

Abstract
Small cell lung cancer (SCLC) is a highly aggressive malignancy with poor outcomes associated with resistance to cisplatin-based chemotherapy. Enhancer of Zeste Homolog 2 (EZH2) is the catalytic subunit of Polycomb Repressive Complex 2 (PRC2), which silences transcription through trimethylation of histone H3 lysine 27 (H3K27me3) and has emerged as an important therapeutic target with inhibitors targeting its methyltransferase activity under clinical investigation. Here, we show that EZH2 has a non-catalytic and PRC2 independent role in stabilizing DDB2 to promote nucleotide excision repair (NER) and govern cisplatin resistance in SCLC. Using a synthetic lethality screen, we identified important regulators of cisplatin resistance in SCLC cells, including EZH2. EZH2 depletion causes cellular cisplatin and UV hypersensitivity in an epistatic manner with DDB1-DDB2. EZH2 complexes with DDB1-DDB2 and promotes DDB2 stability by impairing its ubiquitination independent of methyltransferase activity or PRC2, thereby facilitating DDB2 localization to cyclobutane pyrimidine dimer (CPD) crosslinks to govern their repair. Furthermore, targeting EZH2 for depletion with DZNep strongly sensitizes SCLC cells and tumors to cisplatin. Our findings reveal a non-catalytic and PRC2-independent function for EZH2 in promoting NER through DDB2 stabilization, suggesting a rationale for targeting EZH2 beyond its catalytic activity for overcoming cisplatin resistance in SCLC.
Keywords: EZH2, DNA repair, DNA damage response, checkpoint, nucleotide excision repair, NER, cisplatin, small cell lung cancer, SCLC, DDB1, DDB2, PRC2, non-catalytic, catalytic, siRNA screen, synthetic lethal
INTRODUCTION
SCLC is a highly aggressive malignancy with a 5-year survival rate of only 7 percent [1]. The first-line treatment regimen for SCLC consists of platinum-based EP chemotherapy: cisplatin or carboplatin, DNA crosslinking agents, in combination with etoposide, a topoisomerase II inhibitor. While most SCLC patients will initially respond to EP treatment, the majority will ultimately develop treatment resistance [2]. Response rates for second-line topoisomerase I inhibitors for SCLC are much lower [3], and SCLC currently lacks any FDA-approved targeted therapies. Therefore, novel therapeutic approaches for SCLC treatment are urgently needed.
The DNA damage response (DDR) is critical for responding to DNA damage induced by chemotherapy. Cisplatin primarily induces cytotoxicity by creating 1,2-intrastrand d(GpG) adducts. The major mechanism for repair of these DNA intrastand crosslinks is nucleotide excision repair (NER), which is also involved in the repair of other helix-distorting DNA lesions, including UV-induced CPD and 6–4 pyrimidine-pyrimidone photoproducts (6–4PP) [4]. The Damage Specific DNA Binding Protein 1 and 2 (DDB1-DDB2) heterodimer is a component of the CUL4-RING E3 ubiquitin ligase complex (CRL4), which promotes the repair of NER lesions through the global genome branch of the NER pathway. DDB2 recognizes DNA lesions [5] and recruits downstream NER factors to repair the lesion [6]. Upon lesion detection, DDB2 is ubiquitinated and targeted for degradation by CRL4 [6-8], although the mechanism by which DDB2 ubiquitination is regulated is not fully understood.
EZH2 is the catalytic subunit of the PRC2, which functions together with Embryonic Ectoderm Development (EED) and Suppressor of Zeste 12 (SUZ12) as a histone methyltransferase to silence areas of active transcription through H3K27me3. EZH2 is an oncogene that is overexpressed in many cancer types, including SCLC [9-11], and high EZH2 expression is correlated with tumorigenesis, cancer progression, metastasis, and poor prognoses [10, 12-19]. As such, EZH2 has emerged as an important therapeutic target. Several EZH2 inhibitors targeting its methyltransferase activity have been developed [20-25] and are currently undergoing clinical trial testing. However, it is unclear if inhibiting EZH2’s catalytic activity is sufficient to impair its activities governing cancer cell survival, as non-catalytic and PRC2-independent roles for EZH2, largely involving transcriptional regulation independent of H3K27me3, have also been reported [26-32]. EZH2 has also been shown to directly methylate non-histone proteins to promote their degradation [29, 33].
A role for EZH2 in genome maintenance has previously been described. EZH2 has been reported to mediate resistance to DNA damaging agents, including etoposide in SCLC [34-37] and cisplatin in several other cancer cell types [9, 38-41], localize to DNA damage sites [42, 43], and promote DNA double-strand break (DSB) repair [42, 44], degradation of stalled replication forks [45], and checkpoint signaling [46]; however, these functions have largely been attributed to H3K27Me3, either through transcriptional repression or a recently-described function for H3K27Me3 in the recruitment of MUS81 [45]. Significantly, a role for EZH2 in promoting NER, as well as in promoting genome maintenance independent of its catalytic activity or PRC2, has previously not been shown.
Here, we define a novel role for EZH2 in governing cisplatin resistance in SCLC by promoting NER. We show that EZH2 has a non-catalytic and PRC2-independent role in stabilizing DDB2 by impairing its ubiquitination, thereby facilitating DDB2 localization to CPD crosslinks to govern their repair. Furthermore, targeting EZH2 for depletion with DZNep strongly sensitizes SCLC cells and tumors to cisplatin, revealing a potential rationale-driven approach for overcoming cisplatin resistance in SCLC.
MATERIALS AND METHODS
Cell lines
All cell lines were originally purchased from the American Type Culture Collection (ATCC, Manassas, VA). Human SCLC cell lines (H128, H146, H187, H69, DMS114 and DMS153) were provided by the laboratory of Dr. Taofeek Owonikoko [47]. H128, H187, H69, and H146 cells were grown in RPMI 1640 (Gibco) with 7.5% fetal bovine serum (FBS). DMS114 and DMS153 cells were grown in Waymouth’s medium (Gibco) with 5% FBS. HeLa, U2OS, HEK293T, and HCT116 cells were grown in DMEM (Gibco) with 7.5% FBS. BEAS-2B were cultured in DMEM/F-12 medium with 10% FBS. Primary small airway epithelial cells (HSAEC) were grown in Airway Basal Medium (ATCC PCS-300-030) with one Bronchial Epithelial Growth Kit (ATCC PCS-300-040) according to manufacturer’s instructions. All cell lines were grown at 37°C under humidified conditions with 5% CO2 and 95% air.
Details of additional methodologies can be found in Supplementary Materials and Methods.
RESULTS
A siRNA screen targeting nuclear enzymes identifies genes that mediate cisplatin resistance in SCLC
To identify genes critical for governing platinum resistance in SCLC, we performed a cisplatin sensitivity screen with siRNAs targeting 1,006 genes, biased towards nuclear enzymes for future translational application, in SCLC cells. We have previously used our nuclear enzyme siRNA library to perform drug sensitivity screens in other cancer cell types to identify novel regulators of the DDR [48, 49]. We chose the SCLC cell line H128, a cell line with intact DNA repair pathways from a treatment-refractory tumor. H128 cells are strongly resistant to cisplatin, with over 80 percent viability 72 hours following continuous cisplatin treatment at 5 μM, the upper threshold of cisplatin dose achievable in patients. The screen was conducted in triplicate in 96-well plates using siRNA targeting ERCC1 and ATR as positive controls and a non-targeting (NT) siRNA as a negative control. Cells were transfected with the siRNA library, treated after 48 hours with or without cisplatin for 72 hours, and assayed for cell viability with resazurin reagent (Fig. 1A). Sensitivity results from the screen are shown as a volcano plot of log2-transformed average viability against strictly standardized mean difference (SSMD) (Fig. 1B, Supplementary Table S1). We identified 118 cisplatin sensitization hits based on the following criteria: an average cell viability of <0.6, an average SSMD of <−2, and a two-tailed t-test p-value of <0.05 (Supplementary Table S2). The Z-factor of the screen, an indicator of screen quality, was 0.556, which is in the excellent range, indicating that our screen is robust.
