

Abstract
Calcium and integrin binding protein 1 (CIB1) is an EF-hand-containing, small intracellular protein that has recently been implicated in cancer cell survival and proliferation. In particular, CIB1 depletion significantly impairs tumor growth in triple-negative breast cancer (TNBC). Thus, CIB1 is a potentially attractive target for cancer chemotherapy that has yet to be validated by a chemical probe. To produce a probe molecule to the CIB1 helix 10 (H10) pocket and demonstrate that it is a viable target for molecular intervention, we employed random peptide phage display to screen and select CIB1-binding peptides. The top peptide sequence selected, UNC10245092, was produced synthetically and binding to CIB1 was confirmed by isothermal titration calorimetry (ITC) and a time-resolved fluorescence resonance energy transfer (TR-FRET) assay. Both assays showed that the peptide bound to CIB1 with low nanomolar affinity. CIB1 was co-crystallized with UNC10245092, and the 2.1 Å resolution structure revealed that the peptide binds as an α-helix in the H10 pocket, displacing the CIB1 C-terminal H10 helix and causing conformational changes in H7 and H8. UNC10245092 was further derivatized with a C-terminal Tat-derived cell penetrating peptide (CPP) to demonstrate its effects on TNBC cells in culture, which are consistent with results of CIB1 depletion. These studies provide a first-in-class chemical tool for CIB1 inhibition in cell culture and validate the CIB1 H10 pocket for future probe and drug discovery efforts.
Introduction.
Breast cancer is the most frequently diagnosed cancer and one of the leading causes of cancer death for women worldwide, with 2.1 million new cases and over 620,000 deaths recorded last year.1 In the United States, from the 255,000 cases of breast cancer diagnosed in 2017, approximately 10-20% of all new cases are triple-negative breast cancer (TNBC), a subtype that lacks expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2).2 The lack of these receptors is the primary reason that there are no specific therapeutic agents available for TNBC. TNBC disproportionately affects premenopausal women of African or Hispanic ancestry and progresses aggressively, accounting for 15–20% of breast cancer cases and 25% of deaths.3, 4 Although TNBC is sensitive to chemotherapy, the overall prognosis for TNBC patients is worse than for non-TNBC sub-types: TNBC tumors are frequently larger and less differentiated5, 6 and 2.5-fold more likely to metastasize.4 TNBC patients also have a shorter median time to death (4.2 vs. 6 years) and poorer overall survival rates compared to other breast cancer subtypes.4 Given the lack of validated molecular targets and the poor outcome these patients, there is a clear need for the development of improved therapeutics. The majority of TNBC cases are basal-like, and typically exhibit constitutively activated RAF–MEK–ERK and PI3K–AKT signaling pathways.2, 7 Ongoing research is focused on novel targets for TNBC therapy, and several targeted agents have progressed into clinical trials. Pharmacological inhibition of both ERK and AKT signaling pathways is a promising approach to treat TNBC.7, 8,9 However, preclinical and clinical studies have suggested that combined inhibition of both PI3K and MEK may improve efficacy at the expense of increased toxicity.10-13 Therefore, there is an acute unmet need for development of new targeted therapeutics, with improved safety and efficacy, for TNBC patients.
The calcium and integrin binding protein, CIB1, is a small intracellular protein that has recently been identified as a promising target for TNBC.9, 14-17 Structurally, CIB1 is a 22 kDa protein composed of ten α-helices, eight of which form four helix-loop-helix EF-hand cation binding domains (EF-I to EF-IV).18 While the C-terminal EF-III and -IV domains bind Ca2+ with high affinity (Kd, 1.9 μM and 0.5 μM, respectively), EF-III can also bind Mg2+, albeit at slightly lower affinity (Kd, 120 μM) 19, 20 CIB1 is further organized into an N-terminal and C-terminal lobe displaying a myristoylation site and hydrophobic binding pocket, respectively.16 CIB1 was first discovered as a binding partner of the αIIb integrin cytoplasmic domain 21 and subsequently found to bind a wide variety of proteins that include additional α-integrin cytoplasmic domains 22, p21-activated kinase-1 9, and sphingosine kinase 1 23. The molecular interactions between CIB1 and the αIIb cytoplasmic domain are the most well characterized and biophysical evidence indicate that integrin cytoplasmic tails, and possibly other partners bind within the CIB1 hydrophobic channel 18, 22, 24-26. Previous NMR analyses also suggest a mechanism by which the CIB1 C-terminal helical domain (H10) shields the αIIb binding site in the C-lobe hydrophobic channel and acts as an auto-inhibitory mechanism to regulate ligand binding to CIB1 25, 26 More recent biological studies have shown that CIB1 depletion impairs both ERK and AKT activation and induced cell death in MDA-MB-468 TNBC cells.9,17 CIB1 depletion is tolerated in non-cancerous cell lines 17, 27 and CIB1−/− mice show no apparent developmental defects,16 suggesting that therapeutic inhibition of CIB1 may not require specific targeting of cancerous cells. This hypothesis is also supported by the finding that CIB1 may operate via non-oncogene addiction, in which certain cancer cells become reliant on, or addicted to, a non-oncogene to support proliferation and survival, whereas normal cells can tolerate loss of that same gene17. Thus, a large subset of TNBC cell lines exhibiting elevated AKT activity were found to depend on CIB1 for cell survival and tumor growth and that CIB1 expression levels per se were not predictive of the sensitivity to CIB1 interference.17 Taken together, CIB1 appears to be a safe and effective anti-tumor drug target, but no small molecule inhibitor or peptide capable of engaging CIB1 in a cellular context has been previously reported.
Herein, we report discovery of first-in-class peptide inhibitors of CIB1. Using random peptide phage display, we rapidly discovered numerous peptides that potently bind to CIB1. Isothermal titration calorimetry (ITC) confirmed synthetic peptide binding to CIB1. Moreover, an alanine scan was used to identify key binding determinants of the peptide:protein interaction. We also report the first co-crystal structure of CIB1 in complex with a peptide inhibitor, which correlates well with the alanine scan data. A cell-penetrating peptide (CPP) conjugate of one CIB1 inhibitory peptide, UNC10245092, proved active in cells, thereby validating the helix 10 (H10) binding hydrophobic pocket on CIB1 as a viable target for CIB1 disruption in TNBC. Overall, these results set the stage for further developing next generation cancer therapeutics via CIB1 inhibition.
