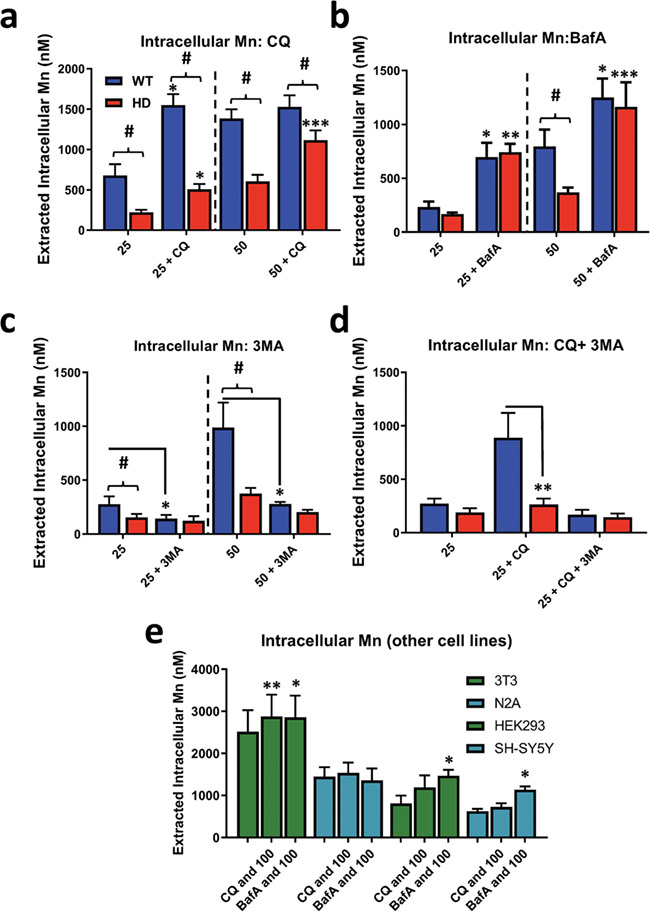
CQ and BafA increase net Mn uptake which is blocked by 3MA. Assessment of intracellular Mn by CFMEA in STHdh Q7/Q7 and Q111/Q111 cells after 24 h exposure with (a) Mn (25/50 uM) and CQ (10 uM), (b) Mn (25/50 uM) and BafA (10 nM), (c) Mn (25/50 uM) and 3MA (5 mM) and (d) Mn (25 uM), CQ (10 uM) and 3MA (5 mM). Note: dashed lines separate experiments where 25 uM or 50 uM Mn was used. N = 3 for all experiments; error bars = SEM. Blue = WT; red = HD. Number sign, significant genotype difference; asterisks, significance by Sidak multiple comparison test. Two-way ANOVA analysis in Supplementary Material, Figure S4. (e) CFMEA measuring intracellular Mn levels in NIH3T3, Neuro2A, HEK293 and SH-SY5Y cells after 24 h exposure with Mn (100 uM), CQ (10 uM) and BafA (10 nM). Asterisks, significance by paired student’s t-test compared to Mn alone at same concentration. *P < 0.05, **P < 0.01, ***P < 0.001.
HD genotype suppression of Mn-induced autophagy markers is also observed in human patient-derived striatal neuroprogenitors
We investigated whether Mn-induced autophagy was also a phenotype in non-transformed neural precursor cells derived from HD patient derived–induced pluripotent stem cells. Utilizing a differentiation protocol developed in our laboratory, we differentiated three control and three HD patient-derived cell lines to 11-day-old Islet-1 expressing striatal-like neuroprogenitors (27). These cells were exposed to 200 μM Mn with or without CQ/BafA for 24 h. Previous reports from our laboratory show 200 uM Mn exposures in these human cells results in nearly the same amount of intracellular Mn (nm/ug DNA) as STHdh cells after 50 uM (27). Mn-induced LC3-II/I expression (with BafA) was similar between control and HD cells (Fig. 4a and b). However, Mn-induced p62 was severely blunted in all Mn-exposed conditions in HD patient-derived cells (Fig. 4a, c and d). We did not detect a difference in basal p62 expression. We also noted that CQ had little effect on LC3 or p62 expression in these cells; in a separate experiment, we used CQ at twice the concentration (20 uM) and still observed no change in the expression of LC3 or p62 (data not shown). The lack of an observable LC3-II band in most conditions may reflect highly efficient autophagy processing in these cells. As CQ and BafA inhibit autophagic degradation somewhat differently (CQ inhibits lysosome-autophagosome binding, BafA primarily prevents acidification of lysosomes; 53–55), this may explain why BafA, but not CQ, resulted in a prominent LC3-II band. These data suggest Mn can activate autophagy in human induced pluripotent stem cell (hiPSC)-derived neuroprogenitors and that defects in Mn-induced autophagy, particularly Mn-induced p62, are present in both HD mouse and human striatal neuroprogenitors.
Figure 4.
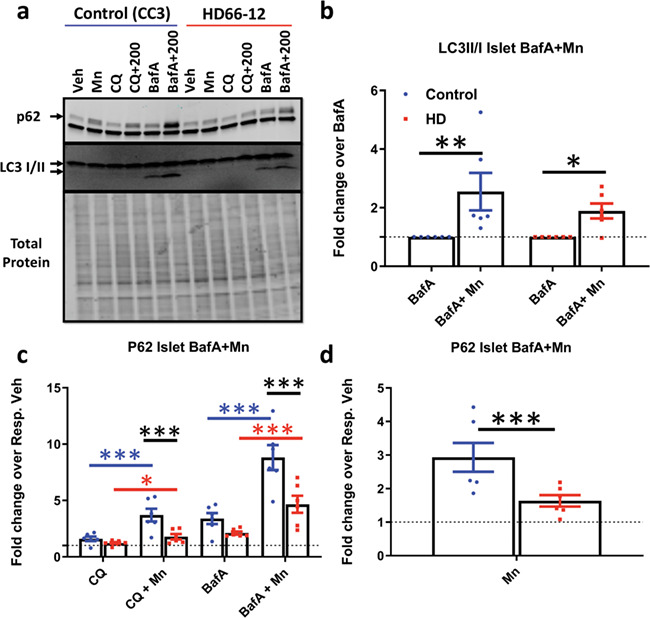
HD patient iPSC-derived neuroprogenitors exhibit defects in Mn-induced autophagy. (a) Representative western blot of p62 and LC3-II/I in hiPSC-derived neuroprogenitors (CC3 = control cells; HD66-12 = HD cells) after 24 h exposure with Mn (200 uM), CQ (10 uM) and BafA (10 nM). p62 = top arrow, (non-specific LC3 band below p62); LC3-I = top arrow, LC3-II = bottom arrow. (b) Quantification of LC3-II/I in cells treated with BafA and BafA+Mn. N = 5. Dotted line: BafA = 1. Two-way ANOVA; F(1, 4) = 20.38; P = 0.0107. (c) Quantification of p62 after treatment with Mn and CQ, and BafA. (d) Quantification of p62 after treatment with Mn alone. Error bars = SEM. Blue = WT; red = HD. N = 6. Dotted line: vehicle = 1. Two-way ANOVA; treatment = F(4, 20) = 65.73; P = 0.0001; genotype = F(1, 5) = 13.40; P = 0.0146; treatment–genotype interaction = F(4, 20) = 3.730; P = 0.0200. Asterisks, significant by Sidak (b) Tukey’s (c) multiple comparison test. Blue asterisks, WT treatment difference; red asterisks, HD treatment difference; black asterisks, genotypes difference. *P < 0.05, **P < 0.01, ***P < 0.001.
HD-dependent defects in Mn-induced autophagy are corroborated by reduced LC3-II puncta
In order to confirm our western blot findings, we also performed LC3-II fluorescent microscopic analyses. To quantify specifically LC3-II we utilized dsRED-LC3-II expressing STHdh cells. Q7 and Q111 cells exhibit similar levels of basal LC3-II puncta (Fig. 5a). CQ induced LC3 puncta in both cell lines, though to a lesser magnitude in HD cells (Fig. 5c; Supplementary Material, Figs S6b and S7a and b). Mn alone increased LC3-II puncta significantly in both cell lines, though this occurred at significantly lower concentrations in WT cells and this genotype and genotype x Mn difference was significant by two-way ANOVA (Fig. 5b; Supplementary Material, Figs S6a and S7a and b). Contrarily, Mn-induced autophagic flux (i.e. with CQ) was similar, if not more sensitive, in HD cells (Fig. 5d; Supplementary Material, Fig. S6a and b), similar to our western blot results, and is likely due to the normalization of net Mn uptake after CQ exposure in STHdh cells. In addition, we measured autophagic severity in Q7 cells via a 4-point, blinded assessment and observed similar results (Supplementary Material, Fig. S7c). Mn-induced p62 puncta similarly to LC3-II puncta, even in presence of CQ (Supplementary Material, Fig. S7a–c); however, we did not detect a genotype difference between Q7 and Q111 cells (data not shown) likely due to limited sensitivity of this assay and analysis. Mn did not increase co-localization between LC3-II and p62 (data not shown). Taken together, these results strongly support the conclusion that reduced Mn-induced autophagy in HD can be attenuated by restoring Mn levels to a non-deficient condition.
Figure 5.