Fig. 1.

A siRNA screen targeting nuclear enzymes identifies genes that mediate cisplatin resistance in SCLC. a Primary screen format: H128 cells were transfected with a siRNA library biased towards targeting nuclear enzymes. 48 hours post-transfection, cells were treated with or without 10 μM cisplatin for 72 hours prior to measuring cell viability. b Primary screen results: the normalized cell viability was plotted against the strictly standardized mean difference (SSMD) for each gene targeted by the library. Normalized viability was calculated as the log2 ratio of treated versus untreated cell viability relative to the non-targeting (NT) siRNA control. c Summary of most enriched categories from Gene Ontology (GO) analysis of cisplatin sensitization hits (n = 118). The top 10 significant processes (p ≤ 0.01) are shown, sorted by the percent associated genes found.
To characterize common pathways and potential interactions of our cisplatin sensitization hits, we performed gene ontology (GO) analysis (Fig. 1C, Supplementary Fig. S1A) and network analysis (Supplementary Fig. S1B). As anticipated, DNA repair pathways, including NER, DNA replication, interstrand cross-link repair, and UV protection, emerged as key processes among the cisplatin sensitization hits, demonstrating that our screen can yield DDR proteins previously defined for mediating cisplatin resistance. We also identified histone modification, ubiquitination, chromosome separation, DNA geometric change, and peptidyl-arginine N-methylation, among other processes associated with the cisplatin sensitization hits.
Indeed, a number of NER factors emerged as cisplatin sensitization hits in our primary screen: ERCC1, ERCC2, ERCC4 and ERCC6. EZH2 was among the most efficient sensitizing hits identified, which when silenced, sensitized H128 cells to cisplatin to a similar magnitude as the NER hits. EZH2 is intriguing because it was the only cisplatin sensitization hit that showed significant overexpression in SCLC tissue compared to normal lung epithelial tissue [50, 51], and EZH2 overexpression has been shown to mediate cisplatin resistance in several other cancer cell types [38-41]. One microarray expression dataset reported that EZH2 mRNA was expressed 21 (p=6.01E-9)* fold greater in SCLC versus normal lung tissue [50], while another reported that EZH2 mRNA was expressed 3.9 (p=3.01E-4)* fold greater in SCLC versus normal lung tissue [51]. We also found that EZH2 protein level was overexpressed in SCLC cell lines compared to nontumorigenic BEAS-2B and HSAEC lung epithelial cells (Supplementary Fig. S2A). Thus, EZH2 may be a promising therapeutic target for overcoming cisplatin resistance in SCLC.
EZH2 mediates cisplatin resistance in SCLC
From the primary screen, 23 hits were selected for further validation based on previous identification in other high-throughput DNA damage sensitivity screens, putative interactions with known DNA repair proteins by mass spectrometry (MS), or potential disease relevance, including in SCLC (Supplementary Table S3). To validate each target, we tested if the cisplatin sensitization phenotype could be achieved across more than one individual siRNA to rule out off-target effects of the pooled siRNA used in the primary screen. Of the 23 hits tested, 13 were validated, including EZH2 (Supplementary Table S4). Two unique siRNAs targeting EZH2 sensitized H128 cells to cisplatin (Fig. 2A, Supplementary Fig. S2B, Supplementary Table S4) and western blot confirmed successful EZH2 knockdown (Fig. 2B). We validated this phenotype in H146 SCLC cells (Fig. 2C-D), though the results were not as strong as H128 cells, perhaps due to the lower EZH2 levels in H146 cells (Fig. 2E). Of note, a correlation between the degree of EZH2 knockdown with cisplatin sensitization was observed between the two siRNAs (Fig. 2A-D and Supplementary Fig. S2B), providing additional support that the cisplatin sensitization phenotype following EZH2 depletion in SCLC cells is not due to an off-target effect.
Fig. 2.

EZH2 mediates cisplatin resistance in SCLC. a H128 and c H146 cells were transfected with siRNA targeting EZH2, ATR, or a NT control. 72 hours after transfection, cells were treated with cisplatin for 72 hours prior to measuring cell viability. b and d Western blot analysis of EZH2 expression in b H128 and d H146 cells, respectively, demonstrating EZH2 knockdown. e Western blot analysis of EZH2 expression across a panel of SCLC cell lines. f Densitometry quantification of EZH2 expression corresponding to e was plotted against cisplatin IC50 [47] across a panel of SCLC cell lines, with a correlation of 0.7355. (For a and c, mean and standard deviation of three replicas is shown.) *** indicates p < 0.001.
EZH2 expression level positively correlates with cisplatin resistance across SCLC
Because EZH2 is highly expressed in SCLC and EZH2 depletion in SCLC cells causes cisplatin hypersensitivity, we hypothesized that EZH2 levels could be an important determinant of cisplatin resistance in SCLC. To this end, we examined a panel of six SCLC cell lines for EZH2 protein expression compared to cisplatin IC50 [47] (Fig. 2E-F). We found a positive correlation between EZH2 expression and cisplatin IC50 trending towards significance, suggesting that as EZH2 expression increases, SCLC cells become more resistant to cisplatin.
EZH2 localizes to UV damage sites and promotes repair of UV-induced CPD lesions
Cisplatin and UV-induced damage can be repaired by multiple DNA repair pathways, including NER. To determine if EZH2 plays a role in NER, we examined if EZH2 knockdown sensitizes SCLC cell lines to UV damage, which is primarily repaired by NER. We found that EZH2 depletion sensitized H128, H146 and U2OS cells to UV (Fig. 3A-D, Supplementary Fig. S3A-E). EZH2 has been reported to localize to DSBs and DNA damage sites induced by laser microirradiation [42, 43]. We similarly found that GFP-EZH2 expressed in cells localizes to DNA damage sites induced by laser microirradiation at 365 nm (Fig. 3E), which causes multiple forms of DNA damage, including CPDs, DSBs, single-strand breaks, crosslinks, and ROS-DNA damage. To determine if EZH2 responds more specifically to lesions repaired by NER, we examined if EZH2 could comparably localize to damage sites induced by a UVC lamp filtered through a micropore membrane overlaying the cells. Indeed, GFP-EZH2 localized to UV damage sites marked by the NER lesion CPD as well as γH2AX (Fig. 3F, Supplementary Fig. S3F), suggesting that EZH2 may have a role in the repair of UV damage requiring its direct localization. A number of factors involved in the NER pathway are established to localize to the chromatin soluble fraction of internucleosomal DNA in response to UV, including DDB1, DDB2, and XPC [52]. As expected, EZH2 was associated with a chromatin insoluble fraction both before and after UV damage, likely due to its canonical role in PRC2 (data not shown). Interestingly, we found that a population of EZH2 was recruited to the chromatin soluble internucleosomal fraction in response to UV damage (Fig. 3G). Given that EZH2 co-localizes with CPD at UV damage sites, we then examined if EZH2 could promote the resolution of CPD lesions. Using slot blot analysis, we found that EZH2 knockdown impaired the resolution of CPD lesions in response to UV (Fig. 3H, 3I). More directly, we observed that EZH2 knockdown caused a reduction in NER repair capacity using a fluorescence based multiplex host cell reactivation (FM-HCR) assay [53] (Fig. 3J). Collectively, these data indicate that EZH2 mobilizes to and governs the repair of CPD crosslinks induced by UV irradiation.
