

Introduction
Migraine is a highly prevalent, disabling chronic-episodic and progressive disorder affecting up to a fifth of the entire world population. It has tremendous socioeconomic impact in loss of productivity and suffering. In 2016, migraine has become the second largest cause of disability in every corner of the world [12]. We have made great strides in our understanding of migraine neurobiology over the past decade. Yet, existing treatments still have limited efficacy. Considering the complex pathophysiology and genetics of migraine, novel therapeutic modalities with favorable tolerability are an urgent unmet need.
Invasive VNS (iVNS) using an implanted device has serendipitously improved migraines in patients treated for intractable epilepsy and treatment-resistant depression [7; 35]. Subsequent studies directly examined the efficacy of noninvasive cervical transcutaneous VNS (nVNS) in migraine [9; 39; 40]. In a recent randomized trial in episodic migraine with or without aura, nVNS delivered within 20 minutes after the onset of an attack tripled and doubled pain-free rates at 30 and 60 minutes, respectively, compared with sham controls [40]. More recently, the PREMIUM study of nVNS in the prevention of episodic migraine showed a significant reduction in number of migraine days in nVNS group compared with sham in treatment-adherent patients [9]. Taken together, these studies led to the FDA-approval of nVNS for treatment of acute migraine in adults. Despite the clinical progress, however, the mechanisms of action of VNS in migraine are still unknown.
We have recently reported that VNS inhibits cortical spreading depression (CSD) [8], an intense depolarization wave that is widely accepted as the electrophysiological substrate of migraine aura and a putative headache trigger [4; 48]. Experimental CSD susceptibility is a robust platform to screen for migraine therapies [3; 6]. Our previous data showed that VNS suppresses CSD comparable in magnitude to migraine prophylactic drugs [8]. The effect developed within thirty minutes after VNS and persisted for at least three hours. Moreover, unilateral VNS had bilateral efficacy.
While vagus is best known as a parasympathetic efferent nerve, 80% of vagus nerve is visceral sensory afferent fibers projecting to the nucleus tractus solitarius (NTS). Hence, the efficacy of cervical VNS in any given disorder could be via stimulation of either the afferent or the efferent fibers. Indeed, VNS activates both the efferent and the afferent vagal fibers, and has shown efficacy in both central (e.g. epilepsy, major depression) and peripheral disorders (e.g. sepsis, glucose intolerance, lung injury) [19]. Here, we dissected the contribution of afferent and efferent vagal fibers to CSD suppression and examined the mediators. Our data suggest that CSD suppression by VNS is mediated exclusively by central vagal afferents to NTS activating serotonergic and norepinephrinergic projections to the cortex.
Materials and methods
Animals and general surgical preparation
We used a total of 194 Sprague-Dawley rats (male, 230–400 grams, Charles River Laboratories, Raleigh, NC, USA, or BioLasco, Taipei City, Taiwan) in this study (Supplemental Table 1). Only males were used in this mechanistic study to minimize variability due to female ovarian hormones. Animals were housed under controlled conditions (22 ± 1 °C, 40–70% humidity and 12-hour light/dark cycle) and had access to food and water ad libitum. Femoral artery was cannulated, and a tracheostomy tube was placed under isoflurane anesthesia (4% induction, 1.5% maintenance, in 70% N2O / 30% O2), for mechanical ventilation (SAR-830; CWE, Ardmore, PA). Rectal core temperature was maintained at 37 °C via a thermostatically controlled heating pad. Blood pressure (BP) was continuously recorded via the femoral artery catheter. Arterial pH, pCO2 and O2 were periodically measured (Rapidlab 248 blood gas/pH analyzer, Siemens HealthCare, Eschborn, Germany) and maintained within physiological range by adjusting the ventilation parameters (Supplemental Table 2). All experiments were approved by the Massachusetts General Hospital or National Yang-Ming University Institutional Animal Care and Use Committees following the NIH Guide for Use and Care of Laboratory Animals.
Vagus nerve stimulation
Animals were randomly assigned to VNS and sham groups. Stimulations were delivered with the clinically used gammaCore device (electroCore LLC, Basking Ridge, NJ) modified with custom-made bipolar hook (1 mm thick, 2 mm tip separation, for iVNS) or disk (6 mm diameter, 5 mm tip separation, for nVNS) electrodes to allow stimulation of rat vagus. VNS consisted of a 1-millisecond pulse of 5-kHz sine waves, repeated at 25 Hz, for 2 minutes, delivered twice with an inter-stimulus interval of 5 minutes, identical to the clinically used protocol (Fig. 1A). For iVNS, after a midline neck incision, the right cervical vagus nerve was located near the carotid artery and the trachea, surrounding connective tissue gently dissected, and a bipolar hook electrode was placed around the middle segment. Sham controls had identical surgery and the electrode placement without stimulation. For nVNS, anterior cervical skin was shaved over the right vagus nerve and bipolar disc electrodes placed in firm contact with the skin using conductive gel, approximately 5–8 mm lateral from midline at the level of the larynx. Sham controls had the same procedure without stimulation. During VNS, BP was monitored for the transient drop typically observed with successful VNS. iVNS was used only for vagotomy experiments to ensure selective afferent or efferent stimulation. All subsequent experiments used nVNS as the FDA approved intervention for migraine, where both afferent and efferent paths are stimulated in the clinical setting.
Figure 1: Proximal, but not distal, vagotomy abrogates VNS efficacy on CSD.
