

Abstract
Therapeutic delivery selectively to lymph nodes has the potential to address a variety of unmet clinical needs. However, owing to the unique structure of the lymphatics and the size-restrictive nature of the lymph node reticular network, delivering cargo to specific cells in the lymph node cortex and paracortex is difficult. Here, we describe a delivery system to overcome lymphatic and intra-lymph node transport barriers by combining nanoparticles that are rapidly conveyed to draining lymph nodes after administration in peripheral tissues with programmable degradable linkers. This platform enables the controlled release of intra-lymph-mobile small molecular cargo, which can reach vastly more immune cells throughout the lymph node than either the particles or free compounds alone. The release rate can be programmed, allowing access to different lymph node structures and thus, specific lymphocyte subpopulations. We are thereby able to alter the subtypes of drugged lymph node cells to improve immunotherapeutic effects.
Introduction
Organised lymphocyte accumulation occurs in lymph nodes (LNs), several hundred of which exist in humans, constituting a significant fraction of the total lymphocyte population of the body. Accordingly, LNs are attractive therapeutic targets for the treatment of a variety of unmet clinical needs, including the elimination of B and T cell malignancies,1 viral reservoirs consisting of latently infected cells,2 and sentinel LN metastases3 as well as improving vaccines4 and promoting immune tolerance.5 LN localisation has been shown to confer augmented efficacy in a variety of therapeutic contexts, including cancer6 and transplantation.7 However, due to the nature and anatomy of LNs,8 high concentrations of therapeutics are difficult to achieve in these tissues through the administration of free drugs or by conventional formulations. In principle, LN-targeted drug delivery vehicles can improve the efficiency of LN delivery allowing for lower overall doses and reduced off-target effects and toxicities.
The anatomical and microstructural organization of the LN is an important contributor to the tight orchestration of intracellular signalling underpinning the adaptive immune response.9–11 Lymphatics are the channel by which antigen presenting cells traffic to local draining lymph nodes (dLNs) to mediate T cell priming within the LN paracortex. Antigen, cytokine, and/or pathogen-laden interstitial fluid are also transported as lymph out of peripheral tissues to LNs by lymphatics,12, 13 the rapid timescale of which facilitates the expedited initiation of B cell priming14 and elicitation of T cell help.12, 13 These phenomena are regulated by the opposing effects of solute hydrodynamic size on peripheral tissue retention versus interstitial transport.8 Accordingly, LN delivery via these lymphatic transport mechanisms is particularly facile using nanoparticles tens of nanometers in hydrodynamic diameter, avoiding both the rapid clearance of small molecules into the circulation and the entrapment of large particles within the tissue extracellular matrix.8
The LN reticular network, on the other hand, restricts access of lymph-borne solutes to the cortical parenchyma. This is important not only in preserving the naïve state of lymphocyte microenvironments,15 but also in protecting from a wide variety of immunogenic molecules that would adversely influence an immune response occurring within the cortex, such as certain soluble agents produced during microbial infections.16 This exclusion barrier is also highly size-dependent: particles as well as solutes 70 kDa and greater are restricted from the LN conduits and cortex by the subcapsular sinus (SCS),13, 14 molecules less than 70 kDa have access to the conduits, and lower molecular weight solutes experience graduated but not absolute exclusion from the extra-conduit spaces.12, 16
While smart delivery in the context of tumour targeting is a longstanding goal with multiple established paradigms for tumour-enhanced delivery and triggered release,17, 18 such principles are less developed for LN targeting.19 For example, akin to the size-dependent enhanced permeability and retention effect for tumour targeting, lymphatic uptake is highly sensitive to molecule and particle hydrodynamic size.20, 21 However, physiological deviations in redox, pH, and enzymatic microenvironments often exploited for tumour-triggered release are not currently known to exist for lymphatic vessels and LNs. Benefits of controlled release after uptake by phagocytic immune cells within lymphatic tissues have been documented but limited in application to linkers responsive to intracellular triggers.22–25 With these factors as well as the potential benefits of direct delivery to LN cortex- and paracortex-resident immune cells in mind, we suggest that a multistage delivery system that facilitates lymphatic uptake as well as intra-LN transport could greatly augment the number and change the “flavor” of drugged lymphocytes.
We describe here a synthetic nanoparticle carrier system for enhanced lymphatic uptake and transport to the LN that can release its payload at different rates, thereby altering access to LN structures and tuning the amount of cargo delivered at the site of interest. This approach begins with thiolated poly(propylene sulfide) (PPS) nanoparticles (NPs)26, 27 that are efficiently taken up by lymphatics and accumulate in dLNs after peripheral administration.28, 29 To this platform we bring thiol-reactive oxanorbornadiene (OND) linkers that fragment by a first-order retro-Diels-Alder mechanism in a pH- and solvent-insensitive manner with programmable half-lives ranging from hours to days.30–32 We demonstrate in vivo that this two-stage system facilitates improved lymphatic delivery and access of small and medium-sized molecules to lymphocyte subpopulations poorly accessed with conventional drug formulations. Large increases were observed in the total number of LN-resident cells receiving the cargo, as well as in the delivered dose to those cells, with timing controlled by the half-life of intralymphatic release. This multistage delivery principle, which we extended to other lymph-accessing nanoformulations, results in augmented therapeutic activity within dLNs to combat LN tumours.
Results
Multistage NP-OND spatiotemporal effects in vitro and in vivo
Living anionic emulsion polymerization of PPS inside Pluronic F-127 micelles (Fig. 1a–c) generates stable polymer NPs with drug loading capacity defined by their synthesis parameters.26, 27, 33 These particles are highly amenable to conjugation of a diverse array of molecules33, 34 due to their amphiphilic nature, creating multiple commutable points for derivatization. The reactivity of thiols remaining after post-polymerization oxidative crosslinking of the PPS NP core was assessed by addition of the OND dansyl (Dn) derivatives 1-Dn through 5-Dn (Extended Data Fig. 1). The resulting Michael addition generates a fluorescent signal35 that makes it easy to monitor this step (Fig. 1d), which occurred at rates (Supplementary Table 1) approximately equivalent to those previously measured with small-molecule and protein-derived thiol nucleophiles.30, 32, 35
Figure 1: Nanoparticle-OND preparation and properties.
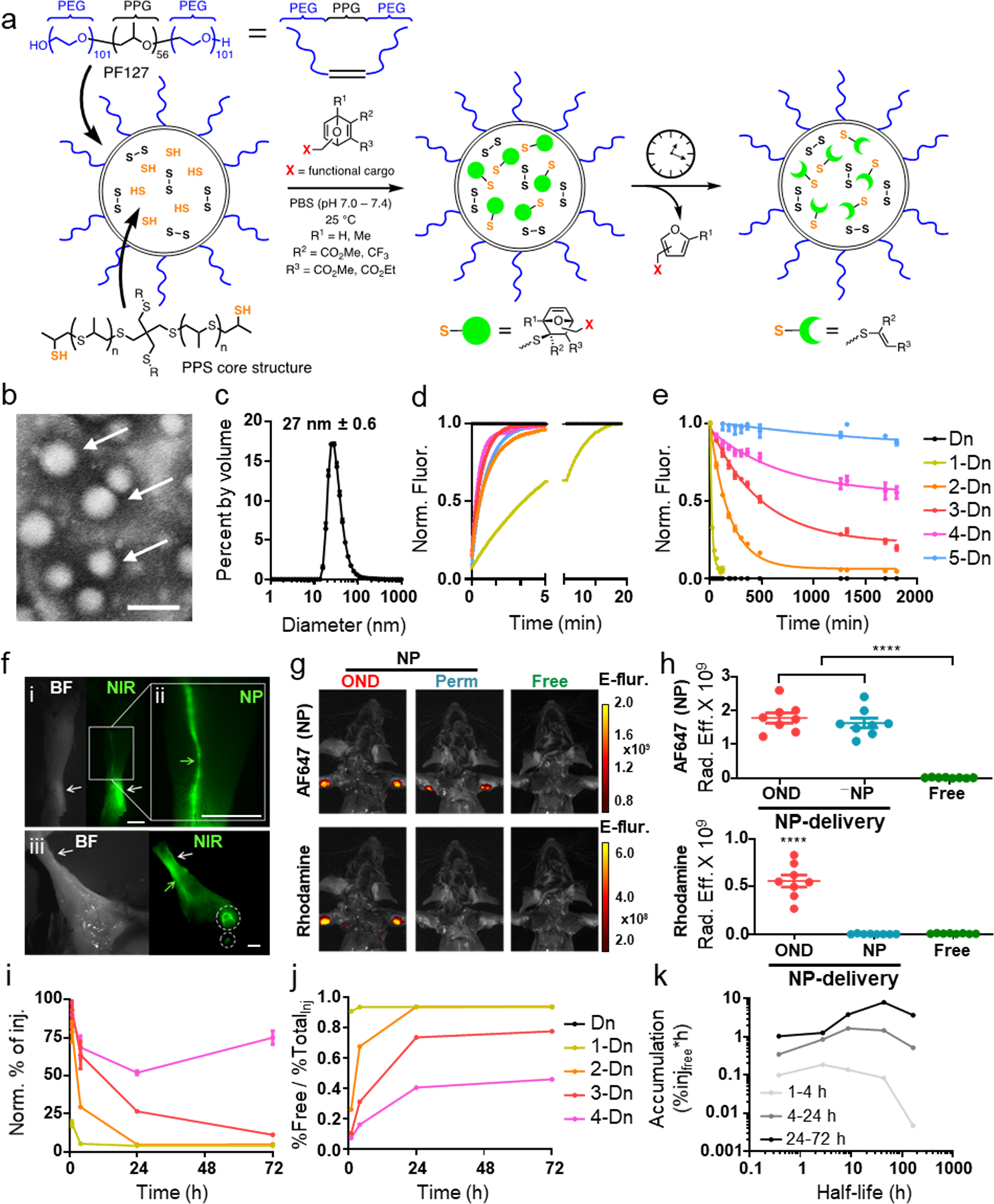
a) Schematic representation of poly(propylene sulfide) (PPS) nanoparticle (NP) preparation, conjugation with OND electrophiles, and retro-Diels-Alder release of furan-tagged cargo. b) Representative TEM image of NPs; scale bar = 100 nm. n=3 independent experiments. c) Overlaid DLS traces of the starting NPs and PDS-NPs (described in Figure 5). n=3 independent experiments. Ave NP size is 27 nm. d) Addition rates of OND derivatives, each bearing a dansyl (Dn) group, to NP represented as normalized fluorescence (Norm. Fluor.). This experiment was repeated twice with similar results. e) Release curves of the Dn-furan fragment from OND linkers 1-5 at 37°C. Linker structures and kinetic parameters of addition and fragmentation are given in Supporting Information. n=6 independent samples per group. Data is shown as normalized fluorescence (Norm. Fluor.). f) Representative bright-field (BF) and near infrared (NIR) images showing NP transport after 10 min from site of i.d. injection (white arrows) via C57Bl6 mouse forearm lymphatic vessel (green arrows) to draining LN (white circles). All images are anterior views of the right distal forelimb with i and ii focusing in on the site of injection at the anterior region of the wrist and iii showing the proximal medial lymphatic drainage to the brachial and axillary LN of same i.d. injection. Scale bar = 0.25 cm. This experiment was repeated twice with similar results. g) Representative IVIS images of mice after i.d. injection of: (OND) NPs labeled with both Alexa647-maleimide (AF647, non-cleavable linker) and 3-Rhod (Suppl. Fig. 1), NPs labeled with only Alexa647, and (Free) Rhod-F (Suppl. Fig. 1). Top row = 720 nm emission channel; bottom row = 580 nm emission channel. Data is in Epi-fluorescence: fluorescent emission normalized to incident excitation intensity (unitless). This experiment was repeated once with similar results. Representative images of n=8 biological replicates. h) Quantification of IVIS images of the type shown in panel g. n=8 biological replicates. i) Dn signal (both NP-bound and released) accumulated in LN vs. time after injection of NP loaded with the indicated OND-Dn reagent normalized to the signal from non-cleavable 5-Dn conjugate. Key is the same as in panels e and j. n=3 biological replicates per group. j) Percent of free Dn cargo delivered to dLN vs. time after injection of the same NPs shown in panel I; “TotalInj” represents total percent of injection as measured by Dn signal. n=3 biological replicates per group. k) AUC (area under the curve, representing total amounts) of OND released cargo for different time periods: 1–4, 4–24, and 24–72 h vs. half-life of rDA cleavage for the indicated OND-Dn derivatives. n=3 biological replicates per group. For all graphs, the columns/points and error bars represent the mean + SEM. Statistics were performed by ordinary one-way ANOVA with Tukey’s multiple comparisons test. ****=p<.0001, ***=p<.005, *=p<.05. ****=p<.0001, ***=p<.005, *=p<.05.
