

Abstract
Open access to 3D structure information from the Protein Data Bank (PDB) facilitated discovery and development of >90% of the 79 new antineoplastic agents (54 small molecules, 25 biologics) with known molecular targets approved by the FDA 2010–2018. Analyses of PDB holdings, the scientific literature and related documents for each drug–target combination revealed that the impact of public-domain 3D structure data was broad and substantial, ranging from understanding target biology (~95% of all targets) to identifying a given target as probably druggable (~95% of all targets) to structure-guided lead optimization (>70% of all small-molecule drugs). In addition to aggregate impact assessments, illustrative case studies are presented for three protein kinase inhibitors, an allosteric enzyme inhibitor and seven advanced-stage melanoma therapeutics.
Keywords: Protein Data Bank, structure-guided drug discovery, FDA oncology approvals, small-molecule drug, biologic drug, open access
Teaser:
Open access to 3D macromolecular structure information managed by the Protein Data Bank facilitated discovery and development of >90% of new antineoplastic agents approved by the FDA 2010–2018.
Introduction
Over the past two decades, protein crystallography and structure-guided drug discovery have become established tools used throughout the biopharmaceutical industry [1,2]. 3D structures of biological macromolecules can inform our understanding of target biology (reviewed in [3]). They can confirm that a given protein target is likely to be druggable using small-molecule and/or biologic agents (reviewed in [4]). In the most favorable cases, protein crystallography can enable structure-guided optimization of affinity of small-molecule leads [1]. 3D structural data have also proven useful in overcoming some of the other challenges (e.g., avoiding unwanted off-target binding) inherent in turning biochemically active compounds into potent drug-like molecules suitable for safety and efficacy testing in animals and humans [5]. In the realm of biologics (~20% of approved drugs in the current era) [6], 3D structural information is also used routinely to inform engineering of monoclonal antibodies and other protein-based therapeutics [7,8].
Public-domain 3D structure information is distributed on an open-access basis by a single, global data resource known as the Protein Data Bank (PDB) [9]. Since 2008, publication of new macromolecular structures in most scientific journals has been contingent on mandatory deposition to the PDB of the 3D atomic coordinates constituting the structure together with experimental data and related metadata. Many governmental and nongovernmental research funders also require PDB deposition of macromolecular structural data by their grantees. When the PDB was established in 1971 as the first open-access digital data resource in biology, it housed only seven protein structures [9]. Today, the PDB is regarded as a ‘global public good’, vital to research and education or training across the biological and biomedical sciences. At the time of this publication, the PDB housed >160 000 experimentally determined, atomic-level 3D structures of biological macromolecules (i.e., proteins, DNA and RNA), many of which have been visualized in the act of binding one or more small-molecule ligands including FDA-approved drugs. Since 2003, the PDB has been managed jointly according to the FAIR principles: findability, accessibility, interoperability, reusability [10], by the Worldwide Protein Data Bank (wwPDB) partnership [11,12] – including the US Research Collaboratory for Structural Bioinformatics Protein Data Bank or RCSB PDB [13,14], Protein Data Bank in Europe [15], Protein Data Bank Japan [16] and BioMagResBank [17].
The RCSB PDB (rcsb.org) recently published a quantitative overview of the impact of PDB structures on 210 new medical entities (NMEs or new drugs) approved by the FDA 2010–2016 [18]. This work built on previously published analyses of lessons learned from 20 years of anticancer drugs [19] and went beyond individual case studies [20] and presentations at scientific meetings that described the impact of structure-guided drug discovery and of protein crystallographers working in industry. In all, we documented that nearly 6000 atomic-level 3D structures of molecular targets stored in the PDB archive facilitated discovery and development of ~88% of the 210 new drugs approved 2010–2016 across all therapeutic areas.
Given the large number of recent FDA drug approvals for oncology indications, we now review the ways that open access to PDB data facilitated discovery and development of 79 antineoplastic agents with known molecular targets (54 small molecules, 25 biologics) approved 2010–2018. In addition to an aggregate review of PDB impact on new drug approvals, we review three case studies illustrating the impact of PDB data, including three hinge-binding CDK4/CDK6 inhibitors (palbociclib, ribociclib and abemaciclib), an isocitrate dehydrogenase 2 (IDH2) allosteric inhibitor (enasidenib) and seven therapeutic agents that have transformed clinical management of advanced-stage melanoma (protein kinase inhibitors: vemurafenib, dabrafenib, trametinib and encorafenib; antibodies: ipilumumab, pertuzumab and nivolumab).
Antineoplastic drugs approved by the FDA 2010–2018
A total of 81 antineoplastic NMEs were approved by the FDA 2010–2018. Two of these newly approved drugs: trabectedin and ingenol mebutate, were not considered in this review because their molecular targets are unknown. All but three of the 79 remaining antineoplastic agents target human proteins. Nearly three-quarters (54/79, ~68%) of the NMEs are of low molecular weight (<1000 Da, denoted LMW-NMEs), all targeting human proteins. We classified the remaining NMEs (25/79, ~32%; ≥1000 Da) as biologic-NMEs. Three of these biologic-NMEs have small-molecule targets: dinutuximab, which is a monoclonal antibody that recognizes glycolipid GD2; and two l-asparaginases that hydrolyze l-asparagine yielding l-aspartate and ammonia. The remaining 22 biologic-NMEs (22/25, 88%) target extracellular human proteins.
Impact of PDB structures on antineoplastic drug approvals
We searched the entire PDB archive using corresponding reference amino acid sequences from UniProt (uniprot.org) [21] to identify 3D structures that include all or part of the known macromolecular target for each of the 79 antineoplastic NMEs (Table 1). As of September 2019, the archive contained one or more structures for 74 of the 79 NME targets (~95%). Every LMW-NME has at least one target structure present in the PDB (54/54, 100%). The LMW-NMEs themselves are also well represented in the PDB. For more than three-quarters of the LMW-NMEs (41/54, ~76%), one or more public-domain PDB structures reveal at the atomic level precisely how the drug binds to the corresponding NME target protein and, in some cases, to other so-called off-target proteins. Eighty percent of the biologic-NMEs (20/25) have one or more target structures in the PDB. For approximately one-half of the biologic-NMEs (13/25, 52%), the PDB archive houses one or more 3D structures of the drug itself and/or the drug–target complex. For the LMW-NMEs and the biologic NMEs, >95% of the target structures were deposited to the PDB at least a decade before the drug was approved for clinical use by the FDA.
Table 1.
Overview of PDB holdings for antineoplastic NMEs and their known molecular targets approved 2010–2018
| NME class | Number | Number with target structure(s) in PDB (95% identity) | Number with NME structure(s) in PDB | Number with NME target complex structure(s) in PDB |
|---|---|---|---|---|
| Antineoplastic agents with known molecular targets | 79 | 74 (~94%) | 53 (~67%) | 47 (~59%) |
| LMW-NMEs | 54 | 54 (100%) | 40 (~74%) | 38 (~70%) |
| Biologic-NMEs | 25 | 20 (80%) | 13 (52%) | 9 (36%) |
Small-molecule NMEs
The 54 LMW-NMEs target 13 distinct classes of proteins (Table 2). Without exception, known protein targets of every one of the 54 LMW-NMEs were represented in the PDB archive. In all, we identified 2115 ‘relevant structures’, which include PDB structures containing the following: (i) a reference or a mutant or variant form of the target protein; (ii) a LMW-NME bound to a reference or mutant or variant form of its target protein; (iii) a LMW-NME bound to a potential alternative target protein; or (iv) a LMW-NME bound to a possible off-target protein. The number of relevant structures identified for each target or target class ranges from <10 for IDH2 to 1136 for the protein kinases.
Table 2.
PDB holdings for antineoplastic LMW-NMEs approved 2010–2018
| LMW-NME target class | Number in target class | Number with target structure(s) in PDB | Total unique PDB IDs for NME target structures (95% identity) | Number with target NME complex structure(s) in PDB |
|---|---|---|---|---|
| Protein kinasesa | 33 | 33 (100%) | 1136 | 26 (~76%) |
| IDH1 | 1 | 1 (100%) | 40 | 0 (0%) |
| IDH2 | 1 | 1 (100%) | 9 | 1 (100%) |
| BCL-2 | 1 | 1 (100%) | 26 | 1 (100%) |
| PARPs | 4 | 4 (100%) | 89 | 4 (100%) |
| Androgen receptor | 2 | 2 (100%) | 96 | 0 (0%) |
| HDACs | 2 | 2 (100%) | 92 | 2 (100%) |
| Smoothened | 3 | 3 (100%) | 11 | 1 (~33%) |
| CYP17A1 | 1 | 1 (100%) | 13 | 1 (100%) |
| E3 ubiquitin ligase | 1 | 1 (100%) | 50 | 1 (100%) |
| Proteasome | 2 | 2 (100%) | 62 | 2 (100%) |
| Tubulin | 2 | 2 (100%) | 249 | 1 (50%) |
| Ribosome A site | 1 | 1 (100%) | 134 | 1 (100%) |
| All | 54 | 54 (100%) | 2007 | 41 (~74%) |
Bold italics indicate the targets or target classes for which 3D structure information and structure-guided drug discovery facilitated approval of 39/54 (~72%) newly approved LMW-NMEs.
Abbreviations: IDH1, isocitrate dehydrogenase 1; IDH2, isocitrate dehydrogenase 2; BCL-2, B cell lymphoma 2; PARP, poly ADP-ribose polymerase; HDAC, histone deacetylase; and CYP17A1, cytochrome p450 17A1.
