

Abstract
Manganese oxidation is an important biogeochemical process that is largely regulated by bacteria through enzymatic reactions. However, the detailed mechanism is poorly understood due to challenges in isolating and characterizing these unknown enzymes. A manganese oxidase, Mnx, from Bacillus sp. PL-12 has been successfully overexpressed in active form as a protein complex with a molecular mass of 211 kDa. We have recently used surface induced dissociation (SID) and ion mobility - mass spectrometry (IM-MS) to release and detect folded subcomplexes for determining subunit connectivity and quaternary structure. The data from the native mass spectrometry experiments led to a plausible structural model of this multicopper oxidase which has been difficult to study by conventional structural biology methods. Because each Mnx subunit binds a variable number of copper ions, ambiguities in assigning some of the observed peaks remained as a barrier to fully understanding the role of metals and potential unknown ligands in Mnx due to the heterogeneity of the system and limited mass resolution. In this study, we performed SID in a modified Fourier transform – ion cyclotron resonance (FTICR) mass spectrometer. The high mass accuracy and resolution offered by FTICR unveiled unexpected artificial modifications on the protein that had been previously thought to be iron bound species based on lower resolution spectra. Additionally, isotopically resolved spectra of the released subcomplexes revealed the metal binding stoichiometry at different structural levels. This method holds great potential for in-depth characterization of metalloproteins and protein-ligand complexes.
Keywords: native mass spectrometry, high resolution mass spectrometry, protein complex, surface induced dissociation, metalloprotein, protein-ligand interaction
Graphical Abstract
Introduction
Mass spectrometry (MS) has been extensively used for characterization of proteins.[1] Most of the routine analyses of proteins are carried out at the peptide level from enzymatically digested proteins. However, there is a growing interest in analyzing proteins using a combination of analytical techniques to gain more insight into higher order structure, especially with respect to how the different proteins interact with other proteins.[2] In native MS,[3, 4] proteins are ionized from volatile aqueous buffer at physiological pH to preserve the native-like state in the gas phase, where noncovalent interactions are largely maintained. Subunit stoichiometry and protein ligand/metal interactions can be studied by measuring the mass of the intact protein-protein or protein-ligand/metal complexes.[5, 6] The ability of MS for simultaneously detecting coexisting populations of a protein with various binding partners at different masses has provided critical insights for understanding the dynamics of heterogeneous protein complexes[7–10] and allosteric mechanisms of bound ligands[11]. Recent developments in high resolution MS instrumentation [12–14] have significantly expanded the structural details that can be obtained because even relatively small differences in ligands/modifications on high mass native proteins can be detected with high mass accuracy.[15–17] Spectra collected on high resolution mass spectrometers such as Orbitrap and Fourier Transform Ion Cyclotron Resonance (FTICR) MS for membrane protein assemblies have also been reported with promising results.[18–21] In addition, intact protein complexes can be activated in the gas phase to release subcomplexes for mapping the subunit connectivity and characterizing the quaternary structure.[4, 22, 23]
Multicopper oxidases (MCOs) have been implicated as the Mn oxidase in several Mn-oxidizing bacteria. These and other microbes contribute significantly to Mn redox cycling in a range of terrestrial and aquatic environments including soils, sediments, freshwater and marine systems.[24] However, the roles of manganese oxidases in the geochemical processes of transforming soluble forms of Mn to minerals remain uncharacterized. An active Mn oxidase from Bacillus sp. PL-12, Mnx, has been successfully heterologously overexpressed and purified from E. coli. The overexpression construct contained four of the genes in the polycistronic mnx operon.[25] Previous bottom-up and top-down liquid chromatography – mass spectrometry (LC-MS) experiments have shown that the protein complex consists of three subunits: MnxE (12.2 kDa), MnxF (11.2 kDa), and MnxG (138 kDa).[25, 26] The presence of accessory proteins was surprising because no MCO has been previously purified as a heteromultimeric protein complex.[25] While MnxE and MnxF both have homologs in other sporulating bacteria, they lack homology to any proteins with known structures or predicted function. Mnx is unusual in that it mediates a two-electron oxidation of a metal while all known MCOs with metal substrates only oxidize those metals by one electron.[27] Furthermore, other known MCOs do not require accessory proteins to function. The Mnx complex has not been successfully crystallized, thus its high-resolution structure remains elusive.
Although the intact mass of the Mnx complex determined by native MS (211.2 ± 0.8 kDa) [26] restricts the stoichiometry to a maximum of one MnxG and six copies of MnxE and/or MnxF, the exact copy numbers of MnxE and MnxF cannot be confidently determined because they have similar mass. The mass difference between MnxE and MnxF represents only about 0.5% of the mass of the intact complex, and the broad peak detected for the native complex generates about 1% of uncertainty. Additionally, the protein is known to bind 6–15 copper atoms, depending on preparation conditions, as determined by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) [28]. This variation further contributes to the uncertainty of the composition assignment. Breaking down the intact complex into smaller subunits that can be better resolved by MS would allow confident determination of the stoichiometry and metal binding ratios.
We recently performed collision induced dissociation (CID) and surface induced dissociation (SID) of Mnx [26] on a modified ion mobility - time-of-flight (IM-TOF) mass spectrometer (Waters Synapt G2s, Manchester, UK), an instrument type that has been extensively used for studying protein quaternary structure.[22, 29] In contrast to the commonly used CID which often causes subunit unfolding,[4, 22, 30] SID typically generates folded subcomplexes that better represent the native structure.[22, 29] CID of Mnx resulted in ejection of the MnxE and MnxF monomers from the complex, providing limited information on the inter-subunit connectivity within the protein complex. Instead, at the available collision energy, SID yielded a variety of species, primarily the MnxE/F monomers, MnxE3F3 hexamer, and MnxG. Based on the pattern of the SID products, and specifically the MnxE/F multimers, we proposed the symmetrical structure of MnxE3F3 with alternating MnxE and MnxF subunits. This discovery led to a first plausible structural model for the uncharacterized Mnx complex, preceding any successful characterization by conventional structural biology techniques.[26] In addition, MnxE is shown to bind strongly to at least one copper, while MnxF binds 0–2 Cu atoms with weaker affinity than MnxE. Metal binding by the two accessory proteins suggest they may play a more active role (e.g. serving as Cu chaperones) rather than being simply a structural part of the enzyme. However, several questions remained. Particularly, many suspected MnxE and MnxF peaks could not be confidently assigned due to unidentifiable extra mass based on expected combinations of different subunits and numbers of Cu bound. Because Mnx is known to bind other metal ions, these peaks could correspond to different metal bound MnxEF species.[31] Our ability to confidently identify detected species was limited by the resolution achievable with TOF mass analyzer. However, FTICR MS, with added SID capability, [32] provided the high resolution needed to resolve the uncertainties in peak assignment. The results demonstrate the unique potential of SID combined with high resolution MS for characterization of large heterogeneous protein complexes.
Experimental
The SID data presented in this work were collected on a modified Bruker (Billerica, MA) SolariX XR 15T FTICR [32] equipped with a dynamically harmonized cell (ParaCell).[33] Briefly, the original collision cell was replaced with a custom collision cell, which consist of an SID device followed by a short rectilinear quadrupole for trapping (Ardara Technologies, Ardara, PA). The voltages on the electrodes of the custom cell were controlled both by an external power supply (static DC voltages) and by the existing power supply connections in the instrument (RF and pulsed DC voltages). The instrument performance has been examined in several experiments with well characterized protein complexes, which yielded SID data similar to previous studies in terms of types of fragments but with higher resolution and mass accuracy.[32]
The wild-type Mnx protein was expressed in E. coli with the mnxDEFG operon following methods described previously.[25] The purified protein complex was buffer exchanged into 100 mM ammonium acetate with a Micro Bio-Spin 6 column (Bio-Rad, Hercules, CA) twice before native MS analysis. The protein was sprayed at 2.6 mg/mL (12 µM of Mnx complex) from a glass capillary pulled in-house, with a platinum wire inserted at ground potential. A voltage of −1.25 kV was applied in the inlet capillary for electrospray with dry gas flowing at 4 L/min at 180 °C. The spectrum shown in the figures was collected from m/z 500 – 20000 at 8M resolution (transient length 9.2 s), with 300 spectrum averages each at an ion accumulation time of 0.5 s. RF voltages were set to 1.4 MHz, 2000 V in the collision cell, and 1MHz, 450 V on the transfer guide. The collision cell gas flow was set to 100% for increasing trapping efficiency. In source fragmentation was set to 70 V for optimal desolvation and transmission for the intact Mnx complex. The mass calibration was performed by using perfluoroheptanoic acid (PHFA) up to the m/z of 8000. The absorption mode processing was calibrated with 0.1mg/mL sodium formate and lithium formate, and the baseline was corrected using the CUSUM method with negative intensity trimmed in the spectra shown.
