

SUMMARY
DNA replication stress can stall replication forks leading to genome instability. DNA damage tolerance pathways assist fork progression, promoting replication fork reversal, translesion DNA synthesis (TLS) or repriming. In the absence of the fork remodeler HLTF, forks fail to slow following replication stress, but underlying mechanisms and cellular consequences remain elusive. Here, we demonstrate that HLTF-deficient cells fail to undergo fork reversal in vivo and rely on PRIMPOL for repriming, unrestrained replication and S-phase progression upon limiting nucleotide levels. By contrast, in an HLTF-HIRAN mutant, unrestrained replication relies on the TLS protein REV1. Importantly, HLTF-deficient cells also exhibit reduced DSB formation and increased survival upon replication stress. Our findings suggest that HLTF promotes fork remodeling, preventing other mechanisms of replication stress tolerance in cancer cells. This remarkable plasticity of the replication fork may determine the outcome of replication stress in terms of genome integrity, tumorigenesis and response to chemotherapy.
Graphical Abstract
eTOC Blurb
Under replication stress, cells deficient in the fork remodeler HLTF fail to slow DNA replication. Here, Bai et al. report that when HLTF is disrupted, replication is completed by alternative, PRIMPOL- or REV1-dependent mechanisms. Both replication modes are potentially mutagenic and lead to replication stress resistance.
INTRODUCTION
A variety of DNA damaging agents, protein-DNA complexes and DNA secondary structures can threaten genome stability by slowing replication fork progression, a condition defined as replication stress (Zeman and Cimprich, 2014). Nucleotide depletion induced by oncogene activation or hydroxyurea (HU) treatment also causes replication stress (Kotsantis et al., 2018). Cells initiate a complex response to replication fork stalling that allows them to maintain fork stability and ultimately complete DNA replication (Cortez, 2019). This response is tightly regulated and coordinated by the checkpoint kinase ATR, which is activated by ssDNA-containing DNA structures that form when replication forks stall (Saldivar et al., 2017). Unresolved or persistent stalled forks are vulnerable structures susceptible to nucleolytic processing and double-strand break (DSB) formation, and ultimately cause genome instability (Cortez, 2019; Pasero and Vindigni, 2017).
DNA damage tolerance (DDT) pathways are another crucial response to replication stress (Branzei and Szakal, 2017). Replication fork reversal is one form of DDT proposed to protect fork integrity during replication stress (Neelsen and Lopes, 2015). By reannealing the nascent DNA strands on each sister chromatid to form a fourth regressed arm, fork reversal actively converts the three-armed fork into a Holliday junction (HJ)-like structure. Different kinds of genotoxic stress can lead to helicase-polymerase uncoupling and ssDNA accumulation, but fork reversal restrains replication fork progression and is thought to prevent ssDNA accumulation at the fork (Neelsen and Lopes, 2015; Ray Chaudhuri et al., 2012; Zellweger et al., 2015). Fork reversal may also promote template switching and error-free lesion bypass (Cortez, 2019; Neelsen and Lopes, 2015; Saugar et al., 2014). Thus, it is proposed to protect and resolve stalled replication forks.
Two other forms of DDT are also possible in mammalian cells. Specialized translesion synthesis (TLS) polymerases can directly bypass DNA lesions in order to resume DNA synthesis, preventing persistent replication fork stalling and ultimately DSB formation (Sale, 2013; Saugar et al., 2014). Alternatively, repriming can restart DNA synthesis downstream of a stalled polymerase. In higher eukaryotes, a central effector of this process is the primase-polymerase, PRIMPOL, which can utilize its DNA primase activity to reprime DNA synthesis downstream of the lesion, leaving a ssDNA gap behind the fork (Bianchi et al., 2013; Garcia-Gomez et al., 2013; Keen et al., 2014; Kobayashi et al., 2016; Mouron et al., 2013; Pilzecker et al., 2016; Schiavone et al., 2016; Svikovic et al., 2019; Wan et al., 2013). After PRIMPOL extends the DNA primer by a few nucleotides using its polymerase activity, the replicative polymerase can continue nascent DNA synthesis. How mammalian cells choose between the alternative forms of DDT - fork reversal, TLS and repriming - is not clear, although several proteins have been implicated in regulating these processes.
PCNA is a central regulator of DDT. In yeast and higher eukaryotes, PCNA monoubiquitination promotes TLS polymerase recruitment and lesion bypass in a potentially error-prone manner (Hoege et al., 2002; Sale, 2013). PCNA polyubiquitination, mediated by the E3 ligase Rad5 in yeast, promotes template switching, which uses the sister chromatid as a template for error-free lesion bypass (Branzei and Szakal, 2017; Hoege et al., 2002). In mammalian cells, the E3 ubiquitin ligases, HLTF and SHPRH contribute to PCNA polyubiquitination, although polyubiquitination is still observed upon loss of both proteins (Saugar et al., 2014; Unk et al., 2010). This implies that additional factors are likely involved and that DDT processes are more complex in mammalian cells.
In higher eukaryotes, multiple proteins participate in fork remodeling via replication fork reversal, although the distinct contributions of each are not known (Neelsen and Lopes, 2015). Three regulators of the process, SMARCAL1, ZRANB3 and HLTF, are members of the SWI/SNF2 family. Each of these remodelers is capable of fork reversal in vitro (Achar et al., 2011; Bansbach et al., 2009; Betous et al., 2012; Blastyak et al., 2010; Ciccia et al., 2009; Ciccia et al., 2012; Couch et al., 2013; Yuan et al., 2012; Yusufzai et al., 2009), and each is recruited to the replication fork through distinct interactions (Poole and Cortez, 2017). Electron microscopy (EM) studies also indicate SMARCAL1 and ZRANB3 are required for fork reversal in vivo (Kolinjivadi et al., 2017; Vujanovic et al., 2017; Zellweger et al., 2015), but whether HLTF is needed in vivo has not been addressed.
HLTF, like its yeast orthologue Rad5, contains an ATPase domain and an E3 ubiquitin ligase domain (Unk et al., 2010). Both proteins also contain a HIRAN domain, which binds specifically to 3’-OH ssDNA ends. HLTF’s ATPase and 3’-ssDNA binding activity are needed for fork reversal in vitro (Achar et al., 2015; Chavez et al., 2018; Hishiki et al., 2015; Kile et al., 2015). In vivo, HLTF slows replication fork progression upon nucleotide depletion, and in its absence forks fail to slow and progress unrestrained. As the HIRAN domain is needed to restrain replication fork progression, fork reversal and fork slowing may be linked (Kile et al., 2015). Indeed, the loss of two other proteins involved in fork reversal, RAD51 and ZRANB3, also leads to unrestrained fork progression upon replication stress (Vujanovic et al., 2017; Zellweger et al., 2015). How unrestrained replication fork progression is sustained in the absence of HLTF is unknown.
Increased endogenous replication stress is a hallmark of cancer cells and can be induced by nucleotide depletion or conditions that perturb DNA replication, including oncogene activation and deregulation of origin firing (Kotsantis et al., 2018). Interestingly, HLTF is frequently silenced in colorectal cancer (Moinova et al., 2002), and its deficiency accelerates tumorigenesis in a mouse model (Sandhu et al., 2012). This suggests HLTF is a tumor suppressor (Dhont et al., 2016). Given HLTF’s ability to restrain DNA replication and its potential role in cancer, we sought to understand how HLTF affects the replication stress response and the role of HLTF-mediated fork remodeling in this process.
Here, we report that HLTF loss limits DSB formation and promotes increased resistance to replication stress, allowing cells to continue DNA replication using PRIMPOL. Surprisingly, a specific defect in HLTF’s HIRAN domain also leads to unrestrained DNA replication and replication stress resistance, but in this case via REV1-mediated TLS. Our results suggest that HLTF’s activities are central to regulate replication fork reversal and to prevent alternative mechanisms of stress-resistant DNA replication that promote DNA synthesis, S phase progression and cellular resistance to replication stress. They also demonstrate the remarkable plasticity of the replication fork in tolerating replication stress when fork reversal is disrupted. We therefore propose that HLTF loss may promote tumorigenesis by unleashing alternative, and potentially more mutagenic, modes of replication stress tolerance.
RESULTS
HLTF promotes fork reversal and restrains fork progression in vivo
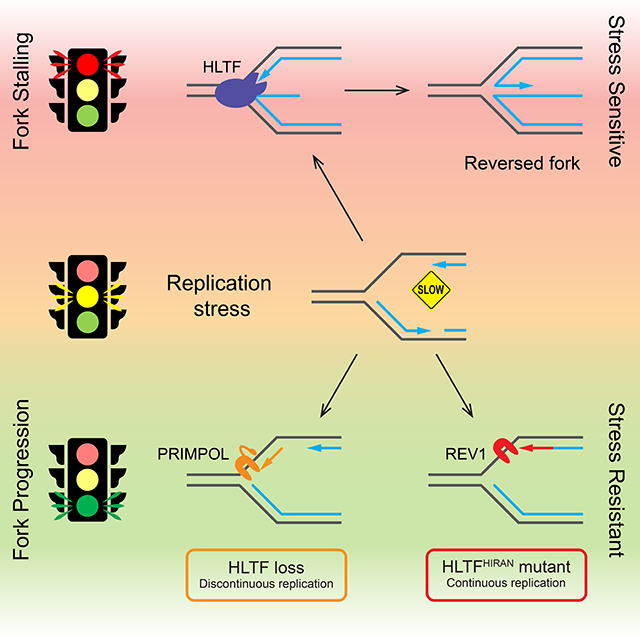
HLTF promotes fork reversal in vitro on model replication fork structures (Achar et al., 2011; Blastyak et al., 2010). To test whether HTLF can also promote fork reversal in vivo, we used electron microscopy (EM) to monitor fork reversal in HLTF-KO cell lines generated using CRISPR targeting (Sup. Fig. 1A) (Kile et al., 2015). After exposing control and HLTF-KO cells to a low dose of HU (50 μM), we isolated replication intermediates and analyzed their structure using in vivo psoralen crosslinking and EM. Reversed fork structures represented approximately 23% of the replication intermediates we observed in HU-treated wild-type (WT) cells (Fig. 1A, B), consistent with the number of reversed forks observed following other types of treatment (Zellweger et al., 2015). By contrast, both HLTF-KO cell lines exhibited a significant 2–3 fold reduction in reversed fork frequency. This finding demonstrates that HLTF is a bona-fide fork reversal protein in human cells.
Figure 1. HLTF promotes fork reversal in vivo and limits PRIMPOL-mediated unrestrained fork progression.
(A) Electron micrographs of representative replication intermediates. Black arrows indicate fork junctions. Scale bars: (main images) 500nm; (insets) 20nm.
(B) Frequency of reversed replication forks in WT or HLTF-KO U2OS cells treated with 50 μM HU for 1h. Means ± SEM (n≥3). ***, p<0.001; ****, p<0.0001, by one-way ANOVA then Dunnett’s test.
(C) Experimental setup for replication fork progression assay. Representative fields of DNA fibers are shown. Scale bar: 15μm.
(D) Dot plot and median of CldU tract lengths for 3 independent experiments (n=3). ns, not significant; ****, p < 0.0001, by Kruskal-Wallis test.
(E and F) Dot plot and median of CldU tract lengths after control or siPRIMPOL-3 knockdown in mock or HU-treated (50 μM) cells (E) and treated with or without S1 nuclease (F). Open circle, no treatment; closed circle, HU treatment (n=3). ns, not significant; *, p < 0.05; ****, p < 0.0001, by Mann-Whitney test. See also Supplemental Figure 1.
A lack of fork reversal in vivo is associated with unrestrained fork progression (Vujanovic et al., 2017; Zellweger et al., 2015), and our previous data suggest HLTF loss also leads to this phenotype (Kile et al., 2015). To confirm and extend this finding, we monitored fork progression using the dose of HU used in the fork reversal assays and a dose of the DNA crosslinker, mitomycin C (MMC), that induces fork reversal in vivo (Vujanovic et al., 2017). Briefly, we pulse-labeled cells with IdU, added the drug during a second CldU pulse, and examined fork progression using DNA spreading (Fig. 1C). In contrast to WT cells in which replication tracts were shortened by about 30% upon drug treatment, replication tracts in both HLTF-KO clones were unaffected and thus exhibited unrestrained fork progression (Fig. 1D). We also observed this phenotype in chronic myelogenous leukemia K562 cells and non-cancerous retina pigmented epithelium RPE1 HLTF-KO cell lines (Sup. Fig. 1A, B and Sup. Table 1). These findings suggest that HLTF’s ability to restrain fork progression is not cell-type specific and occurs in response to multiple types of replication stress.