Methods
Protein purification
Full length human CIB1 and an N-terminal truncation of the first eight disordered residues of CIB1 (CIB1 Δ1–8, residues 9–191) were expressed with an N-terminal hexahistidine tag (His-tag) and purified as described.18 The first 8 disorded residues of CIB1 were removed to facilitate crystallization as previously described.18 Both proteins were expressed in Rosetta2 BL21(DE3)pLysS cells (Novagen, EMD Chemicals). A 6 L culture of each was grown to mid log phase at 37°C, at which time the temperature was lowered to 18°C and protein expression was induced by addition of 0.5 mM IPTG. Cells were harvested by centrifugation and pellets were stored at −80°C. Thawed cell pellets were resuspended in 30 mL of lysis buffer (50 mM sodium phosphate pH 7.2, 500 mM NaCl, 30 mM imidazole, 1X EDTA free protease inhibitor cocktail (Roche Diagnostics) per L of culture. Cells were lysed on ice by sonication with a Branson Digital 450 Sonifier (Branson Ultrasonics) at 40% amplitude for 12 cycles with each cycle consisting of a 20 second pulse followed by a 40 second rest. Cell lysates were clarified by centrifugation and loaded onto a HisTrap FF column (GE Healthcare) preequilibrated with 10 column volumes of binding buffer (50 mM sodium phosphate pH 7.2, 500 mM NaCl, 30 mM imidazole) using an AKTA Purifier (GE Healthcare). The column was washed with 15 column volumes of binding buffer and protein was eluted in a linear gradient to 100% elution buffer (50 mM sodium phosphate pH 7.2, 500 mM NaCl, 500 mM imidazole) over 20 column volumes. Peak fractions containing the desired protein were pooled and concentrated in Amicon Ultra-15 concentrators (10K MW cut-off, Merck Millipore). For crystallization, the N-terminal His-tag was removed from the CIB1 Δ1–8 protein by cleavage with His-tagged TEV protease. To remove His-TEV and uncleaved protein, TEV treated protein was passed back over a HisTrapFF column and the column flow through was collected and concentrated in an Amicon Ultra-15 concentrators (10K MW cut-off, Merck Millipore). Full length His-CIB1 and CIB1 Δ1-8 with the His-tag removed were further purified by size exclusion chromatography. Concentrated protein was loaded onto a HiLoad 26/60 Superdex 75 prep grade column (GE Healthcare) that had been preequilibrated with 1.2 column volumes of sizing buffer (25 mM Tris pH 7.5, 250 mM NaCl, 2 mM DTT, 5% glycerol) using an AKTA Purifier (GE Healthcare). Protein was eluted isocratically in sizing buffer over 1.3 column volumes at a flow rate of 2 mL/min, collecting 3mL fractions. Peak fractions were analyzed for purity by SDS-PAGE and those containing pure protein were pooled and concentrated to less than 2 mL using Amicon Ultra-15 concentrators (10K MW cut-off). Full length CIB1 was dialyzed into a buffer containing 50 mM HEPES pH 7.4, 150 mM NaCl, 0.1 mM CaCl2 for use in ITC and CIB1 Δ1-8 was dialyzed into 20 mM MES pH 6.5, 10 mM NaCl, 2mM DTT and stored at 30 mg/ml for crystallography.
Full length biotinylated CIB1 for use in TR-FRET assays was generated by cloning the CIB1 coding sequence into a modified pET28 vector that includes both an N-terminal His-tag and an N-terminal Avi tag sequence (GLNDIFEAQKIEWHE). The resulting construct was cotransformed into Rosetta2 BL21(DE3)pLysS competent cells (Novagen, EMD Chemicals) along with an expression plasmid for E. coli biotin ligase (BirA pET21a-BirA was a gift from Alice Ting, Addgene plasmid # 20857). A 2 L culture was grown to mid log phase at 37°C, at which time the temperature was lowered to 18°C and protein expression was induced by addition of 0.5 mM IPTG. D-biotin was then added to the culture to a final concentration of 50 μM. Expression was allowed to continue overnight. Cells were harvested by centrifugation and pellets were stored at −80°C.
Biotinylated CIB1 was purified by resuspending thawed cell pellets in 30 mL of lysis buffer (50 mM sodium phosphate pH 7.2, 500 mM NaCl, 30 mM imidazole, 1X EDTA free protease inhibitor cocktail (Roche Diagnostics) per L of culture. Cells were lysed on ice by sonication with a Branson Digital 450 Sonifier (Branson Ultrasonics) at 40% amplitude for 12 cycles with each cycle consisting of a 20 second pulse followed by a 40 second rest. The cell lysate was clarified by centrifugation and loaded onto a HisTrap FF column (GE Healthcare) that had been preequilibrated with 10 column volumes of binding buffer (50 mM sodium phosphate pH 7.2, 500 mM NaCl, 30 mM imidazole) using an AKTA Purifier (GE Healthcare). The column was washed with 15 column volumes of binding buffer and protein was eluted in a linear gradient to 100% elution buffer (50 mM sodium phosphate pH 7.2, 500 mM NaCl, 500 mM imidazole) over 20 column volumes. Peak fractions containing the desired protein were pooled and concentrated to 2 mL in Amicon Ultra-15 concentrators (10K MW cut-off, Merck Millipore). Concentrated protein was loaded onto a HiLoad 26/60 Superdex 75 prep grade column (GE Healthcare) that had been preequilibrated with 1.2 column volumes of sizing buffer (25 mM Tris pH 7.5, 250 mM NaCl, 2 mM DTT, 5% glycerol) using an AKTA Purifier (GE Healthcare). Protein was eluted isocratically in sizing buffer over 1.3 column volumes at a flow rate of 2 mL/min, collecting 3mL fractions. Peak fractions were analyzed for purity by SDS-PAGE and those containing pure protein were pooled and concentrated to less than 2 mL using Amicon Ultra-15 concentrators (10K MW cut-off). Biotinylated CIB1 was isolated by passing concentrated protein over a 2 mL monomeric avidin column by gravity flow following the manufacturer’s recommendations (Pierce Biotechnology/Thermo Scientific). Bound protein was eluted in 12 mL PBS containing 2 mM D-biotin. Biotinylated CIB1 was concentrated in Amicon Ultra-15 concentrators as above and dialyzed into a buffer consisting of 50 mM HEPES, pH 7.4, 150 mM NaCl, 10% glycerol, aliquoted, and stored at −80°C.
Peptide Phage Display
An unbiased, random 10-mer peptide phage display library was constructed and used as previously described.28, 29 Briefly, full length CIB1 protein was immobilized on eight wells of a 96-well immuno plate (Nunc® MaxiSorp™) using 100 μL of a 10 μg/mL solution in Tris-buffered saline pH 7.5 (TBS). TBS alone was added to an additional column of wells (as negative control). After two hours with gentle shaking at room temperature, the CIB1 solution was removed and the wells were washed 3X with TBS and 0.05% tween-20 (Sigma; TBST). The phage library was diluted into casein blocking buffer (CBB; 1% Hammarsten grade casein in 20 mM Na2HPO4 pH 7.1, 200 mM NaCl). The phage dilution was added (100 μL) to each well to yield 6 x 1011 phage particles per well. After two hours at room temperature with gentle shaking, the wells were then washed seven times with 200 μL of TBST. Bound phage were eluted with 100 μL glycine solution 0.2 M glycine, pH 2.2) for 30 min at room temperature. The solution was removed and neutralized with 15 μL 1 M Tris-HCl pH 8.0. The neutralized solution was added to 1 mL of TG1 cells (OD600 ~ 1.0; Lucigen), incubated for 1 h at 37°C and then further diluted into 200 mL 2XYT media containing 10 μg/mL tetracycline and grown overnight. The next day, phage were isolated using standard methods (precipitation with PEG 8000-NaCl and centrifugation). Phage were titered using TG1 cells and tetracycline agar plates. Four selection rounds were completed, with input phage dropped 10-fold at each round, until enrichment (>10,000-fold) was observed compared to blank control wells. For next generation sequencing analysis, phage DNA from amplified round four was extracted using phenol chloroform and PCR amplified using Q5 polymerase. DNA samples from each round of selection were sequenced by the Genewiz Amplicon-EZ next generation sequencing service. A custom MATLAB routine was used to analyze the sequencing reads and determine enrichment of sequences. Individual clones from the round four selection were also selected for culture, phage preparations, dsDNA preparations, and DNA sequencing (Eton Bioscience Inc.). Initially, selected clones were analyzed for CIB1 binding using phage ELISA.30 Briefly, CIB1 protein (50 μL of 10 μg/mL CIB1 stock solution in TBS) was added to wells of a 384-well white Nunc® MaxiSorp™ plate and incubated for two hours at room temperature; buffer alone was used for control wells. Phage were serially diluted 3-fold in CBB and added to the washed CIB1 plate and incubated 2 h at room temperature. Unbound phage were removed by washing 5x with 100 μL TBST. 50 μL of a 1:10,000 dilution of HRP/anti-M13 mAb (GE Healthcare 27-9421-01) in CBB was added and incubated for one hour. Wells were again washed 5x with 100 μL TBS, followed by addition of 20 μL per well of SuperSignal™ ELISA Pico Chemiluminescent Substrate (Thermo Fisher Scientific 37070) and the plate was finally read using on an Envision Multilabel Plate Reader (PerkinElmer; luminescence). Phage ELISA binding curves were analyzed with a four parameter, non-linear curve fit using GraphPad Prism to determine phage concentration EC50.
