

Abstract
Regulatory T cells (Tregs) are required to control immune responses and maintain homeostasis, but are a significant barrier to anti-tumor immunity1. Conversely, Treg instability, characterized by loss of the master transcription factor Foxp3 and acquisition of pro-inflammatory properties2, can promote autoimmunity and/or facilitate more effective tumor immunity3,4. A comprehensive understanding of the pathways that regulate Foxp3 could lead to more effective Treg therapies for autoimmune disease and cancer. Despite improved functional genetic tools that now allow for systematic interrogation, dissection of the gene regulatory programs that modulate Foxp3 expression has not yet been reported. In this study, we developed a CRISPR-based pooled screening platform for phenotypes in primary mouse Tregs and applied this technology to perform a targeted loss-of-function screen of ~490 nuclear factors to identify gene regulatory programs that promote or disrupt Foxp3 expression. We discovered several novel modulators including ubiquitin-specific peptidase 22 (Usp22) and ring finger protein 20 (Rnf20). Usp22, a member of the deubiquitination module of the SAGA chromatin modifying complex, was discovered to be a positive regulator that stabilized Foxp3 expression; whereas the screen suggested Rnf20, an E3 ubiquitin ligase, can serve as a negative regulator of Foxp3. Treg-specific ablation of Usp22 in mice reduced Foxp3 protein and created defects in their suppressive function that led to spontaneous autoimmunity but protected against tumor growth in multiple cancer models. Foxp3 destabilization in Usp22-deficient Tregs could be rescued by ablation of Rnf20, revealing a reciprocal ubiquitin switch in Tregs. These results reveal novel modulators of Foxp3 and demonstrate a screening method that can be broadly applied to discover new targets for Treg immunotherapies for cancer and autoimmune disease.
While unstable Foxp3 expression in Tregs can result in autoimmunity, similar changes that reduce Treg suppressive function can contribute to more effective anti-tumor immune responses4. Understanding the fundamental regulators of Foxp3 is critical, especially as we navigate towards new potential applications for Treg therapies to treat autoimmunity and cancer5.
To discover novel regulators of Foxp3 stability, we developed a pooled CRISPR screening platform in primary mouse Tregs (Fig. 1a). We first designed a targeted library of ~490 nuclear factors based on optimized single guide RNA (sgRNA) sequences from the Brie library6 (Extended Data Fig. 1a) and used a retroviral vector to introduce this library into ex vivo Tregs isolated from Foxp3GFP-CreRosa26LSL-RFPCas9 mice (Extended Data Figs. 1b–1e). We then stained for endogenous Foxp3 protein and sorted the highest Foxp3-expressing cells (Foxp3high) and the lowest (Foxp3low). MAGeCK software7 systematically identified sgRNAs that were enriched or depleted in Foxp3low cells relative to Foxp3high cells (Supplementary Table 1). We were able to maintain high sgRNA coverage of our library (~1000x) and non-targeting control (NTC) sgRNAs showed no effect (Extended Data Figs. 1f, 1g) which provided confidence that our hits identified biological pathways controlling Foxp3 levels.
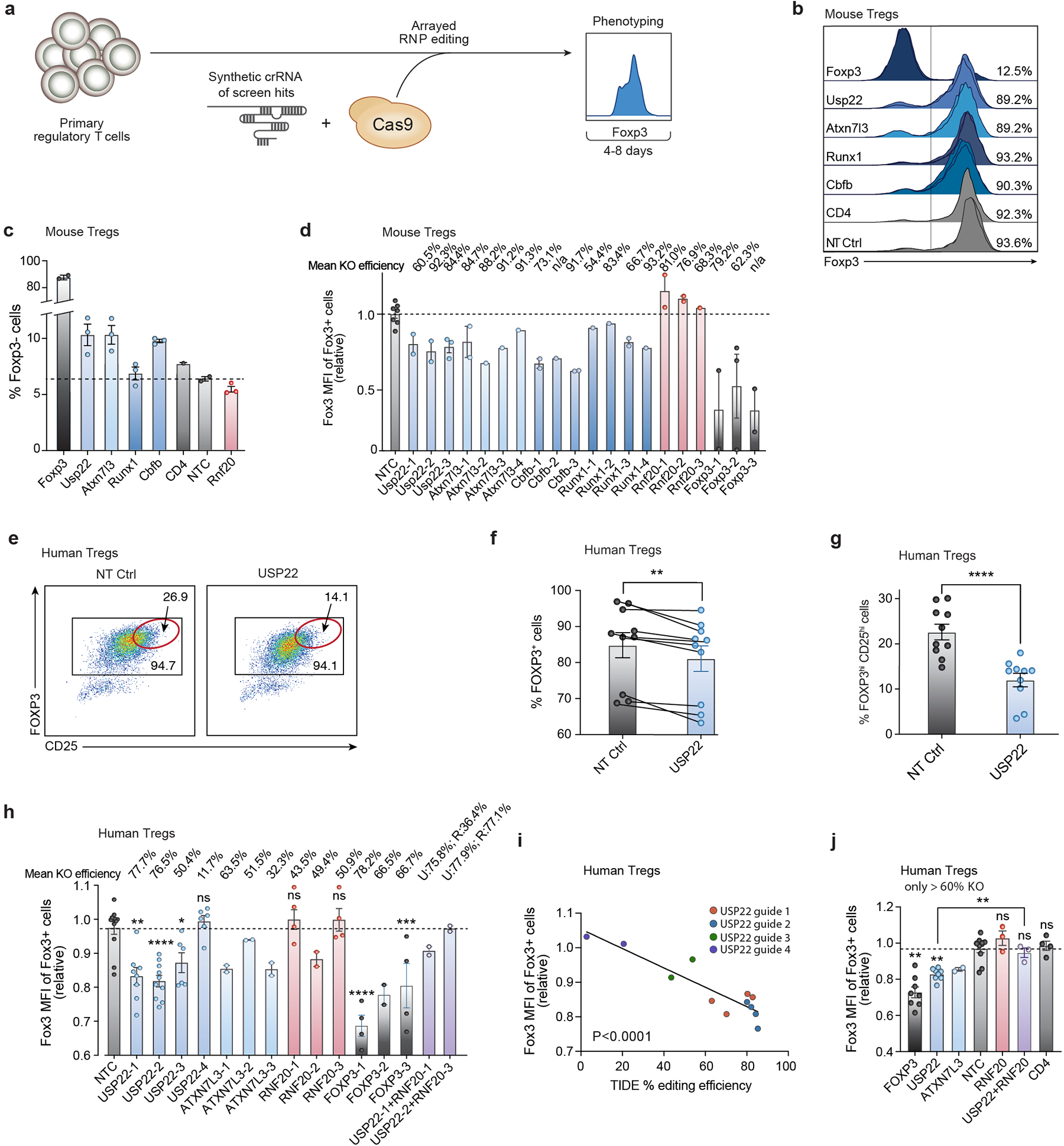
Figure 1. Discovery and Validation of Regulators of Foxp3 in Primary Mouse Tregs Using a Targeted Pooled CRISPR Screen.
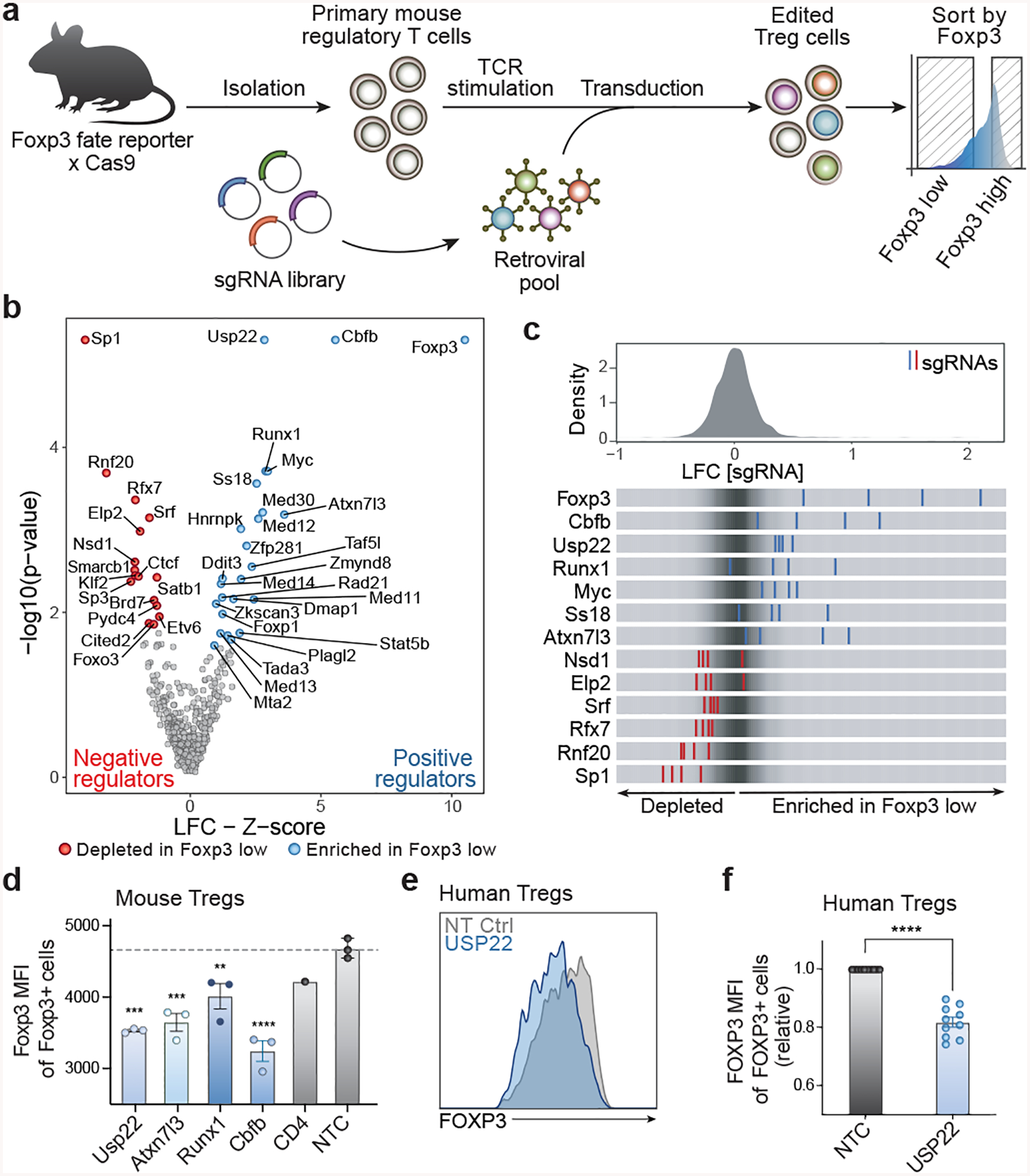
a) Diagram of pooled CRISPR screening platform in primary mouse Treg cells.
b) Volcano plot for hits from the screen. X-axis shows Z-score for gene-level log2 fold-change (LFC); median of LFC for all single guide RNAs (sgRNAs) per gene, scaled. Y-axis shows the p-value as calculated by MAGeCK7. Red are negative regulators (depleted in Foxp3 low cells), while blue dots show all positive regulators (enriched in Foxp3 low cells) defined by FDR < 0.5 and Z-score > 0.5.
c) Top panel: distribution of sgRNA-level LFC values of Foxp3 low over Foxp3 high cells for 2,000 guides. Bottom panel: LFC for all four individual sgRNAs targeting genes enriched in Foxp3 low cells (blue lines) and depleted genes (red lines), overlaid on grey gradient depicting the overall distribution.
d) Mean fluorescence intensity (MFI) of Foxp3 in Foxp3+ cells from data in Extended Data 2b. Each data point represents effects of an independent gRNA for each target gene. Statistics are based on comparison to non-targeting control (NTC).
e) Representative histogram showing FOXP3 MFI (pre-gated on live cells) from human Tregs treated with non-targeting control (NTC) or USP22 Cas9 RNPs.
f) Statistical analysis of FOXP3 MFI in human Tregs from 10 biological replicates. Tregs from each donor here were targeted with the same high efficiency gRNA (USP22–2).
All data are presented as mean ±SEM. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section.
Our screen revealed many novel Foxp3 regulators, with a bias towards identifying positive regulators over negative regulators (Figs. 1b, 1c). sgRNAs enriched in the Foxp3low population reflect positive regulators (blue) that promote Foxp3 expression while sgRNAs depleted in the Foxp3low population reflect negative regulators (red) that inhibit Foxp3 expression. As expected, sgRNAs targeting Foxp3 were enriched in the Foxp3low population. We also identified many established regulators known to be important for maintenance of Foxp3 expression including Cbfb, Runx1 and Stat5b8–14 as positive regulators and Sp3 and Satb1 as negative regulators9,15 providing further confidence in our hits. Importantly, several novel factors and complexes that modulate Foxp3 were identified including positive regulators Usp22, Atxn7l3 and negative regulator Rnf20. The deubiquitinase (DUB) Usp22 and cofactor Atxn7l3, are both members of deubiquitination module of the SAGA chromatin modifying complex16 (Extended Data Fig. 1h).
To validate the effects of our screen hits on Foxp3 levels, we assessed five of the top-ranking positive regulators by individual CRISPR knockout with Cas9 ribonucleoproteins17 (RNPs) Extended Data Fig. 2a). The effects on Foxp3 levels were consistent across multiple guide RNAs (gRNAs) targeting the key candidate regulators (Fig. 1d, Extended Data Figs. 2b–d and Supplementary Table 2). These results strongly confirmed the candidate genes identified from our screen as positive regulators of Foxp3. As our screen indicated that Usp22 is a positive regulator of Foxp3, we next wanted to assess the potential therapeutic relevance of USP22 by knocking it out with RNPs in human Tregs. We saw a significant decrease in FOXP3 mean fluorescence intensity (MFI) (Figs. 1e, 1f) and frequencies of FOXP3+ and FOXP3hiCD25hi cells in USP22-targeted human Tregs (Extended Data Figs. 2e–g) across experiments performed in ten different blood donors. The effects of USP22 targeting in human Tregs on FOXP3 levels were also observed with multiple distinct gRNAs (Extended Data Figs. 2h–j). Together these findings confirm critical regulation of Foxp3 by members of the SAGA complex, especially Usp22.