PRIMPOL is required for unrestrained replication fork progression in HLTF-deficient cells
Intrigued by the nature of the unrestrained fork progression, we next asked whether the replication observed in HLTF-KOs was continuous or whether forks might use another mode of DNA synthesis in these cells. In fact, recent studies suggest the unrestrained replication observed in HLTF-deficient cells may be associated with discontinuous DNA replication (Peng et al., 2018). To test whether replication is discontinuous in our HLTF-KO cells, we treated cells with 50μM HU, and then incubated permeabilized cells with and without S1 nuclease. This ssDNA-specific nuclease cleaves replication intermediates that contain ssDNA formed at gaps or DNA secondary structures (Quinet et al., 2017; Quinet et al., 2016). We found that S1 treatment specifically shortened replication tracts produced in HLTF-KO cells under HU-induced replication stress (Sup. Fig. 1C). This finding strongly suggests that replication proceeds in a discontinuous way when HLTF is lost, with the production of ssDNA gaps.
In higher eukaryotes, de novo priming mediated by PRIMPOL facilitates fork progression by allowing the replisome to skip over barriers, leaving a ssDNA gap behind the fork (Garcia-Gomez et al., 2013; Wan et al., 2013). To determine if PRIMPOL mediates discontinuous replication in HLTF-KO cells under conditions of nucleotide depletion, we knocked down PRIMPOL and monitored fork progression. Replication tracts were significantly shortened specifically in HLTF-KO cells treated with HU after PRIMPOL knockdown, consistent with the idea that PRIMPOL supports replication under these conditions (Fig. 1E and Sup. Fig. 1D). Similar results were obtained with a second PRIMPOL siRNA (Sup. Fig. 1E). Importantly, S1 nuclease treatment only marginally affected tract length when PRIMPOL was knocked down, indicating that replication was no longer discontinuous (Fig. 1F). Moreover, PRIMPOL levels were similar in WT and HLTF-KO cells after HU treatment, suggesting the observed effects are unlikely to reflect PRIMPOL upregulation in the HLTF-KOs (Sup. Fig. 1F). To confirm PRIMPOL’s role in unrestrained fork progression, we knocked out PRIMPOL in our HLTF-KO cells and in WT cells using CRISPR targeting (Sup. Fig. 1G and Sup. Table 1). Consistent with the results obtained using the PRIMPOL siRNAs, knockout of PRIMPOL in the HLTF-KO cells prevented unrestrained fork progression (Sup. Fig. 1H). Taken together, our results strongly suggest that PRIMPOL promotes unrestrained fork progression in HLTF-KO cells under conditions of mild replication stress.
PRIMPOL promotes S phase progression in the absence of HLTF
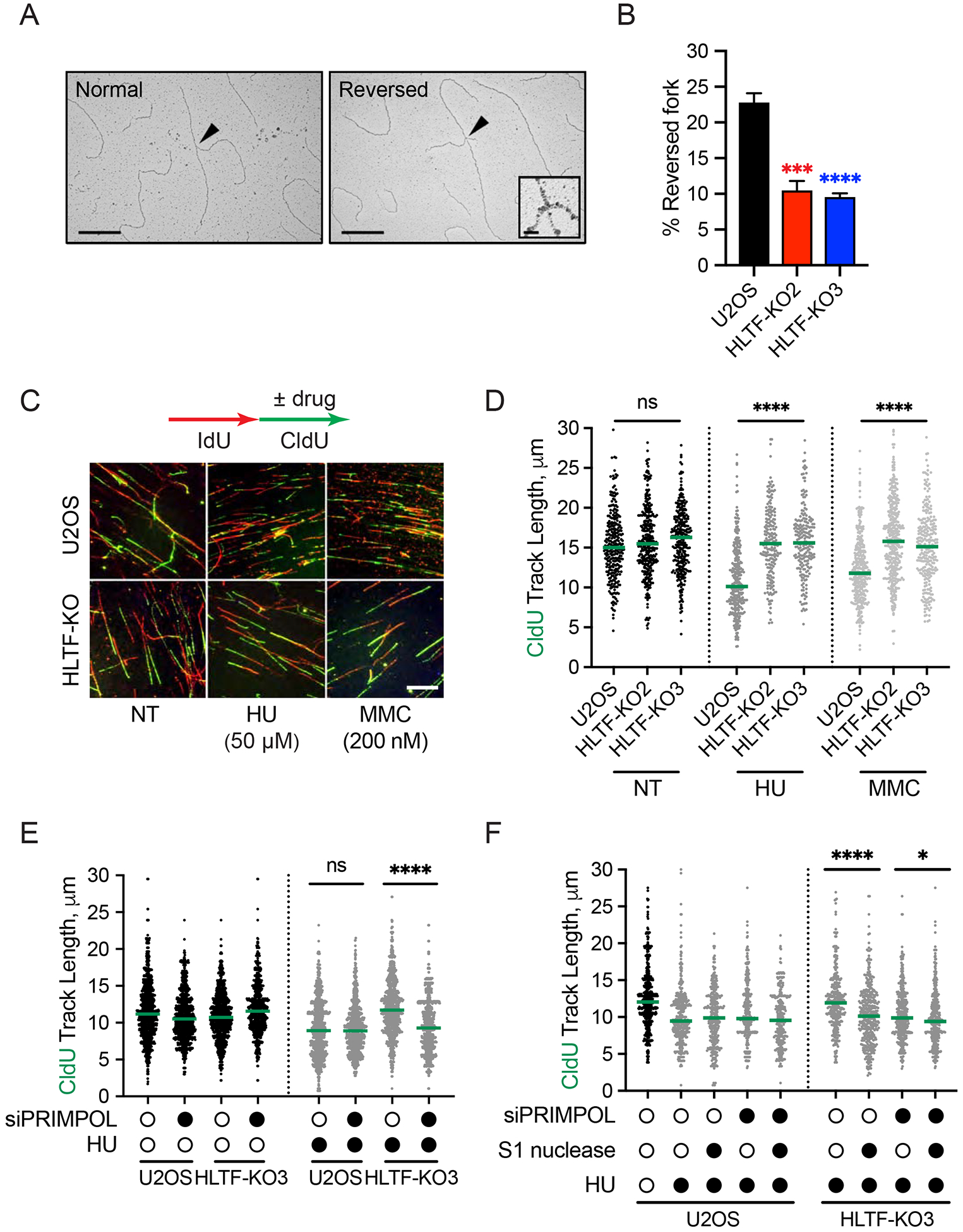
As HLTF loss allows unrestrained fork progression upon replication stress, we next asked how HLTF status affects the cell cycle. HLTF-KO and WT cells have similar cell cycle distributions in the absence of replication stress (Sup. Fig. 2A). By contrast, upon treatment with a low dose of HU for 6 h, we observed significant differences in the cell cycle profiles, with fewer HLTF-deficient cells in S phase and more in G2 phase (Sup. Fig. 2A). As effects on fork progression are immediately observed in DNA fiber experiments (Fig. 1C, D), we reasoned that cell cycle differences were due to increased S phase progression in HLTF-deficient cells, which we measured using a quantitative image-based cytometry (QIBC) assay (Saldivar et al., 2018). Briefly, asynchronous cells were pulsed with EdU, allowed to progress through the cell cycle with or without HU, then pulsed with BrdU and imaged (Fig. 2A). The lack of BrdU staining in EdU-positive cells (red dots) signals successful transition from S to G2 phase (Fig. 2B).
Figure 2. HLTF loss promotes PRIMPOL-dependent S to G2 cell cycle progression.
(A) Experimental setup for S to G2 cell cycle progression assay.
(B) QIBC generated scatter plots. 1500 cells/sample were randomly selected to generate the scatter plot.
(C) Fraction of EdU-positive cells that progressed to G2 phase was determined as described in methods. Mean ± SEM (n=3). ****, p<0.0001, by two-way ANOVA then Dunnett’s test. Test results between HU-treated HLTF-KO vs. U2OS cells are shown.
(D) S-G2 progression assay as described in A and C in indicated cells. Mean ± SEM (n=3). ns, not significant; **, p<0.01; ****, p<0.0001, by two-way ANOVA then Dunnett’s test. Test results between HU-treated PRIMPOL KO or HLTF-PRIMPOL dKO vs. U2OS cells are shown. See also Supplemental Figure 2.
Both WT and HLTF-KO cell lines similarly progressed through S phase in the absence of HU (Fig. 2C and Sup. Fig. 2B). In the presence of HU, WT cells slowed S phase progression as expected, and very few cells entered G2. Strikingly, however, both HLTF-KO clones completed S phase more quickly (Fig. 2B, C). The same results were obtained with RPE1 cells (Sup. Fig. 2C). These findings suggest that the faster fork progression observed in HLTF-KO cells allows them to progress more rapidly through S phase.
To better understand this mechanism, we asked if the accelerated S phase progression observed in HLTF-KO cells was dependent upon PRIMPOL. PRIMPOL-KOs and HLTF-PRIMPOL-dKOs behaved similar to WT cells in the absence of HU treatment (Sup. Fig. 2D), suggesting no significant effect of PRIMPOL loss on normal S phase progression. PRIMPOL-KOs also slowed S phase progression like WT cells when treated with HU (Fig. 2D). By contrast, S phase progression was significant slowed in the HU-treated HLTF-PRIMPOL-dKO cells relative to the HLTF-KOs, although the HLTF-PRIMPOL-dKO cells were still faster than the WT cells (Fig. 2D). Similar results were observed after PRIMPOL knockdown (Sup. Fig. 2E). Taken together, our data suggest that unrestrained fork progression, primarily driven by PRIMPOL, promotes S phase progression in HLTF-deficient cells.
HLTF loss limits DNA damage signaling and DSB formation
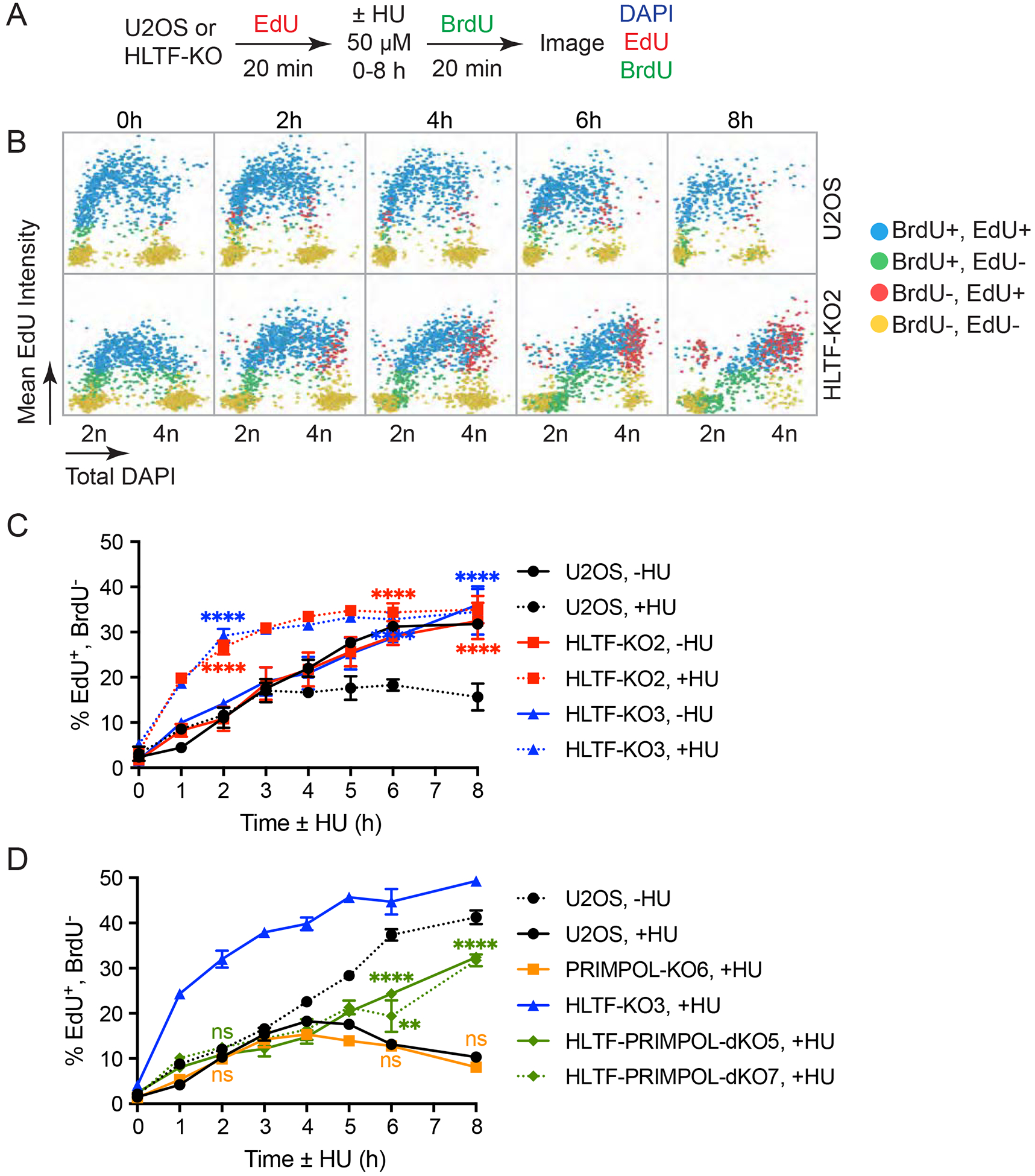
Proper control of replication fork speed can alleviate replication stress and suppress the DNA damage response (Maya-Mendoza et al., 2018). As HLTF-deficient cells fail to slow fork speed and S phase progression under conditions of low dose HU treatment (50 μM), we examined DNA damage signaling in HLTF-KOs using higher HU doses (3 mM) to facilitate detection of damage signaling. Surprisingly, we found that HU-induced markers of DNA damage signaling, including markers of ATR and ATM activation, were reduced in the absence of HLTF (Fig. 3A).