The resulting adducts fragment by retro-Diels-Alder reactions (Fig. 1a), the half-lives of which differ (Fig. 1e) with different OND substituents (Supplementary Table 1).30, 32 We tested variants that reacted with thiols quickly (second-order rate constants between 5 and 60 M−1s−1 at 25 °C) but varied in their first-order fragmentation half-lives from 17 min to 29 h at 37 °C. To explore the potential scope of this mechanism in programming alternative release profiles, we addressed the same thiol-containing NPs with mixtures containing different ratios of three fluorogenic OND reagents. The behaviour of the resulting particles matched that predicted by the linear combination of the individual first-order rate equations (Supplementary Fig. 1), demonstrating significant tunability for the desired application.
NPs of the size range used here (27 ± 0.6 nm diameter) experience rapid lymphatic uptake and selective accumulation in dLNs relative to systemic tissues after injection in the peripheral skin (Supplementary Figs. 2–5).29, 36, 37 We confirmed this for the NPs in this study by irreversibly labelling them with an IRDye800-maleimide that engages the same thiol residues (Supplementary Fig. 6) as OND reagents. Examination of tissue minutes after intradermal (i.d.) administration showed the expected lymphatic trafficking to the dLN (Fig. 1f). Contrariwise, when a small-molecule dye (Rhod-F, Extended Data Fig. 1) alone was administered i.d., it was unable to selectively target the dLN, only reaching this organ when carried by the NP via an OND linkage (Fig. 1g–h). Dansyl fluorescence was found to substantially increase in the dLN over the course of 72 h for OND linkers having longer half-lives (Fig. 1i). As expected, the amount of free Dn as a fraction of the NP-OND conjugate injected was inversely proportional to OND linker half-life (Fig. 1j).
The fact that maximum exposure of the dLN to the freed cargo does not occur quickly after injection for short-lived linkers shows that both the transport and release functions are necessary. The required timescale of NP exposure to freed cargo for most applications will be hours to days, although this may vary by delivered agent, target cell population, and lymphatic drainage basin. Linker half-lives of intermediate duration were thus judged to be more useful (Fig. 1k). We focused on further characterization of linker 3 (retro-Diels-Alder half-life of approximately 10 h) vs. the rapidly cleaved 1 (half-life 17 min) and the slowly cleaved 4 (half-life 29 h). Given lymphatic drainage rates, the roughly 10x difference in rodent versus human lymphatic vessel length38, 39 is unlikely to substantially influence the formulation composition requirements, whereas states of disease, such as lymphedema, may necessitate such modifications.
Enhanced cargo delivery to draining lymph node cortical cells
Partitioning of particles and solutes in the LN is regulated according to size (Fig. 2a),12, 16 inhibiting access of large particulates to lymphocytes, which constitute the vast majority of LN resident cells [Fig. 2a; barrier cells = medullary cord (MCM), medullary sinus (MSM), and SCS macrophages (SSM); paracortex cells = T, B, and dendritic cells (DCs)]. To test the capacity of linker fragmentation to differentially regulate LN spatial distributions of NP and small molecular cargo, we labelled NPs with a small amount of fluorescein via a non-cleavable maleimide linkage (Fluor-Mal, Extended Data Fig. 1 and Supplementary Fig. 6) to the PPS NP core. The resulting NPs were reacted with OND reagents 1-Coum and 3-Rhod (Extended Data Fig. 1), thereby making release of coumarin more rapid than that of rhodamine. These particles were applied to a collagen hydrogel similar in composition to the collagenous structures of LNs as an in vitro model of intra-LN diffusion (Fig. 2b).40 After 12 h at 37°C, the short fragmentation half-life of 1-Coum allowed the coumarin dye to penetrate farther compared to the rhodamine dye (3-Rhod) attached to the longer half-life linker. The NPs migrated only to a very small extent through the collagenous matrix, as expected (Fig. 2b–d). These trends were recapitulated in vivo using NPs fluorescently labelled with a non-cleavable AlexaFluor647 (AF647-Mal, Extended Data Fig. 1) and loaded with cleavable 3-Rhod. One day after i.d. administration into mice, rhodamine was found throughout the dLN, distributed far more deeply and centrally into the tissue than the NPs, which were retained at the periphery (Fig. 2e–f, and Supplementary Fig. 7).
Figure 2. Altered access by NP-OND to draining lymph node-resident cell populations.
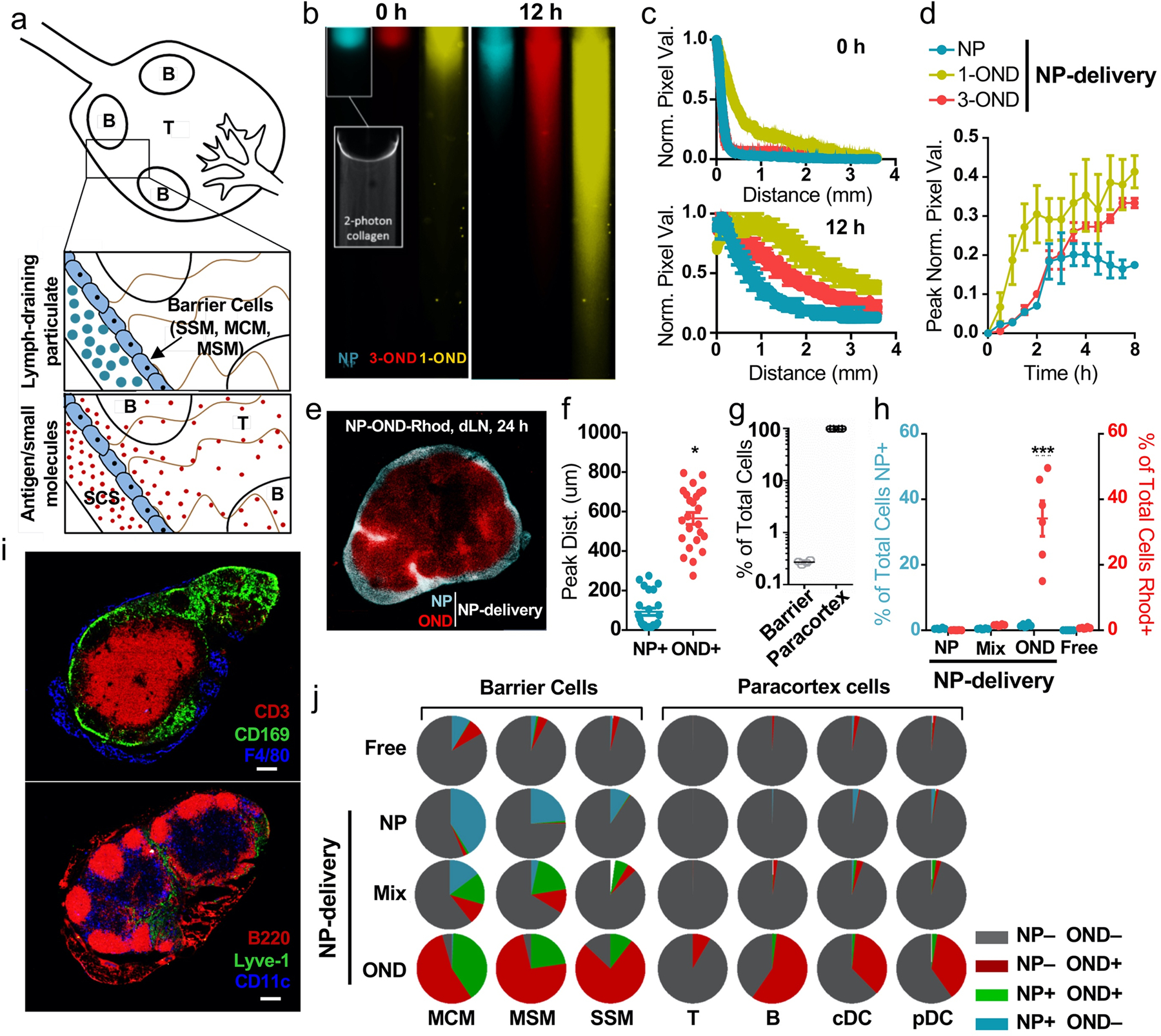
a) Schematic representation of the two-stage delivery mechanism. The cells lining the subcapsular sinus (green) are expected to restrict large particulates (blue) while allowing small molecules (red) into the LN cortex. b) Fluorescence images of collagen hydrogels (7.5 mg collagen/mL) after application of NPs labeled with (NP) Fluorescein-maleimide, (3-OND) 3-Rhod, and (1-OND) 1-Coum. The indicated time represents the period after particle labeling for several minutes followed by application to the top of the gel. Movement through the gel is diffusive, not under flow. Scale bar (white) = 500 μm. Inset = 2-photon image of collagen hydrogel. This experiment was repeated once with similar results. c) Normalized intensity profile across field of view at 0 and 12 h time points for collagen diffusion experiment at 37°C. n=3 samples per group. d) Pixel value (normalized to the 0 h time point) for the half-way point in the field of view at 12 h at 37°C. n=3 samples per group. e) Representative fluorescence image of excised dLN 24 h after i.d. administration of NP labeled with both 3-Rhod and AF647-maleimide. Red = rhodamine OND cargo, blue = false-colored AF647-labeled NP. Scale bar (white) = 500 μm. This experiment was repeated twice with similar results. f) Peak distance for the penetration of (NP+) NP-Alexa and (OND+) dansyl cargo into the LN from the LN boundary, derived from images such as in panel a; n = 24 biological replicates per group. g) Distance of centroid of cell fluorescence from capsule showing deeper lymphocytes (Ly) and more shallow barrier cells (B.C.). h) Percentage of total cells (analyzed by flow cytometry) positive for AF647 (blue) and rhodamine (red) from dLNs 24 h after i.d. administration of: NPs labeled with AF647-maleimide only, (Mix) NPs labeled with AF647-maleimide and mixed with Rhod-F, (OND) NPs labeled with both AF647-maleimide and 3-Rhod, (Free) Rhod-F only. n=6 biological replicates per group. i) Representative images of LN sections stained with markers characteristic of lymphocytes (CD3, B220) vs. barrier cells (CD169, F4/80, Lyve-1) and including CD11c. This experiment was repeated twice with similar results. j) Fraction of total cells positive for OND cargo or NP-AF647 within the dLN. Administration details same as in panel c. n=6 biological replicates per group. For all graphs, the columns/points and error bars represent the mean + SEM. Statistics for f were performed by two-tailed parametric t-test. Statistics for h were performed by two-way ANOVA with Tukey’s multiple comparisons test. ****=p<.0001, ***=p<.005, *=p<.05.