Fig. 3.

EZH2 localizes to and promotes repair of UV-induced CPD lesions. a H128 and c H146 cells were transfected with siRNAs targeting EZH2, ERCC1, or a NT control. 72 hours after transfection, cells were treated with UV. Cell viability was measured 72 hours after UV irradiation. b and d Western blot analysis of EZH2 expression (for b H128; and d H146 cells) demonstrating knockdown. e HeLa cells were transiently transfected with GFP-EZH2 or GFP-RPA1. 72 hours post-transfection, cells were subjected to laser microirradiation at 365 nm wavelength. Representative images of GFP localization as seen before and seconds after laser microirradiation are shown. Scale bars represent 10 μm. f HeLa cells were transfected with GFP-EZH2 and 24 hours post transfection, cells were UV irradiated at 100 J/m2 through a 5 μm micropore membrane. Cells were fixed and stained with the indicated markers after one hour recovery. Representative images are shown. g HeLa cells were left untreated or UV irradiated at 30 J/m2. Cells were lysed and fractionated to obtain the chromatin soluble fraction by salt extraction followed by 10 minutes of MNase digestion at room temperature. This fraction was run on SDS-PAGE and probed for EZH2 and H3 for loading. h and i Slot blot analysis of the repair of CPD lesions over time in H128 cells in response to 30 J/m2 of UV. h Quantitation of percent of CPD lesions remaining over time. i Representative slot blot. SYBR Gold signal indicates total DNA loaded. (For a, c, and h, mean and standard deviation of three replicas is shown. j NER repair capacity as measured by FM-HCR was quantified in cells transfected with siRNAs targeting EZH2, ERCC1, XPA or NT control. Relative NER repair capacity was normalized to a NT control. *** indicates p<0.001. For g and i-j, representative blots from 3 independent experiments (n=3) is shown).
EZH2 interacts with and is epistatic with DDB1-DDB2 in sensitizing SCLC cells to cisplatin and UV
To provide insight into how EZH2 governs cisplatin and UV resistance, we performed MS analysis of GFP-EZH2 purified from cells treated with cisplatin (Fig. 4A). As expected, we found enrichment of EZH2 as well as key members of the PRC2 complex, SUZ12 and EED (Fig. 4B). Interestingly, we also found enrichment of DDB1, a member of the NER pathway. Co-IP of GFP-EZH2 pulled down HA-DDB1 (Fig. 4C), and similarly, reciprocal co-IP of HA-DDB1 pulled down GFP-EZH2 in cells (Fig. 4D). This interaction was preserved following benzonuclease treatment (Supplementary Fig. S4A), suggesting that the interaction is not mediated through DNA. No change in interaction was observed following cisplatin treatment (Supplementary Fig. S4B), implying that the interaction is not regulated by DNA damage in this context. Co-IP of GFP-EZH2 also pulled down both endogenous DDB1 and DDB2, its interacting partner (Fig. 4E). We also validated the endogenous interaction of EZH2 with DDB1-DDB2 in H128 cells by co-IP (Fig. 4F), confirming that EZH2 complexes with DDB1-DDB2 in SCLC cells. To identify the region of DDB2 that interacts with EZH2, we generated FLAG-DDB2 deletion mutants and performed co-IP of FLAG-DDB2 WT and mutants with GFP-EZH2 in HEK293T cells. FLAG-DDB2 1-196 but not DDB2 Δ40 co-IP’d with GFP-EZH2 (Fig. S4C), indicating that DDB2 1-196 is sufficient and DDB2 1-40 is necessary for interaction with EZH2 and suggesting that EZH2 interacts with the N-terminus of DDB2. Because EZH2 and DDB1-DDB2 mediate resistance to UV [4] we performed epistasis experiments to determine if they function together in a common pathway. Indeed, combined knockdown of EZH2 and DDB1/DDB2 caused no further sensitization of SCLC cells to UV and cisplatin compared with knockdown of EZH2 or DDB1/DDB2 alone (Fig. 4G-K), implying that EZH2 and DDB1-DDB2 function together in governing UV and cisplatin resistance in SCLC cells.
Fig. 4.

EZH2 interacts with and is epistatic with DDB1-DDB2 in sensitizing SCLC cells to cisplatin and UV. a HeLa cells were transiently transfected with GFP-EZH2. 72 hours post-transfection, cells were treated with 25 μM cisplatin and harvested 4 hours later, and GFP-EZH2 was immunoprecipitated (IP’d) with protein A agarose beads. Beads were washed and processed for mass spectrometry (MS) analysis for protein-protein interactions. b Summary of IP-MS results for GFP-EZH2. c and d HeLa cells were transfected with HA-DDB1 and GFP-EZH2 or empty vector and IP’d with an anti-GFP antibody (c) or anti-HA antibody (d) respectively, run on SDS-PAGE, and immunoblotted with indicated antibodies. e HeLa cells were transfected with GFP-EZH2. Cells were harvested, lysed, and IP’d with an anti-EZH2 antibody or IgG as indicated. Samples were run on SDS-PAGE and immunoblotted with indicated antibodies. f H128 cells were lysed and IP’d with an anti-EZH2 antibody or IgG as indicated. Samples were run on SDS-PAGE and immunoblotted with indicated antibodies. g, h and j H128 cells were transfected with siRNAs targeting EZH2, DDB1, DDB2, ATR, ERCC1, or a NT control. 72 hours after transfection, cells were treated with cisplatin or UV. Cell viability was measured 72 hours after treatment. i Western blot analysis of samples corresponding to g and h. k Western blot analysis of samples corresponding to j. (For g, h and j, mean and standard deviation of three replicas is shown. For c-f, representative blots from 3 independent experiments (n=3) is shown).
EZH2 promotes DDB2 stability independent of its catalytic activity and PRC2
Given that EZH2 complexes with and is epistatic with DDB1-DDB2 in mediating cisplatin and UV resistance in SCLC and promotes CPD crosslink repair, we sought to determine if EZH2 plays a specific role in the NER pathway. Interestingly, EZH2 knockdown decreased DDB2 but not DDB1 protein levels both at baseline and more prominently in response to UV and cisplatin treatment in H128 and HCT116 cells (Fig. 5A-B, Supplementary Fig. S5A). Quantitative RT-PCR revealed no corresponding significant decrease in DDB2 mRNA levels following EZH2 knockdown (Fig. 5C, Supplementary Fig. S5B), suggesting that the EZH2-mediated decrease in DDB2 protein levels is likely not through EZH2’s canonical role in transcription. Consistently, the DDB2 degradation phenotype was alleviated by proteasomal inhibition with MG132 (Fig. 5D), implying that the decrease in DDB2 levels are a result of proteasomal degradation and that EZH2 promotes DDB2 stability. Overexpression of GFP-EZH2 in HCT116 cells also increased DDB2 protein levels at baseline and in response to UV (Fig. 5E).
Fig. 5.