SID spectra of protein complexes under 150 kDa have been successfully collected with the FTICR by using voltages similar to the previously established SID conditions in TOF mass spectrometers. However, using similar conditions on the modified FTICR, SID of larger complexes suffered from low sensitivity. An unconventional tuning was developed to generate SID spectra that are consistent with previous SID data acquired on a Q-IM-TOF for serum amyloid P pentameric complex (data not shown, precursor charge +16~+19, m/z = 7000 – 8000 similar to the m/z of Mnx precursor). In this tuning, the surface target is at ground with two steering lenses (attractive voltages on the front top and the middle bottom deflectors) bending the ion trajectory off the central axis. We suspect that this tuning causes the precursor ions to collide with a stainless steel electrode on the path, instead of colliding on the gold surface target coated with self-assembled fluorocarbon normally used for SID experiments,[34] based on SIMION simulation (data not shown). It has previously been shown that stainless steel surfaces can successfully generate SID products[35] and they have been used for SID of protonated peptides,[36] proteins,[37] and protein complexes[38]. Optimization of the SID design for high mass ions in FTICR will require additional studies.
The SID spectra shown in this study were acquired with the acceleration voltage (i.e. the potential difference between the quadrupole and the offset on the shortened collision cell) set to 57 V. The front top and middle bottom deflectors of the SID device were set to −40 V and −50 V, respectively. Other SID lenses were set to ground. Within this collision cell, the protein ions presumably hit and generate SID products after being steered off axis by the voltages in the SID device. For collecting mass spectra, all lenses in the SID device were set to near ground and the collision voltage was set to 4 V for transmission with minimal activation. Because the quadrupole in the system cannot isolate ions with m/z above 6000, the m/z in the quadrupole tune method was set to 1000 without isolation and used as a high pass filter to remove unwanted low mass species. The formulas of the three subunits of Mnx protein are obtained from the reported sequences in UniProt (Table S1). The sequences of MnxE and MnxF have both been validated with top-down analysis.[26] It is noted that MnxF has only been observed with the sequence starting after the second methionine in the sequence reported in UniProt. In addition, the top-down data suggested there is a disulfide bond in MnxE that is responsible for a −2Da mass shift from the expected sequence. The MnxG sequence was confirmed by ~70% coverage with bottom-up analysis (data not shown). However, we have not yet been able to detect the intact MnxG under denaturing conditions, possibly due to low ionization efficiency and/or likelihood of precipitation in organic solvent. The theoretical isotopic distributions of the assigned species were generated using Bruker Data Analysis software using the calculated molecular formula, with peak width set to 0.02. We defined a background noise level to be 2×106 (arbitrary unit) by taking the average of the signal in the 16500 < m/z < 18500 where no protein species were detected. All the species assigned in the spectra have intensities significantly above the noise level (S/N > 5).
For bottom-up LC-MS, the higher-energy collisional dissociation (HCD) spectra of Mnx peptides were acquired on a Thermo Orbitrap Elite (data dependent MS/MS of top 6 peaks; HCD normalized collision energy 28) equipped with a Waters NanoAcquity liquid chromatography system (70 cm, 75 µm i.d. C18 reversed phase column; mobile phase 0.1% formic acid in H2O/acetonitrile at 0.3 µL/min; acetonitrile gradient from 5% to 35% over 2 hours). Mnx protein was denatured in urea, reduced by dithiothreitol, alkylated with iodoacetimide and digested with trypsin to obtain the Mnx peptides. The LC-MS data were first analyzed with MassMatrix[39, 40] with a database containing Mnx protein sequences and the E coli. proteome. Carbamidomethyl of cysteine was set as a fixed modification. Several custom variable modifications were included: oxidation on methionine, gluconoylation on the protein N-terminus, phosphogluconoylation on the protein N-terminus, and (2-aminoethyl) benzenesulfonyl fluoride hydrochloride on tyrosine. Peptide mass tolerance was set to 10 ppm and fragments mass tolerance was set to 0.01 Da. Spectra associated with modifications of interest were selected and manually annotated.
Results & Discussion
SID Dissects Mnx into Smaller Building Blocks for Determining the Quaternary Structure
The 211 kDa Mnx complex has been successfully detected in the SID-modified FTICR mass spectrometer[32] as shown in Figure 1 (inset). The resolved charge states of the intact Mnx complex and Mnx dimer are in the m/z range from 7000 to 10000. Note that the charge states have not been isotopically resolved due to sample heterogeneity even with a 15T FTICR. Therefore, it is still necessary to dissect Mnx into smaller subcomplexes for definitive in-depth characterization. The SID spectrum collected on the modified FTICR (Figure 1) is qualitatively the same as the SID spectra obtained on the Q-IM-TOF, with the MnxE/F monomers appearing at m/z < 5000, MnxE3F3 hexamers at m/z 5000 – 7500, and MnxG mixed with undissociated Mnx complex at m/z > 7500. Due to hardware limitations, no quadrupole isolation could be applied above m/z = 6000 resulting in the detection of the full package of precursor ions (Mnx and Mnx dimers) in the SID spectrum.
Figure 1.
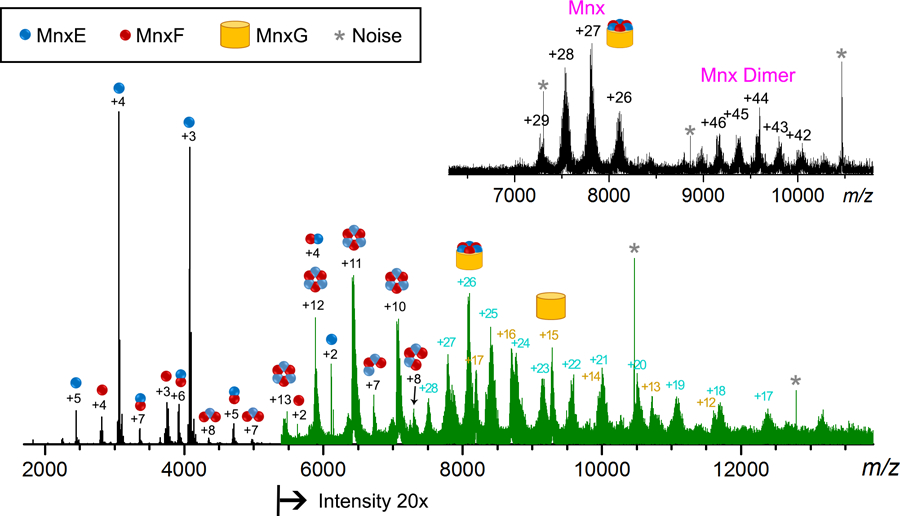
SID spectrum of Mnx complex on a modified FTICR. The assignments of major peaks are illustrated with cartoon structures (legend shown in the box on top) with their charge states annotated. The blue and red circles are MnxE and MnxF, respectively. Released MnxE/F multimers are drawn as clusters red/blue of the circles based on proposed structures. Most of the peaks with m/z > 7500 are assigned to undissociated Mnx complex (charge state series labeled in blue, +28 to +17) and the released MnxG (charge state series labeled in dark yellow, +17 to +12). Intensities of peaks higher than m/z of 5400 are 20x magnified for clarity and plotted in green. The peaks labeled with asterisks are noise in the background (sharp peaks with no isotope distributions). The insert on the top right corner is the MS spectrum of Mnx complex without significant activation.
Several of the MnxE/F multimers at m/z 5000 – 7500, especially the MnxE/F tetramers and pentamers, were only present at relatively low abundance and masked by the other more intense signals (Figure 1). The ion mobility separation afforded by the IM-TOF instrument provided an additional dimension of separation and significantly improved the resolving power for adequately interpreting the highly convoluted multi-subunit SID spectrum.[26] However, with the high resolution offered by the FTICR, most of these underlying species can be isotopically resolved and distinguished from overlapping species. The data acquired with the two instruments are highly complementary: the two-dimensional IM-MS provides an effective survey of all the species in the spectra, and the high resolution FTICR provides a means to confidently assign specific peaks unassignable in the IM-MS data and reveal unexpected modifications as discussed below.