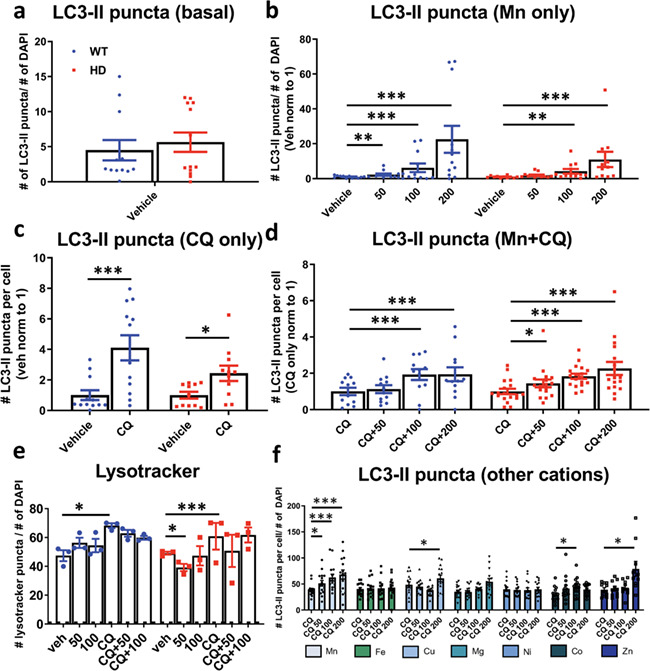
Mn increases LC3-II puncta. Quantification of number of LC3-II puncta over number of DAPI in STHdh Q7/Q7 and Q111/Q111 after 24 h exposures with (a) untreated (basal), (b) Mn (50/100/200 uM), (c) CQ (10 uM) or (d) Mn (50/100/200 uM) + CQ (10 uM). Quantified as (# of LC3-II puncta)/(# of DAPI) per image. N = 3–4 biological replicates; each with five images per condition. Images of the vehicle treatment (or CQ in panel f) per biological set were all averaged, and this value was set to 1. All other data points in each biological set were normalized respective to this value. Error bars = SEM. Two-way ANOVA statistics for panel b–d shown in Supplementary Material, Figure S5. (e) Quantification of number of lysotracker puncta per number of DAPI puncta after 24 h treatment with Mn (25/50/100 uM) and/or CQ (10 uM) in STHdh Q7/Q7 and Q111/Q111. Quantified as (# lysotracker puncta)/(# of DAPI) per image. Two-way ANOVA; treatment = F(5, 70) = 9.456; P ≤ 0.0001. N = 3 biological replicates with 5 images per set. Error = SEM. Blue = WT; red = HD. (f) Quantification of LC3-II puncta after 24 h treatment with Mn, Fe, Cu, Mg, Ni, Co, Zn (0/50/100/200 uM) with CQ (10 uM) for 24 h. Two-way ANOVA; treatment = F(31, 34) = 15.43; P ≤ 0.0001. N = 3 biological replicates, each with 5 images per condition. Quantified as (# LC3-II puncta)/(# of DAPI) per image. Error bars = SEM. Asterisks, significant difference from Veh (b, c and e) or CQ (d and f) by Dunnet’s multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001.
Lysosomes are necessary for the proper degradation of autophagosomes and, thus, modulation of lysosomal processing can be reflected in autophagic measures. Previous reports found Mn decreased lysosome number and increased lysosome size (31). We quantified the number of lysosomes via Lysotracker in dsRED-LC3-II expressing STHdh cells. As predicted, CQ increased Lysotracker puncta in Q7 and Q111 cells. Mn did not affect the number of lysosomal puncta in WT cells, but reduced puncta slightly in HD cells at 50 uM. (Fig. 5e, Supplementary Material, Figs s8 and S9). This suggests Mn-induced autophagy with these cells/conditions is not a secondary effect of an Mn–lysosome interaction.
Mn-induced autophagy is cation-specific and occurs in a dose-dependent manner
Up to this point our data supported the hypothesis that increasing cellular Mn levels activate autophagy, but it has been unclear whether this effect was specific for Mn. Many other metals are capable of modulating autophagy; however, it is unknown whether this is through activation or inhibition of autophagic flux, or whether this can occur at sub-toxic concentrations (53–57). To assess whether other metal cations activate autophagy similarly to Mn at similar concentrations we measured LC3-II puncta in dsRed-LC3-II expressing Q7 cells after 24 h exposures with Mn, Mg, Fe, Cu, Zn, Ni and Co with
CQ. Consistent with an Mn-specific effect, we found that Mn was able to significantly stimulate autophagy at 50 uM, the lowest concentration compared to other metals, and a level which is sub-cytotoxic in these cells. LC3-II puncta induced by other metals (Cu, Co, Zn) trended upward only with higher doses, and this was associated with increased cell death in Zn-treated cells (Fig. 5f, Supplementary Material, Fig. S10). Together, these data suggest that the ability of Mn to modulate autophagy in these cells is cation-specific and occurs in a dose-dependent manner (Fig. 5f). This specificity further supports the hypothesis that reduced bioavailable Mn (but not other essential or heavy metals) in HD cells contributes to the observed autophagic defects.
Mn increases mutHTT aggregate–autophagosome association in 72Q-expressing HEK293 cells in a dose-dependent manner
Poly-Q aggregates are primarily degraded by autophagy. We hypothesized that Mn treatment facilitates mutHTT aggregate association into autophagosomes. Our data suggest Mn can activate autophagy; thus, we posited that Mn promotes mutHTT aggrephagy (autophagy of protein aggregates) via activation of autophagic flux, possibly via upregulation of p62 (as perturbations in p62 cargo recognition have been proposed to underlie autophagy-related HD defects). However, Q111 cells do not produce quantifiable HTT aggregates; thus mutHTT-transfected cell lines are often used (11, 58). In order to examine the effect of Mn on mutHTT aggregate loading into autophagosomes, we utilized HEK293 cells which were transfected to express GFP-72Q-mutHTT. These cells produce robust amounts of aggregates immediately after transfection, which can be easily assessed by fluorescent microscopy. We quantified the degree to which mutHTT aggregates were proximately located with autophagic markers, based upon the expectation from previous work by others that LC3 puncta would be associated with, but not co-localized with, mutHTT aggregates (87, 88). We found some mutHTT aggregates associated closely, but not precisely co-localized, with LC3 puncta after Mn+CQ exposure (Fig. 6a). Because autophagy is a process in flux and HTT aggregates are continuously produced, we utilized CQ to inhibit autophagosome degradation to ascertain whether mutHTT aggregate clearance was autophagy-dependent in these cells. Consistent with previous reports, 10 uM CQ increased the number of aggregates by ~75% (higher concentrations were cytotoxic; Figs 6b and 7). These data suggest mutHTT clearance is, at least, partially autophagy-dependent in 72Q cells. We further investigated whether elevated cellular Mn levels increase aggregate–autophagosome association. We found that a 72 h exposure to 50–100 uM Mn treatment in the presence of CQ significantly increased the number of visible HTT aggregates compared to CQ alone (up to ~2-fold; Figs 6c and 7a). Whereas shorter exposures of 24 h had no effect, with an intermediate effect being seen after 48 h (data not shown). We observed no effect with Mn alone, even at the highest concentrations tested, suggesting Mn is (1) not inhibiting autophagic degradation similar to CQ, (2) not promoting aggregation by itself to increase visible aggregate number and (3) the effect of Mn on aggregate number is autophagy-dependent (Figs 6d and 7b). Furthermore, p62 expression was significantly upregulated in the presence of Mn+CQ compared to CQ alone, mirroring the effects seen on mutHTT aggregate number (Fig. 6e and f). Total LC3 or the LC3-II/I ratio was unaffected by CQ or Mn in 72Q cells (data not shown). Together these data suggest Mn can increase aggregate–autophagosome association in a dose-dependent and time-dependent manner.
Figure 6.
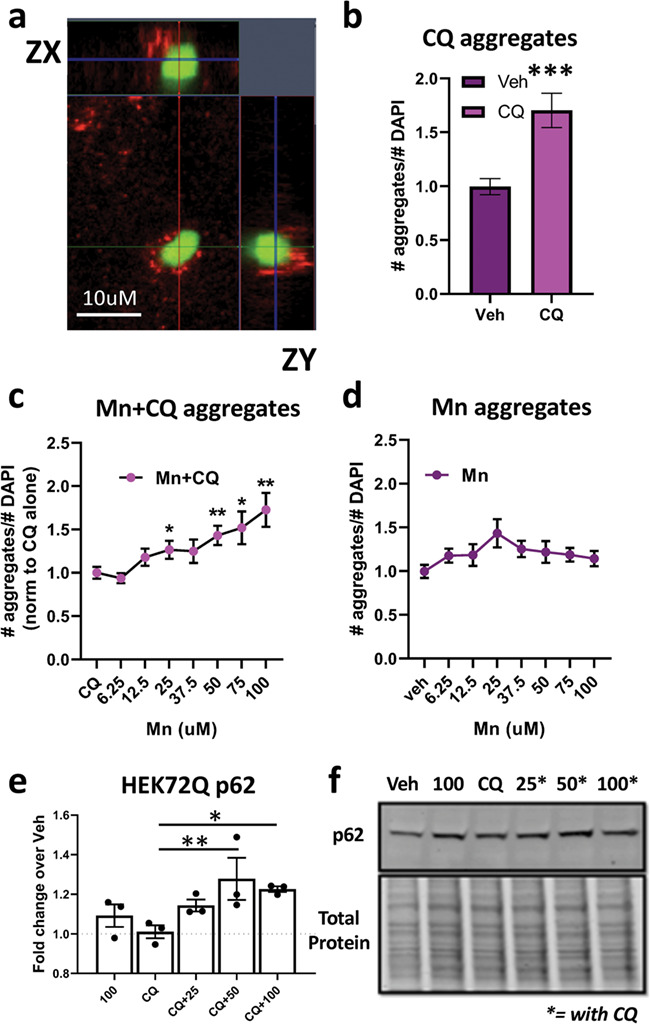
Mn increases the association of autophagosomes with mutant HTT aggregates. (a) Representative 63× confocal image of GFP-HTT-72Q aggregates (green) near LC3 puncta (red) in transfected HEK293 cells after treatment with 75 uM Mn + 10 uM CQ. Includes ZX and ZY view. (b) Quantification of relative number of aggregates after CQ (10 uM) treatment for 72 h. Asterisks, significant difference by student’s t-test. (c) Quantification of number of aggregates after 72 h treatment with increasing amounts of Mn + 10 uM CQ. Univariate ANOVA; F(4.085, 98.04) = 5.552; P = 0.0004. (d) Quantification of number of aggregates after 72 h treatment with increasing amounts of Mn alone. Univariate ANOVA; F(4.046, 89.02) = 1.369; P = 0.2907. Quantification of (c) is normalized to CQ alone and quantification of (d) is normalized to no treatment. N = 3 biological replicates, each with 10 images. (e) Quantification of p62 after 72 h treatment with Mn (25/50/100 uM) and/or CQ (10 uM), in 72Q-GFP HEK293 cells. N = 3. Univariate ANOVA; F (4, 8) = 5.743; P = 0.0177. (f) Representative western blot for HEK72Q p62. Asterisks, exposed with 10 uM CQ. Error bars = SEM; *P < 0.05, **P < 0.01, ***P < 0.001. Asterisks, significance difference by Dunnet’s multiple comparison tests.