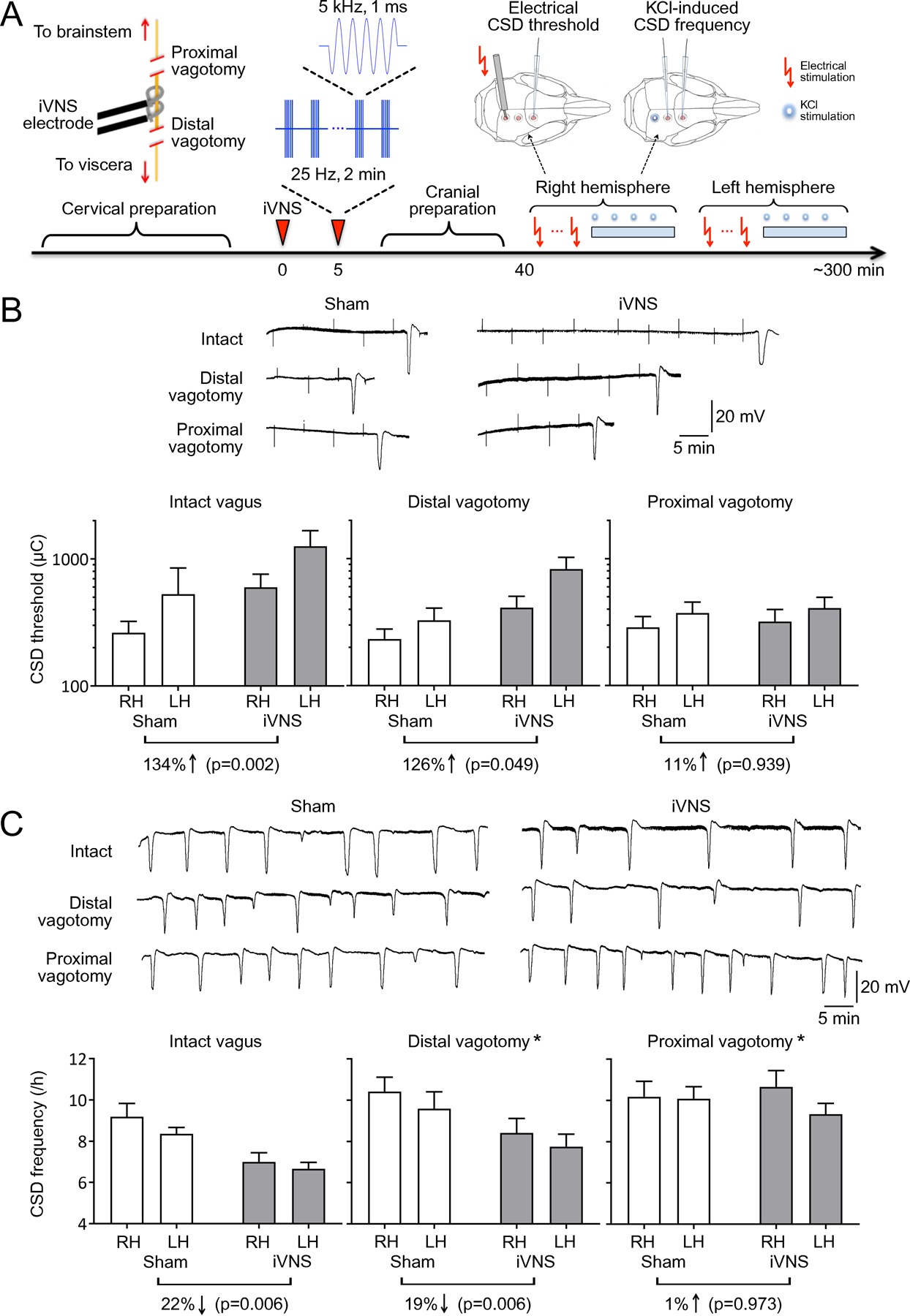
A) Experimental timeline. Cervical preparation involved surgical exposure of the right vagus nerve and either distal or proximal vagotomy. Two 2-minute iVNS trains (1 ms, 5 kHz sine wave repeated at 25 Hz) were delivered 5 minutes apart (arrowhead). After the cranial preparation (burr holes, electrode placement, ~40 minutes), electrical CSD threshold and frequency of topical KCl-induced CSDs were determined first in the right (i.e. ipsilateral to iVNS) and then in the left hemisphere. B) Tracings show electrical CSD threshold determination in representative intact, distal or proximal vagotomy rats. Stimulus artefacts indicate the electrical charge application approximately every 5 minutes, and slow negative potential shifts indicate CSDs triggered at threshold intensities. iVNS elevated the electrical CSD threshold by more than 2-fold in intact vagus or distal vagotomy groups but had no effect after proximal vagotomy. C) Representative tracings show topical KCl-induced recurrent CSDs for 1 hour in representative intact, distal or proximal vagotomy rats. iVNS reduced CSD frequency in intact vagus and distal vagotomy groups only. Vagotomy, either distal or proximal, increased KCl-induced CSD frequency. Data are mean±SE. n=12/group. *p < 0.05 vs. sham; †p < 0.05 vs. intact vagus (general linear mixed model with vagotomy, stimulation and hemisphere as independent variables). RH: Right Hemisphere, LH: Left Hemisphere.
CSD susceptibility testing
Animals were placed on a stereotactic frame (Stoelting, Wood Dale, IL, USA). Craniotomies were created over the occipital (electrical and KCl stimulation site; AP −4.5mm, ML 2mm, 2mm diameter), parietal (recording site; AP −2.0mm, ML 2mm, 1mm diameter) and frontal cortices (recording site; AP 1.5mm, ML 2mm, 1mm diameter). The electrocorticogram and direct current (DC) potential was recorded with glass capillary microelectrodes (~250 μm deep). Signals were amplified (EX1 differential amplifiers, Dagan Corporation, Minneapolis, MN, USA) and continuously recorded for offline analysis (PowerLab, ADInstruments, Colorado Springs, CO, USA). Susceptibility to CSD was evaluated by determining the electrical threshold for CSD and the frequency of KCl-induced repetitive CSDs. The electrical threshold to induce CSD was determined by direct cortical stimulation using a stimulus isolator (WPI, Sarasota, FL) and a bipolar stimulation electrode (400 μm tip diameter, 1 mm tip separation; FHC, Bowdoin, ME, USA) placed on the pial surface. Single square pulses of increasing duration and intensity (50–4000 microcoulombs, μC) were delivered at 5-minute intervals until a CSD was observed. For analysis of CSD frequency, a cotton ball (1.5–2 mm diameter) soaked with KCl (1M) was placed on the cortex and refreshed every 15 minutes for one hour, and the number of recurrent CSDs counted. As per standard experimental protocol in this paradigm [5], electrical threshold always preceded the SD frequency assessment, since 1-hour topical KCl application may cause injury and confound subsequent SD threshold assessment. Slow extracellular potential shifts less than 5mV in amplitude were excluded from analysis according to preset criteria. CSD propagation speed, amplitude and duration at half-maximal amplitude were also quantified. This protocol (electrical threshold followed by KCl-induced CSD frequency) was then repeated in the left hemisphere. We have previously shown that alternating the testing order (i.e. right vs. left hemispheres tested first) after nVNS did not affect efficacy in each hemisphere [8]. Experimental set up and protocol are shown on Fig. 1A.
Vagotomy
To test whether central or peripheral projections mediate CSD suppression upon VNS, we performed distal or proximal vagotomy. Exposure of the vagus nerve, vagotomy and electrode placement for iVNS were carried out in this order prior to intubation. After a midline incision, subcutaneous tissue was gently dissected to reach the right vagus adjacent to the trachea. The nerve was then dissected free, a 5–0 silk suture was passed around and ligated at either the proximal or the distal end of the exposed nerve, and the nerve was cut either proximal (cranial) or distal (caudal) to the ligature and the iVNS site (Fig. 1A). The stimulation electrode was then placed around the mid segment of the vagus as described above. After two 2-minute stimulations 5 minutes apart, electrode was removed, and trachea cannulated as above.
Pharmacological silencing of the nucleus tractus solitarius
The visceral afferent vagal fibers project to NTS, bilaterally. We, therefore, tested whether pharmacological silencing of neurotransmission in NTS affects nVNS suppression of CSD susceptibility. To this end, we used lidocaine as well as CNQX, an AMPA/Kainate ionotropic glutamate receptor antagonist, since vagal afferents to the NTS are glutamatergic. NTS microinjections and subsequent CSD testing were performed under barbiturate anesthesia (pentobarbital sodium, 50 mg/kg, intraperitoneal, followed by femoral intravenous infusion at 15–20 mg/kg/h), as previously reported. Rats were placed in a stereotaxic apparatus (Kopf, Tujunga, USA), and an occipital craniotomy performed to expose the dorsal medulla. A glass micropipette adapted to a Hamilton syringe (Reno, USA) was used to perform microinjections at 0.5 mm anterior, 0.5 mm lateral, 0.5 mm ventral to obex, bilaterally. Vehicle (artificial cerebrospinal fluid (aCSF); 117 mM NaCl, 1.25 mM MgSO4, 4.7 mM KCl, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 2.5 mM CaCl2, 25 mM NaHCO3, 11 mM glucose, adjusted to pH 7.4), 2% lidocaine (AstraZeneca, Cambridge, UK) or 200 pM CNQX (Tocris, Bristol, UK) were injected in 20 nl volume over 40 seconds, as previously described [30; 46]. To confirm the microinjection placement, the brainstem was removed and fixed in 30% sucrose containing 10% paraformaldehyde (PFA) for 72 h after physiological experiment. Coronal sections were cut (20 μm) and then stained with neutral red, and the microinjection site was identified under a microscope.