Ultra-High Resolution Helps to Decipher the Heterogeneity of Mnx by Identifying Multiple Metal Binding Events and Modifications
The most abundant charge states of MnxE and MnxF released in SID are magnified and shown in Figure 2. Although both CID and SID showed the strong binding of one Cu on MnxE, the majority of bound Cu on MnxF was lost in CID but not in SID.[26] Even without SID modification, CID of Mnx in the FTICR instrument did not yield meaningful spectra, presumably due to ineffective trapping of large protein ions with high kinetic energy. Therefore, only SID spectra are discussed in this work. Similar to the previous results on the Q-IM-TOF, the MnxE released in SID showed strong binding of one Cu as indicated by the most abundant peak in Figure 2a (average ~1.2 Cu per full-length, unmodified MnxE); MnxF (full-length, unmodified) revealed variable binding of 1~3 Cu, with an intensity-weighted average of ~1.4 (Figure 2b). Similar distributions were observed for the second most abundant charge states (i.e. +3 for MnxE, and +4 for MnxF). In addition to the major peaks corresponding to different numbers of bound Cu and a truncated form of MnxE (loss of the first 9 residues, Figure 2b), a few other peaks containing significant amounts of extra mass were also observed for both MnxE (m/z > 3083) and MnxF (m/z > 3808). It is interesting to note that the truncated MnxE showed a much lower percentage of Cu incorporation (average ~0.2 Cu, suggesting that only ~20% of truncated MnxE binds Cu, Figure 2b) than the full length MnxE (average ~1.2 Cu, Figure 2a). For the full-length MnxE, ~5% is present in apo form, and the majority is bound to one Cu (~70%), or more than one Cu (~25%). The truncation at the N-terminus of MnxE (cleavage of the first nine residues) is likely to be responsible for the loss of the majority of bound Cu (~80% in apo form). The methionine, histidine, and aspartic acid at positions 1–3 are all known Cu binding sites in Cu binding peptides/proteins.[41, 42] It is possible that there is a major Cu binding site including one or more of the nine residues at the N-terminus of MnxE. The percentage of the full-length MnxE binding more than one Cu (~25%) coincided with the ~20% of remaining bound Cu in the truncated MnxE. This implies that the additional Cu binding site(s) are unaffected by the cleavage of the first nine residues and are probably located in a different region of the protein.
Figure 2.
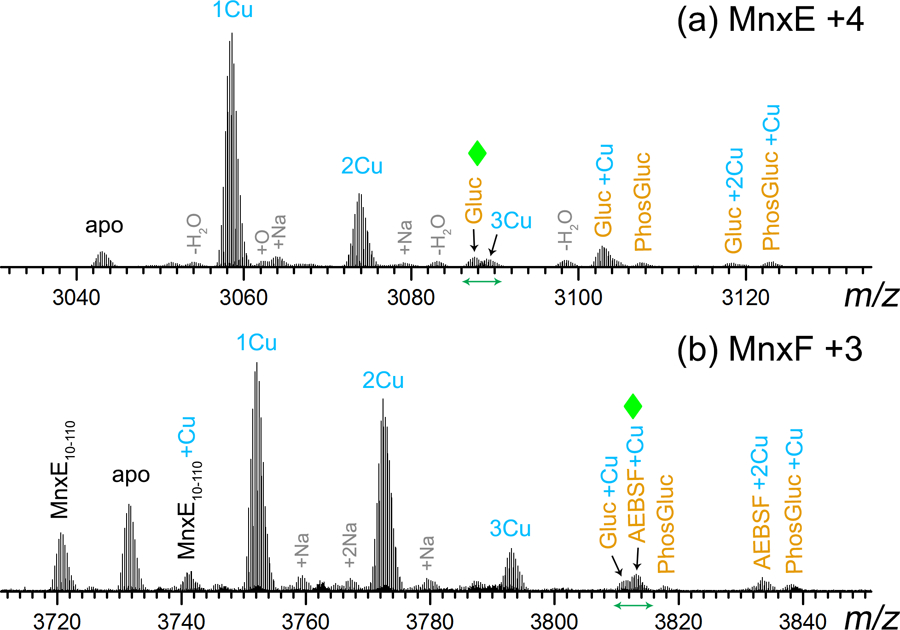
Zoom-in views of the most abundant charge states of (a) MnxE and (b) MnxF monomers released in SID. Abbreviations: MnxE10–110 – truncated MnxE, Gluc – gluconoylation, PhosGluc – phosphogluconoylation, AEBSF - (2-aminoethyl) benzenesulfonyl fluoride hydrochloride. All major peaks are assigned based on their accurate mass. Several species were previously thought to be iron containing species based on data acquired on a TOF mass spectrometer. High resolution data, however, strongly suggest these are post-translationally modified proteins. The peaks highlighted by green diamonds are discussed further in Figure 3 and Figure 4 (zoom-in spectra in the range shown by the green arrows along the m/z axis).
Earlier Q-IM-TOF data revealed a MnxE species with mass higher by ~178 Da from the sequence mass. This species was thought to be an Fe-containing species because the mass could not be explained simply by multiple Cu additions (average mass addition of 61.5 Da for Cu2+ and 53.8 Da for Fe2+, two Cu2+ and one Fe2+ add up to 177 Da). This assignment was further reinforced by the fact that trace iron (~0.04 per complex) was detected in ICP-OES analysis.[31] The high resolution (200–300k) and high mass accuracy (mass error < 1 ppm) FTICR spectra allowed confident identification of all detected species (Table S2). The isotope distributions of the experimental and the theoretical spectra were also closely matched as exemplified by the data for holo MnxE (Figure S1) and apo MnxF (Figure S2). We then generated the theoretical isotope distribution of MnxE bound to two Cu and one Fe (Figure 3b) and compared it to the species detected in high resolution spectra (highlighted by green diamonds in Figure 2a and the zoom-in view in Figure 3a). There was a clear discrepancy implying a wrong assignment. The most abundant monoisotopic peak of the theoretical distribution was also 1 Da lower than the experimental mass. This species cannot be explained by binding of other metals in this mass range (e.g. Ni, Mn, Fe), because the unique mass defects introduced by metal ions do not fit the experimental accurate mass. Thus, it is unlikely to be a metal bound species of MnxE. Instead, a modification of C6H10O6 from apo MnxE (formula based on accurate mass fit) matches the mass shift (178.048 Da) and the isotope distribution with minimal deviation (Figure 3c). This gluconoylation modification with a mass shift of 178 Da has been documented in the literature [43, 44] and it is an artificial modification for proteins expressed in E. coli. Hence, the other major peaks above m/z 3083 can be confidently assigned taking gluconoylation into consideration, including gluconoylated MnxE bound to1~2 Cu and MnxE with phosphogluconoylation (another artificial modification).
Figure 3.
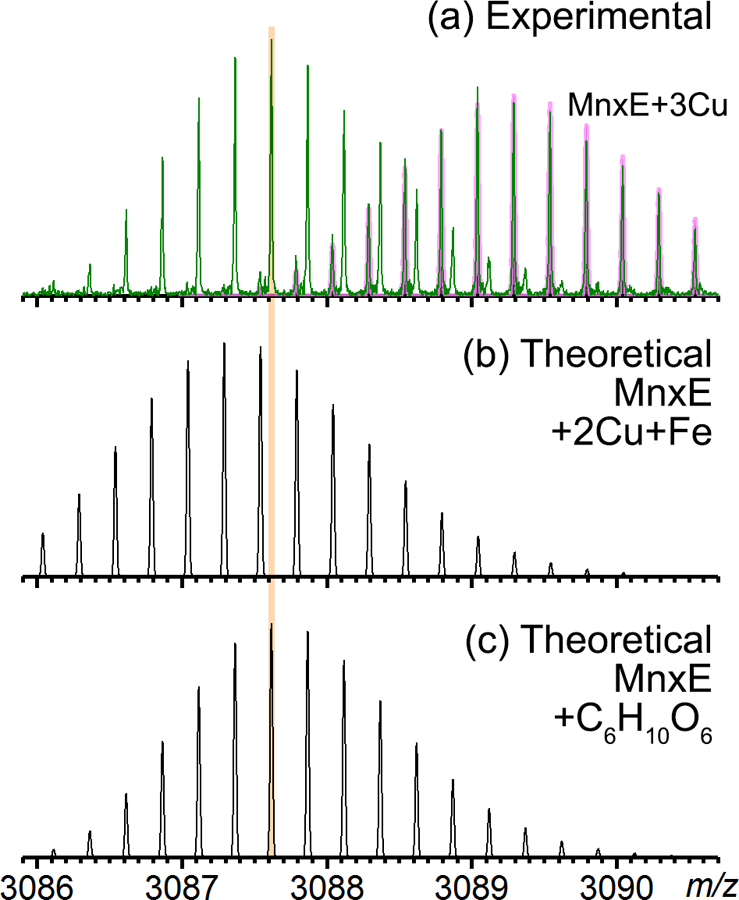
(a) Zoom-in of the SID product peaks highlighted by the green diamond in Figure 2a, previously thought to be iron bound MnxE monomer (charge state +4), centered at the m/z 3087.6. This isotopic distribution partially overlaps with isotopic distribution arising from a different species centered at m/z 3089.3, which can be matched to the theoretical isotopic distribution of MnxE bound to 3 copper (magenta bars). (b) Theoretical isotopic distribution of MnxE bound to 2 copper ions and 1 iron ion that was previously assigned based on TOF data. The mass defect introduced by the transition metal does not match to the experimental spectrum shown on top, as highlighted by the vertical line for visual alignment of the peaks. This discrepancy clearly excluded the presence of transition metals at similar masses. (c) Theoretical isotopic distribution of MnxE with an addition of C6H10O6, which matches to the experimental spectrum with a mass error of 0.17 ppm. This mass shift is assigned to gluconoylation which is a known modification of recombinant proteins expressed in E coli.