The ability of small-molecular cargo to access the LN interior, where the majority of LN-resident cells reside (Fig. 2g), despite NP restriction at the SCS was verified by flow cytometry measurements of cellular uptake of the released dye. Approximately 30–40% of LN-resident cells were found to be labelled with significant amounts of Rhod-F released from the NP (Fig. 2h). This represents dramatic improvements relative to results observed with free Rhod-F (Free, <1%), fluorophore (AlexaFluor647) tethered permanently to NPs using a non-cleavable linkage (NP, <1%), or free Rhod-F mixed with AlexaFluor647-labelled NPs (Mix, <1%) (Fig. 2h). Analysis of component cell types distributed throughout LNs in prescribed locations (Fig. 2i) confirmed the expected location-dependent uptake. Administration of free Rhod-F gave very poor access to all LN cells except the barrier MCM, approximately 10% of which showed detectable levels of dye (Fig. 2j). These and the other two barrier cell types examined (CD169+ SSM and MSM) were moderately targeted by NP alone. Co-administration of non-conjugated dye and NPs did somewhat better in accessing barrier cells (up to approximately one-third of MCM and MSM cells taking up Rhod-F, NP, or both; Fig. 2j). However, very little to none of the cortex and paracortex-positioned cells [T and B cells and both conventional (cDCs) and plasmacytoid DCs (pDCs)] were found to be addressed by these agents (Fig. 2j). Only when delivered into lymph via the NP and released from the OND covalent linkage was the small-molecule dye able to access these cells (Fig. 2j). As expected, a significantly greater fraction of the barrier cells was also labelled with free Rhod-F released from NP-Rhod-OND (Fig. 2j).
Overall, the number of targeted cells was improved by orders of magnitude by the two-stage NP-based method: T cells, from hundreds to tens of thousands; B cells, from several thousand to the hundreds of thousands; DCs, from hundreds to thousands (Fig. 3a–b). These improvements correspond to 10, 60, and 50% of respective cell subtypes resident within targeted LNs (Fig. 3c). Thus, LN macrophage targeting is size-independent whereas T-, B- and dendritic cell lymphocytes are best reached with small molecules that can diffuse through the tissue, in our case made vastly more efficient by being “dropped off” within lymph by the carrying NP, presumably prior to uptake by LN barrier cells.
Figure 3. Increased cargo delivered by NP-OND to lymph node-resident cells.
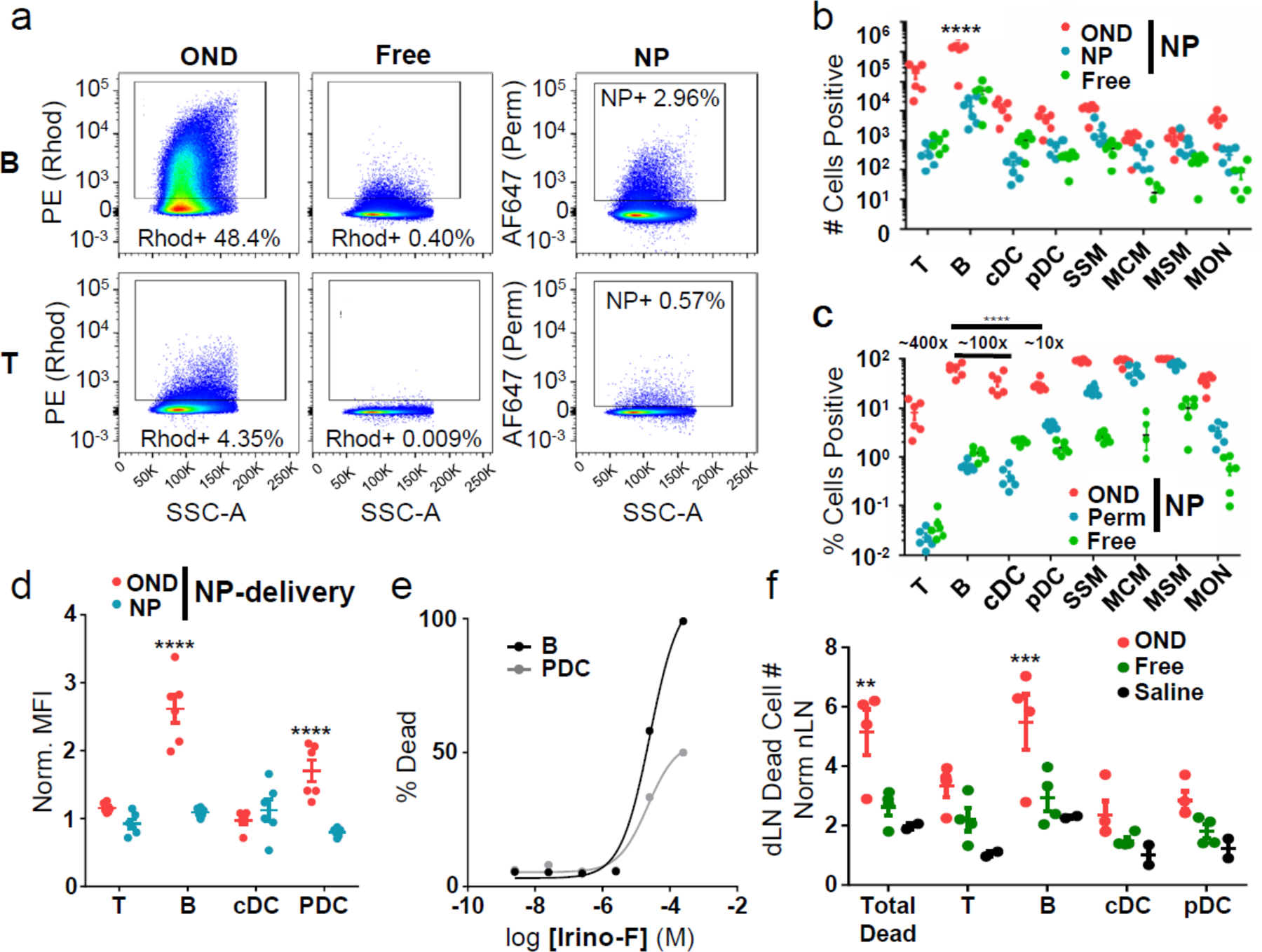
a) Representative flow cytometry scatter plots for T and B cells isolated from the dLN 24 h after i.d. administration of: (OND) NPs labeled with both AF647-maleimide and 3-Rhod, (Free) Rhod-F only, (NP) NPs labeled with AF647-maleimide only. This experiment was repeated twice with similar results. b) Number of dLN cells of various types positive for NP-delivered (OND, red) versus free (Free, green) rhodamine and AF647 (NP, blue) measured by flow cytometry after NP-OND and free rhodamine treatment defined as in panel a. n=6 biological replicates per group. c) Data from panel b, represented as percentages of each cell type. n=6 biological replicates per group. d) Normalized mean fluorescence intensity (MFI) of rhodamine (red) and AF647 (blue) in the indicated dLN immune cells after NP-OND or free rhodamine treatment defined as in panel a, normalized to free rhodamine or signal from plain (non-OND derivatized) NP. n=6 biological replicates per group. e) Toxicity of irinotecan-furan derivative Irino-F toward cultured B cells and pDCs after 24 h exposure in media at 37°C. This experiment was repeated once with similar results. f) Number of dead cells in the dLN normalized to the nLN 24 h after i.d. administration of: (OND) NPs labeled with 3-Irino (4 mg/kg), (Free) Irino-F (4 mg/kg), (Saline) negative control. n=3 biological replicates per group. For all graphs, the columns/points and error bars represent the mean + SEM. Statistics were performed by two-way ANOVA with Tukey’s multiple comparisons test. ****=p<.0001, ***=p<.005, *=p<.05.
In addition to increasing the frequency of targeted cells, release from the NPs resulted in an increase in total cargo delivery per cell to B cells and, to a lesser extent, pDCs, but not to T cells or cDCs (Fig. 3d). These results can be attributed to the asymmetric LN distributions of these cells resulting in differential accessibility to afferent lymph-borne species; molecular percolation proximal to the SCS and confinement within the conduits results in preferential access to lymphatic-sinus lining DCs and B cells compared to cortical DCs and T cells, especially at low concentrations.12, 41, 42
The advantage of two-stage delivery was found with a different dye and NP loading method. Cy5.5-COOH was encapsulated noncovalently in PPS NPs via solvent dispersion, and the NP was separately labeled with AF488 via a maleimide connector. When incubated in buffer, the Cy5.5 dye was released in a first-order process (~11 h half-life, Extended Data Fig. 2a). After i.d. delivery, the NP-bound and diffusible dye exhibited much different profiles of cellular association (Extended Data Fig. 2b). Specifically, the passively encapsulated and released Cy5.5 associated primarily with LN lymphocytes and DCs whereas the NP instead associated at higher levels with barrier cells (Extended Data Fig. 2b–c). Cellular association with Cy5.5 also decayed >4 h post administration (Extended Data Fig. 2c) whereas NP signal for lymphocytes continued to increase, as would be expected given the vastly slower diffusion time of NPs. The success of simple hydrophobic sequestration and release to reach LN cortex/paracortex-resident cell subpopulations shows the generality of the two-stage approach.
OND-based enhancement of LN delivery was also extended to a virus-like particle of similar diameter as the PPS NP.43, 44 In this case, “permanent” and cleavable linkages were made to different dyes via amide and OND-thioether bonds, respectively (Extended Data Fig. 3a–b). After administration, the virus-like particles were found to accumulate at lower total levels compared to PPS NP (Extended Data Fig. 3c–e), but time-dependent control over cargo release and uptake via retro-Diels-Alder cleavage was again observed (Extended Data Fig. 3f–g).
The delivery of a functional cargo by an NP-OND carrier was demonstrated with a cytotoxic molecule. An OND-irinotecan derivative (3-Irino) and the corresponding furan derivative (Irino-F) (Extended Data Fig. 1) were prepared and shown to be moderately toxic to splenocytes, approximately the same as irinotecan itself (Extended Data Fig. 4a–b). When administered to ex vivo cultured primary B cells and pDCs, the former were found to be somewhat more sensitive to this molecule (Fig. 3e). Irino-F was attached to NPs via the OND derivative 3-Irino and delivered by i.d. injection. Selective increases in B cell death within the dLN (but not the contralateral LN, nLN) were observed 24 h post administration; effects far exceeding those of free Irino-F alone administered i.d. (Fig. 3f) or i.p. (Extended Data Fig. 4c).
Finally, a variation on OND-based attachment and release from PPS NP was engineered by chemical modification of the Pluronic corona (Fig. 4a, Supplementary Fig. 8).33 The terminal carboxylate groups of the Pluronic polymer were functionalized to display pyridyl disulfide groups, followed by reduction to unmask reactive thiols on the NP exterior. After conjugation with 3-Rhod or 4-Rhod (Extended Data Fig. 1), the rates of release of furan-derivatised molecules were assessed by dialysis (Fig. 4b). Again, the retro-Diels-Alder fragmentation of the OND-Rhod adducts were found to vary based on chemical composition, with half-lives of ~8.1 h and ~22 h for 3-Rhod and 4-Rhod, respectively. As with particles bearing internally attached cargo, uptake by dLN-resident cells after i.d. administration was controlled by linker cleavage rate, peaking at approximately twice the linker fragmentation half-life in each case (Fig. 4c). The extent of delivery also increased over time for cells farther from the LN SCS (e.g. T cells in contrast to B cells, Fig. 4d).