Confirmation of NTS activation by VNS using c-Fos staining
In a group of rats without CSD testing, c-Fos immunohistochemistry in NTS was performed two hours after nVNS. Rats were deeply anesthetized with barbiturate anesthesia and were transcardiac perfused with 37°C normal saline followed by ice-cold 4% PFA (pH=7.4). The brain was rapidly removed and post-fixed in 4% paraformaldehyde for 24h. After post-fixation, serial dehydration was carried out by soaking the brain in 15% sucrose solution for 24h and then transferred to 30% sucrose solution for 72h. The staining was carried out by free-floating method. Coronal sections of brainstem (20 μm) at the level of NTS were blocked in 3% normal horse serum (containing 0.375% gelatin and 0.2% Triton-X-100) for 30 minutes and then incubated in primary monoclonal mouse anti-c-Fos antibody (1:500, Santa Cruz Biotechnology#sc-8047, Texas, US) for 48h at 4°C. After primary antibody incubation, the sections were incubated in horse anti-mouse biotinylated secondary antibody (antibody from Vector Laboratories#PK-6102, Burlingame, USA) for 1h and then conjugated with avidin-biotin complex for 30 minutes at room temperature (reagents from Vector Laboratories#PK-6102). Finally, c-Fos protein was visualized by a black reaction product using a nickel-enhanced diaminobenzidine substrate kit (Vector Laboratories#PK-4100).
Pharmacological ablation of norepinephrinergic and serotonergic neurotransmission
To test whether norepinephrine (NE) and serotonin (5-HT) mediate CSD suppression by nVNS, we used toxins to deplete these neurotransmitters in the brain. To deplete NE we used DSP4 (N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine hydrochloride), a neurotoxin that induces selective degeneration of locus coeruleus axons in rodents, administered 15 days before the experiment (50 mg/kg, intraperitoneal; Sigma-Aldrich, St. Louis, MO, US) [44]. DSP4 irreversibly binds NE transporter, accumulates in the nerve terminals, and depletes NE [17; 34]. To deplete 5-HT, we used tryptophan hydroxylase inhibitor PCPA (100mg/kg, intraperitoneal; Sigma-Aldrich, St. Louis, MO, US), administered 3 days before the experiment [18; 21]. No locomotor disturbance and general reduction in wellness of animals was observed after injections.
Histochemistry of DSP4 or PCPA-treated animals
A separate group of rats that received DSP4 or PCPA as above, but without the CSD testing, were anesthetized with chloral hydrate (1g/kg body weight i.p.) and were transcardially perfused with 0.9% heparinized saline followed by 4% PFA. Brains were removed, PFA post-fixed for 4h and cryoprotected with 30% sucrose. After cryoprotection, the brains were frozen with isopentane plus dry ice and stored at −80 °C. The brains were coronally cut (30 μm thick) at 3 levels, 1.5 mm apart (AP = +0.5 mm, −1.0 mm and −2.5 mm from bregma). To detect microglia, astrocytes and degenerating neurons a free-floating staining was performed using an anti‐Iba1 Rabbit Antibody (1:200) (WAKO), a Monoclonal Anti-Glial Fibrillary Acidic Protein (GFAP) antibody produced in mouse (1:400) (Sigma) and a Fluoro-Jade® C (Millipore), respectively. Sections from a separate brain with ischemic infarct served as positive control for Fluoro-Jade® C. Pictures were taken using a Nikon Ti-S inverted fluorescence microscope and color and black and white cameras with NIS-elements Basic Research software. Mean gray value was obtained using Image J software.
Study design and statistics
Experiments were carried out and analyzed in a blinded fashion. A priori sample size determinations aimed to detect a 20% effect size (α=0.05, 95% power) for an estimated standard deviation of 15% of the mean for iVNS and 10% of the mean for nVNS based on prior experience using KCl-induced CSD frequency as the main readout. Data were analyzed using general linear mixed model analysis with vagotomy (sham, proximal, distal), stimulation (present, absent), and hemisphere (right, left; repeated measurement) as fixed factors for the vagotomy experiment. Multiple comparisons were adjusted using Bonferroni. For the NTS block experiment, ordinary one-way ANOVA was used followed by Tukey’s post-hoc comparisons. Two-way ANOVA was used to evaluate pharmacological ablation of norepinephrinergic and serotonergic neurotransmission with treatment (vehicle, DSP4, PCPA) and stimulation (present, absent) as fixed factors, followed by Bonferroni post-hoc comparisons. Histological data were analyzed using one-way ANOVA. Statistical analysis was performed using SPSS statistics (v.25, IBM, Armonk, NY) and Prism (v.7, GraphPad, San Diego, CA). A priori exclusion criteria were surgical failure (n=1 in distal vagotomy iVNS group), poor systemic physiology (no exclusions), and poor tissue fixation for histology (n=1 each in DSP4 and PCPA groups). A p-value of <0.05 was considered statistically significant.
Results
Effects of vagotomy
Cervical VNS activates both the visceral motor efferents (i.e. parasympathetic) and visceral sensory afferents projecting to the NTS in brain stem. Therefore, we first tested whether the afferent or efferent vagal fibers (central and peripheral projections, respectively) were responsible for CSD suppression upon VNS by performing a vagotomy either cranial (i.e. proximal) or caudal (i.e. distal) to a surgically implanted iVNS electrode (Fig. 1A). Shortly after cervical preparation, we delivered iVNS and 40 minutes later tested electrical threshold for CSD induction followed by the frequency of topical KCl-induced repetitive CSDs in this order, first in the hemisphere ipsilateral to the iVNS and then in the contralateral hemisphere (Fig. 1A, timeline).
The mixed model analysis (n=12/group) showed that in the presence of an intact vagus nerve, iVNS elevated the electrical stimulation threshold of CSD induction by more than two-fold (F(1,67) = 9.874, p = 0.002; Fig. 1B, left panel), reduced KCl-induced CSD frequency by more than 20% (F(1,66) = 8.013, p = 0.006; Fig. 1C, left panel), and slowed CSD propagation (F(1,60)=8.802, p = 0.004; Supplemental Table 3) compared with sham controls, as previously reported [8]. Distal vagotomy to eliminate visceral motor efferents did not significantly diminish iVNS efficacy on CSD susceptibility. After distal vagotomy, iVNS once again increased the electrical CSD threshold by more than two-fold (F(1,67) = 4.022, p = 0.049; Fig. 1B, middle panel), and decreased KCl-induced CSD frequency by almost 20% (F(1,65)=8.043, p = 0.006; Fig. 1C, middle panel). In contrast, proximal vagotomy completely abolished iVNS efficacy. After proximal vagotomy, iVNS had no effect on electrical CSD threshold (F(1,67) = 0.006, p = 0.939; Fig. 1B, right panel) or KCl-induced CSD frequency (F(1,66)=0.001, p = 0.973; Fig. 1C, right panel). These data indicated that VNS suppressed SD susceptibility via activation of visceral sensory afferents projecting to the brain stem NTS.