All the released MnxF species can also be assigned from the high resolution spectra. As shown in Figure 4a, the peak near m/z 3813 (highlighted by a green diamond in Figure 2b) is actually composed of at least three species resolved by FTICR (Figure 4b–d). All three isotopic distributions can be matched to theoretical formulas with mass error < 0.5 ppm, including the C6H10O6 (same as the gluconoylation observed for MnxE), and another artificial modification of (2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF, Figure 4c), which was assigned based on the UniMod database (http://www.unimod.org/) entry for the detected mass shift of 183 Da (aminoethylbenzenesulfonylation). We confirmed the presence of these unexpected modifications on Mnx by re-analyzing our bottom-up LC-MS data and including the modifications in the database search. High confidence HCD spectra supported the presence of gluconoylated MnxE, phosphogluconoylated MnxE, and AEBSF modified MnxF (Figure 5). The N-terminal MnxF peptide with glycan moieties may not be well retained in reversed phase LC due to its hydrophilicity, and was not identified in our LC-MS data. Additional supporting evidence was provided by top-down fragmentation spectra of these modified intact proteins (Figure S3 and Figure S4). Note that the gluconoylated and phosphogluconoylated intact MnxF were not directly identified in LC-MS, likely due to their low abundance. In addition to the accurate mass and localization of the modifications by the peptide fragmentation data, the fact that a phosphate loss was observed in HCD of the phosphogluconolylated MnxE peptide (i.e., doubly charged peak at m/z 494.231 in Figure 5b) confirmed the assignment of a phosphate containing modification. It is proposed that the acylating reagent, 6-phosphoglucono-1,5-lactone, produced in the normal nucleic acid synthesis pathway of the host cell can non-enzymatically react with primary amines in a protein to generate the phosphogluconoylation modification. Host cell phosphatase then removes the phosphate group yielding the gluconoylated protein.[43] AEBSF was indeed used during the sample preparation as a protease inhibitor. Despite the relatively low abundance of this modification (~5% of MnxF) we were able to localize it near the MnxF N-terminus by bottom-up (on Tyr 4, Figure 5c) and top-down experiments (Figure S4, likely on Tyr 4, Ser 5, or Lys6).
Figure 4.
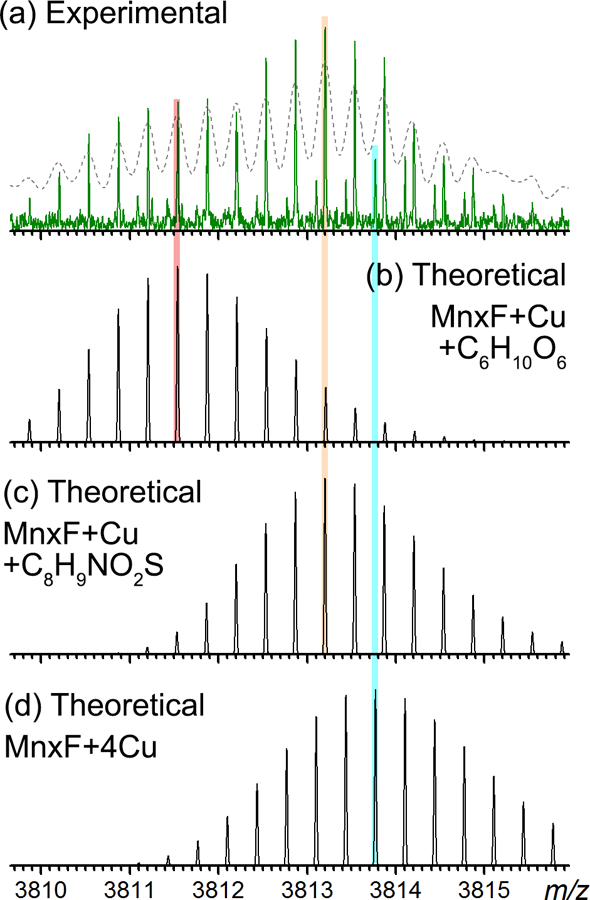
(a) Zoom-in of the SID product peaks highlighted by a green diamond in Figure 2b, indicating presence of multiple MnxF species released in SID (all at +3 charge state). There are at least 3 isotopic distributions in this region, which are matched to the theoretical spectra of (b) MnxF with gluconoylation and 1 bound Cu, (c) MnxF modified by AEBSF and bound to 1 Cu, (d) MnxF bound to 4 Cu. As shown by the overlaid dotted curve in (a), the three species are no longer discernable after smoothing the spectrum with a 0.05 Da window which reduced the resolution from 100k (green curve, original spectrum) to about 10k (dotted curve, smoothed spectrum). The high resolution of the mass analyzer is essential to fully resolve all the species, in particular peaks corresponding to low-abundance species that may easily coalesce at limited resolution.
Figure 5.
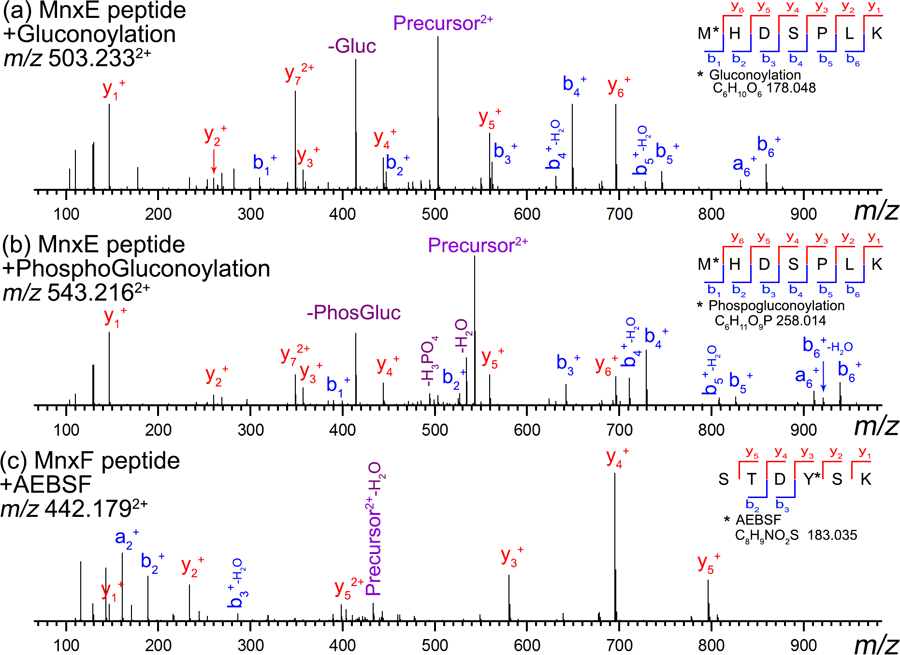
HCD fragmentation spectra for tryptic peptides of Mnx protein identified in a separate LC-MS experiment on Orbitrap instrument: (a) N-terminal MnxE peptide with gluconoylation. (b) N-terminal MnxE peptide with phosphogluconoylation. (c) N-terminal MnxF peptide with AEBSF modification assigned on the Tyr residue. All spectra show good coverage (insert at top right corner of each spectrum) for supporting the assignments of the modifications. In particular, losses of the gluconoylation (“-Gluc” in top spectrum) and phosphogluconoylation (“-PhosGluc” and “-H3PO4” in middle spectrum) were observed and further confirm the presence of these modifications.