Figure 7.
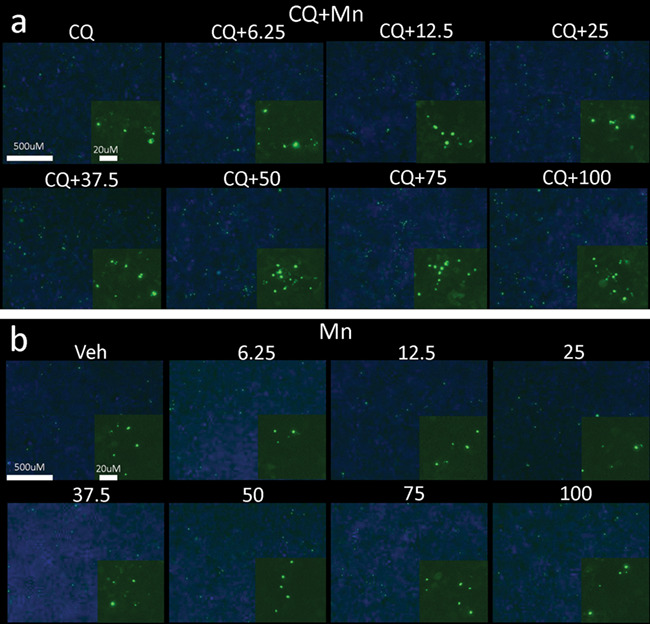
Mn increases the association of autophagosomes with mutant HTT aggregates. (a and b) Representative images in GFP-HTT-72Q transfected HEK293 cells taken at 5× on a Zeiss Fluorescent light microscope of Mn+CQ (a) and Mn alone (b) with GFP-HTT-72Q aggregates (green) and DAPI (blue). Magnified insets of HTT aggregates included in bottom right corner of each condition. 10 uM CQ; 6.25/12.5/25/37.5/50/75/100 uM Mn used. N = 3 biological replicates, each with 10 images.
Acute Mn treatment ameliorates cargo-recognition failure in Q111 cells
Given our evidence that Mn exposure can activate autophagy and promote aggregate–autophagosome association, we posited that Mn exposure also promotes proper autophagic function, particularly in HD cells which exhibit reduced Mn bioavailability. The presence of empty autophagic vacuoles (APVs), lacking osmiophilic, visible cargo, is one of the most striking cellular phenotypes in STHdh Q111 cells. This has been attributed to failed interactions between p62 and mutHTT during autophagosome scaffolding and formation, leading to autophagic ‘cargo recognition failure’ (11). Since Mn-induced p62 is robust in these cell lines, we hypothesized that our acute Mn treatment protocol, which causes the upregulation of autophagic flux, could increase autophagosome recognition and loading. Similar to previous reports by other investigators, we independently confirmed that STHdh Q111 cells exhibit 2–3-fold more empty/partially full APVs than the WT control STHdh Q7 cells (11). Consistent with our hypothesis, 100 μM Mn restoration treatment rescued the number of full APVs to WT levels after 24 h (Fig. 8a and c). This suggests acute, sub-toxic, Mn exposure ameliorates the autophagy cargo sequestration defect in Mn-deficient HD cells. This suggests Mn upregulates the amount of osmophilic cellular constituents incorporated into autophagosomes such as endosomes, ER and mitochondria. We also found that 100 μM Mn decreased the number of empty APVs in WT cells to <5% of total autophagosomes, suggesting that this Mn-induced effect is not necessarily HD-specific (Fig. 8b).
Figure 8.
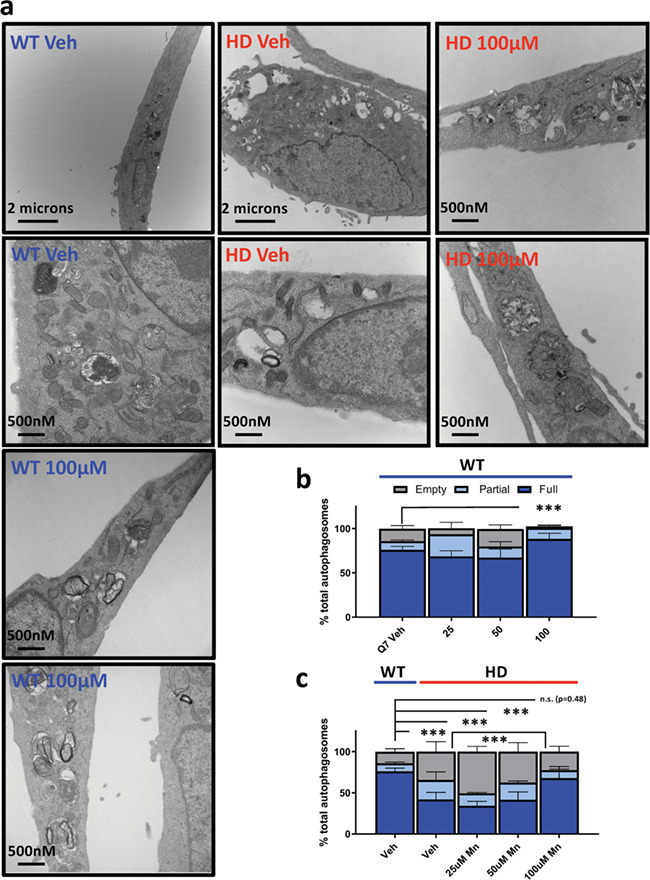
Acute Mn exposure attenuates empty APV phenotype in Q111 STHdh cells. (a) Representative transmission electron microscopy (TEM) images of STHdh WT Q7/Q7 (blue) and HD Q111/Q111 (red) cells with no treatment (Veh) or 100 uM Mn for 24 h. (b and c) Quantification of empty, partial or full autophagosomes after 24 h treatment with 25/50/100 uM Mn in STHdh WT Q7/Q7 (b) and HD Q111/Q111 (c) cells. Note: for panel c, vehicle-treated WT is re-plotted on the left for comparison. A total of 15–20 images per condition were analyzed across two independent TEM sections from two independent biologically experimental sets giving a technical n = ~ 30–40 per condition. Error bars = SEM; Asterisks, significant difference by chi-square analysis. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Numerous studies have observed Mn-induced autophagy but most of these studies based their findings on ‘snapshot’ assessments of autophagy and utilized limited methodology, without pharmacologically manipulating autophagic degradation (30–32, 34, 35). Autophagy is a complex biological process which is constantly in flux (autophagosome synthesis versus degradation). Thus, to properly study the effects of Mn on autophagic flux, we inhibited autophagosome degradation with CQ and BafA, known lysosomal inhibitors. We found that Mn upregulates expression of two key autophagy proteins—LC3-II/I and p62—in the absence or presence of saturating concentrations of CQ or BafA. Given our current understanding of autophagy (12, 59, 60), our data thus demonstrate that Mn increases autophagosomes induction/formation in neuroprogenitor-like cells types. Induction of autophagy appears to be specific to Mn, since eight other metals tested did not increase LC3-II puncta (in the presence of CQ) when applied at sub-cytotoxic concentrations. The mechanism by which Mn increases the LC3-II/I and p62 expression does not involve the upregulation mRNA expression. Given the results of our study, future studies should determine whether basal levels of Mn play a role in modulating normal autophagic function, or whether this effect only occurs under high, exogenous concentrations of Mn.
While our study provides strong evidence that increasing cellular Mn levels can activate autophagy, future studies are needed to explore the mechanism by which Mn activates autophagy and if Mn also plays a role in modulating autophagic function at basal levels. Our study does offer some clues. First, we show that Mn-induced p62 is PI3K-dependent, as LY294002 completely abrogates Mn-induced p62 in STHdh cells. These results are consistent with other published work suggesting LY294002 actively inhibits p62 synthesis and autophagy (61). This has been hypothesized to occur via inhibition on Class III PI3K (VPS34), an early autophagy protein which interacts with Beclin, rather than Class I PI3K which canonically acts upstream of the AKT pathway (61–63). Furthermore, rapamycin had no effect, suggesting Mn-induced autophagy is independent of mTOR, despite of its involvement in the regulation of autophagy (4). Additionally, we observed increased p62 and LC3II/I expression after exposure with NU7441, a DNApk inhibitor which is consistent with previous reports suggesting NU7441 can activate autophagy (though the mechanism is unclear) and causes radiosensitization in cancer cells leading to selective autophagic cell death. When cells were co-treated with Mn, this caused additive increases in p62 and LC3II/I expression, providing additional proof-of-principle that Mn can activate autophagy. Interestingly, addition of NU7441+Mn normalized Mn-induced autophagy in the Q111 cells. However, this interaction between NU7441, Mn and autophagy remains unclear because (1) the mechanism by which NU7441 and DNApk inhibition increase autophagy is unknown, (2) NU7441 inhibits PI3K at concentrations of 5 uM or higher (45, 50, 89), yet exhibits the opposite trend on autophagy as LY294002, a specific PI3K inhibitor and (3) NU7441 can actually decrease Mn uptake in STHdh cell at concentrations of 5 uM or higher because it inhibits PI3K (45, 89). Thus, while this interaction is interesting, specific complexities prevented fruitful follow-up studies.