Isothermal Calorimetry
Isothermal titration calorimetry (ITC) was performed to quantify the thermodynamics of binding between CIB1 and synthetic peptides. Purified full length CIB1 was buffer exchanged into 50 mM HEPES, pH 7.4, 150 mM NaCl, 0.1 mM CaCl2, (NAP-5 column; GE Healthcare Life Sciences) and diluted to a concentration of 30 μM. Peptides were freshly dissolved into 300 μM in the same buffer as CIB1. Protein and peptide final concentrations were verified on a Nano Drop 2000 spectrophotometer (Thermo Scientific) using respective molar absorptivities (https://web.expasy.org/protparam/) prior to ITC setup. All ITC measurements were recorded at 25°C with an AutoITC 200 microcalorimeter (Malvern Instruments). Peptides were titrated 0.2 μL/injection into a 200 μL cell with CIB1 protein, performed with a spacing of 180 seconds and a reference power of 7 μcal/sec. Control experiments were performed by titrating each peptide into buffer alone under identical conditions to determine potential heats of dilution, which were subtracted from the protein-peptide binding curves. The titration data were analyzed using Origin Software (Malvern Instruments) by non-linear least squares, fitting the heats of binding as a function of the peptide:protein ratio to a one site binding model.
Crystallization, data collection and structure determination
CIB1 Δ1–8 was mixed with 1.5 mM UNC10245092 and allowed to stand at 4°C for 2 h. The crystallization screens were performed under similar conditions to those described previously18 using the Mosquito HTS (TTP Labtech). Crystals of CIB1 Δ1–8 complexed with peptide UNC10245092 were obtained by sitting drop vapor diffusion at 21°C for 5–8 days by equilibrating against 137.5 mM calcium acetate, 16 % PEG 3350, 20 mM Bis-Tris propane,pH 6.8) using a microseeding technique from initial needle crystals obtained in 300 mM calcium acetate, 20 % PEG 3350, 20 mM bis-tris propane, pH 5.8. Crystals of CIB1 Δ1–8 in complex with UNC10245109 formed in 0.1 M HEPES:NaOH, pH 7.5, 20 % (w/v) PEG 8000 and were cryoprotected by the addition of ethylene glycol to a final concentration of 15% (w/v), mounted on nylon loops, and flash-cooled in liquid nitrogen. X-ray diffraction data were collected in the protein crystallography ID-22 beamline at The Advanced Photon Source (APS) at Argonne National Laboratory (Argonne, Illinois, USA). Diffraction data from CIB1 bound to UNC10245092 was processed with an HKL2000,31 while data from CIB1 bound to peptide UNC10245109 was processed with XDS via xia2.32, 33 The structures were solved by molecular replacement using the program PHASER34 from CCP4 and CIB1 (PDB code: 1XO5) as a model. The co-crystal model was refined with BUSTER,35 built with COOT,36 and validated using MolProbity.37 Figures were generated and analysis done with PyMOL or Schrodinger. Data collection and refinements statistics are detailed in Table S1 (see Supporting Information).
Time-resolved fluorescence energy transfer (TR-FRET)
A robust TR-FRET assay was developed to drive ongoing peptide optimization efforts, with the goal of improving potency, selectivity, stability, and cell permeability. For this assay, the UNC10245092 peptide was conjugated with Alexa Fluor 647 (Thermo Fisher Scientific; Alexa647-CIB1-peptide) and this fluorescent tracer peptide was used to test competitive binding affinity of new peptides. The TR-FRET assay was performed in 384-well plates in a total volume of 10 μL per well. Assay ready plates were prepared by performing a three-fold dilution of each peptide inhibitor (100X in DMSO, 0.1 μL) was added into a 384-well microplate (Greiner 784904; white, small volume, non-binding) using a Mosquito HTS nanoliter dispensing instrument (TTP Labtech). Biotinylated-CIB1 and the detection reagents were diluted into assay buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM calcium, 1 mM CHAPS) and dispensed to the assay plate using a Multidrop Combi Reagent Dispenser (Thermo Scientific). Five μL of biotinylated-CIB1 (3 nM final concentration) was first added to the assay ready plate and incubated 20 min at room temperature. A detection solution containing Lance Eu-labeled streptavidin-W1024 (Perkin Elmer; AD0063) and Alexa647-CIB1-peptide were added to a final concentration of 2 nM and 30 nM, respectively, in a 10 μL final reaction volume. Following a 30 minute incubation, the plate was read on an Envision Multilabel Plate Reader (PerkinElmer; Eu excitation 320 nm, Eu emission 615 nm; Alexa Dye emission 665 nm). The TR-FRET signal was measured as a ratio (665nm/615nm). Percent inhibition of test peptides was calculated using full column controls containing the detection solution (Lance Eu-streptavidin, Alexa647-CIB1-peptide, and biotinylated-CIB1) plus DMSO only (0% inhibition) and a known competitor peptide (typically UNC10245092; 100% inhibition). Normalized binding curves were analyzed with a four parameter non-linear curve fit using either GraphPad Prism or ScreenAble Solutions software. Z-prime values (>0.9) were monitored for each dose-response plate to assess robustness and reproducibility of the assay.
Cell lines
MDA-MB-468 (MDA-468) and MDA-MB-231 (MDA-231) breast cancer cell lines (UNC Lineberger Tissue Culture Facility, UNC, Chapel Hill, NC, USA) were cultured in DMEM supplemented with 1% non-essential amino acids (Gibco/Invitrogen) and 10% FBS (Gemini). All cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
Cell death analysis and Western blotting
To determine the cellular activity of UNC10245092, MDA-468 and MDA-231 cells were incubated with DMSO or 30 μM UNC10245092 or control UNC10245351 peptide for 6 and 24 h. Adherent and floating cell populations were collected and cell death was quantified using trypan blue dye exclusion and expressed as the mean percentage of dead cells (i.e., trypan blue positive) from both floating and adherent cell populations. Statistical analysis was performed using GraphPad Prism software. To determine the effects on CIB1-dependent signaling pathways, MDA-468 and MDA-231 cells harvested from the above cell death assay were pelleted and lysed with buffer containing 50 mM HEPES, pH 7.4, 10 mM CHAPS, 50 mM sodium fluoride, 10 mM β-glycerophosphate, 0.1 mM sodium pervanadate, and Protease Inhibitor Cocktail Set III (Calbiochem). Equal amounts of protein based on total cell number were separated by SDS-PAGE, transferred to PVDF membranes, and incubated with primary antibodies overnight at 4°C. Immunoblots were incubated with HRP-conjugated secondary antibodies against rabbit, mouse, or chicken and visualized using enhanced chemiluminescence (ECL2, Pierce). Antibodies used in this study: CIB1 chicken polyclonal antibody was produced as previously described,14 antibodies against p-AKT473 (#9271), p-ERK (#9101), p-H2AXSer139 (#9718), and AKT (#4691) were purchased from Cell Signaling, antibodies against ERK (sc-92) and vinculin (V9131) were purchased from Santa Cruz and Sigma, respectively.