The unexpected, covalent modifications are further confirmed after the removal of the majority of bound copper. By treating Mnx with ethylenediaminetetraacetic acid (EDTA) prior to MS, we were able to obtain highly simplified SID spectra of released MnxE and MnxF monomers (Figure S5). Although all of the unexpected modifications were only detected at less than ~10% of the “native”, unmodified proteoforms, these modifications were the source of additional heterogeneity precluding us from confidently defining metal binding stoichiometry of Mnx, especially when these different proteoforms were assembled into multi-subunit complexes. Gluconoylation is one of the known, undesired posttranslational modification for heterologously expressed proteins.[44] AEBSF has also been reported to covalently modify proteins.[45] Optimization of protein expression and purification conditions to minimize such unwanted modifications would be beneficial, in particular for production of proteins for pharmaceutical and medical applications. It is noted that both the gluconoylation (Figure 4b) and AEBSF (Figure 4c) have a similar unit mass to that of multiple Cu bound MnxF (Figure 4d). Hence, high resolution is essential for characterization of highly heterogeneous systems because some components (including unexpected modifications) could easily become indistinguishable at lower resolution. As a proof of principle, the high resolution spectrum was smoothed and the resolution was reduced by a factor of 10 from about 100k to 10k, as shown by the original spectrum (green line) and the smoothed spectrum (dotted line) in Figure 4a. The MnxF+4Cu species became buried and invisible, while the gluconoylation and AEBSF species coalesced into a mixed isotope distribution that is difficult to interpret. For larger MnxE/F multimers (Figure S6), overlapping isotope distributions (consistent with multiple bound Cu and modifications) may not be sufficiently resolved and distinguished from each other even with the high resolution of FTICR. Such sample heterogeneity arising from undesirable artifacts introduced during sample handling remains a significant challenge for achieving the ultimate resolution for large proteins and protein complexes despite the ever increasing resolving power of the mass analyzer. Nonetheless, the ability to confidently identify unexpected modifications is critical for improving the homogeneity of protein samples for other techniques such as X-ray crystallography, which benefit from obtaining highly homogeneous material.[46]
Sample Heterogeneity and Incomplete Desolvation are Major Obstacles for High Resolution Analysis of Native Proteins
The MnxE and MnxF bind to Cu at varying ratios, and are also partially modified to different degrees as shown by the well resolved spectra of the released subunits (Figure 2–4). The heterogeneity in both metal binding and modifications propagate into larger assemblies, resulting in complicated spectra. However, for MnxE/F multimers, most of the species were still isotopically resolved (Figure S6). The regularly spaced isotope peaks can readily be distinguished from the background signal (Figure S7). In low resolution mass spectra of large proteins, multiple charge states have to be identified and fit to a distribution for charge deconvolution. Isotopically resolved spectra enable charge state assignment solely based on the intervals of isotope peaks. Therefore, the high resolution offered by the FTICR remarkably simplifies peak assignment for spectra with many species and hence many (potentially overlapping) charge state envelopes. The pattern of varying Cu binding on the MnxE/F monomers is qualitatively maintained, with an average of about 1.3 Cu per monomer unit consistent throughout all the multimers (estimated based on the weighted average of peak heights of peaks labeled in Figure S6 for qualitative comparison), even for the MnxE3F3 hexamer which showed about 1.4 Cu per monomer (Figure 6). But it becomes increasingly difficult to fully resolve larger species, as the heterogeneity builds up combinatorially from the heterogeneity in each subunit. Figure 6a shows the zoom-in view of the released MnxE3F3 hexamer (charge state +10), where a few distinctly resolved peaks can be observed. A closer look revealed that the spacing between the three major peaks with 7050 < m/z < 7070, all isotopically resolved, corresponds to the mass of Cu (Figure 6b). Cluster of peaks higher in mass by about 180 Da likely corresponds to the addition of gluconoylation (178 Da) or AEBSF (183 Da) to one of the monomeric subunits, resembling the pattern observed for smaller multimers (Figure S6). A similar pattern was observed for MnxE3F3 hexamer at the charge state of +11 (m/z ~6400).
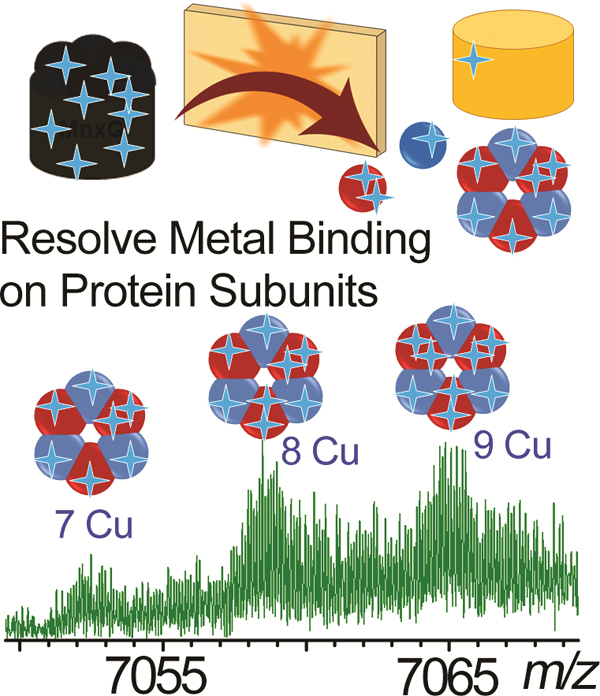
Figure 6.
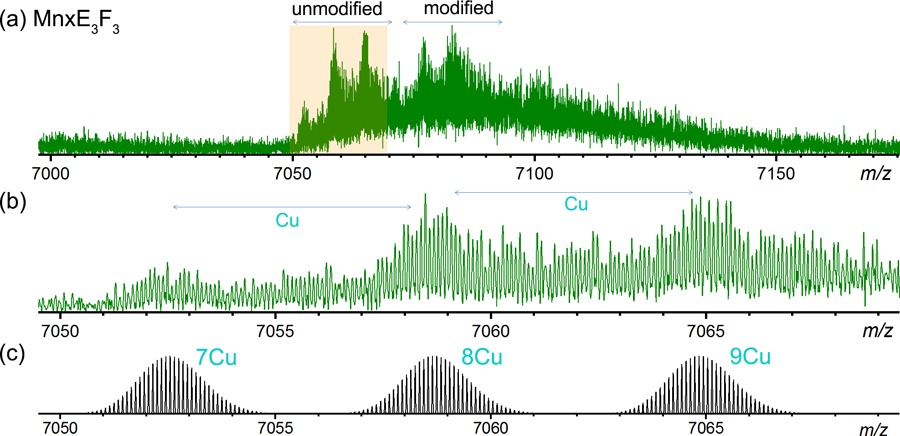
(a) Zoom-in spectrum of the released MnxE3F3 hexamer (charge state 10+) in SID. The uneven baseline originates from incomplete desolvation and salt adduction, which are commonly observed for native proteins and can significantly impact attainable resolution. Nonetheless, the hexamer is resolved into distinct features, with at least two groups of peaks. Cluster of peaks in 7050 < m/z < 7070 range represents unmodified hexamer bound to a varying number of copper (unmodified, highlighted with a gold background), and the cluster higher in mass represents these same species modified by gluconoylation and/or AEBSF (modified). (b) Expanded zoom-in view of the unmodified cluster (shown in peach box in (a)) shows resolved species corresponding to different numbers of bound Cu on the released MnxE3F3 hexamer. The separation between the isotopic peaks is 0.1 m/z, as expected for 10+ charge states. (c) The theoretical isotopic distributions of MnxE3F3 hexamer bound to 7, 8, and 9 copper ions, closely match experimental spectrum shown in (b). These species were not experimentally baseline resolved from each other due to overlapping isotopic envelopes from other unidentified species, likely MnxE3F3 hexamer with salt (e.g. sodium) adducts. The high resolution offered by the FTICR allowed individual Cu binding events to be measured on a 70 kDa native protein subcomplex, despite the fact that the protein is highly heterogeneous.
The theoretical isotope distribution of completely desolvated MnxE3F3 hexamer carrying 7–9 Cu (Figure 6c) can be matched to the three major peaks in the 7050 < m/z < 7070 range. It is interesting to note that the baseline of the peak is increased starting from m/z 7050 to 7150. Apart from the modifications, the elevated baseline likely originates from extra adducts (solvent, salt, etc.) which adds to the heterogeneity that cannot be resolved even with high resolution. The ability to resolve different Cu bound species in Figure 6 is attributed to the sufficient desolvation that occurred upon ionization, SID, and measurement in the FTICR, yielding several relatively well-defined, homogenous species at high abundance above the baseline. Instrument conditions and salts in sample buffer tend to affect desovlation and may hamper differentiation of species bound to varying number of Cu. Fluctuations in such minor experimental details are typically overlooked because the small buffer adducts typically cannot be resolved at low resolution. But they can affect spectral quality to a remarkable extent in high resolution measurements, affecting the ability to resolve metal binding on large protein complexes.