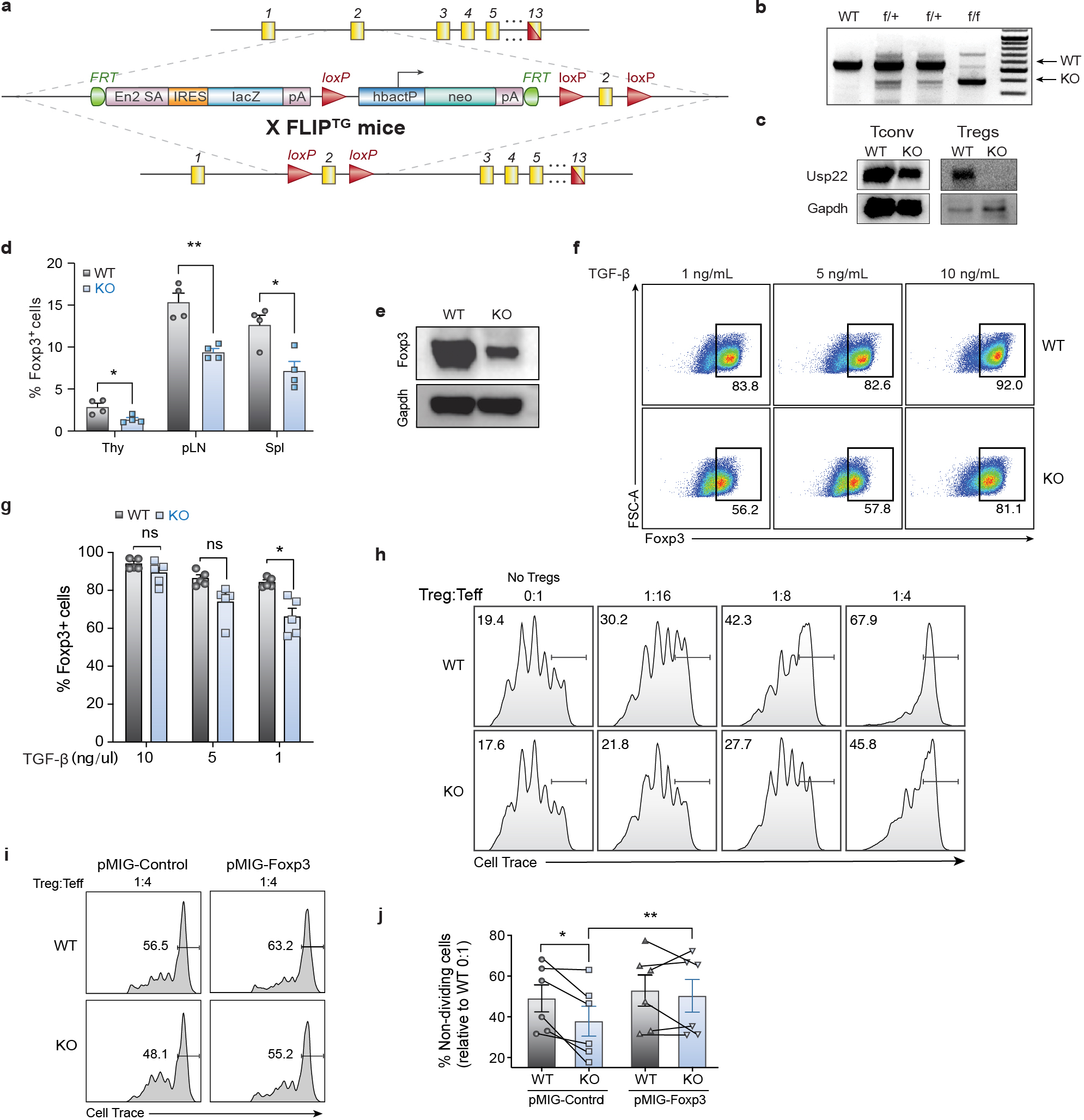
To understand the in vivo significance of Usp22 in Tregs, we generated mice with Treg-specific ablation of Usp22 by creating Usp22fl/fl mice (Extended Data Figs. 3a, 3b) and crossing them with Foxp3YFP-Cre mice18. Western blot analysis confirmed specific deletion of Usp22 in Treg cells, but not in CD4+ conventional T (Tconv) cells (Extended Data Fig. 3c). Usp22fl/flFoxp3YFP-Cre knockout (KO) mice had a marked decrease in Foxp3 MFI in Tregs isolated from spleens, thymus and peripheral lymph nodes (pLN) (Figs. 2a–c) compared to Usp22+/+Foxp3YFP-Cre wild-type (WT) mice, as well as decreased Treg frequencies (Extended Data Fig. 3d). Western blot analysis confirmed a significant reduction in Foxp3 protein in Usp22-null Tregs (Extended Data Fig. 3e). A decrease in Foxp3+ cells was also seen in induced Tregs (iTregs), although less pronounced with increasing levels of TGF-β (Extended Data Figs. 3f, 3g). Given the diminished Foxp3 levels in Usp22 KO Tregs, we reasoned that these cells may exhibit defects in suppressive function. Indeed, Usp22 KO Tregs were less able to suppress T effector cells than WT Tregs from Usp22+/+Foxp3YFP-Cre mice (Fig. 2d, Extended Data Fig. 3h), and this defect could be rescued by heterologous expression of Foxp3 (Extended Data Figs. 3i, 3j). These data substantiate our screen data and suggest Usp22 promotes Foxp3 levels and is critical for Treg function.
Figure 2. Usp22 is Required for Foxp3 Maintenance and Treg Suppressive Function.
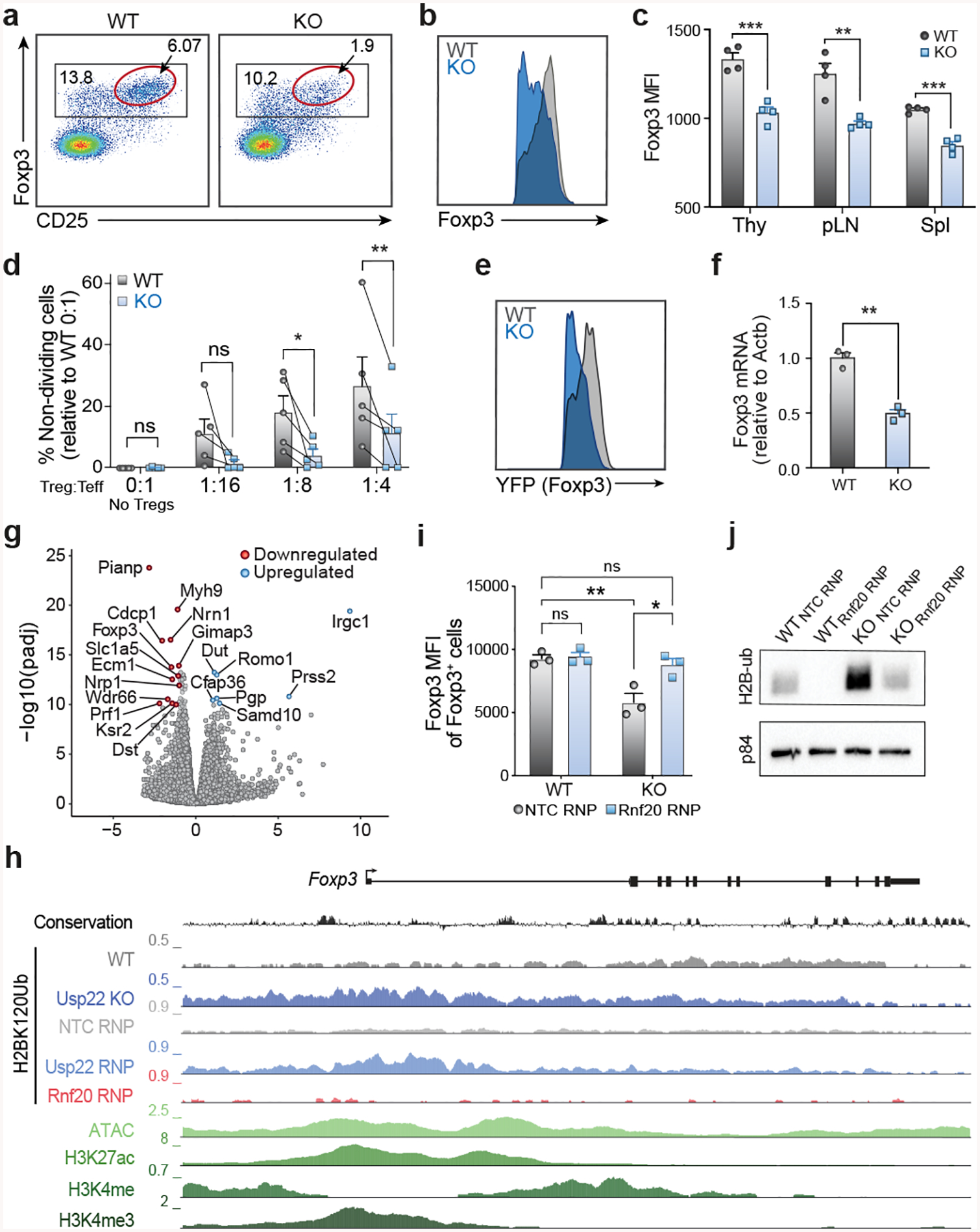
a) Representative flow cytometry analysis of the Treg population (gated on CD4+ cells) from the spleen of Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice. A subset of Tregs with the highest expression of Foxp3 and CD25 is highlighted with a red gate.
b) Histogram of Foxp3 expression in Foxp3+ Tregs from spleens of Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice from panel a.
c) Statistical analysis of Foxp3 MFI from CD4+Foxp3+ Tregs in thymus (Thy), peripheral lymph nodes (pLN) and spleen (Spl) of Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice.
d) Summary data of in vitro suppression experiments, corresponding to Extended Figure 3h. Lines connect paired samples. Data are presented as the frequency of non-dividing cells relative to WT 0:1 No Treg control, with any negative values after normalization replaced with 0.
e) Histogram of YFP expression in Tregs from the spleen and lymph nodes of Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice from Extended Data Fig. 5a.
f) qPCR analysis of Foxp3 mRNA levels in sorted YFP+ cells of spleen from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice.
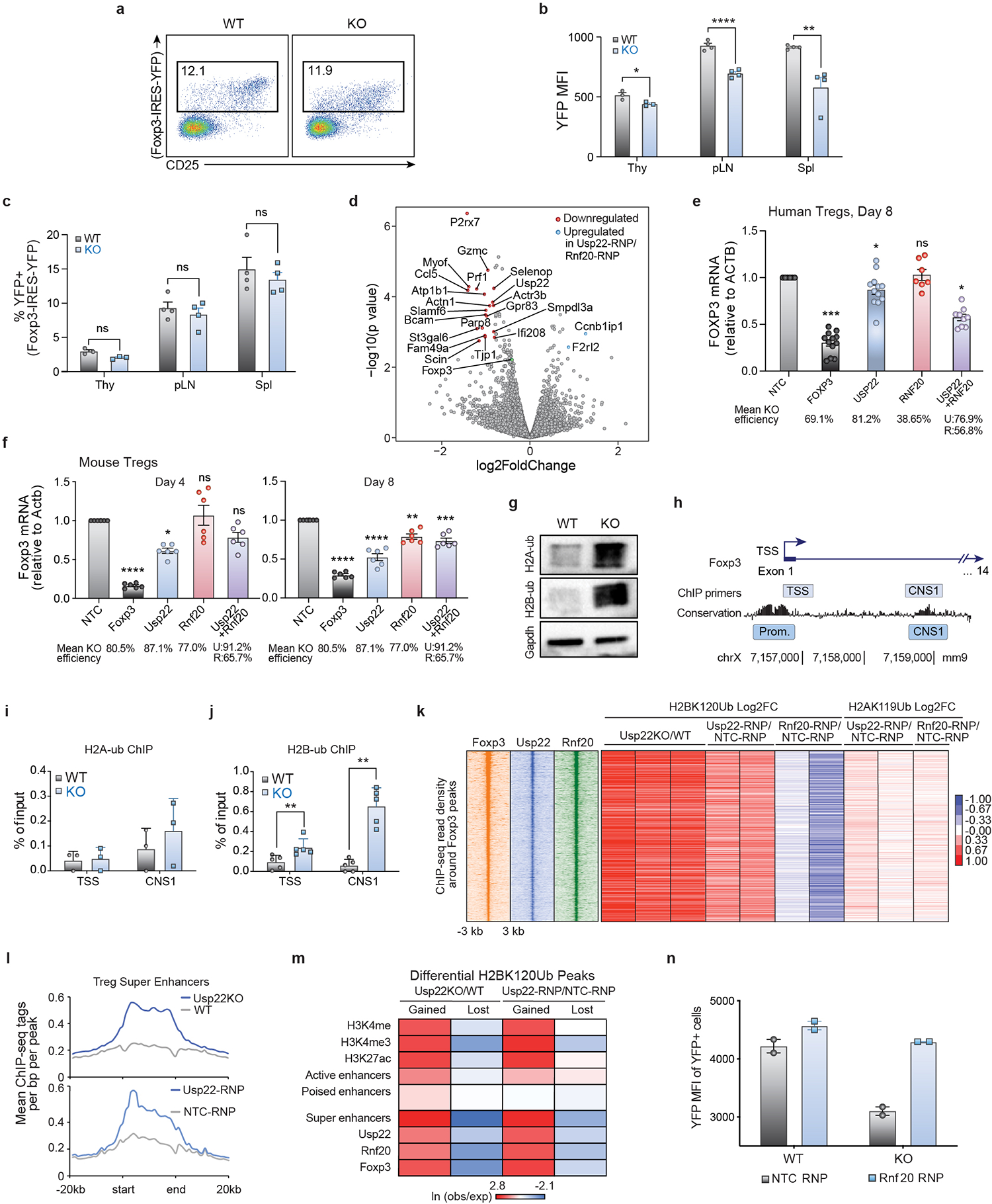
g) Volcano plot for RNA sequencing of YFP+ Tregs sorted from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice. X-axis shows log2FoldChange (LFC). Y-axis shows the –log10 of the adjusted p-value (padj) as calculated by DESeq2. Genes downregulated in the KO are shown in red and genes upregulated are shown in blue defined by padj <1e-10 and LFC > 1.
h) Genome tracks of ChIP-seq for H2BK120Ub at the Foxp3 locus in wild-type (WT), Usp22 KO, non-targeting control (NTC-RNP) treated, Usp22-RNP treated and Rnf20-RNP treated Tregs. Evolutionary conservation, ATAC-seq, and ChIP-Seq for H3K27ac, H3K4me3, and H3K4me in WT Tregs are also shown.
i) Analysis of reciprocal regulation of Foxp3 by deubiquitinase Usp22 and E3 ubiquitin ligase Rnf20. Foxp3 MFI of Tregs sorted from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice and then electroporated with either NT control (NTC-RNP) or Rnf20 RNP.
j) Western blot analysis of H2BK120Ub (H2B-ub) levels in Tregs sorted from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice and then electroporated with either NT control (NTC-RNP) or Rnf20 RNP; corresponding to panel i. p84 was used as a loading control. Source data can be found in Supplementary Figure 1.
All data are presented as mean ±SEM. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section.
Control of Foxp3 stability, and thus Treg function, can occur at the transcriptional and/or post-transcriptional level19. Chromatin modifications at the Foxp3 locus and other key loci can affect Foxp3 transcription20–25. As Usp22 is a component of the chromatin modifying SAGA complex, we hypothesized that Usp22 controls Foxp3 expression through transcriptional regulation. IRES-YFP knock-in to the Foxp3 locus of Usp22fl/flFoxp3YFP-Cre mice allowed us to use YFP as a reporter to assess the effect of Usp22 on Foxp3 transcript levels. Similar to endogenous Foxp3 protein, YFP MFI was significantly decreased in Usp22-null Tregs isolated from the thymus, pLN and spleen in Usp22fl/flFoxp3YFP-Cre mice compared to Usp22+/+Foxp3YFP-Cre mice, despite normal Treg frequencies (Fig. 2e, Extended Data Figs. 5a–c). Furthermore, by qPCR, Foxp3 transcripts were significantly reduced in splenic Tregs from Usp22fl/flFoxp3YFP-Cre compared to Usp22+/+Foxp3YFP-Cre mice (Fig. 2f). RNA sequencing confirmed that Foxp3 transcripts are significantly reduced in Tregs from Usp22fl/flFoxp3YFP-Cre mice relative to WT (Fig. 2g). Foxp3 transcript levels also trended down with acute targeting by Usp22 RNPs in mouse and human Tregs, although results were less consistent perhaps due to variability in knockout efficiency and/or other experimental factors (Extended Data Figs. 5d–f). Foxp3 protein can also be dynamically controlled post-translationally by DUBs or ubiquitin ligases in response to proinflammatory signals26–30. We investigated whether Foxp3 protein can be directly targeted by Usp22. Usp22 loss contributed to increased Foxp3 ubiquitination and degradation (Extended Data Fig. 4). These results are consistent with Usp22 tuning Foxp3 expression at transcriptional and post-transcriptional levels.
Usp22 is required for SAGA-mediated deubiquitination of histones, which regulates transcriptional activity31. We therefore tested if histone ubiquitination was altered in Usp22 KO Tregs. Western blot analysis confirmed that Usp22fl/flFoxp3YFP-Cre mice had increased levels of ubiquinated histone 2A and 2B (H2AK119Ub and H2BK120Ub, respectively) in iTregs compared to Usp22+/+Foxp3YFP-Cre mice (Extended Data Fig. 5g). Chromatin immunoprecipitation followed by qPCR (ChIP-qPCR) showed increased H2BK120Ub in the conserved non-coding sequence 1 (CNS1) region of the Foxp3 locus in Usp22fl/flFoxp3YFP-Cre Tregs, whereas effects on H2AK119Ub levels at the locus were not significant (Extended Data Figs. 5h–j). Further interrogation with ChIP followed by genome-wide sequencing (ChIP-seq) revealed significant increases in H2BK120Ub levels across the Foxp3 locus in Usp22fl/flFoxp3YFP-Cre Tregs compared with control Tregs. A significant accumulation of H2BK120Ub at the locus also was observed in Tregs electroporated with Usp22 Cas9 RNPs compared to those treated with NTC Cas9 RNPs (Fig. 2h). These findings demonstrated that Usp22 is essential for chromatin regulation at the Foxp3 locus.