More than 98% of the 2115 relevant structures were deposited to the PDB well before the LMW-NME was approved for clinical use by the FDA. The median time between PDB deposition and approval exceeded 14 years (data not shown). The vast majority of the 2115 relevant structures (1807/2115; ~85%) were reported in a PubMed-indexed publication around the time of PDB deposition. These 1807 papers had garnered a total of 172 653 literature citations as of September 2019, giving an average of >95 citations per primary publication. For reference, the average number of literature citations per primary publication across the entire PDB archive is ~50 [22].
Review of PDB archival holdings and the scientific literature pertaining to each NME target and/or LMW-NME combination summarized in Tables 2 and 3 revealed that public-domain 3D structure data facilitated discovery and development of all 54 LMW-NMEs in the following ways:
- Target biology: atomic-level 3D structures provide functional insights that are not always readily apparent from amino acid sequence (reviewed in [22]).
- In every case, the PDB provides one or more experimentally determined structure of each unique NME target.
- Target druggability: 3D structures enable visualization of surface features deemed likely to bind small organic compounds for inhibition of enzymatic action or other biochemical or biological function (reviewed in [4]).
- In every case, a PDB structure(s) revealed one or more potential small-molecule binding sites, either on the surface of the NME target or within a protein–protein interface (e.g., the homodimeric enzymes IDH1 and IDH2).
- Small-molecule binding: co-crystal structure studies permit 3D assessment of binding of tool compounds or small-molecule hits coming from biochemical, biophysical [23] or fragment [24] screening campaigns, thereby aiding medicinal chemistry decision making (e.g., [25]).
- For >41 of 54 of cases (>76%) one or more structures of a small molecule bound to the NME target were freely available from the PDB.
- Structure-guided lead optimization: co-crystal structures are widely used across the biopharmaceutical industry to guide optimization of potency (reviewed in [1]). In the most favorable cases, knowledge of co-crystal structures with potential off-targets (e.g., GSK-3β, inhibition of which causes hyperglycemia) can also be employed to help ensure the desired selectivity profile and reduce the likelihood of off-target toxicity. In the absence of experimental co-crystal structures of the target protein, in silico docking tools are typically used to guide lead optimization (reviewed in [26]). Where an experimental 3D structure of the target protein is not available, homology models are routinely combined with these same in silico docking tools. Machine learning approaches are also being used with increasing frequency to drive medicinal chemistry campaigns (reviewed in [27]).
- In 39/54 (~72%) of cases, there is direct or indirect evidence from the PDB archive and the scientific literature that structure-guided lead optimization with the target protein or computational tools with public domain PDB structures have been employed by one or more biopharmaceutical companies prosecuting the NME target (Table 3).
- Not surprisingly, the vast majority of the 39 LMW-NMEs identified as confirmed or probable products of structure-guided discovery correspond to 28 of the LMW-NMEs targeting one or more protein kinases (Table 3).
- Twenty-four of the kinase inhibitors were confirmed as products of structure-guided drug discovery (‘Yes’ in Table 3) on the basis of direct evidence from the scientific literature (or private communications) that the sponsor company or its predecessor (for acquired programs) or a competitor company used crystallography and/or computational modeling to study how each LMW-NME bound to its target protein [28–46].
- Four kinase inhibitors were identified as probable products of structure-guided drug discovery (‘Prob’ in Table 3) on the basis of indirect evidence. In these cases, a PDB structure of the target protein was publicly available ten or more years before approval and an academic research group used crystallography to study each LMW-NME bound to its target protein. We classified these four less clear-cut cases as probable, because we think it highly likely that the sponsor company was in possession of the same or similar data given the ubiquity of expert protein crystallography and computational chemistry teams across the industry [47–49].
- Four kinase inhibitors for which a PDB structure of the target protein was publicly available less than 10 years before approval were identified as possible products of structure-guided drug discovery (‘Poss’ in Table 3). In these cases, we were unable to find direct or indirect evidence that they were products of structure-guided drug discovery.
- Three LMW-NMEs, including two isocitrate dehydrogenase inhibitors (ivosidentib, enasidenib) and venetoclax (targeting BCL-2), were confirmed as products of structure-guided drug discovery (‘Yes’ in Table 3) on the basis of direct evidence from the scientific literature (or private communications with industry experts) that the sponsor company used crystallography to study how each LMW-NME bound to its target protein [50,51].
- Eight LMW-NMEs, including four poly ADP-ribose polymerase (PARP) inhibitors, two nonsteroidal antiandrogens and two histone deacetylase (HDAC) inhibitors, were identified as probable products of structure-guided drug discovery (‘Prob’ in Table 3) on the basis of indirect evidence. In these cases, a PDB structure of the target protein was publicly available at least ten before approval. With the exception of the two antiandrogens, an academic research group used crystallography to study each LMW-NME bound to its target protein. We classified these eight less clear-cut cases as probable, because we think it highly likely that the sponsor company or its predecessor (for acquired programs) was in possession of the same or similar data given the ubiquity of expert protein crystallography and computational chemistry teams across the industry [52,53].
- Six LMW-NMEs were identified as unlikely to be products of structure-guided drug discovery (‘Unl’ in Table 3). In these cases, a PDB structure of the target protein was either not publicly available at the time of approval or only became available shortly before approval.
- Four LMW-NMEs were identified as natural product derivatives (‘Nat Prod’ in Table 3). The breadth and depth of PDB structures and publications coming from industry revealed by our analyses confirm that 3D structures are impacting discovery of LMW-NMEs in real time. Conservative estimates suggest that X-ray crystal structures of proteins held as trade secrets inside company firewalls across the biopharmaceutical industry are comparable in aggregate to PDB archival holdings (i.e., ~160 000 structures). Willingness on the part of industry to share a subset of these data with academic researchers is essential for the long-term health of the experimental and computational ecosystems that support structure-guided drug discovery. It is encouraging that approximately two-thirds (27/41, ~66%) of the PDB structures of the antineoplastic LMW-NMEs bound to their targets enumerated in Table 3 were deposited by industrial protein crystallography teams (NB: given the highly competitive nature of biopharmaceutical industry, PDB deposition of structures from biopharmaceutical companies often lags behind the actual research). Equally encouraging is the fact that several biopharmaceutical companies generously contributed ‘post-competitive’ co-crystal structures and affinity data that enabled blinded computational docking and scoring challenges organized over the past 5 years by the Drug Design Data Resource (D3R, https://drugdesigndata.org) [54–56].
- Optimization of ADME properties: finally, 3D structures of proteins are also used to overcome ADME issues (reviewed in [5]).
- Of particular relevance are PDB structures of cytochrome P450 enzymes (earliest PDB ID: 1og2) [57], the P-glycoprotein multidrug transporter (earliest PDB ID: 3g5u) [58] and the human ether-a-go-go related potassium channel (earliest PDB ID: 5va1) [59]. Notwithstanding availability of 3D structures for these and other ADME-related proteins in the PDB, we were unable to find evidence in the scientific literature that 3D structure was used to overcome ADME issues for any of the 54 LMW-NMEs enumerated in Table 3.
Table 3.