Secondly, previous reports suggest that in microglial cells Mn-induced autophagy is dependent on ATG5 (34). We found that Mn-induced autophagy is unaffected by ATG5 siRNA-mediated knockdown, a protein heavily involved in the formation of autophagosomes, suggesting Mn-induced autophagy is not ATG5-dependent (Supplementary Material, Fig. S12). However, this knockdown was only partially effective (~40–50% free ATG5 knockdown, no knockdown of ATG5–12 complex), so it is unclear whether the remaining pool of ATG5 could compensate for the knockdown. Ma et al. (36) recently demonstrated Mn-induced iNOS increased s-nitrosylation of JNK and Bcl-2, which affects interactions between Bcl-2 and Beclin-1. In our study, we found that a 24 h exposure to 50 um Mn (or CQ/BafA) did not affect Beclin-1 expression (Fig. 2f); thus we don’t believe this mechanism is driving Mn-induced autophagy in our cells. As Mn is a known co-factor for many kinases (PI3K, insulin/insulin growth factor (IGF) receptor, ATM) which are implicated in autophagy, it is possible that Mn activates autophagy via a specific set of these kinases (49, 64, 65). Additionally, while Mn does not seem to greatly affect mRNA expression of autophagy proteins, Mn could help stabilize p62 or LC3 protein expression via interactions with the proteasome (66, 67). Nrf2 and KEAP1 signaling have also been heavily implicated in Mn exposure and are known to be activated by p62 (68–70). Given these results, future studies should investigate whether Mn-induced Nrf2 signaling is mediated via Mn-associated p62 expression. Lastly, Mn is a co-factor for PP2A phosphatase which has been proposed to regulate axonal transport of autophagosomes (74). Furthermore, PP2A also complexes with MID1, a ubiquitin ligase, and can regulate translation of muthtt mRNA (75). Together, these may provide a mechanism by which Mn regulates autophagy and mutHTT protein expression.
This study is the first, to our knowledge, to provide evidence that inhibiting autophagy can increase net Mn uptake. While we cannot distinguish whether CQ and BafA (lysosomal autophagy inhibitors) increase intracellular Mn by upregulating influx or decreasing efflux, the data are consistent with previous reports suggesting lysosomes are a common storage organelle for intracellular Mn (71, 72). The mechanism(s) underlying neuronal Mn homeostasis and transport have long been a ‘black-box,’ and few Mn-specific transport routes have been elucidated (21). Our study suggests that autophagy is a novel and poorly understood mechanism of neuronal Mn transport (and possibly responsible for the Mn uptake defect in HD cells), though further studies assessing basal neuronal Mn levels in different autophagy models are needed.
We set out to explore the impact of reduced bioavailable Mn on autophagic function in HD. We found that Mn-induced autophagy is blunted in cellular models which exhibit reduced Mn uptake (25, 27). Furthermore, we found that lysosomal autophagy inhibitors (CQ and BafA) increase net Mn uptake. In Q111/Q111 HD cells, these inhibitors normalize Mn uptake, thereby ameliorating the defects in Mn-induced autophagy. This demonstrates that reduced Mn bioavailability in these cells hampers activation of Mn-induced autophagy and that normalizing intracellular Mn can effectively attenuate this defect. Perhaps the most notably autophagy-related defect in HD is the presence of empty APVs, devoid of osmophilic cargo, due to ‘cargo-recognition failure’ (11). In this study, we have recapitulated this phenotype in Q111 cells (~50% empty or partially full APVs). Further, we found that rescue of an Mn deficit condition by restorative treatment with 100 uM Mn for 24 h eliminated this defect. Lastly, positing that Mn is facilitating cargo-loading in HD cells, we wanted to know whether Mn also facilitates the packaging of mutHTT aggregates into autophagosomes. As Q111 cells do not produce HTT aggregates, we utilized transiently transfected HEK293 cells with green fluorescent protein (GFP)-tagged, 72Q mutHTT. While these results do not confirm whether Mn can increase aggregate clearance itself, as we did not observe an effect on aggregate number by Mn alone, we found that treatment with CQ and Mn caused a significant (~2-fold) increase in the number of aggregates compared to CQ alone. Our current understanding of autophagic processing and the effects of CQ lead us to hypothesize that Mn facilitates cargo loading and mutHTT aggregate packaging into autophagosomes in HD cells. At present, it is unclear why Mn+CQ exposure is able to increase the total number of visible aggregates. Mn alone does not change aggregate number suggesting that Mn does not promote aggregation. If Mn exposure increases aggregate packing into autophagosomes, Mn+CQ should only change the localization of aggregates (outside autophagosomes to inside autophagosomes) since autophagy-mediated clearance is blocked, but this treatment should not increase the total number of visible aggregates. However, we suggest two possible ways Mn+CQ may result in increased number of visible aggregates: either (1) Mn inhibits the proteasome and shunts aggregates to autophagy, allowing aggregates that would have previously been degraded by the proteasome to be visualized; or (2) Mn helps package small mutHTT ‘seeds,’ precursors to mutHTT aggregates, into autophagosomes (perhaps by stalling autophagic processing), thus increasing GFP signal and concurrently, the visible number of aggregates. Further studies manipulating the proteasome (i.e. using MG132) or investigating aggregates at a higher resolution (which can detect mutHTT seeds) are needed to confirm either of these hypotheses. These studies are needed to address whether elevating cellular Mn levels would increase mutHTT aggregate clearance or merely increase the association between autophagosomes and mutHTT aggregates.
Martinez-Vicente and colleagues posit that the cargo recognition failures in HD may be due to an abnormal association between mutHTT and p62, a key protein for recognizing autophagosomes with polyubiquinated aggregates and organelles. Given the robust increase in p62 expression after Mn treatment (even under saturating CQ conditions), we hypothesize Mn-induced p62 expression may promote cargo recognition. Additionally, p62 directly associates with the adapter protein ALFY, which also facilitates autophagic degradation of protein aggregates. Of particular interest, overexpression of ALFY has led to significant reduction in HTT aggregates in Drosophila HD models (73, 74). Further research should investigate whether Mn also increases expression of ALFY, in addition to p62, which may further explain its effects on cargo sequestration.
At first glance, our results are quite contrary to the recent findings of Harischandra et al., which describe the ability of Mn to promote exosome transmission of alpha synuclein aggregation-inducing miRNAs (75). There could be several reasons for the discrepancy between our findings and Harischandra et al.’s. First, the mechanisms of PD and alpha synuclein pathology compared to HD/mutHTT are similar, but distinct (76–78). Mn could have entirely different effects on alpha synuclein aggregation than mutHTT aggregation, particularly in HD cells which exhibit reduced, brain-bioavailable Mn. Second, these findings were observed under toxic Mn concentrations while ours have focused on high, yet sub-cytotoxic exposures. Third, it is quite possible Mn could have both positive and negative effects on aggregation. Though our results pinpoint a therapeutic facet of Mn replacement therapy, this treatment could very well have off target effects (potentially through exosome-mediated miRNA transmission) which could promote aggregation. Altogether, these data warrant further investigation into the differential effects of Mn on autophagy/proteasome function and PD/HD.
Although our data strongly suggest that Mn activates autophagic vesicle formation, it does not exclude the possibility that Mn could both activate autophagic flux by elevating autophagosome formation while inhibiting the processing of autophagosome maturation and fusion with lysosomes. If this is the case, it is possible that the effect Mn has on filling autophagosomes is actually driven by the slowing of autophagosome processing. As seen in Supplementary Material, Fig. S11, exposure with CQ or BafA alone in Q7, or BafA alone in Q111, significantly increases the number of full autophagosomes by EM, similar to Mn exposure. We hypothesize that Mn, CQ and BafA are inhibiting autophagosome degradation, stalling autophagosome processing long enough for HD cells to package more cargo into them. This may explain why Mn+CQ exposure increases the number of mutHTT aggregates, by slowing down autophagy processing and allowing for greater accumulation of mutHTT aggregates into autophagosomes.
Autophagy activators have long been studied in the context of HD as a potential therapy for the disease—primarily via upregulation of autophagy-mediated degradation of mutHTT aggregates. Fasting, IGF-treatment, upregulation of AMPK and, most commonly, modulation of mTOR have all been investigated as potential HD therapies with promising results on a variety of cellular and in vivo pathologies (12, 13, 17, 20, 79–82, 83–86). Our study suggests that Mn treatment may also be a viable therapy by modulation of autophagy and autophagy-related HD pathologies. Furthermore, as our laboratory has shown, there is reduced bioavailable Mn in HD models, and Mn treatment could target a contributing cause of defective autophagy (reduced net Mn uptake), rather than merely upregulating the autophagy process, to combat disease pathology and at the same time benefit other Mn enzymatic pathways affect by HD, such as arginase in the urea cycle (28).
Conclusions
We demonstrate that the essential metal, Mn, is capable of activating autophagy in mouse and human-derived neuroprogenitors at sub-toxic concentrations via the use of pharmacological inhibitors and complementary autophagy methodologies. Furthermore, Mn treatment attenuates the well-characterized autophagic cargo recognition failure phenotype in HD cells and can increase autophagosome–aggregate association, suggesting Mn can facilitate cargo sequestration, including autophagic sequestration of HTT aggregates. Together these findings provide novel evidence that Mn-centric therapies could remedy autophagy-related phenotypes in HD and warrant further investigation.
Materials and Methods
Immortalized cell culture
The immortalized, murine striatal cell lines (STHdhQ7/Q7 and STHdh Q111/Q111) were obtained from Coriell Cell Repository (Cambden, NJ). Cells were cultured in Dulbecco’s Modified Eagle Medium (D6546, Sigma-Aldrich, St. Louis MO) supplemented with 10% FBS (Atlanta Biologicals, Flowery Branch, GA), 2 mM GlutaMAX (Life Technologies, Carlsbad, CA), Penicillin-Streptomycin, 0.5 mg/mL G418 Sulfate (Life Technologies), MEM non-essential amino acids solution (Life Technologies) and 14 mM HEPES (Life Technologies). They were incubated at 33°C and 5% CO2. Cells were passaged before reaching greater than 90% confluency and were never passaged past the recommended 14th passage. The cells were split by trypsinization using 0.05% Trypsin-EDTA solution (Life Technologies) incubated for 5 min. One day prior to exposure, cells were plated in the appropriate cell culture plate type at 8×104 cells/mL for WT and 1×105 cells/mL for HD. HEK293 cells were a generous gift from William Tansey. HEK293, 3T3, SH-SY5Y and Neuro2A cells were maintained at 37°C and treated in high glucose DMEM with L-glutamine and (Corning CV-0013) and Penicillin/Streptomycin, and cells plated at 100 000 cells/mL for experiments.
hiPSC cell culture
Islet-1 positive hiPSC-derived neuroprogenitor cells were differentiated as previously described using iPSCs from three separate control and HD patients (27). HD patient mutant alleles were 58, 66 and 70 CAG repeats. All hiPSC lines were confirmed to be pluripotent (PluriTest- Expression Analysis, Durham, NC) have normal karyotypes (Genetics Associates, Nashville, TN). Additionally, a subset of cells was fixed with 4% paraformaldehyde for 15 min, and immunocytochemistry was performed to ensure all cultures expressed Islet-1. For experiments, differentiated cells from the Islet-1 positive cells were plated at 300 000 cells/mL at day 10 of differentiation. At day 11, rock inhibitor containing media was taken off of cells are replaced with fresh media containing purmorphamine (0.65 uM). For 24 h exposures, cells were exposed to Mn and/or small molecules in day 11 differentiation media containing purmorphamine.