We next analyzed effects of Usp22 loss on chromatin states across the Treg genome. First, we found evidence that Usp22 can co-occupy many Foxp3-bound regions in Tregs (Extended Data Fig. 5k). Foxp3-bound regions tended to gain H2BK120Ub in Usp22-deficient cells compared to control cells and increases in H2BK120Ub were more pronounced than effects on H2AK119Ub, suggesting that H2KB120Ub is likely the more relevant chromatin target of Usp22 in Tregs (Fig. 2h, Extended Data Fig. 5k). Looking more broadly across the genome, we found that sites that significantly gained H2BK120Ub in both Usp22KO and Usp22-RNP targeted Tregs were enriched for activating histone modifications (H3K4me3, H3K4me and H3K27ac) suggesting that changes occurred at gene regulatory elements, including at Treg-specific super enhancers (Extended Data Figs. 5l, 5m). These data revealed a critical role for Usp22 in control of H2KB120Ub across the Treg chromatin landscape.
Our screen nominated E3-ubiquitin ligase Rnf20 as a candidate negative regulator of Foxp3. We hypothesized that the DUB Usp22 and E3-ubiquitin ligase Rnf20 might have an epistatic relationship given their reciprocal effects on histone ubiquitination. To test this, we used RNPs to knockout Rnf20 in Usp22 KO or WT Tregs. Although Cas9 RNP-mediated loss of Rnf20 alone did not significantly increase Foxp3 levels in WT Tregs (Extended Data Fig. 2d and Fig. 2h), Rnf20 RNP knockout was able to rescue the impairment in Foxp3 transcript levels (assessed by YFP levels) in Usp22-deficient Tregs (Extended Data Fig. 5n). Double RNP knockout of both USP22 and RNF20 in mouse and human Tregs also rescued FOXP3 protein levels relative to USP22 RNP treatment alone, although effects on transcript levels were less consistent (Fig. 2i, Extended Data Figs. 2h, 2j, 5e, 5f). Consistent with a model where the ubiquitin ligase Rnf20 and DUB Usp22 have reciprocal functional roles, we found that Rnf20 co-occupies Foxp3-bound regions (Extended Data Fig. 5k). Although Rnf20 ablation did not affect already low levels of H2BK120Ub at the Foxp3 locus, targeting Rnf20 tended to reduce H2BK120Ub levels at these Foxp3-bound regions genome-wide whereas Usp22 deficiency increased them (Fig. 2h, Extended Data Fig. 5k). Western blot analysis confirmed that targeting Rnf20 in Usp22-deficient cells restored H2BK120Ub levels back to those of control Tregs (Fig. 2j). Taken together, these results revealed reciprocal regulation of Foxp3 and key chromatin regions in Tregs by Usp22 and Rnf20.
To determine the in vivo functional relevance of Usp22 deficiency in Tregs, we characterized the spontaneous autoimmune symptoms of Usp22fl/flFoxp3YFP-Cre mice. While Usp22fl/flFoxp3YFP-Cre mice were born at normal size, their body weights were lower than those of age and sex matched Usp22+/+Foxp3YFP-Cre mice after 5 weeks of age (Fig. 3a, Extended Data Fig. 6a). We next assessed whether this body weight reduction might be due to chronic inflammation as is observed with impaired Treg function1. Indeed, flow cytometry analysis detected greater frequencies of CD4+ and CD8+ effector memory T cells (CD44hiCD62Llo) and corresponding lower percentages of naïve T cells (CD44loCD62Lhi) in 7-month-old KO mice compared to WT (Extended Data Figs. 6b, 6c). Additionally, histological analysis of aged mice detected lymphocyte infiltration in multiple organs, including kidney, lung, colon and liver (Fig. 3b). Importantly, ablation of Usp22 in all T cells in Usp22fl/flLckCre mice largely phenocopied the reduced levels of Foxp3 in Tregs and lymphoproliferation observed in the Usp22fl/flFoxp3YFP-Cre mice (Extended Data Fig. 7). These findings underscored a relatively selective role of Usp22 in Foxp3 regulation and Treg suppressive function, rather than global requirement for Usp22 in T cell function.
Figure 3. Treg-Specific Ablation of Usp22 Results in Autoimmunity and Enhances Anti-Tumor Immunity.
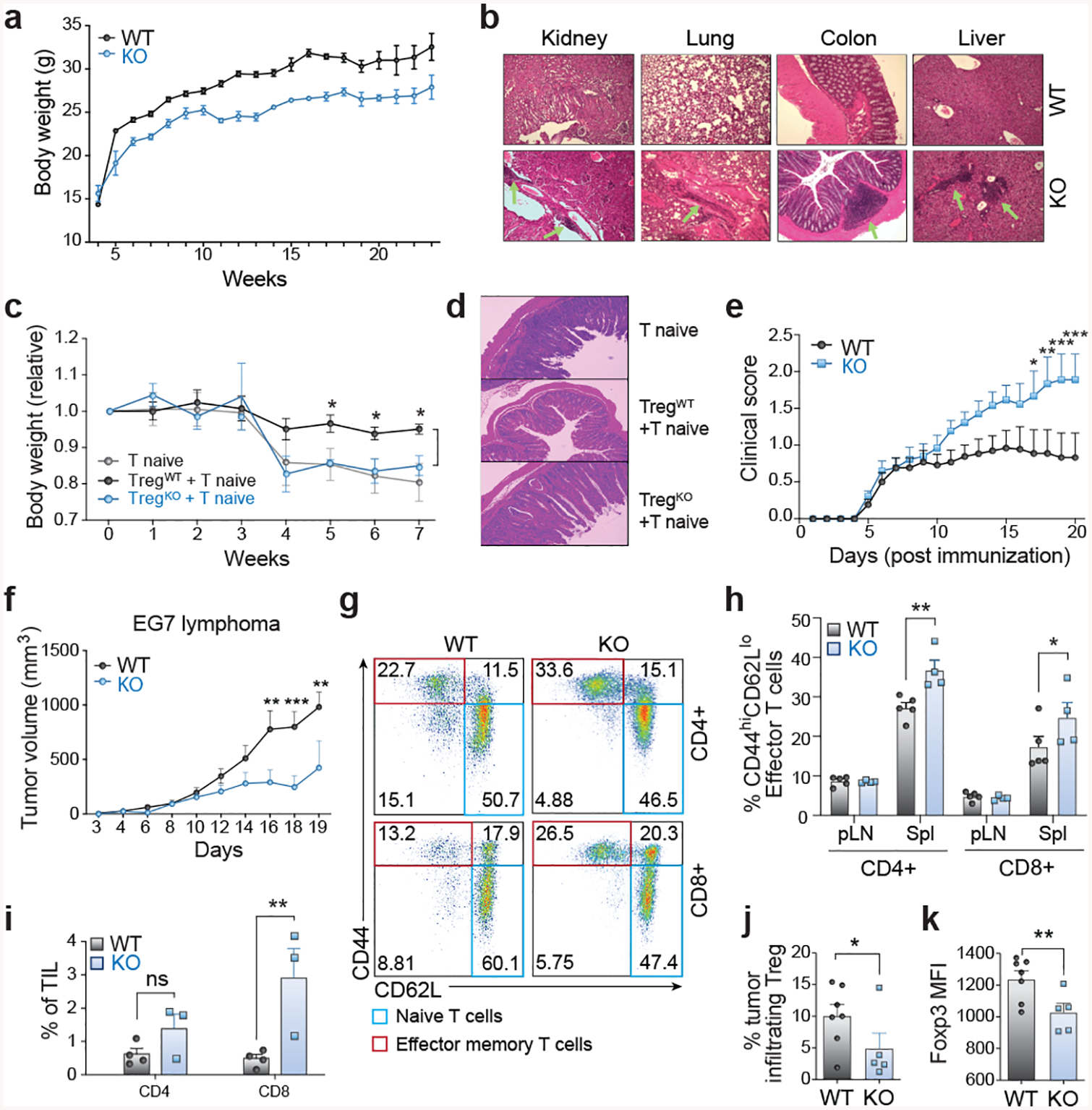
a) Body weight differences between Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO littermate mice.
b) Hematoxylin-and-eosin (H&E) staining of kidney, lung, colon and liver sections from 7-month-old Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice. Original magnification at 100x (fold).
c) Body weight of Rag−/− recipient mice over time after adoptive transfer of CD4+CD25−CD44loCD62hi (CD45.1+) naïve T cells sorted from SJL mice alone or together with CD4+YFP+ (CD45.2+) Treg cells from 9-week-old Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice, presented relative to weight at day 0.
d) H&E staining of colon tissues from the Rag−/− recipient mice shown in panel c, 7 weeks post-transfer. Original magnification at 100x.
e) Clinical severity of EAE in Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice was monitored for 20 days post immunization with MOG peptide.
f) EG7 lymphoma tumor volume in Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice. Mice were subcutaneously inoculated with 1×106 EG7 cells. Tumor volume was measured every 1–2 days by scaling along 3 orthogonal axes (x, y, and z) and calculated as (xyz)/2.
g) Representative flow cytometry analysis of the expression of CD44 and CD62L in both CD4+ and CD8+ T cells of spleen from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO EG7 tumor-bearing mice.
h) The frequency of effector T cells (CD44hiCD62Llo) from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO EG7 tumor-bearing mice summarized.
i) Statistical analysis of tumor-infiltrating lymphocyte (TIL) percentages from EG7-bearing Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice collected 19 days after tumor inoculation.
j) Statistical analysis of tumor-infiltrating Treg percentages from EG7 tumor-bearing Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice collected 19 days after tumor inoculation.
k) Foxp3 MFI of the CD4+Foxp3+ EG7 tumor-infiltrating Treg population in Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice summarized.
All data are presented as mean ±SEM. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section.
We further validated the in vivo requirements for Usp22 in Treg suppressive function using multiple models of autoimmune disease. We assessed Treg suppressive activity in vivo using an adoptive transfer model of colitis and a MOG-induced experimental autoimmune encephalomyelitis (EAE) model. In the colitis model, mice that received defective Usp22 KO Tregs were not protected against colitis, in contrast to those that received WT Tregs (Figs. 3c, 3d). Similarly, in the EAE model, Usp22fl/flFoxp3YFP-Cre mice showed worse clinical scores compared to WT mice suggesting an inability of the Usp22-deficient Tregs to limit autoimmunity (Fig. 3e).
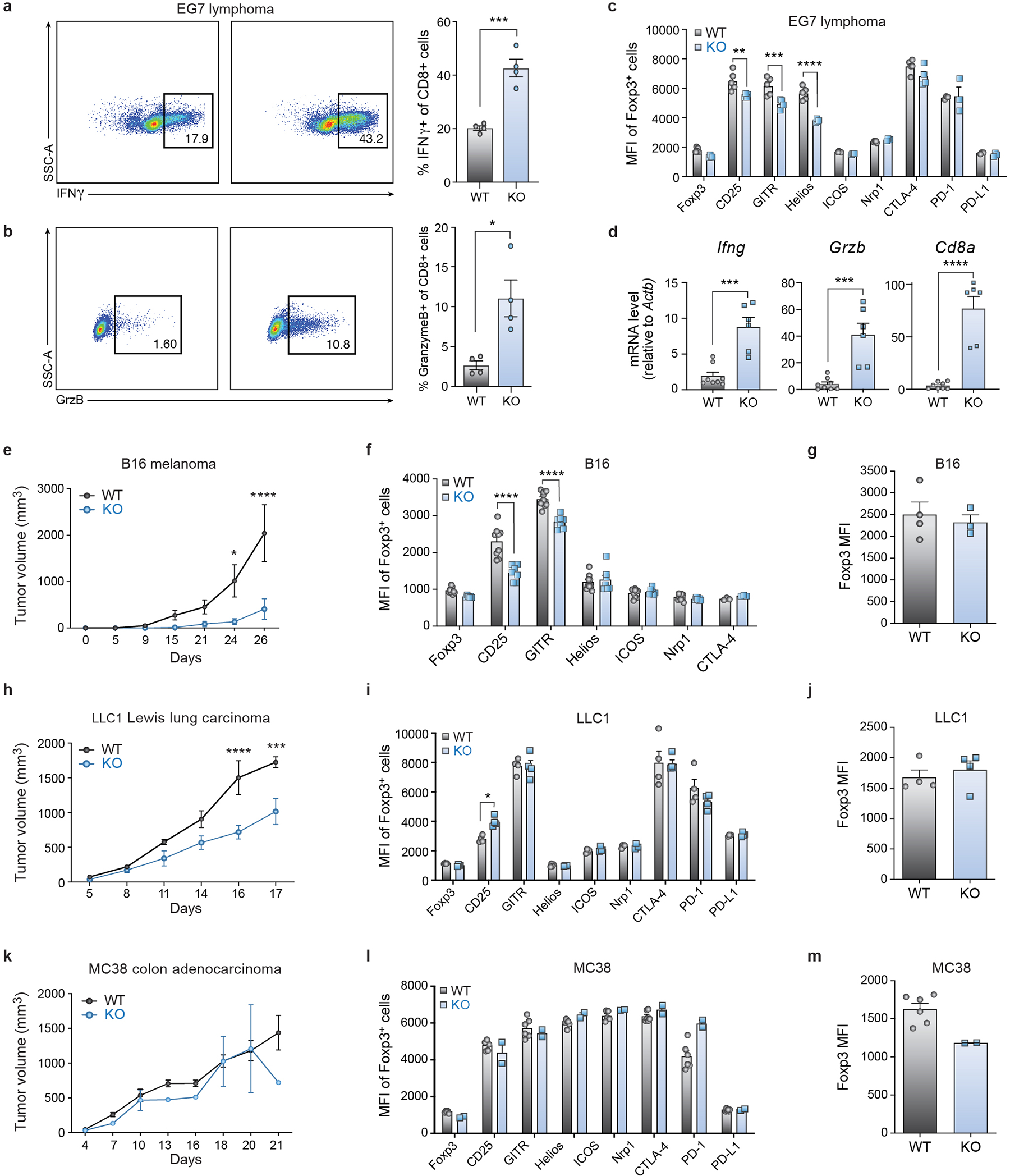
Since these data suggest that Usp22 deficiency reduces Foxp3 stability and impairs Treg suppressive function, we next tested whether Usp22fl/flFoxp3YFP-Cre mice would exhibit increased anti-tumor immunity using syngeneic tumor models. As expected, growth of EG7 lymphoma tumors was significantly inhibited by Treg-specific Usp22 gene deletion (Fig. 3f). We next examined the immune responses in these tumor-bearing mice and found greater proportions of effector-memory CD4+ and CD8+ T cells in the spleens of Usp22fl/flFoxp3YFP-Cre mice compared to WT (Figs. 3g, 3h). We also found increased frequencies of interferon-γ (IFNγ) and granzyme B producing CD8+ T cells, as well as increased mRNA levels of Ifng, Gzmb and Cd8a from tumor tissue (Extended Data Fig. 8d), suggesting an increased cytotoxic lymphocyte response due to impaired Treg suppressive function. Splenic Tregs from these mice showed reduced MFIs of Foxp3 target genes important for Treg function including CD25 (Extended Data Figs. 8a–c). Further analysis of tumor-infiltrating lymphocytes indicated a significant increase in CD8+ T cell frequencies and decreased percentages of intratumoral Foxp3+ Tregs in EG7 tumor-bearing Usp22fl/flFoxp3YFP-Cre mice (Fig. 3i–j). Consistent with the lymphoid organs, we found that the Foxp3 MFI was significantly decreased in the intratumoral Treg cells from EG7 tumor-bearing KO mice (Fig. 3k). Taken together, these data indicate Usp22 KO impairs Treg suppressive function and reduces Treg abundance in EG7 tumors, consequently enhancing the anti-tumor immune response. We also showed that Usp22fl/flFoxp3YFP-Cre mice exhibit increased anti-tumor immunity in additional tumor models (Extended Data Figs. 8e–m). These results highlight Usp22 in Tregs as a new potential target for anti-tumor immunotherapies.