Interestingly, the mixed model analysis also revealed that vagotomy, both distal and proximal, increased KCl-induced CSD frequency compared with intact vagus nerve (F(2,66)=10.376, *p < 0.001), implicating tonic vagal suppression of cortical excitability; electrical CSD threshold was not altered (F(2,67)=0.588, p = 0.558) and CSD duration was shorter after vagotomy (Supplemental Table 3). Moreover, KCl-induced SD frequencies obtained in the first hemisphere were higher than the second hemisphere (F(1,65)= 4.036, p = 0.049), although CSD thresholds did not statistically differ between the hemispheres (F(1,66)=2.148, p = 0.147).
Effects of NTS block
The visceral afferent vagal fibers entering the brain stem project to NTS, bilaterally. We tested whether pharmacological blockade of neurotransmission in NTS affects nVNS suppression of CSD susceptibility (Fig. 2A). In the vehicle group (i.e. intact NTS), nVNS reduced KCl-induced CSD frequency by a third compared with non-stimulated controls (one-way ANOVA, F(3,19) = 6.844, p = 0.003, Fig. 2C). Stereotactic microinjection of either lidocaine or CNQX into the NTS prevented nVNS from suppressing CSD susceptibility. Electrophysiological properties of CSD did not differ among the groups (Supplemental Table 4). We also confirmed that nVNS indeed activates NTS by using c-Fos immunoreactivity, a widely used surrogate for neuronal activation [26]. We found an increase in c-Fos immunoreactivity in NTS 2 hours after nVNS suggesting that nVNS indeed activates NTS (Fig. 2D). Altogether, these data indicated that NTS is a critical relay for VNS to suppress CSD susceptibility.
Figure 2: NTS block abrogates VNS efficacy on CSD.
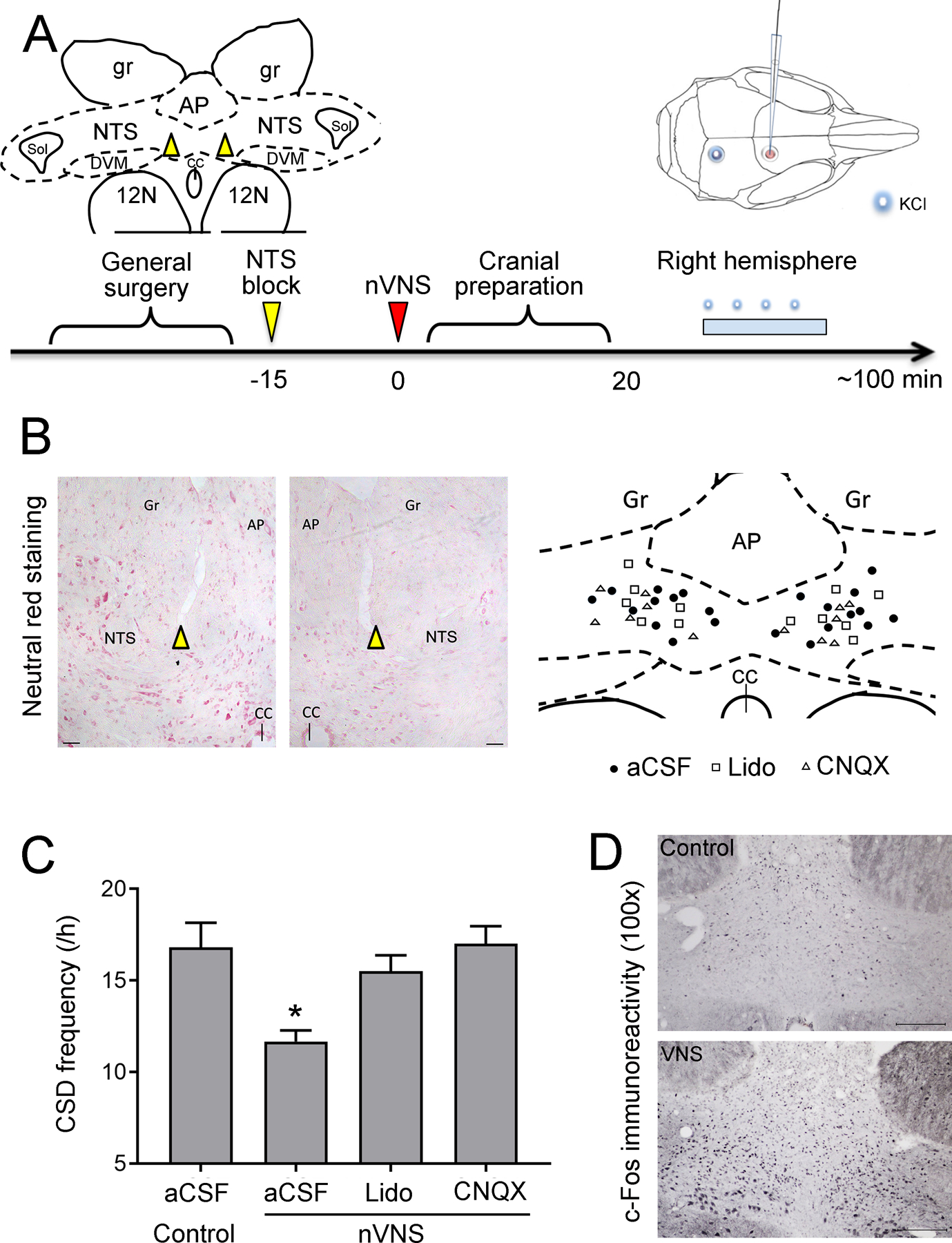
A) Experimental timeline. Dorsal cervical area was prepared and stereotaxic NTS injections (artificial CSF, lidocaine or CNQX) performed 15 minutes prior to nVNS. Inset shows the approximate location of NTS injections. After the cranial preparation (burr holes, electrode placement, ~20 minutes, the frequency of topical KCl-induced CSDs were determined in the right hemisphere (i.e. ipsilateral to nVNS) for one hour. AP: area postrema; gr: gracile nucleus; NTS: nucleus tractus solitarii; CC: central canal; Sol: solitary tract; DMV: dorsal motor nucleus of vagus; 12N: hypoglossal nucleus. B) Representative histological images (neutral red staining) and schematic diagrams illustrate the anatomical locations of microinjection. Individual microinjection sites are presented as circles (aCSF), squares (Lidocaine), or triangles (CNQX). Scale bar: 50 μm; AP: area postrema; Gr: gracile nucleus; NTS: nucleus tractus solitarii; CC: central canal. C) nVNS reduced CSD frequency by 31% in aCSF injected animals (Control: N=5, nVNS: N=6). This effect was abolished when NTS was blocked by lidocaine (N=6) or CNQX (N=6). Of note, overall CSD frequency was higher in these experiments compared with the other cohorts because of the difference in anesthetic regimen (isoflurane/N2O vs. barbiturate). D) Coronal brainstem sections through NTS showing increased c-Fos immunostaining 2 hours after a single 2-minute nVNS compared with control, confirming NTS activation after nVNS. Scale bar = 200 μm. Data are mean±SE. *p < 0.05 vs. all other groups (one-way ANOVA).