Inhibitors/metals
CQ diphosphate, BafA, KB-R7943, 3MA and LY294002 were purchased from Tocris Biologicals (Minneapolis, MN). Inhibitors were used at following concentrations unless otherwise stated: CQ diphosphate = 10 uM, BafA = 10 nM, KB-R7943 = 10 uM, 3MA = 5 mM and LY294002 = 7 uM. The following metallic compounds were used as sources for the metal exposures: Mn = MnCl2-4H20 (Sigma-Aldrich), Fe = FeCl3 (Sigma-Aldrich), Cu = CuCl2-2H20 (Alfa Aesar, Ward Hill, MA), Mg = MgCl2-6H20 (Alfa Aesar), Ni = NiCl2-6H20 (Alfa Aesar), Co = CoCl2-6H20 (MP Biomedicals, Solon, OH) and Zn = ZnCl2 (Acros Organics, Morris, NJ).
CFMEA was performed as described previously (27).
Western blot
Protein samples were prepared by scraping cells into ice-cold phosphate buffered saline (PBS), centrifuging and adding radioimmunoprecipitation assay (RIPA) buffer containing protease (Sigma-Aldrich) and phosphatase inhibitor cocktails 2 and 3 (Sigma, Sigma-Aldrich) to the pellet. After gentle homogenization, cells were centrifuged at 4°C for 10 min at 20 000g. Protein concentration was quantified using the BCA assay (Peirce Technologies, Thermo Fisher Scientific, Waltham, MA) with a bovine serum albumin (BSA) standard curve. Samples were mixed with 5× SDS loading buffer containing 1% 2-mercaptoethanol and boiled for 5 min. A total of 15 μg of protein was loaded for each sample onto a 4–15% pre-cast gel SDS-PAGE gel (Bio-Rad, Hercules, CA) and run at 90 V for 120 min. The protein bands were then transferred onto nitrocellulose membranes using iBlot Gel Transfer Device (Life Technologies). The remaining gel was stained with IRDye Blue protein stain (LI-COR, Lincoln, NE). Since the gels retained ~1/3 of the original protein after transferring with the iBlot, we imaged the stained gel on the Li-Cor Odyssey Imaging System and quantified the intensity entire lane from ~150 to 20 kDa. This value was used to normalize the values of immunostained bands. The membrane was blocked in Odyssey Blocking Buffer for 1 h prior to the addition of the primary antibodies. The primary antibodies were diluted 1:1000 in Odyssey Blocking Buffer containing 0.1% TWEEN and incubated overnight. After washing five times for 5 min in TBST, membranes were incubated with secondary antibodies at 1:10 000 (LI-COR) for 1 hour. Membranes were imaged using the Li-Cor Odyssey Imaging System, and quantification was performed using Image Studio Lite (LI-COR). LC3A/B (4108), p62 (5114) antibodies were purchased from cell signaling technologies and used at 1:1000 dilution except LC3A/B was used at 1:500. LC3 was quantified at the ratio of LC3-II divided LC3-I. Note that the doublet for p62 in Figure 2A is consistent with previous reports in neuronal cell types (51), but this is different from the non-specific band near p62 in Figure 4A which appears after probing for LC3 in the hiPSC-derived neuroprogenitors.
Gene expression analysis
STHDH Q7/Q7 (referred to as Q7) and Q111/Q111 (referred to as Q111) cells were plated in 6 well plates and treated with Mn and/or lysosomal autophagy inhibitors. After 24 h, cells were lysed in 1 mL of TRIzol® Reagent (CatNo. 15596) and stored at −80°C until use. Total RNA was extracted using TRIzol® Reagent, according to the manufacturer’s user guide (Invitrogen, Carlsbad, CA). RNA was DNase I treated, following the New England Biosciences (NEB) DNase I Reaction Protocol (M0303; New England BioLabs® Inc., Ipswich, MA). cDNA was generated on Bio-Rad Laboratories’ S1000™ Thermal Cycler, using 50 uM Random Hexamers (P/N 100026484, Invitrogen), 50 U MuLV Reverse Transcriptase (P/N 100023379, Applied Biosystems, Foster City, CA), 10 mM dNTPs (CatNo. N0446S, NEB), 20 U RNase, Inhibitor (CatNo. N8080119, Applied Biosystems), 25 mM MgCl2 (P/N100020476, Applied Biosystems), 10× PCR Buffer II (Ref No. 4486220 Applied Biosystems by Life Technologies, Austin, TX), and 1 ug of extracted RNA and using the following cycling times: 25°C for 10 min, 42°C for 60 min, 99°C for5 min, 5°C for 5 min and 4°C forever. Quantitative Reverse Transcriptase PCR (qRT-PCR) was performed on Eppendorf’s Mastercycler® epGradientS Realplex (2), using KAPA SYBR® FAST qPCR Master Mix (2×) Universal according to the manufacturer’s recommendations (KM4101, KAPA Biosystems, Wilmington, MA). Sequences of primers used are below. Expression of LC3B and p62 gene products were normalized to levels of PGK1 and plotted as induction over vehicle-treated samples. Primer sequences are as follows: mpgk1_F2 GCTATCTTGGGAGGCCTAA; mpgk1_R2 AAAGGCCATTCCACCACCAA; p62 (SQSTM1) forward 5′-GATGACATCTTCCGCATCTACA-3′; p62 (SQSTM1) reverse 5′-TGCAACCATCACAGATCACA-3′; LC3B (Map 1lc3b) forward 5′-CGTCCTGGACAAGACCAAGT-3′; LC3B (Map 1lc3b) reverse 5′-CCATTCACCAGGAGGAAGAA-3′; mATG5 FWD GATGGACAGCTGCACACACT; mATG5 REV TTGGCTCTATCCCGTGAATC.
pQCXI DsRED-LC3-II Retrovirus Production and Transduction
HEK 293T cells were plated at 2.5 × 10 (6) cells per 6 cm dish and maintained in DMEM supplemented with 10% FBS and 100 IU/ml Penicillin. The next day, HEK 293Ts were transfected using 8 μg pQCXI Puro DsRED-LC3-GFP plasmid (from Addgene#31182) and 4 μg pCL10A (from Imgenex, San Diego, CA), 168 μL sterile water, 20 μL 2.5 M CaCl2 and 200 μL HBS. The following day, the medium was changed on transfected HEK 293Ts to standard STHDH Q7/Q7 and Q111/Q111 cell medium. Q7 and Q111 were also plated with 40 000/50 000 cells/mL, respectively, into 6 well plates. Twenty-four hours later, viral supernatant was collected from HEK293 cells, 0.45 μM sterile filtered and 8 μg/mL final concentration of Hexadimethrine bromide was added—viral supernatant was incubated with Hexadimethrine bromide at room temperature for 5 min. The medium was then removed from Q7 and Q111 cells and replaced with virus-containing media. Q7 and Q111 cells were transduced following this same procedure two more times over the next 2 days. After three total days/rounds of transduction, the medium was replaced with standard media and cells were given 24 h to recover before selection with Puromycin at 1 ug/mL for roughly 48 h. The population was maintained in low dose Puromycin for expansion, then frozen down. For all LC3-II IF experiments (aside from severity score experiment), cells were thawed in normal media and immediately used. Though this plasmid should express LC3I in GFP and LC3II in dsRed, we found that the GFP signal after transduction was very weak and diffuse and did not offer quantificational benefit. However, as only LC3II is expressed in dsRed (not LC3I), this methodology allows for specific localization of LC3II which cannot be picked up via an LC3 antibody (as both forms are detected). Once these images were quantified, the images were decoded to reveal exposure conditions and averaged together for final values. For p62 immunofluorescence, cells were permeabilized in 0.2% triton in PBS for 20 min following fixation with 4% paraformaldehyde for 15 min. Cells were incubated overnight at 4°C in 1:100 primary antibody (Abcam #ab56416) in 5% normal donkey serum (Jackson Immunoresearch #3017-000-121) with 0.05% triton. The following day, cells were washed with 0.05% triton and incubated with anti-mouse-488 secondary for 3 h. Cells were washed three more times and incubated with Hoecst (1:2000) for 10 min. Cells were washed three more times and imaged. For LC3-II and p62 puncta quantification, Cell Profiler was used to detect the number of LC3-II puncta and cells per image.
For the LC3 severity score assay, STHdh Q7/Q7 and Q111/Q111 cells were transfected with the pQCXI Puro DsRED-LC3-GFP plasmid (Addgene#31182) using Mirus TransIT-LT transfection reagent (Madison, WI) following manufacturer’s protocol. Plasmid was transformed into DH5 alpha Escherichia coli, colonies were picked and inoculated in LB broth and plasmid DNA was isolated via Qiagen Maxi Prep kit. Cells were plated at 100 000 cells/mL 18–24 h prior to transfection in a 6 well plate. A total of 2.5 μg of plasmid and 7.5 ul of TransIT-LT were combined in 250 ul of serum-free Opti-MEM per well. This was added dropwise into the media in the well. Twenty-four hours after transfection, cells were selected with 2.5 μg/μL of puromycin for 3 days. These cells were plated at 50 000 cells/mL and 16–24 hours later, the cells were exposed with manganese or small molecules. Cells were then fixed with 4% paraformaldehyde for 15 min after 24 h and nuclei were stained with Hoecst stain. Images were taken using a Zeiss ObserverZ1 Light microscope by an investigator blinded to the exposures. For LC3-II severity scores, a second, blinded individual designated each image with an arbitrary code name. A third person then blindly scored each cell by severity (none–low, mild, medium, high or severe) using the representative scale in Supplementary Material, Figure S6c.