Here, we developed the first CRISPR-based pooled screening platform for primary mouse Tregs and applied this technology for systematic identification of gene modifications that control Foxp3 levels. We discovered several novel regulators of Foxp3 including Usp22 and Rnf20. We developed a Treg-specific Usp22 KO mouse and showed that Usp22 is critical to stabilize Foxp3 and maintain suppressive functions in vivo. We demonstrate that Usp22 is a regulator of Foxp3 transcript levels, likely through deubiquitination of H2B at Foxp3 and other key loci, and that Usp22 can also regulate Foxp3 post-translationally. Mice with Usp22-null Tregs showed impaired ability to resolve autoimmune inflammation and an enhanced anti-tumor immune response. Usp22 could be a particularly attractive cancer immunotherapy target because in addition to its role in Tregs that can limit anti-tumor immune responses, over-expression of Usp22 in cancer cells is associated with poor prognosis in a variety of tumor types32 and Usp22 knockdown in cancer cells can induce their apoptosis33. This study provides a resource of novel Foxp3 regulators that can be perturbed to fine tune Treg function and specifically defines the function of Usp22 and Rnf20 as important modulators of Foxp3 and potential targets for Treg immunotherapies.
Methods
Mice for Screen and RNP Validation
B6 Foxp3GFP-Cre mice36 were crossed with B6 Rosa26LSL-RFP reporter mice37 as previously described38 to generate the Foxp3 fate reporter mice. These mice were then crossed to B6 constitutive Cas9-expressing mice39 to generate the Foxp3GFP-CreRosa26LSL-RFPCas9 mice used for the CRISPR screen. For the arrayed validation experiments, B6 Foxp3EGFP knock-in mice40 that were obtained from Jackson Laboratories (Strain No. 006772) were used. These mice were maintained in the UCSF specific-pathogen-free animal facility in accordance with guidelines established by the Institutional Animal Care and Use Committee and Laboratory Animal Resource Center.
Isolation and Culture of Primary Mouse Tregs for Screen and Validation
Spleens and peripheral lymph nodes were harvested from mice and dissociated in 1x PBS with 2% FBS and 1 mM EDTA. The mixture was then passed through a 70-μm filter. CD4+ T cells were isolated using the CD4+ Negative Selection Kit (StemCell Technologies, Cat# 19752) followed by fluorescence-activated cell sorting. For the prescreen sort, Tregs were gated on lymphocytes, live cells, CD4+, CD62L+, RFP+, Foxp3-GFP+ cells. For the arrayed validation experiments, Tregs were gated on lymphocytes, live cells, CD4+, Foxp3-GFP+ cells. Sorted Tregs were cultured in complete DMEM, 10% FBS, 1% pen/strep + 2000U hIL-2 in 24 well plates at 1 million cells/mL. Tregs were stimulated using CD3/CD28 Mouse T-Activator Dynabeads (Thermo Fisher, Cat# 11456D) at a ratio of 3:1 beads to cells for 48 hours. Cells were split and media was refreshed every 2–3 days.
Pooled sgRNA Library Design and Construction
For the cloning of the targeted library, we followed the custom sgRNA library cloning protocol as previously described41. We utilized a MSCV-U6-sgRNA-IRES-Thy1.1 backbone (gifted from the Bluestone Lab). To optimize this plasmid for cloning the library, we first replaced the sgRNA with a 1.9kb stuffer derived from the lentiGuide-Puro plasmid (Addgene, plasmid# 52963) with flanking BsgI cut sites. This stuffer was excised using the BsgI restriction enzyme (NEB, Cat# R0559) and the linear backbone was gel purified (Qiagen, Cat# 28706). We designed a targeted library to include all genes matching Gene Ontology for “Nucleic Acid Binding Transcription Factors”, “Protein Binding Transcription Factors”, “Involved in Chromatin Organization” and “Involved in Epigenetic Regulation.” Genes were then selected based on those that have the highest expression levels across any mouse CD4 T cell subset as defined by Stubbington et al42. In total, we included 489 targets with 4 guides per gene, GFP and RFP controls with 8 guides for each, and 28 non-targeting controls. Guides were subsetted from the Brie sgRNA library6, and the pooled oligo library was ordered from Twist Bioscience (San Francisco, CA) to match the vector backbone. Oligos were PCR amplified and cloned into the modified MSCV backbone by Gibson assembly as described by Joung et al41. The library was amplified using Endura ElectroCompetent Cells following the manufacturer’s protocol (Endura, Cat# 60242–1). All oligos included in the library and primer sequences are listed in Supplementary Table 3.
Retrovirus Production
Platinum-E (Plat-E) Retroviral Packaging cells (Cell Biolabs, Inc., Cat# RV-101) were seeded at 10 million cells in 15 cm poly-L-Lysine coated dishes 16 hours prior to transfection and cultured in complete DMEM, 10% FBS, 1% pen/strep, 1 μg/mL puromycin and 10 μg/mL blasticidin. Immediately before transfection, the media was replaced with antibiotic free complete DMEM, 10% FBS. The cells were transfected with the sgRNA transfer plasmids (MSCV-U6-sgRNA-IRES-Thy1.1) using TransIT-293 transfection reagent per the manufacturer’s protocol (Mirus, Cat# MIR 2700). The following morning, the media was replaced with complete DMEM, 10% FBS, 1% pen/strep. The viral supernatant was collected 48 hours post transfection and filtered through a 0.45 μm, polyethersulfone sterile syringe filter (Whatman, Cat# 6780–2504), to remove cell debris. The viral supernatant was aliquoted and stored until use at −80°C.
Retroviral Transduction
Tregs were stimulated as described above for 48–60 hours. Cells were counted and seeded at 3 million cells in 1 mL of media with 2x hIL-2 into each well of a 6 well plate that was coated with 15 μg/mL of RetroNectin (Takara, Cat# T100A) for 3 hours at room temperature and subsequently washed with 1x PBS. Retrovirus was added at a 1:1 v/v ratio (1 mL) and plates were centrifuged for 1 hour at 2000g at 30°C and placed in the incubator at 37°C overnight. The next day, half (1 mL) of the 1:1 retrovirus to media mixture was removed from the plate and 1 mL of fresh retrovirus was added. Plates were immediately centrifuged for 1 hour at 2000g at 30°C. After the second spinfection, cells were pelleted, washed, and cultured in fresh media.
Foxp3 Intracellular Stain and Post-Screen Cell Collection
Tregs were collected from their culture vessels 8 days after the second transduction and centrifuged for 5 min at 300g. Cells were first stained with a viability dye at a 1:1,000 dilution in 1× PBS for 20 min at 4°C, then washed with EasySep Buffer (1× PBS, 2% FBS, 1 mM EDTA). Cells were then resuspended in the appropriate surface staining antibody cocktail and incubated for 30 min at 4°C, then washed with EasySep Buffer. Cells were then fixed, permeabilized, and stained for transcription factors using the Foxp3 Transcription Factor Staining Buffer Set (eBioscience, Cat# 00-5523-00) according to the manufacturer’s instructions. Antibodies used in this study are listed in Supplementary Table 4. For the CRISPR screen, Foxp3high and Foxp3low populations were isolated using fluorescence-activated cell sorting by gating on lymphocytes, live cells, CD4+ and gating on the highest 40% of Foxp3-expressing cells (Foxp3high) and lowest 40% of Foxp3-expressing cells (Foxp3low) by endogenous Foxp3 intracellular staining. Over 2 million cells were collected for both sorted populations to maintain a library coverage of at least 1,000 cells per sgRNA (1000x).
Isolation of Genomic DNA from Fixed Cells
After cell sorting and collection, genomic DNA (gDNA) was isolated using a protocol specific for fixed cells. Cell pellets were resuspended in cell lysis buffer (0.5% SDS, 50 mM Tris, pH 8, 10 mM EDTA) with 1:25 v/v of 5M NaCl to reverse crosslinking and incubated at 66°C overnight. RNase A (10 mg/mL) was added at 1:50 v/v and incubated at 37°C for 1 hour. Proteinase K (20 mg/mL) was added at 1:50 v/v and incubated at 45°C for 1 hour. Phenol:Chloroform:Isoamyl Alcohol (25:24:1) was added to the sample 1:1 v/v and transferred to a phase lock gel light tube (QuantaBio, Cat# 2302820), inverted vigorously and centrifuged at 20,000g for 5 mins. The aqueous phase was then transferred to a clean tube and NaAc at 1:10 v/v, 1 μl of GeneElute-LPA (Sigma, Cat# 56575), and isopropanol at 2.5:1 v/v were added. The sample was vortexed, and incubated at −80°C until frozen solid. Then thawed and centrifuged at 20,000g for 30 mins. The cell pellet was washed with 500 μl of 75% EtOH, gently inverted and centrifuged at 20,000g for 5 mins, aspirated, dried, and resuspended in 20 μl TE buffer.
Preparation of Genomic DNA for Next Generation Sequencing
Amplification and bar-coding of sgRNAs was performed as previously described43 with some modifications. Briefly, after gDNA isolation, sgRNAs were amplified and barcoded with TruSeq Single Indexes using a one-step PCR. TruSeq Adaptor Index 12 (CTTGTA) was used for the Foxp3low population and TrueSeq Adaptor Index 14 (AGTTCC) was used for the Foxp3high population. Each PCR reaction consisted of 50μL of NEBNext Ultra II Q5 Master Mix (NEB, Cat# M0544), 1μg of gDNA, 2.5μL each of the 10μM forward and reverse primers, and water to 100μL total. The PCR cycling conditions were: 3 minutes at 98°C, followed by 10 seconds at 98°C, 10 seconds at 62°C, 25 seconds at 72°C, for 26 cycles; and a final 2 minute extension at 72°C. After the PCR, the samples were purified using Agencourt AMPure XP SPRI beads (Beckman Coulter, Cat #A63880) per the manufacturer’s protocol, quantified using the Qubit ssDNA high sensitivity assay kit (Thermo Fisher Scientific, Cat #Q32854), and then analyzed on the 2100 Bioanalyzer Instrument. Samples were then sequenced on an Illumina MiniSeq using a custom sequencing primer. Primer sequences are listed in Supplementary Table 3.
Pooled CRISPR Screen Pipeline
Primary Tregs were isolated from the spleen and lymph nodes of three male Foxp3-GFP-Cre/Rosa26-RFP/Cas9 mice aged 5–7 months old, pooled together, and stimulated for 60 hours. Cells were then retrovirally transduced with the sgRNA library and cultured at a density of 1 million cells/ml continually maintaining a library coverage of at least 1,000 cells per sgRNA. Eight days after the second transduction, cells were sorted based on Foxp3 expression defined by intracellular staining. Genomic DNA was harvested from each population and the sgRNA-encoding regions were then amplified by PCR and sequenced on an Illumina MiniSeq using custom sequencing primers. From this data, we quantified the frequencies of cells expressing different sgRNAs in each in each population (Foxp3high and Foxp3low) and quantified the phenotype of the sgRNAs, which we have defined as Foxp3 stabilizing (enriched in Foxp3high) or Foxp3 destabilizing (enriched in Foxp3low).
Analysis of Pooled CRISPR Screen
Analysis was performed as previously described44. To identify hits from the screen, we used the MAGeCK software to quantify and test for guide enrichment7. Abundance of guides was first determined by using the MAGeCK “count” module for the raw fastq files. For the targeted libraries, the constant 5’ trim was automatically detected by MAGeCK. To test for robust guide and gene-level enrichment, the MAGeCK “test” module was used with default parameters. This step included median ratio normalization to account for varying read depths. We used the non-targeting control guides to estimate the size factor for normalization, as well as to build the mean-variance model for null distribution, which was used to find significant guide enrichment. MAGeCK produced guide-level enrichment scores for each direction (i.e. positive and negative) which were then used for alpha-robust rank aggregation (RRA) to obtain gene-level scores. The p-value for each gene was determined by a permutation test, randomizing guide assignments and adjusted for false discovery rates by the Benjamini–Hochberg method. Log2 fold change (LFC) was also calculated for each gene, defined throughout as the median LFC for all guides per gene target. Where indicated, LFC was normalized to have a mean of 0 and standard deviation of 1 to obtain the LFC Z-score. MAGeCK analysis for sgRNA and gene level enrichment and normalized and raw count files can be found in Supplementary Table 1.
Arrayed Cas9 Ribonucleotide Protein (RNP) Preparation and Electroporation
RNPs were produced by complexing a two-component gRNA to Cas9, as previously described17. In brief, crRNAs and tracrRNAs were chemically synthesized (IDT), and recombinant Cas9-NLS were produced and purified (QB3 Macrolab). Lyophilized RNA was resuspended in Nuclease-free Duplex Buffer (IDT, Cat# 1072570) at a concentration of 160 μM, and stored in aliquots at −80°C. crRNA and tracrRNA aliquots were thawed, mixed 1:1 by volume, and annealed by incubation at 37°C for 30 min to form an 80 μM gRNA solution. Recombinant Cas9 was stored at 40 μM in 20 mM HEPES-KOH, pH 7.5, 150 mM KCl, 10% glycerol, 1 mM DTT, were then mixed 1:1 by volume with the 80 μM gRNA (2:1 gRNA to Cas9 molar ratio) at 37°C for 15 min to form an RNP at 20 μM. RNPs were electroporated immediately after complexing. RNPs were electroporated 3 days after initial stimulation. Tregs were collected from their culture vessels and centrifuged for 5 min at 300g, aspirated, and resuspended in the Lonza electroporation buffer P3 using 20 μl buffer per 200,000 cells. 200,000 Tregs were electroporated per well using a Lonza 4D 96-well electroporation system with pulse code EO148 (mouse) or EH115 (human). Immediately after electroporation, 80 μL of pre-warmed media was added to each well and the cells were incubated at 37°C for 15 minutes. The cells were then transferred to a round-bottom 96-well tissue culture plate and cultured in either complete DMEM, 10% FBS, 1% pen/strep + 2000U hIL-2 at 200,000 cells/well in 200 μl of media (mouse) or X-VIVO 15 media (Lonza, Cat# 04–418Q), supplemented with 5% FBS, 50uM 2-mercaptoethanol, 10uM N-acetyl L-cysteine and 1% pen/strep with hIL-2 at 300U/mL at 200,000 cells/well in 200 μl of media (human).
PCR Amplification of Target Regions and TIDE Analysis
Editing of the DNA was confirmed by Tracking of Indels by DEcomposition (TIDE) analysis 4–8 days post-electroporation and performed as previously described45. A total of 5*10^4 to 1*10^5 cells were re-suspended in 30μL of QuickExtract DNA Quick Extraction solution (Epicentre) to lyse the cells and extract genomic DNA. The cell lysate was incubated at 65°C for 15 min, 95°C for 5 min, and then stored at −20°C until PCR could be performed across the CRISPR/Cas9 target sites. Unique genomic primers to amplify across the proposed cut sites were designed using the Primer3 online web tool (http://bioinfo.ut.ee/primer3/), chemically synthesized (IDT), and suspended at 100μM. Each PCR reaction contained 2μl 10x High-fidelity PCR buffer (Life Technologies), 3μl 2mM dNTPs (Bioline), 0.8μl 50mM MgCl2 (Life Technologies), 0.6μl 10μM forward primer, 0.6μl 10μM reverse primer, 0.2μl 5U/μl Platinum HIFI Taq (Life Technologies), 1μl extracted DNA, and 11.8μl H2O. The primer sets used for each crRNA can be found in Supplementary Table 2. The thermocycler setting consisted of one step at 95°C for 5 minutes, followed by 14 cycles at 94°C for 20 seconds, 65°C for 20 seconds, and 72°C for 1 minute (wherein the annealing temperature was decreased by 0.5°C per cycle), followed by 35 cycles at 94°C for 20 seconds, 58°C for 20 seconds, and 72°C for 1 minute with one final step at 72°C for 10 minutes. PCR cleanup and capillary sequencing was performed by Quintarabio (San Francisco, CA). Sequencing traces were analyzed with the TIDE webtool (http://tide.nki.nl/)46.
