

Abstract
Background/Aim:
Steatohepatitis drives fibrogenesis in alcoholic liver disease. However, recent studies have suggested that hepatic stellate cells (HSC) may regulate the parenchymal cell injury and inflammation that precedes liver fibrosis, although the mechanism remains incompletely defined. Neuropilin-1 (NRP-1) and synectin are membrane proteins implicated in HSC activation. In this study, we disrupted NRP-1 and synectin as models to evaluate the role of HSC activation on the development of steatohepatitis in response to alcohol feeding in mice.
Methods:
Mice with HSC-selective deletion of NRP (ColCre/NRP-1loxP) or synectin (ColCre/synectinloxP) vs. paired NRP-1loxP or synectinloxP mice were fed a control diet or the chronic/binge alcohol feeding model. Several markers of steatosis and inflammation were evaluated.
Results:
ColCre/NRP-1loxP mice showed less fibrosis, as expected, but also less inflammation and steatosis as assessed by Oil-Red O and Bodipy staining and hepatic triglyceride content. Similar results were observed in the synectin model. Hepatocytes treated with supernatant of HSC from ColCre/NRP-1loxP mice compared to supernatant from NRP-1loxP mice were protected against ethanol-induced lipid droplet formation. An adipokine and inflammatory protein array from the supernatant of HSC with NRP-1-knockdown showed a significant reduction in Igfbp3 (a major insulin-like growth factor binding protein with multiple metabolic functions) and an increase in SerpinA12 (a serine-protease inhibitor) secretion compared to wildtype HSC. Recombinant Igfbp3 induced lipid droplets, triglyceride accumulation, and lipogenic genes in hepatocytes in vitro, while SerpinA12 was protective against ethanol-induced steatosis. Finally, Igfbp3 was increased, and SerpinA12 was decreased in serum and liver tissue from patients with alcoholic hepatitis.
Conclusion:
Selective deletion of NRP-1 from HSC attenuates alcohol-induced steatohepatitis through regulation of Igfbp3 and SerpinA12 signaling.
Keywords: alcohol, steatosis, steatohepatitis, neuropilin-1, Igfbp3, hepatic stellate cell, alcoholic liver disease, alcoholic hepatitis, insulin like growth factor binding protein 3, SerpinA12, vaspin, src-kinase, integrin
Graphical Abstract

LAY SUMMARY
Hepatic stellate cells are known for their role in fibrosis (scarring of the liver). In this study, we describe their role and mechanisms in the modulation of fat deposit and inflammation in the liver, which occurs secondary to alcohol damage.
INTRODUCTION
Alcohol-related liver disease (ALD) is a major cause of morbidity and mortality in the United States [1, 2]. It encompasses a clinico-histologic spectrum of disease including fatty liver, alcoholic hepatitis (AH), and alcoholic cirrhosis [3–5]. Steatosis is an early pathogenic lesion in the spectrum of ALD [6]. Steatosis is characterized by the accumulation of triglycerides, phospholipids, and cholesterol esters in hepatocytes. Alcohol consumption regulates lipid metabolism by regulation of hepatic lipogenesis [7]. Understanding the pathophysiological mechanisms occurring early in the disease process are needed to develop new and innovative therapies to treat patients in the early stages of ALD [8, 9].
Hepatic stellate cells (HSC) are non-parenchymal cells of the liver with an established role in fibrosis and tissue repair [10]. In response to steatohepatitis, quiescent HSC undergo activation that drives fibrosis in the canonical injury to fibrosis pathway [11, 12]. However, recent studies have suggested that HSC may also reciprocally regulate the parenchymal cell injury and inflammation that precedes liver fibrosis [11]. This mechanism suggests bidirectional hepatocyte-HSC crosstalk [13]. Thus, while the role of HSC in fibrogenesis is well established, their contributions to hepatocyte lipid droplets and liver steatohepatitis are not well understood.
Neuropilin-1 (NRP-1) is a growth factor co-receptor implicated in HSC activation [14], and its inhibition has been shown to ameliorate the recruitment and migration of HSC and block liver fibrosis [14]. Therefore, we used mice with selective deletion of NRP-1 in HSC to test the hypothesis that HSC activation through NRP-1 may regulate hepatocyte steatohepatitis in response to alcohol feeding in mice. We also used an animal model with selective deletion of synectin, as a second model of impaired HSC activation. Synectin, also known as GIPC, is a scaffold protein, which links the transport of cell-surface receptors, including NRP-1, to their intracellular signaling pathways. Like NRP-1, synectin has been implicated in HSC activation [15]. Our results demonstrated that HSC are not only regulators of the fibrogenic process but also mediate steatohepatitis in a model of ALD. Furthermore, we showed specific mechanistic pathways involving insulin-like growth factor binding protein-3 (Igfbp3, a major IGF-1 binding protein with multiple metabolic functions) and SerpinA12 (a serine-protease inhibitor) by which HSC exert their lipogenic effects on hepatocytes.
MATERIALS AND METHODS
See the Supporting Methods for more details.
Generation of Col1Cre/NRP-1loxP mice
Neuropilin-1 floxed mice (NRP-1loxP) and synectin floxed mice (synectinloxP) mice [16, 17] were crossed with ColCre mice [18, 19] to generate offspring with HSC selective deletion of NRP-1 (ColCre/NRP-1loxP) or synectin (ColCre/synectinloxP). In all cases, control mice were littermates carrying floxed genes (NRP-1loxP or synectinloxP) (Figure 1A–C and Supplementary Figure 1A–B). The efficiency of Cre recombinase in the ColCre mice was assessed by crossing ColCre with Rosa26LacZ reporter mouse, where the LacZ gene follows a STOP codon inserted between two loxP sites. In the ColCre/Rosa26LacZ offsprings, in Cre+ cells, Cre recombinase deletes the STOP codon leading to the LacZ gene expression and subsequent β-galactosidase protein expression. We then isolated hepatic stellate cells from control or ColCre/Rosa26LacZ mice and performed X-gal assay to examine Cre efficiency through β-galactosidase expression. HSC isolated from Cre− control mice contained no blue cells (Supplementary Figure 1C). However, HSC isolated from ColCre/Rosa26LacZ mice were 74.24% blue, confirming a good recombination efficiency of the Cre (Supplementary Figure 1D). Primary mouse hepatic stellate cells (mHSC) from ColCre/Rosa26LacZ mice were isolated, as described previously [20]. β-galactosidase (β-gal) staining was performed as described previously [21]. Cells were washed with PBS and observed through an optical microscope. β-gal staining was quantified using ImageJ.
Fig. 1. Impaired HSC activation by selective NRP-1 deletion reduces EtOH-induced steatosis and inflammation in vivo.

Panel (A), Hepatic stellate cells isolated from ColCre/NRP-1loxP mice showed the absence of signal for NRP-1 by western blot compared to NRP-1loxP mice (n=3–6; p<0.05). Panels (B) and (C), represent α-smooth muscle actin (α-SMA) and collagen type I alpha 1 (Col1α1) mRNA expression respectively, in response to Transforming Growth Factor-β (TGF-β) and Cre recombinase containing adenovirus (AdCRE) in NRP-1loxP and ColCre/NRP-1loxP mice (n=3–6; p<0.05). (D) Alcohol fed ColCre/NRP-1loxP mice exhibit less steatosis compared to matched NRP-1loxP mice as assayed by Oil-Red O staining (intensity quantification in the adjacent panel; n=6–9; 5x magnification; p<0.0001; one-way ANOVA). (E) HSC selective deletion of neuropilin-1 inhibits triglyceride accumulation in the liver in response to chronic/binge alcohol feeding (n=6–9; p<0.05; one-way ANOVA). (F-H) ColCre/NRP-1loxP mice exhibit lower expression of inflammatory markers in response to alcohol as evidenced by reduced TNFα, IL1β, and MCP-1 levels (n=6–9; p<0.05; one-way ANOVA). (I) Alcohol fed ColCre/NRP-1loxP mouse livers exhibit less infiltrative macrophages (arrowheads) compared to matched NRP-1loxP mice based on CD68 staining (intensity quantification adjacent to representative images; n=6–9; 20X magnification; area 1.3mm2, p<0.05, one-way ANOVA). (J) Alcohol fed ColCre/NRP-1loxP mouse livers exhibit less number of inflammatory foci (arrowhead) per field compared to matched NRP-1loxP mice (representative images; n=6–9; 10X magnification, p<0.05, one-way ANOVA).