Figure 3. HLTF loss limits DNA damage signaling, RPA chromatin binding and DSB formation.
(A) Western blot of indicated proteins in WT and HLTF-KO U2OS cells treated with 3 mM HU for the time shown.
(B) Cells were treated with 3 mM HU for the indicated time. Total DAPI as well as mean RPA and γH2AX intensities were measured at the single cell level by QIBC after pre-extraction. Scatter plot of single cells with RPA intensity (y-axis) vs DAPI intensity (x-axis) shown. Mean γH2AX intensity/cell is shown for each cell using a color scale. Box indicates the gated RPA positive population used for analyses shown in (C and E). ~1500 cells were randomly selected for each sample.
(C) Cells are treated as in (B), and data are presented as a scatter plot with mean RPA intensity (x-axis) vs mean γH2AX/pRPA (S4/8 or T21) intensity (y-axis). Individual cells with different RPA, γH2AX and pRPA intensities are colored as follows: RPA negative cells are in green, and RPA positive cells are in red (RPA+, in red boxes), unless they also stain positive for pRPA (pRPA-S4/8+ or pRPA-T21+, colored in light blue and in light blue boxes). Total intensities (mean intensity × nuclear area) were calculated to account for differences in the nuclear size of isogenic WT and HLTF-KO cell lines. Population medians of total cellular RPA or γH2AX intensities and percentage (%) of pRPA-S4/8+ or pRPA-T21+ cells among RPA-positive cells from each experiment were averaged to generate the plot, ± SEM. (n≥3). Statistics: ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001 by one-way ANOVA then Dunnett’s test. Test results for each HLTF-KO clone vs. WT are shown.
(D) Neutral comet assay results of WT and HLTF-KO U2OS cells after 24 h HU (3 mM) treatment (n=3). Statistics: ****, p<0.0001 by one-way ANOVA then Dunnett’s test.
(E) Cells were treated with 5 μM ATRi for 80min and after washout with 3mM HU for the indicated times. Total RPA, γH2AX intensities and % of pRPA S4/8+ or T21 + cells among RPA-positive cells are plotted as described in (C). (n>3). Statistics: ns, not significant; **, p<0.01; ***, p<0.001; ****, p<0.0001 by one-way ANOVA then Dunnett’s test. Test results between each HLTF-KO clone vs. WT are shown. See also Supplemental Figure 3.
Replication stress can lead to helicase-polymerase uncoupling, activating ATR through the accumulation of RPA-coated ssDNA (Saldivar et al., 2017). Prolonged replication stress induced by nucleotide depletion leads to excessive ssDNA accumulation, resulting in depletion of the RPA available for ssDNA protection (“RPA exhaustion”). Unprotected ssDNA leads to DNA breaks and ATM activation, a condition described as replication catastrophe (Couch et al., 2013; Toledo et al., 2017; Toledo et al., 2013). To further investigate the impact of HLTF loss on DNA damage signaling, we used QIBC to quantitatively monitor RPA chromatin-binding and phospho-H2AX (γH2AX), analyzing RPA and γH2AX levels in the subset of cells with increased RPA chromatin binding (RPA-positive cells) (Fig. 3B, C) as previously reported (Toledo et al., 2013). This single-cell analysis allowed us to avoid biases that might arise from cell cycle differences. HU treatment increased RPA chromatin binding in both WT and HLTF-KO S-phase cells. Remarkably, however, HLTF-KO clones exhibited significantly less RPA chromatin binding than the WT cells throughout the HU treatment (Fig. 3B, C). Furthermore, RPA-positive cells exhibited reduced H2AX phosphorylation at the single-cell level in HLTF-KO clones (Fig. 3C). H2AX can be phosphorylated by ATR, ATM and DNA-PK in response to multiple types of DNA damage and replication stress. Therefore, we also monitored RPA phosphorylation on T21 and S4/8. These sites are phosphorylated by the DSB-activated kinases ATM and/or DNA-PK, and both sites are phosphorylated during replication catastrophe (Marechal and Zou, 2015; Toledo et al., 2013). Surprisingly, RPA phosphorylation at both sites was significantly reduced in the RPA-positive HLTF-KO cells (Fig. 3C), strongly suggesting that DSB formation is reduced. To further test this idea, we directly monitored DSB formation using the neutral comet assay. Both HLTF-KO U2OS clones showed reduced DSB formation (Fig. 3D), as did HLTF-deficient K562 cell lines (Sup. Fig. 3A). Thus, we conclude that HLTF loss reduces DNA damage signaling and DSB formation under conditions of replication stress.
Upon nucleotide depletion, DSB formation occurs when RPA is no longer available to protect exposed ssDNA, resulting in replication catastrophe (Toledo et al., 2017; Toledo et al., 2013). Because we observed less RPA chromatin binding in HLTF-deficient cells, we asked whether the reduced damage signaling observed was simply a consequence of reduced RPA chromatin binding and therefore increased availability of free RPA. If true, WT and HLTF-KO cells would have similar levels of damage signaling under conditions in which RPA chromatin binding was the same. By contrast, if HLTF has another role in promoting DNA damage, HLTF loss would still reduce damage signaling when RPA chromatin binding is equal.
To distinguish between these possibilities, we repeated the QIBC experiments in cells pretreated with an ATR inhibitor (ATRi) and then exposed to HU. ATR inhibition causes excessive origin firing, accelerating ssDNA accumulation, RPA chromatin binding and DSB formation (Couch et al., 2013; Toledo et al., 2013). The absence of ATRi during HU treatment allows ATR to remain active under conditions of replication stress, preventing disruption of other ATR functions that could lead to DSB formation. Importantly, under these conditions, HLTF loss dramatically reduced H2AX phosphorylation even when RPA chromatin binding was similar (Fig. 3E, left). Furthermore, RPA phosphorylation on T21 and S4/8 was reduced (Fig. 3E, right). Similar damage signaling effects were observed when ATRi and HU were combined to increase replication stress (Sup. Fig. 3B). These findings strongly suggest that HLTF’s absence reduces break formation even when the pool of free RPA is exhausted, thereby uncoupling RPA exhaustion from DSB formation. They also indicate that HLTF’s effects on DNA damage are not a direct result of RPA exhaustion, and HLTF has a separate role in promoting DSB formation. Taken together, our results demonstrate that HLTF loss promotes a different replication mode upon stress, which is associated with reduced DSB formation and reduced DNA damage signaling.
HLTF loss promotes resistance to replication stress
HLTF-deficient cells exhibit less DNA breaks and less DNA damage signaling, progressing more rapidly through S phase than WT cells. We therefore tested the long-term impact of HLTF loss on cells by performing colony survival assays. Surprisingly, under a range of HU doses including those used to assess DNA damage and signaling, HLTF-KO cells exhibited increased resistance to HU-induced replication stress (Fig. 4A). ATR inhibitors are under investigation for the treatment of cancer (Lecona and Fernandez-Capetillo, 2018). As HLTF loss protects cells from DNA damage induced by ATR inhibition combined with HU treatment (Sup. Fig. 3B), we tested the impact of this combination on survival and found HLTF-KO cells were more resistant (Fig. 4B). Remarkably, increased resistance was also observed in HLTF-KO cells treated with MMC (Fig. 4C). Collectively, these findings suggest that HLTF loss protects cells from replication stress and fork collapse induced by a variety of treatments.
Figure 4. HLTF loss promotes resistance to replication stress.
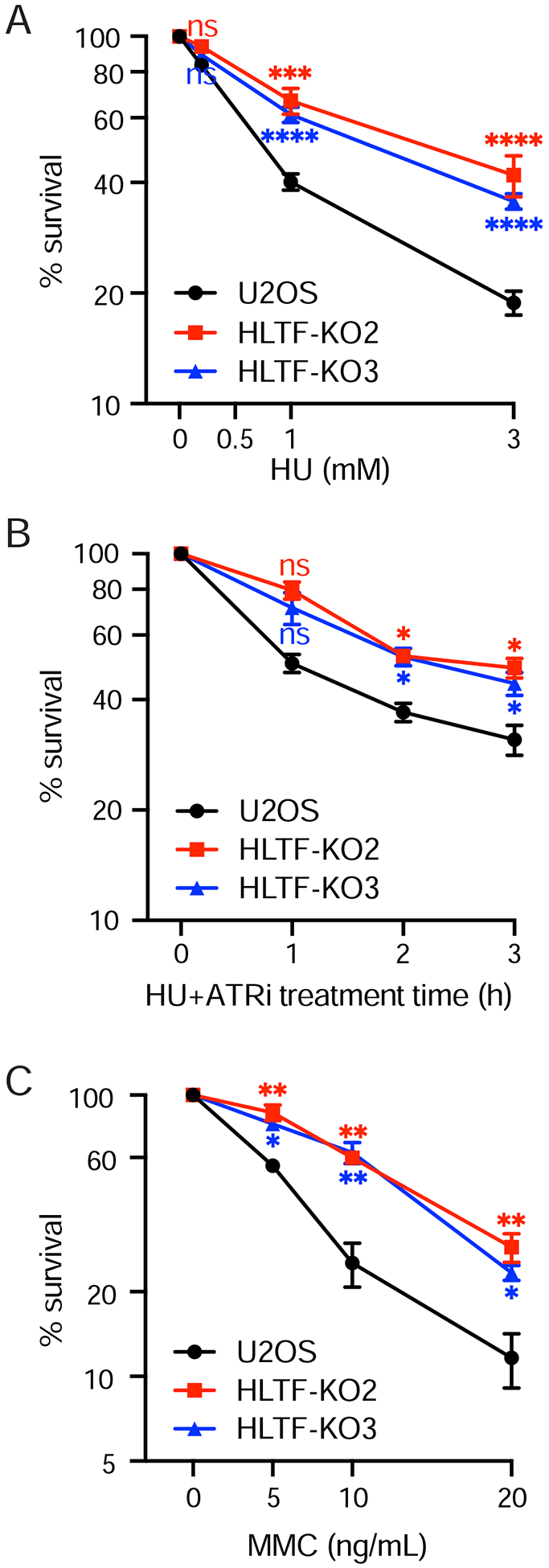
(A to C) Clonogenic survival assay of WT and HLTF-KO U2OS cells after HU (A), 3 mM HU and 5 μM ATRi (B) or MMC (C) treatment. Mean ± SEM (n=3). Statistics: ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001 by one-way ANOVA then Dunnett’s test. Test results between each HLTF-KO clone vs. WT are shown.
HLTF loss protects cells from replication stress in a PRIMPOL-independent manner
PRIMPOL sustains unrestrained replication in HLTF’s absence while promoting S phase progression, raising the possibility that HLTF-deficient cells may depend on PRIMPOL for their replication stress response (Fig. 1E, Fig. 2D). Therefore, we asked how PRIMPOL affects the accumulation of DNA breaks, DNA damage signaling and survival in HU-treated WT and HTLF-KO cells. We found that PRIMPOL-KOs exhibited significantly increased RPA chromatin binding, H2AX and RPA phosphorylation (Fig. 5A). Consistent with the increased DNA damage signaling, PRIMPOL-KO cells were sensitive to HU (Fig. 5B), as previously reported (Kobayashi et al., 2016). Furthermore, neutral comet assays indicate elevated DSB formation in PRIMPOL-KO cells treated with HU (Fig. 5C). These observations suggest that PRIMPOL reduces RPA chromatin binding and DSB formation, likely by reducing ssDNA exposure through repriming and DNA synthesis or by simply binding to ssDNA. More importantly, HLTF loss in PRIMPOL-KO cells (HLTF-PRIMPOL-dKOs) reduced γH2AX and pRPA to WT levels (Fig. 5A), suggesting HLTF loss limits DSB formation even when PRIMPOL is absent. Comet assays also suggest that HLTF loss reduces DSB formation in a PRIMPOL-deficient background (Fig. 5C). Moreover, HLTF-PRIMPOL-dKO cells were less sensitive to HU than PRIMPOL-KOs in a survival assay (Fig. 5B). Similar results were obtained upon knockdown of PRIMPOL (Sup. Fig. 4A, B). Taken together, these findings demonstrate that HLTF loss can protect cells from replication stress in a process that is independent of PRIMPOL.