Isolation, Culture and FOXP3 Intracellular Staining of Human Treg Cells
Primary human Treg cells for all experiments were sourced from healthy donors from leukoreduction chamber residuals after Trima Apheresis (Vitalant, formerly Blood Centers of the Pacific) or from freshly drawn whole blood under a protocol approved by the UCSF Institutional Review Board (IRB# 13–11950). Peripheral blood mononuclear cells (PBMCs) were isolated from samples by Lymphoprep centrifugation (StemCell, Cat #07861) using SepMate tubes (StemCell, Cat# 85460). CD4+ T cells were isolated from PBMCs by magnetic negative selection using the EasySep Human CD4+ T Cell Isolation Kit (StemCell, Cat# 17952) and Tregs were then isolated using fluorescence-activated cell sorting by gating on CD4+, CD25+, CD127low cells. After isolation, cells were stimulated with ImmunoCult Human CD3/CD28/CD2 T Cell Activator (StemCell, Cat# 10970) per the manufacturer’s protocol and either electroporated after 48h of stimulation or expanded for 9 days. If expanded, Tregs were restimulated on day 9; 48h before RNP electroporation. Cells were cultured in X-VIVO 15 media (Lonza, Cat# 04–418Q), supplemented with 5% FBS, 50uM 2-mercaptoethanol, 10uM N-acetyl L-cysteine and 1% pen/strep with hIL-2 at 300U/mL at 1 million cells/mL. For intracellular staining, cells were collected and centrifuged at 300xg for 5mins. Cells were resuspended with a viability dye at a 1:1,000 dilution in 1× PBS for 20 min at 4°C, then washed with EasySep Buffer (1× PBS, 2% FBS, 1 mM EDTA). Cells were then resuspended in the appropriate surface staining antibody cocktail and incubated for 30 min at 4°C, then washed with EasySep Buffer. Cells were then fixed, permeabilized, and stained for transcription factors using the True-Nuclear Transcription Factor Buffer Set (BioLegend, Cat# 424401) according to the manufacturer’s instructions. Antibodies used in this study are listed in Supplementary Table 4.
Generation of Usp22 Knockout Mice
Usp22 floxed mice were generated and used as recently reported47. The Usp22 target mouse embryonic stem cells from C57BL/6 mice were purchased from the Wellcome Trust Sanger Institute. Blastocyst injections resulted in several chimeric mice with the capacity for germline transmission. Breeding of heterozygous mice yielded Usp22+/+, Usp22+/targeted but not Usp22targeted/targeted mice due to the obligation of Usp22 expression by the neomycin selection and β-gal reporter cassette, which causes embryonic lethality33. We then bred Usp22+/targeted mice with Flp recombinase transgenic mice to delete the selection cassette, leading to the generation of Usp22+/fl mice, further breeding of which produced Usp22+/+, Usp22+/fl and Usp22fl/fl mice without phenotypic abnormalities in expected Mendelian ratios (Extended Data Figs. 3a, 3b). Treg-specific Usp22-null mice were generated by breeding Usp22fl/fl mice with Foxp3YFP-Cre mice18. T cell-specific Usp22-null mice were generated by breeding Usp22fl/fl mice with LckCre mice. Additionally, C57BL/6 Rag−/−mice, SJL CD45.1 congenic mice were purchased from Jackson Laboratories. These mice were maintained and used at the Northwestern University mouse facility under pathogen-free conditions according to institutional guidelines and using animal study proposals approved by the institutional animal care and use committees. Unless stated otherwise, all figures are representative of experiments with healthy 6–8 week-old mice.
Cell lines, Plasmids, Antibodies, and Reagents
Platinum-E (Plat-E) Retroviral Packaging cells (Cell Biolabs, Inc., Cat# RV-101) were provided by the Bluestone and Cyster Labs and cultured per the manufacturer’s instructions. Human embryonic kidney 293 cells (HEK293) were stored in the Fang lab and were cultured in DMEM containing 10% FBS. EG7 lymphoma, MC38 colon cancer, LLC1-OVA lung carcinoma and B16-SIY melanoma cell lines were provided by Dr. Bin Zhang and used for tumor models as previously reported48. Cell lines were not genetically authenticated. HEK293 and cancer cell lines were tested for mycoplasma using LookOut Mycoplasma PCR detection kit (Sigma, Cat# MP0035–1KT). Myc-Usp22, Myc-Usp22(C185A), FLAG-Foxp3 and HA-ubiquitin expression plasmids and their tagged vectors were constructed and stored in the Fang lab. Antibodies used for Western blots, Co-IPs and flow cytometry are listed in Supplementary Table 4. PMA (phorbol 12-myristate 13-acetate), ionomycin, and cycloheximide (CHX) were purchased from Sigma. Monesin was from eBioscience.
Cell Isolation and Flow Cytometry for Analysis of Usp22 KO Mice
Peripheral T cells were isolated from mouse spleen by a CD4+ T-cell negative (Stem Cell) or positive selection kit (Invitrogen). Enriched CD4+ T cells were further sorted for either YFP+ (Foxp3+) T cells or CD25−CD44loCD62Lhi naïve T cells by FACSAria (BD Bioscience). Purity of sorted cells was > 99%. Lymphocytes isolated from the intestinal lamina propria were acquired by following previously described methods49. To isolate tumor-infiltrating lymphocytes, subcutaneous tumors were cut into small fragments and digested by collagenase D (Sigma) and DNase (Sigma) for 1h at room temperature. Flow cytometry was done with a FACSCanto II. Samples were initially incubated with anti-CD16/32 antibodies to block antibody binding to Fc receptor. Single-cell suspensions were stained with relevant antibodies (Supplementary Table 4) and then washed twice with cold PBS containing 3% FBS. For intracellular staining, cells were then fixed, permeabilized, and stained for transcription factors using the Foxp3 Transcription Factor Staining Buffer Set (eBioscience, Cat# 00-5523-00) according to the manufacturer’s instructions. For cytokine staining, cells were first stimulated for 4–5 h with 20 ng/ml PMA plus 0.5μM ionomycin in the presence of monesin (10 μg/ml) before staining. Data were analyzed with FlowJo software.
Tumor Models
Cultured cancer cells were trypsinized and washed once with PBS. 1×106 tumor cells (for EG7 cells, LLC1 cells and MC38 cells) or 5×104 tumor cells (for B16 melanoma) in suspension were subcutaneously injected into WT or Usp22fl/flFoxp3YFP-Cre 8–10 week-old mice. Tumors were measured every 2–3 days by measuring along 3 orthogonal axes (x, y, and z) and calculated as (xyz)/2 as recently reported48. To isolate tumor-infiltrating lymphocytes, tumors were cut into small fragments and digested with 50 mg/ ml collagenase D (Sigma) and 20 mg/ml DNase (Sigma) for 1h at room temperature. Tumors were then washed and subsequently strained through a 70 micron filter to achieve single cell suspensions. The tumor size limit agreed by IRB was 2cm3.
In Vitro Treg Suppression Assay
Naïve CD4+CD25− T cells (5×104) labeled with eFluor 670 cell proliferation dye were used as responder T cells and cultured in 96-well U-bottom plate for 72h together with increasing ratio of sorted YFP+ Treg cells from WT or Usp22fl/fl Foxp3YFP-Cre mice in the presence of irradiated splenocytes depleted of T cells (5×104) plus anti-CD3 (2 μg/ml). The suppressive function of Treg cells was determined by measurement of the proliferation of activated CD4+ and CD8+ effector T cells on the basis of eFluor 670 cell proliferation dye dilution as reported50.
Rescue Experiment with Foxp3 Overexpression
CD4+ cells were isolated from harvested LN and spleens of 8-week-old Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice with the Invitrogen CD4+ purification kit (ThermoFisher Cat# 11331D). YFP+ Tregs were sorted and subsequently stimulated with 3:1 beads to cells using CD3/CD28 dynabeads in complete DMEM with 2000U hIL-2 at a culture density of ~1 million cells/mL in a 24 well plate for 48 hours. After 48h stimulation, cells were transferred to RetroNectin coated 6-well plates at a density of 3 million cells/mL with a 1:1 ratio of virus to media. RetroNectin coating was done at room temperature for 3hr with 1mL of 15ug/mL in PBS in each of the wells in the 6-well plate. The cells were spinfected for 1hr at 2000xg, then left overnight in the plate at 37°C. The following day, 1mL of new virus was added to the cells for a second spinfection for 1hr at 2000xg. Once spinfection was complete, the cells were plated in a 24-well plate at a density of 1million/well in T cell media in complete RPMI 1640 medium containing 10% FBS, 1% penicillin/streptomycin (MediaTech), 50 μM β-mercaptoethanol (Gibco), and 1% L-glutamine (Gibco) for 72 hours. After rest, the cells were sorted for YFP+GFP+ viral infected Tregs. Naïve CD4+CD25− T cells (5×104) labeled with APC CFSE cell proliferation dye were used as responder T cells and cultured in 96-well u-bottom plate for 72h together with increasing ratio of the sorted GFP+YFP+ Treg cells with anti-CD3 and anti-CD28 (2 μg/ml). The suppressive function of Treg cells was assessed by flow cytometry measurement of the proliferation of activated CD4+ effector T cells on the basis of APC CFSE cell proliferation dye dilution.
Induced Treg (iTreg) Differentiation
0.5×106 spenic CD4+CD25− naïve T cells were isolated from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice and cultured in 24-well plates coated with 3 μg/ml anti-CD3 and 5μg/ml anti-CD28 antibodies for 5 days. For iTreg cell polarization, the cultures were supplemented with IL-2 (5 ng/ml), anti-IFN-γ (2 μg/ml), anti-IL-4 (2 μg/ml) and TGF-β (at 2, 5 or 10 ng/ml). Cytokines were purchased from Peprotech.
Th1, Th2 and Th17 In Vitro Differentiations
0.5×106 splenic CD4+CD25− naïve T cells were isolated from Usp22fl/flLckCre KO or Usp22+/+LckCre WT mice and cultured in 24-well plates coated with 3 μg/ml anti-CD3 and 5μg/ml anti-CD28 antibodies for 5 days. For skewing towards different T cells subsets, the cultures were supplemented with the following cytokines and antibodies. Th1, IL-2 (5 ng/ml) + IL-12 (5 ng/ml) + anti-IL-4 (1 μg/ml); Th2, IL-2 (5ng/ml) + IL-4 (30 ng/ml); Th17, IL-6 (50 ng/ml) + TGF-β (10 ng/ml) + anti-IFN-γ (1μg/ml)+anti-IL-4 (1μg/ml).
Quantitative PCR (qPCR)
RNA was extracted using a RNeasy Micro Kit (Qiagen, Cat# 74004) from sorted Foxp3+ (GFP+ or YFP+) Tregs and qPCR was performed following the manufacturer’s protocol using gene-specific primer sets (Supplementary Table 3).
Histology
Mouse tissues were fixed in 10% formalin and embedded in paraffin. 4μm sections were stained with hematoxylin and eosin. The images were viewed on an olympus CX31 microscope and taken with a PixelLink camera.
Co-Immunoprecipitation and Western blot
Co-IPs and Western blots were performed as previously described51. Cells were collected and resuspended in RIPA buffer (Millipore, Cat# 20–188) with protease inhibitors (Roche, Cat# 36363600) and incubated on ice for 30 min. Cells were centrifuged (12000g for 10 min) at 4°C and the cell debris was discarded. The supernatant was incubated with protein G-sepharose beads at 4°C for 30 min and then with the indicated antibody (1 μg/test) for 2 h followed by incubation with protein G-sepharose beads overnight with rotation at 4°C. The cells were then washed 5 times with RIPA buffer and the protein G-sepharose beads were dissolved with loading buffer and boiled for 5 min. Supernatants were subjected to SDS-PAGE gel and transferred to nitrocellulose membrane. With blocking with 5% (w/v) skim milk in TBS-T buffer, the membrane was incubated overnight with indicated primary antibodies at 4°C followed by HRP-conjugated secondary antibody or with HRP conjugated primary antibodies (Supplementary Table 4). Membranes were then developed with enhanced chemiluminescence (ECL).
Ubiquitination Assay
Flag-Foxp3 and HA-ubiquitin plasmids were co-transfected into HEK293 cells using Turbofect Transfection Reagent (Cat# R0532) along with either Myc-empty vector, Myc-Usp22, or the catalytically inactive mutant Myc-Usp22C185A (C>A), where the conserved cysteine (C) residue in the C19 peptidase domain was replaced by an alanine (A) residue. After 48 hours, cells were collected, immunoprecipitated with anti-Flag to pull down Foxp3, and immunoblotted for HA-ubiquitin to assess Foxp3 ubiquitination in the presence or absence of functional Usp22. Whole cell lysate (WCL) controls were immunoblotted with HRP-conjugated Myc and HRP-conjugated Flag to show transfection efficiency.
ChIP-qPCR sample preparation
T cells were polarized using Treg polarizing conditions described above in a 24-well plate for 3 days, and 3 million cells were used per immunoprecipitation. Cells were fixed in 37% formaldehyde for 10 min at 37°C. Glycine was added to a final concentration of 0.125 M, and the incubation was continued for an additional 5 min at room temperature. Cells were washed twice with ice-cold phosphate-buffered saline with 1x Protease Inhibitor cocktail (Roche, Cat# 36363600). Millipore ChIP Assay Kit (Lot 3154126) was used for the remainder of the protocol. Cells were resuspended in 1 ml of SDS lysis buffer (Millipore, Cat# 20–163) with protease inhibitors and set on ice for 15 minutes. Samples were then sonicated at the medium setting (308/608) for 7 minutes. Samples were centrifuged at 14 000 rpm at 4°C for 10 min. After removal of an input control (whole-cell extract), supernatants were diluted 10-fold in ChIP dilution buffer (Millipore, Cat# 20–153), and 1x protease inhibitor. 40 uL of Salmon Sperm DNA/Protein A Agarose-50% (Millipore, Cat #16–157C) for 30 min spinning at 4°C. Agarose pelleted out with brief centrifugation and supernatant moved to a new tube. Samples were incubated with either 4 ul of antibody rabbit anti-IgG (Cell Signaling, Cat# 2729), rabbit anti-USP22 (Abcam, ab195289), ub-Histone H2A Lys119 (Cell Signaling, Cat# 8204S), and ub-Histone H2B Lys120 (Cell Signaling, Cat# 5546P) overnight at 4°C rotation. Samples then incubated with 30 ul of of Salmon Sperm DNA/Protein A Agarose-50% for 1 hour at 4°C with rotation. Agarose was pelleted and placed at rotation for five minutes with Low Salt Immune Complex Wash Buffer (Millipore, Cat #20–154), then pelleted. High Salt Immune Complex Wash Buffer (Millipore, Cat #20–155) was added to the pellet and the sample was spun for five minutes at 4°C then pelleted. The agarose was re-suspended in LiCl Immune Complex Wash Buffer and placed at rotation for five minutes at 4°C. The samples were then spun down and re-suspended in 1X TE (Millipore, Cat #20–157) and placed at rotation at room temperature for 5 minutes (repeated once more). The sample was then pelleted and resuspended in 100uL of elution buffer (1%SDS, 0.1M NaHCO3 in water) and placed at rotation for 10 minutes at room temperature. Sample was spun down and supernatant was saved, and step was repeated. 10uL of 5M NaCl was added to the combined eluates and to the input starting material and heated at 65°C overnight. 0.5 M EDTA, 1M Tris-HCl (pH 6.5) and 10mg/mL of protein kinase were added to the samples and incubated at 45°C for one hour. DNA was recovered using a PCR purification kit (Qiagen, Cat #28004).