Animal studies
All animal experiments were approved by IACUC, carried out in accordance with institutional guidelines, and were conform to ARRIVE guidelines. Female C57BL/6J control mice (NRP-1loxP or synectinloxP; n=6–9 each) and ColCre/NRP-1loxP or ColCre/synectinloxP (n=6–9 each) mice were divided into four experimental groups receiving either the Lieber–deCarli diet for 10 days followed by alcohol gavage (chronic/binge alcohol feeding model or NIAAA model) [22] or control diet. Each group of animals were housed in transparent polycarbonate cages subjected to 12 h light/darkness cycles under a temperature of 21°C and relative humidity of 50%. After ending the feeding course, mice were euthanized, and serum and liver samples were collected and processed or stored at −80°C until analyses. Female mice were used because they are more susceptible to alcohol-related liver injury [23].
Human Cells
Human primary HSC (ScienCell Research Laboratories, CA, USA) and HepG2 hepatocyte cell line overexpressing ethanol-metabolizing enzyme cytochrome P450 2E1 (HepG2Cyp2E1) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. Experiments were performed after overnight serum starvation. Small interfering RNA (siRNA) for NRP-1 (SI00066787 and SI02663206), integrin beta1, synectin, and SerpinA12 were purchased from Qiagen (Netherlands) or Thermo-Fisher (USA).
Biochemical determinations and compounds
Hepatic triglyceride content was assessed according to Carr et al. [24]. For the evaluation of cellular triglyceride content, cells were grown in monolayer, resuspended in PBS with 0.5% Triton X-100 and lysed with an ultrasound tip for evaluation with a commercial kit (Zen-bio, USA), according to Beller M. (personal communication). Compounds used were murine or human recombinant Igfbp3 (R&D systems, 0.3 μg/mL), Cytochalasin D (Sigma-Aldrich, 1:1000), PP2 (Sigma-Aldrich, 10 uM), recombinant SerpinA12 (R&Dsystems, 100 ng/mL), Akt inhibitor (Tocris, Akti-1/2, 250 nM), PQ-401 (Tocris, 10uM) and AMPK inhibitor (Tocris, Dorsomorphin dihydrochloride, 20 uM).
Inflammatory proteins and adipokine arrays
Supernatant and lysate from control or NRP-1 knockdown (KD) human HSC samples were subjected to human cytokines and adipokines antibody-based array (ARY024; R&D Systems, USA) according to manufacturer’s instructions and quantitated using ImageJ-software (NIH).
Statistical analysis
Results are expressed as the mean ± SD from three or more independent experiments. Two-tailed Student’s t-test or ANOVA was used to test the statistical significance between groups as appropriate. A P-value of less than 0.05 was considered as statistically significant.
RESULTS
Impaired activation of HSC by selective deletion of NRP-1 reduces EtOH-induced fibrosis, inflammation and steatosis in vivo.
HSC are established key regulators in hepatic fibrogenesis [25]. We previously showed that NRP-1 is implicated in HSC activation and its blockage by a neutralizing antibody ameliorated liver fibrosis in mice [14]. In this study, we used a genetic model of impaired HSC activation by selectively deleting NRP-1 from HSC (Figure 1A–C and Supplementary Figure 1) to test whether impaired HSC activation could affect steatohepatitis in a model of alcohol-induced liver disease. To validate the reduction in fibrosis in this model, we performed Sirius Red staining from liver tissue sections. As expected, fibrosis was lower in ColCre/NRP-1loxP mice fed with alcohol compared with the alcohol-fed control mice (Supplementary Figure 2A). Also supporting this finding, the evaluation of fibrosis markers, α-smooth muscle actin (α-SMA), collagen type I alpha 1 (Col1α1), and tissue inhibitor of metalloproteinases-1 (TIMP-1) mRNA levels were lower in ColCre/NRP-1loxP mice fed with alcohol compared with the alcohol-fed control mice (Supplementary Figure 2B–D). Based on the hypothesis that HSC may regulate steatosis and inflammation that precedes liver fibrosis, we determined if selective deletion of NRP-1 in HSC could protect against steatosis and inflammation in response to alcohol feeding. Control mice fed with the chronic/binge alcohol model developed significant steatosis compared with those fed with a control diet, as observed by Oil-Red O staining, while ColCre/NRP-1loxP mice were protected against alcohol-induced steatosis (Figure 1D). These observations were consistent with a 50% reduction in hepatic triglyceride content after alcohol feeding in ColCre/NRP-1loxP mice compared to controls (p<0.05) (Figure 1E). Furthermore, the inflammatory markers tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and monocyte chemoattractant protein-1 (MCP-1) mRNA levels increased in response to alcohol feeding in control mice but not in ColCre/NRP-1loxP mice (Figure 1F–H). Concordantly, CD68 staining from liver tissue sections showed an increase in macrophage infiltration in response to alcohol in NRP-1loxP mice, but not in the ColCre/NRP-1loxP mice (Figure 1I). More specifically, macrophage markers CD68 and F4/80 increased with alcohol-feeding in NRP-1loxP mice, but not in the ColCre/NRP-1loxP mice (Supplemental Figure 2F–G). Additionally, we assessed inflammatory foci quantification with consistent results (Figure 1J). We validated these results in a second model of impaired HSC activation, produced by selective deletion of synectin in HSC (ColCre/synectinloxP) obtaining similar results (Supplementary Figure 3A–B). Thus, these approaches indicate that impairing the activation of HSC can protect against alcohol-induced fibrosis, steatosis, and inflammation in mice models.
HSC supernatant from ColCre/NRP-1loxP mice decreases lipid droplet formation in hepatocytes exposed to ethanol in vitro.
Based on the observation that selective deletion of NRP-1 from HSC protects from alcohol-induced steatosis, we hypothesized that HSC could produce a NRP-1-dependent paracrine signal to regulate hepatocyte steatosis. To test this hypothesis, we isolated and cultured HSC from ColCre/NRP-1loxP and NRP-1loxP mice. The cell supernatant was collected and used to treat primary murine hepatocytes in combination with different concentrations of EtOH (25 and 50 mM). Hepatocytes treated with HSC supernatant isolated from ColCre/NRP-1loxP mice had fewer lipid droplets as compared to NRP-1loxP mice, as shown by quantitative BODIPY staining (Figure 2A). We next used siRNA-mediated approaches to test our hypothesis in human cells further. We performed indirect co-culture using a Transwell device. We plated hepatocytes on the bottom plate and HSC on the insert (top). Previously, we inhibited or not the expression of NRP-1 in HSC by using specifics siRNAs. Then, we exposed them to alcohol and assessed lipid droplets by Bodipy staining. Hepatocytes co-cultured with NRP-1 siRNA HSC developed fewer lipid droplets (7.7-fold decrease in Bodipy staining intensity comparing siRNA control vs. siRNA for NRP-1) (Figure 2B). Next, we isolated and cultured primary human HSC to obtain supernatant from control HSC and with NRP-1 KD. Treatment of HepG2Cyp2E1 cells with supernatant from NRP-1 KD HSC decreased lipid droplet formation in response to ethanol (Figure 3A). In summary, these experiments indicate that one or more soluble molecules secreted by HSC modulate the development of steatosis in hepatocytes and support a bi-directional model of HSC-hepatocyte crosstalk.