Figure 5. HLTF loss protects cells from replication stress in a PRIMPOL-independent manner.
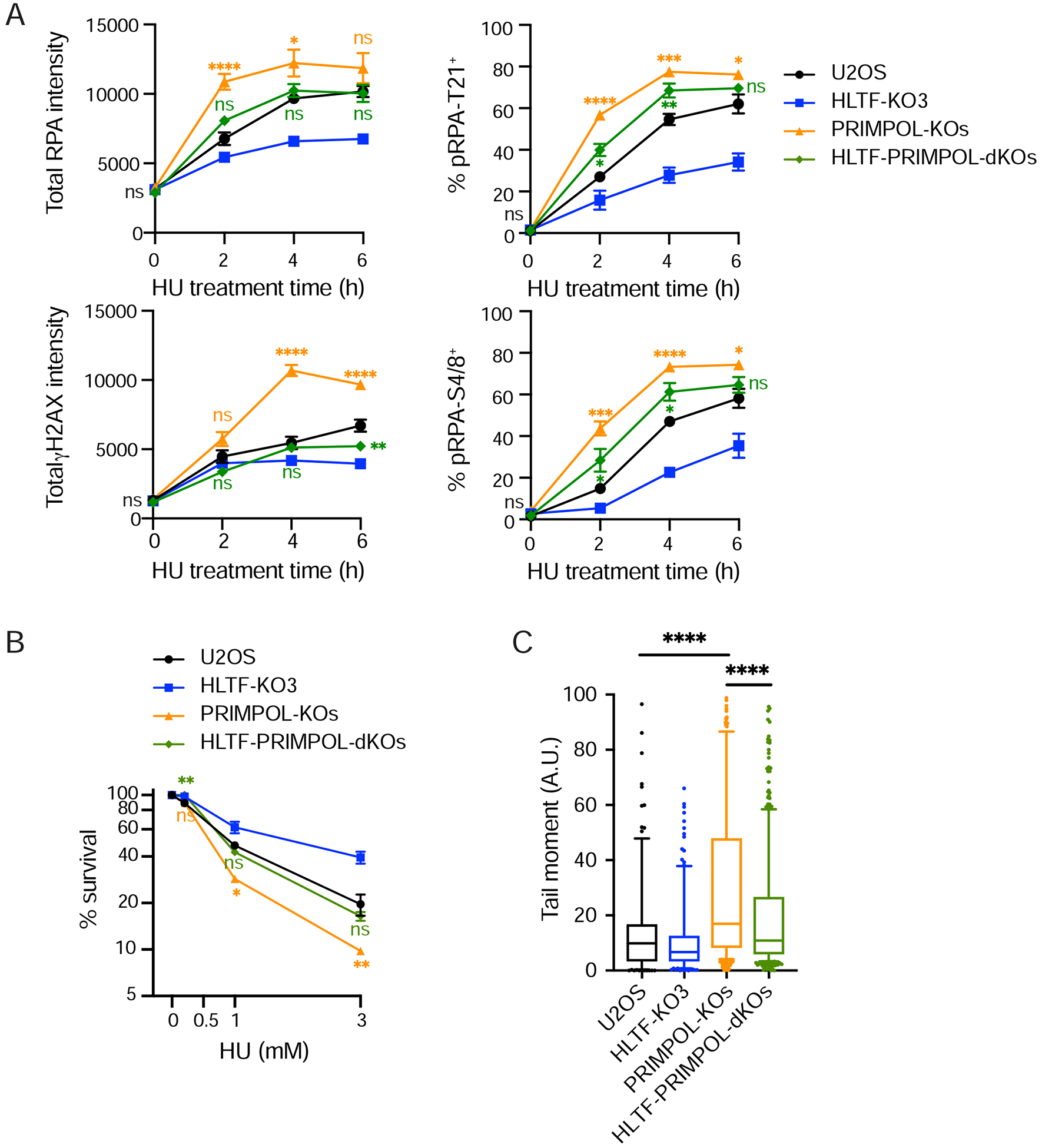
(A) Total RPA and γH2AX intensities and percentage of pRPA-T21+ and pRPA S4/8+ cells were measured in RPA-positive cells as in Figure 3C. For PRIMPOL-KOs or PRIMPOL-HLTF dKOs, 3 clones of each genotype were analyzed individually and averaged for each independent experiment (A to C). Mean ± SEM (n=4). Statistics: ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001 by one-way ANOVA then Dunnett’s test. Test results between PRIMPOL-KO or HLTF-PRIMPOL dKO vs. WT are shown.
(B) Clonogenic survival assay in indicated cells after 24 hr HU (3 mM) treatment. Mean ± SEM (n=2). PRIMPOL-KO or PRIMPOL-HLTF-dKO results are compared to WT cells in a statistical test. ns, not significant; *, p<0.05; **, p<0.01, by two tailed t-test.
(C) Neutral comet assay in indicated cells after 24 hr HU (3 mM) treatment. Mean ± SEM (n=2). ****, p < 0.0001, by two-tailed Mann-Whitney test. See also Supplemental Figure 4.
The HLTF HIRAN mutant promotes an alternative mechanism of stress-resistant DNA replication
HLTF’s ATPase and HIRAN domains are required for replication fork reversal activity in vitro. Moreover, transient expression of a HIRAN mutant incapable of fork reversal in vitro failed to restrain fork progression in HLTF-KOs (Kile et al., 2015). To explore the HIRAN domain’s impact on the HLTF-dependent functions described herein, we first expressed and purified the HLTF R71E mutant protein and tested ATPase, fork regression, and ubiquitin ligase activities. As expected, this mutant retained its ATPase activity but had significantly impaired fork reversal activity (Fig. 6A, B). In addition, this mutation did not impact HLTF’s ability to mediate PCNA ubiquitination (Fig. 6C). Thus, the R71E HIRAN mutant has lost its ability to reverse replication forks, but its ATPase and ubiquitin ligase activities remain intact.
Figure 6. The HLTF HIRAN mutant promotes an alternative mechanism of stress-resistant DNA replication.
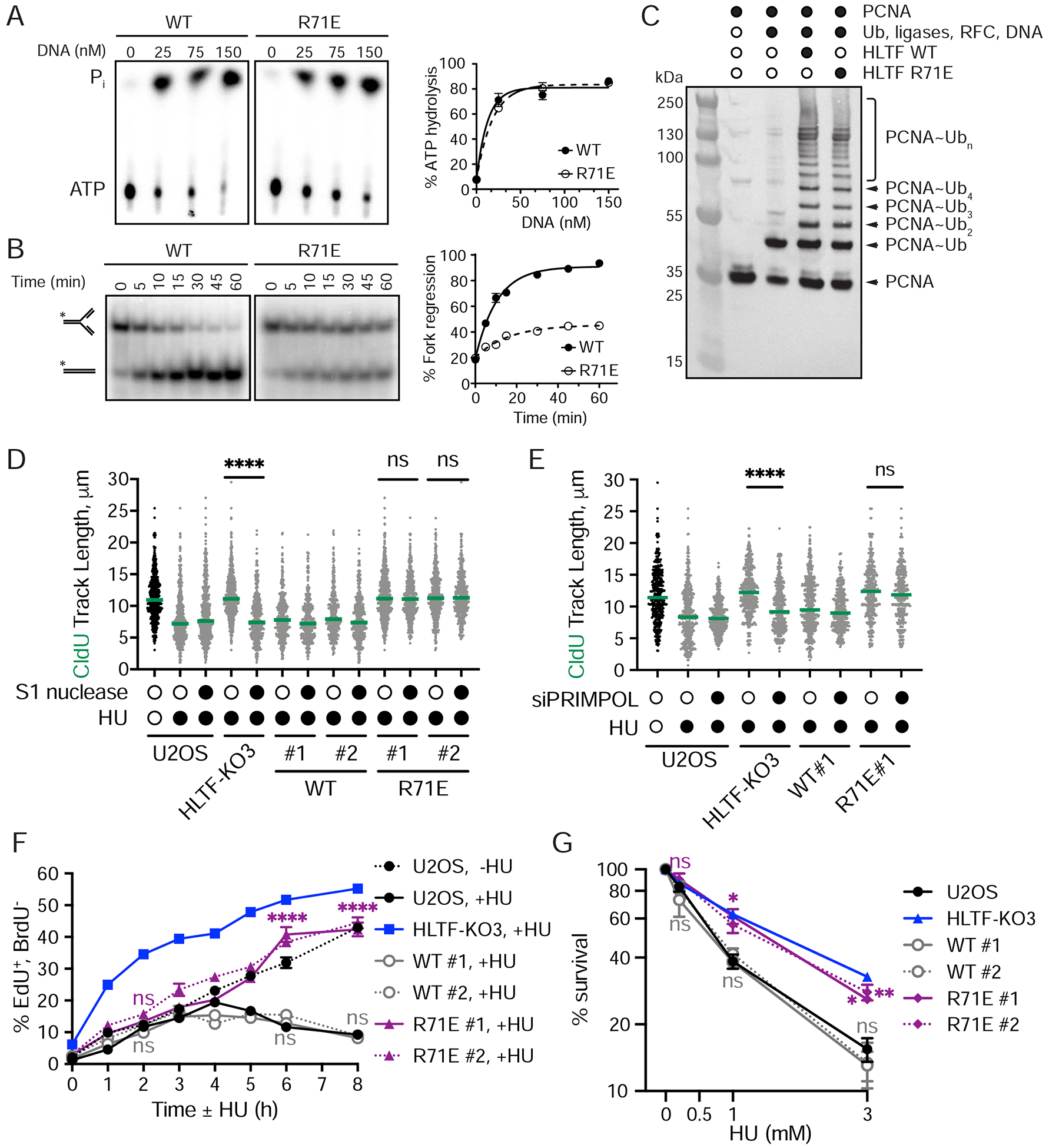
(A) Left, representative TLC plates. Right, quantification of DNA-dependent ATPase activity of indicated protein. Mean ± SEM (n=3).
(B) Left, representative native PAGE results showing fork regression experiment. Model DNA forks were incubated with WT or HIRAN mutant HLTF proteins. The * represents the position of the 5’−32P-labeled oligonucleotide in the fork structure and product. Right, quantification of the results shown on the left. Mean ± SEM (n=3).
(C) Western blot (α-PCNA) analysis of HLTF-dependent PCNA polyubiquitylation using either WT or R71E mutant HLTF.
(D) Dot plot and median CldU tract length in indicated cells with or without S1 nuclease treatment after mock or HU (50 μM) treatment during CldU labeling. (n=3). ns, not significant; ****, p < 0.0001, by two-tailed Mann-Whitney test.
(E) Dot plot and median CldU tract length in indicated cells after control or siPRIMPOL-3 knockdown. Cells were labeled and HU treated as described in (D). (n=3). ns, not significant; ****, p < 0.0001, by two-tailed Mann-Whitney test.
(F) S-G2 progression as described in Figure 2. Mean ± SEM (n=3). ns, not significant; **, p<0.01; ****, p<0.0001, by two-way ANOVA then Dunnett’s test. Test results between WT or R71 E-rescue vs. U2OS are shown.
(G) Colony formation following 24hr HU treatment. Mean ± SEM (n=3). ns, not significant; *, p<0.05; **, p<0.01, by one-way ANOVA then Dunnett’s test. Test results between WT or R71E- rescue vs. U2OS are shown. See also Supplemental Figure 5.
Next, we generated HLTF-KO cells stably expressing WT or the R71E mutant HLTF. Although designed to be doxycycline-inducible, HLTF was expressed in the absence of induction, and doxycycline addition only modestly increased protein expression (Sup. Fig. 5A). Nevertheless, we carried out all experiments in the presence of doxycycline. WT HLTF was expressed in all selected clones at levels slightly higher than the endogenous protein, while the R71E mutant was expressed at levels similar to the endogenous protein.
First, we asked if the WT-HLTF or R71E mutant could restrain fork progression upon HU treatment. HU-treated cells expressing WT-HLTF slowed fork progression, while cells expressing the R71E mutant did not, consistent with our previous results (Kile et al., 2015) (Sup. Fig. 5B). Next, we sought to determine if the unrestrained fork progression observed in the HIRAN mutant was discontinuous, as observed in HLTF-KOs, by testing S1-sensitivity of the fibers. To our surprise, S1 nuclease treatment did not shorten replication tracts produced in R71E HLTF cells, while it did so in the HLTF-KO cells (Fig. 6D). By contrast, HLTF-KO cells expressing the WT-HLTF protein slowed fork progression and were insensitive to S1 treatment, as observed in control U2OS cells (Fig. 6D). These findings suggest that the unrestrained replication observed in cells expressing the HIRAN mutant is mechanistically distinct from that observed in the HLTF-KO cells. To further characterize this phenotype, we tested the impact of knocking down PRIMPOL in these cells. Consistent with the lack of S1 sensitivity, PRIMPOL knockdown in cells expressing the R71E mutant had no effect on replication fork progression (Fig. 6E). These findings demonstrate that the HIRAN mutant cells are still capable of unrestrained fork progression, and that this fork progression occurs in a PRIMPOL-independent manner.