Effects of serotonin and norepinephrine depletion
NTS neurons project to the LC and DRN, which in turn provide NE and 5-HT innervation of the cortex, respectively. VNS indeed stimulates NE and 5-HT release [25], both of which may suppress CSD susceptibility [3]. We, therefore, tested whether NE and 5-HT mediate CSD suppression by nVNS, by depleting these neurotransmitters using DSP4 (NE depletion) and PCPA (5-HT depletion), alone or in combination (Fig. 3). nVNS showed a significant main effect on KCl-induced CSD frequency (F(3,53) = 5.732, p = 0.020; two-way ANOVA). Post-hoc analysis showed that nVNS decreased CSD frequency by 23% compared with sham in vehicle-treated animals (p = 0.008). Either DSP4 or PCPA alone diminished the CSD suppression by nVNS (11% and 10% reduction, p = 0.912 and p = 0.061, respectively). In combination, these drugs completely abolished nVNS effect on CSD susceptibility (2% reduction, p = 0.999). These data suggested that both NE and 5-HT are important central mediators for VNS suppression of cortical CSD susceptibility. Interestingly, NE and 5-HT depletion also affected resting state CSD susceptibility (F(3,53) = 24.690, *p < 0.001). DSP4 decreased (p = 0.005) whereas PCPA increased (p = 0.005) CSD frequency compared with saline controls. Electrophysiological properties of CSD did not differ among the groups (Supplemental Table 4).
Figure 3: NE and 5-HT depletion abrogates VNS efficacy on CSD.
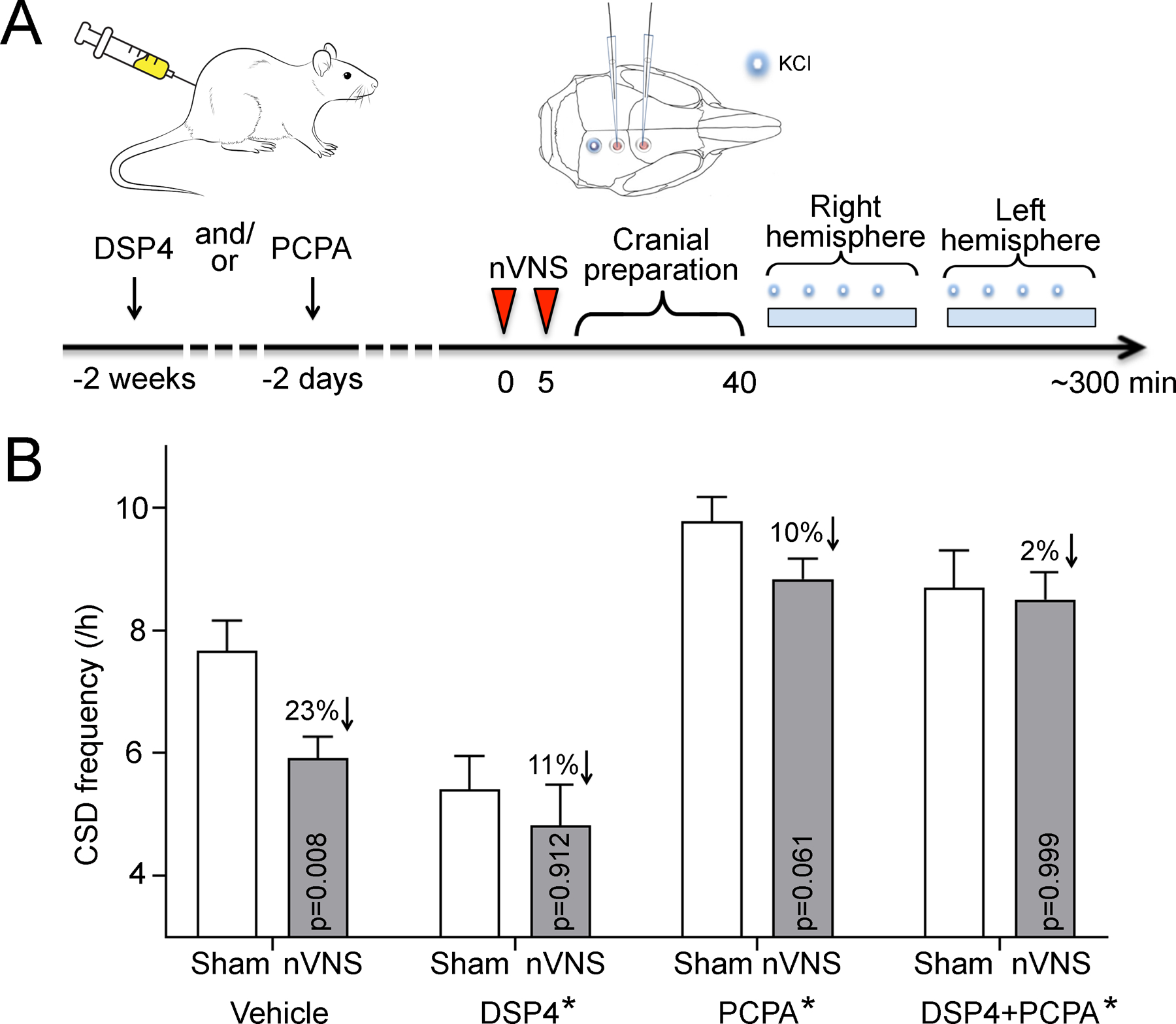
A) Experimental timeline. NE and/or 5-HT were depleted by systemic injection of DSP4 and/or PCPA. On the experiment day (i.e. 2 weeks after DSP4 and/or 2 days after PCPA injection), two 2-minute nVNS were delivered 5 minutes apart (arrowheads). After the cranial preparation (burr holes, electrode placement, ~40 minutes, the frequency of topical KCl-induced CSDs were determined first in the right (i.e. ipsilateral to nVNS) and then in the left hemisphere. B) nVNS reduced CSD frequency by 23% in vehicle injected animals (Sham: N=12, nVNS: N=13). NE or 5-HT depletion by DSP4 (Sham: N=6, nVNS: N=6) or PCPA (Sham: N=7, nVNS: N=6) partially prevented nVNS effect on CSD. When both neurotransmitters were depleted, nVNS effect was completely blocked. Each toxin alone also affected the CSD frequency. Right and left hemispheres did not differ; therefore, data from the two hemispheres were averaged and shown as mean±SE. p < 0.05 vs. sham, *p < 0.05 vs. vehicle (two-way ANOVA with stimulation and treatment as independent variables).
To test whether the effect of NE and 5-HT depletion on CSD susceptibility was associated with reactive changes in the tissue, we examined microglia and astrocytes using Iba1 (F(2,25) = 0.085, p = 0.919) and GFAP (F(2,25) = 0.048, p = 0.954) immunoreactivity, respectively, and did not observe any change after treatment with either toxin (Fig. 4A). Because DSP4 has been reported to cause neuronal loss, we performed Fluoro-Jade® C staining and did not find any evidence of cell death after DSP4 treatment at the time of CSD susceptibility testing (i.e. 2 weeks; Fig. 4B).
Figure 4: DSP4 and PCPA does not alter microglia and astrocytes in the cortex.