ChIP-seq sample preparation
Treg cells were collected and either cross-linked in 1% formaldehyde for ten minutes or cross-linked first in 3mM disuccinimidyl glutarate (DSG) in 1X PBS for thirty minutes then in 1% formaldehyde for another ten minutes, both at room temperature. After quenching the excess formaldehyde with a final concentration of 125 mM glycine, the fixed cells were washed in 1X PBS, pelleted, flash-frozen in liquid nitrogen, and stored at −80°C. The cells were thawed on ice and incubated in lysis solution (50 mM HEPES-KOH pH 8, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP40, 0.25% Triton X-100) for ten minutes. The isolated nuclei were washed with wash solution (10 mM Tris-HCl pH 8, 1 mM EDTA, 0.5 mM EGTA, 200 mM NaCl) and shearing buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris-HCl pH 8) then sheared in a Covaris E229 sonicator for ten minutes to generate DNA fragments between ~ 200–1000 base pairs (bp). After clarification of insoluble material by centrifugation, the chromatin was immunoprecipitated overnight at 4°C with antibodies against Usp22 (1:100 v/v), Rnf20 (1:100 v/v), H2BK120Ub (1:100 v/v), and H2AK119Ub (1:100 v/v). The next day, the antibody bound DNA was incubated with Protein A+G Dynabeads (Invitrogen) in ChIP buffer (50 mM HEPES-KOH pH 7.5, 300 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% DOC, 0.1% SDS), washed and treated with Proteinase K and RNase A. Cross-linking was reversed by incubation at 65°C overnight. Purified ChIP DNA was used for library generation (NuGen Ovation Ultralow Library System V2) according to manufacturer’s instructions for subsequent sequencing. ChIP-seq samples were performed with at least 2 biological replicates; with the exception of Usp22 ChIP which has 1 biological replicate with 2 technical replicates (due to poor IP efficiency in the second biological replicate) and H2AK119Ub ChIP performed on Rnf20-RNP cells which has only 1 biological replicate (due to limiting cell number, H2BK120Ub ChIP was prioritized).
ChIP-seq analysis
Single-end 50 base pair (bp) or paired-end 42 bp reads were aligned to mouse genome mm10 using STAR alignment tool (V2.5)52. ChIP-seq peaks were called using findPeaks within HOMER using parameters for histone (-style histone) or transcription factor (-style factor) (Christopher Benner, HOMER, http://homer.ucsd.edu/homer/index.html, 2018). Peaks were called when enriched > four-fold over input and > four-fold over local tag counts, with Benjamin-Hochberg false discovery rate (FDR) 0.001. For histone ChIP, peaks within a 1000 bp range were stitched together to form regions. ChIP-Seq peaks or regions were annotated by mapping to the nearest TSS using the annotatePeaks.pl command. Differential ChIP peaks were found by merging peaks from control and experiment groups and called using getDiffExpression.pl with fold change ≥ 1.5 or ≤ −1.5, Poisson p value < 0.0001. Significance of peak overlap was determined by calculating the number of peaks co-occurring across the entire genome using the HOMER mergePeaks program. For the heatmaps of ChIP-seq read densities at sites bound by Foxp3, publicly available Foxp3 ChIP-seq data was used35. For enhancer enrichment analysis, we defined the different enhancer classes using publicly available Treg ChIP-seq data for histone modifications H3K4me, H3K4me3, and H3K27ac53. All enhancers were called by identifying all H3K4me-positive regions that are at least 1 kb away from the nearest TSS or H3K4me3 mark54. These were sub-divided as either active (H3K27ac-positive) or poised (H3K27ac-negative)55. To call super enhancers, we used the findPeaks program in HOMER with the style option super54. This was performed with the two H3K27ac ChIP-seq replicates and the sites common between the two were used for further analysis. H2BK120Ub ChIP-seq read density histograms at Treg super enhancers were generated by partitioning each super enhancer into 20 bins and also considering 20 kb upstream and downstream, which were also binned similarly. The number of peaks per kb per bin was calculated and averaged across all super enhancers in the genome. To compare across samples with different number of peaks, the final averaged values were normalized by the number of peaks in each data set. Genome browser tracks for H2BK120Ub ChIP-seq data were generated by combining the tag directories from replicate experiments and using the makeBigWig command in HOMER.
RNA sequencing
For RNA-sequencing of Usp22 KO vs WT Tregs, YFP+ cells were sorted from spleen and LN of Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice (n=5) and total RNA was isolated from 1×106 cells per sample using a RNeasy Mini Kit (Qiagen, Cat# 74104) as previously described56. For RNA sequencing of Usp22 RNP KO and Rnf20 RNP KO Tregs, GFP+ Tregs were sorted from Foxp3EGFP mice, stimulated for 48h and electroporated with Cas9 RNPs (n=2). The cells were collected at day 5 post-electroporation and RNA was isolated as described above. ERCC ExFold RNA Spike-In Mixes (Thermo Fisher, Cat# 4456739) were then added to total RNA for each sample. WT and NTC RNP samples were spiked with Mix #1, while KO and RNP KO samples were spiked with Mix# 2. Total RNA was then provided to the Functional Genomics Laboratory at UC Berkeley where RNA-seq libraries were prepared by Oligo dT enrichment followed by a stranded Illumina library prep protocol with the KAPA mRNA HyperPrep kit (Kapa Biosystems, KK8580). Libraries were checked for quality on an AATI Fragment analyzer (Agilent, DNF-935–1000), quantified using the Illumina Quant Universal qPCR Mix (Kapa Biosystems, KK4824), and pooled evenly at 3nM. The Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley then performed one lane of 150bp paired-end Illumina, HiSeq4000 sequencing followed by demultiplexing and bclfile to fastq conversion using Illumina bcl2fastq v2.19 software (Illumina). Reads were mapped to the GRCm38.p6 assembly (Ensembl annotation) using kallisto v0.45 with default parameters57. Transcript-level abundance estimates were collapsed to create gene-level count matrices and Usp22 KO vs WT samples were normalized to the ERCC spike-ins using loess regression58. Differentially expressed genes were then detected using DESeq2 with default parameters59. Pseudogenes beginning with “Gm” were excluded from volcano plots; RNP data was batch corrected and genes with read counts < 10 were removed. Data from this experiment is included in Supplementary Table 5.
Adoptive transfer model of colitis
Naïve T cells (CD4+CD25−CD44loCD62Lhi) were sorted from congenic CD45.1 B6.SJL mice and YFP+ Treg cells were sorted from WT or Usp22fl/flFoxp3YFP-Cre mice.
Rag1−/− mice were given intraperitoneal injection of naïve T cells (4×105) alone or in combination with WT or Usp22 KO Treg cells (2×105). After T cell reconstitution, mice were weighed weekly and monitored for clinical of signs of disease. Mice were sacrificed when their body weight decreased 20%. At cessation, colons were harvested for measurement and histology and flow cytometry.
Induced experimental autoimmune encephalomyelitis (EAE)
8–10 week-old WT or Usp22fl/flFoxp3YFP-Cre mice were subcutaneously injected with 200μg of MOG33–55 peptide (Genemed Synthesis). The MOG33–55 peptide was emulsified in complete Feund’s adjuvant (CFA) which contained 200μg of Mycobacterium tuberculosis H37Ra (Difco). The mice were then subsequently intraperitoneally injected with 200 ng of pertussis toxin (List Biological Laboratories) on day 0 and day 2. Clinical signs of EAE were assessed daily. Scores were given as follows: 0, no sign of disease; 2, limp tail, 3, hind leg weakness or limp; 3, partial back limb paralysis; 4, complete hind limb paralysis; 5, total limb paralysis.
Data availability
Data from the screen (Fig. 1) and RNA sequencing (Fig. 2, Extended Data Fig. 5) are provided here in Supplementary Tables 1 and 5. ChIP-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus under the accession code GSE140102 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140102]. Publicly available ChIP-seq and ATAC-seq data were downloaded from the indicated repositories and processed using HOMER v4.8 (Christopher Benner, HOMER, http://homer.ucsd.edu/homer/index.html, 2018). Foxp3 ChIP-seq GEO accession code GSE40684; ATAC-seq and ChIP-seq for H3K4me, H3K27ac, H3K4me3 SRA accession number DRP003376.
Code availability
All code used for data visualization in this manuscript will be made available on request.
Extended Data
Extended Data Fig. 1. Design and Quality Control of Targeted Pooled CRISPR Screen in Primary Mouse Tregs.
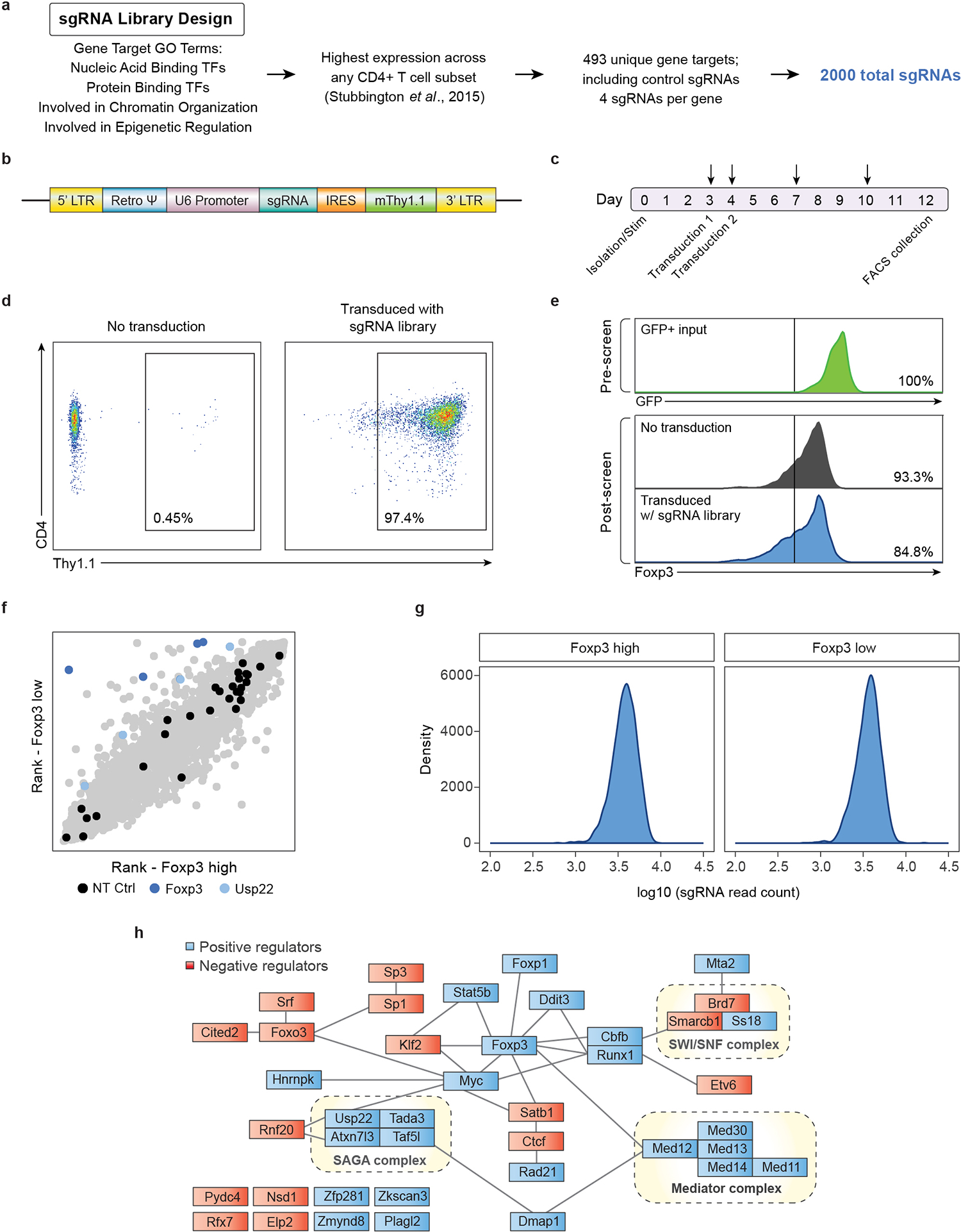
a) Design strategy for selection of genes for unbiased targeted library of 493 targets, including 490 nuclear factors and 3 control targets (NT, GFP, and RFP). Genes were selected based on gene ontology (GO) annotation and then sub-selected based on highest expression across any CD4 T cell subset for a total of 2,000 sgRNAs.
b) Diagram of MSCV expression vector with Thy1.1 reporter used for retroviral transduction of the sgRNA library.
c) Detailed timeline schematic of the 12-day targeted screen pipeline. Arrows indicate when the cells were split, and media was replenished.
d) Retroviral transduction efficiency of the targeted library in primary mouse Tregs shown by Thy1.1 surface expression measured by flow cytometry. The infection was scaled to achieve a high efficiency multiplicity of infection.
e) Foxp3 expression from screen input, output and control cells measured by flow cytometry. Top: Foxp3 expression from input Foxp3+ purified Tregs as measured by GFP expression on Day 0. Middle: Foxp3 expression as measured by endogenous intracellular staining from control Tregs (not transduced with library) on Day 12. Bottom: Foxp3 expression as measured by endogenous intracellular staining from screen Tregs (transduced with library) on Day 12.
f) Targeted screen (2,000 guides) shows that sgRNAs targeting Foxp3 and Usp22 were enriched in Foxp3 low cells (blue). Non-targeting control (NT Ctrl) sgRNAs were evenly distributed across the cell populations (black).
g) Distribution of read counts after next generation sequencing of sgRNAs of sorted cell populations, Foxp3high and Foxp3low.
h) Schematic of experimentally determined and predicted protein-protein interactions between top hits, 16 negative regulators (red) and 25 positive regulators (red), generated by STRING-db34. Black lines connect interacting proteins and dotted lines outline selected known protein complexes.
All data are presented as mean ±SEM. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section.
Extended Data Figure 2. Validation of Foxp3 Modulators in Primary Mouse and Human Tregs with Cas9 RNP Electroporation.
a) Overview of orthogonal validation strategy using arrayed electroporation of Cas9 RNPs in Tregs.
b) Foxp3 expression 4 days post electroporation of Cas9 RNPs in mouse Tregs as measured by flow cytometry of top screen hits. Each row shows 3 histograms layered on top of one another (1–2 for controls) with each representing effects of independent gRNAs for each target gene. Percentages shown on the right depict the average frequency of Foxp3+ cells across gRNAs targeting each gene.
c) Percentage of Foxp3- cells of live, CD4+ cells 4 days post electroporation of Cas9 RNPs in mouse Tregs as measured by flow cytometry of top screen hits. Each data point represents an independent sgRNA for each target gene.
d) Foxp3 MFI of Foxp3+ mouse Tregs for 3–4 distinct gRNAs targeting each gene paired with the mean KO efficiency (top) for each guide as determined by TIDE analysis.
e) Representative flow plots depicting FOXP3 and CD25 expression 7 days post electroporation of Cas9 RNPs targeting USP22 or NT Ctrl in human Tregs. The subpopulation of cells with the highest expression of FOXP3 and CD25 (FOXP3hiCD25hi) is highlighted with a red gate.
f) Percentage of FOXP3+ cells from human Tregs electroporated with Cas9 RNPs targeting USP22 or NT Ctrl in 10 biological replicates. Lines connect paired samples.
g) Percentage of FOXP3hiCD25hi cells from human Tregs electroporated with Cas9 RNPs targeting USP22 or NT Ctrl in 10 biological replicates.
h) FOXP3 MFI of human Tregs for 3–4 distinct gRNAs targeting each gene paired with the mean KO efficiency (top) for each guide as determined by TIDE analysis.
i) Simple linear regression of FOXP3 MFI (y-axis) by percentage of editing efficiency determined by TIDE analysis (x-axis) for 4 gRNAs targeting USP22 in 2–4 biological donors.
j) FOXP3 MFI of human Tregs electroporated with Cas9 RNPs with 2–3 distinct sgRNAs each in 2–4 biological donors; corresponding to panel h. Data points with less than 60% editing efficiency KO by TIDE analysis were excluded from the graph.
All data are presented as mean ±SEM. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section.