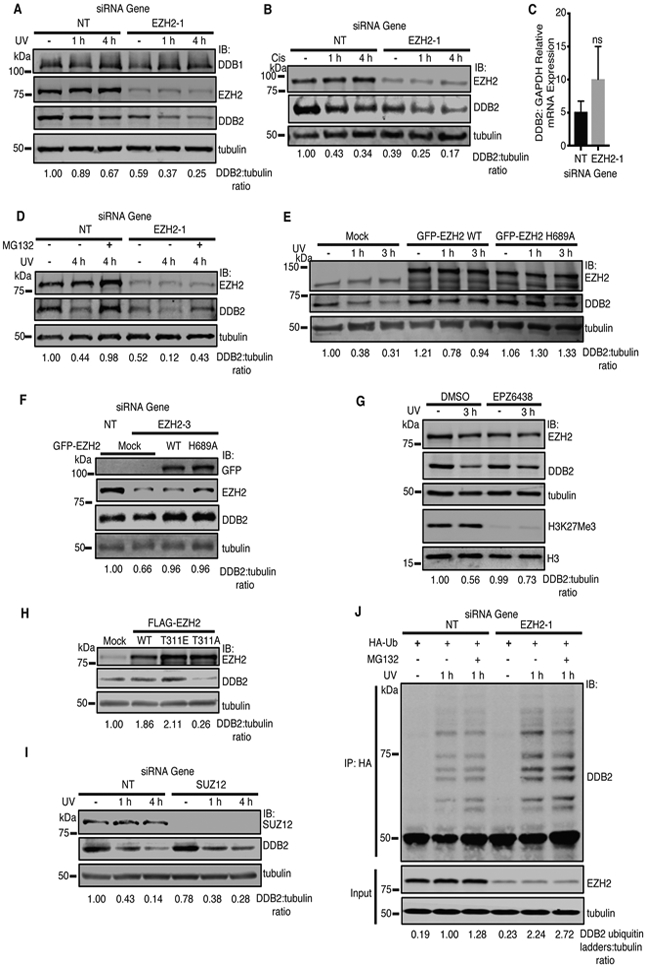
EZH2 promotes the stability of DDB2 independent of its catalytic activity and PRC2. a-d EZH2 promotes the stability of DDB2. H128 cells were transfected with siRNAs targeting EZH2 or a NT control. 72 hours after transfection, cells were pre-treated with cycloheximide (CHX) for 2 hours, and UV irradiated at 30 J/m2 (a), treated with 15 μM cisplatin (b), or left untreated (−). Cells were harvested after treatment as indicated. Whole cell lysates were run on SDS-PAGE and immunobloted with the indicated antibodies. c EZH2 knockdown does not alter mRNA levels of DDB2. H128 cells were transfected with siRNAs targeting EZH2 or a NT control and RT-PCR analysis was performed to measure DDB2 and GAPDH mRNA levels. The relative ratio of DDB2 to GAPDH mRNA level is represented. Mean and standard deviation of three replicas is shown. d H128 cells were transfected with siRNAs targeting EZH2 or a NT control. 72 hours post transfection, cells were pre-treated with 5 μM MG132 for 4 hours, and CHX for 2 hours for all groups. Cells were then UV irradiated at 30 J/m2 and harvested at the indicated timepoints prior to SDS-PAGE and western blot analysis with indicated antibodies. e-i EZH2 stabilization of DDB2 is independent of its PRC2 function. e HCT116 cells were transfected with GFP-EZH2 WT or GFP-EZH2 catalytic inactive mutant (H689A) plasmids. 24 hours post transfection, cells were pre-treated with CHX for 2 hours, and left untreated (−) or UV irradiated at 30 J/m2, harvested after UV treatment as indicated, run on SDS PAGE, and probed with indicated antibodies. f H128 cells were knocked down with siRNAs targeting the EZH2 5’UTR (EZH2-3) or a NT control, and the following day, transfected with GFP-EZH2 WT or GFP-EZH2 catalytic inactive mutant (H689A) or mock transfected. 48 hours post-transfection, groups were pre-treated with CHX for 2 hours, harvested, run on SDS PAGE, and probed with the indicated antibodies. g HCT116 cells were pre-treated with 1 μM SAM-competitive EZH2 inhibitor EPZ-6438 or DMSO for 4 days. Cells were then pre-treated with CHX for 2 hours. Cells were left untreated (−) or UV irradiated at 30 J/m2 and harvested after 3 hours recovery. Whole cell lysates were run on SDS-PAGE and immunobloted with the indicated antibodies. h HeLa cells were transfected with with FLAG-EZH2 WT or PRC2 mutants (FLAG-EZH2 T311E/A). 48 hours post transfection, cells were pre-treated with CHX for 2 hours, and harvested. Whole cell lysates were run on SDS-PAGE and immunobloted with the indicated antibodies. i HCT116 Cells were transfected with siRNAs targeting SUZ12 or a NT control. 72 hours after transfection, cells were pre-treated with CHX for 2 hours, and left untreated (−) or UV irradiated at 30 J/m2, and harvested after UV treatment as indicated. Whole cell lysates were run on SDS-PAGE and immunobloted with the indicated antibodies. j His-Ubi HEK-293T cells were transfected with indicated siRNA and 72 hours post transfection, cells were pre-treated with CHX and MG132 where indicated. Ubiquitinated DDB2 was measured. (For a-b and d-j, representative blots from 3 independent experiments (n=3) is shown).
To determine if EZH2 methyltransferase activity promotes DDB2 stability, we overexpressed catalytically inactive GFP-EZH2 H689A and found a similar increase in DDB2 levels (Fig. 5E, Supplementary Fig. S5C). Furthermore, the decrease in DDB2 levels resulting from EZH2 depletion in H128 cells was rescued by expression of GFP-EZH2 WT and H689A (Fig. 5F), suggesting that EZH2 has a non-catalytic role in promoting DDB2 stability. Consistently, treatment of H128 and HCT116 cells with EPZ-6438, a S-Adenosyl-l-methionine (SAM) competitive EZH2 inhibitor, which targets its catalytic activity but does not deplete EZH2 levels, did not decrease DDB2 levels at baseline or in response to UV (Fig. 5G, Supplementary Fig. S5D). We also found that the cisplatin hypersensitivity of H128 cells depleted of EZH2 is alleviated by expression of GFP-EZH2 WT and to a lesser extent H689A (Supplementary Fig. S5E), suggesting that EZH2 also has a catalytically inactive role in mediating cisplatin resistance; however, because GFP-EZH2 H689A only partially rescued the cisplatin hypersensitivity of EZH2 depletion, EZH2’s noncatalytic role in promoting DDB2 stability may not fully account for its effects on mediating cisplatin resistance.
To determine if EZH2 promotes DDB2 stability in association with PRC2, we overexpressed phospho-mimetic FLAG-EZH2 T311E, which impairs its interaction with SUZ12 and its methyltransferase activity [54]. Overexpression of FLAG-EZH2 T311E but not non-phosphorylatable FLAG-EZH2 T311A strongly increased DDB2 levels, indicating that when EZH2 is decoupled from PRC2, it is able to participate in stabilizing DDB2 and promoting NER. (Fig. 5H). Moreover, SUZ12 knockdown in H128 cells failed to decrease DDB2 levels at baseline or in response to UV (Fig. 5I). Given that DDB2 is targeted for degradation by ubiquitination, we tested the effect of EZH2 depletion on DDB2 ubiquitination and found that it was enhanced in response to UV damage (Fig. 5J). DDB2 has been reported to negatively regulate p21 levels in NER [55, 56] so, we examined if EZH2 depletion results in p21upregulation. Indeed, EZH2 knockdown also caused a corresponding increase in p21 levels with a decrease in DDB2 levels at baseline and in response to cisplatin treatment (Fig. S5F), providing further support that EZH2 promotes DDB2 stability and that EZH2 may promote NER through DDB2. Collectively, our findings suggest that EZH2 promotes DDB2 stability by impairing its ubiquitination independent of its catalytic activity and PRC2.