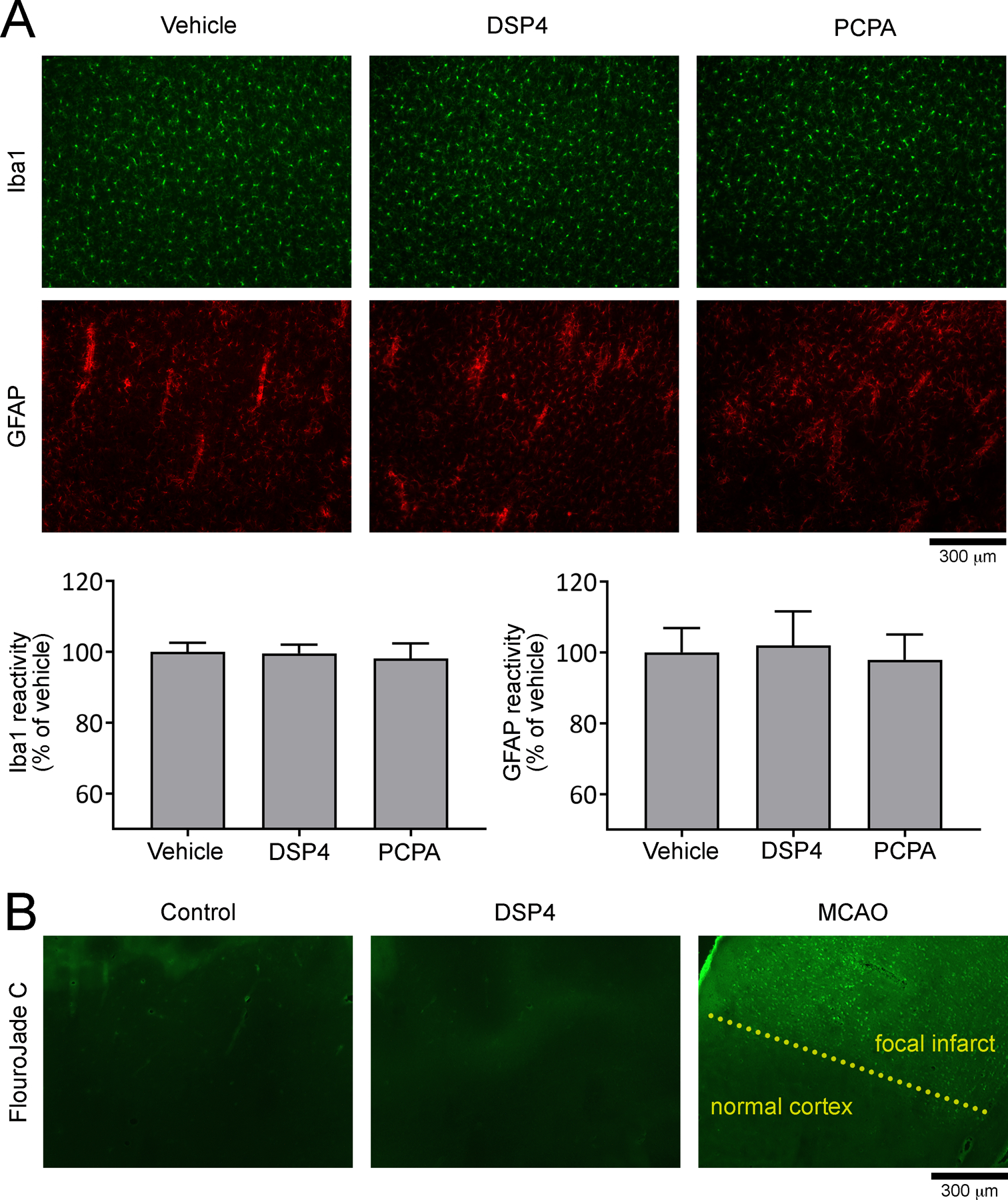
A) Representative coronal sections of cerebral cortex show Iba1 and GFAP immunostaining to examine microglia and astrocytes, respectively, after vehicle DSP4 or PCPA treatment at the time of CSD susceptibility testing (i.e. 2 weeks after DSP4 and/or 2 days after PCPA injection) but without VNS or CSD induction. Immunofluorescence intensity imaged under identical settings did not significantly differ among the groups. Scale bar = 300 μm. B) Representative coronal sections of cerebral cortex stained with fluoro-Jade C show that vehicle (N=5) or DSP4 (N=4) treatment was not associated with cell death detectable at 2 weeks. A representative section from a similar cortical region from a mouse brain after middle cerebral artery occlusion (MCAO) is also shown (bright green cells in upper half of the image) as positive control stained simultaneously. The scale bar indicates 300 μm. Iba1 and GFAP expression data showed normal distribution (D’Agostino-Pearson omnibus test K2, p = 0.836 and p = 0.529, respectively).
Discussion
Our data indicate that CSD suppression by VNS is mediated by activation of the vagal visceral sensory afferents to NTS (Aβ fibers), which in turn project to the LC and DRN providing NE and 5-HT innervation, respectively, throughout the brain. The data provide a mechanism of action of VNS in the prevention and treatment of migraine, especially with aura [13; 39; 40] Indeed, the mechanism overlaps with the anti-epileptic and anti-depressant mechanisms of action of VNS [11; 22; 45], not surprising given the clinical association between migraine with aura, epilepsy [32] and depression [47]. Several anti-epileptic and anti-depressant drugs are among the commonly used migraine prophylactic medications, and our data adds VNS among this class of treatments.
There is ample electrophysiological and functional MRI evidence in humans showing that nVNS is capable of activating target fibers in vagus nerve much like iVNS [10; 23; 27; 43]. It is also well established that NTS is the immediate relay of vagal afferents in the brainstem [20]. Blockade of neurotransmission in NTS by lidocaine completely blocked nVNS efficacy on CSD, confirming the critical relay role of NTS in this paradigm. The AMPA/Kainate receptor antagonist CNQX also blocked nVNS efficacy, consistent with the fact that vagal afferents to the NTS are glutamatergic [1; 29]. The NTS, major recipient of vagal sensory afferents, sends projections to and activates diffuse regions of the brain, including LC and DRN [2; 42]. Indeed, VNS increases the firing of NE neurons in LC and 5-HT neurons in DRN, presumably via α1 postsynaptic adrenoceptors [24]. The consequent rapid rise in the extracellular NE levels has been detected in cortex, hippocampus and amygdala after VNS [15; 25; 33]. We did not attempt unilateral NTS inhibition to dissect laterality, because: (i) it is well established that NTS receives bilateral vagal projections [20], (ii) unilateral nVNS induced bilateral c-Fos activation in NTS in our experiments, and (iii) unilateral nVNS inhibits CSD susceptibility bilaterally [8].