Evidence summary for structure-guided drug discovery (SGDD) of LMW-NMEs approved 2010–2018
| Generic drug name | Sponsor company | Target protein class | Target protein | Earliest >95% identical target (domain) PDB ID/year | FDA approval year | Target LMW-NME complex PDB ID | Source of target–drug complex PDB ID (Academia, industry) | SGDD (Yes, Prob, Poss, Unl, Nat Prod) |
|---|---|---|---|---|---|---|---|---|
| Vemurafeniba | Roche | Kinase | BRAF | 1uwh/2004 [83] | 2011 | 3og7 [43] | Industry | Yes |
| Dabrafeniba | GSK | Kinase | BRAF | “ | 2013 | 4xv2 [34] | Industry | Yes |
| Encorafeniba | Novartis | Kinase | BRAF | “ | 2018 | Yes | ||
| Vandetanib | AstraZeneca | Kinase | VEGFRs | 1vr2/1999 [116] | 2011 | 2ivu [49] | Academia | Prob |
| Axitinib | Pfizer | Kinase | VEGFRs | “ | 2012 | 4ag8 [38] | Industry | Yes |
| Lenvatinib | Esai | Kinase | VEGFRs | “ | 2015 | 5zv2 [29] | Industry | Yes |
| Bosutinib | Pfizer | Kinase | SRC | 1fmk/1997 [117] | 2012 | 4mxo[48] | Academia | Prob |
| Regorafenib | Bayer | Kinase | KIT | 1pkg/2003 [118] | 2012 | Poss | ||
| Ponatinib | Ariad | Kinase | T315I ABL | 1iep/2001 [119] | 2012 | 3ik3[44] | Academia/industry | Yes |
| Neratinib | Puma | Kinase | EGFRs | 1m14/2002 [120] | 2017 | 2jiv [121] | Academe | Prob |
| Dacomitinib | Pfizer | Kinase | EGFRs | “ | 2018 | 4i23 [37] | Industry | Yes |
| Osimertinib | AstraZeneca | Kinase | EFGRs | “ | 2015 | 4zau [47] | Academia | Prob |
| Afatinib | Boehringer Ingelheim | Kinase | EGFRs | “ | 2013 | 4g5j [39] | Industry | Yes |
| Crizotinib | Pfizer | Kinase | ALK | 2yt2/2007 [122] | 2011 | 2xp2 [41] | Industry | Yes |
| Ceritinib | Novartis | Kinase | ALK | “ | 2014 | 4mkc [36] | Academia/industry | Yes |
| Alectinib | Roche | Kinase | ALK | “ | 2015 | 3aox [42] | Industry | Yes |
| Brigatinib | Ariad | Kinase | ALK | “ | 2107 | 6mx8 [33] | Industry | Yes |
| Lorlatinib | Pfizer | Kinase | ALK | “ | 2018 | 4cli [46] | Industry | Yes |
| Palbociclib | Pfizer | Kinase | CDK4/6 | 1bi8/1998 [76] | 2015 | 5l2i [31] | Industry | Yes |
| Ribociclib | Novartis | Kinase | CDK4/6 | “ | 2017 | 5lt2 [31] | Industry | Yes |
| Abemaciclib | Lilly | Kinase | CDK4/6 | “ | 2017 | 5l2s [31] | Industry | Yes |
| Cobimetinib | Exelixis | Kinase | MEK | 1s9j/2004 [90] | 2015 | 4an2 [40] | Industry | Yes |
| Binimetinib | Array Biopharma | Kinase | MEK | “ | 2018 | Yes | ||
| Trametiniba | JapanTobacco | Kinase | MEK | “ | 2013 | Poss | ||
| Cabozantinib | Bristol Myers Squibb | Kinase | MET | 1r0p/2003 [123] | 2016 | 3lq8 [45] | Industry | Yes |
| Idelalisib | Gilead | Kinase | PI3Ks | 2rd0/2007 [124] | 2014 | 4xe0 [35] | Industry | Yes |
| Copanlisib | Bayer | Kinase | PI3Ks | “ | 2017 | 5g2n [32] | Industry | Yes |
| Duvelisib | Intellikine | Kinase | PI3Ks | “ | 2018 | Poss | ||
| Ibrutinib | Celera | Kinase | BTK | 1btk/1997 [125] | 2013 | 5p9i [30] | Industry | Yes |
| Acalabrutinib | Acerta | Kinase | BTK | “ | 2017 | Yes | ||
| Larotrectinib | Array Biopharma | Kinase | TRKs | 5jfw/2016 [126] | 2018 | Poss | ||
| Gilteritinib | Astellas | Kinase | FLT3 | 1rjb/2004 [127] | 2018 | 6jqr [28] | Industry | Yes |
| Ivosidenib | Agios | Enzyme | IDH1 | 1t09/2004 [128] | 2018 | Yes | ||
| Enasideniba | Agios | Enzyme | IDH2 | 1lwd/2002 [77] | 2017 | 5i96 [50] | Industry | Yes |
| Venetoclax | Abbott | Programmed cell death | BCL-2 | 1g5m/2000 [129] | 2016 | 6o0k [51] | Industry | Yes |
| Olaparib | AstraZeneca | Enzyme | PARPs | 1uk0/2004 [130] | 2014 | 4tvj [52] | Academia | Prob |
| Rucaparib | Clovis Oncology | Enzyme | PARPs | “ | 2016 | 4rv6 [52] | Academia | Prob |
| Niraparib | GSK | Enzyme | PARPs | “ | 2017 | 4r6e [52] | Academia | Prob |
| Talazoparib | Pfizer | Enzyme | PARPs | “ | 2018 | 4und [52] | Academia | Prob |
| Enzalutamide | Medivation | Nuclear hormone receptor | Androgen receptor | 1e3g/2000 [131] | 2012 | Prob | ||
| Apalutamide | Janssen | Nuclear hormone receptor | Androgen receptor | “ | 2018 | Prob | ||
| Belinostat | Spectrum Pharmaceuticals | Epigenetic | HDACs | 1t64/2004 [132] | 2014 | 5een [53] | Academia | Prob |
| Panobinostat | Novartis | Epigenetic | HDACs | “ | 2015 | 5ef8 [53] | Academia | Prob |
| Vismodegib | Roche | GPCR | Smoothened | 4jkv/2013 [132] | 2012 | 5l7i [133] | Academia | Unl |
| Sonidegib | Sun Pharma | GPCR | Smoothened | “ | 2015 | Unl | ||
| Glasdegib | Pfizer | GPCR | Smoothened | “ | 2018 | Unl | ||
| Abiraterone acetate | Apotex | Cytochrome P450 | CYP17A1 | 3ruk/2012 [134] | 2011 | 3ruk [134] | Academia | Unl |
| Pomalidomide | Celgene | Protein degradation | E3 ubiquitin ligase | 2hye/2006 [135] | 2013 | 6h0f [136] | Academia | Unl |
| Ixazomib citrate | Millennium | Protein degradation | Proteasome | 4r3o/2015 [137] | 2015 | 5lf7 [138] | Academia | Unl |
| Carfilzomib | Amgen | Protein degradation | Proteasome | “ | 2012 | 4r67 [137] | Academia | Nat Prod |
| Cabazitaxel | Sanofi-Aventis | Cell division | Tubulin | 1z5v/2005 [139] | 2010 | Nat Prod | ||
| Eribulin | Eisaai | Cell division | Tubulin | “ | 2010 | Nat Prod | ||
| Omacetaxine mepesuccinate | Teva | Ribosome | A site | 3j7y/2014 [140] | 2012 | 3g6e [141] | Academia | Nat Prod |
| Midostaurin | Millennium | Kinase | Multiple kinases | N/A | 2017 | 4nct [142] | Academia | Nat Prod |
Indicates LMW-NMEs featured in the three case studies described at the end of this review.
Abbreviations: VEGFR, vascular endothelial growth factor receptor; EGFR, epidermal growth factor receptor; ALK, anaplastic lymphoma kinase; CDK, cyclin-dependent kinase; PI3K, phosphoinositide 3-kinase; BTK, Bruton’s tyrosine kinase; TRK, tropomyosin receptor kinase.
Biologic-NMEs
The 25 biologic-NMEs approved by the FDA in 2010–2018 were divided into three types (Table 4). Most (20/25, 80%) are either monoclonal antibodies (16/25, 64%; 12 unique targets) or antibody–drug conjugates (ADCs: 4/25, 16%; three unique targets), which consist of a monoclonal antibody linked to either a protein toxin (moxetumomab pasudotox-tdfk) or a small-molecule drug (brentuximab vedotin, adotrastuzumab emtansine and inotuzumab ozogamicin). Five ‘other’ biologic-NMEs (Table 4) include two l-asparaginases (calaspargase pegol-mknl, asparaginase Erwinia chrysanthemi), extracellular portions of vascular endothelial growth factor receptor (VEGFR)1 and VEGFR2 fused to human IgG1 immunoglobulin Fc domains (Ziv-aflibercept, targeting VEGF1 and VEGF2), human interleukin (IL)3 fused to diphtheria toxin (tagraxofusp-erzs, targeting the IL3 receptor) and a radiolabeled oligopeptide that targets somatostatin receptors (lutetium Lu 177 dotatate).
Table 4.
PDB holdings for antineoplastic biologic-NMEs approved 2010–2018
| Types of biologic-NMEs | Number of biologic-NMEs of each type | Number of biologic-NMEs with PDB target structure(s) | Unique PDB IDs for biologic-NME target structures (95% identity) | Biologic-NME structure(s) in PDB |
|---|---|---|---|---|
| Antibodies | 16 | 14 (~88%) | 395 | 8 (50%) |
| ADCs | 4 | 4 (100%) | 30 | 1 (25%) |
| Othera | 5 | 2 (40%) | 48 | 4 (80%) |
| All | 25 | 20 (80%) | 405 | 13 (52%) |
Other: calaspargase pegol-mknl, asparaginase Erwinia chrysanthemi, ziv-aflibercept, tagraxofusp-erzs, lutetium Lu 177 dotatate.
For 24 of the 25 biologic-NMEs (96%), we identified >479 ‘relevant structures’ in the PDB, which include the following: (i) the reference or mutant or variant form of the protein targeted by the biologic-NME; (ii) all or part of the biologic-NME itself; or (iii) all or part of the biologic-NME bound to a reference or mutant or variant form of its target protein. All 479 relevant structures were deposited to the PDB well before the NME was approved by the FDA for clinical use (data not shown). The median time between PDB deposition and FDA approval exceeded 9.5 years. The vast majority of the 479 relevant structures (415/479, 87%) were reported in a PubMed-indexed publication around the time of PDB deposition. These 415 papers had garnered 42 115 literature citations as of September 2019, giving an average of >101 citations per primary publication versus the average across the entire PDB archive of ~50 [22].
Review of PDB archival holdings and the scientific literature pertaining to each NME target and/or biologic-NME combination summarized in Table 4 revealed that public domain 3D structural data facilitated discovery and development of >90% of the 25 biologic-NMEs as follows:
- Target biology: atomic-level 3D structures provide functional insights that are not always readily apparent from amino acid sequence (reviewed in [22]).
- For the 22 biologic-NMEs with known protein targets, the PDB archive contains one or more experimentally determined structure(s) for all but two of the targets (20/22, 90%).
- Protein engineering: across the biopharmaceutical industry, antibody engineering depends on our knowledge of 3D structures (reviewed in [7,8,60]). X-ray crystallographic studies of human and mouse antibodies began to bear fruit as early as the 1970s and continue to do so. The first human protein structure in the PDB was that of a Bence–Jones immunoglobulin light-chain dimer (PDB ID: 1rei) [61]. The first PDB structure of a Fab fragment was that of McPC603, a phosphocholine-binding mouse myeloma protein (PDB ID: 1mcp) [62], deposited in 1985. The first PDB structure of single-chain Fv was that of Se155–4 bound to a trisaccharide ligand (PDB ID: 1mfa) [63], deposited in 1994. Today, thousands of antibody structures are represented in the PDB, ranging from entire immunoglobulins to Fab fragments and single-chain Fvs.