Fig. 2. Supernatant from HSC with selective deletion or knockdown of NRP-1 prevents lipid droplets formation in primary hepatocytes treated with ethanol.
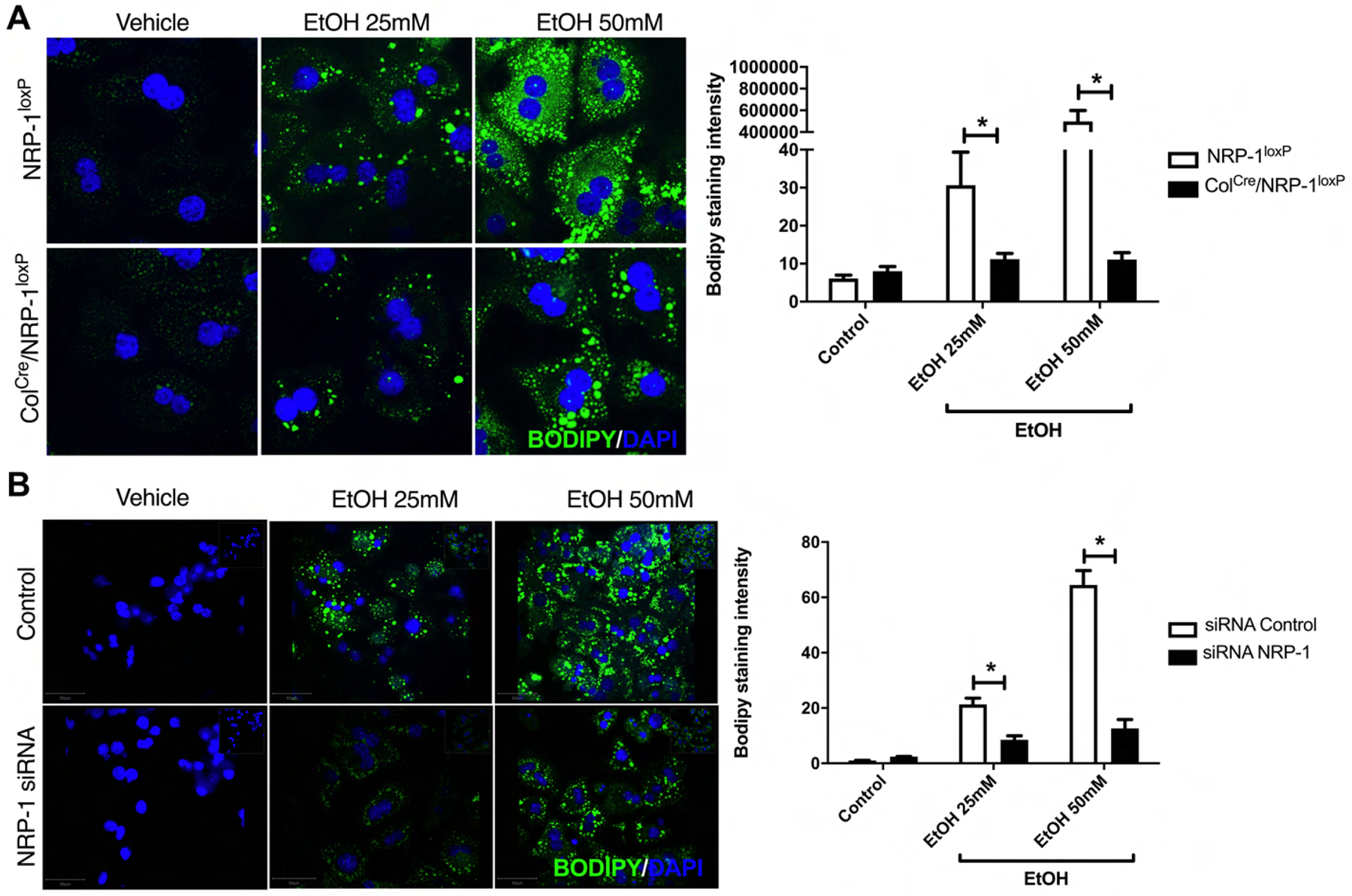
(A) Supernatant from HSC from NRP-1loxP and Col1Cre/NRP-1loxP mice prevent lipid droplets formation in primary hepatocytes treated with ethanol. Representative images of primary mouse hepatocytes treated with supernatant of HSC from ColCre/NRP-1loxP or NRP-1loxP and different concentrations of ethanol (EtOH) (0, 25 and 50mM). (n=5–9, 63x). Quantification of lipid droplets by BODIPY® 493/503 intensity evaluation. (n=6–9, *p<0.05, one-way ANOVA). Hepatocytes treated with supernatant of ColCre/NRP-1loxP HSC are prevented from the development of lipid droplets in response to EtOH. (B) Supernatant from HSC with knockdown of NRP-1 by siRNA prevents lipid droplets formation in primary hepatocytes treated with ethanol. Representative images of hepatocytes treated with supernatant from NRP-1 KD HSC and different concentrations of ethanol (EtOH) (0, 25 and 50mM). (n=5–9, 40x). Quantification of lipid droplets by BODIPY® 493/503 intensity evaluation. (n=6–9, *p<0.05, one-way ANOVA).
Fig. 3. NRP-1 knockdown in human-derived primary HSC increases Igfbp3 while decreases SerpinA12 levels.
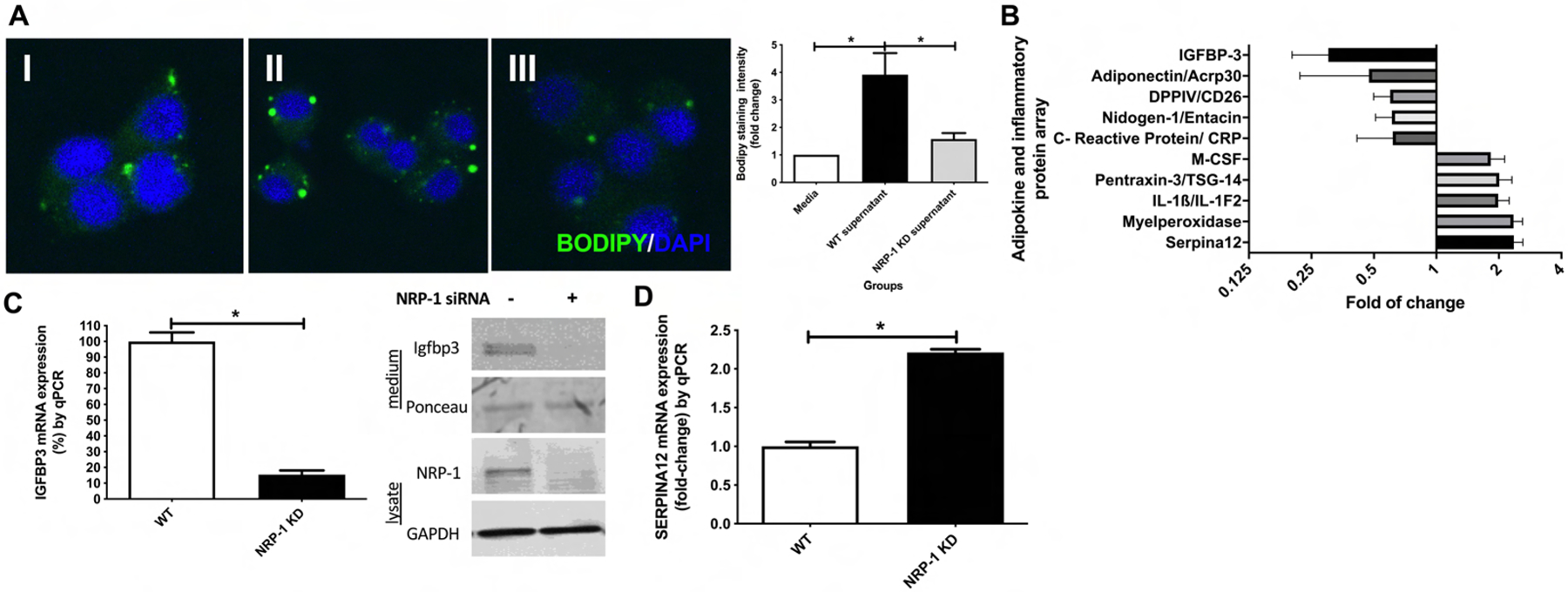
(A) Representative BODIPY® staining images and quantification from HepG2Cyp2E1 cells treated with (I) medium, (II) control HSC supernatant, (III) siRNA mediated NRP-1 knockdown HSC supernatant. (III) shows decreased lipid droplet formation in vitro (n=6–9, 63X, one-way ANOVA). (B) Top ten down/upregulated molecules from adipokine and inflammatory protein array from NRP-1 KD HSC supernatant compared to controls. IGFBP3 and SerpinA12 were the most significantly decreased and increased proteins respectively (n=3, p<0.05, unpaired t-test). (C) Igfbp3 mRNA and protein expression in NRP-1 KD HSC is lower than human WT HSC on qPCR and WB (n=6–9, p<0.05, unpaired t-test). (D) SerpinA12 mRNA expression in NRP-1 KD HSC is higher than human WT HSC (n=6–9, p<0.05, unpaired t-test).