Results
Discovery of UNC10245092, a high affinity peptide inhibitor of CIB1
We employed phage display of a fully randomized 10-amino acid peptide library to screen and select CIB1-binding peptides (Figure 1). This library was displayed as a fusion to the pIII coat protein of filamentous M13 phage via a standard phage vector as a polyvalent display system (typically 3 to 5 peptides per phage particle). Rounds of selection were monitored by colony formation units and phage ELISA to confirm enrichment, and promising pools of selectants were sent for sequencing. Significant enrichment was observed after only three rounds and by the fourth round, the selections converged on a small suite of peptides with a conserved, hydrophobic C-terminal tail. Interestingly, although none of the peptides selected from this unbiased library showed significant similarity to a known, relatively weak and naturally occurring CIB1 -binding sequence, the αIIb integrin peptide, they did display short regions of conserved hydrophobicity, reminiscent of the consensus motif for the CIB1 binding epitope.38 Phage ELISA was used to rank order the peptides based on CIB1 binding affinity and the clone with the selected insert FWYGAMKALY proved to be the most potent (Figure 1B). This top candidate sequence was then synthesized as NH2-EDGGSFWYGAMKALYG-amide (UNC10245092; where non bolded residues are derived from the flanking regions of the phage displayed peptide) for validation and further characterization. ITC showed that UNC10245092 bound CIB1 with 1:1 stoichiometry and exhibited a Kd = 29.4 ± 9.5 nM (Figure 1D and Table 1). Neither N-acetylation of the N-terminal amine, nor removal of the N-terminal EDGGS motif, which is a spacer sequence between the signal peptide and randomized region, significantly affected the binding affinity of UNC10245092 (Table 1). To determine position-specific contributions to binding energy, we generated alanine and/or glycine-mutants of UNC10245092 by solid phase peptide synthesis (SPPS). As shown in Table 1, mutation of peptide residues F1, W2, L9, and Y10 at the N-terminal and C-terminal ends of UNC10245092 resulted in an approximately 100-fold reduction in binding affinity, indicating these residues contribute the majority of binding energy. The central amino acids, A5 and M6, proved to be moderately important for binding affinity. Alanine substitution at Y3 and glycine substitution at A8 were well tolerated relative to other residues. Alanine substitutions at G4 and K7 significantly reduced peptide solubility such that these mutant peptides could not be evaluated by ITC. Overall, these data suggest that the peptide may adopt an α-helix since placement of the key binding residues is consistent with a helical i, i + 3 and i, i + 4 pattern. Thus, we sought to confirm this structural hypothesis and gain additional insights into the mode of peptide binding by co-crystallography with CIB1.
Figure 1. CIB1 phage display selection and ITC analysis.

A) Schematic of random peptide phage library selection process. B) Phage ELISA binding curve to immobilized CIB1 (black circles) and no coat control (gray squares) for the most prevelant hit peptide with the 10-mer insert sequence of FWYGAMKALY. Data shows an average of ten replicant experiments and line shows fit using four parameter analysis (EC50 = 2.2 x 1010 phage/mL). C) Top peptide sequences from sequence analysis from the fourth round of selection. Approximately 80,000 sequences were obtained from a next generation sequencing run and shown are the number of counts and percent of the total for the top ten hits. D) Example ITC binding isotherm for the synthetic peptide UNC10245092 injection into CIB1 protein (black circles) and buffer control (gray squares).
Table 1.
Binding analysis of synthetic peptides and variants to CIB1 measured by ITC*.
| Description | Peptide Sequence | Kd (nM) | n | ΔH (kcal/mol) | Kd fold |
|---|---|---|---|---|---|
| UNC10245092 | NH3-EDGGSFWYGAMKALYG | 29.4 ± 9.5 | 1.1 ± 0.1 | −14.4 ± 2.2 | --- |
| Acetyl long | Ac-EDGGSFWYGAMKALYG | 45.0 ± 14.4 | 1.1 ± 0.1 | −16.1 ± 0.4 | 1.5 |
| Acetyl short | Ac-FWYGAMKALYG | 52.1 ± 19.2 | 1.1 ± 0.2 | −14.0 ± 1.1 | 1.8 |
| F1A | NH3-EDGGSAWYGAMKALYG | 2650 | 0.94 | −9.8 | 90.1 |
| W2A | NH3-EDGGSFAYGAMKALYG | 3910 | 0.67 | −5.9 | 132.9 |
| Y3A | NH3-EDGGSFWAGAMKALYG | 104 | 1.0 | −16.1 | 3.6 |
| A5G | NH3-EDGGSFWYGGMKALYG | 1990 | 1.0 | −7.7 | 67.8 |
| M6A | NH3-EDGGSFWYGAAKALYG | 1120 | 1.0 | −8.1 | 38.0 |
| A8G | NH3-EDGGSFWYGAMKGLYG | 112 | 1.0 | −13.8 | 3.8 |
| L9A | NH3-EDGGSFWYGAMKAAYG | 2810 | 0.99 | −8.8 | 95.5 |
| Y10A | NH3-EDGGSFWYGAMKALAG | 3690 | 1.1 | −7.1 | 125.5 |
Data shown for UNC10245092. Acetyl long, and acetyl short represent average and standard deviations for five, four, and five replicate experiments, respectively. Data shown for mutant peptides represent either a single ITC titration or the average of two experiments (for F1A and M6A).
UNC10245092 binds in the C-lobe hydrophobic pocket of CIB1
To gain a better sense of how UNC10245092 interacts with CIB1, we solved the crystal structure of the protein:peptide complex (Figure 2). The 2.1 Å resolution structure was solved by molecular replacement in the P21 space group and shows good geometric and crystallographic parameters with no residues in disallowed regions of the Ramachandran plot (see Supporting Information; PDB: 6OD0). Moreover, there were numerous similarities between the CIB1-UNC10245092 complex and prior CIB1 crystal structures (PDB: 1XO5, 1Y1A). As in the previous solid state structures, CIB1 crystallized as a dimer, although it is known to be monomeric in solution as revealed by a number of studies.18, 39 CIB1 in complex with UNC10245092 also exhibits 10 α-helices arrayed in two distinct N- and C-terminal globular domains, each comprised of two helix-loop-helix, EF-hand motifs capable of divalent cation binding18 (Figure 3). Notably, the orientation of the two lobes relative to one another recapitulates that observed in the crystal structure by Gentry et al. (PDB: 1XO5) and the NMR structure by Huang et al. (PDB: 2L4H). Two of the four EF-hand domains, EF-III and EF-IV in the N-lobe, bound divalent calcium ions. An additional calcium ion is found at the surface of the CIB1-UNC10245092 complex but is most likely due to the high calcium concentration (137.5 mM) used in the crystallization conditions and is probably not biologically relevant. Importantly, UNC10245092 is bound to the hydrophobic pocket in the C-lobe.
Figure 2. CIB1-peptide co-crystal structure.

A) Crystal structure of CIB1 (blue) in complex with peptide UNC10245092 (red); PDB: 6OD0. Protein is aligned with native CIB1 showed in green (PDB 1XO5) B) 2Fobs-Fcalc electron density map of peptide UNC10245092 bound to CIB1 (sigma of 1.0). C) Surface view of UNC10245092 peptide bound to CIB1. Surface is colored according to protein electrostatic potential calculated in APBS. D) Details of contacts between UNC10245092 (yellow) and CIB1 (blue). E) Summary of the crystal structure contacts of UNC10245092 peptide residues (numbered 1-10) with CIB1 C-lobe pocket residues (green).
Figure 3. UNC10245351 induces cell death and inhibits AKT and ERK signaling in CIB1-depletion sensitive MDA-468 TNBC cells.