DDB2 functions downstream of EZH2 in promoting NER
To determine if EZH2 facilitates DDB2 function in NER, we examined DDB2 localization to UV micropore-generated CPD foci. EZH2 knockdown significantly impaired DDB2 localization to CPD foci (Fig. 6A-B), suggesting that EZH2 promotes DDB2 localization to CPD lesions. Furthermore, the expression of FLAG-DDB2 rescued the impairment in resolution of CPD lesions following EZH2 knockdown (Fig. 6C-D), showing unequivocally that EZH2 promotes the repair of CPD lesions through DDB2. The expression of FLAG-DDB2 also alleviated the cisplatin hypersensitivity of EZH2 depletion (Fig. 6E-F), suggesting that EZH2 mediates cisplatin resistance at least in part through DDB2.
Fig. 6.

DDB2 functions downstream of EZH2 in NER and in mediating cisplatin resistance in SCLC. a and b EZH2 knockdown impairs DDB2 recruitment to CPD lesions. HeLa cells were transfected with siRNAs targeting EZH2 or a NT control. The following day, both groups were transfected with FLAG-DDB2. Cells were UV-irradiated at 100J/m2 through a micropore membrane, fixed, and stained for CPD lesions and FLAG-DDB2. Relative percent of CPD stained lesions that were positive for DDB2 staining, normalized to NT, was quantified (a). Representative images are shown (b). c and d FLAG-DDB2 rescues impaired resolution of CPD lesions that occur through EZH2 knockdown. H128 cells were transfected with siRNAs targeting EZH2 or a NT control. The following day, both groups were transfected with FLAG-DDB2. Cells were UV irradiated, and harvested immediately (0h) or after 12h of repair time. Slot blot analysis was performed. c Quantitation of percent of CPD lesions remaining over time as normalized to 0 hrs of repair time. d Representative slot blot. SYBR Gold signal indicates total DNA loaded. e FLAG-DDB2 alleviates the sensitization of SCLC to cisplatin upon EZH2 knockdown. H128 cells were transfected with siRNAs corresponding to EZH2, ERCC1 or a NT control. The following day, groups were transfected with FLAG-DDB2 or mock control. 48 hours post overexpression, cell viability analysis was performed. f Western blot of FLAG-DDB2 expression achieved in H128 corresponding to panels c-e. For a-b, quantification was achieved through 3 independent experiments counting at least 50 independent CPD lesion events per group (n=50) and representative images are shown. c-d, representative blots from 3 independent experiments (n=3) is shown). *** indicates p<0.001.
EZH2 depletion with DZNep sensitizes SCLC cells and tumors to cisplatin
Given that siRNA mediated depletion but not catalytic inhibition of EZH2 decreases DDB2 levels, we tested the effect of DZNep, an EZH2 inhibitor that depletes EZH2 [21], on cisplatin sensitization and found that DZNep strongly sensitized H128 cells to cisplatin (Fig. 7A). In contrast, we observed no significant effect on cisplatin sensitization when cells were treated with EPZ-6438. In addition, we tested the impact of DZNep on DDB2 levels in SCLC cells. DZNep treatment depleted EZH2 levels in H128 cells, leading to decreased DDB2 levels, phenocopying EZH2 depletion by knockdown (Fig. 7B). Furthermore, the expression of GFP-EZH2 WT and H689A partially rescued the DZNep mediated decrease in DDB2 (Fig. S6A), suggesting that DZNep targets a noncatalytic function of EZH2 in depleting DDB2 levels. DZNep treatment was also epistatic with DDB2 knockdown in sensitizing SCLC cells to cisplatin and UV damage (Fig. S6B-C). Together, these data indicate that targeting DDB2 stability can be achieved by depleting EZH2 through DZNep to sensitize SCLC cells to cisplatin.
Fig. 7.

EZH2 depletion with DZNep sensitizes SCLC to cisplatin in vitro and in vivo and model for EZH2 in NER. a H128 cells are sensitized to cisplatin through EZH2 depletion but not through S-Adenosyl-l-methionine (SAM) competitive inhibition. H128 cells were treated with DZNep, EPZ-6438, or DMSO for 72 hours followed by cisplatin treatment for 72 hours prior to assaying for cell viability. b DZNep-mediated EZH2 depletion destabilizes DDB2. H128 cells were treated with DZNep or DMSO for 6 days followed by 2 hours of CHX pretreatment and 30 J/m2 UV damage, and allowed to recover at the times indicated. Cells were then harvested, lysed, and run on SDS-PAGE. c-e The combination of DZNep and cisplatin synergistically suppresses SCLC tumor growth in vivo. c and d Nu/Nu mice with H128 lung cancer xenografts were treated with DZNep (2.5mg/kg; 2 times per week), cisplatin (2.5mg/kg; 2 times per week), or the combination i.p. for 28 days. Each group included 5 mice. Tumor volumes were measured once every 2 days. The error bars indicate ± SD. e Weights corresponding to figure c were measured once every 2 days. The error bars indicate ± SD. * indicates p<0.05, by 2-tailed t test. **indicates p<0.01, by 2-tailed t test. f Model for EZH2 function in NER. Following cisplatin or UV damage, EZH2 mobilizes to DNA damage sites where it complexes with DDB1-DDB2 and promotes DDB2 stability by preventing its ubiquitination independent of its methyltransferase activity or PRC2. EZH2 stabilization of DDB2 may facilitate DDB2 binding and assembly of CRL4 at the site of the NER lesion, which in turn promotes the ubiquitination of critical downstream NER targets to facilitate NER. In the absence of EZH2 or with EZH2 depletion by DZNep, there is increased ubiquitination of DDB2 leading to its degradation and impaired NER and thereby causing sensitization to cisplatin and UV damage. (For b, a representative blot from 3 independent experiments (n=3) is shown).
To determine if DZNep sensitizes SCLC tumors to cisplatin in vivo, we generated tumor xenografts using H128 cells in Nu/Nu mice. Treatment of the mice with the combination of DZNep and cisplatin strongly suppressed the growth of tumors compared with treatment with DZNep or cisplatin alone (Fig. 7C-D), providing in vivo validation that DZNep sensitizes SCLC tumors to cisplatin. Consistent with findings in other tumor types [57, 58] treatment of the mice with DZNep alone also significantly delayed tumor growth but to a lesser extent than combined treatment with DZNep and cisplatin. No significant difference in body weight was observed in treatment groups compared to controls, indicating treatments were well tolerated. (Fig. 7E).