To interrogate the specific monoamine systems, we used DSP4, which leads to a rapid and long-lasting loss of NE and a slower decrease in dopamine-β-hydroxylase activity and immunoreactivity in the regions innervated by LC [34], and PCPA, which is a tryptophan hydroxylase inhibitor that depletes 5-HT [21]. Depletion of either NE or 5-HT reduced VNS efficacy on CSD, and combined depletion completely abolished the VNS effect, consistent with data showing that NE and 5-HT suppress CSD susceptibility [3; 31; 44]. Indeed, as expected, 5-HT depletion by PCPA enhanced resting state CSD susceptibility (i.e. in the absence of VNS) [36]. Contrary to our expectations, however, NE depletion by DSP4 led to a decrease in resting state CSD susceptibility. Our data also differed from a recent study showing elevated CSD susceptibility after DSP4 using very similar paradigms, albeit under propofol anesthesia [44]. DSP4 did not appear to activate microglia or astrocytes in our study, which can suppress CSD susceptibility [37; 38; 41], as a potential explanation. Alternatively, DSP4-induced inhibition of NE transporter in the cortex may limit the ability of the tissue to buffer NE influx from sources outside the CNS (i.e. sympathetics), thus leading to a paradoxical steady state rise in cortical NE levels [34]. Such a mechanism would reduce resting-state CSD susceptibility but still block the NE release from LC projections upon VNS. Regardless, however, DSP4-induced suppression of CSD frequency did not create a floor effect since CSD frequency can be pharmacologically suppressed down to zero [5].
In conclusion, our data elucidate the mechanism of action of VNS on CSD that overlaps with its anti-epileptic and anti-depressant mechanisms, with relevance in the management of not only migraine but also brain injury where numerous CSDs occur over many days and may exacerbate the outcome [14]. Needless to say, multiple parallel mechanisms of action likely exist for VNS in migraine, such as inhibition of mechanical nociception, repressed expression of proteins associated with peripheral and central trigeminal sensitization, and decreased trigeminal nociception by suppressing the rise in glutamate after nitric oxide treatment [16; 28]. It remains to be tested whether the NTS-LC/DRN axis also plays a role in these potentially complementary mechanisms, which may be important for VNS efficacy in cluster headache and abortive treatment of migraine.
Supplementary Material
Acknowledgment
This work was funded by the National Institute of Health (R01NS102969) and the Ministry of Science and Technology (MOST 104-2314-B-010-017-MY2).
Footnotes
Competing interests: Dr. Bruce Simon is an employee of electroCore, Inc. The other authors declare no competing financial interests.
Competing interests
Dr. Simon is a consultant for electroCore, Inc. Dr. Ayata has received research grants from electroCore, Inc. The other authors declare no competing financial interests.
References
- [1].Andresen MC, Yang MY. Non-NMDA receptors mediate sensory afferent synaptic transmission in medial nucleus tractus solitarius. The American journal of physiology 1990;259(4 Pt 2):H1307–1311. [DOI] [PubMed] [Google Scholar]
- [2].Aston-Jones G, Shipley MT, Chouvet G, Ennis M, van Bockstaele E, Pieribone V, Shiekhattar R, Akaoka H, Drolet G, Astier B. Afferent regulation of locus coeruleus neurons: anatomy, physiology and pharmacology. Prog Brain Res, Vol. 88, 1991. pp. 47–75. [DOI] [PubMed] [Google Scholar]
- [3].Ayata C. Spreading depression: from serendipity to targeted therapy in migraine prophylaxis. Cephalalgia 2009;29(10):1095–1114. [DOI] [PubMed] [Google Scholar]
- [4].Ayata C. Cortical spreading depression triggers migraine attack: pro. Headache 2010;50(4):725–730. [DOI] [PubMed] [Google Scholar]
- [5].Pearls Ayata C. and pitfalls in experimental models of spreading depression. Cephalalgia 2013;33(8):604–613. [DOI] [PubMed] [Google Scholar]
- [6].Ayata C, Jin H, Kudo C, Dalkara T, Moskowitz MA. Suppression of cortical spreading depression in migraine prophylaxis. Ann of Neurol 2006;59(4):652–661. [DOI] [PubMed] [Google Scholar]
- [7].Basic S, Sporis D, Chudy D, Grahovac G, Nevajda B. The effect of vagus nerve stimulation on migraine in patient with intractable epilepsy: Case report. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology 2013;34(5):797–798. [DOI] [PubMed] [Google Scholar]
- [8].Chen SP, Ay I, Lopes de Morais A, Qin T, Zheng Y, Sadeghian H, Oka F, Simon B, Eikermann-Haerter K, Ayata C. Vagus nerve stimulation inhibits cortical spreading depression. Pain 2016;157(4):797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Diener H-C, Goadsby PJ, Ashina M, Al-Karagholi MA-M, Sinclair A, Mitsikostas D, Magis D, Pozo-Rosich P, Sieira PI, Làinez MJA, Gaul C, Silver N, Hoffmann J, Marin J, Liebler E, Ferrari MD, Group oBotPS. Non-invasive vagus nerve stimulation (nVNS) for the preventive treatment of episodic migraine: the multicentre, double-blind, randomised, sham-controlled PREMIUM trial. Cephalalgia 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Frangos E, Komisaruk BR. Access to Vagal Projections via Cutaneous Electrical Stimulation of the Neck: fMRI Evidence in Healthy Humans. Brain Stimul 2017;10(1):19–27. [DOI] [PubMed] [Google Scholar]
- [11].Furmaga H, Shah A, Frazer A. Serotonergic and noradrenergic pathways are required for the anxiolytic-like and antidepressant-like behavioral effects of repeated vagal nerve stimulation in rats. Biol Psychiatry 2011;70(10):937–945. [DOI] [PubMed] [Google Scholar]
- [12].GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017;390(10100):1211–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Goadsby PJ, de Coo IF, Silver N, Tyagi A, Ahmed F, Gaul C, Jensen RH, Diener HC, Solbach K, Straube A, Liebler E, Marin JCA, Ferrari MD. Non-invasive vagus nerve stimulation for the acute treatment of episodic and chronic cluster headache: A randomized, double-blind, sham-controlled ACT2 study. Cephalalgia 2018;38(5):959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hartings JA, Shuttleworth CW, Kirov SA, Ayata C, Hinzman JM, Foreman B, Andrew RD, Boutelle MG, Brennan KC, Carlson AP, Dahlem MA, Drenckhahn C, Dohmen C, Fabricius M, Farkas E, Feuerstein D, Graf R, Helbok R, Lauritzen M, Major S, Oliveira-Ferreira AI, Richter F, Rosenthal ES, Sakowitz OW, Sanchez-Porras R, Santos E, Scholl M, Strong AJ, Urbach A, Westover MB, Winkler MK, Witte OW, Woitzik J, Dreier JP. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leao’s legacy. J Cereb Blood Flow Metab 2017;37(5):1571–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hassert DL, Miyashita T, Williams CL. The effects of peripheral vagal nerve stimulation at a memory-modulating intensity on norepinephrine output in the basolateral amygdala. Behavioral neuroscience 2004;118(1):79–88. [DOI] [PubMed] [Google Scholar]
- [16].Hawkins JL, Cornelison LE, Blankenship BA, Durham PL. Vagus nerve stimulation inhibits trigeminal nociception in a rodent model of episodic migraine. Pain rep 2017;2(6):e628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jaim-Etcheverry G, Zieher LM. DSP-4: a novel compound with neurotoxic effects on noradrenergic neurons of adult and developing rats. Brain research 1980;188(2):513–523. [DOI] [PubMed] [Google Scholar]