- Design of the limited repertoire of molecular scaffolds used across the biopharmaceutical industry utilized knowledge of PDB structures, making all 20 of the recently approved antibodies or ADCs indirect products of 3D structure. It was not possible from public domain information to determine whether project-specific structural data directly drove engineering of a particular antibody or ADC. Consultation with industry experts revealed that proprietary 3D structures held inside company firewalls are used in a substantial number of cases but by no means the majority (private communications). In a limited number of cases, structures of these antibody frameworks have been publicly disclosed and deposited to the PDB (e.g., PDB ID: 4kmt [64], 5i15, 5i16, 5i17, 5i18, 5i19, 5i1a, 5i1c, 5i1d, 5i1e, 5i1g, 5i1h, 5i1i, 5i1j, 5i1k and 5i1l [65]).
- Going beyond conventional antibody scaffolds, PDB structural data are also in routine use across the biopharmaceutical industry to guide design of various bispecific and trispecific agents. Three such molecules were approved by the FDA 2010–2018.
- Blinatumomab approximates T cells to the surfaces of malignant B cells by simultaneously targeting CD3 on the T cell and CD19 on the B cell using two antibody variable region heterodimers (VL–VH) fused together by a linker between the two VH segments [66].
- Tagraxofusp-erzs consists of IL3 fused to a truncated form of the diphtheria toxin (DT) protein. First PDB structures of IL3 (PDB ID: 1jli) [67] and DT (PDB ID: 1ddt) [68] were made publicly available in the mid-1990s, well before approval in 2018. Frankel et al. [69] described use of PDB ID: 1ddt [68] to guide design of various IL3–DT fusions.
- Ziv-aflibercept is a triple-fusion protein consisting of extracellular portions of VEGFR1 fused to corresponding extracellular portions of VEGFR2 fused to an IgG1-Fc domain. The dimeric assembly targets free VEGF1 and VEGF2 growth factors for internalization and degradation by cells bearing Fc receptors. The first PDB structures of IgG Fc (PDB ID: 1fc1) [70] and VEGFR1 (PDB ID: 1flt) [71] were made publicly available in 1981 and 1995, respectively, well before FDA approval in 2012.
Molecular recognition: use of 3D structures to understand how antibodies bind their target proteins has contributed to biologic drug discovery in various ways. For example, structural studies of anti-HER2 antibodies showed that they bind to distinct antigenic epitopes, revealing the molecular underpinnings of effective combination antibody therapy for breast cancer (reviewed in [72]) with pertuzumab (PDB ID: 1s78) [73] and trastuzumab (PDB ID: 1n8z) [74]. Co-crystal structures of monoclonal antibodies recognizing their targets and other biophysical findings also provide detailed maps of target-binding sites. This information is frequently used in patent applications to strengthen intellectual property protection claims for biologic-NMEs [75].
PDB structures provide insights into how eight of 16 (50%) antibodies and one of four (25%) antibody–drug conjugates bind to their protein targets.
Case studies
Going beyond these aggregate analyses, we now review three case studies illustrating the impact of PDB data: (i) three CDK4/CDK6 inhibitors; (ii) an IDH2 inhibitor; and (iii) seven therapeutic agents for treatment of advanced-stage melanoma. The following considerations influenced selection of NMEs for inclusion in these three case studies.
The CDK4/CDK6 inhibitors were selected because they exemplify parallel use of structure-guided drug discovery by three large biopharmaceutical companies that competed head-to-head on targeting precisely the same binding site in two closely related protein kinases (i.e., the hinge regions), building on PDB structure data for many protein kinases including one of the targets (human CDK6) [76] that entered the public domain ~17 years before FDA approval.
The IDH2 allosteric inhibitor was selected because it exemplified use of structure-guided drug discovery by a small biotechnology company, building on a lone PDB structure of a highly similar mammalian homolog of human IDH2 (i.e., porcine IDH2) [77] that entered the public domain ~15 years before FDA approval. The PDB structure of the LMW-NME bound to IDH2 was deposited by the sponsor company ~1 year in advance of approval [50].
Finally, seven NMEs that have transformed clinical management of advanced-stage melanoma were selected for detailed review, including four protein kinase inhibitors and three monoclonal antibodies targeting five distinct human proteins in aggregate. They were discovered and developed by six companies competing intensively in the same clinical arena to address very considerable unmet medical needs. These seven discovery and development efforts were built on understanding of target biology and target druggability in 3D, which was facilitated by open access to thousands of PDB structures of cellular signaling proteins and the four target proteins and their complexes with proteins and small-molecule ligands. Three of the four LMW-NMEs were the product of structure-guided drug discovery campaigns targeting BRAF. PDB structure data preceded FDA approval by only 7 years in the earliest instance (i.e., vemurafenib, approved in 2011). The relatively short timeline reflects the combined impact of understanding target biology and target druggability on target selection (i.e., mutant BRAF), structure-guided lead compound discovery and optimization, and highly focused clinical trial design that, together with a companion diagnostic, supported accelerated approval by the FDA. Protein engineering of the three biologic-NMEs was, at a minimum, indirectly facilitated by open access to the extensive collection of antibody structures housed within the PDB archive. Co-crystal structures of all three biologic-NMEs with their target proteins are also freely available from the PDB.
CDK4/CDK6 case study
Two closely related cyclin-dependent kinases, CDK4 and CDK6, are responsible for controlling progression through the G1 phase of the cell cycle, playing central parts in cell proliferation and tumorigenesis. The first PDB structures of human CDK4 and human CDK6 entered the public domain more than a decade ago (CDK4: PDB ID: 2w96 [78]; CDK6: PDB ID: 1bi8 [76]). Efforts to discover and develop CDK4 and CDK6 inhibitors as targeted cancer therapies began in the early 1990s (reviewed in [79]), culminating in FDA approval of three dual CDK4/CDK6 inhibitors for treatment of breast cancer (palbociclib, ribociclib and abemaciclib). All three of these LMW-NMEs came from structure-guided drug discovery efforts carried out independently by different sponsor companies. Each discovery team could rely on open access to tens of CDK structures and thousands of other protein kinase structures previously archived in the PDB. Co-crystal structures of each new drug bound to CDK6 were generously deposited in the PDB by Pfizer. Figure 1 compares the earliest structures of CDK4 (PDB ID: 2w96) and CDK6 (PDB ID: 1bi7) with co-crystal structures for palbociclib (PDB ID: 5l2i [31]), ribociclib (PDB ID: 5l2t) [31] and abemaciclib (PDB ID: 5l2s) [31] bound to CKD6. Close inspection of the modes of inhibitor binding reveals common (e.g., H-bonding engagement of the hinge region) and disparate features of CDK6–ligand interactions for the three inhibitors (Figure 1d).
Figure 1.
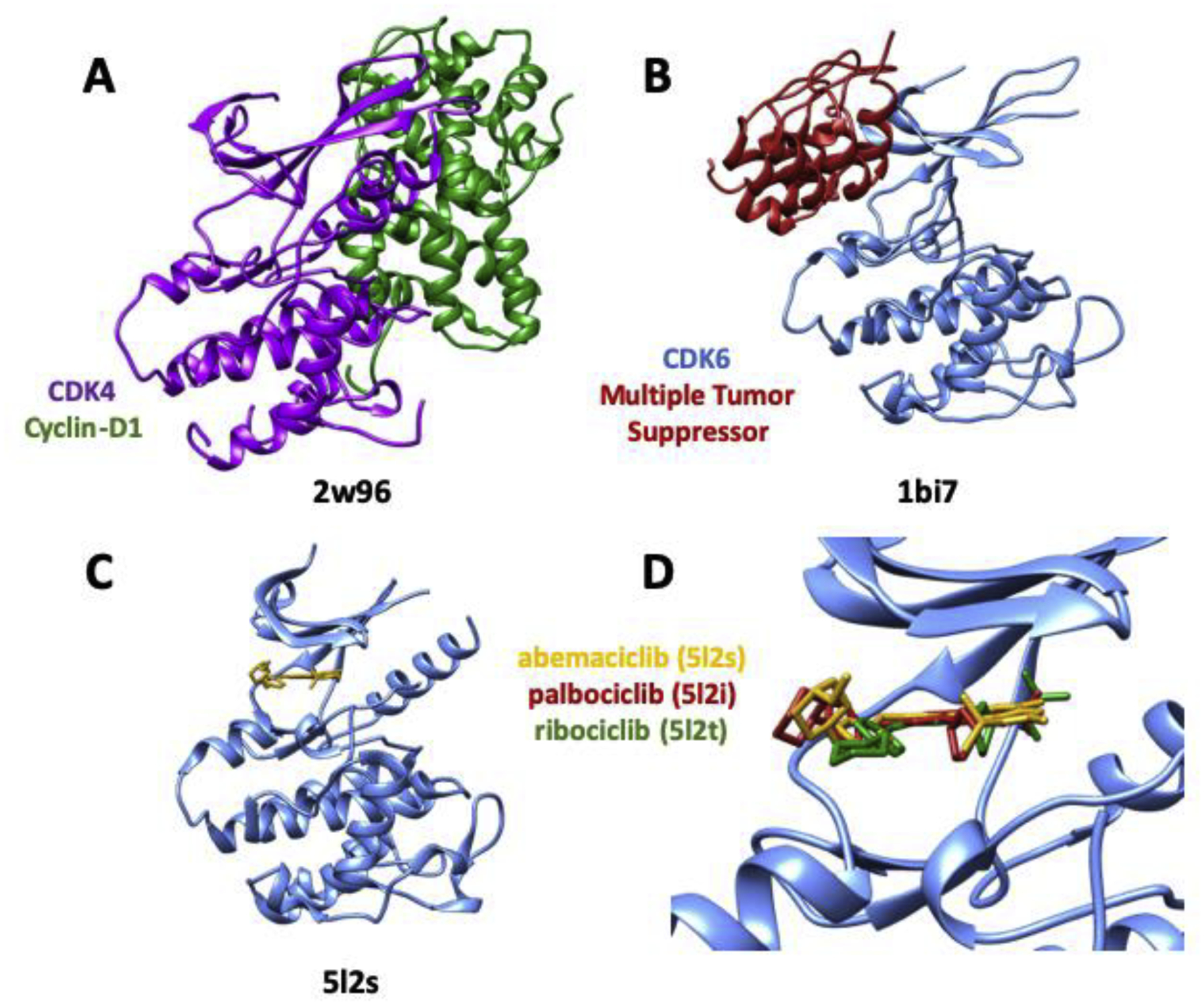
Hinge-binding inhibitors targeting two cyclin-dependent kinases. (a) CDK4 (purple) bound to cyclin-D1 (green) (PDB ID: 2w96). (b) CDK6 (blue) bound to multiple tumor suppressor (red) (PDB ID: 1bi7). (c) CDK6 (blue) bound to abemaciclib (yellow) (PDB ID: 5l2s). (d) Active site of CDK6 (PDB ID: 5l2s) showing bound abemaciclib (yellow; PDB ID: 5l2s), overlaid with palbociclib (red; PDB ID: 5l2i) and ribociclib (green; PDB ID: 5l2t).