Figure 4. Temporal effects of multistage NP-OND delivery.
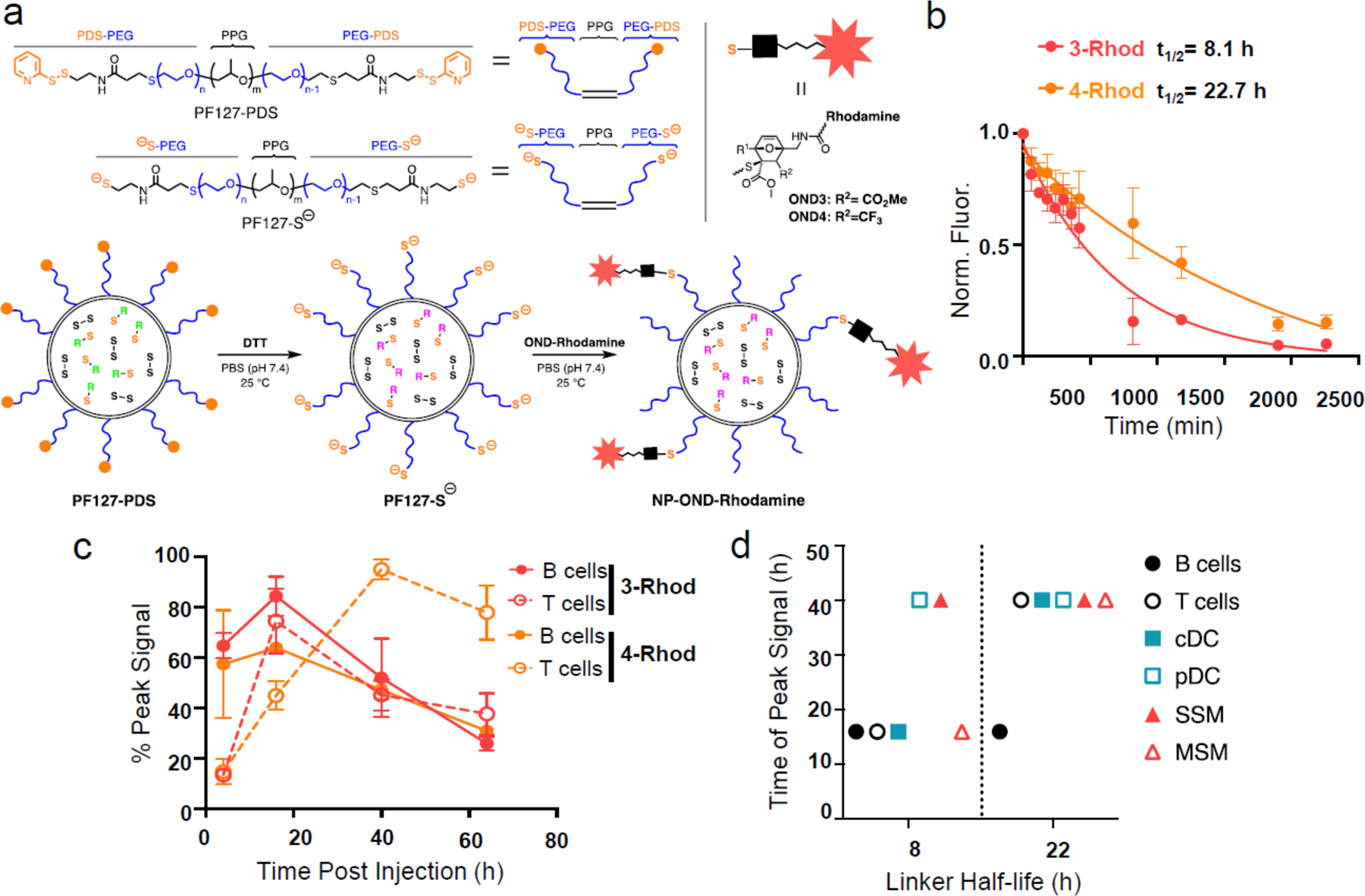
a) Schematic representation of the preparation of NPs carrying Rhodamine via OND linkers 3 and 4. b) Release curves of the Rhodamine-furan fragment from OND linkers 3 and 4 at 37°C. n=3 per group. c) Half-life of Rhodamine- OND 3 and 4 at 37°C. n=3 samples per group. This experiment was repeated once with similar results. d) Peak uptake of Rhodamine-furan cargo released from OND linkers 3 (red) and 4 (orange) in the indicated dLN immune cells after NP-OND treatment defined as in panel a. n=5 biological samples per group. For all graphs, the columns/points and error bars represent the mean + SEM.
To explore the use of this chemistry to augment the bioactivity of immunotherapeutic agents, we employed a thiol-terminated CpG oligonucleotide as a Toll-like receptor 9 (TLR9) ligand. This molecule (denoted “OND-CpG”, Extended Data Fig. 1) was coupled to a PPS NP displaying OND electrophiles on its exterior. The subsequent retro-Diels-Alder reaction of the adduct, assessed by size exclusion chromatography, was found to proceed at a similar rate (half-life ≈ 24 h) to that of the analogous small-molecule OND-rhodamine analogue (Supplementary Fig. 9). NP-OND-CpG was administered i.d. and the effects in dLNs and nLNs were assessed 24 h later, paying particular attention to CD40+ and CD86+ cortex-resident B cells, cDCs, and paracortex-resident pDCs, all TLR9-expressing LN-resident cells. Increases of at least three-fold were observed in the total number of these cells as well as T cells (Fig. 5a) and in each subtype (Fig. 5b–d) only in response to administration of the NP-OND-CpG but not free CpG, and only in dLN but not nLNs (Supplementary Fig. 10). An analogous CpG-decorated NP was prepared in which the TLR9 agonist was attached to the NP corona by a disulfide linkage (SS), which requires cellular internalization of the NP to trigger release by intracellular glutathione.33 This agent elicited no additional activation or proliferation compared to vehicle- and CpG-only controls (Fig. 5a–d), and levels of CpG in systemic tissues were found to be equivalent between conjugation methods (Supplementary Fig. 11). Thus, both NP trafficking to the LN and timed release in the extracellular intralymphatic space were required for activity at the low dose tested (0.05 mg/kg).45
Figure 5. Augmented immunotherapeutic effects of adjuvant delivery with NP-OND system.
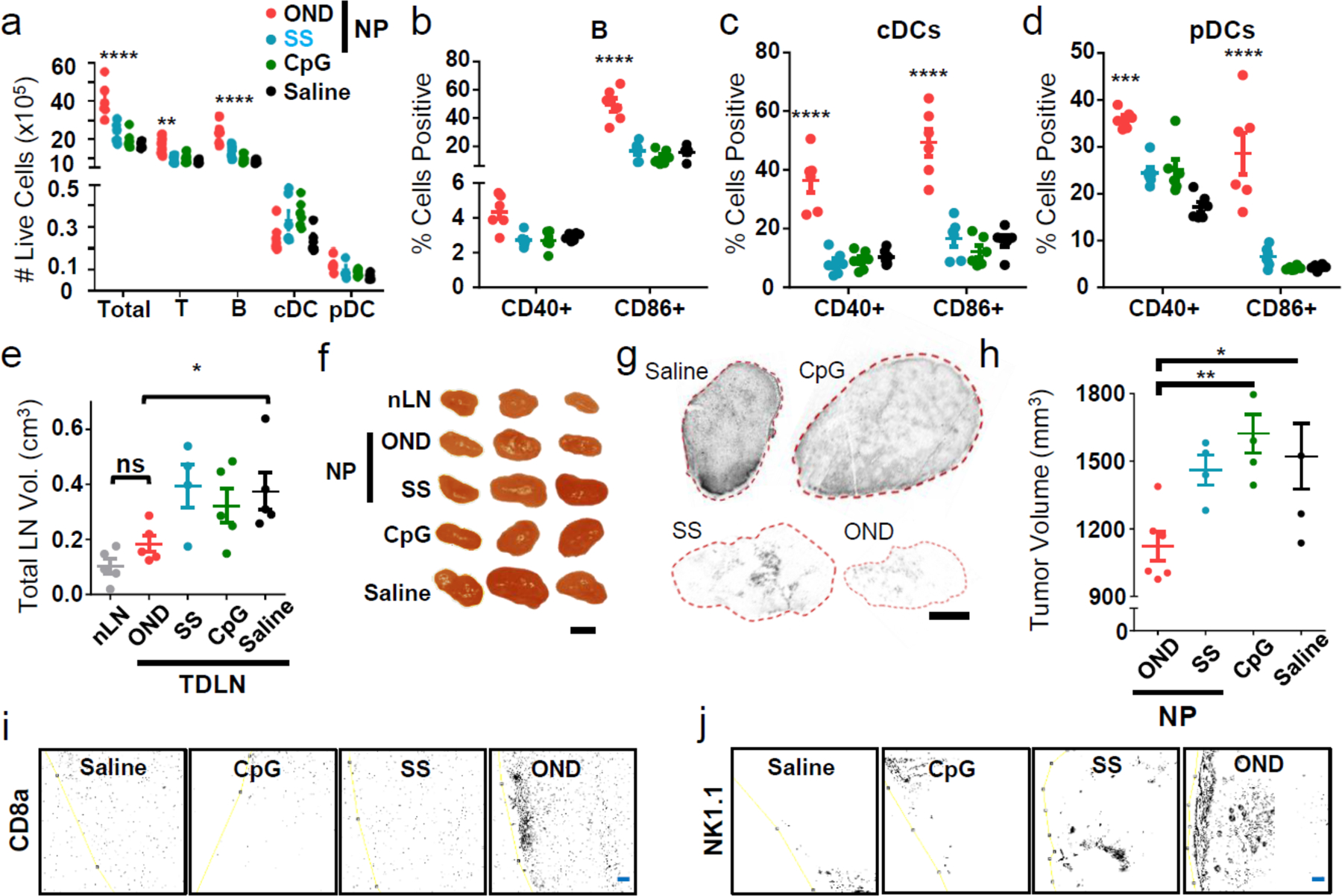
a) Total numbers of dLN cells of various types 24 h after i.d. treatment with (OND) NP-OND-CpG, (SS) CpG disulfide bonded to NP, (CpG) free CpG oligonucleotide, and saline as negative control. n=6 biological replicates per group. b-d) Maturation of (b) B cells, (c) cDCs, and (d) pDCs from dLNs 24 h after i.d. treatment with NP-OND-CpG, determined by flow cytometry analysis for the indicated markers. n=6 biological replicates per group. e-j) Results at day 12 after tumor implantation, following treatment for five days (starting at day 4) with: (OND) NP-OND-CpG, (SS) NP-SS-CpG, (CpG) free CpG oligonucleotide, or saline. n=5 biological replicates per group. e) EL4 LN tumor sizes. f) Representative LN images. Black bar = 0.5 cm. This experiment was repeated once with similar results. g) Representative TCRVβ12 staining of sectioned LN tumors. Black bar = 1.0 mm. This experiment was repeated once with similar results. h) Primary tumor size d12 post treatment. Immunohistochemistry staining of primary EL4 tumors for CD8a (i) and NK1.1 (j) cells. n=5 biologic replicates per group. Yellow line is the tumor border. Blue bar = 100 um. For all graphs, the columns/points and error bars represent the mean + SEM. Statistics for a-d were performed by two-way ANOVA with Tukey’s multiple comparisons test. Statistics for e and h were performed by ordinary one-way ANOVA with Tukey’s multiple comparisons test. ****=p<.0001, ***=p<.005, *=p<.05.