Insulin-like growth factor-binding protein 3 (Igfbp3) expression is decreased and SerpinA12 is increased in supernatant and cell lysate from NRP-1 KD HSC.
To evaluate possible mechanisms by which NRP-1 KD could influence the steatotic process, we performed an adipokine and inflammatory protein array (Supplementary Figure 4). This array showed a 4.4-fold reduction in Igfbp3 expression and a 2.4-fold increase of SerpinA12 from supernatant from NRP-1 KD HSC compared to WT HSC (Figure 3B). We confirmed this finding by qPCR analyses for Igfbp3 and SerpinA12 from cells extracts and Western Blotting (WB) for Igfbp3 in the conditioned medium. (Figure 3C–D). These experiments confirmed that knockdown of NRP-1 induces Igfbp3 downregulation with a concomitant SerpinA12 upregulation.
Igfbp3 is the main insulin-like growth factor (IGF) binding protein found in human serum and has multiple metabolic functions either dependent upon, or independent from IGF [26]. Meanwhile, SerpinA12 is a serine-protease inhibitor which has been shown to modulate insulin resistance and endoplasmic reticulum stress [27]. Since these metabolism-related molecules were the top positively and negatively regulated hits from the protein array, we mechanistically tested their role in the development of steatosis in our model.
Recombinant Igfbp3 increases lipid droplet formation, triglyceride content, and lipogenic gene expression in hepatocytes in vitro.
To obtain further mechanistic insight regarding Igfbp3, we returned to our primary murine hepatocyte model. The use of murine recombinant Igfbp3 in primary mouse hepatocytes increased lipid droplet formation, similar to that observed with EtOH, with a synergistic effect at low doses of EtOH (10 mM and 25 mM), as assessed by quantitative BODIPY staining (Figure 4A). We next confirmed these results using a biochemical assay for cellular triglyceride content. Cultured hepatocytes from wildtype (WT) mice were treated with 50 mM EtOH, Igfbp3 alone, EtOH plus Igfbp3, or vehicle. Hepatocytes treated with Igfbp3 had an average of 104.3 ± 10.4 μg of triglyceride/mg of protein (p<0.05 compared to controls), similar to hepatocytes treated with EtOH alone and to those treated with Igfbp3 plus EtOH (Figure 4B). In summary, these experiments showed that recombinant Igfbp3 increases lipid droplet formation and cellular triglyceride content in primary mouse hepatocytes.
Fig. 4. Recombinant murine-Igfbp3 and ethanol (EtOH) increase lipid droplet formation, triglycerides, and lipogenic gene expression in primary mouse hepatocytes.

(A) Representative images and BODIPY staining from hepatocytes treated with basal medium ± EtOH in different concentrations ± murine recombinant Igfbp3 (BODIPY® + DAPI staining, n=6, 63x, one-way ANOVA). Treatment of primary mouse hepatocytes with murine recombinant Igfbp3 and ethanol significantly increases lipid droplets formation in vitro. (B) Quantification of triglycerides. Cultured primary mouse hepatocytes were treated with Igfbp3, EtOH 50mM, Igfbp3 plus EtOH, or vehicle. All treated groups showed higher cellular triglyceride content compared to control (n=6, *p<0.05 compared to control, one way ANOVA). (C) Expression of lipogenic genes in response to recombinant Igfbp3 administration in primary mouse hepatocytes. Igfbp3 administration induces key lipogenic genes involved in hepatic lipid homeostasis (SREBP-1c, ACC, FASN, SCD1, and DGAT1) (n=6, *p<0.05 compared to control, one way ANOVA). (D) Protein expression of SREBP-1c and FASN by WB in response to Igfbp3. Igfbp3 administration increases both protein expression (n=6, *p<0.05 compared to control, unpaired t-test). (E) In vitro human recombinant IGFBP3 treatment to human hepatocytes induces the main lipogenic genes involved in triglyceride de novo synthesis viz. SREBP-1c, ACC, FASN, SCD1, DGAT1 (n=6–9, *p<0.05, one-way ANOVA). (F) Supernatant from siRNA-mediated NRP-1 knockdown HSC reduces the protein expression of SREBP-1c by WB (n=6, *p<0.05, unpaired t-test). (G) In vitro reversal of protection against alcohol of NRP-1 siRNA-mediated knockdown in HSC. Hepatocytes treated with supernatant and alcohol +/− recombinant Igfbp3. Hepatocytes treated with supernatant from NRP-1 KD HSC developed fewer lipid droplets, and this protective effect was reverted in the presence of Igfbp3 (n=6, *p<0.05 compared to control siRNA + Igfbp3, one way ANOVA).