IDH2 case study
The first 3D structure of a mammalian IDH2 (porcine, 96% identical in amino acid sequence to human) was deposited in the PDB in 2002 by academic researchers (PDB ID: 1lwd) [77]. IDH2 is a homodimeric, NADP(+)-dependent, mitochondrial enzyme responsible for catalyzing oxidative decarboxylation of isocitrate to 2-oxoglutarate. Certain IDH2 gene mutations confer a gain-of-function on malignant cells, resulting in accumulation and secretion of the oncometabolite (R)-2-hydroxyglutarate (reviewed in [80]). As of September 2019, the PDB archive housed six X-ray structures of human IDH2, all of which were contributed by biopharmaceutical companies (Novartis or Agios). Agios deposited the earliest human IDH2 structure (PDB ID: 4ja8) [81], which revealed the allosteric mechanism by which one of their proprietary compounds (AGI-6780) inhibited the R140Q form of IDH2 by binding within the dimer interface (data not shown). A structure-guided drug discovery campaign at Agios subsequently yielded enasidenib (Figure 2, PDB ID: 5i96) [50]. This LMW-NME was approved by the FDA in 2017 for relapsed or refractory acute myeloid leukemia in individuals with specific mutations of the IDH2 gene confirmed by an FDA-approved diagnostic test.
Figure 2.

Allosteric inhibitor enasidenib (yellow) targeting homodimeric IDH2 (green and blue) (PDB ID: 5i96).
Advanced-stage melanoma case study
Changing clinical management paradigms for advanced-stage (stages 3 and 4) melanoma provide compelling evidence for the transformative impact of 3D structure information and structure-guided drug discovery on FDA drug approvals. Ten years ago, treatment options for this disease were decidedly limited and of marginal benefit (i.e., median overall survival ~9 months). Today, median overall survival exceeds 2 years [82], and is expected to increase further with optimization of standard-of-care using these and other recently approved agents.
Seven NMEs were approved for treatment of unresectable or metastatic melanoma by the FDA 2010–2018. Four of these new drugs are LMW-NMEs that inhibit protein kinases (vemurafenib, approved 2011; dabrafenib, 2013; trametinib, 2013; encorafenib, 2018). The remainder are biologic-NMEs that target cytotoxic T lymphocyte protein 4 (CTLA-4) (ipilimumab, approved 2011) or programmed death receptor 1 (PD-1) (pembrolizumab, 2014; nivolumab, 2014) and block downregulation of T cell function by tumor cells.
The first PDB structure of the catalytic domain of wild-type human BRAF (PDB ID: 1uwh) [83] was deposited by academic researchers in 2004. At that time, >30 BRAF gene mutations had been associated with human cancers. Most of these mutations mapped to the activation segment or the P-loop within the catalytic domain, where they were thought to destabilize the inactive conformation of the enzyme. A second PDB structure of mutant BRAF (PDB ID: 1uwj) [83] contributed by the same group revealed a tool compound (BAY43–9006) binding to the inactive conformation of the enzyme. It was subsequently documented that the V600E mutant form of BRAF is present in ~50% of late-stage, metastatic melanomas (reviewed in [84]), making this mutant enzyme a highly attractive discovery target.
Vemurafenib was discovered by Plexxikon in the course of a well-publicized fragment-based, structure-guided lead-optimization campaign targeting V600E BRAF (PDB ID: 3c4c, 3c4d, 3c4e, 3c4f and 4fk3) [85]. The Plexxikon structure of vemurafenib bound to V600E BRAF (PDB ID: 3og7) [43] is illustrated in Figure 3a. Dabrafenib was discovered by GSK with the aid of computational docking into one of the PDB structures of V600E BRAF determined by Plexxikon [86]. Two PDB structures of dabrafenib bound to other mutant forms of BRAF were subsequently contributed to the PDB by Boehringer Ingelheim (PDB ID: 5cs2) [87] and Genentech (PDB ID: 5hie) [88]. Vemurafenib binding to V600E BRAF is compared in Figure 3b to that of dabrafenib (PDB ID: 4xv2) [34]. Close inspection of the modes of inhibitor binding reveals common (e.g., H-bonding engagement of the hinge region) and disparate features of BRAF–ligand interactions for the two inhibitors (Figure 3b).
Figure 3.

Hinge-binding inhibitors targeting mutant BRAF. (a) V600E BRAF kinase (blue) bound to vemurafenib (yellow) (PDB ID: 3og7). (b) Active site of V600E BRAF kinase (PDB ID: 3og7) showing vemurafenib (yellow; PDB ID: 3og7) overlaid with dabrafenib (red; PDB ID: 4xv2).
BRAF V600E mutations result in constitutive activation of the signaling pathway that includes the mitogen-activated protein kinases MEK1 and MEK2 (reviewed in [89]), making these enzymes attractive drug discovery targets for advanced-stage melanoma. The earliest public-domain human MEK1 (PDB ID: 1s9j) [90] and MEK2 (PDB ID: 1s9i) [90] structures were deposited to the PDB in 2004 by Pfizer. Trametinib was discovered by Japan Tobacco using medicinal chemistry optimization of an HTS hit [91]. Trametinib is not represented in the PDB. Unlike the three ATP-competitive inhibitors of BRAF (vemurafenib, dabrafenib and encorafenib), trametinib is an allosteric inhibitor of MEK1/MEK2 [92]. Trametinib inhibits BRAF V600 mutation positive melanoma cell growth in vitro and in vivo (NB: trametinib was approved for the treatment of patients who have not received prior BRAF inhibitor therapy).
Vemurafenib and dabrafenib were each initially approved for single-agent treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation confirmed by an FDA-approved test. Initial results were promising, with objective tumor responses in approximately half of patients with advanced-stage melanoma. However, the duration of responses proved limited in most patients, with progression-free survival of ~6 months as a result of emergence of acquired resistance following activation of MEK1 and/or MEK2 [93]. In 2015, cobimetinib, an additional MEK inhibitor, was approved in combination with vemurafenib for treatment of unresectable or metastatic melanoma with a BRAF V600E or V600K mutation. Cobimetinib was discovered by Exelixis during the course of a structure-guided drug discovery campaign (PDB ID: 4an2) [40].
The remaining LMW-NME targeting BRAF (encorafenib) was approved in 2018 for use in combination with the MEK inhibitor binimetinib for treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation confirmed by an FDA-approved diagnostic test. Encorafenib was discovered by the Novartis Institutes for Biomedical Research during the course of a structure-guided drug discovery campaign [94]. Encorafenib is not represented in the PDB. Binimetinib is a mitogen-activated protein kinase 1/2 (MEK1/2) inhibitor that was discovered by Array Biopharma during the course of a structure-guided drug discovery campaign (private communication). The encorafenib–binimetinib combination showed significant clinical benefit versus encorafenib or vemurafenib used as single agents [95] (see [96] for a comprehensive review of the structural biology of small-molecule BRAF inhibitors).
Modulation of T-cell-mediated immunity is a medically important phenomenon that has been significantly impacted by structural biologists and the PDB. The archive currently houses >750 related PDB structures, which together reveal the molecular mechanisms underpinning antigen presentation to T cell receptors and explain much of the T cell regulation in 3D. The earliest such contribution was the landmark crystal structure of the major histocompatibility complex (MHC) (PDB ID: 1hla) [97]. Subsequently, deposited PDB structures revealed how MHC presents linear peptide antigens to T cells (e.g., PDB ID: 1hsa) [98] and, in turn, how MHC-peptide-antigen complexes are recognized by T cell receptors (e.g., PDB ID: 1ao7) [99]. Thereafter, structural biologists revealed at the atomic level many of the protein–protein interactions responsible for regulating T cells. Various biopharmaceutical companies acted on these insights by successfully targeting immune checkpoints leading to FDA approval of seven antibody therapeutics 2010–2018.