For lysotracker imaging, LC3-II expressing STHdh cells were exposed for 24 h and then stained with 50 nM lysotracker Deep Red (L12492, Invitrogen) and DAPI for 30 min. Cells were washed 3× with fresh media and immediately imaged. Cell profiler was used to detect the number of lysotracker puncta and cells (DAPI) per image.
Electron Microscopy
Electron microscopy was carried about by the Vanderbilt Cell Imaging Shared Resource (CISR). Samples were washed, fixed and prepared as designated by the core. Methods are as follows: samples for electron microscopy were rinsed in 0.1 M cacodylate buffer, fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer and post-fixed in 1% osmium tetroxide. Following fixation, the samples were dehydrated through a graded series of alcohol and embedded in Embed 812 resin. Thin sections were cut and observed at 100 keV with a Tecnai 12 electron microscope (FEI/Thermo Fisher).Transmission Electron Microscopy (TEM) images were taken by a CISR technician who was uninformed of any hypotheses. Roughly 8–15 images were taken for each condition across two separate biological replicates, each with two separate sections. Autophagosome loading status (i.e. empty, partial, full) was assessed by a single blinded individual after all images were randomized by a separate individual. Empty < 10% full, partial > 10%, <90%, full > 90% as assessed by eye.
GFP-HTT 72Q HEK293 cells
The exon-1-72Q-HTT-GFP-pCDNA3.1 plasmid was prepared in Dr Vittorio Maglione’s laboratory (Italy). Plasmid was transformed into DH5 alpha E. coli and grown on ampicillin plates. Colonies were picked and inoculated in LB broth, and plasmid DNA was isolated via Qiagen Midi Prep kit. HEK293 cells were transfected with 1 μg/μl plasmid using Mirus LT. Twenty-four hours after transfection cells were split. Forty-eight hours after transfection, cells were assessed to confirm aggregate production and were frozen down so that a new vial could be used for each set of experiments. Cells were plated at 100 000 cells/mL and treated 16–24 h after plating. Cells were treated for 24 h with Mn and/or CQ and fixed with 4% paraformaldehyde for 15 min at room temperature. Cells were later stained with Hoecst stain prior to imaging. GFP-tagged HTT was visualized using a Zeiss ObserverZ1 Light Microscope. Cell Profiler was used to quantify the number of aggregates (GFP puncta) and cells (Hoecst) per image. Confocal imaging was performed on LSM 710 META Inverted microscope by a Vanderbilt CISR technician, and images were viewed using Ziess Zen Black Software. For confocal imaging of LC3 puncta, an LC3A/B antibody (Cell signaling 4108) was used which detects both LC3I and LC3II. This is different from the virally transduced pQCXI dsRED-LC3-II expressing STHdh which only express the lipidated LC3-II form under dsRED.
siRNA ATG5 knockdown
The transfection was conducted using Lipofectamine® RNAiMAX Transfection Reagent from ThermoFisher Scientific and ON-TARGETplus Mouse ATG5 siRNA (CatNo. L-064838-00-0005) from Dharmacon, with the scrambled condition as Silencer™ Negative Control No. 1 siRNA (CatNo. AM4611; ThermoFisher Scientific). For each transfection, 9 μL RNAiMax and 15 μL of 5 μM scrambled siRNA, or 15 μL of 5 μM ATG5 siRNA, in a total volume of 250 μL of TE buffer were used per well of a 6 well plate. Q7/Q7 STHdh ds-RED-LC3II cells were plated at normal plating density into 6 well plates and grown for 16–24 h prior to siRNA transfection. After transfection, cells were cultured for an additional 24 h. The following day these cells were split once more for experiments into 6 well plates for western blot or 96 well plates for immunofluorescence. Sixteen to twenty-four hours later, cells were exposed with Mn and/or CQ/BafA for 24 h. A total of 72 h after siRNA transfection, cells were then processed for western blot or immunofluorescence to detect LC3/p62 or LC3-II puncta, respectively.
Statistical analysis
For most statistical tests, GraphPad Prism v8.0.1 was used. Most data are shown as linear fold change versus vehicle controls to ease data interpretation. However, all data that were analyzed by analysis of variance (ANOVA) was converted to log values to better approximate a normal distribution prior to statistical analysis. For experiments in which data are graphed in comparison to a unitary value of a vehicle control (=1), raw, unnormalized data were first converted to log values and underwent ANOVA analysis. When data sets were significant by ANOVA, post-hoc multiple comparisons tests were used to determine specific differences within data sets. For analyses between all possible treatment and genotype groups, Tukey’s test was performed. For analyses comparing data back to a specific condition only, Dunnet’s or Sidak’s tests were performed. For the majority of data sets, paired analyses were used as all samples were collected as full sets. For a few data sets which two groups were specifically compared, a paired student’s t-test was used. Microscopy data including LC3 puncta, lysotracker and HEK293 72Q were all quantified using CellProfiler 3.0.0. Most representative sets of microscopy images were brightened so that they could be more easily interpreted by the reader, but these images were not adjusted prior to quantification. If this was performed, all images were equally brightened.
Funding:
National Institute of Health/National Institute of Environmental Health Sciences (RO1 ES010563 to A.B.B. and M.A. ES016931 to A.B.B. F31 ES028084 to M.R.B. and T32 ES007028 to A.B.B. and P.J.).
Authors’ contributions
MRB and ABB designed all experiments. MRB performed most data analyses on western blot and microscopy data. MRB, MO, KN, DR, RN, and MU carried out cell culture and experiments for all figures. AMF generated LC3-expressing STHdh cell lines and performed all qRT-PCR. PJ carried out hiPSC-derived cell culture and differentiations. AP and VM generated and shared the 72Q-GFP plasmid. ZZ and MA assisted in experimental design and interpretation. All authors read and approved the final manuscript.
Supplementary Material
Acknowledgments
Experiments/data analysis/presentation of all Electron Microscopy studies were performed in part through the use of the Vanderbilt CISR (supported by NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126). CRISPR sequencing was done by GeneWiz (South Plainfield, NJ) and single-cell sorting was performed by the Vanderbilt Cell Imaging Resource Center. The graphic in Supplementary Material, Figure S1 was designed in BioRender. We would also like to thank many other members of the laboratory including Dr Anna Pfalzer, Dr Diana Neely, Dr Terry Jo Bichell, Dr Bingying Han, Dr Emily Warren, Kyle Horning, Jordyn Wilcox, Yueli Zhang and Ilyana Ilieva for technical expertise and for thoughtful assistance with experimental design and interpretation.
References
- 1. Bano D., Zanetti F., Mende Y. and Nicotera P. (2011) Neurodegenerative processes in Huntington's disease. Cell Death Dis., 2, e228. doi: 10.1038/cddis.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bates G., Tabrizi S. and Jones L. (2014) Huntington's disease. Huntington's disease. [Google Scholar]
- 3. Kumar A., Singh S., Kumar V., Kumar D., Agarwal S. and Rana M. (2015) Huntington's disease: an update of therapeutic strategies. Gene, 556, 91–97. doi: 10.1016/j.gene.2014.11.022. [DOI] [PubMed] [Google Scholar]
- 4. Ravikumar B. and Rubinsztein D.C. (2006) Role of autophagy in the clearance of mutant huntingtin: a step towards therapy? Mol. Aspects Med., 27, 520–527. doi: 10.1016/j.mam.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 5. Sarkar S. and Rubinsztein D.C. (2008) Huntington's disease: degradation of mutant huntingtin by autophagy. FEBS J., 275, 4263–4270. doi: 10.1111/j.1742-4658.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- 6. Truant R., Atwal R., Desmond C., Munsie L. and Tran T. (2008) Huntington's disease: revisiting the aggregation hypothesis in polyglutamine neurodegenerative diseases. FEBS J., 275, 4252–4262. doi: 10.1111/j.1742-4658.2008.06561.x. [DOI] [PubMed] [Google Scholar]
- 7. Arrasate M. and Finkbeiner S. (2011) Protein aggregates in Huntington's disease. Exp. Neurol., 238, 1–11. doi: 10.1016/j.expneurol.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ochaba J., Lukacsovich T., Csikos G., Zheng S., Margulis J., Salazar L., Mao K., Lau A.L., Yeung S.Y., Humbert S. et al. (2014) Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci., 111, 16889–16894. doi: 10.1073/pnas.1420103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gelman A., Rawet-Slobodkin M. and Elazar Z. (2015) Huntingtin facilitates selective autophagy. Nat. Cell Biol., 17, 214–215. doi: 10.1038/ncb3125. [DOI] [PubMed] [Google Scholar]
- 10. Rui Y.-N.N., Xu Z., Patel B., Chen Z., Chen D., Tito A., David G., Sun Y., Stimming E.F., Bellen H.J. et al. (2015) Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol., 17, 262–275. doi: 10.1038/ncb3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martinez-Vicente M., Talloczy Z., Wong E., Tang G., Koga H., Kaushik S., de Vries R., Arias E., Harris S., Sulzer D. et al. (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat. Neurosci., 13, 567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamamoto A., Cremona M.L. and Rothman J.E. (2006) Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol., 172, 719–731. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Floto A.R., Sarkar S., Perlstein E.O., Kampmann B., Schreiber S.L. and Rubinsztein D.C. (2007) Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington's disease models and enhance killing of mycobacteria by macrophages. Autophagy, 3, 620–622. doi: 10.4161/auto.4898. [DOI] [PubMed] [Google Scholar]