To further investigate the role of the HIRAN domain in the cellular replication stress response, we monitored S phase progression in these cells. In the absence of HU, cells expressing either the WT or R71E mutant HLTF protein progress through S phase at a rate similar to WT or HLTF-KO U2OS cells (Sup. Fig. 5C). Intriguingly, however, upon HU treatment HLTF-KO cells expressing R71E-HLTF progressed through S-phase more rapidly than HLTF-KO cells expressing WT-HLTF or normal U2OS cells, yet slower than HLTF-KO cells (Fig. 6F). Thus, the unrestrained fork progression observed in the HIRAN mutant sustains faster S phase progression, although to a lesser extent than that resulting from HLTF loss.
Finally, we addressed the impact of the HIRAN domain on cell survival and DNA damage signaling. HLTF-KO cells expressing either WT-HLTF or the R71E mutant exhibited levels of H2AX and RPA phosphorylation (T21 and S4/8) similar to those observed in control U2OS cells (Sup. Fig. 5D). As expected, expression of WT-HLTF fully restored cellular sensitivity to HU in the HLTF-KOs. Surprisingly, however, expression of the R71E HLTF mutant did not significantly change the HU sensitivity of HLTF-KOs (Fig. 6G). Taken together, these findings suggest that HLTF loss and expression of a mutant HLTF incapable of fork reversal enable different mechanisms of unrestrained fork progression, both leading to increased cellular resistance to replication stress.
REV1 is required for unrestrained replication fork progression in the HIRAN mutant
Next, we asked how cells expressing the R71E mutant sustain replication fork progression independent of PRIMPOL. The yeast orthologue of HLTF, Rad5, interacts with REV1, a BRCT-domain-containing Y family polymerase that facilitates the recruitment of TLS polymerases to stressed replication forks to continue DNA synthesis (Gallo et al., 2019; Kuang et al., 2013; Pages et al., 2008; Xu et al., 2016). Because PRIMPOL is not required for unrestrained DNA replication in the R71E mutant, we hypothesized that TLS might sustain fork progression in this scenario. To test this idea, we knocked down REV1 in the HLTF-KO cells expressing either WT HLTF or the R71E mutant (Sup. Fig. 6A). Although REV1 knockdown had no effect on fork progression in either the HLTF-KO cells or KO cells expressing the WT protein, it prevented unrestrained fork progression in the R71E mutant cell lines (Fig. 7A).
Figure 7. REV1 is required for unrestrained replication fork progression in the HIRAN mutant.
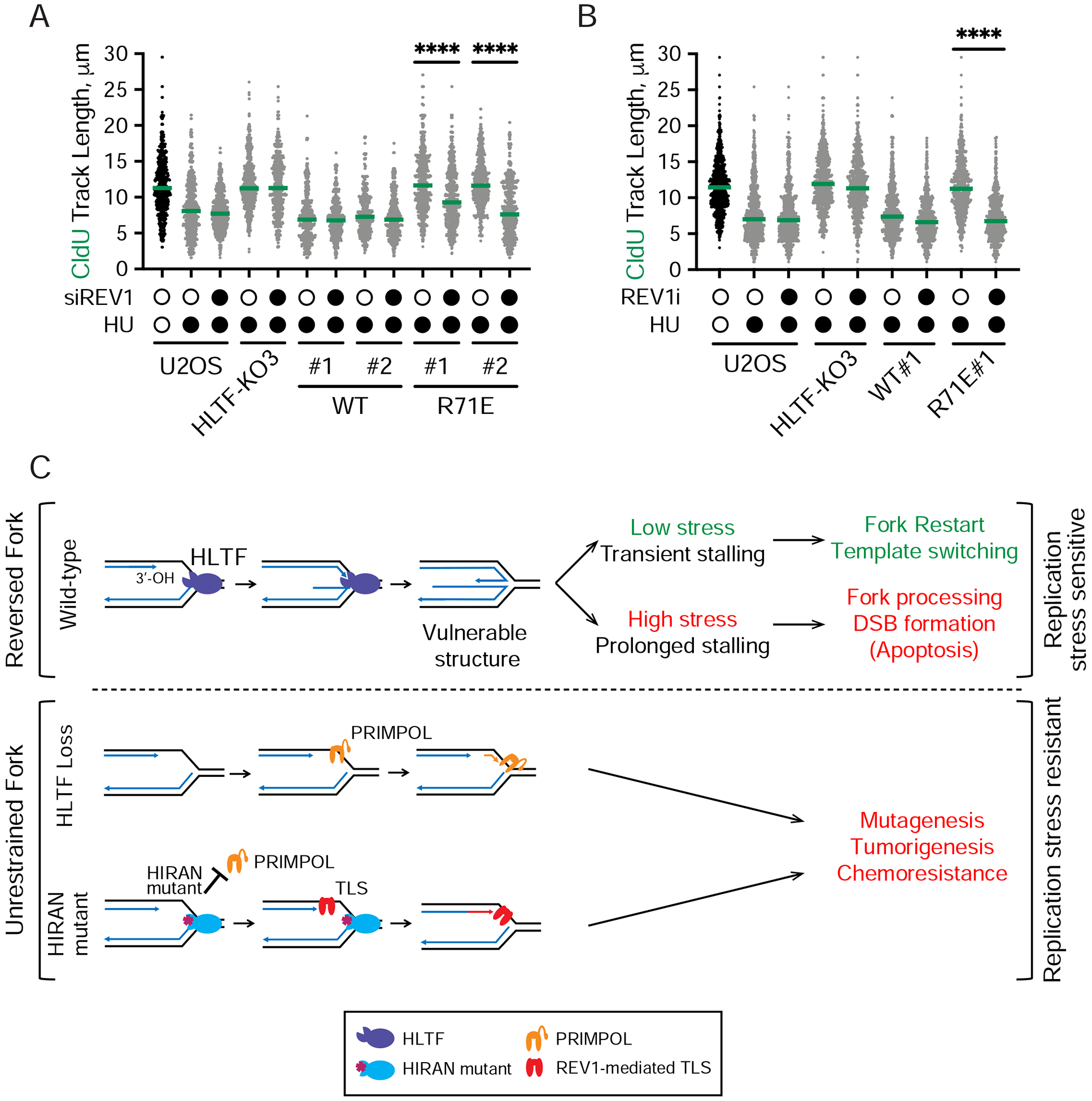
(A) Dot plot and median CldU tract length in indicated cells after control or REV1 knockdown (siREVI). (n=3). ****, p < 0.0001, by two-tailed Mann-Whitney test.
(B) Dot plot and median CldU tract length in indicated cells after control or REV1 inhibitor treatment (REV1i, 15 μM). REVi was added 30 min prior to labeling and remained throughout the experiment. (n=3). ****, p < 0.0001, by two-tailed Mann-Whitney test.
(C) Proposed model for how HLTF prevents stress-resistant DNA replication. At forks stalled by replication stress, HLTF uses its HIRAN domain to engage the free 3’-OH group at the stalled fork to promote fork reversal and restrain fork progression (top). In response to transient stalling induced by low level replication stress, HLTF-mediated fork remodeling facilitates template switching and fork restart. At high levels of replication stress, fork stalling is prolonged and the HLTF remodeled replication fork is susceptible to nucleolytic processing and DSB formation. These events contribute to the sensitivity of WT cells to replication stress. When HLTF is lost (middle), fork progression is unrestrained and depends on PRIMPOL-mediated repriming, leading to discontinuous replication, and S1-sensitive gaps in the DNA. Mutation in HLTF’s HIRAN domain (bottom) disrupts its ability to engage the 3’-OH group at the stalled fork and prevents fork reversal, while the HIRAN mutant protein prevents PRIMPOL-mediated replication. Extension of the free 3’-OH group by REV1-mediated TLS sustains unrestrained fork progression. Both PRIMPOL-dependent and REV1-dependent fork progression contribute to replication stress resistance and potentially promote mutagenesis. Cancer cells might utilize these stress-resistant mechanisms of DNA replication to enhance tumorigenesis and chemoresistance. See also Supplemental Figure 6.
To further validate this result, we monitored replication fork progression in R71E cells treated with a REV1 inhibitor that directly binds to REV1’s C-terminal domain and disrupts its ability to recruit TLS polymerases (Sail et al., 2017). Treatment with this inhibitor had minimal effect on fork progression in the HLTF-KO or WT HLTF cells (Fig. 7B). However, the inhibitor significantly slowed replication fork progression in the R71E mutant, consistent with REV1 knockdown. Taken together, these results demonstrate that cells expressing the R71E HLTF mutant, but not WT-HLTF or HLTF-KO cells, rely on REV1 for replication fork progression under conditions of replication stress.
DISCUSSION
Here, we show that HLTF mediates a complex replication stress response, promoting replication fork reversal, and as a result, suppressing alternative, stress-resistant mechanisms of DNA replication. When HLTF is lost, as occurs by silencing in some cancers, cells become resistant to replication stress (Fig. 7C). We identify at least two HLTF functions that may contribute to replication stress sensitivity. First, HLTF restrains replication fork and S phase progression by promoting fork reversal and suppressing alternative mechanisms of DNA synthesis. Second, HLTF exposes cells to DSB formation upon prolonged replication stress. We hypothesize that unless promptly restarted, reversed forks or other Holliday junction-like structures generated by HLTF are targeted by structure-specific nucleases to produce DSBs. We also suggest that HLTF regulates the balance between fork reversal, repriming and TLS, and that human cells initially attempt fork reversal under stress conditions, likely to minimize mutation. In the absence of, or as an alternative to fork reversal, replication forks demonstrate remarkable plasticity in their ability to continue DNA synthesis, through either discontinuous PRIMPOL-dependent replication or continuous REV1-dependent replication.
HLTF promotes fork reversal in vivo and suppresses multiple mechanisms of stress- resistant replication fork progression
Here, we provide direct evidence that HLTF is a bona-fide fork reversal protein in vivo, using EM, the gold standard in this field (Fig. 1A, B). Intriguingly, we also identify multiple mechanisms by which forks progress when HLTF is disrupted. Upon complete loss of HLTF, replication fork progression is mediated by PRIMPOL (Fig. 1E), and the S1 sensitivity of this fork progression suggests that PRIMPOL promotes discontinuous replication (Fig. 1F). Importantly, disrupting HIRAN domain binding to the 3’-OH of nascent DNA also impairs fork reversal and enables unrestrained fork progression via a distinct, REV1-mediated process (Fig. 7A, B). As PRIMPOL is no longer needed for unrestrained replication (Fig. 6E) in HIRAN mutant cells, other activities of HLTF, such as its ATPase or ubiquitin ligase activities, or the simple presence of HLTF on the chromatin, may prevent the action of PRIMPOL. In HIRAN mutant cells, we envision that the 3’-nascent DNA end may be extended by TLS polymerases recruited by REV1 (Fig. 7C). Thus, the HIRAN domain may prevent REV1-mediated DNA synthesis by blocking the 3’-nascent DNA end and promoting fork remodeling. In support of this model, replication in HU-treated cells expressing the HIRAN mutant is unrestrained but insensitive to S1 nuclease activity (Fig. 6D). Why then doesn’t REV1 act in the HLTF-KOs? In yeast, Rad5 interacts with REV1 and recruits it to replication forks under conditions of stress (Gallo et al., 2019; Kuang et al., 2013; Pages et al., 2008). Thus, one possibility is that HLTF recruits REV1 to stalled forks regardless of whether the HIRAN domain binds DNA, but that TLS polymerases only extend the nascent DNA strand when the 3’-DNA end is not shielded by the HIRAN domain. In wild-type cells, HIRAN domain engagement could therefore be a regulated switch controlling the balance between TLS and other processes that occur at the stalled fork.
Continued DNA synthesis during S phase appears to be important for replication stress resistance. Indeed, PRIMPOL- or Rev1-mediated DNA synthesis in the HLTF-KO’s and HIRAN mutants respectively, drives continued replication and S phase progression (Fig. 2, 6F). Surprisingly, in HLTF’s absence, S phase progression is faster than in untreated WT cells, and PRIMPOL loss does not fully suppress S phase progression (Fig. 2D), suggesting other factors may contribute to this process. As HLTF loss reduces DNA breaks and checkpoint signaling (Fig. 3), one possibility is that a less active checkpoint contributes to cell cycle progression. In either case, efficient DNA replication in HLTF’s absence could give HLTF-deficient tumors a proliferative advantage.