Since Igfbp3 stimulates lipid droplets formation histologically, we next sought to evaluate the effect of direct administration of recombinant Igfbp3 into in vitro primary mouse hepatocytes to further assess canonical genes implicated in triglycerides biosynthesis [28]. Treatment of primary mouse hepatocytes with murine recombinant Igfbp3 increased expression of key genes for hepatic lipid homeostasis. Sterol regulatory element-binding protein 1c (SREBP-1c), a key transcription factor in sterol and fatty acid biosynthesis, showed a 4.2 ± 0.5 fold-increase in mRNA expression compared to control. We observed similar results with other key genes associated with triglyceride synthesis (Figure 4C) including acetyl-CoA carboxylase (ACC, 2.7 ± 0.5 fold), fatty acid synthase (FASN, 2.1 ± 0.2 fold), stearoyl-CoA desaturase 1 (SCD1, 6.4 ± 1.6 fold-increase), and diglyceride acyltransferase 1 (DGAT1, 2.6 ± 0.2 fold-increase). These findings were consistent with our evaluation of SREBP1-c and FAS protein levels by WB in response to Igfbp3 (Figure 4D). Similar to primary mouse hepatocytes, treatment of HepG2Cyp2E1 cells with human recombinant-Igfbp3 increased expression of key genes implicated in de novo hepatic lipogenesis. SREBP-1c showed a 1.5 ± 0.2 fold-increase compared to control mRNA (Figure 4E). We observed similar results with ACC (1.9 ± 0.5 fold), FASN (1.4 ± 0.1 fold), SCD1 (1.8 ± 0.4 fold), and DGAT1 (1.6 ± 0.2 fold). Consistently, SREBP1-c protein levels by WB showed a reduction in hepatocytes treated with supernatant from HSC with siRNA mediated KD of NRP-1 (Figure 4F). Additionally, we assessed genes implicated in fatty acid oxidation observing a reduction in the enzymes carnitine-palmitoyl-transferase 1 (CPT1) and the chain length-specific acyl-CoA dehydrogenases (long-chain [LCAD] and medium-chain acyl-CoA dehydrogenase [MCAD]) by qPCR in hepatocytes treated with supernatant from NRP-1 KD HSC (Supplementary Figure 5A). To assess reversal of protection against alcohol-induced steatohepatitis by NRP-1 siRNA-mediated KD in HSC, we treated hepatocytes with conditioned medium and alcohol in the presence or absence of recombinant Igfbp3. Conditioned medium from NRP-1 KD HSC protected hepatocytes from alcohol injury, and Igfbp3 abolished this protection (Figure 4G). Together, these data suggest that Igfbp3 increases steatosis in hepatocytes by activation of key lipogenic genes and facilitates the accumulation of triglycerides.
Igfbp3 increases p-Akt through Src-kinase signaling in primary hepatocytes.
To evaluate how Igfbp3 exerts its pro-steatotic effects, we aimed to explore intracellular pathways related to hepatic lipogenesis and growth (Akt, Erk, and IGF). We found that the phosphorylated form of Akt (p-Akt) was induced 6.2-fold compared to control by the administration of murine recombinant Igfbp3 in vitro in primary hepatocytes (p<0.05) (Figure 5A). As a specificity control, no significant changes were seen in the phosphorylation of the insulin-like growth factor 1 (IGF-1) receptor (IGFR) or the extracellular signal-regulated kinase, Erk (Figure 5A). Additionally, we observed an upregulation of lipogenic proteins (SREBP-1c [3.9 ± 0.4-fold comparing control vs. Igfbp3 + PQ-401; p=0.0007] and FAS [6.3 ± 1.5-fold comparing control vs. Igfbp3 + PQ-401; p=0.009]) in the presence of Igfbp3 regardless of pharmacologic IGF-1R inhibition (PQ-401) (Supplemental Figure 6A). In order to determine whether Igfbp3-induced lipogenesis depends upon its interaction with IGF1, we transduced primary mouse hepatocytes with either wildtype (WT)-Igfbp3 adenovirus or adenovirus containing a construct of an Igfbp3-GGG mutant protein (I56G, L80G and L81G, mutant IGF binding sites), which expresses full-length Igfbp3 but has no binding affinity to IGF1 nor IGF1R. Either Igfbp3 or Igfbp3 mutant construct induced key lipogenic genes expression (Figure 5B).
Fig. 5. Igfbp3 increases p-Akt through integrin receptor/Src-kinase signaling in primary hepatocytes.

(A) The phosphorylated form of Akt is induced by the administration of murine recombinant Igfbp3 in vitro in primary hepatocytes (n=6, p<0.05, one way ANOVA). (B) Transfection with either WT Igfbp3 and Igfbp3 GGG mutant adenovirus (I56G, L80G and L81G, mutant IGF binding sites), which expresses full-length Igfbp3 but has no binding affinity to IGF1 and does not bind to IGF1R) induced key lipogenic genes expression (n=3, p<0.05, one way ANOVA). (C) Igfbp3-induced phosphorylation of p-Akt is blunted by Akt inhibitor pretreatment (triplicates, n=9, p<0.05, one way ANOVA). (D) Igfbp3-induced phosphorylation of p-Akt is blunted by Cytochalasin D (Cyt D) pretreatment (n=6, p<0.05, one way ANOVA). (E) Igfbp3-induced phosphorylation of p-Akt is blunted by RGD peptide pretreatment (n=6, p<0.05, one way ANOVA). (F) Igfbp3-induced phosphorylation of p-Akt is blunted by PP2 pretreatment (duplicates, n=6, p<0.05, one way ANOVA). (G) Igfbp3-induced phosphorylation of p-Akt is not affected by IGF-1R inhibitor (PQ401) pretreatment (n=6, p<0.05, one way ANOVA). (H) Representative microscopy images of lipid droplets and Bodipy staining quantification from primary mouse hepatocytes treated with vehicle or Igfbp3 ± PP2 or Akt inhibitor. Pretreatment with PP2 or Akt inhibitor protects against lipid droplet formation induced by Igfbp3 (n=6, p<0.05, 63x, one way ANOVA). (I) mRNA expression of lipogenic genes by qPCR. Igfbp3 administration to in vitro primary mouse hepatocytes induces key lipogenic genes involved in hepatic lipid homeostasis and this effect is blunted by Cytochalasin D, Akt inhibitor, and PP2 (SREBP-1c, ACC, FASN, SCD1, and DGAT1) (n=6, *p<0.05 compared to control/vehicle, #p<0.05 compared to Igfbp3 alone, one way ANOVA).
The effect of Igfbp3 on Akt phosphorylation was blunted by the use of a specific Akt inhibitor (Akti-1/2) (Figure 5C). Since Igfbp3 has been reported to signal in target cells through integrin receptors [29], we tested p-Akt signaling in response to Igfbp3 in the presence of a direct integrin receptor inhibitor (RGD peptide) and downstream inhibitors of integrin receptors (Cytochalasin D, an inhibitor of actin polymerization; and PP2, a Src-kinase inhibitor). Cytochalasin D, RGD peptide and PP2 blunted phosphorylation of Akt in response to Igfbp3 (Figures 5D–F), while IGF1-R inhibitor (PQ-401) didn’t affect Akt phosphorylation (Figure 5G), similar to the transfection with mutant Igfbp3-GGG protein (Supplemental Figure 6C) suggesting that this effect is not mediated by IGF1. This effect on p-AKT was correlated with an inhibition of the lipid droplet formation induced by Igfbp3, as revealed by Bodipy staining (Figures 5H). Additionally, pretreatment of hepatocytes with Cytochalasin D, Akt inhibitor, RGD peptide, and PP2 blunted the Igfbp3-mediated induction of key lipogenic genes (SREBP-1c, ACC, FASN, SCD1, DGAT1) by qPCR (Figure 5I) and as evidenced by WB for SREBP-1c by RGD peptide, PP2 and an Akt inhibitor (Supplemental Figures 6B and 7B). In total, these experiments indicate that Igfbp3 may increase lipogenesis through integrin receptor and p-Akt signaling.
SerpinA12 treatment protects against ethanol-induced steatosis through the p-AMPK pathway.