- 14. Sarkar S., Perlstein E.O., Imarisio S., Pineau S., Cordenier A., Maglathlin R.L., Webster J.A., Lewis T.A., O'Kane C.J., Schreiber S.L. et al. (2007) Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat. Chem. Biol., 3, 331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang L., Yu J., Pan H., Hu P., Hao Y., Cai W., Zhu H., Yu A.D., Xie X., Ma D. et al. (2007) Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci., 104, 19023–19028. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Williams A., Sarkar S., Cuddon P., Ttofi E.K., Saiki S., Siddiqi F.H., Jahreiss L., Fleming A., Pask D., Goldsmith P. et al. (2008) Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol., 4, 295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sarkar S., Ravikumar B., Floto R.A. and Rubinsztein D.C. (2008) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ., 16, 46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- 18. King M.A., Hands S., Hafiz F., Mizushima N., Tolkovsky A.M. and Wyttenbach A. (2008) Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol. Pharmacol., 73, 1052–1063. doi: 10.1124/mol.107.043398. [DOI] [PubMed] [Google Scholar]
- 19. Fox J.H., Connor T., Chopra V., Dorsey K., Kama J.A., Bleckmann D., Betschart C., Hoyer D., Frentzel S., DiFiglia M. et al. (2010) The mTOR kinase inhibitor Everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington's disease. Mol. Neurodegener., 5, 1–12. doi: 10.1186/1750-1326-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee J.H., Tecedor L., Chen Y., Monteys A., Sowada M.J., Thompson L.M. and Davidson B.L. (2014) Reinstating aberrant mTORC1 activity in Huntington's disease mice improves disease phenotypes. Neuron, 85, 303–315. doi: 10.1016/j.neuron.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horning K.J., Caito S.W., Tipps K.G., Bowman A.B. and Aschner M. (2015) Manganese is essential for neuronal health. Annu. Rev. Nutr., 35, 71–108. doi: 10.1146/annurev-nutr-071714-034419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tidball A.M., Bichell T. and Bowman A.B. (2015) Manganese in health and disease. RSC, 540–573, 22. doi: 10.1039/9781782622383-00540. [DOI] [Google Scholar]
- 23. Madison J.L., Wegrzynowicz M., Aschner M. and Bowman A.B. (2012) Disease-toxicant interactions in manganese exposed Huntington disease mice: early changes in striatal neuron morphology and dopamine metabolism. PLoS One, 7, e31024. doi: 10.1371/journal.pone.0031024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Williams B.B., Kwakye G.F., Wegrzynowicz M., Li D., Aschner M., Erikson K.M. and Bowman A.B. (2010) Altered manganese homeostasis and manganese toxicity in a Huntington's disease striatal cell model are not explained by defects in the iron transport system. Toxicol. Sci., 117, 169–179. doi: 10.1093/toxsci/kfq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Williams B.B., Li D., Wegrzynowicz M., Vadodaria B.K., Anderson J.G., Kwakye G.F., Aschner M., Erikson K.M. and Bowman A.B. (2010) Disease-toxicant screen reveals a neuroprotective interaction between Huntington's disease and manganese exposure. J. Neurochem., 112, 227–237. doi: 10.1111/j.1471-4159.2009.06445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwakye G.F., Li D. and Bowman A.B. (2011) Novel high-throughput assay to assess cellular manganese levels in a striatal cell line model of Huntington's disease confirms a deficit in manganese accumulation. Neurotoxicology, 32, 630–639. doi: 10.1016/j.neuro.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tidball A.M., Bryan M.R., Uhouse M.A., Kumar K.K., Aboud A.A., Feist J.E., Ess K.C., Neely D.M., Aschner M. and Bowman A.B. (2015) A novel manganese-dependent ATM-p53 signaling pathway is selectively impaired in patient-based neuroprogenitor and murine striatal models of Huntington's disease. Hum. Mol. Genet., 24, 1929–1944. doi: 10.1093/hmg/ddu609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bichell T.V., Wegrzynowicz M., Tipps G.K., Bradley E.M., Uhouse M.A., Bryan M., Horning K., Fisher N., Dudek K., Halbesma T. et al. (2017) Reduced bioavailable manganese causes striatal urea cycle pathology in Huntington's disease mouse model. Biochim. Biophys. Acta Mol. Basis Dis., 1863, 1596–1604. doi: 10.1016/j.bbadis.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ngwa H., Kanthasamy A., Gu Y., Fang N., Anantharam V. and Kanthasamy A.G. (2011) Manganese nanoparticle activates mitochondrial dependent apoptotic signaling and autophagy in dopaminergic neuronal cells. Toxicol. Appl. Pharmacol., 256, 227–240. doi: 10.1016/j.taap.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J., Cao R., Cai T., Aschner M., Zhao F., Yao T., Chen Y., Cao Z., Luo W. and Chen J. (2013) The role of autophagy dysregulation in manganese-induced dopaminergic neurodegeneration. Neurotox. Res., 24, 478–490. doi: 10.1007/s12640-013-9392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gorojod R.M., Alaimo A., Porte Alcon S., Pomilio C., Saravia F. and Kotler M.L. (2015) The autophagic–lysosomal pathway determines the fate of glial cells under manganese-induced oxidative stress conditions. Free Radic. Biol. Med., 87, 237–251. doi: 10.1016/j.freeradbiomed.2015.06.034. [DOI] [PubMed] [Google Scholar]
- 32. Tai Y., Chew K.C.M., Tan B.W.Q., Lim K.-L. and Soong T. (2016) Iron mitigates DMT1-mediated manganese cytotoxicity via the ASK1-JNK signaling axis: implications of iron supplementation for manganese toxicity. Sci. Rep., 6, 21113. doi: 10.1038/srep21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z., Miah M., Culbreth M. and Aschner M. (2016) Autophagy in neurodegenerative diseases and metal neurotoxicity. Neurochem. Res., 41, 409–422. doi: 10.1007/s11064-016-1844-x. [DOI] [PubMed] [Google Scholar]
- 34. Wang D., Zhang J., Jiang W., Cao Z., Zhao F., Cai T., Aschner M. and Luo W. (2017) The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy. 13, 914–927. doi: 10.1080/15548627.2017.1293766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou Q., Fu X., Wang X., Wu Q., Lu Y., Shi J., Klaunig J.E. and Zhou S. (2018) Autophagy plays a protective role in Mn-induced toxicity in PC12 cells. Toxicology, 394, 45–53. doi: 10.1016/j.tox.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 36. Ma Z., Wang C., Liu C., Yan D.Y., Deng Y., Liu W., Yang T.Y., Xu Z.F. and Xu B. (2017) The role S-nitrosylation in manganese-induced autophagy dysregulation in SH-SY5Y cells. Environ. Toxicol., 32, 2428–2439. doi: 10.1002/tox.22457. [DOI] [PubMed] [Google Scholar]
- 37. Hirata Y., Adachi K. and Kiuchi K. (1998) Activation of JNK pathway and induction of apoptosis by manganese in PC12 cells. J. Neurochem., 71, 1607–1615. [DOI] [PubMed] [Google Scholar]
- 38. Bae J.-H., Jang B.-C., Suh S.-I., Ha E., Baik H., Kim S.-S. and Lee M.-y. & Shin D.-H. (2006) Manganese induces inducible nitric oxide synthase (iNOS) expression via activation of both MAP kinase and PI3K/Akt pathways in BV2 microglial cells. Neurosci. Lett., 398, 151–154. doi: 10.1016/j.neulet.2005.12.067. [DOI] [PubMed] [Google Scholar]
- 39. McDougall S.A., Der-Ghazarian T., Britt C.E., Varela F.A. and Crawford C.A. (2011) Postnatal manganese exposure alters the expression of D2L and D2S receptor isoforms: relationship to PKA activity and Akt levels. Synapse, 65, 583–591. doi: 10.1002/syn.20877. [DOI] [PubMed] [Google Scholar]
- 40. Cordova F.M., Aguiar A.S., Peres T.V., Lopes M.W., Gonçalves F.M., Remor A.P., Lopes S.C., Pilati C., Latini A.S., Prediger R.D.S. et al. (2012) In vivo manganese exposure modulates Erk, Akt and Darpp-32 in the striatum of developing rats, and impairs their motor function. PLoS One, 7. doi: 10.1371/journal.pone.0033057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kanninen K.M., Grubman A., Meyerowitz J., Duncan C., Tan J.-L., Parker S.J., Crouch P.J., Paterson B.M., Hickey J.L., Donnelly P.S. et al. (2013) Increased zinc and manganese in parallel with neurodegeneration, synaptic protein changes and activation of Akt/GSK3 signaling in ovine CLN6 neuronal ceroid lipofuscinosis. PLoS One, 8, e58644. doi: 10.1371/journal.pone.0058644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dearth R.K., Hiney J.K., Srivastava V.K., Hamilton A.M. and Dees W.L. (2014) Prepubertal exposure to elevated manganese results in estradiol regulated mammary gland ductal differentiation and hyperplasia in female rats. Exp. Biol. Med., 239, 871–882. doi: 10.1177/1535370214531865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Exil V., Ping L., Yu Y., Chakraborty S., Caito S.W., Wells K.S., Karki P., Lee E. and Aschner M. (2014) Activation of MAPK and FoxO by manganese (Mn) in rat neonatal primary astrocyte cultures. PLoS One, 9, e94753. doi: 10.1371/journal.pone.0094753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Srivastava V.K., Hiney J.K. and Dees W.L. (2016) Manganese stimulated Kisspeptin is mediated by the insulin-like growth factor-1/Akt/mammalian target of rapamycin pathway in the prepubertal female rat. 157, 3233–41. Endocrinology. doi: 10.1210/en.2016-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bryan M.R., Uhouse M.A., Nordham K.D., Joshi P., Rose D., O’Brien M.T., Aschner M. and Bowman A.B. (2017) Phosphatidylinositol 3 kinase (PI3K) modulates manganese homeostasis and manganese-induced cell signaling in a murine striatal cell line. Neurotoxicology. 64, 185–194. doi: 10.1016/j.neuro.2017.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheng H., Xia B., Su C., Chen K., Chen X., Chen P., Zou Y. and Yang X. (2018) PI3K/Akt signaling pathway and Hsp70 activate in hippocampus of rats with chronic manganese sulfate exposure. J. Trace Elem. Med. Biol. 50, 332–338. doi: 10.1016/j.jtemb.2018.07.019. [DOI] [PubMed] [Google Scholar]
- 47. Wong V., Wu A., Wang J., Liu L. and Law B. (2015) Neferine attenuates the protein level and toxicity of mutant Huntingtin in PC-12 cells via induction of autophagy. Molecules, 20, 3496–3514. doi: 10.3390/molecules20033496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sato T., Nakashima A., Guo L. and Tamanoi F. (2009) Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J. Biol. Chem., 284, 12783–12791. doi: 10.1074/jbc.m809207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chauhan V., Singh S.S., Chauhan A. and Brockerhoff H. (1995) Phosphatidylinositol 3-kinase: inhibition of intrinsic protein-serine kinase activity by phosphoinositides, and of lipid kinase activity by Mn2+. Biochim. Biophys. Acta Mol. Cell Res., 1267, 139–144. doi: 10.1016/0167-4889(95)00032-N. [DOI] [PubMed] [Google Scholar]