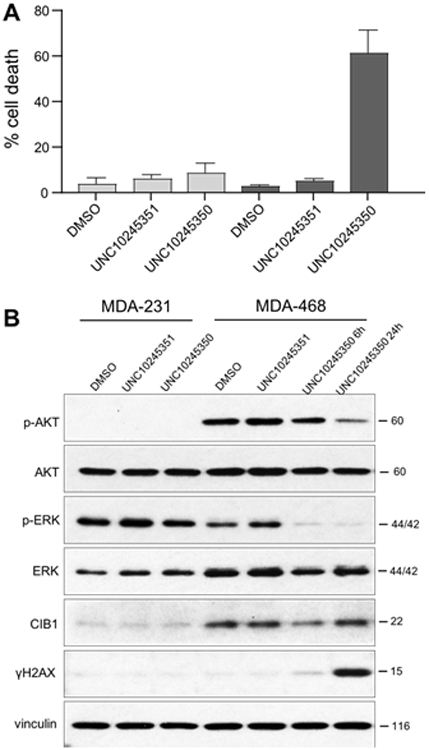
CIB1-depletion sensitive (MDA-468) and insensitive (MDA-231) TNBC cells were treated with DMSO vehicle or 30 μM UNC10245351 or UNC10245350 for 24 h. A) Cell death was assessed by trypan blue exclusion and results are expressed as the mean % of dead cells (i.e., trypan blue positive) from adherent and floating cell populations. Data represent mean +/− SD (n = 4). B) Lysates from MDA-231 and MDA-468 cells were analyzed with the indicated antibodies to determine the activity of AKT, ERK, and γH2AX. Vinculin was used as an additional loading control. MDA-468 cells treated with 30 μM UNC10245350 were also harvested at 6 h to assess early effects of CIB1 inhibition on CIB1-dependent signaling.
The relative positioning of UNC10245092 and auto-inhibitory helix-10 (H10) are perhaps the most salient features of the crystal structure. In previous CIB1 crystal and NMR structures, H10 shields the αIIb-binding site in the C-lobe hydrophobic channel 18, 38, 40. The CIB1-UNC10245092 co-crystal structure shows that the inhibitor peptide displaces H10 completely and binds as a well-formed α-helix in this same hydrophobic channel (Figure 2). Thus, the structure confirms a previously proposed mechanism of action of CIB1, wherein H10 interacts weakly with the C-terminal domain and is readily displaced when CIB1 interacts with the αIIb integrin and other binding partners.26, 40 Interestingly, circular dichroism shows that peptide UNC10245092 is a random coil in solution, but apparently adopts an α-helical confirmation on binding. Again, this is in agreement with prior data that supports a model in which the hydrophobic crevices of many EF-hand containing proteins nucleate helical interactions, for example, the binding of Kv channel-interacting proteins (e.g. KChIP1) to the Kv4.2 channel41 or calcineurin A binding to calcineurin B.42 Additionally, somewhat subtler conformational changes can be seen in H7 and H8 of CIB1 in the CIB1-UNC10245092 complex. Specifically, these two helices appear to have opened up upon peptide binding to provide a deeper, more well-defined binding crevice (Figure 2A). Together, these conformational changes in H7, H8, and H10 of CIB1 facilitate the binding of UNC10245092. Full electron density for UNC10245092 can be seen between residues 5 to 15, revealing that much of the binding surface is made up of hydrophobic interactions. Importantly, the side chains of UNC10245092 residues F1 and W2 and L9 and Y10 are appropriately spaced along the UNC10245092 peptide backbone to serve as large hydrophobic ligand sets for two hydrophobic pockets within the larger cleft. Peptide residues A5 and M6 also make similar interactions along the side of the cleft. Additionally, two key hydrogen bonding interactions, one between the side chains of W2 of UNC10245092 and S160 of CIB1 (3.1 Å) and another between the side chain of Y10 of UNC10245092 and the backbone of K107 (2.7 Å), provide a “charge clamp” to help position the peptide in the hydrophobic channel (Figure 2D and 2E). Interestingly, the side chain of S160 is largely buried in the structure of native CIB1 (PDB: 1XO5, 1Y1A) and only becomes exposed to UNC10245092 hydrogen bonding because of the degree to which H10 is displaced from the binding cleft. Gratifyingly, all of these peptide-protein interactions are in good agreement with the alanine scanning data originally collected after the phage peptide library screen and validate both the structure and the ITC results. Thus, UNC10245092 and the CIB1-UNC10245092 co-crystal structure provide potential starting points for small molecule inhibitor development.
Rational design and screening of peptide analogs using a TR-FRET assay
To further probe the structure-activity relationships of UNC10245092 analogs, we established a robust time-resolved fluorescence energy transfer (TR-FRET) assay. We anticipated that this assay would prove faster and easier than ITC for evaluating analogs. To this end, UNC10245092 was synthesized with an N-terminal cysteine to facilitate conjugation with maleimide AlexaFluor 647 (see Supporting Information); this fluorophore could act to efficiently accept FRET from biotinylated CIB1 bound with europium-labeled streptavidin. Competition assays with unlabeled-UNC10245092 showed that it exhibits an IC50 of 46 ± 13 nM (Table 2). We validated this assay in a 384-well format, which demonstrated good dynamic range and a Z’-factor >0.9, suitable for high-throughput determination of IC50s of additional peptide analogs.
Table 2.
Inhibitory activity of UNC10245092 analogs as determined by TR-FRET assay.
| Description | Peptide Sequence | Description | Kd (nM) | Rep |
|---|---|---|---|---|
| UNC10245092 | NH3-EDGGSFWYGAMKALYG | WT | 46 ± 13 | 17 |
| UNC10245121 | FWYGAMKALYG | cyclic | 32 ± 10 | 3 |
| UNC10245106 | NH3-EDGGSFWYGAibMKALYG | A5Aib | 17 ± 8 | 6 |
| UNC10245109 | NH3-EDGGSFWYRAMKALYG | G4R | 16 ± 7 | 8 |
| UNC10245107 | NH3-EDGGSFWYGAMKAibLYG | A8Aib | 50 ± 2 | 2 |
| UNC10245098 | GDAbuLPHKLEDGGSFWYGAMKALYG | mauro | 33 ± 8 | 6 |
| UNC10245350 | NH3-GRKKRRQRRRPQEDGGSFWYGAMKALYG | HIV TAT (+) | 192 ± 17 | 3 |
| UNC10245351 | NH3-GRKKRRQRRRPQEDGGSFAYGAMKALAG | HIV TAT (−) | >10000 | 1 |
| UNC10245104 | NH3-RQIKIWFQNRREDGGSFWYGAMKALYG | penetratin | >10000 | 2 |
A strong body of work has demonstrated the benefits of peptide stapling to stabilize binding conformations and improve peptide affinity and activity.43-45 With a TR-FRET assay in hand, we next set out to synthesize and test a small series of UNC10245092 analogs with modifications designed to stabilize a helical conformation similar to the one in the CIB1-UNC10245092 co-crystal structure. In the first analog, UNC10245121, a bridge was installed between two solvent exposed residues, G4 and K7, which do not appear to make significant contacts with CIB1 in the crystal structure. In this case, G4 was changed to a glutamic acid, which could be cyclized chemically (see Supporting Information) onto the K7 side-chain amine to yield an amide bond “staple.” A second design, UNC10245106, replaced A5 of UNC10245092 with α-aminoisobutyric acid (Aib), which is a non-proteinogenic, but naturally-occurring helix-inducing amino acid. In the third and final design, we sought to take advantage of a strategically positioned hydrogen bond acceptor to stabilize the helix in solution. Thus, we prepared UNC10245109, exhibiting a G4R substitution which we anticipated might form a hydrogen bond with the downstream serine residue (S0, Figure 2E) in a pre-formed helical peptide. Results from the TR-FRET assay show that the cyclic peptide, UNC10245121, exhibits an IC50 similar to that of the parent UNC10245092 (32 nM and 46 nM, respectively, Table 2), whereas both the Aib-containing UNC10245106 and G9R UNC10245109 had only slightly improved IC50s (17 nM and 16 nM, respectively).