HLTF loss promotes resistance to replication stress-inducing agents
Our data suggest that the ability of HLTF-deficient cells to avoid fork remodeling and to continue replication ultimately promotes cell cycle progression and survival under replication stress. Moreover, when fork progression is robustly inhibited by high HU concentrations, or when fork collapse is accelerated by addition of ATRi and HU, HLTF loss still protects cells from some of the deleterious consequences of replication stress. Consistent with this, DNA damage signaling and neutral comet assays suggest that HLTF loss reduces DSB formation when the replication fork is acutely blocked (Fig. 3). Moreover, survival is enhanced upon recovery from such stress (Fig. 4). This enhanced survival may be due to reduced DNA damage and fork collapse/processing in HLTF-KO cells, avoiding cell death and/or apoptosis upon acute replication stress.
Our studies with PRIMPOL support the idea that HLTF loss protects cells from fork collapse and DNA break formation. PRIMPOL loss sensitized cells to replication stress (Fig. 5B), consistent with previous reports and with its role in replication fork restart (Kobayashi et al., 2016). Furthermore, RPA chromatin loading, DNA damage signaling and DSB formation are enhanced in PRIMPOL’s absence (Fig. 5A, C). This suggests that repriming by PRIMPOL reduces ssDNA exposure at stalled forks and delays fork collapse. Nevertheless, HLTF loss suppresses DSB formation in PRIMPOL-KOs and improves cell survival. Thus, HLTF loss can protect cells in at least two ways - first by allowing for PRIMPOL-mediated DNA replication, and second by reducing replication fork collapse, independent of PRIMPOL. We hypothesize that reduced fork collapse is due to the inability of HLTF-deficient cells to form a reversed fork or related structure, which upon prolonged stress and/or fork deprotection is susceptible to processing (Fig. 7C). In fact, several studies implicate reversed fork processing by both exonucleases and structure-specific nucleases, upon acute replication stress or specific genetic defects (Kolinjivadi et al., 2017; Lemacon et al., 2017; Mijic et al., 2017; Neelsen et al., 2013; Porebski et al., 2019; Taglialatela et al., 2017). Alternatively, HLTF might serve as a scaffold for nucleases that process the fork into DSBs. Interestingly, HLTF loss reduced HU-induced RPA chromatin binding, and we uncoupled this effect from HLTF’s effects on replication catastrophe (Fig. 3E). We speculate that HLTF-mediated fork remodeling allows some resection of the reversed fork that leads to RPA loading.
HIRAN mutant cells exhibit considerable replication stress resistance (Fig. 6F, G). This is consistent with their continued replication using REV1-mediated TLS and with the idea that HLTF-mediated fork remodeling could be deleterious for cell survival upon prolonged stalling. Nevertheless, near wild-type levels of DNA damage signaling are observed in the HIRAN mutants (Sup. Fig. 5D), even though these mutants do not reverse the fork and continue replication. One possibility is that the DNA damage associated with the deregulated HIRAN mutant, which retains its ubiquitin ligase and ATPase activities, results from processing a distinct DNA structure from that processed in WT cells and is less toxic for the cell. Regardless, the enhanced replication stress resistance of HLTF-KOs and HIRAN mutants could have significant consequences in cancer cells, where HLTF is commonly silenced, and the resistance is consistent with HLTF’s role as a tumor suppressor (Dhont et al., 2016). These data and our studies with the ATR inhibitor (Fig. 4B) identify HLTF as a candidate biomarker for resistance to ATR inhibitors and other treatments increasing replication stress (Lecona and Fernandez-Capetillo, 2018).
The costs and benefits of replication fork reversal
The efficiency of replication observed when HLTF is absent or the HIRAN domain is dysfunctional raises the question of why cells rely on fork reversal, especially if it has deleterious consequences. We speculate that reversed forks may act as “sensors” of replication stress levels. Upon mild treatments and transient fork stalling, they may in fact help cells tolerate lesions and stress, providing time for template repair and error-free damage tolerance, via template switching. However, by offering entry points for nucleolytic degradation, upon prolonged fork stalling or in genetic backgrounds that fail to protect them, such as loss of BRCA1/2 (Kolinjivadi et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017), reversed forks may trigger processing events that mediate chemosensitivity and eliminate cells dangerously prone to genome instability. We also hypothesize that PRIMPOL or REV1 use may allow cells to continue replication under conditions of stress, but with the likely cost of increased mutagenesis. Indeed, PRIMPOL is error-prone (Guilliam et al., 2015a; Guilliam et al., 2015b), and HLTF loss promotes mutation accumulation (Frizzell et al., 2014; Lin et al., 2011; Seelinger and Otterlei, 2020). REV1-mediated TLS is also mutagenic (Lawrence et al., 2000; Vaisman and Woodgate, 2017). Consistent with this, HLTF-KOs accumulate in G2 phase after HU treatment (Sup. Fig.2A), suggesting they undergo some type of post-replicative repair. In fact, yeast Rad5 promotes TLS polymerases use for mutagenic repair of undamaged DNA templates, predominantly in G2 cells (Gallo et al., 2019). While beneficial in yeast, this mutagenic process could be risky in mammalian cells and contribute to explain the observed association between HLTF silencing and tumorigenesis.
Our results raise interesting questions about how cells prioritize replication fork reversal versus error-prone TLS pathways. In yeast, cells frequently resolve replication stress behind the fork using template switching or TLS in G2 (Gallo et al., 2019; Wong et al., 2020). Indeed, except for topoisomerase I inhibition (Ray Chaudhuri et al., 2012), yeast do not utilize fork reversal to respond to replication stress unless Rad53 is inactivated (Cotta-Ramusino et al., 2005; Sogo et al., 2002). Our data and that of others suggest that in mammalian cells the priorities are different, since fork reversal is a frequently observed response (Zellweger et al., 2015). Consistent with this idea, there are several differences in how loss of Rad5 versus HLTF affects the response to HU. In yeast, Rad5 loss promotes sensitivity to HU, ssDNA accumulation, checkpoint activation and slowed cell cycle progression (Gallo et al., 2019). By contrast, we show that loss of HLTF in mammalian cells has the opposite effects. The preferred use of fork reversal may in fact prioritize replication fidelity for higher eukaryotes. It could also help eliminate cells with excessive levels of replication stress by promoting DSB formation and cell death, ultimately protecting the organism. Thus, loss of fork reversal and utilization of PRIMPOL or REV1-mediated TLS could ultimately drive cancer progression and resistance.
Recent work suggests that PRIMPOL upregulation may be an adaptive response in BRCA-mutant cancer cells treated repeatedly with cisplatin, which allows these cells to avoid nucleolytic fork degradation by promoting PRIMPOL-mediated replication (Quinet et al., 2020). Similar to our work, PRIMPOL also plays a role in replication when the remodeler SMARCAL1 is lost (Quinet et al., 2020). However, in that case the mechanism may be distinct from what we observe in HLTF-KOs as forks continue progression in the absence of PrimPol when SMARCAL1 is lost. Interestingly, PrimPol also promotes continued fork progression in UV-treated cells after Rad51 depletion (Vallerga et al., 2015). These findings taken together with our results imply a balance between PrimPol functions and fork reversal, as recently discussed (Quinet et al., 2020). They also suggest that the replication fork has remarkable ability to adapt in different contexts.
HLTF is unique among other fork remodelers
The impact of HLTF loss on cells during nucleotide depletion is also intriguing when considered in the context of SMARCAL1 and ZRANB3, as losing each of the three proteins can rescue phenotypes associated with fork reversal, such as the fork protection defect associated with BRCA-deficiency (Kolinjivadi et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017). However, our data suggest that there are differences in the impacts of losing these proteins on cellular survival. SMARCAL1 or ZRANB3 loss leads to increased sensitivity to a variety of replication stress-inducing agents and ATR inhibitors, supporting the idea that fork reversal is beneficial (Bansbach et al., 2009; Ciccia et al., 2009; Ciccia et al., 2012; Couch et al., 2013; Weston et al., 2012; Yuan et al., 2012). By contrast, HLTF loss leads to increased HU and MMC resistance and increased resistance to ATR inhibitors (Fig. 4). While EM data suggest all three proteins contribute to fork reversal in vivo, these findings raise the possibility that the specific products formed by each protein is distinct and that these differences cannot be distinguished or detected by EM. A related possibility is that these proteins might act together. For example, if HLTF acts upstream of SMARCAL1 and ZRANB3, its loss could prevent the generation of intermediates that are more susceptible to processing than others and ultimately more detrimental to cell viability. This will need to be tested with appropriate experiments.
Summary
In summary, our data suggest that HLTF is a key regulator of the cellular replication stress response, providing an opportunity for error-free resolution of transiently stalled forks and minimizing continued, potentially mutagenic forms of replication. However, an indirect consequence of this pathway choice may be detrimental fork processing. Although this processing could cause cell death, it may also be viewed as a protective mechanism that limits the proliferation of cells experiencing high levels of replication stress. Nucleotide depletion can be induced by oncogene activation and deregulation of origin firing in cancer cells, and HLTF is frequently silenced in cancer. It will therefore be interesting to determine if HLTF loss under these conditions promotes survival and increased mutation, contributing to cancer progression.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Karlene Cimprich (cimprich@stanford.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
This study did not generate/analyze datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and RNA Interference
U2OS cells were maintained in DMEM (Life Technologies) supplemented with 10% FBS, 2 mM L-glutamine, and 100 U/mL penicillin/streptomycin in 5% CO2, at 37°C. U2OS rescue cells were maintained in tetracycline-free FBS (Clontech) to minimize HLTF protein expression from the doxycycline-inducible promoter. K562 cells were maintained in RPMI (Life Technologies) in suspension supplemented with 10% FBS, 100 U/mL penicillin/streptomycin in 5% CO2, at 37°C. RPE1 cells were maintained in DMEM/F12 (Life Technologies) supplemented with 10% FBS, 100 U/mL penicillin/streptomycin and 10 μ/mL hygromycin B. siRNAs against HLTF, PRIMPOL (CCDC111) and REV1 (smart pool) were purchased from Dharmacon and transfected using Dharmafect 1 (Thermo Fisher Scientific) according to the manufacturer’s directions.
CRISPR/Cas9-mediated Knockouts
The single-guide RNAs (sgRNA) used to knockout HLTF or PRIMPOL have been previously described (Kile et al., 2015; Quinet et al., 2020). sgRNAs were cloned into the pX458 and pX459 plasmid (Ran et al., 2013)(Addgene). RPE1 cells were transfected with the pX458-sgRNA-HLTF. U2OS cells were transfected with pX459-sgRNA-PRIMPOL or pX459-sgRNA-HLTF plasmids, and K562 cells were transfected with pX459-sgRNA-HLTF. To generate U2OS HLTF-PRIMPOL dKO cells, we transfected WT and HLTF-KO#3 cells with pX459-sgRNA-PRIMPOL. For RPE1 cells, single GFP positive cells were sorted into a 96-well plate 48h post-transfection. For K562 cells, 24h post-transfection, cells were selected with 1μg/mL puromycin for 48h to enrich for positive transfectants then propidium iodide (PI) staining was performed and single PI-negative cells were sorted into a 96-well plate. For U2OS cells, ~300 cells were plated in 150mm dishes. For all cell types, cells were allowed to form colonies in the incubator. For RPE1 and K562 cells, well-isolated single colonies were further expanded and screened by western blot analysis for the loss of HLTF expression. For U2OS cells, single colonies were isolated using cloning rings and further expanded to screen for loss of HLTF and/or PRIMPOL expression by western blot. All knockout clones were verified by sequencing (see Sup. Table 1).
Generation of HLTF-rescue cell lines
FLAG-tagged WT and HIRAN mutant HLTF (R71E) were cloned from pcDNA3.1(+) backbone (Kile et al., 2015) into pCW57.1 using NEB HiFi assembly. Cloned fragments were sequenced verified. pCW57.1-HLTF vectors were packaged into lentivirus particles using the 3rd generation lentiviral packaging system (pMD2.G, pMDLg & pRSV-Rev) in HEK293T cells, with TransIT®- LT1 Transfection Reagent (Mirus). Virus-containing media was harvested 48 & 72h posttransfection and filtered through 0.45μm PES membrane syringe filter to eliminate packaging cells. Lentivirus particles were further concentrated using Lenti-X™ Concentrator (Clontech) according to the manufacturer’s instructions. U2OS HLTF-KO3 cells were infected with the purified lentivirus particles with the presence of Polybrene (1μg/mL, Millipore) overnight. 48h post-infection, 1μg/mL Puromycin was added to the media to start the selection of infected cells. The resistant cells were clonally isolated using cloning cylinders and maintained in Tetracycline-free FBS with 1μg/mL Puromycin throughout.
Drugs and Cell Culture Supplements
HU (Sigma-Aldrich), MMC (Sigma-Aldrich) or ATR inhibitor (VE821, Selleckchem) were applied as indicated. dNTP analogs BrdU, CldU, IdU (Sigma-Aldrich), and EdU (Life Technologies) were used as indicated. REV1 inhibitor (REV1i) was previously described (compound 4 in (Sail et al.,2017)) and used as indicated.