In contrast to Igfbp3, SerpinA12 was the most upregulated molecule in NRP-1 KD HSC from the adipokine and inflammatory protein array (Figure 3B). To investigate the effect of SerpinA12 on EtOH-induced steatosis, we treated primary mouse hepatocytes with murine recombinant SerpinA12, with or without EtOH. As shown in Figure 6A, SerpinA12 was protective against the lipid droplet formation induced by EtOH. With the aim to evaluate how SerpinA12 exerts its anti-steatotic effect, we assessed intracellular pathways related to hepatic lipogenesis and growth. We found no differences in the phosphorylated forms of Akt or Erk (Supplementary Figure 8B). Since the phosphorylated form of the 5’ adenosine monophosphate-activated protein kinase (p-AMPK) has been shown to be reduced in the presence of EtOH [30], we tested the protein levels of p-AMPK in response to EtOH with and without SerpinA12. Indeed, EtOH decreased p-AMPK compared to control, but this effect was lost in the presence of recombinant SerpinA12 (Figure 6B). Importantly, the effect of SerpinA12 on AMPK phosphorylation and the protection against EtOH-induced lipid droplet formation was blunted by the use of an AMPK inhibitor (Dorsomorphin dihydrochloride) (Figure 6B–C). These observations correlated with reduced expression of several key lipogenic genes (SREBP-1c, ACC, FASN, SCD1, DGAT1) as measured by qPCR (Figure 6D). Concordantly, the siRNA-mediated knockdown of SerpinA12 in primary human HSC followed by the use of this supernatant in HepG2Cyp2E1 cells in the presence of alcohol correlated with increased expression of lipogenic genes compared to control (Figure 6E). In summary, these results indicate that SerpinA12 inhibits ethanol-induced steatosis in hepatocytes and supports a model whereby Igfbp3 and SerpinA12 released by HSC have opposing effects on hepatocyte steatosis.
Fig. 6. SerpinA12 treatment protects against ethanol-induced steatosis in primary mouse hepatocytes and correlates with the p-AMPK pathway.

(A) Representative images and BODIPY® staining quantification show pretreatment with SerpinA12 protects against lipid droplet formation induced by EtOH (n=6, 63x, p<0.05, one way ANOVA). (B) WB and densitometry quantification shows that EtOH induces a reduction in the phosphorylation of AMPK compared to control. SerpinA12 induces phosphorylation of AMPK, even in the presence of EtOH, and this effect is blunted by the presence of AMPK inhibitor (duplicates, n=6, *p<0.05, one way ANOVA). (C) Representative images and BODIPY staining quantification show protection of SerpinA12 against EtOH-induced lipid droplet formation on treatment with SerpinA12 ± EtOH. This effect was blunted by using an AMPK inhibitor (n=6, 63x, *p<0.05, one way ANOVA). (D) EtOH induces key lipogenic genes involved in hepatic lipid homeostasis, and this effect is reduced by SerpinA12. The protective effect of SerpinA12 is attenuated by an AMPK inhibitor (n=6, *p<0.05 compared to control, #p<0.05 compared to EtOH alone, one way ANOVA). (E) Human hepatocytes treated with supernatant from human HSC with a siRNA-mediated knockdown of SerpinA12 ± EtOH. EtOH ± supernatant from siRNA-mediated knockdown of SerpinA12 increases expression of SREBP-1c, FASN, and SCD1 compared to EtOH in siControl cells (n=9, *p<0.05 compared to control, #p<0.05 compared to EtOH alone, one way ANOVA).
Igfbp3 is increased and SerpinA12 is decreased in serum and liver tissue from patients with alcoholic liver disease.
To show disease relevance in humans, we evaluated the protein levels of Igfbp3 and SerpinA12 from the serum of patients with alcoholic hepatitis (AH) and controls by WB. Indeed, Igfbp3 expression was 5.5-fold higher in patients with AH compared with controls (p<0.05), while SerpinA12 was 77% lower (p<0.05) (Figure 7A). Similar findings were obtained by ELISA for Igfbp3 from NRP-1loxP and ColCre/NRP-1loxP mouse serum. We observed an increase in Igfbp3 serum levels in NRP-1loxP alcohol-fed mice, which was attenuated in ColCre/NRP-1loxP mice (Supplementary Figure 7C). Additionally, isolated HSC from mice fed with control diet or ethanol showed consistent Igfbp3 and SerpinA12 mRNA expression levels (Supplementary Figure 7D). These data support our model, suggesting that high Igfbp3 levels and low SerpinA12 levels correlate with human alcoholic liver disease.
Fig. 7. Igfbp3 is increased while SerpinA12 is decreased in serum and liver tissue from patients with alcoholic liver disease.

(A) Representative WB and densitometry quantification for Igfbp3 and SerpinA12 from plasma from control subjects and with alcoholic hepatitis. Subjects with alcoholic hepatitis have significative higher levels of Igfbp3 and significative lower levels of SerpinA12 compared to controls (n=9, p<0.05, unpaired t-test). (B) Schematic representation of the proposed mechanism of hepatic stellate cells (HSC)-activation-induced steatosis. Activated HSC increase Insulin-like growth factor-binding protein 3 (Igfbp3) and reduces SerpinA12 secretion, which have a paracrine effect over hepatocytes. Igfbp3 increases p-Akt signaling through integrin receptor leading to lipid droplets formation, triglyceride content, and lipogenic gene expression; EtOH reduces p-AMPK, increasing lipogenesis. SerpinA12 protects against ethanol-induced steatosis through increasing p-AMPK.
DISCUSSION
The present study includes novel observations that advance our understanding of how HSC interact with hepatocytes. These observations demonstrate, for the first time, that HSC activation modulates EtOH-induced steatohepatitis. We find that the HSC-secreted molecules, Igfbp3 and SerpinA12, mediate lipid droplet formation in hepatocytes. This finding highlight novel signaling between HSC and hepatocytes. Mechanistically, we define a previously unknown role for Igfbp3 in which it increases p-Akt through integrin receptor/Src-kinase signaling in hepatocytes and promotes lipid droplet formation, triglyceride content, and lipogenic gene expression. Furthermore, SerpinA12 treatment protects against ethanol-induced steatosis through the p-AMPK pathway. Lastly, we show that Igfbp3 is increased in serum from patients with alcoholic hepatitis while SerpinA12 is decreased, demonstrating relevance to human ALD. Thus, these studies establish HSC as regulatory cells of the hepatocyte steatotic process in ALD, as summarized in Figure 7C.
HSC are activated during liver injury to orchestrate the fibrogenic response. However, HSC not only play a role in fibrosis [31] but also, as suggested by our data, modulate steatohepatitis [32]. Recently, it has been suggested that HSC produce cytokines and chemokines that act as antigen-presenting cells [33]. It has also been shown that activated HSC secrete IL-6 and TNFα, which further suppress the expression of HNF1α and SHP-1 in hepatocytes and lead to more inflammation and fibrogenesis [34]. Thus, activated HSC may be considered as initiating cells that regulate crosstalk with hepatocytes rather than simple target cells of liver injury in fibrogenesis.
Neuropilins (NRPs) are single-spanning transmembrane glycoproteins related to neuronal development, angiogenesis, tumor growth, and progression [35, 36]. Additionally, NRPs have shown to have a role in liver disease, through their actions as co-receptors for the vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β) [37, 38]. NRPs are mainly expressed in HSC and liver sinusoidal endothelial cells (LSECs) [39, 40], and are expressed in hepatocytes in malignant conditions [41]. NRP-1, with its synectin binding partner, plays a crucial role in fibronectin fibril assembly in tumor-associated myofibroblasts [42] and in the regulation of PDGF and TGF-β signaling, which are involved in canonical pathways of HSC activation and fibrosis [15]. Our data using both NRP-1 and synectin as models supports a role for HSC activation for driving the steatotic process through paracrine crosstalk with hepatocytes.