Extended Data Fig. 3. Design and Validation of Treg-specific Usp22 Knockout Mice.
a) Diagram of the murine Usp22 locus. Targeting vector contains IRES-lacZ and a neo cassette inserted into exon 2.
b) Genotyping by PCR showed a 600-bp band for the wild-type allele and a 400-bp band for mutant allele, simultaneously in the homozygous floxed (f/f) mice.
c) Western blot analysis of Usp22 in CD4+CD25− conventional T cells (Tconv) and CD4+CD25+ Treg cells isolated from Usp22+/+Foxp3YFP-Cre WT and Usp22fl/flFoxp3YFP-Cre KO mice. Gapdh was used as a loading control.
d) Statistical analysis of CD4+Foxp3+ Treg frequencies, corresponding to Figure 2c.
e) Western blot analysis of Foxp3 protein level from Tregs isolated from spleen and LN of Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice. Gapdh was used as a loading control.
f) iTreg differentiation of naïve CD4+ T cells from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice with titration of TGF-β (as indicated).
g) Summary of iTreg differentiation of naïve CD4+ T cells from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice with titration of TGF-β (as indicated).
h) In vitro suppressive activity of Tregs assessed by the division of naïve CD4+CD25− T cells. Naïve T cells were labeled with cytosolic cell proliferation dye and activated by anti-CD3 and antigen presenting cells (irradiated splenocytes from wild-type mice, depleted of CD3+ T cells), then co-cultured at various ratios (as indicated above) with YFP+ Treg cells sorted from 8-week-old Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice. Numbers indicate the percentage of non-dividing cells for each ratio.
i) In vitro suppressive activity of control (pMIG-Control) or Foxp3+ (pMIG-Foxp3) transduced YFP+ Tregs sorted from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice. Naïve T cells were labeled with cytosolic cell proliferation dye and activated then co-cultured at 1:4 transduced YFP+ Treg cells to naïve T effectors (Teff). Numbers indicate the percentage of non-dividing cells for each ratio.
j) Summary data of in vitro suppression experiments represented as frequency of non-dividing cells relative to WT 0:1 No Treg control, corresponding to panel i. Lines connect paired samples.
All data are presented as mean ±SEM. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section. Source data for gels and blots can be found in Supplementary Figure 1.
Extended Data Fig. 4. Usp22 Acts as Deubiquitinase to Control Post-Translational Foxp3 Expression.
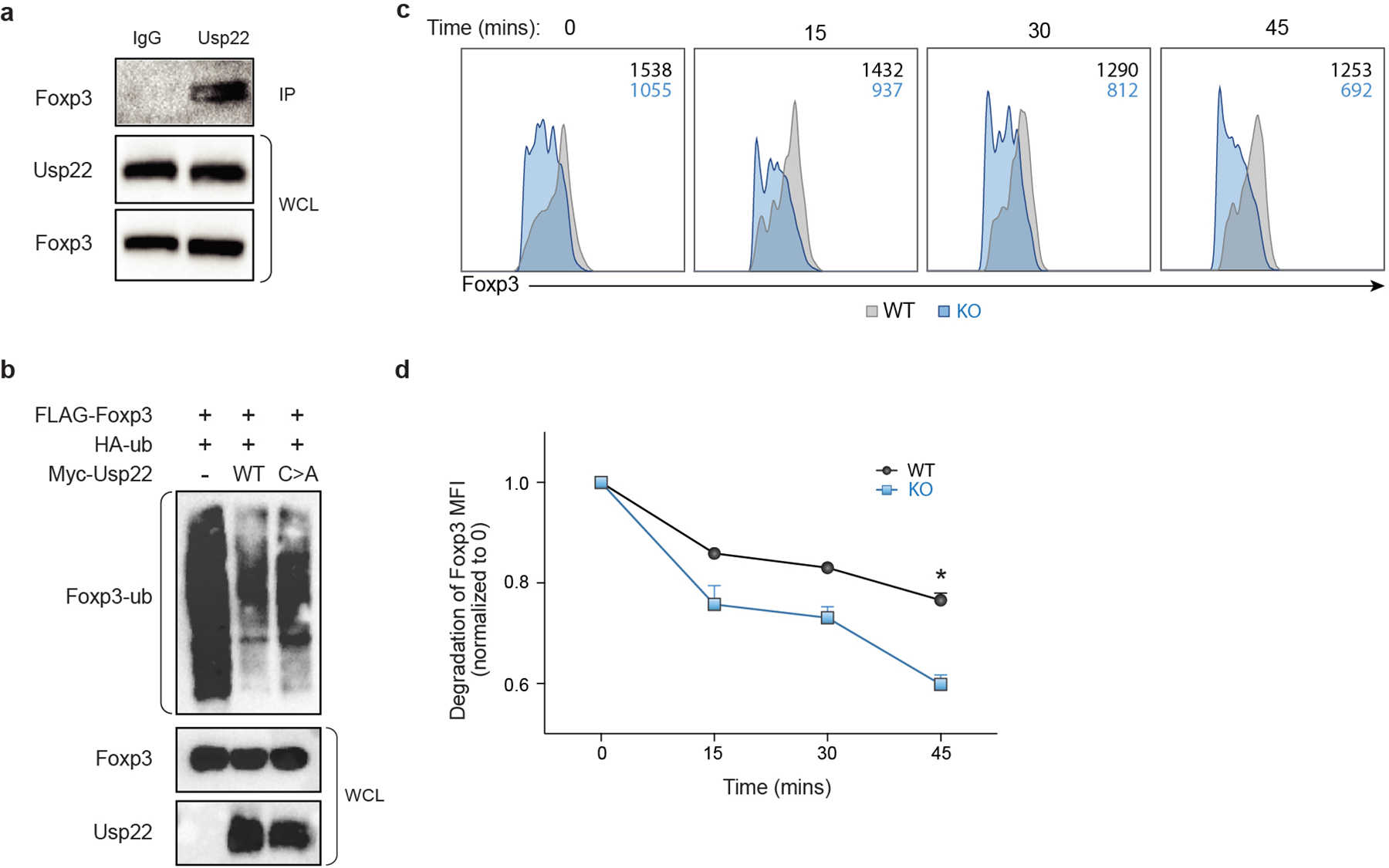
a) Endogenous interaction of Usp22 and Foxp3 in murine iTreg cells from WT mice. Rabbit anti-Usp22 antibody was used to perform the immunoprecipitation and mouse anti-Foxp3 antibody was used to detect the bound Foxp3. Normal rabbit IgG was used as control. Whole cell lysates (WCL) were used as sample processing controls.
b) Ubiquitination assay of Foxp3. HEK293 cells were co-transfected with Flag-Foxp3 and HA-ubiquitin (HA-ub) along with either Myc-empty vector, Myc-Usp22, or the catalytically inactive mutant Myc-Usp22C185A (C>A), and then immunoprecipitated with anti-Flag and immunoblotted for HA-ubiquitin (Foxp3-ub). Whole cell lysates (WCL) were used as sample processing controls.
c) Splenocytes isolated from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice were treated with 200 μg/ml cycloheximide (CHX) for the indicated time course. Inset numbers for each histogram indicate the MFI of Foxp3 in Tregs (black=WT, blue=KO).
d) Foxp3 MFI from splenic CD4+CD25+Foxp3+ Treg population treated with 200 μg/ml cycloheximide (CHX) for the indicated time course, n=3; corresponding to panel c. All data are presented as mean ±SEM. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section. Source data for blots can be found in Supplementary Figure 1.
Extended Data Fig. 5. Usp22 Regulates Foxp3 through Transcriptional Mechanisms.
a) Representative flow cytometry analysis of the YFP+ Treg population (gated on CD4+ cells) from the spleen and lymph nodes of Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice.
b) Statistical analysis of YFP MFI in CD4+YFP+ Tregs from the thymus (Thy), peripheral lymph nodes (pLN), and spleen (Spl) of Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice.
c) Statistical analysis of CD4+YFP+ Treg frequencies in Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice, corresponding to panel b.
d) Volcano plot for RNA sequencing of Usp22 RNP KO Tregs vs Rnf20 RNP KO murine Tregs. X-axis shows log2FoldChange (LFC). Y-axis shows the –log10 of the p-value as calculated by DESeq2. Genes downregulated in the Usp22 RNP KO compared to Rnf20 RNP KO are shown in red and genes upregulated are shown in blue defined by p-value <5e-3 and LFC > 0.8. Foxp3 (shown in green) trended down but did not reach significance.
e) qPCR analysis of FOXP3 mRNA levels in human Tregs from 2 donors 8 days post-electroporation with Cas9 RNPs targeting NTC , FOXP3, USP22, RNF20 or both USP22 and RNF20. Normalized to the expression of β-ACTIN transcripts. Data are presented as mean ±SEM and are representative of at least two independent experiments.
f) qPCR analysis of Foxp3 mRNA levels in mouse Tregs 4 and 8 days post-electroporation with Cas9 RNPs targeting NTC, Foxp3, Usp22, Rnf20 or both Usp22 and Rnf20. Normalized to the expression of β-actin transcripts.
g) Western blot analysis of ubiquitinated histone 2A (H2AK119Ub; H2A-ub) and ubiquitinated histone 2B (H2BK120Ub; H2B-ub) from iTregs from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice. Gapdh was used as a loading control. Source data can be found in Supplementary Figure 1.
h) Schematic of Foxp3 locus depicting PCR products used for ChIP-qPCR data shown in panel i and panel j.
i) ChIP-qPCR data analysis for H2AK119Ub (H2A-ub) where primers amplified across the transcriptional start site (TSS) and the CNS1 enhancer region of the Foxp3 locus. Data are normalized to the input and are presented as mean ±SD.
j) ChIP-qPCR data analysis for H2BK120Ub (H2B-ub) for PCR across the transcriptional start site (TSS) and across the CNS1 enhancer region of the Foxp3 locus. Data are normalized to the input and are presented as mean ±SD.
k) Heatmap of ChIP-seq read density for Foxp3, Usp22, and Rnf20 at sites bound by Foxp3 (using previously published Foxp3 ChIP data35), ranked by highest to lowest Foxp3 binding signal. The corresponding log2 fold change (log2fc) for either H2BK120Ub or H2AK119Ub upon Usp22 or Rnf20 deletion at these sites are plotted on the right, with each biological replicate shown as an individual column.
l) Average ChIP-seq read density of H2BK120Ub at Treg super enhancers in control versus Usp22-deficient Tregs.
m) Co-occurrence analysis showing the natural log of the ratio of the observed number of overlapping regions over the expected values for sites that either gain or lose H2BK120Ub in Usp22-deficient Tregs against publicly available histone modifications H3K4me, H3K4me3 and H3K27ac as well as enhancer classes, as described in the Methods.
n) Analysis of reciprocal regulation of Foxp3 by deubiquitinase Usp22 and E3 ubiquitin ligase Rnf20. YFP MFI of Tregs sorted from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice and then electroporated with either NT control (NTC-RNP) or Rnf20 RNP, corresponding with Figure 2j where Foxp3 MFI from the same experiment is shown.
All data are presented as mean ±SEM, unless otherwise stated. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section.
Extended Data Fig. 6. Autoimmune Inflammation in Treg-specific Usp22 Knockout Mice.
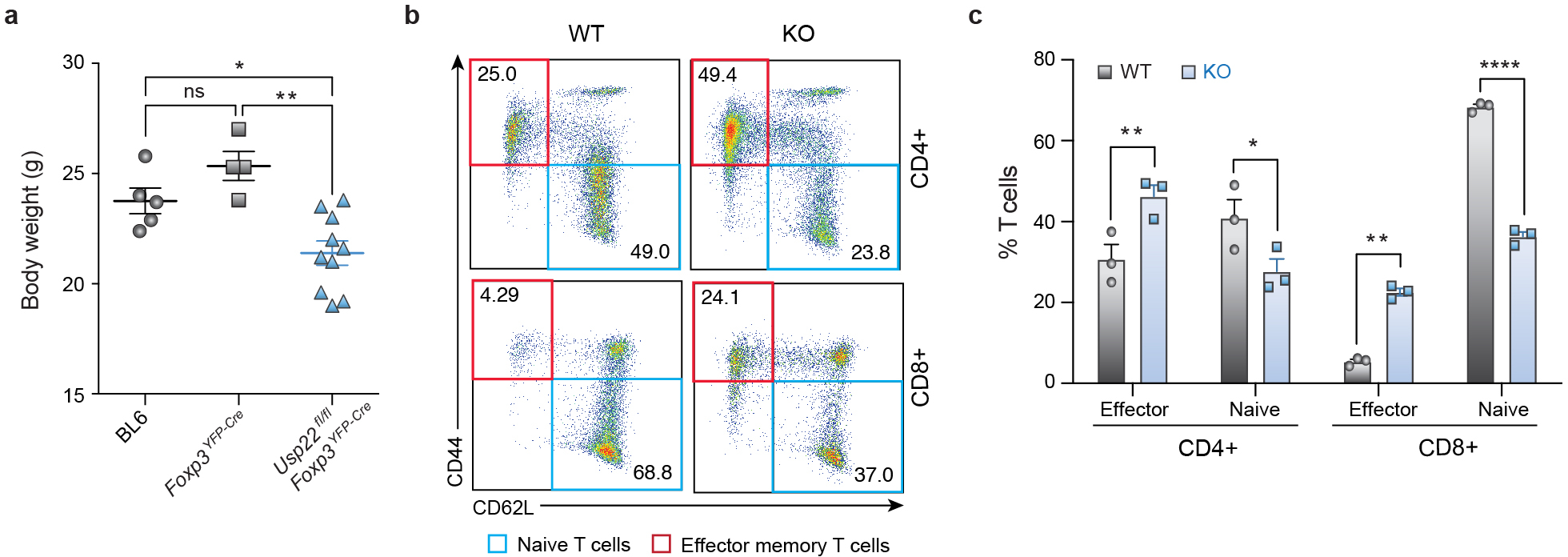
a) Body weight differences (in grams, g) between 8-week-old, sex-matched C57BL/6 WT (BL6), Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice.
b) Representative flow cytometry analysis of CD44 and CD62L expression in splenic CD4+ and CD8+ T cells from aged 7-month-old Usp22+/+Foxp3YFP-Cre WT and Usp22fl/flFoxp3YFP-Cre KO mice. Numbers in quadrants indicate percentage of each cell population.
c) The frequency of splenic CD4+ and CD8+ effector T cells (CD44hiCD62Llo) and naïve T cells (CD44loCD62Lhi) of aged 7-month-old Usp22+/+Foxp3YFP-Cre WT and Usp22fl/flFoxp3YFP-Cre KO mice summarized, corresponding to panel b.
All data are presented as mean ±SEM. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section.
Extended Data Figure 7. T Cell-Specific Ablation of Usp22 Resulted in Decreased Foxp3 and Increased T Cell Activation.