For an initial functional test, we chose the EL4 model of murine lymphoma, which exhibits strong immunosuppressive features46 and reliably induces secondary tumours in tumour dLNs. This progression results in increased LN size (Supplementary Fig. 12) and can be followed by antibody staining for flow cytometry (Supplementary Fig. 13) and immunohistochemistry (Supplementary Fig. 14). I.d. administration of NP-OND-CpG carrying a low CpG dose (0.05 mg/kg)45 upstream of the diseased LNs resulted in substantially reduced dLN size relative to administration of either the disulfide-linked NP-SS-CpG or CpG in its free form (Fig. 5e–f). Indeed, the NP-OND-CpG treated diseased LNs were no larger than the non-diseased nLN in the same animals (Fig. 5e–f), and a nearly complete loss of LN tumour burden was observed by immunohistochemistry (Fig. 5g, Supplementary Fig. 15). These results suggest a substantial elimination and further prevention of lymphomas in this model. This treatment also resulted in a reduction of the size of the primary tumour (Fig. 5h, Supplementary Fig. 16). Since EL4 lymphoma cells lack TLR9 expression,47, 48 we suggest that this therapeutic effect results from immune stimulation augmented by LN cortex- and paracortex-accessible CpG. Within the tumour dLN that is bathed in tumour antigen by virtue of both lymphatic transport29 and resident lymphoma cells, CpG delivery has previously been shown to induce anti-tumour adaptive immunity.29, 46 Such an effect is evidenced here by the observed abscopal effect in the primary tumour (Fig. 5h) and by corresponding increases in CD8 and NK cell infiltration into primary tumours (Fig. 5i–j). Additionally, TLR-stimulated pDCs can elicit direct tumour cytotoxicity49, a mechanism that can explain the elimination of LN tumours with NP-OND-CpG treatment that we are exploring in future work.
Conclusions
While not protected to the same extent, LNs present some of the same types of problems for drug delivery as the brain. Possessed of no intrinsic ability to concentrate systemically administered molecules and protected by a barrier layer of cells that block access of particle agents, the interior of the LN is difficult to address. We show here the development of a two-stage approach inspired by the way particulate antigen is processed by the immune system. First, antigen particles are transported by the lymphatics with great efficiency to dLNs without significant systemic exposure. Second, OND-thiol chemistry is used to attach small-molecule cargo to these particles, to be released in a programmable manner for passive diffusion after arrival within lymph. With this system we achieved orders-of-magnitude improvements in total delivery to cells resident within dLNs, dramatically increasing delivery to normally inaccessible lymphocyte subpopulations. Furthermore, the timing of that delivery could be controlled by the use of linkers with different fragmentation half-lives.
LN resident cells are extremely sensitive to antigen dose based on their differential locations within the organ, with SCS-lining cells receiving greater exposure than deeper cortical cells. Our two-stage system represents an unprecedented way to adjust for this physiological structure in order to reach currently difficult-to-access DCs, T cells, and cancer cells at desired times post administration for improved therapeutic outcomes. Such an outcome was demonstrated here in preliminary fashion in the amelioration of LN tumour burden. The concept of multistage delivery, both specifically and broadly, resembles strategies that have been long explored in the context of drug targeting to tumours, but heretofore have been underutilized in immunoengineering and immunotherapy.
Methods
Synthesis of NP-OND
NPs were synthesized as previously described.27, 28, 33 Briefly, 500 mg of Pluronic F127 (Sigma-Aldrich, St. Louis, MO, USA, P2443) was added to 10 mL of degassed Milli-Q water, allowed to dissolve for 30 min with stirring, and was again degassed. To this solution, 400 μL (5.0 mmol.) of propylene sulfide (Sigma-Aldrich, P53209) was added under argon and stirred for 30 min. S,S’-(2,2-bis((acetylthio)methyl)propane-1,3-diyl) diethanethioate (28.8 mg, 0.04 mmol.) was treated with sodium methoxide (322 μL of 25 wt% solution in methanol, Sigma-Aldrich, 156256) and the mixture was added under Ar. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) 0.4 mmol, Sigma-Aldrich, 139009) was added under Ar to the solution 15 min later and the entire reaction was stirred for 24 h. The mixture was then exposed to air for 2 h with stirring, and was subsequently dialyzed for 3 d against 4×5 L of Milli-Q water using 100 kDa molecular weight cut off cellulose membrane dialysis tubing (Spectrum Lab., Rancho Dominguez, CA, USA, 131414). NP size was measured by dynamic light scattering using a Zetasizer Nano ZS (Malvern Panalytical, Malvern, UK). Ellman’s assay (Thermo Fisher Scientific, Rockford, IL, USA, 22582) was used to determine the concentration of NP free thiols. NPs were rendered into NP-OND conjugates by reacting OND (typically 5 × 10−4 mmol) with excess NP thiols (typically 1 mL containing approximately 0.02 mmol of reactive thiol) in buffer at pH 7.4 for 1 h. For OND-Dn variants, fluorescence was only present after conjugation and no further purification was required. For OND variants bearing rhodamine and coumarin cargos, excess or biproduct small molecules were separated via PD-10 column chromatography (GE Healthcare, Pittsburg, PA, USA , 17–0851-01) followed by concentration with 10,000 Da spin filters (Millipore, Bedford, MA, USA, 42407) to achieve pure NP-OND adducts at the starting NP concentration. Alternatively, PDS-NP were synthesized as previously described33. PDS-NP were thiol-activated by adding tris(2-carboxyethyl)phosphine (TCEP), 10 mM, for 20 min at RT. NP were then cleaned of TCEP and PDS on three consecutive 5 mL 7kDa MWCO Zeba columns at 1000 x g for 2 min in 1x −/− PBS. OND-Rhodamine, maleimide-Rhodamine, or OND-CpG were added to excess (activated) NP (30 mg/mL) and reacted for 1 h at RT. Upon completion, reactions were purified by PD-10 size exclusion chromatography in 1x −/− PBS and concentrated to original volume using 30 kDa MWCO Amicon Ultra 4 centrifugal filters at 4000 x g for 30 min. Because retro-Diels-Alder fragmentation reaction is initiated by NP thiol addition to the OND electrophile, all biological experiments were either initiated immediately after this rapid reaction or the NPs were stored briefly (<2 h) at 4°C, under which conditions the retro-Diels-Alder reaction is greatly slowed.31
In vitro collagen hydrogel penetration
Collagen hydrogels at 7.5 mg/mL at physiological pH were prepared as reported elsewhere.17, 40 Briefly, bovine collagen (144 μL of 8.63 mg/mL stock) was mixed with NaOH (1 M, 3.7 μL) and EDTA (19.2 μL of 0.17 M solution). The resulting collagen solution was drawn into 100 μm capillary tubes (World Precision Instruments, Inc., Sarasota, FL, USA, 1B100–3) and then allowed to gel for 24 h at 37 °C. Fluorescein-labelled NPs were conjugated to two OND derivatives with different dye cargo (rhodamine and coumarin), purified and concentrated as described above, and then added to the collagen gel using a 31-gauge syringe. Fluorescence images were taken over the course of 12 h at 37 °C using a Zeiss Axio Observer XLmulti S1 to measure the penetration of OND cargo relative to NPs. Fluorescein-labelled NPs were measured on the GFP channel, 3-Rhod was measured on the RHOD channel, and 1-Coum was measured on the DAPI channel. 16-bit grey scale intensity profiles were drawn from the loaded NP-OND solution through the length of the gel.
In vivo experiments
All animal studies were approved by and adhered to guidelines set forth by the Georgia Institute of Technology Institutional Animal Care and Use Committee. All mice were purchased from Charles River Laboratory (Cambridge, MA) as C57BL6/J 6 week old females. Mice were euthanised by CO2 asphyxiation. NP conjugations were dose-matched based on relative fluorescence prior to injection.
Flow cytometry
Excised LNs were placed in 900 μL +/+ 1x PBS on ice; axillary and brachial LNs are pooled for most experiments, unless otherwise indicated. The cells were extracted from the LN as previously reported.28 Briefly, 100 μL of 10 mg/mL Collagenase D was added and the LN were incubated for 1 h at 37°C. The LN were then mechanically disrupted on a 70 um cell strainer and cells were plated at a concentration of 10M/1mL. In all experiments cells were blocked with 2.4G2 (1:200) followed by Live Dead staining with Zombie Aqua (1:100). Marker staining and fluorophore selection will be described in each experiment. For all experiments samples were run on a BD Fortessa flow cytometer. Cells were gated as follows: B cells – B220+CD3-CD11b-; T cells – CD3+B220-; conventional dendritic cells (CDCs) – CD11c+B220-; plasmacytoid dendritic cells (PDCs) – CD11c+B220+; subcapsular sinus macrophages (SSM) – B220-CD3-CD11b+CD11c+CD169+F4/80-; medullary sinus macrophages (MSM) – B220-CD3-CD11b+CD11c+CD169+F4/80+; and medullary cord macrophages (MCM) – B220-CD3-CD11b+CD11c+CD169-F4/80+; and monocytes (MON) – CD11b+F4/80+.
IVIS imaging
Fluorescent imaging was performed with an IVIS® Spectrum instrument (Perkin Elmer) according to previously published protocols.28 Animals were injected i.d. in the forearm with a solution of AlexaFluor647-NP or of OND-NP (3-Rhod) either with or without AlexaFluor647 labelling. The animals were sacrificed 24 h later. To assess AlexaFluor647-NP drainage to LN, animals were imaged at 720 nm with 640 nm excitation, exposure time of 0.1 s. To assess 3-Rhod drainage to the LN, animals were imaged using a modified Rhodamine Red-X channel (Ex: 540 nm, Em: 580 nm) at an exposure time of 0.1 s. Total fluorescent counts and radiant efficiency (p/s·sr·μW) were evaluated for each experiment and treatment group using elliptical regions of interest in Living Image Software (Perkin Elmer, Waltham, MA, USA).
Confocal microscopy of whole LNs
AlexaFluor647-labelled NP were conjugated to 3-Rhod, purified and concentrated, and then 30 μL of the resulting solution, containing 1.8 mg of NPs, was injected i.d. in both forearms of three 6 wk old female C57BL6/J mice. After 24 h the mice were sacrificed and the axillary and brachial LN excised, cleaned under stereoscopic observation, and plated on a glass-bottom 96-well plate for confocal imaging (Zeiss NLO 710). Using a 10x objective lens, 16-bit fluorescence imaging was taken for AlexaFluor647 and rhodamine. Z-stacks approximately 6.4 μm in depth were taken through the depth of the LN (approx. 160 μm). In order to acquire the entire LN, convex hull tiling was performed and the resulting images were stitched together.
LN cellular distribution experiments
C57BL6 mice were injected into both forearms id. with 30 μL OND-labelled (3-Rhod) AF647-NPs. The mice were sacrificed after 24 h and the LNs excised and stained as follows: CD11c – BV421 (1:80), B220 – BV650 (1:40), CD3 – BV711 (1:40), F4/80 – BV785 (1:40), CD169 – FITC (1:100), CD11b – AF700 (1:200). Rhodamine and NP were measured on the PE and APC channels, respectively. Cells were gated as described above.
LN size and immunofluorescence quantification
Pictures of LNs were taken using either IVIS or a Celestron 44360 digital microscope (Celestron, Torrance, CA). Images were processed using ImageJ. Briefly, the picture scale was set using a known length. The images were converted to 8-bit and then thresholded. LN areas were taken using the Analyze Particles macro. LN volumes were estimated by assuming an ellipsoidal shape, taking the LN height to be equal to the smallest ellipse dimension.