The first of these biologic-NMEs (ipilimumab, Bristol-Myers Squibb, approved 2011) targets CTLA-4, thereby blockading negative regulation of T cells by B7–1 or B7–2 proteins found on the surface of tumor cells. The PDB houses multiple structures of CTLA-4 (earliest PDB ID: 1ah1) [100], including those of CTLA-4 binding to B7–1 (PDB ID: 1il8) [101] and CTLA-4 binding to B7–2 (PDB ID: 1ah1) [102]. Publication of the co-crystal structure of the Fab fragment of ipilumumab recognizing CTLA-4 followed some years after drug approval (PDB ID: 5tru) [103].
Nivolumab (Bristol-Myers Squibb) and pembrolizumab (Merck) were both approved in 2014. These antibodies target PD-1, thereby blockading downregulation of T cells as a result of PD-1 binding to programmed death receptor ligand 1 (PD-L1) or PD-L2 found on the surface of tumor cells (Figure 4). The PDB houses multiple structures of PD-1 (earliest PDB ID: 1npu) [104], PD-L1 (earliest PDB ID: 3bis) [105] and PD-L2 (earliest PDB ID: 3bov) [106]. In addition, the PDB contains structures of PD-1–PD-L1 complexes (earliest PDB ID: 3bik; Figure 4a) [105] and PD-1–PD-L2 complexes (earliest PDB ID: 3bp5; Figure 4b) [106]. Structures of nivolumab (earliest PDB ID: 5ggq) [107] and pembrolizumab (earliest PDB ID: 5dk3) [108] are similarly available from the PDB. The PDB also contains multiple structures of nivolumab–PD-1 complexes (earliest PDB ID: 5ggr) [107] and pembrolizumab–PD-1 complexes (earliest PDB ID: 5jxe; Figure 4c) [109].
Figure 4.

Immune checkpoint blockade. (a) PD-1(red) bound to PD-L1 (blue) (PDB ID: 3bik). (b) PD-1 (red) bound to PD-L2 (light blue) (PDB ID: 3bp5). (c) PD-1 (red) recognition by pembrolizumab Fab (green) (PDB ID: 5ggs). (d) PD-1 (red) recognition by nivolumab Fab (green) (PDB ID: 5ggr).
Current standard-of-care for advanced-stage melanoma [110,111] begins with either pembrolizumab or nivolumab, particularly in individuals where tumor cells do not possess mutant BRAF. Both of these biologic-NMEs can shrink tumors for long periods of time in favorable cases (former US president, Jimmy Carter benefited from pembrolizumab). Ipilimumab is not typically used as a first-line treatment, although it can be combined with nivolumab or pembrolizumab to improve the likelihood of a tumor response. If a BRAF gene mutation is detected in the affected individual’s tumor, combination therapy with a small-molecule BRAF inhibitor plus a MEK inhibitor (e.g., vemurafenib–cobimetinib, dabrafeninb–trametinib or encorafenib–binimetinib) can be used as an alternative first-line treatment strategy. At present, optimal choices as to first-line treatment, combinations of antibodies and combinations of antibodies with targeted agents are being evaluated in clinical trials. Prognoses for individuals with advanced-stage melanoma appear likely to improve further as clinical oncologists and dermatologists gain more experience using these new agents.
Concluding remarks
This review documents that PDB structural data contribute broadly to oncology drug discovery and development in the biopharmaceutical industry (and to a lesser extent in academia). For the 54 LMW-NMEs analyzed, all of which have known protein targets, there is evidence from the PDB and/or the scientific literature that discovery and development of every one of these new drugs was facilitated by the availability of public-domain 3D structure information. In >70% of cases, the LMW-NMEs were the product of biopharmaceutical company structure-guided drug discovery efforts, involving co-crystal structural studies and/or computational docking into crystal structures, among others. For the 25 biologic-NMEs analyzed there is again evidence from the PDB and/or the scientific literature that discovery and development of >90% of these new drugs was facilitated directly or indirectly by the availability of public-domain 3D structural information.
With year-on-year growth in the number of structures in the PDB approaching 10%, the impact of the resource and structure-guided approaches on drug discovery and development is destined to remain significant. Moreover, the growing number of PDB structures coming from cryoelectron microscopy since the advent of the ‘resolution revolution’ [112] promises even broader 3D structural coverage of the human proteome. We can expect deposition of new PDB structures of many of the integral membrane proteins and other macromolecular machines that are currently being suboptimally targeted with relatively nonspecific agents or are considered to be undruggable [113].
The long-standing requirement for PDB deposition of 3D atomic coordinates and experimental data and metadata upon publication ensures that this valuable information is made immediately available to basic and applied researchers around the world without limitations on usage. Moreover, expert biocuration and standardized validation of the experimental data and the atomic coordinates across the PDB help to ensure that the archive as a whole can be mined for new knowledge using statistical tools [114,115] or machine learning approaches [27].
As custodian of the PDB Core Archive, the wwPDB partnership is committed to the FAIR principles [10], which help ensure the broadest possible use of public domain biomedical research data. The PDB has been recognized as a Core Certified Repository by CoreTrustSeal (coretrustseal.org). This international, community-based, nongovernmental, non-profit organization promotes sustainable and trustworthy data infrastructures of which the PDB is widely regarded as a gold-standard exemplar.
Supplementary Material
Highlights:
Protein Data Bank (PDB) provides open access to >150K 3D structures of biomolecules
79 anticancer NMEs with known molecular targets were approved by the FDA 2010–2018
PDB provides target structures for >90% of anticancer NMEs approved 2010–2018
PDB provides target–NME co-structures for >50% anticancer NMEs approved 2010–2018
PDB facilitated discovery and development of >90% anticancer NMEs approved 2010–2018
Acknowledgments
We thank the >30 000 structural biologists who have deposited structures to the PDB Core Archive since 2000. We also thank Drs James (Jay) Bradner, Kirk L. Clark, Marc C. Deller, Dashyant Dhanak, Jose Duarte, Gary L. Gilliland, Fred D. Ledley, Catherine E. Peishoff, Alexander Rose, Joan Segura, Paul A. Sprengeler, Jonathan H. Watanabe, Christine Zardecki and X.F. Steven Zheng for their insightful comments, and we gratefully acknowledge contributions from all members of the Research Collaboratory for Structural Bioinformatics PDB and our Worldwide Protein Data Bank partners. RCSB PDB is jointly funded by the National Science Foundation (DBI-1832184), the US Department of Energy (DE-SC0019749) and the National Cancer Institute, National Institute of Allergy and Infectious Diseases and National Institute of General Medical Sciences of the National Institutes of Health under grant R01GM133198. J.D.W., R.S, B.P.H. and S.K.B. assembled and analyzed the data and wrote the manuscript.
Biographies
John D. Westbrook
John D. Westbrook PhD is a computational chemist and the lead data and software architect for the RCSB Protein Data Bank at Rutgers, State University of New Jersey. He serves on data standards committees for the International Union of Crystallography, the American Crystallographic Association and Research Data Alliance. Awards and prizes include Biocuration Career Award from the International Biocuration Society, Rutgers University Supercomputer Fellowship, Rutgers University Johnson Fellowship, Raymond Davis Memorial Fellowship from the Society of Photographic Science and Engineering, and Minolta Corporation Fellowship in Imaging Science.

Brian P. Hudson
Brian P. Hudson PhD is an expert in structural biology and biological database curation. He currently serves as a Senior Biocurator at the RCSB Protein Data Bank at Rutgers, State University of New Jersey.

Stephen K. Burley
Stephen K. Burley MD, DPhil is an expert in the areas of molecular biophysics, structural biology, bioinformatics, data science, structure/fragment-based drug discovery and clinical medicine/oncology. Burley currently serves as University Professor and Henry Rutgers Chair, Founding Director of the Institute for Quantitative Biomedicine, and Director of the RCSB Protein Data Bank at Rutgers, State University of New Jersey. He is a Member of the Rutgers Cancer Institute of New Jersey, where he co-leads Cancer Pharmacology. Previous work experience includes senior leadership roles at Lilly Research Laboratories, SGX Pharmaceuticals, The Rockefeller University and the Howard Hughes Medical Institute.

Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary material is provided describing the assembly and analysis of the dataset described in this review.
Conflicts of interest
The authors declare no competing financial interests.