- 50. Yu L., Shang Z.-F., Hsu F.-M., Zhang Z., Tumati V., Lin Y.-F., Chen B.P.C. and Saha D. (2014) NSCLC cells demonstrate differential mode of cell death in response to the combined treatment of radiation and a DNA-PKcs inhibitor. Oncotarget, 6, 3848–3860. doi: 10.18632/oncotarget.2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Waguri S. and Komatsu M. (2009) Methods in enzymology. Methods Enzymol., 453, 181–196. doi: 10.1016/s0076-6879(08)04009-3. [DOI] [PubMed] [Google Scholar]
- 53. Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C.M., Adams P.D., Adeli K. et al. (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy, 12, 1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mauthe M., Orhon I., Rocchi C., Zhou X., Luhr M., Hijlkema K.-J., Coppes R.P., Engedal N., Mari M. and Reggiori F. (2018) Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy, 14, 1–21. doi: 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yoshimori T., Yamamoto A., Moriyama Y., Futai M. and Tashiro Y. (1991) Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem., 266, 17707–17712. [PubMed] [Google Scholar]
- 56. Klionsky D.J., Elazar Z., Seglen P.O. and Rubinsztein D.C. (2008) Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Does bafilomycin A1 block the fusion of autophagosomes with lysosomes?. Autophagy, 4, 849–850. doi: 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- 57. Fung F.K.C., Law B.Y.K. and Lo A.C.Y. (2016) Lutein attenuates both apoptosis and autophagy upon cobalt (II) chloride-induced hypoxia in rat Műller cells. PLoS One, 11, e0167828. doi: 10.1371/journal.pone.0167828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen R., Jiang T., She Y., Xu J., Li C., Zhou S., Shen H., Shi H. and Liu S. (2017) Effects of cobalt chloride, a hypoxia-mimetic agent, on autophagy and atrophy in skeletal C2C12 myotubes. Biomed. Res. Int., 2017, 7097580. doi: 10.1155/2017/7097580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee S.-J. and Koh J.-Y. (2010) Roles of zinc and metallothionein-3 in oxidative stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Mol. Brain, 3, 30. doi: 10.1186/1756-6606-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Trejo-Solís C., Jimenez-Farfan D., Rodriguez-Enriquez S., Fernandez-Valverde F., Cruz-Salgado A., Ruiz-Azuara L. and Sotelo J. (2012) Copper compound induces autophagy and apoptosis of glioma cells by reactive oxygen species and jnk activation. BMC Cancer, 12, 156. doi: 10.1186/1471-2407-12-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kang Y.-T., Hsu W.-C., Wu C.-H., Hsin I.L., Wu P.-R., Yeh K.-T. and Ko J.-L. (2014) Metformin alleviates nickel-induced autophagy and apoptosis via inhibition of hexokinase-2, activating lipocalin-2, in human bronchial epithelial cells. Oncotarget, 5, 105536–105552. doi: 10.18632/oncotarget.22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shibata M., Lu T., Furuya T., Degterev A., Mizushima N., Yoshimori T., MacDonald M., Yankner B. and Yuan J. (2006) Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem., 281, 14474–14485. doi: 10.1074/jbc.m600364200. [DOI] [PubMed] [Google Scholar]
- 63. Ravikumar B., Duden R. and Rubinsztein D.C. (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet., 11, 1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 64. Yamamoto A., Tagawa Y., Yoshimori T., Moriyama Y., Masaki R. and Tashiro Y. (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct., 23, 33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 65. Seglen P.O. and Gordon P.B. (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci., 79, 1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Puissant A., Fenouille N. and Auberger P. (2012) When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res., 2, 397–413. [PMC free article] [PubMed] [Google Scholar]
- 67. Liu W., Ye L., Huang W., Guo L., Xu Z., Wu H., Yang C. and Liu H. (2016) p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett., 21, 29. doi: 10.1186/s11658-016-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Swanson M.L. and Pessin J.E. (1989) High affinity insulin binding in the human placenta insulin receptor requires αβ heterodimeric subunit interactions. J. Membr. Biol., 108, 217–225. doi: 10.1007/bf01871736. [DOI] [PubMed] [Google Scholar]
- 69. Mooney R.A. and Green D.A. (1989) Insulin receptor dephosphorylation in permeabilized adipocytes is inhibitable by manganese and independent of receptor kinase activity. Biochem. Biophys. Res. Commun., 162, 1200–1206. doi: 10.1016/0006-291x(89)90801-2. [DOI] [PubMed] [Google Scholar]
- 70. Cai T., Yao T., Li Y., Chen Y., Du K., Chen J. and Luo W. (2007) Proteasome inhibition is associated with manganese-induced oxidative injury in PC12 cells. Brain Res., 1185, 359–365. doi: 10.1016/j.brainres.2007.09.075. [DOI] [PubMed] [Google Scholar]
- 71. Li H., Wu S., Shi N., Lian S. and Lin W. (2011) Nrf2/HO-1 pathway activation by manganese is associated with reactive oxygen species and ubiquitin–proteasome pathway, not MAPKs signaling. J. Appl. Toxicol., 31, 690–697. doi: 10.1002/jat.1654. [DOI] [PubMed] [Google Scholar]
- 72. Jain A., Lamark T., Sjøttem E., Larsen K., Awuh J., Øvervatn A., McMahon M., Hayes J.D. and Johansen T. (2010) p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem., 285, 22576–22591. doi: 10.1074/jbc.m110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fan W., Tang Z., Chen D., Moughon D., Ding X., Chen S., Zhu M. and Zhong Q. (2010) Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy, 6, 614–621. doi: 10.4161/auto.6.5.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jiang T., Harder B., de la Vega M., Wong P.K., Chapman E. and Zhang D.D. (2015) p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med., 88, 199–204. doi: 10.1016/j.freeradbiomed.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Krauss S., Griesche N. and Jastrzebska E. (2013) Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1–PP2A protein complex. Nature Comm., 4, 1511. doi: 10.1038/ncomms2514. [DOI] [PubMed] [Google Scholar]
- 76. Neisch A.L., Neufeld T.P. and Biol H.-T.S.J. (2017) A STRIPAK complex mediates axonal transport of autophagosomes and dense core vesicles through PP2A regulation. J. Cell Biol. 216, 441–461. doi: 10.1083/jcb.201606082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Suzuki H., Wada O., Inoue K., Tosaka H. and Ono T. (1983) Role of brain lysosomes in the development of manganese toxicity in mice. Toxicol. Appl. Pharmacol., 71, 422–429. doi: 10.1016/0041-008x(83)90030-3. [DOI] [PubMed] [Google Scholar]
- 78. Okamoto Y., Oshima R., Inagaki K., Aita S., Nisioka H., Kondo Y., Ishizuka H., Takada J. and Nishida M. (1997) The presence of a manganese-rich particle in lysosome of rat pancreas due to excess manganese treatment. IUBMB Life, 41, 389–394. doi: 10.1080/15216549700201401. [DOI] [PubMed] [Google Scholar]
- 79. Isakson P., Holland P. and Simonsen A. (2012) The role of ALFY in selective autophagy. Cell Death Differ., 20, 12–20. doi: 10.1038/cdd.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Filimonenko M., Isakson P., Finley K.D., Anderson M., Jeong H., Melia T.J., Bartlett B.J., Myers K.M., Birkeland H., Lamark T. et al. (2010) The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell, 38, 265–279. doi: 10.1016/j.molcel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Harischandra D.S., Ghaisas S., Rokad D., Zamanian M., Jin H., Anantharam V., Kimber M., Kanthasamy A. and Kanthasamy A.G. (2018) Environmental neurotoxicant manganese regulates exosome-mediated extracellular miRNAs in cell culture model of Parkinson's disease: relevance to α-synuclein misfolding in metal neurotoxicity. Neurotoxicology, 64 267–277. doi: 10.1016/j.neuro.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ross C.A. and Poirier M.A. (2005) What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol., 6, 891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 83. Ravikumar B. and Rubinsztein D.C. (2004) Can autophagy protect against neurodegeneration caused by aggregate-prone proteins? Neuroreport, 15, 2443. doi: 10.1097/00001756-200411150-00001. [DOI] [PubMed] [Google Scholar]
- 84. Rubinsztein D.C. (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature, 443, 780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 85. Ehrnhoefer D.E., Martin D.D.O., Schmidt M.E., Qiu X., Ladha S., Caron N.S., Skotte N.H., Nguyen Y.T.N., Vaid K., Southwell A.L. et al. (2018) Preventing mutant huntingtin proteolysis and intermittent fasting promote autophagy in models of Huntington disease. Acta Neuropathol. Commun., 6, 16. doi: 10.1186/s40478-018-0518-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sarkar S., Davies J.E., Huang Z., Tunnacliffe A. and Rubinsztein D.C. (2007) Trehalose, a novel mTOR-independent autophagy enhancer accelerates the clearance of mutant Huntingtin and α-synuclein. J. Biol. Chem., 282, 5641–5652. doi: 10.1074/jbc.m609532200. [DOI] [PubMed] [Google Scholar]
- 87. Mealer R.G., Murray A.J., Shahani N., Subramaniam S. and Snyder S.H. (2013) Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J. Biol. Chem., 289, 3547–3554. doi: 10.1074/jbc.m113.536912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pircs K., Petri R., Madsen S., Brattås P., Vuono R., Ottosson D.R., St-Amour I., Hersbach B.A., Matusiak-Brückner M., Lundh S. et al. (2018) Huntingtin aggregation impairs autophagy, leading to Argonaute-2 accumulation and global microRNA dysregulation. Cell Rep., 24, 1397–1406. doi: 10.1016/j.celrep.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 89. Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Øvervatn A., Stenmark H. and Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol., 171, 603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Leahy J.J.J., Golding B.T., Griffin R.J., Hardcastle I.R., Richardson C., Rigoreau L. and Smith G.C.M. (2004) Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg. Med. Chem. Lett., 14, 6083–6087. doi: 10.1016/j.bmcl.2004.09.060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.