Neither the released MnxG nor the undissociated Mnx complex was isotopically resolved in magnitude mode in the full spectrum with SWEEP excitation. Absorption mode processing [47, 48] significantly improved the resolution and yielded isotopic resolution for MnxG (139 kDa) as shown in Figure 7, and even for the undissociated Mnx complex (210 kDa) shown in Figure S8. Note that the MnxG spectrum was only obtained as a fragmentation product of SID – intact MnxG alone has not been successfully analyzed by MS likely due to precipitation in solution. The broad distribution of isotopes detected in the m/z range of 8150–8250 for MnxG in Figure 7a showed some barely resolved “features” (spikes on the peak) but they are not as well-defined as those for the MnxE3F3 hexamer shown in Figure 6a. Such broad distributions of isotopically resolved peaks for large proteins (as shown in Figure 6b and Figure 7b) are reminiscent of previous reports from Li et al.[49, 50] and Valeja et al.[51]. Lössl et al.[52] reasoned that the overlapping isotopic distributions from sodium and ammonium adducts can decrease the apparent resolution for native proteins, resulting in a practical limit of resolving sodium adducts to 65 kDa. It is also noted that the theoretical mass of MnxG plus four Cu (predicted minimum Cu load based on homology) is lower than the apex of the experimental peak by ~ 1 kDa. This mass difference cannot be explained by a reasonable number of extra Cu binding, and is likely due to salt adducts and unknown modifications. Additional top-down or middle-down experiments are necessary to explain this extra mass on the MnxG subunit. Even with isotopic resolution attainable in the absorption mode, the metal binding on the MnxG cannot be determined due to heterogeneity. However, for native protein samples that are sufficiently homogenous and clean (such as monoclonal antibodies, ~150 kDa), it is possible to obtain isotopically resolved native high resolution mass spectra similar to the predicted isotopic distributions (unpublished data).
Figure 7.
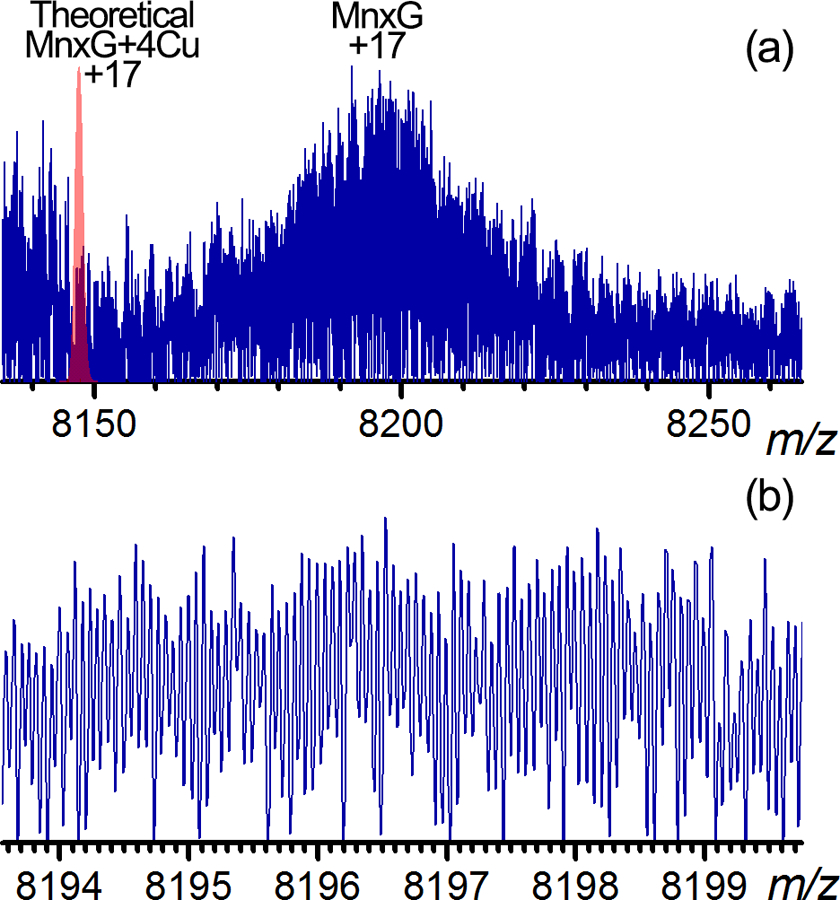
(a) Zoom-in spectrum of the high mass region for the +17 charge state of released MnxG in absorption mode. The red peak at average m/z = 8148 corresponds to the theoretical m/z calculated for the 17+ charge state of MnxG bound to 4 Cu. The significant mass shift observed indicates the MnxG is either not completely desolvated, or heavily modified. (b) Zoom-in near the apex of the MnxG peak in (a), showing isotopic resolution with a clear charge state determination of +17.
Conclusion
In summary, a heterogeneous protein complex Mnx, previously characterized only by native MS on Q-IM-TOF and refractive to characterization by other structural biology tools, has been interrogated by SID coupled with high resolution MS. Dissecting the intact complex into smaller subunits by SID reduces the complexity and allowed the heterogeneity of each subunit to be readily determined. The noncovalently bound metals were maintained in the released subunits for further characterization because of the minimal unfolding introduced by SID in the activation process. Two of the protein subunits, MnxE and MnxF, released from the intact complex were shown to bind Cu at varying ratios (average Cu load 1.2 and 1.4 per monomer, respectively), and were partially modified to varying degrees. The Cu load on the MnxE3F3 hexamer was directly measured to be 7–9 with the isotopically resolved hexamer peaks despite the heterogeneity of the protein, and is qualitatively consistent with an average of about 1.3 Cu per monomer unit. The metal binding stoichiometry derived from the most abundant charge states of the same species was similar. Previously, the Cu load in Mnx was estimated to be at least 8 by electron paramagnetic resonance (EPR)[53] and a variable value of 10–15 by ICP-OES depending on the dialysis buffer.[54] Apart from the four canonical Cu in MnxG that are essential for catalytic activity for MCOs, the majority of the remaining 6–11 Cu are most likely located in MnxE and MnxF as suggested by the SID spectra. In addition, Cu can be partially removed from Mnx with metal chelators, resulting in change of catalytic activity.[26, 53] Recent SID experiments also showed that the Cu bound to MnxF are labile and could be the primary cause for the varying Cu load of Mnx under different buffer conditions.[26] While our goal of this manuscript is to demonstrate the value of high resolution MS for studying metalloprotein complexes, future work will utilize SID to characterize Mnx under different conditions to quantitatively monitor the dynamics of metal binding within individual subunits.
Combining SID and FTICR allowed “high resolution” dissection of the Mnx complex at each substructure level, from MnxE/F monomers up to MnxE3F3 hexamer. Furthermore, unexpected modifications, gluconoylation and AEBSF, were identified based on the accurate mass. These modified species can easily be misinterpreted as iron-containing species at lower resolution. High resolution was critical for eliminating the ambiguity of the peak assignment for species with very similar masses. Information regarding metal binding and modification on individual subunits within a protein complex cannot be easily obtained using techniques that only examine large ensembles. The native MS and SID method described here offer a unique opportunity for structural analysis of individual building blocks through dissecting intact protein complexes in the gas phase.
There are still significant challenges in resolving isotopic distributions of very large proteins and protein complexes due to heterogeneity, incomplete desolvation, and decreased sensitivity for detection of high mass ions. Future instrument developments are expected to increase ion transmission in the high m/z range and reduce the acquisition time (i.e. number of averages) needed for high resolution native MS, thus making it more amenable to coupling with online separations for high throughput analysis. A combination of different methods, in particular effective ion activation methods such as SID, is indispensable for thorough characterization of heterogeneous protein complexes. While both CID and SID primarily induce cleavage of noncovalent interactions in protein complexes, other activation methods, including ultraviolet photodissociation (UVPD)[55], electron capture dissociation (ECD),[56, 57] electron transfer dissociation,[58] and electron ionization dissociation (EID)[59] have been applied for native proteins. These methods generate protein backbone fragments for structural elucidation at the residue level. Ideally, the folded subunits released by SID can be examined by these methods in order to probe metal/ligand binding sites and potentially further break down the complexity of large proteins. Development of “top-down” workflows for intact proteins [60–62] and native protein complexes is anticipated to provide an alternative structural biology tool for in-depth and rapid characterization of proteins.
Supplementary Material
Acknowledgement
The authors thank Yang Song and Arpad Somogyi at The Ohio State University for helping with the SID experiments; Jeremy Wolff and Michael Easterling at Bruker Corporation, Randy Pedder at Ardara Technologies for helping with the instrument modification. This work was funded by the National Science Foundation (NSF DBI 1455654; SID development and installation) and National Institute of Health (NIH 1S10OD018507; FTICR purchase) to V.H.W., NSF CHE-1410688 to B.M.T, and an NSF Postdoctoral Research Fellowship in Biology Award ID: DBI-1202859 to C.A.R. A portion of the research was supported by the Environmental and Molecular Sciences Laboratory (EMSL), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory.