To better understand the structural basis for the change in affinity or lack thereof, we sought to co-crystallize these analogs with CIB1, in manner similar to UNC10245092. Unfortunately, while we could obtain a structure for the complex with UNC10245109 (see Supporting Information, Figure S3), neither UNC10245106 nor cyclic UNC10245121 yielded diffractable crystals. The UNC10245109 co-complex reveals that the G4R peptide adopts a similar binding mode to UNC10245092 and that the G4R mutation does in fact seem to stabilize the peptide by an extra hydrogen bond between UNC10245109 residues R9 and S0 (2.8 Å; PDB 6OCX). This extra interaction may explain why the additional residues 2-5 are visible in this structure as opposed to the UNC10245092 structure. Upon observation of the crystal structures, one possibility for the binding mode of UNC10245106 may be that the A5Aib could contribute additional hydrophobic interactions with the cleft formed by F115, L131, and L135, which may combine with added helicity to improved its activity. Taken together, these results indicate that little is gained in terms of affinity by strictly enforcing the pre-adopted helix as in cyclic UNC10245121, but that some amount of ‘breathable’ or non-covalent helicity may facilitate binding of UNC10245109 and UNC10245106 to CIB1.
Design of cell-penetrating peptide fusions for CIB1 inhibitor peptides
To facilitate the testing of UNC10245092 biological activity in cells, we synthesized a series of peptides with a selection of N-terminal cell-penetrating peptide sequences (CPPs). We chose to place the CPPs at the N-terminus of UNC10245092, since this would allow a divergent SPPS synthetic strategy. Moreover, the crystal structure indicates that the N-terminus could readily accommodate the added steric bulk and thereby minimize effects on the binding interaction. Three different CPP-conjugates of UNC10245092 were synthesized: 1) an HIV-Tat conjugate (UNC10245350), 2) a short penetratin conjugate (UNC10245104), and 3) a conjugate with a newly identified small cell penetrating peptide sequence derived from the scorpion toxin, maurocalcine (UNC10245098). The latter, short-maurocalcine sequence was developed by Poillot et al.46 and contains 2-aminobutyric acid (Abu) in place of an internal cysteine present in the native peptide. While UNC10245908 binds with similar affinity to the original phage peptide (IC50 = 33 nM vs. 46 nM, Table 2), the addition of the HIV-Tat sequence slightly decreased the binding affinity (IC50 = 192 nM, Table 2). In contrast, the penetratin conjugate, UNC10245104, did not appear to bind. The CIB1-UNC10245092 co-crystal structure shows that there is an additional hydrophobic cleft near where S0 of UNC10245092 is positioned that could accommodate additional amino acid residues. As a potential negative control, we tested a mutant peptide that likely disrupts the charge-clamp interaction (N-terminal HIV Tat conjugate W2A, Y10A; UNC10245351) and confirmed that binding was essentially ablated by alanine subsititution of these residues (Table 1) . These results confirm that UNC10245092 can accommodate N-terminal CPP tags, allowing us to or develop reagents and controls for testing CIB1 disruption in cellulo.
Cellular activity of UNC10245350 conjugated to a cell-penetrating peptide
Having confirmed that several CPP-conjugates of UNC10245092 could still bind CIB1, we next sought to validate the cellular impact of molecular blockade at the C-terminal pocket of CIB1. A preliminary screen for cytotoxicity in CIB1 depletion-sensitive MDA-MB-468 cells showed minimal effects of UNC10245098 (maurocalcine) and UNC10245104 (penetratin), but a moderate effect of UNC10245350 (HIV TAT). Therefore, we chose the N-terminal HIV Tat conjugate, UNC10245350, and a double alanine mutant, UNC10245351, as positive and negative controls, respectively, for further cell-based assays. CIB1 depletion-sensitive MDA-468 and -insensitive MDA-231 TNBC cell lines were separately treated with each peptide and cell death was assessed using trypan blue exclusion. Because UNC10245350 is a relatively large, linear peptide we anticipated that it would be active in cells well above its measured in vitro IC50. Consistent with this prediction, 30 μM UNC10245350 induced significant and selective cell death in CIB1 depletion-sensitive MDA-468, but not–insensitive MDA-231 cells (Figure 3A). In contrast, the double mutant control peptide, UNC10245351 had minimal effect on cell death, suggesting that UNC10245350 activity is dependent on its interaction with CIB1. To further confirm the biological activity of UNC10245350, we examined the status of AKT and ERK phosphorylation, as well as the induction of the DNA damage marker γH2AX as previously reported in CIB1 depleted MDA-468 cells.9, 17 Interestingly, UNC1025350 but not the control peptide UNC10245351, rapidly inhibited ERK activity by 6h, with inhibition of AKT activity, and induction of the DNA damage response marker γH2AX occurring by 24h specifically in MDA-468 cells (Figure. 3B). Thus, the CIB1 peptide inhibitor UNC1025350 closely replicates the effects of CIB1 depletion on MDA-468 TNBC cell death and inhibition of CIB1-dependent signaling pathways.9, 17 Taken together, these results confirm that UNC10245350, which binds the CIB1 C-terminal pocket in vitro, selectively induces cell death in CIB1 depletion-sensitive MDA-468 cells and promotes phospho-proteome alterations consistent with CIB1 disruption.
Discussion
To date, cellular functions and cell signaling roles of CIB1 have been investigated using biochemical and genetic methods. Importantly, strong evidence has accumulated that CIB1 is a viable target for intervention against TNBC. This is largely attributed to its ability to regulate AKT and ERK oncogenic pathways in transformed as opposed to non-transformed cells. This effect has been repeatedly confirmed by RNA interference studies. In vitro studies indicate that CIB1 interacts with a number of protein partners that include the cytoplasmic portion of integrin α-chains,22 kinases ASK1,47 PAK1,14 SPHK1,23 Src,48 and transcription factor PAX3,49 among others. While the CIB1 C-lobe hydrophobic pocket has been implicated in some of these interactions, it was not entirely clear whether this same pocket represents the prime and tractable biochemical target in CIB1 cancer biology. The structure and activity of UNC10245092 now provide a model for testing the role of this CIB1 pocket in pharmacological interventions.
CIB1 appears to present a conserved interface to many of its binding partners, via key residues within the C-lobe hydrophobic pocket.22 UNC10245092 takes advantage of several of these common interacting residues, but also interacts with additional residues to attain its pronounced affinity. In total, UNC10245092 makes varying degrees of contact with 17 different residues in the hydrophobic pocket (occupied by C-terminal H10 in apo structure PDB: 1XO5), 8 of which are shared with the integrins and other CIB1 binding partners (Figure 2). For example, CIB1 residues Ile-Phe115, Phe117, Leu-Ile153, and Phe173 were previously reported to be important for αIIb binding and are also found to interact with UNC10245092.26 Additionally, a combination of docking, co-immunoprecipitation, and binding assays have been used to show that these same residues are important for binding additional α-integrin cytoplasmic tails with Kds in the range of 0.5-20 μM.22 UNC10245092 mimics the same hydrophobic armament of these α-integrin cytoplasmic tail peptides but supplements it with the W2-Y10 charge clamp and additional contacts, giving it a steep gain in affinity. Since K107 and S160, the CIB1 counterparts to this binding motif, had not previously been implicated in the native interactions in substantive ways, the current binding mode may have been challenging to predict. Indeed, UNC10245092 is slightly shifted relative to the binding pose of H10 (Figure 2A), facilitating its interaction with these new residues. Moreover, the newly exposed S160 pocket may prove advantageous in future drug discovery efforts based on fragments of UNC10245092 or other peptide binding partners.