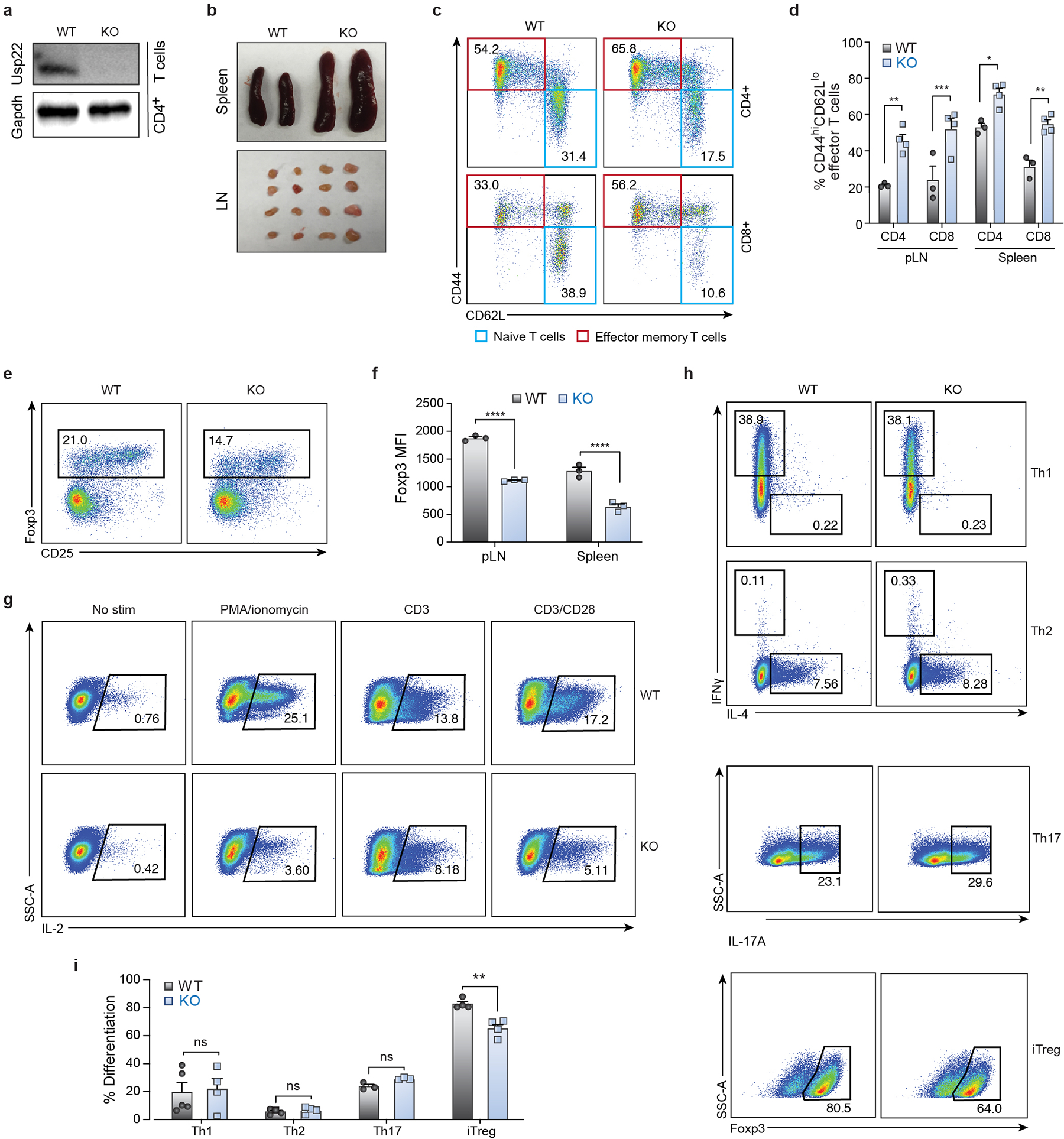
a) Western blot analysis of Usp22 protein levels in CD4+ T cells isolated from spleens of Usp22fl/flLckCre KO and Usp22+/+LckCre WT mice. Gapdh was used as a loading control. Source data can be found in Supplementary Figure 1.
b) Representative macroscopic images of spleens and peripheral lymph nodes (pLN) from 10-month-old Usp22fl/flLckCre KO and Usp22+/+LckCre WT mice.
c) Representative flow cytometry plots showing CD44 and CD62L expression in CD4+ and CD8+ T cells from spleens of 10-month-old Usp22fl/flLckCre KO and Usp22+/+LckCre WT mice.
d) Frequency of effector-memory T cells (CD44hiCD62Llo) in peripherallymph nodes (pLN) and spleens from 10-month-old Usp22fl/flLckCre KO and Usp22+/+LckCre WT mice.
e) Representative flow cytometry plots showing the splenic CD4+Foxp3+ Treg population from 10-month-old Usp22fl/flLckCre KO and Usp22+/+LckCre WT mice.
f) Foxp3 MFI of the CD4+Foxp3+ Treg population in the spleen and pLN from 10-month-old Usp22fl/flLckCre KO and Usp22+/+LckCre WT mice.
g) IL-2 production by CD4+CD25− T cells under various stimulation conditions (as indicated) for 3 days was assessed by flow cytometry in Usp22fl/flLckCre KO and Usp22+/+LckCre WT mice. Although the dominant effect of Usp22-deficiency in T cells was increased T cell activation and lymphoproliferation, we found some evidence of impaired IL-2 production in conventional T cells.
h) Usp22-deficiency in T cells led to a selective defect in iTreg differentiation. In vitro differentiation of CD4+ naïve T cells cultured under Th1, Th2, Th17 or sub-optimal TGF-β (1ng/mL) iTreg conditions from Usp22fl/flLckCre KO and Usp22+/+LckCre WT mice was assessed by flow cytometry.
i) Summary of in vitro differentiation experiments showing percent differentiation, corresponding to panel h.
All data are presented as mean ±SEM. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section.
Extended Data Figure 8. Tumor Growth is Inhibited in Treg-specific Usp22 Knockout Mice in Multiple Cancer Models.
a) Left: Representative flow cytometric analysis of splenic IFNγ in CD8+ T cells from EG7 tumor-bearing Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO. Right: Statistical analysis of IFNγ production by splenic CD8+ T cells from EG7 tumor-bearing Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO.
b) Left: Representative flow cytometric analysis of splenic Granzyme B (GrzB) in CD8+ T cells from EG7 tumor-bearing Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO. Right: Statistical analysis of Granzyme B production by splenic CD8+ T cells from EG7 tumor-bearing Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO.
c) The MFI of various Treg markers (as indicated) from splenic CD4+Foxp3+ Tregs from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO EG7 tumor-bearing mice, assessed by flow cytometry.
d) qPCR analysis of Ifng, Gzmb and Cd8a mRNA levels in the tumor tissue of Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO EG7 tumor-bearing mice.
e) Tumor volumes from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice subcutaneously inoculated with 5×104 B16 melanoma cells. For e, h, k, tumor volumes were measured every 2–3 days by scaling along 3 orthogonal axes (x, y, and z) and calculated as (xyz)/2.
f) The MFI of various Treg markers (as indicated) from splenic CD4+Foxp3+ Tregs in Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO B16 tumor-bearing mice, assessed by flow cytometry.
g) Foxp3 MFI of Foxp3+ cells from tumor-infiltrating Tregs in Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO B16 tumor-bearing mice.
h) Tumor volumes from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice subcutaneously inoculated with 1×106 LLC1 Lewis lung carcinoma cells.
i) The MFI of various Treg markers (as indicated) from splenic CD4+Foxp3+ Tregs in Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO LLC1 tumor-bearing mice, assessed by flow cytometry.
j) Foxp3 MFI of Foxp3+ cells from tumor-infiltrating Tregs in Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO LLC1 tumor-bearing mice.
k) Tumor volumes from Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO mice subcutaneously inoculated with 1×106 MC38 colon adenocarcinoma cells.
l) The MFI of various Treg markers (as indicated) from splenic CD4+Foxp3+ Tregs in Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO MC38 tumor-bearing mice, assessed by flow cytometry.
m) Foxp3 MFI of Foxp3+ cells from tumor-infiltrating Tregs in Usp22+/+Foxp3YFP-Cre WT or Usp22fl/flFoxp3YFP-Cre KO MC38 tumor-bearing mice.
All data are presented as mean ±SEM. ns indicates no significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. The exact sample sizes (n), p-values, statistical tests and number of times the experiment was replicated can be found in the “Statistics and Reproducibility” section.
Supplementary Material
Acknowledgements
We thank all members of the Marson lab as well as Mark S. Anderson, K. Mark Ansel, Chun J. Ye, Kathrin Schumann and Luke Gilbert for helpful suggestions and technical advice. We thank Jacob Freimer, Siddharth Raju and Eric Guo for helpful advice and assistance with the RNA-seq analysis pipeline. We thank Vinh Nguyen, Victoria Tobin, Ryan Apathy, Michelle Nguyen, the UCSF Flow Cytometry Core, and Nasun Hah and Grace Chou in the Salk NGS Core Facility for technical assistance. We thank Sarah Pyle for assistance with graphics. We thank David Nguyen for critical reading of the manuscript. J.T.C. is supported by the National Science Foundation Graduate Research Fellowship Program grant 1650113. J.G. was supported by the Salk Institute T32 Cancer Training Grant T32CA009370 and the NIGMS NRSA F32 GM128377-01. D.C.H. is supported by the National Institutes of Health (NIH) (GM128943-01, CA184043-03), the V Foundation for Cancer Research V2016-006, the Pew-Stewart Foundation for Cancer Research, and the Leona M. and Harry B. Helmsley Charitable Trust. The Marson lab has received gifts from J. Aronov, G. Hoskin, K. Jordan, B. Bakar, the Caufield family and funds from the Innovative Genomics Institute (IGI) and the Parker Institute for Cancer Immunotherapy (PICI). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, is an investigator at the Chan Zuckerberg Biohub and is a recipient of a The Cancer Research Institute (CRI) Lloyd J. Old STAR grant. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant, the UCSF Flow Cytometry Core, supported by the Diabetes Research Center grants NIH P30 DK063720 and NIH S10 1S10OD021822-01, and the Salk NGS Core Facility, supported by the NIH-NCI CCSG: P30 014195, the Chapman Foundation, and the Helmsley Charitable Trust. D.F. is supported by NIH R01 grants (AI079056, AI108634 and CA232347). E.M. is supported by NIH F31 CA220801-03.
Footnotes
Declaration of Interests
The authors declare competing financial interests: T.L.R. is a co-founder of Arsenal Biosciences. A.M. is a co-founder of Spotlight Therapeutics and Arsenal Biosciences. A.M. has served as an advisor to Juno Therapeutics, is a member of the scientific advisory board at PACT Pharma, and is an advisor to Trizell. A.M. owns stock in Arsenal Biosciences, Spotlight Therapeutics and PACT Pharma. The Marson lab has received sponsored research support from Juno Therapeutics, Epinomics, Sanofi, and a gift from Gilead. J.A.B. is a co-founder of Sonoma BioTherapeutics; a consultant for Juno, a Celgene company; a stock holder and member of the Board of Directors on Rheos Medicines; and a stock holder and member of the Scientific Advisory Boards of Pfizer Center for Therapeutic Innovation, Vir Therapeutics, Arcus Biotherapeutics, Quentis Therapeutics, Solid Biosciences, and Celsius Therapeutics. J.A.B. owns stock in MacroGenics Inc., Vir Therapeutics, Arcus Biotherapeutics, Quentis Therapeutics, Solid Biosciences, Celsius Therapeutics, and Kadmon Holdings. A patent application has been filed based on the screen data described here.
References
- 1.Sakaguchi S, Yamaguchi T, Nomura T & Ono M Regulatory T Cells and Immune Tolerance. Cell 133, 775–787 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Zhou X et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nature Immunology 10, 1000–1007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey-Bucktrout SL & Bluestone JA Regulatory T cells: stability revisited. Trends in Immunology 32, 301–306 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Overacre-Delgoffe AE & Vignali DA A. Treg Fragility: A Prerequisite for Effective Antitumor Immunity? Cancer Immunol Res 6, 882–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bluestone JA & Tang Q Treg cells—the next frontier of cell therapy. Science 362, 154–155 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biology 15, 554(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beyer M & Schultze JL Plasticity of T(reg) cells: is reprogramming of T(reg) cells possible in the presence of FOXP3? Int. Immunopharmacol 11, 555–560 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Maruyama T, Konkel JE, Zamarron BF & Chen W The molecular mechanisms of Foxp3 gene regulation. Semin Immunol 23, 418–423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitoh A et al. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity 31, 609–620 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Rudra D et al. Runx-CBFβ complexes control Foxp3 expression in regulatory T cells. Nat Immunol 10, 1170–1177 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ono M et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446, 685–689 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Yao Z et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood 109, 4368–4375 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams LM & Rudensky AY Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol 8, 277–284 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Beyer M et al. Repression of SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat Immunol 12, 898–907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koutelou E, Hirsch CL & Dent SYR Multiple faces of the SAGA complex. Curr Opin Cell Biol 22, 374–382 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schumann K et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. PNAS 112, 10437–10442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubtsov YP et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558 (2008). [DOI] [PubMed] [Google Scholar]
- 19.da S. Martins M & Piccirillo CA Functional stability of Foxp3+ regulatory T cells. Trends in Molecular Medicine 18, 454–462 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Feng Y et al. Control of the Inheritance of Regulatory T Cell Identity by a cis Element in the Foxp3 Locus. Cell 158, 749–763 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, Liang Y, LeBlanc M, Benner C & Zheng Y Function of a Foxp3 cis-Element in Protecting Regulatory T Cell Identity. Cell 158, 734–748 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Y et al. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463, 808–812 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DuPage M et al. The Chromatin-Modifying Enzyme Ezh2 Is Critical for the Maintenance of Regulatory T Cell Identity after Activation. Immunity 42, 227–238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei G et al. Global Mapping of H3K4me3 and H3K27me3 Reveals Specificity and Plasticity in Lineage Fate Determination of Differentiating CD4+ T Cells. Immunity 30, 155–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Josefowicz SZ et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu X et al. SENP3 maintains the stability and function of regulatory T cells via BACH2 deSUMOylation. Nature Communications 9, 3157(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Z et al. The Ubiquitin Ligase Stub1 Negatively Modulates Regulatory T Cell Suppressive Activity by Promoting Degradation of the Transcription Factor Foxp3. Immunity 39, 272–285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Loosdregt J et al. Stabilization of the Transcription Factor Foxp3 by the Deubiquitinase USP7 Increases Treg-Cell-Suppressive Capacity. Immunity 39, 259–271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dang EV et al. Control of TH17/Treg Balance by Hypoxia-Inducible Factor 1. Cell 146, 772–784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y et al. USP21 prevents the generation of T-helper-1-like Treg cells. Nature Communications 7, 13559(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henry KW et al. Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 17, 2648–2663 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melo-Cardenas J, Zhang Y, Zhang DD & Fang D Ubiquitin-specific peptidase 22 functions and its involvement in disease. Oncotarget 7, 44848–44856 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin Z et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell 46, 484–494 (2012). [DOI] [PubMed] [Google Scholar]
Method References
- 34.Szklarczyk D et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samstein RM et al. Foxp3 Exploits a Pre-Existent Enhancer Landscape for Regulatory T Cell Lineage Specification. Cell 151, 153–166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou X et al. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. The Journal of Experimental Medicine 205, 1983(2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luche H, Weber O, Nageswara Rao T, Blum C & Fehling HJ Faithful activation of an extra-bright red fluorescent protein in ‘knock-in’ Cre-reporter mice ideally suited for lineage tracing studies. Eur. J. Immunol 37, 43–53 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Bailey-Bucktrout SL et al. Self-antigen driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity 39, 949–962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Platt RJ et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 159, 440–455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haribhai D et al. Regulatory T cells dynamically control the primary immune response to foreign antigen. J. Immunol 178, 2961–2972 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Joung J et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 12, 828–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stubbington MJ et al. An atlas of mouse CD4+ T cell transcriptomes. Biology Direct 10, 14(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilbert LA et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shifrut E et al. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hultquist JF et al. A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep 17, 1438–1452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brinkman EK, Chen T, Amendola M & van Steensel B Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melo-Cardenas J et al. USP22 deficiency leads to myeloid leukemia upon oncogenic Kras activation through a PU.1 dependent mechanism. Blood blood-2017-10-811760 (2018) doi: 10.1182/blood-2017-10-811760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen S et al. Host miR155 Promotes Tumor Growth through a Myeloid-Derived Suppressor Cell–Dependent Mechanism. Cancer Res 75, 519–531 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiu J et al. The Aryl Hydrocarbon Receptor Regulates Gut Immunity through Modulation of Innate Lymphoid Cells. Immunity 36, 92–104 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee S-M, Gao B & Fang D FoxP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA-binding activity of AP-1. Blood 111, 3599–3606 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Gao B, Kong Q, Kemp K, Zhao Y-S & Fang D Analysis of sirtuin 1 expression reveals a molecular explanation of IL-2–mediated reversal of T-cell tolerance. PNAS 109, 899–904 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kitagawa Y et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat. Immunol 18, 173–183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whyte WA et al. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Creyghton MP et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A 107, 21931–21936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simeonov DR et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bray NL, Pimentel H, Melsted P & Pachter L Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Lovén J et al. Revisiting global gene expression analysis. Cell 151, 476–482 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from the screen (Fig. 1) and RNA sequencing (Fig. 2, Extended Data Fig. 5) are provided here in Supplementary Tables 1 and 5. ChIP-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus under the accession code GSE140102 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140102]. Publicly available ChIP-seq and ATAC-seq data were downloaded from the indicated repositories and processed using HOMER v4.8 (Christopher Benner, HOMER, http://homer.ucsd.edu/homer/index.html, 2018). Foxp3 ChIP-seq GEO accession code GSE40684; ATAC-seq and ChIP-seq for H3K4me, H3K27ac, H3K4me3 SRA accession number DRP003376.