DISCUSSION
Our findings reveal a non-catalytic and PRC2 independent function for EZH2 in promoting NER through DDB2 stabilization to govern cisplatin resistance in SCLC. Using a synthetic lethality screen of a siRNA library biased towards nuclear enzymes, we identified a number of important regulators of cisplatin resistance in SCLC cells, including EZH2. We found that EZH2 depletion in SCLC cells causes cellular cisplatin and UV hypersensitivity in an epistatic manner with DDB1-DDB2, and moreover that EZH2 mobilizes to damage sites to govern the repair of CPD crosslinks, supporting a novel role for EZH2 in promoting NER. Mechanistically, we showed that EZH2 complexes with DDB1-DDB2 and promotes DDB2 stability by impairing its ubiquitination independent of its methyltransferase activity or PRC2, thereby facilitating DDB2 localization to CPD lesions, defining a novel paradigm for EZH2 in promoting the stability of proteins independent of its canonical role in H3K27Me3. Furthermore, we found that targeting EZH2 for depletion with DZNep but not inhibiting its catalytic activity with EPZ-6438 strongly sensitizes SCLC cells and tumors to cisplatin. Thus, our findings reveal a non-catalytic and PRC2 independent function for EZH2 in promoting NER through DDB2 stabilization, suggesting a rationale for targeting EZH2 beyond its catalytic activity to overcome cisplatin resistance in SCLC.
Based on our findings, we propose a model whereupon following genotoxic insults by cisplatin or UV, EZH2 complexes with DDB1-DDB2 and promotes DDB2 stability by preventing its ubiquitination independent of its methyltransferase activity or PRC2, thereby facilitating DDB2 localization to CPD crosslinks to govern their repair (Fig. 7F). Since EZH2 interacts with the N-terminus of DDB2 where the majority of its autoubiquitination sites are located [59], EZH2 interaction in this region may prevent DDB2 autoubiquitination. EZH2 stabilization of DDB2 may facilitate its binding and assembly of CRL4 at the site of the NER lesion. There, the E3 ligase complex promotes ubiquitination of critical downstream NER targets, including XPC, H3, H4, and DDB2 itself before DDB2 is degraded and the E3 ligase complex disassembles. Our data support a PRC2 independent function for EZH2 in stabilizing DDB2 as SUZ12 knockdown failed to decrease DDB2 levels similarly to EZH2, and overexpression of FLAG-EZH2 T311E, which has impaired interaction with SUZ12 [54], but not FLAG-EZH2 T311A, stabilizes DDB2 levels. Our data suggest that when EZH2 is constitutively phosphorylated, it is able to more efficiently participate in stabilizing DDB2 and promote NER, potentially by becoming decoupled from SUZ12/PRC2. In contrast, overexpression of FLAG-EZH2 T311A, which is functional in its role with PRC2, destabilized DDB2, potentially acting as a dominant negative when EZH2 is not uncoupled from PRC2.
Our data suggest a non-catalytic role for EZH2 in promoting NER by stabilizing DDB2 that is distinct from its canonical role in H3K27Me3-mediated transcriptional silencing. Indeed, EZH2 depletion did not decrease DDB2 mRNA levels and inhibiting the catalytic activity but not total levels of EZH2 with EPZ-6438 decreased H3K27Me3 but failed to decrease DDB2 protein levels or sensitize SCLC cells to cisplatin. Furthermore, the decrease in DDB2 levels following EZH2 knockdown was rescued by expression of GFP-EZH2 H689A. However, because GFP-EZH2 H689A and FLAG-DDB2 only partially rescued the cisplatin hypersensitivity of EZH2 depletion, our results suggest that the noncatalytic role of EZH2 in promoting DDB2 stability may not fully account for its effects on mediating cisplatin resistance. Interestingly, it has been reported that EZH2 is capable of silencing XPA at its promoter through H3K27Me3, thereby impairing NER in nasopharyngeal carcinoma cells [60]. It is possible that EZH2 could have roles in both activating and inactivating NER that are cancer subtype dependent, which will be critical in assessing whether EZH2 should be targeted to overcome cisplatin resistance.
Consistent with our findings, increased DDB2 levels were associated with cisplatin resistance in melanoma cells [61]. On the other hand, DDB2 overexpression has been reported to lead to cisplatin sensitivity in ovarian cancer, and DDB2 has been reported not to be required for the repair of cisplatin-induced DNA damage [62]. Thus, it is possible that the role of DDB2 in responding to cisplatin may also be cancer cell type specific, perhaps depending on dysregulation of other DDR genes. In addition, these contrasting findings may, in part be attributed to the idea that to facilitate NER, DDB2 levels must be tightly and dynamically regulated, as DDB2 must first be stabilized and then degraded in order to promote NER lesion resolution [63]. Thus, while EZH2 knockdown causes cisplatin hypersensitivity in SCLC cells at least in part through DDB2 degradation, it is also possible that too much DDB2 could also impair NER and thus paradoxically lead to cisplatin hypersensitivity. In addition, while our data show that EZH2 promotes DDB2 stability, DDB2 has been reported to recruit EZH2 to the promoters of NEDD4L and RNF43 [64, 65] suggesting a possible feed-forward mechanism of EZH2 in regulating DDB2 stability, which in turn regulates EZH2’s function in transcriptional repression.
EZH2 has been reported to have a non-canonical role in directly methylating non-histone proteins to promote their degradation [29, 33] Our data suggest a new role for EZH2 in promoting protein stability independent of its methyltransferase activity or PRC2. DDB2 ubiquitination has previously been shown to be regulated by the COP9 signalosome (CSN), XPC, Ku, PARP1, and USP24 [59, 66-69]. Our data indicate that EZH2 also impairs DDB2 ubiquitination, highlighting the importance of tight regulation of DDB2 levels in controlling its functions in NER. Of note, EZH2 was recently reported to recruit USP7 to mediate neuronal gene expression [70]; however, whether this is mediated through its methyltransferase activity, PRC2, or H3K27Me3 such as it is for MUS81 [45] was not explored. It is possible that the non-catalytic and PRC2 independent function of EZH2 in stabilizing DDB2 may be a more generalized mechanism in control of the stability of other proteins in addition to DDB2.
Beyond our main findings with EZH2, many hits from the primary screen were largely consistent with the existing literature, however, there were also some surprising results. Arginine methylation, for example, was the most enriched pathway among the cisplatin sensitization hits. Arginine methylation is emerging as a key pathway involved in cell cycle regulation [71], and dysregulation of the cell cycle can render cancers vulnerable to cisplatin treatment. In addition, many reports have demonstrated that BRCA1 and POLQ are promising therapeutic targets that, when depleted, sensitize cancer to cisplatin or other DNA damaging agents. This has been predominantly explored in pancreatic, breast and ovarian cancers [72-75]. Data from our H128 screen, however, indicates that BRCA1 and POLQ depletion promotes cisplatin resistance in H128 cells. Taken together, our data suggest that targeting BRCA1 and POLQ may be advantageous but only in specific cancer subtypes or molecular contexts.
When viewed through a therapeutic lens, our finding that EZH2 has a non-catalytic and PRC2 independent role in promoting NER has important implications in the rationale design and application of EZH2 inhibitors that are currently being investigated for cancer therapy. While EZH2 does govern the repair of some types of DNA damage through H3K27Me3 [34, 35, 37] our data suggest that catalytic inhibition of EZH2 may not fully suppress its role in governing cisplatin resistance. Our assembled data provide rationale for the therapeutic potential of novel anti-cancer strategies including targeting EZH2 with depleting agents such as with DZNep, or disrupting the EZH2-DDB2 interaction with small molecule inhibitors, to overcome cisplatin resistance.