METHOD DETAILS
DNA Spreading
U2OS, K562, RPE1 and derived cell lines were used to monitor DNA replication tracts essentially as described (Jackson and Pombo, 1998). IdU / CldU pulse labeling is 20min each in Figures 1C–F and 30min each in Figures 6D, E and 7A, B, or as stated in supplementary figures legends. For S1 nuclease experiments, cells were trypsinized and collected after labelling, split into two samples, permeabilized and either mock-treated (open circle) or S1-nuclease treated (closed circle, 20U/mL). DNA spreading was then performed and replication tracts were visualized using a Zeiss OBSERVER.Z1 INVERTED microscope and a Plan-APO 40x/1.4 Oil DIC (UV) VIS-IR objective. Fluorescent images were acquired using an Axiocam 506 mono camera (conversion=0.1135) connected to the microscope. ssDNA was also stained to make sure that DNA tracts are not broken. Tracts that represent replication fork termination events (red-green-red or red only tracts) or new origin firing during the CldU labeling (green only) are excluded from the analysis. In all experiments, CldU tract lengths were measured only when preceded by IdU labeling to quantify ongoing replication forks (green followed by red staining pattern). For quantification, at least 2 slides per sample were prepared for each experimental repeat. To avoid bias, after immunodetection, each pair of slides was blinded and we randomly selected 10 fields of view from each slide and acquired images. Hence, for each experiment, we acquired 20 images for each sample. DNA fiber length was measured using an ImageJ plug-in. We randomly score similar number of fibers (~15) from each image. At least 200 replication tracts per sample were measured for each replicate.
Immunofluorescent (IF) staining
For RPA chromatin binding experiments, cells were pre-extracted with CSK100 (100mM NaCl, 300mM sucrose, 3mM MgCl2, 10mM MOPS & 0.5% Triton X-100) buffer at 4°C for 5 min before fixation. Otherwise, cells were immediately fixed with 4% PFA/PBS for 20 min, permeabilized with 0.5% Triton-X 100 for 5 min, blocked in 1% BSA/PBS for 20 min at RT. For EdU staining, the Click-iT reaction was carried out following permeabilization using the Click-iT Cell Reaction Buffer kit (Thermo Fisher C10269) and Alexa Fluor 488 Azide (Thermo Fisher A10266) according to the manufacturer’s guidelines. For BrdU staining, DNA was denatured with 2 N HCl for 30 min and neutralized for 10 min with PBS prior to the blocking step. The primary antibodies were diluted in 1% BSA/PBS and incubated overnight at 4°C: mouse anti-RPA34 (Millipore NA19L, 1:500), mouse anti-RPA34 (Millipore MABE285, 1:500), rabbit anti-phospho-Histone H2A.X (ser139) (Cell Signaling Technology 9718, 1:500), rabbit anti-phospho-RPA32 (Thr21) (Abcam ab61065, 1:500), rabbit anti-phospho-RPA32 (Ser4/8) (Bethyl Laboratories A300–245A), mouse anti-BrdU (BD Biosciences clone B44, 1:100), rabbit anti-phospho-H3 (Ser10) (Millipore 06–570, 1:400). Cells were washed 3x with PBS. Secondary antibodies (diluted 1:1000) and DAPI (5 μg/mL) were diluted in 1% BSA and incubated for 1 h at RT. Cells were washed 3x with PBS and then submerged in PBS during image acquisition.
Quantitative Image-Based Cytometry (QIBC)
Images were acquired in an unbiased fashion with the Molecular Devices ImageXpress Micro automated inverted epifluorescence microscope. Acquisition times for different channels were adjusted to obtain images in non-saturating conditions for all the treatments analyzed. After acquisition, the images were analyzed with automated MetaXpress image analysis software. At least 3000 cells were analyzed per condition, and each experiment was repeated at least 3 times. DAPI signal was used for generating a mask that identified each individual nucleus as an individual object. This mask was then applied to quantify pixel intensities in the different channels for each individual cell/object. After quantification, the quantified values for each cell (mean and total intensities, area, perimeter) were extracted and exported to the proprietary Spotfire software. Spotfire was used to visualize key features of replication stress and DNA damage signaling for thousands of cells and quantify percentages and average values in cell populations. Spotfire filtered data was then used to generate plots using Prism8 (GraphPad Prism version 8.0.2 (159) for Mac OS X, GraphPad Software, La Jolla California USA, www.graphpad.com) software.
S to G2 cell cycle progression assay
Asynchronously grown U2OS cells were pulse labeled with EdU (10 μM) for 20min, then washed with warm PBS and chased with or without HU (50 μM) for different times, up to 8h. At the end of the chase, cells were pulse labeled again with (50 μM) BrdU for another 20min. Cells are then fixed with 4% PFA before immunofluorescent staining to detect EdU and BrdU incorporation. DNA content is determined by DAPI counterstaining. QIBC was used to acquire fluorescent images and determine Mean EdU, BrdU fluorescent intensities and total DAPI intensity at the single cell level. Cells stain positive or negative for either or both nucleotide labeling were further determined. To determine the percentage (%) of EdU+, BrdU− cells corresponding to cells that progressed from S to G2 during the chase time, number of EdU+, BrdU− cells with G2/4N DNA content were combined with EdU+, BrdU− cells with G1/2N DNA content divided by 2 (1 G2 cell gives rise to 2 G1 cells), then normalized to the total cell number.
Neutral comet assay
U2OS or K562 cells were mock treated or treated with 3mM HU for 24h to induce DSB formation before harvest. For U2OS cells, trypsin was applied to obtain a single cell suspension. For K562 cells, cells were directly harvested from suspension culture. Cells were then processed using Trevigen’s CometAssay kit (Cat# 4250–050) following the manufacturer’s instructions. Electrophoresis was performed at 1V/cm for 45min at 4°C. SYBR-Gold stained single cells were visualized using a Zeiss OBSERVER.Z1 INVERTED microscope and a Plan-APO 40x/1.4 Oil DIC (UV) VIS-IR objective. Fluorescent images were acquired using an Axiocam 506 mono camera (conversion=0.1135) connected to the microscope. For each experimental repeat, at least 60 individual cells were imaged per sample. Comet tail moment was determined using OpenComet v1.3.1 (www.cometbio.org) as an ImageJ plugin.
Clonogenic survival assay
U2OS cells were mock treated or treated with drug as indicated in the figure legends. For HU and MMC treatment, treatment time was 24h. For HU+ATRi treatment, increased treatment time was applied to the cells. After drug treatment, cells were washed twice with warm PBS and released into fresh growth medium for 10–14 days to allow for colony formation. Colonies were visualized by crystal violet staining and counted. Percentage of survival (% survival) is calculated by normalizing the number of colonies from treated samples to mock treated samples.
Western blot
Equal number of cells were collected for each sample and lysed in Laemmli sample buffer supplemented with protease inhibitor cocktail (Sigma) and beta-mercaptoethanol (5%) by heating at 95°C for 5 min. Proteins were separated by SDS-PAGE, and transferred to a PVDF membrane (Millipore). Primary antibodies were: rabbit anti-HLTF (Abcam ab183042), mouse anti-alpha-tubulin (Sigma T9026), rabbit anti-phospho-Chk1 (Ser345) (Cell Signaling Technology 2348), mouse anti-Chk1 (sc-8408, G4, Santa Cruz), rabbit anti-phospho-RPA32 (Ser33) (Bethyl Laboratories A300–246A), rabbit anti-phospho-RPA32 (Ser4/8) (Bethyl Laboratories A300–245A), rabbit anti-phospho-Histone H2A.X (ser139) (Cell Signaling Technology 9718), mouse anti-RPA34 (Millipore NA19L), mouse anti-PCNA (sc-56, Santa Cruz), rabbit anti-PRIMPOL (Mouron et al., 2013), rabbit anti-Histone H3 (Abcam ab1791), mouse anti-REV1 (sc-393022, Santa Cruz), and mouse anti-GAPDH (Abcam ab8245). Secondary antibodies were goat anti-rabbit HRP (Molecular Probes G21234) and goat anti-mouse HRP (Invitrogen 81–6520). Chemi-luminescence was carried out using the Immobilon HRP substrate (Millipore WBKLS0500), and blots were imaged with a FluorChem HD2 from Alpha Innotech.
Protein purification
Human HLTF was expressed in Sf-9 insect cells and purified as described (Chavez et al.,2018). UBA1 (Soss et al., 2011), ubiquitin (Brzovic et al., 2006), RFC (Yao et al., 2003), RAD6/RAD18 (Masuda et al., 2010) and UBC13/MMS2 (Campbell et al., 2012), which were expressed and purified as previously described, were kindly provided by Brian Kelch (University of Massachusetts Medical School, yeast RFC), Yuji Masuda (Nagoya University, Japan, human Rad6/Rad18), Mark Glover (University of Alberta, human Ubc13/Mms2) and John Pascal (University of Montreal, human PCNA.
ATPase assays
ATPase reactions were carried out in ATPase buffer (40 mM Tris pH 7.76, 50 mM NaCl, 5 mM MgCl2, and 1 mM TCEP). Wild type and the R71E mutant were assayed at 25 nM in the presence of the indicated amount of fork DNA (annealed oligos DC40, DC20:40, DC20lead, DC20lag) and 1 mM γ32P-ATP. Reactions were incubated at 37°C for 30 min and quenched with EDTA. 1μL was spotted onto a TLC PEI cellulose F chromatography plate (Millipore Sigma) and free phosphate separated from non-hydrolyzed ATP using a mobile phase of 1 M formic acid and 0.25 M LiCl. Results were visualized using autoradiography and quantified with GelAnalyzer.
Fork regression assay
Fork regression was performed as previously described (Chavez et al., 2018). Briefly, 5’−32P-labeled DC48 and DC50 were annealed in 1X SSC buffer (15 mM sodium citrate pH 7, 150 mM NaCl), DC52 and DC53 were annealed in a separate reaction. Forked substrates were formed by mixing the annealed DC48/DC50 in a 1:1.5 ratio with annealed DC52/DC53. Reactions were carried out at 37 °C in ATPase buffer containing 10 nM HLTF, 2 mM ATP, 0.1 mg/mL BSA, and 1 nM of forked substrate. Reactions were stopped at each time point through the addition of one unit of Proteinase K (Sigma) to 10 μL of sample. The samples were resolved by native PAGE and visualized by autoradiography. Quantification was carried out with GelAnalyzer.
Ubiquitin ligase assays
DNA oligonucleotides DC31 and DC75 were annealed in X SSC buffer (15 mM sodium citrate pH 7, 150 mM NaCl) to produce a duplex with a 5’-ssDNA overhang. Reactions were carried out in ubiquitylation buffer (40 mM Tris pH 7.76, 50 mM NaCl, 8 mM MgCl2, 10% glycerol, 0.5 mM ATP, and 0.1 mg/mL BSA. Unless otherwise noted, each reaction contained 0.1 μM Uba1, 0.01 μM yRFC, 0.2 μM Rad6/Rad18, 0.2 μM Ubc13/Mms2, 0.05 μM annealed DC 31/DC75, 50 μM ubiquitin, 0.1 μM PCNA, and 0.2 μM HLTF. Reactions were incubated at 30°C for 60 min and stopped by the addition of 2X Laemmli buffer. Samples were analyzed by Western blot using an anti-PCNA antibody (PC10, Invitrogen).