References
- 1.Blundell TL (2017) Protein crystallography and drug discovery: recollections of knowledge exchange between academia and industry. IUCrJ 4, 308–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klebe G, ed. (2013) Drug design: methodology, concepts, and mode-of-action, Berlin: Springer [Google Scholar]
- 3.Burley SK et al. (2018) RCSB Protein Data Bank: sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci 27, 316–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown KK et al. (2018) Approaches to target tractability assessment – a practical perspective. MedChemComm 9, 606–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stoll F et al. (2011) Utility of protein structures in overcoming ADMET-related issues of drug-like compounds. Drug Discov. Today 16, 530–538 [DOI] [PubMed] [Google Scholar]
- 6.Mullard A (2016) 2015 FDA drug approvals. Nat. Rev. Drug Discov 15, 73–76 [DOI] [PubMed] [Google Scholar]
- 7.Gilliland GL et al. (2012) Leveraging SBDD in protein therapeutic development: antibody engineering. Methods Mol. Biol 841, 321–349 [DOI] [PubMed] [Google Scholar]
- 8.Chiu ML and Gilliland GL (2016) Engineering antibody therapeutics. Curr. Opin. Struct. Biol 38, 163–173 [DOI] [PubMed] [Google Scholar]
- 9.Protein Data Bank (1971) Crystallography: Protein Data Bank. Nature New Biol 233, 22320480989 [Google Scholar]
- 10.Wilkinson MD et al. (2016) The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berman HM et al. (2003) Announcing the worldwide Protein Data Bank. Nat. Struct. Biol 10, 980. [DOI] [PubMed] [Google Scholar]
- 12.wwPDB Consortium (2019) Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res 47, D520–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berman HM et al. (2000) The Protein Data Bank. Nucleic Acids Res 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burley SK et al. (2019) RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res 47, D464–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mir S et al. (2018) PDBe: towards reusable data delivery infrastructure at protein data bank in Europe. Nucleic Acids Res 46, D486–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinjo AR et al. (2017) Protein Data Bank Japan (PDBj): updated user interfaces, resource description framework, analysis tools for large structures. Nucleic Acids Res 45, D282–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ulrich EL et al. (2008) BioMagResBank. Nucleic Acids Res 36, D402–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westbrook JD and Burley SK (2019) How structural biologists and the Protein Data Bank contributed to recent FDA New Drug Approvals. Structure 27, 211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z et al. (2017) Lessons learned from two decades of anticancer drugs. Trends Pharmacol. Sci 38, 852–872 [DOI] [PubMed] [Google Scholar]
- 20.Hu T et al. (2018) The impact of structural biology in medicine illustrated with four case studies. J. Mol. Med 96, 9–19 [DOI] [PubMed] [Google Scholar]
- 21.The UniProt Consortium (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res 45, D158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burley SK et al. (2018) RCSB Protein Data Bank: sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci 27, 316–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Renaud JP et al. (2016) Biophysics in drug discovery: impact, challenges and opportunities. Nat. Rev. Drug Discov 15, 679–698 [DOI] [PubMed] [Google Scholar]
- 24.Erlanson DA et al. (2016) Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov 15, 605–619 [DOI] [PubMed] [Google Scholar]
- 25.Burns MC et al. (2018) High-throughput screening identifies small molecules that bind to the RAS:SOS:RAS complex and perturb RAS signaling. Anal. Biochem 548, 44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin JH (2016) Review structure- and dynamics-based computational design of anticancer drugs. Biopolymers 105, 2–9 [DOI] [PubMed] [Google Scholar]
- 27.Lo YC et al. (2018) Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 23, 1538–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawase T et al. (2019) Effect of Fms-like tyrosine kinase 3 (FLT3) ligand (FL) on antitumor activity of gilteritinib, a FLT3 inhibitor, in mice xenografted with FL-overexpressing cells. Oncotarget 10, 6111–6123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuki M et al. (2018) Lenvatinib inhibits angiogenesis and tumor fibroblast growth factor signaling pathways in human hepatocellular carcinoma models. Cancer Med 7, 2641–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bender AT et al. (2017) Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B-cell receptor signaling. Mol. Pharmacol 91, 208–219 [DOI] [PubMed] [Google Scholar]
- 31.Chen P et al. (2016) Spectrum and degree of CDK drug interactions predicts clinical performance. Mol. Cancer Ther 15, 2273–2281 [DOI] [PubMed] [Google Scholar]
- 32.Scott WJ et al. (2016) Discovery and SAR of novel 2,3-dihydroimidazo[1,2-c]quinazoline PI3K inhibitors: identification of copanlisib (BAY 80–6946). ChemMedChem 11, 1517–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang WS et al. (2016) Discovery of brigatinib (AP26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J. Med. Chem 59, 4948–4964 [DOI] [PubMed] [Google Scholar]
- 34.Zhang C et al. (2015) RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 526, 583–586 [DOI] [PubMed] [Google Scholar]
- 35.Somoza JR et al. (2015) Structural, biochemical, and biophysical characterization of idelalisib binding to phosphoinositide 3-kinase delta. J. Biol. Chem 290, 8439–8446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friboulet L et al. (2014) The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov 4, 662–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gajiwala KS et al. (2013) Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure 21, 209–219 [DOI] [PubMed] [Google Scholar]
- 38.McTigue M et al. (2012) Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. U. S. A 109, 18281–18289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solca F et al. (2012) Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther 343, 342–350 [DOI] [PubMed] [Google Scholar]
- 40.Rice KD et al. (2012) Novel carboxamide-based allosteric MEK inhibitors: discovery and optimization efforts toward XL518 (GDC-0973). ACS Med. Chem. Lett 3, 416–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cui JJ et al. (2011) Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem 54, 6342–6363 [DOI] [PubMed] [Google Scholar]
- 42.Sakamoto H et al. (2011) CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 19, 679–690 [DOI] [PubMed] [Google Scholar]
- 43.Bollag G et al. (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Hare T et al. (2009) AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16, 401–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qian F et al. (2009) Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res 69, 8009–8016 [DOI] [PubMed] [Google Scholar]
- 46.Johnson TW et al. (2014) Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem 57, 4720–4744 [DOI] [PubMed] [Google Scholar]
- 47.Yosaatmadja Y et al. (2015) Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J. Struct. Biol 192, 539–544 [DOI] [PubMed] [Google Scholar]
- 48.Levinson NM and Boxer SG (2014) A conserved water-mediated hydrogen bond network defines bosutinib’s kinase selectivity. Nat. Chem. Biol 10, 127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knowles PP et al. (2006) Structure and chemical inhibition of the RET tyrosine kinase domain. J. Biol. Chem 281, 33577–33587 [DOI] [PubMed] [Google Scholar]
- 50.Yen K et al. (2017) AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov 7, 478–493 [DOI] [PubMed] [Google Scholar]
- 51.Birkinshaw RW et al. (2019) Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat. Commun 10, 2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thorsell AG et al. (2017) Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J. Med. Chem 60, 1262–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hai Y and Christianson DW (2016) Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol 12, 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gathiaka S et al. (2016) D3R grand challenge 2015: evaluation of protein-ligand pose and affinity predictions. J. Comput. Aided Mol. Des 30, 651–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaieb Z et al. (2018) D3R Grand Challenge 2: blind prediction of protein-ligand poses, affinity rankings, and relative binding free energies. J. Comput. Aided Mol. Des 32, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gaieb Z et al. (2019) D3R Grand Challenge 3: blind prediction of protein-ligand poses and affinity rankings. J. Comput. Aided Mol. Des 33, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams PA et al. (2003) Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 424, 464–468 [DOI] [PubMed] [Google Scholar]
- 58.Martinez L et al. (2014) Understanding polyspecificity within the substrate-binding cavity of the human multidrug resistance P-glycoprotein. FEBS J 281, 673–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang W and MacKinnon R (2017) Cryo-EM structure of the open human ether-a-go-go-related K(+) channel hERG. Cell 169, 422–430; e410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiu ML et al. (2019) Antibody structure and function: the basis for engineering therapeutics. Antibodies 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Epp O et al. (1975) The molecular structure of a dimer composed of the variable portions of the Bence–Jones protein REI refined at 2.0-A resolution. Biochemistry 14, 4943–4952 [DOI] [PubMed] [Google Scholar]
- 62.Satow Y et al. (1986) Phosphocholine binding immunoglobulin Fab McPC603. An X-ray diffraction study at 2.7 A. J. Mol. Biol 190, 593–604 [DOI] [PubMed] [Google Scholar]
- 63.Zdanov A et al. (1994) Structure of a single-chain antibody variable domain (Fv) fragment complexed with a carbohydrate antigen at 1.7-A resolution. Proc. Natl. Acad. Sci. U. S. A 91, 6423–6427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Teplyakov A et al. (2014) Antibody modeling assessment II. Structures and models. Proteins 82, 1563–1582 [DOI] [PubMed] [Google Scholar]
- 65.Teplyakov A et al. (2016) Structural diversity in a human antibody germline library. MAbs 8, 1045–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burt R et al. (2019) Blinatumomab, a bispecific B-cell and T-cell engaging antibody, in the treatment of B-cell malignancies. Hum. Vaccin. Immunother 15, 594–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maier W et al. (1996) [Lermoyez syndrome--electrocochleographic studies]. Laryngorhinootologie 75, 372–376 [DOI] [PubMed] [Google Scholar]
- 68.Bennett MJ et al. (1994) Refined structure of dimeric diphtheria toxin at 2.0 A resolution. Protein Sci 3, 1444–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frankel AE et al. (2000) Characterization of diphtheria fusion proteins targeted to the human interleukin-3 receptor. Protein Eng 13, 575–581 [DOI] [PubMed] [Google Scholar]
- 70.Deisenhofer J (1981) Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 20, 2361–2370 [PubMed] [Google Scholar]
- 71.Wiesmann C et al. (1997) Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 91, 695–704 [DOI] [PubMed] [Google Scholar]