Supplementary Material
ACKNOWLEDGMENTS
EmGFP-EZH2 WT plasmid was a gift of Damian Yap (British Colombia Cancer Research Center) [76]. EZH2 PRC2 mutant plasmids were a gift of Lixin Wan (Moffit Cancer Center) [54]. FLAG-DDB2 plasmid was a gift of Qi-En Wang (The Ohio State University) [77].
FUNDING
This work was supported by NIH/NCI [R01CA178999 to D.S.Y., F31CA225119 to A.E.K, R01CA193828 to X.D., P01CA092584 to Z.D.N.]; NIH/NIEHS [U01ES029520 to Z.D.N.]; Lung Cancer Research Foundation [51347 and 60208 to D.S.Y]; Conquer Cancer Foundation of ASCO Young Investigator Award, supported by GO2 Foundation for Lung Cancer [15212 to N.T.P.]. Any opinions, findings, and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of the ASCO®, Conquer Cancer®, or GO2 Foundation for Lung Cancer.
Footnotes
CONFLICT OF INTEREST
The authors declare no competing interests.
REFERENCES
- 1.Program SEER (National Cancer Institute (U.S.)), National Center for Health Statistics (U.S.), National Cancer Institute (U.S.). Surveillance Program., National Cancer Institute (U.S.). Cancer Statistics Branch., National Cancer Institute (U.S.). Cancer Control Research Program. SEER cancer statistics review. NIH publication. U.S Dept. of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute: Bethesda, Md, 1993, p volumes. [Google Scholar]
- 2.Chute JP, Chen T, Feigal E, Simon R, Johnson BE. Twenty years of phase III trials for patients with extensive-stage small-cell lung cancer: perceptible progress. J Clin Oncol 1999; 17: 1794–1801. [DOI] [PubMed] [Google Scholar]
- 3.Horita N, Yamamoto M, Sato T, Tsukahara T, Nagakura H, Tashiro K et al. Topotecan for Relapsed Small-cell Lung Cancer: Systematic Review and Meta-Analysis of 1347 Patients. Sci Rep 2015; 5: 15437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 2014; 15: 465–481. [DOI] [PubMed] [Google Scholar]
- 5.Keeney S, Chang GJ, Linn S. Characterization of a human DNA damage binding protein implicated in xeroderma pigmentosum E. J Biol Chem 1993; 268: 21293–21300. [PubMed] [Google Scholar]
- 6.Fitch ME, Nakajima S, Yasui A, Ford JM. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J Biol Chem 2003; 278: 46906–46910. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Zhang Y, Douglas L, Zhou P. UV-damaged DNA-binding proteins are targets of CUL-4A-mediated ubiquitination and degradation. J Biol Chem 2001; 276: 48175–48182. [DOI] [PubMed] [Google Scholar]
- 8.Fischer ES, Scrima A, Bohm K, Matsumoto S, Lingaraju GM, Faty M et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 2011; 147: 1024–1039. [DOI] [PubMed] [Google Scholar]
- 9.Puppe J, Opdam M, Schouten PC, Jozwiak K, Lips E, Severson T et al. EZH2 Is Overexpressed in BRCA1-like Breast Tumors and Predictive for Sensitivity to High-Dose Platinum-Based Chemotherapy. Clin Cancer Res 2019; 25: 4351–4362. [DOI] [PubMed] [Google Scholar]
- 10.Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov 2012; 2: 798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato T, Kaneda A, Tsuji S, Isagawa T, Yamamoto S, Fujita T et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci Rep 2013; 3: 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nienstedt JC, Schroeder C, Clauditz T, Simon R, Sauter G, Muenscher A et al. EZH2 overexpression in head and neck cancer is related to lymph node metastasis. J Oral Pathol Med 2018; 47: 240–245. [DOI] [PubMed] [Google Scholar]
- 13.Coe BP, Thu KL, Aviel-Ronen S, Vucic EA, Gazdar AF, Lam S et al. Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PLoS One 2013; 8: e71670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hubaux R, Thu KL, Coe BP, MacAulay C, Lam S, Lam WL. EZH2 promotes E2F-driven SCLC tumorigenesis through modulation of apoptosis and cell-cycle regulation. J Thorac Oncol 2013; 8: 1102–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie H, Peng C, Huang J, Li BE, Kim W, Smith EC et al. Chronic Myelogenous Leukemia- Initiating Cells Require Polycomb Group Protein EZH2. Cancer Discov 2016; 6: 1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Cai Q, Godwin AK, Zhang R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol Cancer Res 2010; 8: 1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rao ZY, Cai MY, Yang GF, He LR, Mai SJ, Hua WF et al. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis 2010; 31: 1576–1583. [DOI] [PubMed] [Google Scholar]
- 18.Murai F, Koinuma D, Shinozaki-Ushiku A, Fukayama M, Miyaozono K, Ehata S. EZH2 promotes progression of small cell lung cancer by suppressing the TGF-beta-Smad-ASCL1 pathway. Cell Discov 2015; 1: 15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang FZ, He YY, Wang HH, Zhang HL, Zhang J, Yan XF et al. Mutant p53 induces EZH2 expression and promotes epithelial-mesenchymal transition by disrupting p68-Drosha complex assembly and attenuating miR-26a processing. Oncotarget 2015; 6: 44660–44674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther 2014; 13: 842–854. [DOI] [PubMed] [Google Scholar]
- 21.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther 2009; 8: 1579–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen YT, Zhu F, Lin WR, Ying RB, Yang YP, Zeng LH. The novel EZH2 inhibitor, GSK126, suppresses cell migration and angiogenesis via down-regulating VEGF-A. Cancer Chemother Pharmacol 2016; 77: 757–765. [DOI] [PubMed] [Google Scholar]
- 23.Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A 2012; 109: 21360–21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012; 492: 108–112. [DOI] [PubMed] [Google Scholar]
- 25.Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1 -(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J Med Chem 2016; 59: 9928–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev 2012; 26: 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanulli S, Justin N, Teissandier A, Ancelin K, Portoso M, Caron M et al. Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol Cell 2015; 57: 769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013; 23: 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vasanthakumar A, Xu D, Lun AT, Kueh AJ, van Gisbergen KP, Iannarella N et al. A non-canonical function of Ezh2 preserves immune homeostasis. EMBO Rep 2017; 18: 619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan J, Li B, Lin B, Lee PT, Chung TH, Tan J et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 2016; 128: 948–958. [DOI] [PubMed] [Google Scholar]
- 31.Kim J, Lee Y, Lu X, Song B, Fong KW, Cao Q et al. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep 2018; 25: 2808–2820 e2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012; 338: 1465–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JM, Lee JS, Kim H, Kim K, Park H, Kim JY et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell 2012; 48: 572–586. [DOI] [PubMed] [Google Scholar]
- 34.Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017; 31: 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamaguchi H, Du Y, Nakai K, Ding M, Chang SS, Hsu JL et al. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene 2018; 37: 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia H, Yu CH, Zhang Y, Yu J, Li J, Zhang W et al. EZH2 silencing with RNAi enhances irradiation-induced inhibition of human lung cancer growth in vitro and in vivo. Oncol Lett 2012; 4: 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu L, Tang H, Wang K, Zheng Y, Feng J, Dong H et al. Pharmacological inhibition of EZH2 combined with DNAdamaging agents interferes with the DNA damage response in MM cells. Mol Med Rep 2019; 19: 4249–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu S, Yu L, Li Z, Shen Y, Wang J, Cai J et al. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol Ther 2010; 10: 788–795. [DOI] [PubMed] [Google Scholar]