Immunohistochemistry
LNs and tumours were frozen in optimum cutting temperature (OCT) compound (Sakura Finetek USA, Inc., Torrance, CA, USA) in 2-methylbutane (Sigma-Aldrich) chilled by liquid nitrogen and frozen tissue blocks were immediately stored at −20°C. OCT tissue blocks were sliced using a Cryostar NX70 Cryostat (Thermo Fisher Scientific, Rockford, IL, USA) at −20°C to a thickness of 5–12 um. Sections were directly mounted on a glass slide and stored at −20°C before staining. Tissue sections were left unfixed or fixed with pre-chilled acetone for 10 min at 4°C and subjected to standard immunofluorescence protocols using the following antibodies, which were obtained from Thermo Fisher Inc unless otherwise specified: rabbit anti-mouse Lyve-1 (1:250), Alexa Fluor 633 goat anti-rabbit (1:300), Armenian hamster anti-mouse CD3e (1:50), Alexa Fluor 647 goat anti-hamster (1:300, Abcam plc., Cambridge, MA, USA), Alexa Fluor 488 conjugated rat anti-mouse CD169 (1:100, BioLegend Inc, San Diego, CA, USA), biotinylated rat anti-mouse F4/80 (1:200, Life Technologies), streptavidin-Alexa Fluor 555 (1:400, Life Technologies), biotinylated rat anti-mouse B220 (1:250), and Alexa Fluor 488 conjugated Armenian hamster anti-mouse CD11c (1:50, BioLegend Inc), mouse anti-mouse TCRvβ12 (Abcam, ab171114, 1:50, Cambridge, MA), AlexaFluor 647 goat anti-mouse IgG2b (1:300), AlexaFluor 488 rabbit polyclonal anti-GFP tag antibody 1:300), rat anti-mouse CD8a (1:50), AlexaFluor 647 goat anti-rat (1:300), mouse anti-mouse NK1.1 (1:50), and AlexaFluor 488 goat-anti-mouse IgG2a (1:300). Blocking and antibody dilutions were performed with 10% donkey serum (Sigma-Aldrich) with or without 3.6% goat anti-mouse IgG (Abcam, ab6668, Cambridge, MA) in D-PBS. Slides were washed with 0.1% Tween 20 (Sigma-Aldrich) in D-PBS, counter stained with DAPI (VWR International Inc), and mounted with VECTASHIELD® mounting media (Vector Laboratories, H-1000, Burlingame, CA). Samples were imaged using a 710 NLO confocal microscope (Carl Zeiss Microscopy Ltd, Jena, Germany) with a 20x magnification objective and a Zeiss LSM 780 confocal microscope system with a 10x magnification objective (Carl Zeiss Microscopy Ltd, Jena, Germany).
In vivo toxicity in draining LNs
C57BL6 mice were injected i.d. in the forearm with NP-OND-Irinotecan (4mg/kg eq.), furan-irinotecan (4mg/kg eq.), or saline on one side of the mouse in order to assess systemic effects of the treatment. Injections i.p. of log order doses of furan-irinotecan (100mg/kg, 10mg/kg, and 1mg/kg) in 5% DMSO/PBS were also included to reference clinically relevant dosing and delivery. After 24 h, the mice were sacrificed and dLN and nLN taken for flow cytometry analysis as previously described. Cell staining was performed using the following markers: CD11c – BV421 (1:80), CD169 – FITC (1:100), B220 – PE (1:80), CD3 – PerCp-Cy5.5 (1:40), and CD11b – AF700 (1:200).
In vivo CpG function experiments
C57BL6 mice were injected into both forearms i.d. with 30 μL of CpG in either OND, disulfide-linked (SS), or free form (0.4–0.8 μg CpG) under isoflurane anaesthesia. After 24 h, the mice were sacrificed and the LNs excised and stained as follows: B220 – BV650 (1:40), CD40 – FITC (1:100), CD45 – PerCP (1:100), CD86 – PE (1:100), MHCII – APC (1:200), CD11b – AF700 (1:80), CD11c – APCeFluor 780 (1:80), and then fixed in 2% PFA. Maturation was gated according to the following markers: MHCII vs. CD40 or CD86. The plots were divided into six divisions: MHCII HI-CD40+, MHCII HI – CD40-, MHCII MID - CD40+, MHCII MID CD40-, MHCII LOW CD40+, and MHCII LOW CD40. The same divisions were created for CD86. Reported markers were for MHCII MID CD40+/CD86+.
LN EL4 lymphoma model
EL4 cells expressing green fluorescent protein (EL4-GFP) were a generous gift from Tyler Curiel and were authenticated using flow cytometry by staining for TCRvβ12, GFP, and CD3. C57BL6 mice were implanted i.d. in the left dorsal flank with 250,000 EL4-GFP cells. The progression of the tumour was monitored daily using IVIS imaging and with calipers recording the tumour volume measurements. On day 12, as was done in the therapeutic experiment, the mice were sacrificed and the tumours and LNs removed. dLN as well as nLN on the i.l. side were also taken along with their c.l. counterparts and IVIS imaging was performed on the tissues (Ex: 500 nm, Em: 540 nm, exposure time: 1 s). Mice without tumours were also sacrificed and their tissues taken as controls. Total fluorescent counts and radiant efficiency (p/s·sr·μW) were evaluated for each tissue and treatment group using elliptical regions of interest in Living Image Software (Perkin Elmer). Some of the LN tissues and tumours were processed for flow cytometry as previously described. Cell staining was performed using the following markers: CD3 – BV711 (1:80), and TCRVβ12 – previously published marker found extensively on EL4 cells and in low concentration on C57 lymphocytes50– PE (1:200), CD8 – APC (1:100), CD4 – APC/Cy7 (1:40), and then fixing in 2% PFA. Samples were run on a BD Fortessa flow cytometer. Cells were gated (Supplementary Figure 6) according to the following markers: C57BL6 T cells: FSCLOWSSCLOWCD3+GFP- and EL4-GFP: FSCHISSCHICD3+GFP+TCRVβ12+.
In vivo tumour immunotherapy
Tumour treatment experiments were conducted by implanting 250K EL4 lymphoma cells i.d. These cells were authenticated using flowcytometry staining for TCRvβ12 and CD3. LN tumours were allowed to form until palpable, ~4d, when mice were subsequently treated daily for 5 d i.d. in the upstream forearm lymphatic drainage basin with 0.75 μg of CpG in either OND, SS, or free form. LN sizes were recorded at endpoint and assessed grossly with photographs. Primary tumour sizes were recorded and the tumour volume was modeled as an ellipsoid.
Statistical analysis
Statistical Analysis: All data is expressed as mean ± standard error. Statistical tests were performed in Prism. ****p<0.0001, ***p<0.001, **p<0.01 and *p < 0.05 by unpaired two-tailed t-tests or one- or two-way ANOVA followed by Tukey post-hoc test for multiple comparisons.
Data Availability
The data that support the plots within this paper and other findings of this study are available from the corresponding authors upon reasonable request.
Code Availability
The custom code used in the analysis of LN IHC image’s fluorescence distribution is available from the corresponding author upon reasonable request.
Extended Data
Extended Data Figure 1. Compound structures.
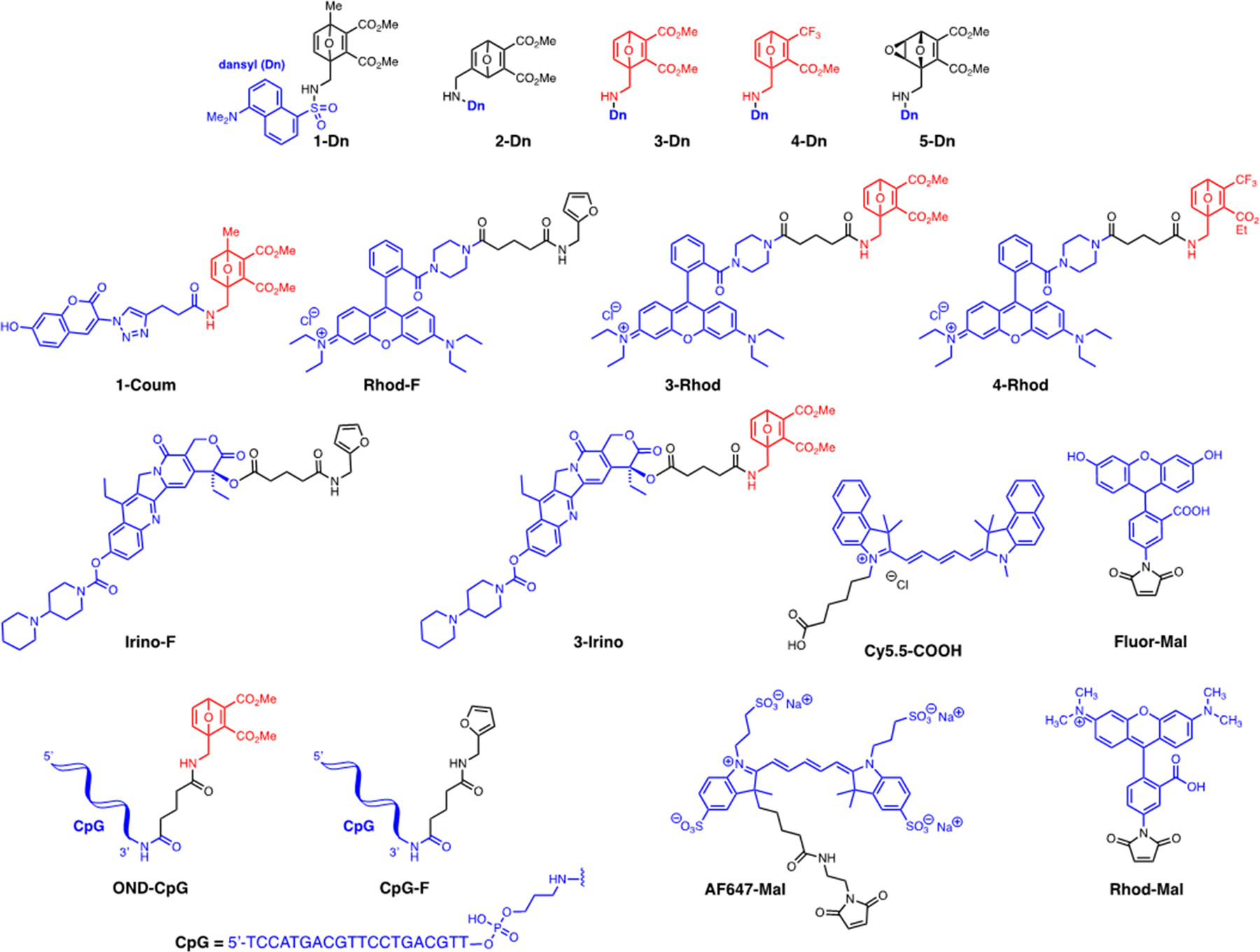
OND (1–4 Dn) and epoxyoxanorbornene (5-Dn) electrophiles, model fluorophore (Fluor-Mal, Rhod-Mal, Cy5.5-COOH, 1-Coum, Rhod-F, 3-Rhod, 4-Rhod) and drug cargos (Irino-F, 3-Irino, OND-CpG) presented in this study.
Extended Data Figure 2. Spatiotemporal effects of NP encapsulation-and-release delivery platform.