References
- 1.Domon B, Aebersold R: Mass Spectrometry and Protein Analysis. Science. 312, 212–217 (2006) [DOI] [PubMed] [Google Scholar]
- 2.Robinson CV, Sali A, Baumeister W: The molecular sociology of the cell. Nature. 450, 973–982 (2007) [DOI] [PubMed] [Google Scholar]
- 3.Heck AJ: Native mass spectrometry: a bridge between interactomics and structural biology. Nat. Methods 5, 927–933 (2008) [DOI] [PubMed] [Google Scholar]
- 4.Benesch JLP, Ruotolo BT, Simmons DA, Robinson CV: Protein Complexes in the Gas Phase: Technology for Structural Genomics and Proteomics. Chem. Rev 107, 3544–3567 (2007) [DOI] [PubMed] [Google Scholar]
- 5.Kitova EN, El-Hawiet A, Schnier PD, Klassen JS: Reliable Determinations of Protein–Ligand Interactions by Direct ESI-MS Measurements. Are We There Yet? J. Am. Soc. Mass Spectrom 23, 431–441 (2012) [DOI] [PubMed] [Google Scholar]
- 6.Flick TG, Merenbloom SI, Williams ER: Effects of Metal Ion Adduction on the Gas-Phase Conformations of Protein Ions. J. Am. Soc. Mass Spectrom 24, 1654–1662 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baldwin Andrew J., Lioe H, Hilton Gillian R., Baker Lindsay A., Rubinstein John L., Kay Lewis E., Benesch Justin L.P.: The Polydispersity of αB-Crystallin Is Rationalized by an Interconverting Polyhedral Architecture. Structure. 19, 1855–1863 (2011) [DOI] [PubMed] [Google Scholar]
- 8.Ihms EC, Zhou M, Zhang Y, Kleckner IR, McElroy CA, Wysocki VH, Gollnick P, Foster MP: Gene regulation by substoichiometric heterocomplex formation of undecameric TRAP and trimeric anti-TRAP. Proceedings of the National Academy of Sciences. 111, 3442–3447 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seeger F, Quintyn R, Tanimoto A, Williams GJ, Tainer JA, Wysocki VH, Garcin ED: Interfacial Residues Promote an Optimal Alignment of the Catalytic Center in Human Soluble Guanylate Cyclase: Heterodimerization Is Required but Not Sufficient for Activity. Biochemistry. 53, 2153–2165 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stengel F, Baldwin AJ, Painter AJ, Jaya N, Basha E, Kay LE, Vierling E, Robinson CV, Benesch JLP: Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. Proceedings of the National Academy of Sciences. 107, 2007–2012 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyachenko A, Gruber R, Shimon L, Horovitz A, Sharon M: Allosteric mechanisms can be distinguished using structural mass spectrometry. Proceedings of the National Academy of Sciences. 110, 7235–7239 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw JB, Lin T-Y, Leach FE, Tolmachev AV, Tolić N, Robinson EW, Koppenaal DW, Paša-Tolić L: 21 Tesla Fourier Transform Ion Cyclotron Resonance Mass Spectrometer Greatly Expands Mass Spectrometry Toolbox. J. Am. Soc. Mass Spectrom 27, 1929–1936 (2016) [DOI] [PubMed] [Google Scholar]
- 13.Nagornov KO, Gorshkov MV, Kozhinov AN, Tsybin YO: High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry with Increased Throughput for Biomolecular Analysis. Anal. Chem 86, 9020–9028 (2014) [DOI] [PubMed] [Google Scholar]
- 14.Rose RJ, Damoc E, Denisov E, Makarov A, Heck AJR: High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat Meth 9, 1084–1086 (2012) [DOI] [PubMed] [Google Scholar]
- 15.Rosati S, Yang Y, Barendregt A, Heck AJR: Detailed mass analysis of structural heterogeneity in monoclonal antibodies using native mass spectrometry. Nat. Protocols 9, 967–976 (2014) [DOI] [PubMed] [Google Scholar]
- 16.Dyachenko A, Wang G, Belov M, Makarov A, de Jong RN, van den Bremer ETJ, Parren PWHI, Heck AJR: Tandem Native Mass-Spectrometry on Antibody–Drug Conjugates and Submillion Da Antibody–Antigen Protein Assemblies on an Orbitrap EMR Equipped with a High-Mass Quadrupole Mass Selector. Anal. Chem 87, 6095–6102 (2015) [DOI] [PubMed] [Google Scholar]
- 17.Debaene F, Bœuf A, Wagner-Rousset E, Colas O, Ayoub D, Corvaïa N, Van Dorsselaer A, Beck A, Cianférani S: Innovative Native MS Methodologies for Antibody Drug Conjugate Characterization: High Resolution Native MS and IM-MS for Average DAR and DAR Distribution Assessment. Anal. Chem 86, 10674–10683 (2014) [DOI] [PubMed] [Google Scholar]
- 18.Marty MT, Zhang H, Cui W, Blankenship RE, Gross ML, Sligar SG: Native Mass Spectrometry Characterization of Intact Nanodisc Lipoprotein Complexes. Anal. Chem 84, 8957–8960 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campuzano IDG, Li H, Bagal D, Lippens JL, Svitel J, Kurzeja RJM, Xu H, Schnier PD, Loo JA: Native MS Analysis of Bacteriorhodopsin and an Empty Nanodisc by Orthogonal Acceleration Time-of-Flight, Orbitrap and Ion Cyclotron Resonance. Anal. Chem 88, 12427–12436 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gault J, Donlan JAC, Liko I, Hopper JTS, Gupta K, Housden NG, Struwe WB, Marty MT, Mize T, Bechara C, Zhu Y, Wu B, Kleanthous C, Belov M, Damoc E, Makarov A, Robinson CV: High-resolution mass spectrometry of small molecules bound to membrane proteins. Nat. Methods 13, 333–336 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lippens JL, Nshanian M, Spahr C, Egea PF, Loo JA, Campuzano IDG: Fourier Transform-Ion Cyclotron Resonance Mass Spectrometry as a Platform for Characterizing Multimeric Membrane Protein Complexes. J. Am. Soc. Mass Spectrom, (2017) [DOI] [PMC free article] [PubMed]
- 22.Zhou M, Wysocki VH: Surface Induced Dissociation: Dissecting Noncovalent Protein Complexes in the Gas phase. Acc. Chem. Res 47, 1010–1018 (2014) [DOI] [PubMed] [Google Scholar]
- 23.Benesch JLP, Aquilina JA, Ruotolo BT, Sobott F, Robinson CV: Tandem Mass Spectrometry Reveals the Quaternary Organization of Macromolecular Assemblies. Chem. Biol 13, 597–605 (2006) [DOI] [PubMed] [Google Scholar]
- 24.Tebo BM, Bargar JR, Clement BG, Dick GJ, Murray KJ, Parker D, Verity R, Webb SM: Biogenic manganese oxides: properties and mechanisms of formation. Annu. Rev. Earth Planet. Sci 32, 287–328 (2004) [Google Scholar]
- 25.Butterfield CN, Soldatova AV, Lee S-W, Spiro TG, Tebo BM: Mn(II,III) oxidation and MnO2 mineralization by an expressed bacterial multicopper oxidase. Proceedings of the National Academy of Sciences. 110, 11731–11735 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romano CA, Zhou M, Song Y, Wysocki VH, Dohnalkova AC, Kovarik L, Pasa-Tolic L, Tebo BM: Biogenic manganese oxide nanoparticle formation by a multimeric multicopper oxidase Mnx. Nat Commun. 8, 746 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soldatova AV, Butterfield C, Oyerinde OF, Tebo BM, Spiro TG: Multicopper oxidase involvement in both Mn (II) and Mn (III) oxidation during bacterial formation of MnO2. JBIC Journal of Biological Inorganic Chemistry. 17, 1151–1158 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butterfield CN, Tao L, Chacón KN, Spiro TG, Blackburn NJ, Casey WH, Britt RD, Tebo BM: Multicopper manganese oxidase accessory proteins bind Cu and heme. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 1854, 1853–1859 (2015) [DOI] [PubMed] [Google Scholar]
- 29.Quintyn RS, Yan J, Wysocki VH: Surface-Induced Dissociation of Homotetramers with D 2 Symmetry Yields their Assembly Pathways and Characterizes the Effect of Ligand Binding. Chem. Biol 22, 583–592 (2015) [DOI] [PubMed] [Google Scholar]
- 30.Popa V, Trecroce DA, McAllister RG, Konermann L: Collision-Induced Dissociation of Electrosprayed Protein Complexes: An All-Atom Molecular Dynamics Model with Mobile Protons. The Journal of Physical Chemistry B. 120, 5114–5124 (2016) [DOI] [PubMed] [Google Scholar]
- 31.Butterfield C: Characterizing the Mn(II) oxidizing enzyme from the marine bacillus sp. PL-12 spore. Oregon Health & Science University; PhD Thesis, (2014) [Google Scholar]