- 72.Olson EM (2012) Maximizing human epidermal growth factor receptor 2 inhibition: a new oncologic paradigm in the era of targeted therapy. J. Clin. Oncol 30, 1712–1714 [DOI] [PubMed] [Google Scholar]
- 73.Franklin MC et al. (2004) Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5, 317–328 [DOI] [PubMed] [Google Scholar]
- 74.Cho HS et al. (2003) Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760 [DOI] [PubMed] [Google Scholar]
- 75.Deng X et al. (2018) Enhancing antibody patent protection using epitope mapping information. MAbs 10, 204–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Russo AA et al. (1998) Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395, 237–243 [DOI] [PubMed] [Google Scholar]
- 77.Ceccarelli C et al. (2002) Crystal structure of porcine mitochondrial NADP+-dependent isocitrate dehydrogenase complexed with Mn2+ and isocitrate. Insights into the enzyme mechanism. J. Biol. Chem 277, 43454–43462 [DOI] [PubMed] [Google Scholar]
- 78.Day PJ et al. (2009) Crystal structure of human CDK4 in complex with a D-type cyclin. Proc. Natl. Acad. Sci. U. S. A 106, 4166–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sherr CJ et al. (2016) Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov 6, 353–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Molenaar RJ et al. (2018) Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 37, 1949–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang F et al. (2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626 [DOI] [PubMed] [Google Scholar]
- 82.Luke JJ et al. (2017) Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol 14, 463–482 [DOI] [PubMed] [Google Scholar]
- 83.Wan PT et al. (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867 [DOI] [PubMed] [Google Scholar]
- 84.Gray-Schopfer V et al. (2007) Melanoma biology and new targeted therapy. Nature 445, 851–857 [DOI] [PubMed] [Google Scholar]
- 85.Tsai J et al. (2008) Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. U. S. A 105, 3041–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rheault TR et al. (2013) Discovery of dabrafenib: a selective inhibitor of Raf kinases with antitumor activity against B-Raf-driven tumors. ACS Med. Chem. Lett 4, 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Waizenegger IC et al. (2016) A novel RAF kinase inhibitor with DFG-out-binding mode: high efficacy in BRAF-mutant tumor xenograft models in the absence of normal tissue hyperproliferation. Mol. Cancer Ther 15, 354–365 [DOI] [PubMed] [Google Scholar]
- 88.Foster SA et al. (2016) Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell 29, 477–493 [DOI] [PubMed] [Google Scholar]
- 89.Acosta AM and Kadkol SS (2016) Mitogen-activated protein kinase signaling pathway in cutaneous melanoma: an updated review. Arch. Pathol. Lab. Med 140, 1290–1296 [DOI] [PubMed] [Google Scholar]
- 90.Ohren JF et al. (2004) Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol 11, 1192–1197 [DOI] [PubMed] [Google Scholar]
- 91.Abe H et al. (2011) Discovery of a highly potent and selective MEK inhibitor: GSK1120212 (JTP-74057 DMSO Solvate). ACS Med. Chem. Lett 2, 320–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gilmartin AG et al. (2011) GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res 17, 989–1000 [DOI] [PubMed] [Google Scholar]
- 93.Eroglu Z and Ribas A (2016) Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Ther. Adv. Med. Oncol 8, 48–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Drahl C (2013) LGX818, made to fight melanoma. Chem. Eng. News 91, 14 [Google Scholar]
- 95.Dummer R et al. (2018) Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised Phase 3 trial. Lancet Oncol 19, 603–615 [DOI] [PubMed] [Google Scholar]
- 96.Agianian B and Gavathiotis E (2018) Current insights of BRAF inhibitors in cancer. J. Med. Chem 61, 5775–5793 [DOI] [PubMed] [Google Scholar]
- 97.Bjorkman PJ et al. (1987) Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329, 506. [DOI] [PubMed] [Google Scholar]
- 98.Madden DR et al. (1992) The three-dimensional structure of HLA-B27 at 2.1 A resolution suggests a general mechanism for tight peptide binding to MHC. Cell 70, 1035–1048 [DOI] [PubMed] [Google Scholar]
- 99.Garboczi DN et al. (1996) Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature 384, 134–141 [DOI] [PubMed] [Google Scholar]
- 100.Metzler WJ et al. (1997) Solution structure of human CTLA-4 and delineation of a CD80/CD86 binding site conserved in CD28. Nat. Struct. Biol 4, 527–531 [DOI] [PubMed] [Google Scholar]
- 101.Stamper CC et al. (2001) Crystal structure of the B7–1/CTLA-4 complex that inhibits human immune responses. Nature 410, 608–611 [DOI] [PubMed] [Google Scholar]
- 102.Schwartz JC et al. (2001) Structural basis for co-stimulation by the human CTLA-4/B7–2 complex. Nature 410, 604–608 [DOI] [PubMed] [Google Scholar]
- 103.Ramagopal UA et al. (2017) Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab. Proc. Natl. Acad. Sci. U. S. A 114, E4223–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang X et al. (2004) Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 20, 337–347 [DOI] [PubMed] [Google Scholar]
- 105.Lin DY et al. (2008) The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl. Acad. Sci. U. S. A 105, 3011–3016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lazar-Molnar E et al. (2008) Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc. Natl. Acad. Sci. U. S. A 105, 10483–10488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee JY et al. (2016) Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun 7, 13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Scapin G et al. (2015) Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat. Struct. Mol. Biol 22, 953–958 [DOI] [PubMed] [Google Scholar]
- 109.Na Z et al. (2017) Structural basis for blocking PD-1-mediated immune suppression by therapeutic antibody pembrolizumab. Cell Res 27, 147–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rozeman EA et al. (2018) Advanced melanoma: current treatment options, biomarkers, and future perspectives. Am. J. Clin. Dermatol 19, 303–317 [DOI] [PubMed] [Google Scholar]
- 111.Domingues B et al. (2018) Melanoma treatment in review. Immunotargets Ther 7, 35–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kuhlbrandt W (2014) Biochemistry. The resolution revolution. Science 343, 1443–1444 [DOI] [PubMed] [Google Scholar]
- 113.Goodsell DS et al. (2020) RCSB Protein Data Bank: enabling biomedical research and drug discovery. Protein Sci 29, 52–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shao C et al. (2018) Outlier analyses of the Protein Data Bank archive using a probability-density-ranking approach. Sci. Data 5, 180293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shao C et al. (2017) Multivariate analyses of quality metrics for crystal structures in the Protein Data Bank archive. Structure 25, 458–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.McTigue MA et al. (1999) Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: a key enzyme in angiogenesis. Structure 7, 319–330 [DOI] [PubMed] [Google Scholar]
- 117.Xu W et al. (1997) Three-dimensional structure of the tyrosine kinase c-Src. Nature 385, 595–602 [DOI] [PubMed] [Google Scholar]
- 118.Mol CD et al. (2003) Structure of a c-kit product complex reveals the basis for kinase transactivation. J. Biol. Chem 278, 31461–31464 [DOI] [PubMed] [Google Scholar]
- 119.Nagar B et al. (2002) Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res 62, 4236–4243 [PubMed] [Google Scholar]
- 120.Stamos J et al. (2002) Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem 277, 46265–46272 [DOI] [PubMed] [Google Scholar]
- 121.Yun CH et al. (2008) The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A 105, 2070–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Koshiba S et al. (2010) Structural basis for the recognition of nucleophosminanaplastic lymphoma kinase oncoprotein by the phosphotyrosine binding domain of Suc1-associated neurotrophic factor-induced tyrosine-phosphorylated target-2. J. Struct. Funct. Genomics 11, 125–141 [DOI] [PubMed] [Google Scholar]
- 123.Schiering N et al. (2003) Crystal structure of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microbial alkaloid K-252a. Proc. Natl. Acad. Sci. U. S. A 100, 12654–12659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huang CH et al. (2007) The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 318, 1744–1748 [DOI] [PubMed] [Google Scholar]
- 125.Hyvonen M and Saraste M (1997) Structure of the PH domain and Btk motif from Bruton’s tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia. EMBO J 16, 3396–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Skerratt SE et al. (2016) The discovery of a potent, selective, and peripherally restricted Pan-Trk inhibitor (PF-06273340) for the treatment of pain. J. Med. Chem 59, 10084–10099 [DOI] [PubMed] [Google Scholar]
- 127.Griffith J et al. (2004) The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol. Cell 13, 169–178 [DOI] [PubMed] [Google Scholar]
- 128.Xu X et al. (2004) Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem 279, 33946–33957 [DOI] [PubMed] [Google Scholar]
- 129.Petros AM et al. (2001) Solution structure of the antiapoptotic protein bcl-2. Proc. Natl. Acad. Sci. U. S. A 98, 3012–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kinoshita T et al. (2004) Inhibitor-induced structural change of the active site of human poly(ADP-ribose) polymerase. FEBS Lett 556, 43–46 [DOI] [PubMed] [Google Scholar]
- 131.Matias PM et al. (2000) Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J. Biol. Chem 275, 26164–26171 [DOI] [PubMed] [Google Scholar]
- 132.Somoza JR et al. (2004) Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12, 1325–1334 [DOI] [PubMed] [Google Scholar]
- 133.Byrne EFX et al. (2016) Structural basis of Smoothened regulation by its extracellular domains. Nature 535, 517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.DeVore NM and Scott EE (2012) Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 482, 116–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Angers S et al. (2006) Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593 [DOI] [PubMed] [Google Scholar]
- 136.Sievers QL et al. (2018) Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362, 6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Harshbarger W et al. (2015) Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 23, 418–424 [DOI] [PubMed] [Google Scholar]
- 138.Schrader J et al. (2016) The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 353, 594–598 [DOI] [PubMed] [Google Scholar]
- 139.Aldaz H et al. (2005) Insights into microtubule nucleation from the crystal structure of human gamma-tubulin. Nature 435, 523–527 [DOI] [PubMed] [Google Scholar]
- 140.Brown A et al. (2014) Structure of the large ribosomal subunit from human mitochondria. Science 346, 718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gurel G et al. (2009) U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J. Mol. Biol 389, 146–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Alexeeva M et al. (2015) The structure of a dual-specificity tyrosine phosphorylation-regulated kinase 1A-PKC412 complex reveals disulfide-bridge formation with the anomalous catalytic loop HRD(HCD) cysteine. Acta Crystallogr. D Biol. Crystallogr 71, 1207–1215 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.