Mechanistic data from our study suggests that HSC activation through NRP-1 exerts its downstream effect through two molecules: Igfbp3 and SerpinA12. Igfbp3 is the main insulin-like growth factor (IGF) binding protein found in human serum and has the potential to either inhibit or enhance IGF actions in many cell types [26]. Our study highlights a novel role for HSC-derived Igfbp3 directly promoting lipid droplet formation and de novo lipogenesis and contributing to ethanol-induced steatosis. Moreover, we were able to show that Igfbp3-induced steatosis, independent of IGF1, and p-Akt signaling can be blunted with the inhibition of the integrin receptor/Src-kinase pathway, opening new possibilities for therapeutic modulation. A recent study has pointed out HSC as the top source of Igfbp3 in the liver [43]. A decrease in plasma Igfbp3 levels has been shown in patients with nonalcoholic fatty liver disease (NAFLD) [44], and lower levels have been associated with poor outcomes in hospitalized patients with cirrhosis [45]. Another study has shown that increased levels of Igfbp3 are a risk factor for steatosis and elevated ALT [46]. Globally, in vivo data suggests that Igfbp3 is associated with hepatic insulin resistance and decreased peripheral glucose sensitivity [47]. In summary, clinical data is mixed, and further studies are needed to establish associations of Igfbp3 with steatosis and to clarify the biological roles of Igfbp3 in glucose and lipid metabolism.
In our study, SerpinA12 was the most upregulated protein from the adipokine and inflammatory protein array, comparing the supernatant from NRP-1 KD HSC and WT HSC. Interestingly, we described a novel function for SerpinA12 as a protective factor for ethanol-induced steatosis through phosphorylation of AMPK. SerpinA12, also known as Vaspin, is a serine protease inhibitor mainly produced by adipose tissue [48]. It has been previously shown that SerpinA12 increases AMPK phosphorylation [27, 49]. The mechanism is not completely clear, but it has been demonstrated that SerpinA12 acts in HepG2 cells by binding to GRP78, a master regulator of the unfolded protein response [27]. SerpinA12 has been shown to modulate insulin action by specifically inhibiting its target protease, kallikrein-related peptidase 7 (KLK7) [50]. In murine models of NAFLD, a high-fat diet induces downregulation of SerpinA12 [51], and low SerpinA12 levels have been shown in patients with NASH [52].
Finally, our data from human serum samples showed that levels of Igfbp3 and SerpinA12 correlate with alcoholic hepatitis, suggesting that they could play a role in human alcohol-induced liver disease. This is a very exciting finding because it provides insight into the pathophysiology of ALD and opens the possibility for potential therapeutic targets aimed at the aforementioned pathways. This finding has potential clinical relevance as it could be speculated that increased levels of Igfbp3, as shown in patients with alcoholic hepatitis, may serve as a marker of severity of the alcohol-induced liver injury, as has been proposed for NAFLD [53, 54]. Also, modulation of serine protease inhibitor family, such as SerpinA12, could be tested as a treatment option in in vivo models of ALD.
This study has limitations, most notably, the difficulty in proving causal-effect relationships of Igfbp3 and SerpinA12 in the attenuation of alcoholic steatohepatitis development. In addition, Col-CRE deletion strategy could be non-specific for HSC. In conclusion, our data support the concept that HSC are not only paracrine target cells of hepatocytes but also regulate steatosis through the production of molecules that act on hepatocytes. Indeed, activated HSC are able to modulate steatohepatitis through two key pathways that we identified here, involving Igfbp3 and SerpinA12.
Supplementary Material
HIGHLIGHTS.
HSC regulate hepatocyte steatosis that precedes liver fibrosis in ALD.
HSC’s derived Igfbp3 & SerpinA12 mediate lipid droplet formation in hepatocytes.
These results are recapitulated in samples from patients with alcoholic hepatitis.
Financial Support Statement:
This work was supported by grant(s) NIH DK59615 and AA021171 (VHS), project FONDECYT #1200227 (JPA), #1150357 & 1191145 (MA) and #11171001 (DC), the Clinical Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567, Dr. L. Boardman). Arab JP was founded by an award from AASLD Foundation (AASLD/LIFER Clinical and Translational Research Fellowship in Liver Diseases). Col1Cre mice were kindly provided by Drs. Tatiana Kisseleva and David A. Brenner (UCSD), and mutant Igfbp3 by Dr. Youngman Oh (VCU). The authors want to thank Mrs. Theresa (Terri) J. Johnson and M. Donna Felmlee Devine, M.S. for their logistic support.
Abbreviations:
- α-SMA
α-smooth muscle actin
- ACC
acetyl-CoA carboxylase
- AH
alcoholic hepatitis
- ALD
alcoholic liver disease
- AMPK
5’ adenosine monophosphate-activated protein kinase
- ASCL4
long-chain-fatty-acid-CoA ligase 4
- Col1α1
Collagen type I alpha 1
- DGAT1
diglyceride acyltransferase 1
- EtOH
ethanol
- FASN
fatty acid synthase
- HepG2
hepatocellular carcinoma cells
- HSC
hepatic stellate cell
- IGF-1
insulin-like growth factor 1
- Igfbp3
Insulin-like growth factor-binding protein 3
- IGFR
insulin-like growth factor 1 receptor
- IL-1β
interleukin-1β
- KD
knockdown
- KO
knockout
- MCP-1
monocyte chemoattractant protein-1
- NAFLD
nonalcoholic fatty liver disease
- NRP-1
neuropilin-1
- NRP-1loxP
Neuropilin-1 floxed mice
- LSECs
liver sinusoidal endothelial cells
- PDGF-β
platelet-derived growth factor-β
- RT-PCR
reverse transcription PCR
- SCD1
stearoil-CoA desaturase 1
- siRNA
small interfering RNA
- SREBP-1c
sterol regulatory element-binding transcription factor 1c
- TGF-β
transforming growth factor β
- TIMP-1
tissue inhibitor of metalloproteinases-1
- TNF-α
tumor necrosis factor-α
- VEGF
vascular endothelial growth factor
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors have nothing to disclose.