The CIB1-UNC10245092 structure also suggests that this pattern of interaction may allow specificity within the broader EF-hand domain family. This potential is evident in an alignment of the 67 sequences in the UniProt human reference genome annotated as EF-hand domain containing proteins. We used the CONSURF web server to visualize the residue-by-residue evolutionary conservation and map it onto the CIB1 structure (PDBID: 1XO5, Figure 4). This map shows that there is a considerable level of conservation in the C-terminal portion of the protein. However, the in the region which accommodates L9 and Y10 of UNC10245092 exhibits somewhat lower homology coverage. Moreover, the loop where S160 sits appears quite variable in the EF-hand proteins sampled. Thus, the bidentate charge clamp exploited by UNC10245092 may be a promising strategy for selective targeting of CIB1 relative to the other EF-hand domain proteins.
Figure 4. Structural analysis of CIB1 and homologs.

A) ConSurf analysis for CIB1 presented as surface model. The amino-acids are colored by their conservation grades using the color-coding bar, with turquoise-through-maroon indicating variable-through-conserved. The figure reveals that the peptide binding site is highly conserved. The run was carried out using PDB code 1XO5, with C-terminal helix (residues S180-V190) omitted from the model, and the figure was generated using the PyMol script output by ConSurf.50 B) Alignment of C-lobe pockets for top six CIB1 homologs based on similarity (similarity scores at left). Highlighted residues are involved in key contacts with UNC10245092 only (red), αIIb integrin tails only (blue), or both (grey).
Conclusion
Previous studies have suggested that CIB1 is a novel target for discovery of new therapeutics for TNBC. There is an immediate need for validated inhibitors, i.e. chemical probes, to further explore this possibility. Due to the rather large binding site in the hydrophobic pocket of CIB1 which might pose a challenge for small molecules, we chose to pursue a peptide inhibitor discovery effort via phage display. As shown, we rapidly identified several CIB1 inhibitor peptides and characterized them biochemically. Importantly, we report the first co-crystal structure of CIB1 in complex with an inhibitor peptide. This structural information has provided the means to design additional peptide analogs with constrained conformations and unnatural amino acids. Overall, this work provides essential information to help to elucidate the mechanisms of action of CIB1 and supports the further development of chemical probes and therapeutic candidates. Additionally, the in cellulo activity of the CPP-linked UNC10245350 may make UNC10245092 a good starting point for further inhibitor development.
Supplementary Material
Funding
We thank the UNC Lineberger Comprehensive Cancer Center for general support of facilities and resources. A.C.P. and K.H.P were supported in part by grants from the NIDDK (NIH 1R01DK103746 and NIH 1R01DK101645). A.A.B., J.W.B., and V.A.H. were supported in whole or in part by a grant from the NIGMS (NIH 1R35GM125005). L.V.P and T.M.L are supported by grants from the NIH (NIH 1R01GM133107) and North Carolina Biotech Center (NCBC) 2017-BIG-6515. A.S.G. is supported by Sao Paulo Research Foundation -FAPESP (2016/19712-9).
Footnotes
Conflict of Interest
L.V.P. and T.M.L. are co-founders of Reveris Therapeutics, LLC.
ASSOCIATED CONTENT
Materials, experimental methods, and supplementary figures can be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org. Coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 6OD0 (UNC10245092 bound) and 6OCX (UNC10245109 bound).
REFERENCES
- 1.Bray F; Ferlay J; Soerjomataram I; Siegel RL; Torre LA; Jemal A, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018, 68 (6), 394–424. [DOI] [PubMed] [Google Scholar]
- 2.Blows FM; Driver KE; Schmidt MK; Broeks A; van Leeuwen FE; Wesseling J; Cheang MC; Gelmon K; Nielsen TO; Blomqvist C; Heikkilä P; Heikkinen T; Nevanlinna H; Akslen LA; Bégin LR; Foulkes WD; Couch FJ; Wang X; Cafourek V; Olson JE; Baglietto L; Giles GG; Severi G; McLean CA; Southey MC; Rakha E; Green AR; Ellis IO; Sherman ME; Lissowska J; Anderson WF; Cox A; Cross SS; Reed MW; Provenzano E; Dawson SJ; Dunning AM; Humphreys M; Easton DF; García-Closas M; Caldas C; Pharoah PD; Huntsman D, Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med 2010, 7 (5), e1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris GJ; Naidu S; Topham AK; Guiles F; Xu Y; McCue P; Schwartz GF; Park PK; Rosenberg AL; Brill K; Mitchell EP, Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute's Surveillance, Epidemiology, and End Results database. Cancer 2007, 110 (4), 876–84. [DOI] [PubMed] [Google Scholar]
- 4.Lee A; Djamgoz MBA, Triple negative breast cancer: Emerging therapeutic modalities and novel combination therapies. Cancer Treat Rev 2018, 62, 110–122. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann BD; Bauer JA; Chen X; Sanders ME; Chakravarthy AB; Shyr Y; Pietenpol JA, Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011, 121 (7), 2750–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dent R; Trudeau M; Pritchard KI; Hanna WM; Kahn HK; Sawka CA; Lickley LA; Rawlinson E; Sun P; Narod SA, Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007, 13 (15 Pt 1), 4429–34. [DOI] [PubMed] [Google Scholar]
- 7.Mirzoeva OK; Das D; Heiser LM; Bhattacharya S; Siwak D; Gendelman R; Bayani N; Wang NJ; Neve RM; Guan Y; Hu Z; Knight Z; Feiler HS; Gascard P; Parvin B; Spellman PT; Shokat KM; Wyrobek AJ; Bissell MJ; McCormick F; Kuo WL; Mills GB; Gray JW; Korn WM, Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res 2009, 69 (2), 565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoeflich KP; O'Brien C; Boyd Z; Cavet G; Guerrero S; Jung K; Januario T; Savage H; Punnoose E; Truong T; Zhou W; Berry L; Murray L; Amler L; Belvin M; Friedman LS; Lackner MR, In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res 2009, 15 (14), 4649–64. [DOI] [PubMed] [Google Scholar]
- 9.Leisner TM; Moran C; Holly SP; Parise LV, CIB1 prevents nuclear GAPDH accumulation and non-apoptotic tumor cell death via AKT and ERK signaling. Oncogene 2013, 32 (34), 4017–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gordon V; Banerji S, Molecular pathways: PI3K pathway targets in triple-negative breast cancers. Clin Cancer Res 2013, 19 (14), 3738–44. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu T; Tolcher AW; Papadopoulos KP; Beeram M; Rasco DW; Smith LS; Gunn S; Smetzer L; Mays TA; Kaiser B; Wick MJ; Alvarez C; Cavazos A; Mangold GL; Patnaik A, The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res 2012, 18 (8), 2316–25. [DOI] [PubMed] [Google Scholar]
- 12.De Luca A; Maiello MR; D'Alessio A; Pergameno M; Normanno N, The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets 2012, 16 Suppl 2, S17–27. [DOI] [PubMed] [Google Scholar]
- 13.Rodon J; Dienstmann R; Serra V; Tabernero J, Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 2013, 10 (3), 143–53. [DOI] [PubMed] [Google Scholar]