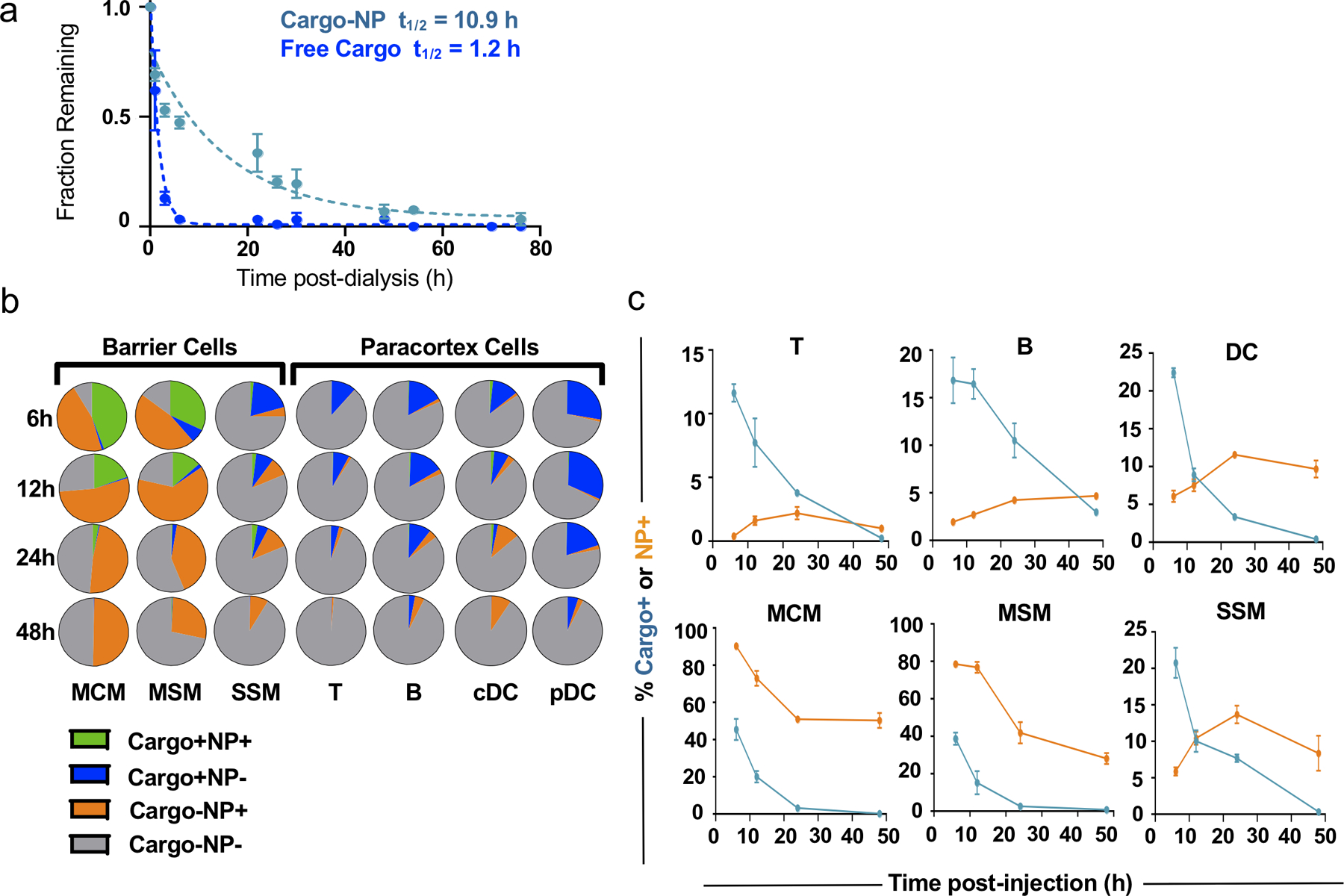
a) In vitro extended release of encapsulated Cy5.5 Cargo from NP (teal; half-life = 10.9h) versus free dye (dark blue; half-life = 1.2h). n=6 samples per group. Fraction (b) and percentage of total (c) dLN cells of various types positive for encapsulated Cy5.5 cargo (blue) and AF488-NPs (orange) measured by flow cytometry 6, 12, 24, and 48h after i.d. administration Cy5.5-encapsulating NPs. n=6 biological replicates per group. For all graphs, the columns/points and error bars represent the mean + SEM.
Extended Data Figure 3. Delivery of OND cargo using virus like particle carriers.
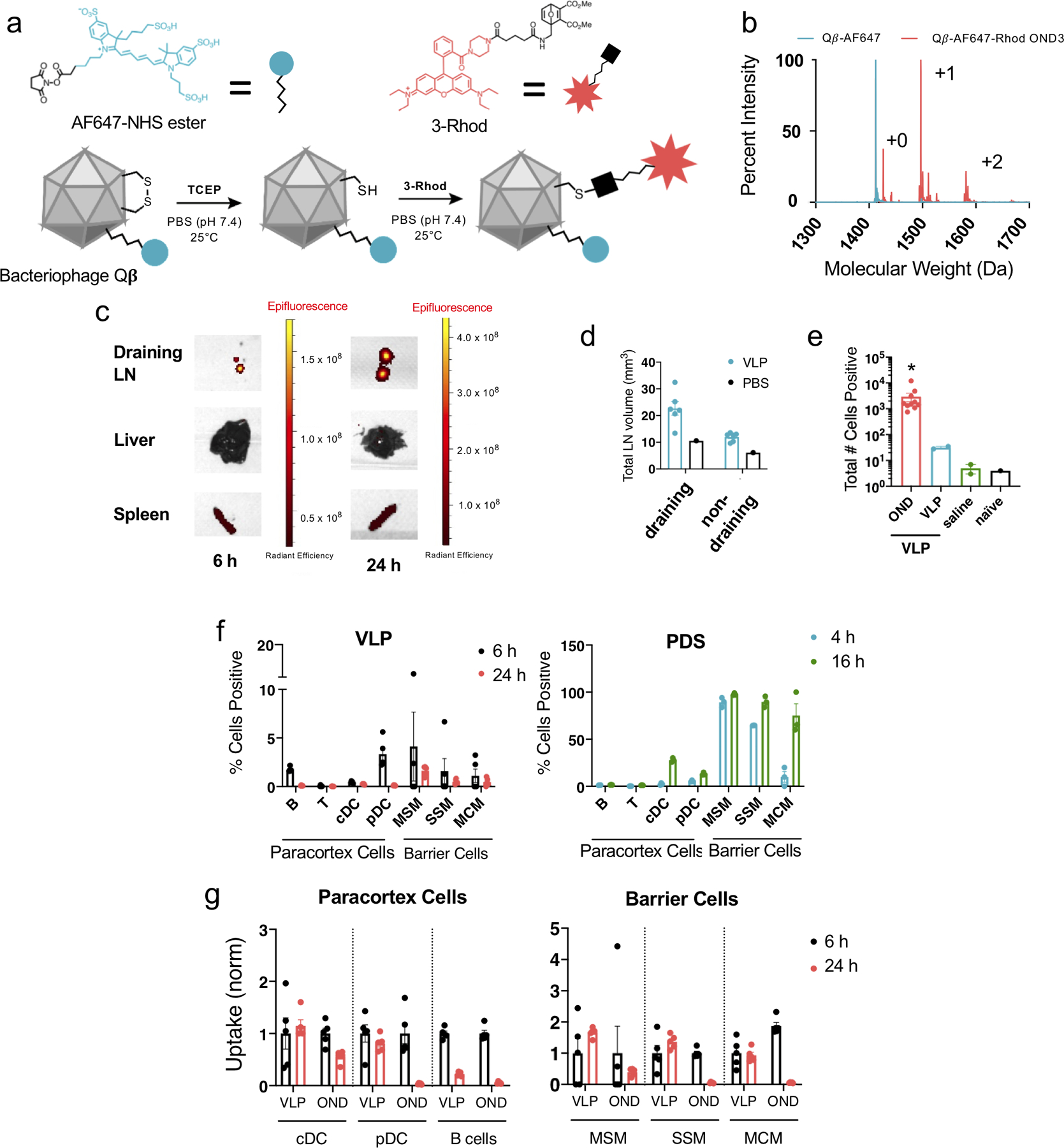
a) Schematic representation of the preparation of VLPs conjugated to 3-Rhod. b) Liquid chromatography time-of-flight mass spectrometry (C3 column) of VLP-OND conjugates. Blue trace represents VLP coat protein associated with AF647-NHS via lysine residues, red trace represents species associated with 3-Rhod. This experiment was repeated once with similar results. c) VLPs covalently labeled with NIR-NHS ester were injected i.d. into mice, followed by organ extraction and IVIS imaging after 6 and 24 h. This experiment was repeated once with similar results. d) dLN size after i.d. injection of VLP. Axial and brachial LN from draining and contralateral (non-draining) sides were photographed and measured using ImageJ. n=6 biological replicates per group. e) Total numbers of AF647 positive and Rhodamine positive dLN cells 24 h after i.d. treatment with VLPs labelled with AF647-NHS (VLP, non-cleavable linker) and 3-Rhod (OND). Saline injected and naïve (uninjected) animals served as Rhodamine channel negative controls. n=6 biological replicates per group. f) Percent of Rhodamine positive cells in dLN 4 h (teal), 6 h (black), 16 h (green), or 24 h (red) after i.d. injection with either VLP (VLP) or pyridyl disulfide PPS NP (PDS) conjugated with 3-Rhod measured by flow cytometry. n=3 biological replicates per group. g) Percent of positive lymphocytes and barrier cells in dLN 6 h (black) or 24 h (red) after i.d. injection of VLP labeled with both Alexa647-NIH (VLP, non-cleavable linker) and 3-Rhod (OND) measured by flow cytometry, normalized to average uptake at 6 h. n=5 biological replicates per group. For all graphs, the columns/points and error bars represent the mean + SEM. Statistics were performed by ordinary one-way ANOVA with Tukey’s multiple comparisons test. ****=p<.0001, ***=p<.005, *=p<.05.
Extended Data Figure 4. Toxicity of OND derivatives of irinotecan.