- [18].Jequier E, Lovenberg W, Sjoerdsma A. Tryptophan hydroxylase inhibition: the mechanism by which p-chlorophenylalanine depletes rat brain serotonin. Molecular pharmacology 1967;3(3):274–278. [PubMed] [Google Scholar]
- [19].Johnson RL, Wilson CG. A review of vagus nerve stimulation as a therapeutic intervention. Journal of Inflammation Research 2018;11:203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kalia M, Sullivan JM. Brainstem projections of sensory and motor components of the vagus nerve in the rat. J Comp Neurol 1982;211(3):248–265. [DOI] [PubMed] [Google Scholar]
- [21].Koe BK, Weissman A. p-Chlorophenylalanine: a specific depletor of brain serotonin. The Journal of pharmacology and experimental therapeutics 1966;154(3):499–516. [PubMed] [Google Scholar]
- [22].Krahl S, Clark K, Smith DC, Browning R. Locus coeruleus lesions suppress the seizure-attenuating effects of vagus nerve stimulation. Epilepsia 1998;39(7):709–714. [DOI] [PubMed] [Google Scholar]
- [23].Lewine JD, Paulson K, Bangera N, Simon BJ. Exploration of the Impact of Brief Noninvasive Vagal Nerve Stimulation on EEG and Event-Related Potentials. Neuromodulation : journal of the International Neuromodulation Society 2018. [DOI] [PubMed] [Google Scholar]
- [24].Manta S, Dong J, Debonnel G, Blier P. Enhancement of the function of rat serotonin and norepinephrine neurons by sustained vagus nerve stimulation. J Psychiatry Neurosci 2009;34(4):272–280. [PMC free article] [PubMed] [Google Scholar]
- [25].Manta S, El Mansari M, Debonnel G, Blier P. Electrophysiological and neurochemical effects of long-term vagus nerve stimulation on the rat monoaminergic systems. The international journal of neuropsychopharmacology 2013;16(2):459–470. [DOI] [PubMed] [Google Scholar]
- [26].Morgan JI, Curran T. Stimulus-transcription coupling in the nervous system: involvement of the inducible proto-oncogenes fos and jun. Annual review of neuroscience 1991;14:421–451. [DOI] [PubMed] [Google Scholar]
- [27].Nonis R, D’Ostilio K, Schoenen J, Magis D. Evidence of activation of vagal afferents by non-invasive vagus nerve stimulation: An electrophysiological study in healthy volunteers. Cephalalgia 2017;37(13):1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Oshinsky ML, Murphy AL, Hekierski H, Cooper M, Simon BJ. Noninvasive vagus nerve stimulation as treatment for trigeminal allodynia. Pain 2014;155(5):1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Perrone MH. Biochemical evidence that L-glutamate is a neurotransmitter of primary vagal afferent nerve fibers. Brain research 1981;230(1–2):283–293. [DOI] [PubMed] [Google Scholar]
- [30].Rashidy-Pour A, Razvani ME. Unilateral reversible inactivations of the nucleus tractus solitarius and amygdala attenuate the effects of bombesin on memory storage. Brain research 1998;814(1–2):127–132. [DOI] [PubMed] [Google Scholar]
- [31].Richter F, Mikulik O, Ebersberger A, Schaible HG. Noradrenergic agonists and antagonists influence migration of cortical spreading depression in rat - A possible mechanism of migraine prophylaxis and prevention of postischemic neuronal damage. J Cereb Blood Flow Metab 2005;25(9):1225–1235. [DOI] [PubMed] [Google Scholar]
- [32].Rogawski MA. Common pathophysiologic mechanisms in migraine and epilepsy. Archives of neurology 2008;65(6):709–714. [DOI] [PubMed] [Google Scholar]
- [33].Roosevelt RW, Smith DC, Clough RW, Jensen RA, Browning RA. Increased extracellular concentrations of norepinephrine in cortex and hippocampus following vagus nerve stimulation in the rat. Brain research 2006;1119(1):124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ross SB, Stenfors C. DSP4, a selective neurotoxin for the locus coeruleus noradrenergic system. A review of its mode of action. Neurotox Res 2015;27(1):15–30. [DOI] [PubMed] [Google Scholar]
- [35].Sadler RM, Purdy R, Rahey S. Vagal Nerve Stimulation Aborts Migraine in Patient with Intractable Epilepsy. Cephalalgia 2002;22(6):482–484. [DOI] [PubMed] [Google Scholar]
- [36].Saengjaroentham C, Supornsilpchai W, Ji-Au W, Srikiatkhachorn A, Maneesri-Le Grand S. Serotonin depletion can enhance the cerebrovascular responses induced by cortical spreading depression via the nitric oxide pathway. Int J Neurosci 2015;125(2):130–139. [DOI] [PubMed] [Google Scholar]
- [37].Seidel JL, Escartin C, Ayata C, Bonvento G, Shuttleworth CW. Multifaceted roles for astrocytes in spreading depolarization: A target for limiting spreading depolarization in acute brain injury? Glia 2016;64(1):5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shibata M, Suzuki N. Exploring the role of microglia in cortical spreading depression in neurological disease. J Cereb Blood Flow Metab 2017;37(4):1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Silberstein SD, Calhoun AH, Lipton RB, Grosberg BM, Cady RK, Dorlas S, Simmons KA, Mullin C, Liebler EJ, Goadsby PJ, Saper JR. Chronic migraine headache prevention with noninvasive vagus nerve stimulation: The EVENT study. Neurology 2016;87(5):529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tassorelli C, Grazzi L, de Tommaso M, Pierangeli G, Martelletti P, Rainero I, Dorlas S, Geppetti P, Ambrosini A, Sarchielli P, Liebler E, Barbanti P, Tassorelli C. Noninvasive vagus nerve stimulation as acute therapy for migraine The randomized PRESTO study. Neurology 2018;91(4):e364–e373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Torrente D, Cabezas R, Avila MF, García-Segura LM, Barreto GE, Guedes RCA. Cortical spreading depression in traumatic brain injuries: Is there a role for astrocytes? Neurosci Lett 2014;565:2–6. [DOI] [PubMed] [Google Scholar]
- [42].Van Bockstaele EJ, Peoples J, Telegan P. Efferent projections of the nucleus of the solitary tract to peri-locus coeruleus dendrites in rat brain: evidence for a monosynaptic pathway. J Comp Neurol 1999;412(3):410–428. [DOI] [PubMed] [Google Scholar]
- [43].Vecchio E, Bassez I, Ricci K, Tassorelli C, Liebler E, de Tommaso M. Effect of Non-invasive Vagus Nerve Stimulation on Resting-State Electroencephalography and Laser-Evoked Potentials in Migraine Patients: Mechanistic Insights. Frontiers in human neuroscience 2018;12:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vila-Pueyo CM, Strother JL, Kefel RM, Goadsby RP, Holland RP. Divergent influences of the locus coeruleus on migraine pathophysiology. Pain 2019;160(2):385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Walker BR, Easton A, Gale K. Regulation of Limbic Motor Seizures by GABA and Glutamate Transmission in Nucleus Tractus Solitarius. Epilepsia 1999;40(8):1051–1057. [DOI] [PubMed] [Google Scholar]
- [46].Wang WZ, Yuan WJ, Pan YX, Bai J, Liao MY, Tang CS. Non-NMDA receptors within caudal ventrolateral medulla are involved in transmission of baroreflex of rats. Acta pharmacologica Sinica 2003;24(8):783–789. [PubMed] [Google Scholar]
- [47].Zarcone D, Corbetta S. Shared mechanisms of epilepsy, migraine and affective disorders. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology 2017;38(Suppl 1):73–76. [DOI] [PubMed] [Google Scholar]
- [48].Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol 2011;69(5):855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.