- 39.Sun S, Zhao S, Yang Q, Wang W, Cai E, Wen Y et al. Enhancer of zeste homolog 2 promotes cisplatin resistance by reducing cellular platinum accumulation. Cancer Sci 2018; 109: 1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang JW, Gwak SY, Shim GA, Liu L, Lim YC, Kim JM et al. EZH2 is associated with poor prognosis in head-and-neck squamous cell carcinoma via regulating the epithelial-to-mesenchymal transition and chemosensitivity. Oral Oncol 2016; 52: 66–74. [DOI] [PubMed] [Google Scholar]
- 41.Liu H, Li W, Yu X, Gao F, Duan Z, Ma X et al. EZH2-mediated Puma gene repression regulates non-small cell lung cancer cell proliferation and cisplatin-induced apoptosis. Oncotarget 2016; 7: 56338–56354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campbell S, Ismail IH, Young LC, Poirier GG, Hendzel MJ. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell Cycle 2013; 12: 2675–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A 2010; 107: 18475–18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeidler M, Varambally S, Cao Q, Chinnaiyan AM, Ferguson DO, Merajver SD et al. The Polycomb group protein EZH2 impairs DNA repair in breast epithelial cells. Neoplasia 2005; 7: 1011–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rondinelli B, Gogola E, Yucel H, Duarte AA, van de Ven M, van der Sluijs R et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol 2017; 19: 1371–1378. [DOI] [PubMed] [Google Scholar]
- 46.Wu Z, Lee ST, Qiao Y, Li Z, Lee PL, Lee YJ et al. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ 2011; 18: 1771–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Owonikoko TK, Zhang G, Deng X, Rossi MR, Switchenko JM, Doho GH et al. Poly (ADP) ribose polymerase enzyme inhibitor, veliparib, potentiates chemotherapy and radiation in vitro and in vivo in small cell lung cancer. Cancer Med 2014; 3: 1579–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith SC, Petrova AV, Madden MZ, Wang H, Pan Y, Warren MD et al. A gemcitabine sensitivity screen identifies a role for NEK9 in the replication stress response. Nucleic Acids Res 2014; 42: 11517–11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colbert LE, Petrova AV, Fisher SB, Pantazides BG, Madden MZ, Hardy CW et al. CHD7 expression predicts survival outcomes in patients with resected pancreatic cancer. Cancer Res 2014; 74: 2677–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A 2001; 98: 13790–13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A 2001; 98: 13784–13789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fei J, Kaczmarek N, Luch A, Glas A, Carell T, Naegeli H. Regulation of nucleotide excision repair by UV-DDB: prioritization of damage recognition to internucleosomal DNA. PLoS Biol 2011; 9: e1001183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagel ZD, Margulies CM, Chaim IA, McRee SK, Mazzucato P, Ahmad A et al. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc Natl Acad Sci U S A 2014; 111: E1823–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wan L, Xu K, Wei Y, Zhang J, Han T, Fry C et al. Phosphorylation of EZH2 by AMPK Suppresses PRC2 Methyltransferase Activity and Oncogenic Function. Mol Cell 2018; 69: 279–291 e275. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Stoyanova T, Roy N, Kopanja D, Bagchi S, Raychaudhuri P. DDB2 decides cell fate following DNA damage. Proc Natl Acad Sci U S A 2009; 106: 10690–10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stoyanova T, Yoon T, Kopanja D, Mokyr MB, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 activates nucleotide excision repair by regulating the level of p21Waf1/Cip1. Mol Cell Biol 2008; 28: 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crea F, Hurt EM, Mathews LA, Cabarcas SM, Sun L, Marquez VE et al. Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol Cancer 2011; 10: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Z, Wang Y, Qiu J, Li Q, Yuan C, Zhang W et al. The polycomb group protein EZH2 is a novel therapeutic target in tongue cancer. Oncotarget 2013; 4: 2532–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsumoto S, Fischer ES, Yasuda T, Dohmae N, Iwai S, Mori T et al. Functional regulation of the DNA damage-recognition factor DDB2 by ubiquitination and interaction with xeroderma pigmentosum group C protein. Nucleic Acids Res 2015; 43: 1700–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang Y, Wang X, Niu X, Wang X, Jiang R, Xu T et al. EZH2 suppresses the nucleotide excision repair in nasopharyngeal carcinoma by silencing XPA gene. Mol Carcinog 2017; 56: 447–463. [DOI] [PubMed] [Google Scholar]
- 61.Barckhausen C, Roos WP, Naumann SC, Kaina B. Malignant melanoma cells acquire resistance to DNA interstrand cross-linking chemotherapeutics by p53-triggered upregulation of DDB2/XPC-mediated DNA repair. Oncogene 2014; 33: 1964–1974. [DOI] [PubMed] [Google Scholar]
- 62.Barakat BM, Wang QE, Han C, Milum K, Yin DT, Zhao Q et al. Overexpression of DDB2 enhances the sensitivity of human ovarian cancer cells to cisplatin by augmenting cellular apoptosis. Int J Cancer 2010; 127: 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.El-Mahdy MA, Zhu Q, Wang QE, Wani G, Praetorius-Ibba M, Wani AA. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J Biol Chem 2006; 281: 13404–13411. [DOI] [PubMed] [Google Scholar]
- 64.Zhao R, Cui T, Han C, Zhang X, He J, Srivastava AK et al. DDB2 modulates TGF-beta signal transduction in human ovarian cancer cells by downregulating NEDD4L. Nucleic Acids Res 2015; 43: 7838–7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang S, Fantini D, Merrill BJ, Bagchi S, Guzman G, Raychaudhuri P. DDB2 Is a Novel Regulator of Wnt Signaling in Colon Cancer. Cancer Res 2017; 77: 6562–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takedachi A, Saijo M, Tanaka K. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol Cell Biol 2010; 30: 2708–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang L, Lubin A, Chen H, Sun Z, Gong F. The deubiquitinating protein USP24 interacts with DDB2 and regulates DDB2 stability. Cell Cycle 2012; 11: 4378–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol 2012; 199: 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R et al. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 2003; 113: 357–367. [DOI] [PubMed] [Google Scholar]
- 70.Lei A, Chen L, Zhang M, Yang X, Xu L, Cao N et al. EZH2 Regulates Protein Stability via Recruiting USP7 to Mediate Neuronal Gene Expression in Cancer Cells. Front Genet 2019; 10: 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B et al. Arginine methylation regulates the p53 response. Nat Cell Biol 2008; 10: 1431–1439. [DOI] [PubMed] [Google Scholar]
- 72.Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M et al. Response to neo-adjuvant chemotherapy in women with BRCA1-positive breast cancers. Breast Cancer Res Treat 2008; 108: 289–296. [DOI] [PubMed] [Google Scholar]
- 73.Byrski T, Huzarski T, Dent R, Marczyk E, Jasiowka M, Gronwald J et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat 2014; 147: 401–405. [DOI] [PubMed] [Google Scholar]
- 74.Stefansson OA, Villanueva A, Vidal A, Marti L, Esteller M. BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer. Epigenetics 2012; 7: 1225–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015; 518: 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011; 117: 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang QE, Praetorius-Ibba M, Zhu Q, El-Mahdy MA, Wani G, Zhao Q et al. Ubiquitylation-independent degradation of Xeroderma pigmentosum group C protein is required for efficient nucleotide excision repair. Nucleic Acids Res 2007; 35: 5338–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.