- 14.Leisner TM; Liu M; Jaffer ZM; Chernoff J; Parise LV, Essential role of CIB1 in regulating PAK1 activation and cell migration. J Cell Biol 2005, 170 (3), 465–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan W; Leisner TM; McFadden AW; Wang Z; Larson MK; Clark S; Boudignon-Proudhon C; Lam SC; Parise LV, CIB1 is an endogenous inhibitor of agonist-induced integrin alphaIIbbeta3 activation. J Cell Biol 2006, 172 (2), 169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leisner TM; Freeman TC; Black JL; Parise LV, CIB1: a small protein with big ambitions. FASEB J 2016, 30 (8), 2640–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Black JL; Harrell JC; Leisner TM; Fellmeth MJ; George SD; Reinhold D; Baker NM; Jones CD; Der CJ; Perou CM; Parise LV, CIB1 depletion impairs cell survival and tumor growth in triple-negative breast cancer. Breast Cancer Res Treat 2015, 152 (2), 337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gentry HR; Singer AU; Betts L; Yang C; Ferrara JD; Sondek J; Parise LV, Structural and biochemical characterization of CIB1 delineates a new family of EF-hand-containing proteins. J Biol Chem 2005, 280 (9), 8407–15. [DOI] [PubMed] [Google Scholar]
- 19.Yamniuk AP; Nguyen LT; Hoang TT; Vogel HJ, Metal ion binding properties and conformational states of calcium- and integrin-binding protein. Biochemistry 2004, 43 (9), 2558–68. [DOI] [PubMed] [Google Scholar]
- 20.Yamniuk AP; Vogel HJ, Calcium- and magnesium-dependent interactions between calcium- and integrin-binding protein and the integrin alphaIIb cytoplasmic domain. Protein Sci 2005, 14 (6), 1429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naik UP; Patel PM; Parise LV, Identification of a novel calcium-binding protein that interacts with the integrin alphaIIb cytoplasmic domain. J Biol Chem 1997, 272 (8), 4651–4. [DOI] [PubMed] [Google Scholar]
- 22.Freeman TC; Black JL; Bray HG; Dagliyan O; Wu YI; Tripathy A; Dokholyan NV; Leisner TM; Parise LV, Identification of novel integrin binding partners for calcium and integrin binding protein 1 (CIB1): structural and thermodynamic basis of CIB1 promiscuity. Biochemistry 2013, 52 (40), 7082–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarman KE; Moretti PA; Zebol JR; Pitson SM, Translocation of sphingosine kinase 1 to the plasma membrane is mediated by calcium- and integrin-binding protein 1. J Biol Chem 2010, 285 (1), 483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barry WT; Boudignon-Proudhon C; Shock DD; McFadden A; Weiss JM; Sondek J; Parise LV, Molecular basis of CIB binding to the integrin alpha IIb cytoplasmic domain. J Biol Chem 2002, 277 (32), 28877–83. [DOI] [PubMed] [Google Scholar]
- 25.Hwang PM; Vogel HJ, Structures of the platelet calcium- and integrin-binding protein and the alphaIIb-integrin cytoplasmic domain suggest a mechanism for calcium-regulated recognition; homology modelling and NMR studies. J Mol Recognit 2000, 13 (2), 83–92. [DOI] [PubMed] [Google Scholar]
- 26.Yamniuk AP; Ishida H; Vogel HJ, The interaction between calcium- and integrin-binding protein 1 and the alphaIIb integrin cytoplasmic domain involves a novel C-terminal displacement mechanism. J Biol Chem 2006, 281 (36), 26455–64. [DOI] [PubMed] [Google Scholar]
- 27.Chung AH; Leisner TM; Dardis GJ; Bivins MM; Keller AL; Parise LV, CIB1 depletion with docetaxel or TRAIL enhances triple-negative breast cancer cell death. Cancer Cell Int 2019, 19, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng SJ; Pearce KH; Dixon EP; Hartley KA; Stanley TB; Lobe DC; Garvey EP; Kost TA; Petty RL; Rocque WJ; Alexander KA; Underwood MR, Identification of peptides that inhibit the DNA binding, trans-activator, and DNA replication functions of the human papillomavirus type 11 E2 protein. J Virol 2004, 78 (5), 2637–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iannone MA; Simmons CA; Kadwell SH; Svoboda DL; Vanderwall DE; Deng SJ; Consler TG; Shearin J; Gray JG; Pearce KH, Correlation between in vitro peptide binding profiles and cellular activities for estrogen receptor-modulating compounds. Mol Endocrinol 2004, 18 (5), 1064–81. [DOI] [PubMed] [Google Scholar]
- 30.Pearce KH; Potts BJ; Presta LG; Bald LN; Fendly BM; Wells JA, Mutational analysis of thrombopoietin for identification of receptor and neutralizing antibody sites. J Biol Chem 1997, 272 (33), 20595–602. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski Z; Minor W, Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–26. [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W, XDS. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winter G, xia2: an expert system for macromolecular crystallography data reduction. Journal of Applied Crystallography 2009, 43 (1), 186–190. [Google Scholar]
- 34.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. J Appl Crystallogr. 2007, 40 (Pt4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smart OS; Womack TO; Flensburg C; Keller P; Paciorek W; Sharff A; Vonrhein C; Bricogne G, Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 2012, 68 (Pt 4), 368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallographica Section D 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang H; Vogel HJ, Structural basis for the activation of platelet integrin αIIbβ3 by calcium- and integrin-binding protein 1. J Am Chem Soc 2012, 134 (8), 3864–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sobczak A; Blazejczyk M; Piszczek G; Zhao G; Kuznicki J; Wojda U, Calcium-binding calmyrin forms stable covalent dimers in vitro, but in vivo is found in monomeric form. Acta Biochim Pol 2005, 52 (2), 469–76. [PubMed] [Google Scholar]
- 40.Huang H; Ishida H; Yamniuk AP; Vogel HJ, Solution structures of Ca2+-CIB1 and Mg2+-CIB1 and their interactions with the platelet integrin alphaIIb cytoplasmic domain. J Biol Chem 2011, 286 (19), 17181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou W; Qian Y; Kunjilwar K; Pfaffinger PJ; Choe S, Structural insights into the functional interaction of KChIP1 with Shal-type K(+) channels. Neuron 2004, 41 (4), 573–86. [DOI] [PubMed] [Google Scholar]
- 42.Griffith JP; Kim JL; Kim EE; Sintchak MD; Thomson JA; Fitzgibbon MJ; Fleming MA; Caron PR; Hsiao K; Navia MA, X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 1995, 82 (3), 507–22. [DOI] [PubMed] [Google Scholar]
- 43.Ali AM; Atmaj J; Van Oosterwijk N; Groves MR; Dömling A, Stapled Peptides Inhibitors: A New Window for Target Drug Discovery. Comput Struct Biotechnol J 2019, 17, 263–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moiola M; Memeo MG; Quadrelli P, Stapled Peptides-A Useful Improvement for Peptide-Based Drugs. Molecules 2019, 24 (20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verdine GL; Hilinski GJ, Stapled peptides for intracellular drug targets. Methods Enzymol 2012, 503, 3–33. [DOI] [PubMed] [Google Scholar]
- 46.Poillot C; Bichraoui H; Tisseyre C; Bahemberae E; Andreotti N; Sabatier JM; Ronjat M; De Waard M, Small efficient cell-penetrating peptides derived from scorpion toxin maurocalcine. J Biol Chem 2012, 287 (21), 17331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoon KW; Cho JH; Lee JK; Kang YH; Chae JS; Kim YM; Kim J; Kim EK; Kim SE; Baik JH; Naik UP; Cho SG; Choi EJ, CIB1 functions as a Ca(2+)-sensitive modulator of stress-induced signaling by targeting ASK1. Proc Natl Acad Sci U S A 2009, 106 (41), 17389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naik MU; Pham NT; Beebe K; Dai W; Naik UP, Calcium-dependent inhibition of polo-like kinase 3 activity by CIB1 in breast cancer cells. Int J Cancer 2011, 128 (3), 587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hollenbach AD; McPherson CJ; Lagutina I; Grosveld G, The EF-hand calcium-binding protein calmyrin inhibits the transcriptional and DNA-binding activity of Pax3. Biochim Biophys Acta 2002, 1574 (3), 321–8. [DOI] [PubMed] [Google Scholar]
- 50.Ashkenazy H; Abadi S; Martz E; Chay O; Mayrose I; Pupko T; Ben-Tal N, ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 2016, 44 (W1), W344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.