- 32.Yan J, Zhou M, Gilbert JD, Wolff JJ, Somogyi Á, Pedder RE, Quintyn RS, Morrison LJ, Easterling ML, Paša-Tolić L, Wysocki VH: Surface-Induced Dissociation of Protein Complexes in a Hybrid Fourier Transform Ion Cyclotron Resonance Mass Spectrometer. Anal. Chem 89, 895–901 (2017) [DOI] [PubMed] [Google Scholar]
- 33.Boldin IA, Nikolaev EN: Fourier transform ion cyclotron resonance cell with dynamic harmonization of the electric field in the whole volume by shaping of the excitation and detection electrode assembly. Rapid Communications in Mass Spectrometry. 25, 122–126 (2011) [DOI] [PubMed] [Google Scholar]
- 34.Galhena AS, Dagan S, Jones CM, Beardsley RL, Wysocki VH: Surface-Induced Dissociation of Peptides and Protein Complexes in a Quadrupole/Time-of-Flight Mass Spectrometer. Anal. Chem 80, 1425–1436 (2008) [DOI] [PubMed] [Google Scholar]
- 35.Dongré AR, Somogyi Á, Wysocki VH: Surface‐induced Dissociation: An Effective Tool to Probe Structure, Energetics and Fragmentation Mechanisms of Protonated Peptides. J. Mass Spectrom 31, 339–350 (1996) [DOI] [PubMed] [Google Scholar]
- 36.Sun W, May JC, Russell DH: A novel surface-induced dissociation instrument for ion mobility-time-of-flight mass spectrometry. International Journal of Mass Spectrometry. 259, 79–86 (2007) [Google Scholar]
- 37.Chorush RA, Little DP, Beu SC, Wood TD, McLafferty FW: Surface-Induced Dissociation of Multiply-Protonated Proteins. Anal. Chem 67, 1042–1046 (1995) [DOI] [PubMed] [Google Scholar]
- 38.Zhou M: Incorporation of Surface Induced Dissociation into a Commercial Ion Mobility-Tandem Mass Spectrometer and Application of Mass Spectrometry Methods for Structural Analysis of Non-covalent Protein Complexes. Thesis, (2013)
- 39.Xu H, Freitas MA: A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics. 8, 133 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu H, Freitas MA: MassMatrix: A database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. PROTEOMICS. 9, 1548–1555 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaufman Katz A, Shimoni-Livny L, Navon O, Navon N, Bock CW, Glusker JP: Copper-Binding Motifs: Structural and Theoretical Aspects. Helv. Chim. Acta 86, 1320–1338 (2003) [Google Scholar]
- 42.Viles JH: Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer’s, Parkinson’s and prion diseases. Coord. Chem. Rev 256, 2271–2284 (2012) [Google Scholar]
- 43.Geoghegan KF, Dixon HBF, Rosner PJ, Hoth LR, Lanzetti AJ, Borzilleri KA, Marr ES, Pezzullo LH, Martin LB, LeMotte PK, McColl AS, Kamath AV, Stroh JG: Spontaneous α-N-6-Phosphogluconoylation of a “His Tag” inEscherichia coli:The Cause of Extra Mass of 258 or 178 Da in Fusion Proteins. Anal. Biochem 267, 169–184 (1999) [DOI] [PubMed] [Google Scholar]
- 44.Aon JC, Caimi RJ, Taylor AH, Lu Q, Oluboyede F, Dally J, Kessler MD, Kerrigan JJ, Lewis TS, Wysocki LA: Suppressing posttranslational gluconoylation of heterologous proteins by metabolic engineering of Escherichia coli. Applied and environmental microbiology. 74, 950–958 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuchard MD, Mehigh RJ, Cockrill SL, Lipscomb GT, Stephan1 JD, Wildsmith J, Valdes-Camin R, Kappel WK, Rai AJ, Scott GBI: Artifactual isoform profile modification following treatment of human plasma or serum with protease inhibitor, monitored by 2-dimensional electrophoresis and mass spectrometry. BioTechniques. 39, 239–247 (2005) [DOI] [PubMed] [Google Scholar]
- 46.Chait BT, Cadene M, Olinares PD, Rout MP, Shi Y: Revealing Higher Order Protein Structure Using Mass Spectrometry. J. Am. Soc. Mass Spectrom 27, 952–965 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qi Y, Witt M, Jertz R, Baykut G, Barrow MP, Nikolaev EN, O’Connor PB: Absorption-mode spectra on the dynamically harmonized Fourier transform ion cyclotron resonance cell. Rapid Communications in Mass Spectrometry. 26, 2021–2026 (2012) [DOI] [PubMed] [Google Scholar]
- 48.Qi Y, Barrow MP, Li H, Meier JE, Van Orden SL, Thompson CJ, O’Connor PB: Absorption-Mode: The Next Generation of Fourier Transform Mass Spectra. Anal. Chem 84, 2923–2929 (2012) [DOI] [PubMed] [Google Scholar]
- 49.Li H, Wolff JJ, Van Orden SL, Loo JA: Native Top-Down Electrospray Ionization-Mass Spectrometry of 158 kDa Protein Complex by High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem 86, 317–320 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Wongkongkathep P, Van Orden S, Ogorzalek Loo R, Loo J: Revealing Ligand Binding Sites and Quantifying Subunit Variants of Noncovalent Protein Complexes in a Single Native Top-Down FTICR MS Experiment. J. Am. Soc. Mass Spectrom 25, 2060–2068 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valeja SG, Kaiser NK, Xian F, Hendrickson CL, Rouse JC, Marshall AG: Unit Mass Baseline Resolution for an Intact 148 kDa Therapeutic Monoclonal Antibody by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem 83, 8391–8395 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lössl P, Snijder J, Heck AJ: Boundaries of mass resolution in native mass spectrometry. J. Am. Soc. Mass Spectrom 25, 906–917 (2014) [DOI] [PubMed] [Google Scholar]
- 53.Tao L, Stich TA, Liou S-H, Delgadillo DA, Romano CA, Spiro TG, Goodin DB, Tebo BM, Casey WH, Britt RD: Copper Binding Sites in the Manganese-Oxidizing Mnx Protein Complex Investigated by Electron Paramagnetic Resonance Spectroscopy (in preparation). (2017) [DOI] [PubMed]
- 54.Butterfield CN, Tebo BM: Substrate specificity and copper loading of the manganese-oxidizing multicopper oxidase Mnx from Bacillus sp. PL-12. Metallomics. 9, 183–191 (2017) [DOI] [PubMed] [Google Scholar]
- 55.O’Brien JP, Li W, Zhang Y, Brodbelt JS: Characterization of native protein complexes using ultraviolet photodissociation mass spectrometry. J. Am. Chem. Soc 136, 12920–12928 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML: Native Electrospray and Electron-Capture Dissociation in FTICR Mass Spectrometry Provide Top-Down Sequencing of a Protein Component in an Intact Protein Assembly. J. Am. Soc. Mass Spectrom 21, 1966–1968 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Breuker K, McLafferty FW: Native Electron Capture Dissociation for the Structural Characterization of Noncovalent Interactions in Native Cytochrome c. Angewandte Chemie International Edition. 42, 4900–4904 (2003) [DOI] [PubMed] [Google Scholar]
- 58.Lermyte F, Konijnenberg A, Williams JP, Brown JM, Valkenborg D, Sobott F: ETD Allows for Native Surface Mapping of a 150 kDa Noncovalent Complex on a Commercial Q-TWIMS-TOF Instrument. J. Am. Soc. Mass Spectrom 25, 343–350 (2014) [DOI] [PubMed] [Google Scholar]
- 59.Li H, Sheng Y, McGee W, Cammarata M, Holden D, Loo JA: Structural Characterization of Native Proteins and Protein Complexes by Electron Ionization Dissociation-Mass Spectrometry. Anal. Chem 89, 2731–2738 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Belov ME, Damoc E, Denisov E, Compton PD, Horning S, Makarov AA, Kelleher NL: From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry. Anal. Chem 85, 11163–11173 (2013) [DOI] [PubMed] [Google Scholar]
- 61.Skinner OS, Havugimana PC, Haverland NA, Fornelli L, Early BP, Greer JB, Fellers RT, Durbin KR, Do Vale LHF, Melani RD, Seckler HS, Nelp MT, Belov ME, Horning SR, Makarov AA, LeDuc RD, Bandarian V, Compton PD, Kelleher NL: An informatic framework for decoding protein complexes by top-down mass spectrometry. Nat Meth 13, 237–240 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Belov AM, Viner R, Santos MR, Horn DM, Bern M, Karger BL, Ivanov AR: Analysis of Proteins, Protein Complexes, and Organellar Proteomes Using Sheathless Capillary Zone Electrophoresis - Native Mass Spectrometry. J. Am. Soc. Mass Spectrom, (2017) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.