REFERENCES
- [1].Paula H, Asrani SK, Boetticher NC, Pedersen R, Shah VH, Kim WR. Alcoholic liver disease-related mortality in the United States: 1980–2003. The American journal of gastroenterology 2010;105:1782–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liangpunsakul S, Haber P, McCaughan GW. Alcoholic Liver Disease in Asia, Europe, and North America. Gastroenterology 2016;150:1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009;360:2758–2769. [DOI] [PubMed] [Google Scholar]
- [4].Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Arab JP, Roblero JP, Altamirano J, Bessone F, Chaves Araujo R, Higuera-De la Tijera F, et al. Alcohol-related liver disease: Clinical practice guidelines by the Latin American Association for the Study of the Liver (ALEH). Ann Hepatol 2019;18:518–535. [DOI] [PubMed] [Google Scholar]
- [6].Dunn W, Shah VH. Pathogenesis of Alcoholic Liver Disease. Clinics in liver disease 2016;20:445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ji C, Chan C, Kaplowitz N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. Journal of hepatology 2006;45:717–724. [DOI] [PubMed] [Google Scholar]
- [8].Donckier V, Lucidi V, Gustot T, Moreno C. Ethical considerations regarding early liver transplantation in patients with severe alcoholic hepatitis not responding to medical therapy. Journal of hepatology 2014;60:866–871. [DOI] [PubMed] [Google Scholar]
- [9].Liangpunsakul S, Gao B. Alcohol and fat promote steatohepatitis: a critical role for fat-specific protein 27/CIDEC. J Investig Med 2016;64:1078–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 2015;61:1066–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Puche JE, Lee YA, Jiao J, Aloman C, Fiel MI, Munoz U, et al. A novel murine model to deplete hepatic stellate cells uncovers their role in amplifying liver damage in mice. Hepatology 2013;57:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tacke F, Trautwein C. Mechanisms of liver fibrosis resolution. Journal of hepatology 2015;63:1038–1039. [DOI] [PubMed] [Google Scholar]
- [13].Coulouarn C, Corlu A, Glaise D, Guenon I, Thorgeirsson SS, Clement B. Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer research 2012;72:2533–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cao S, Yaqoob U, Das A, Shergill U, Jagavelu K, Huebert RC, et al. Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-beta signaling in hepatic stellate cells. The Journal of clinical investigation 2010;120:2379–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Drinane MC, Yaqoob U, Yu H, Luo F, Greuter T, Arab JP, et al. Synectin promotes fibrogenesis by regulating PDGFR isoforms through distinct mechanisms. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bataller R, Brenner DA. Liver fibrosis. The Journal of clinical investigation 2005;115:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008;88:125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nature reviews Molecular cell biology 2012;13:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proceedings of the National Academy of Sciences of the United States of America 2012;109:9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc 2015;10:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kostallari E, Baba-Amer Y, Alonso-Martin S, Ngoh P, Relaix F, Lafuste P, et al. Pericytes in the myovascular niche promote post-natal myofiber growth and satellite cell quiescence. Development 2015;142:1242–1253. [DOI] [PubMed] [Google Scholar]
- [22].Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nature protocols 2013;8:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gao B, Xu MJ, Bertola A, Wang H, Zhou Z, Liangpunsakul S. Animal Models of Alcoholic Liver Disease: Pathogenesis and Clinical Relevance. Gene Expr 2017;17:173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Carr TP, Andresen CJ, Rudel LL. Enzymatic determination of triglyceride, free cholesterol, and total cholesterol in tissue lipid extracts. Clinical biochemistry 1993;26:39–42. [DOI] [PubMed] [Google Scholar]
- [25].Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;134:1655–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer 2014;14:329–341.24722429 [Google Scholar]
- [27].Nakatsuka A, Wada J, Iseda I, Teshigawara S, Higashio K, Murakami K, et al. Vaspin is an adipokine ameliorating ER stress in obesity as a ligand for cell-surface GRP78/MTJ-1 complex. Diabetes 2012;61:2823–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. The Journal of biological chemistry 2002;277:9520–9528. [DOI] [PubMed] [Google Scholar]
- [29].Burrows C, Holly JM, Laurence NJ, Vernon EG, Carter JV, Clark MA, et al. Insulin-like growth factor binding protein 3 has opposing actions on malignant and nonmalignant breast epithelial cells that are each reversible and dependent upon cholesterol-stabilized integrin receptor complexes. Endocrinology 2006;147:3484–3500. [DOI] [PubMed] [Google Scholar]
- [30].You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004;127:1798–1808. [DOI] [PubMed] [Google Scholar]
- [31].Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut 2015;64:830–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kocabayoglu P, Zhang DY, Kojima K, Hoshida Y, Friedman SL. Induction and contribution of beta platelet-derived growth factor signalling by hepatic stellate cells to liver regeneration after partial hepatectomy in mice. Liver Int 2016;36:874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Weiskirchen R, Tacke F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg Nutr 2014;3:344–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Qian H, Deng X, Huang ZW, Wei J, Ding CH, Feng RX, et al. An HNF1alpha-regulated feedback circuit modulates hepatic fibrogenesis via the crosstalk between hepatocytes and hepatic stellate cells. Cell Res 2015;25:930–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kitsukawa T, Shimono A, Kawakami A, Kondoh H, Fujisawa H. Overexpression of a membrane protein, neuropilin, in chimeric mice causes anomalies in the cardiovascular system, nervous system and limbs. Development 1995;121:4309–4318. [DOI] [PubMed] [Google Scholar]
- [36].Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, et al. A requirement for neuropilin-1 in embryonic vessel formation. Development 1999;126:4895–4902. [DOI] [PubMed] [Google Scholar]
- [37].Staton CA, Kumar I, Reed MW, Brown NJ. Neuropilins in physiological and pathological angiogenesis. The Journal of pathology 2007;212:237–248. [DOI] [PubMed] [Google Scholar]
- [38].Eichmann A, Makinen T, Alitalo K. Neural guidance molecules regulate vascular remodeling and vessel navigation. Genes & development 2005;19:1013–1021. [DOI] [PubMed] [Google Scholar]
- [39].Zhuang PY, Wang JD, Tang ZH, Zhou XP, Yang Y, Quan ZW, et al. Peritumoral Neuropilin-1 and VEGF receptor-2 expression increases time to recurrence in hepatocellular carcinoma patients undergoing curative hepatectomy. Oncotarget 2014;5:11121–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Panigrahy D, Adini I, Mamluk R, Levonyak N, Bruns CJ, D’Amore PA, et al. Regulation of soluble neuropilin 1, an endogenous angiogenesis inhibitor, in liver development and regeneration. Pathology 2014;46:416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Berge M, Allanic D, Bonnin P, de Montrion C, Richard J, Suc M, et al. Neuropilin-1 is upregulated in hepatocellular carcinoma and contributes to tumour growth and vascular remodelling. Journal of hepatology 2011;55:866–875. [DOI] [PubMed] [Google Scholar]
- [42].Yaqoob U, Cao S, Shergill U, Jagavelu K, Geng Z, Yin M, et al. Neuropilin-1 stimulates tumor growth by increasing fibronectin fibril assembly in the tumor microenvironment. Cancer research 2012;72:4047–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 2018;9:4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Min HK, Maruyama H, Jang BK, Shimada M, Mirshahi F, Ren S, et al. Suppression of IGF binding protein-3 by palmitate promotes hepatic inflammatory responses. FASEB J 2016;30:4071–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Correa CG, Colombo Bda S, Ronsoni MF, Soares ESPE, Fayad L, Silva TE, et al. Circulating insulin-like growth factor-binding protein 3 as prognostic biomarker in liver cirrhosis. World J Hepatol 2016;8:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Volzke H, Nauck M, Rettig R, Dorr M, Higham C, Brabant G, et al. Association between hepatic steatosis and serum IGF1 and IGFBP-3 levels in a population-based sample. Eur J Endocrinol 2009;161:705–713. [DOI] [PubMed] [Google Scholar]
- [47].Yamada PM, Lee KW. Perspectives in mammalian IGFBP-3 biology: local vs. systemic action. Am J Physiol Cell Physiol 2009;296:C954–976. [DOI] [PubMed] [Google Scholar]
- [48].Kloting N, Berndt J, Kralisch S, Kovacs P, Fasshauer M, Schon MR, et al. Vaspin gene expression in human adipose tissue: association with obesity and type 2 diabetes. Biochem Biophys Res Commun 2006;339:430–436. [DOI] [PubMed] [Google Scholar]
- [49].Jung CH, Lee MJ, Kang YM, Lee YL, Yoon HK, Kang SW, et al. Vaspin inhibits cytokine-induced nuclear factor-kappa B activation and adhesion molecule expression via AMP-activated protein kinase activation in vascular endothelial cells. Cardiovasc Diabetol 2014;13:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Heiker JT, Kloting N, Kovacs P, Kuettner EB, Strater N, Schultz S, et al. Vaspin inhibits kallikrein 7 by serpin mechanism. Cell Mol Life Sci 2013;70:2569–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Richards VE, Chau B, White MR, McQueen CA. Hepatic gene expression and lipid homeostasis in C57BL/6 mice exposed to hydrazine or acetylhydrazine. Toxicol Sci 2004;82:318–332. [DOI] [PubMed] [Google Scholar]
- [52].Polyzos SA, Kountouras J, Polymerou V, Papadimitriou KG, Zavos C, Katsinelos P. Vaspin, resistin, retinol-binding protein-4, interleukin-1alpha and interleukin-6 in patients with nonalcoholic fatty liver disease. Ann Hepatol 2016;15:705–714. [DOI] [PubMed] [Google Scholar]
- [53].Ichikawa T, Nakao K, Hamasaki K, Furukawa R, Tsuruta S, Ueda Y, et al. Role of growth hormone, insulin-like growth factor 1 and insulin-like growth factor-binding protein 3 in development of non-alcoholic fatty liver disease. Hepatol Int 2007;1:287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Takahashi Y The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.