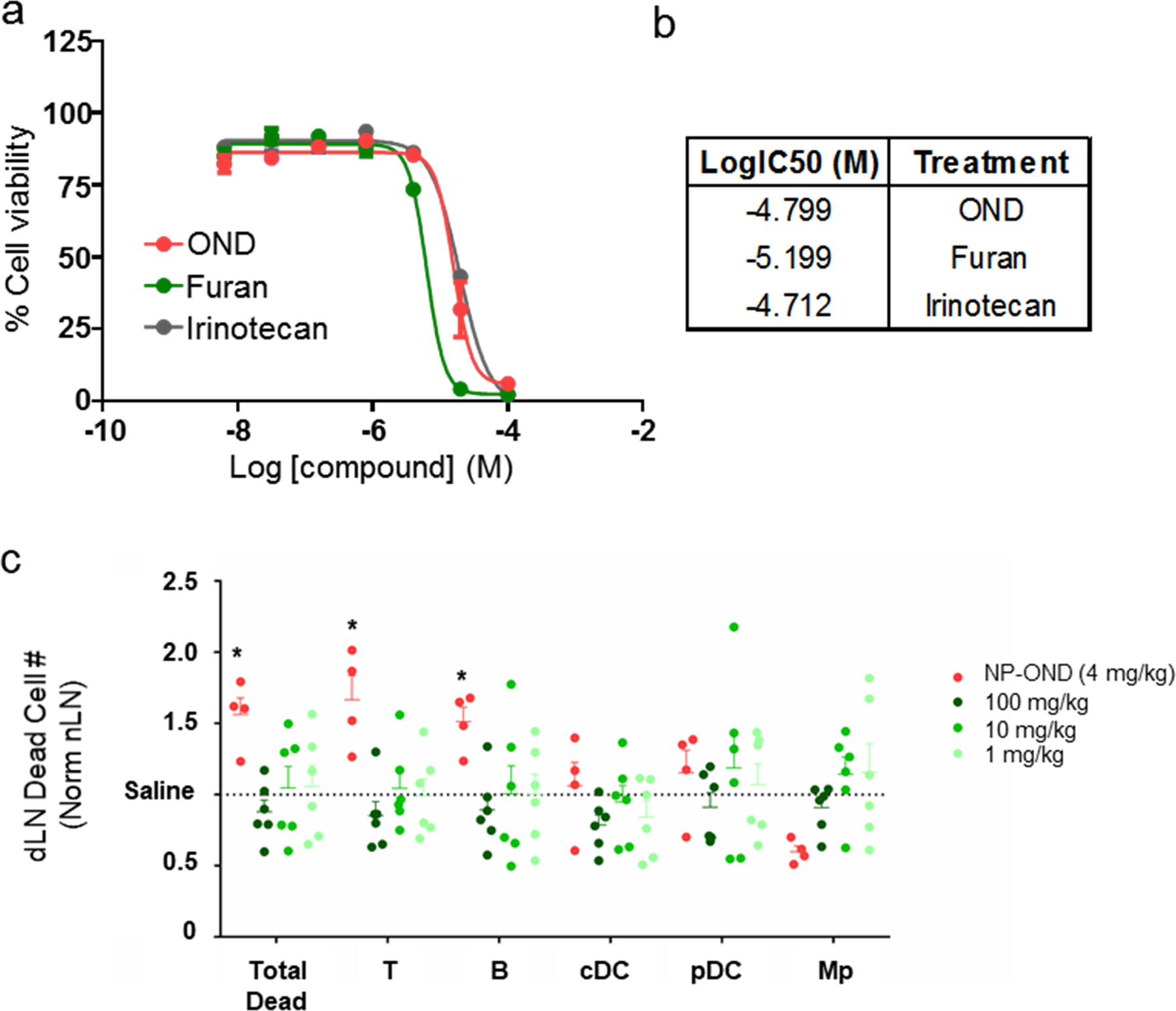
a) Log dose testing of 3-Irino (OND), Irino-F (Furan), and unmodified irinotecan against C57Bl6 splenocytes after 24 h in IMDM at 37°C. n=5 samples per group. b) Log IC50 values derived from panel a. c) Effects of cargo delivered to lymph node-resident cells by lymph accessing NP-OND compared to free drug. Number of dead LN cells normalized to saline-treated animals 24 h after i.d. administration of (NP-OND) NPs labeled with 3-Irino (4 mg/kg) or i.p. administration of free Irino-F at the indicated doses. n=5 biological replicates per group. For all graphs, the columns/points and error bars represent the mean + SEM. Statistics were performed by two-way ANOVA with Tukey’s multiple comparisons test. ****=p<.0001, ***=p<.005, *=p<.05 relative to each dose of free Irino-F. For all graphs, the columns/points and error bars represent the mean + SEM.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R01CA207619), the National Science Foundation (CHE 1011796), grants from the Parker H. Petit Institute for Bioengineering and Bioscience at the Georgia Institute of Technology and Georgia CORE/It’s the Journey, and the National Institutes of Health Training Grants T32EB021962 and T32GM008433. A.S. was an American Heart Association Predoctoral Fellow. C.J.H. and M.P.M. gratefully acknowledge support by the National Science Foundation Graduate Research Fellowship Program.
Footnotes
Financial and Non-Financial Competing Interest Statement
The authors declare no conflict of or cometing interest.
Supplementary information is available in the online version of the paper. Reprints and permission information is available online at www.nature.com/reprints. Correspondence and requests for materials should be addressed to S.N.T. and M.G.F..
References
- 1.Babar IA et al. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proceedings of the National Academy of Sciences 109, E1695–E1704 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lorenzo-Redondo R et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 530, 51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuerer HM et al. Residual metastatic axillary lymph nodes following neoadjuvant chemotherapy predict disease-free survival in patients with locally advanced breast cancer. Am J Surg 176, 502–509 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Liu H et al. Structure-based Programming of Lymph Node Targeting in Molecular Vaccines. Nature 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee J-W et al. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nature Immunology 8, 181 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Kreiter S et al. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer research 70, 9031–9040 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Komori J, Boone L, DeWard A, Hoppo T & Lagasse E The mouse lymph node as an ectopic transplantation site for multiple tissues. Nat. Biotechnol 30, 976 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas SN & Schudel A Overcoming transport barriers for interstitial-, lymphatic-, and lymph node-targeted drug delivery. Curr Opin Chem Eng 7, 65–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qi H, Kastenmuller W & Germain RN Spatiotemporal basis of innate and adaptive immunity in secondary lymphoid tissue. Annual review of cell and developmental biology 30, 141–167 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Chang JE & Turley SJ Stromal infrastructure of the lymph node and coordination of immunity. Trends in immunology 36, 30–39 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Kastenmuller W, Torabi-Parizi P, Subramanian N, Lammermann T & Germain RN A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell 150, 1235–1248 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sixt M et al. The Conduit System Transports Soluble Antigens from the Afferent Lymph to Resident Dendritic Cells in the T Cell Area of the Lymph Node. Immunity 22, 19–29 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Gerner Michael Y., Torabi-Parizi P & Germain Ronald N. Strategically Localized Dendritic Cells Promote Rapid T Cell Responses to Lymph-Borne Particulate Antigens. Immunity 42, 172–185 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Junt T et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 450, 110–114 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Gretz JE, Kaldjian EP, Anderson AO & Shaw S Sophisticated strategies for information encounter in the lymph node: the reticular network as a conduit of soluble information and a highway for cell traffic. The Journal of Immunology 157, 495–499 (1996). [PubMed] [Google Scholar]
- 16.Gretz JE, Norbury CC, Anderson AO, Proudfoot AE & Shaw S Lymph-borne chemokines and other low molecular weight molecules reach high endothelial venules via specialized conduits while a functional barrier limits access to the lymphocyte microenvironments in lymph node cortex. J Exp Med 192, 1425–1440 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong C et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proceedings of the National Academy of Sciences 108, 2426–2431 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pridgen EM, Langer R & Farokhzad OC Biodegradable, polymeric nanoparticle delivery systems for cancer therapy. Nanomedicine (Lond) 2, 669–680 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Colson YL & Grinstaff MW Biologically responsive polymeric nanoparticles for drug delivery. Advanced materials (Deerfield Beach, Fla.) 24, 3878–3886 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Reddy ST, Berk DA, Jain RK & Swartz MA A sensitive in vivo model for quantifying interstitial convective transport of injected macromolecules and nanoparticles. Journal of applied physiology (Bethesda, Md. : 1985) 101, 1162–1169 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Porter CJH & Charman SA Lymphatic transport of proteins after subcutaneous administration. Journal of pharmaceutical sciences 89, 297–310 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Nuhn L et al. pH-degradable imidazoquinoline-ligated nanogels for lymph node-focused immune activation. Proceedings of the National Academy of Sciences of the United States of America 113, 8098–8103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Titta A et al. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proceedings of the National Academy of Sciences 110, 19902–19907 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olson ES et al. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proceedings of the National Academy of Sciences of the United States of America 107, 4311–4316 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwon YJ, James E, Shastri N & Frechet JM In vivo targeting of dendritic cells for activation of cellular immunity using vaccine carriers based on pH-responsive microparticles. Proceedings of the National Academy of Sciences of the United States of America 102, 18264–18268 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rehor A, Hubbell JA & Tirelli N Oxidation-Sensitive Polymeric Nanoparticles. Langmuir 21, 411–417 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Schudel A, Kassis T, Dixon JB & Thomas SN S-Nitrosated Polypropylene Sulfide Nanoparticles for Thiol-Dependent Transnitrosation and Toxicity Against Adult Female Filarial Worms. Adv Healthc Mater 4, 1484–1490, 1423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schudel A, Sestito LF & Thomas SN Winner of the society for biomaterials young investigator award for the annual meeting of the society for biomaterials, April 11–14, 2018, Atlanta, GA: S-nitrosated poly(propylene sulfide) nanoparticles for enhanced nitric oxide delivery to lymphatic tissues. Journal of Biomedical Materials Research Part A, n/a-n/a (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas SN, Vokali E, Lund AW, Hubbell JA & Swartz MA Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 35, 814–824 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Higginson CJ, Eno MR, Khan S, Cameron MD & Finn MG Albumin-Oxanorbornadiene Conjugates Formed ex Vivo for the Extended Circulation of Hydrophilic Cargo. ACS chemical biology 11, 2320–2327 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higginson CJ, Kim SY, Pelaez-Fernandez M, Fernandez-Nieves A & Finn MG Modular degradable hydrogels based on thiol-reactive oxanorbornadiene linkers. J Am Chem Soc 137, 4984–4987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kislukhin AA, Higginson CJ, Hong VP & Finn MG Degradable conjugates from oxanorbornadiene reagents. J Am Chem Soc 134, 6491–6497 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Vlies AJ, O’Neil CP, Hasegawa U, Hammond N & Hubbell JA Synthesis of pyridyl disulfide-functionalized nanoparticles for conjugating thiol-containing small molecules, peptides, and proteins. Bioconjug Chem 21, 653–662 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Hirosue S, Kourtis IC, van der Vlies AJ, Hubbell JA & Swartz MA Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation. Vaccine 28, 7897–7906 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Hong V, Presolski SI, Ma C & Finn MG Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angewandte Chemie (International ed. in English) 48, 9879–9883 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy ST et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nature Biotechnology 25, 1159 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Kourtis IC et al. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PloS one 8, e61646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nathanson SD, Nelson L & Karvelis KC Rates of flow of technetium 99m--labeled human serum albumin from peripheral injection sites to sentinel lymph nodes. Annals of surgical oncology 3, 329–335 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Charman WN & Stella VJ Lymphatic Transport of Drugs. (Taylor & Francis, 1992). [Google Scholar]
- 40.Hettiaratchi MH, Schudel A*, Rouse T, Garcia AJ, Thomas SN, Guldberg RE, McDevitt TC.. A Rapid Method for Determining Protein Diffusion through Hydrogels for Regenerative Medicine Applications. In review. APL Bioengineering (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerner MY, Casey KA, Kastenmuller W & Germain RN Dendritic cell and antigen dispersal landscapes regulate T cell immunity. J Exp Med 214, 3105–3122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roozendaal R et al. Conduits Mediate Transport of Low Molecular Weight Antigen to Lymph Node Follicles. Immunity 30, 264–276 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiedler JD et al. Engineered mutations change the structure and stability of a virus-like particle. Biomacromolecules 13, 2339–2348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Golmohammadi R, Fridborg K, Bundule M, Valegard K & Liljas L The crystal structure of bacteriophage Q beta at 3.5 A resolution. Structure 4, 543–554 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Beaudette TT et al. In vivo studies on the effect of co-encapsulation of CpG DNA and antigen in acid-degradable microparticle vaccines. Mol. Pharm 6, 1160–1169 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeanbart L et al. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol Res 2, 436–447 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Goldstein MJ et al. A CpG-loaded tumor cell vaccine induces antitumor CD4(+) T cells that are effective in adoptive therapy for large and established tumors. Blood 117, 118–127 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi X-F et al. CpG oligodeoxynucleotide induces apoptosis and cell cycle arrest in A20 lymphoma cells via TLR9-mediated pathways. Molecular Immunology 54, 327–337 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Drobits B et al. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. The Journal of Clinical Investigation 122, 575–585 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y et al. Biphasic rapamycin effects in lymphoma and carcinoma treatment. Cancer Res. 77, 520–531 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the plots within this paper and other findings of this study are available from the corresponding authors upon reasonable request.