EM analysis of DNA replication intermediates (RIs) in human cells
EM analysis of DNA replication intermediates (RIs) was performed as previously described (Zellweger and Lopes, 2018). Briefly, asynchronous cells were treated with 50μM HU for 1h and harvested. In vivo crosslinking was performed twice by addition of 4,5’,8-trimethylpsoralen (Sigma T6137) to a final concentration of 10 μg/mL and UV irradiation at 365nm for 3min (UV Stratalinker 1800; Agilent Technologies). Cells were then lysed with lysis buffer (1.28 M sucrose, 40 mM Tris-Cl, pH 7.5, 20 mM MgCl2, and 4% Triton X-100) for 10min on ice. Nuclei were collected by centrifugation and washed. Digestion was performed with digestion buffer (800 mM guanidine-HCl, 30 mM Tris-HCl, pH 8.0, 30 mM EDTA, pH 8.0, 5% Tween 20, 0.5% Triton X-100, and 1mg/ml proteinase K) at 50°C for 2h. Genomic DNA was extracted from these cell lysates by chloroform: isoamylalcohol phase separation and isopropanol precipitation. The extracted genomic DNA was washed and briefly air-dried before resuspension in TE (Tris-EDTA) overnight. The obtained genomic DNA was digested with PvuII-HF (NEB, R3151L) and Rnase A (Sigma-Aldrich, R5503). The digested DNA was concentrated used for electron microscopy analysis. The digested DNA was mixed with benzyldimethylalkylammonium chloride (BAC, Sigma Aldrich B6295), spread on a water surface and loaded onto carbon-coated 400-mesh magnetic nickel grids. The DNA-loaded grids were coated with 13nm of platinum by platinum-carbon rotary shadowing (Leica BAF060) and analyzed using a transmission electron microscope (Tecnai G2 Spirit; FEI; LaB6 filament; high tension ≤ 120 kV). Images were taken at different magnifications using a side mount charge-coupled device camera (2,600 × 4,000 pixels; Orius 1000; Gatan, Inc.). The images were processed with DigitalMicrograph Version 1.83.842 (Gatan, Inc.) and analyzed using ImageJ.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using Prism8 (GraphPad Software). Details of how data is presented, including the definition of center (mean or median) and error bars can be found in the figure legends. Details of statistical test for each experiment, including the type of statistical tests used and the number of repeats, can be found in the figure legends. Statistical test results, presented as levels of significance, are shown in the figures. In all cases: ns, not significant; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Statistical differences in DNA fiber tract lengths were determined by Mann-Whitney test when two samples were compared. When multiple groups were compared, Kruskal-Wallis test was used. Statistical differences for all other grouped analyses, i.e., frequency of fork reversal (EM), cell survival, chromatin-bound RPA and γH2AX intensities in RPA positive cells (QIBC), percentage of cells positive for chromatin-bound pRPA (QIBC) were assessed by one-way ANOVA followed by Dunnett’s test using WT U2OS cells as control. Statistical differences in percentage of cells positive for EdU, negative for BrdU (QIBC) in the S to G2 progression assay were determined by two-way ANOVA followed by Dunnett’s test using WT U2OS cells as control.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-HLTF | Abcam | Cat# ab183042 |
| Mouse monoclonal anti-Chk1 | Santa Cruz Biotechnology | Cat# sc-8408; RRID:AB_627257 |
| Rabbit polyclonal anti-Histone H3 | Abcam | Cat# ab1791; RRID:AB_302613 |
| Mouse anti-GAPDH | Abcam | Cat# ab8245; RRID: AB_2107448 |
| Rabbit monoclonal anti-phospho-Chk1 (Ser345) | Cell Signaling Technology | Cat# 2348; RRID:AB_331212 |
| Rabbit monoclonal anti-phospho-Histone H2A.X (Ser139) | Cell Signaling Technology | Cat# 9718; RRID:AB_2118009 |
| Rabbit polyclonal anti-phospho-Histone H3 (Ser10) | Millipore | Cat# 06–570; RRID:AB_310177 |
| Rabbit polyclonal anti-phospho-RPA32 (Ser33) | Bethyl | Cat# A300–246A; RRID:AB_2180847 |
| Rabbit polyclonal anti-phospho-RPA32 (Ser4/8) | Bethyl | Cat# A300–245A; RRID:AB_210547 |
| Rabbit polyclonal anti-phospho-RPA32 (Thr21) | Abcam | Cat# ab61065; RRID:AB_946322 |
| Mouse monoclonal anti-Replication Protein A | Millipore | Cat# NA19L; RRID:AB_565123 |
| Mouse monoclonal anti-Replication Protein A | Millipore | Cat# MABE285; RRID:AB_11213221 |
| Mouse monoclonal anti-PCNA | Santa Cruz Biotechnology | Cat# sc-56; RRID:AB_628110 |
| Rabbit polyclonal anti-PRIMPOL | (Mouron et al., 2013) | N/A |
| Mouse monoclonal anti-REV1 (sc-393022, Santa Cruz) | Santa Cruz Biotechnology | Cat# sc-393022 |
| Mouse monoclonal anti-α-Tubulin | Sigma-Aldrich | Cat# T9026; RRID:AB_477593 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# G-21234; RRID:AB_2536530 |
| Anti-Mouse IgG (H+L) Polyclonal Antibody, HRP | Innovative Research | Cat# 81–6520; RRID:AB_87763 |
| Goat polyclonal anti-Mouse IgG (H+L), Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11001; RRID:AB_2534069 |
| Goat polyclonal anti-Rabbit IgG (H+L), Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21244; RRID:AB_2535812 |
| Goat polyclonal anti-Rabbit IgG (H+L), Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-11012; RRID:AB_2534079 |
| Goat polyclonal anti-Mouse IgG1, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-21124; RRID:AB_2535766 |
| Goat polyclonal anti-Rat IgG (H+L), Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11006; RRID:AB_2534074 |
| Goat polyclonal anti-Mouse IgG2a, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21241; RRID:AB_2535810 |
| Mouse monoclonal anti-BrdU | BD Biosciences | Cat# 347580; RRID:AB_400326) |
| Rat monoclonal anti-BrdU | Novus Biologicals | Cat# NB500–169; RRID:AB_10002608 |
| Mouse monoclonal anti-ssDNA | Millipore | Cat# MAB3034; RRID:AB_94645 |
| Bacterial and Virus Strains | ||
| E.coli: DH5α | New England BioLabs | Cat# C2987 |
| E.coli: NEB® Stable | New England BioLabs | Cat# C3040 |
| E.coli: ccdB Survival™ 2 | Thermo Fisher Scientific | Cat# A10460 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EdU (5-ethynyl-2’-deoxyuridine) | Click Chemistry Tools | Cat# 1149–100 |
| DAPI (4’,6-Diamidino-2-phenylindole dihydrochloride) | Sigma-Aldrich | Cat# 32670 |
| BrdU (5-Bromo-2’-deoxyuridine) | Sigma-Aldrich | Cat# B5002 |
| CldU (5-Chloro-2’-deoxyuridine) | Sigma-Aldrich | Cat# C6891 |
| IdU (5-Iodo-2’-deoxyuridine) | Sigma-Aldrich | Cat# I7125 |
| DMSO (Dimethyl Sulfoxide) | Millipore | Cat# MX1458–6 |
| Propidium iodide (PI) | Sigma-Aldrich | Cat# 4170 |
| Ampicillin sodium salt | Sigma-Aldrich | Cat# A0166 |
| Chloramphenicol | Sigma-Aldrich | Cat# C0378 |
| Puromycin | InvivoGen | Cat# ant-pr |
| Hydroxyurea (HU) | Sigma-Aldrich | Cat# H8627 |
| Mitomycin C (MMC) | Sigma-Aldrich | Cat# M4287 |
| VE-821 | Selleckchem | Cat# S8007 |
| Rev1 inhibitor (Rev1i) | (Sail et al., 2017) | Compound #4 |
| Alexa Fluor™ 488 Azide | Thermo Fisher Scientific | Cat# A10266 |
| Alexa Fluor™ 647 Azide | Thermo Fisher Scientific | Cat# A10277 |
| Trioxsalen (4,5′,8-trimethylpsoralen) | Sigma-Aldrich | Cat# T6137 |
| PvuII-HF | New England BioLabs | Cat# R3151L |
| RNase A | Sigma-Aldrich | Cat# R5503 |
| Benzalkonium chloride | Sigma-Aldrich | Cat# B6295 |
| Critical Commercial Assays | ||
| Click-iT™ Cell Reaction Buffer Kit | Thermo Fisher Scientific | Cat# C10269 |
| NEBuilder® HiFi DNA Assembly Master Mix | New England BioLabs | Cat# E2621 |
| Q5® High-Fidelity DNA Polymerase | New England BioLabs | Cat# M0491 |
| Monarch® PCR & DNA Cleanup Kit | New England BioLabs | Cat# T1030 |
| QIAquick Gel Extraction Kit | Qiagen | Cat# 28706 |
| QIAprep Spin Miniprep Kit | Qiagen | Cat# 27106 |
| QIAGEN Plasmid Midi Kit | Qiagen | Cat# 12143 |
| TransIT®-LT1 Transfection Reagent | Mirus | Cat# MIR2300 |
| DharmaFECT 1 Transfection Reagent | Dharmacon | Cat# T-2001–03 |
| Lipofectamine™ 3000 Transfection Reagent | Invitrogen | Cat# L3000008 |
| Lenti-X™ Concentrator | Clontech | Cat# 631231 |
| CometAssay® | R&D Systems | Cat# 4250–050 |
| Experimental Models: Cell Lines | ||
| Human: U2-OS | ATCC | HTB-96 |
| Human: hTERT-RPE1 | ATCC | CRL-4000 |
| Human: K562 | ATCC | CCL-243 |
| Human: HEK293T | GenHunter Corporation | Cat# Q401 |
| Oligonucleotides | ||
| HLTF sgRNA_Fwd: CACCGTTGGACTACGCTATTACAC |
This paper | N/A |
| HLTF sgRNA_Rev: AAACGTGTAATAGCGTAGTCCAAC |
This paper | N/A |
| PRIMPOL sgRNA Fwd: CACCGGATAGCGCTCCAGAGACAAC |
(Quinet et al., 2020) | N/A |
| PRIMPOL sgRNA Rev: AAACGTTGTCTCTGGAGCGCTATCC |
(Quinet et al., 2020) | N/A |
| HLTF HiFi_Fwd: CAGATCGCCTGGAGAATTGGATGGACTATAAAGATGACGATG |
This paper | N/A |
| HLTF HiFi_Rev: TGGTGGTGGTGGTGGACCGGTTATAAGTCAATTAATGTTCTGATTTC |
This paper | N/A |
| DC40: CTCAGGACTCAGTTCGTCAGCCCTTGACAGCGATGGAAGC |
This paper | N/A |
| DC20:40: CGAAGGTAGCGACAGTTCCCCTGACGAACTGAGTCCTGAG |
This paper | N/A |
| DC20lead: GCTTCCATCGCTGTCAAGGG |
This paper | N/A |
| DC20lag: GGGAACTGTCGCTACCTTCG |
This paper | N/A |
| DC35: CGACGATGCTCCGGTACTCCAGTGTAGGCAT |
This paper | N/A |
| DC75: AGCTACCATGCCTGCCTCAAGAATTCCCATTATGCCTACACTGGAGTACCGGAG |
This paper | N/A |
| DC48: ACGCTGCCGAATTCTACCAGTGCCTTGCTAGGACATCTTTGCCCACCTGCAGGTTCACCC |
This paper | N/A |
| DC50: GGGTGAACCTGCAGGTGGGCAAAGATGTCC |
This paper | N/A |
| DC52: GGGTGAACCTGCAGGTGGGCAAAGATGTCCCAGCAAGGCACTGGTAGAATTCGGCAGCGTC |
This paper | N/A |
| DC53: GGACATCTTTGCCCACCTGCAGGTTCACCC |
This paper | N/A |
| siPRIMPOL-3: GAGGAAAGCUGGACAUCGA | Dharmacon | J-016804-17-0002 |
| siPRIMPOL-4: AAGAUGUUUCUGACGAAUA | Dharmacon | J-016804-20-0002 |
| SMARTpool siREV1: GAAGUUAAUUGAUGGGUUU CAUAUCAGCUGUACACCAA GUGGAGACUUGCAGUAUAU CAUCAGAGCUGUAUAAUGC |
Dharmacon | Cat# L-008234–00 |
| Recombinant DNA | ||
| Plasmid: pSpCas9(BB)-2A-Puro (PX459) backbone | Addgene | Plasmid # 48139; RRID:Addgene_48139 |
| Plasmid: pX458-sgRNA-HLTF | This study | N/A |
| Plasmid: pX459-sgRNA-HLTF | This study | N/A |
| Plasmid: pX459-sgRNA-PRIMPOL | This study | N/A |
| Plasmid: pCW57.1 backbone | Addgene | Plasmid # 41393; RRID:Addgene_41393 |
| Plasmid: pCW57.1-HLTF | This study | N/A |
| Plasmid: pCW57.1-DEAA | This study | N/A |
| Plasmid: pCW57.1-R71E | This study | N/A |
| Plasmid: pCW57.1-ΔRING-LINKER | This study | N/A |
| Plasmid: pMD2.G | Addgene | Plasmid # 12259; RRID:Addgene_12259 |
| Plasmid: pMDLg | Addgene | Plasmid # 12251; RRID:Addgene_12251 |
| Plasmid: pRSV-Rev | Addgene | Plasmid # 12253; RRID:Addgene_12253 |
| Plasmid: pcDNA3.1(+) | Invitrogen | Cat# V79020 |
| Plasmid: pcDNA3.1-Flag-HLTF | (Kile et al., 2015) | N/A |
| Plasmid: pcDNA3.1-R71E | (Kile et al., 2015) | N/A |
| Software and Algorithms | ||
| Fiji (ImageJ) | NIH | RRID:SCR_002285 |
| OpenComet v1.3.1 | www.cometbio.org | |
| TIBCO Spotfire | Perkin Elmer | RRID:SCR_008858 |
| GraphPad Prism8 | Graphpad | RRID:SCR_002798 |
| MetaXpress | Molecular Devices | RRID:SCR_016654 |
| Other | ||
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11873580001 |
| PhosSTOP™, Phosphatase Inhibitor Tablets | Roche | Cat# 4906845001 |
| Greiner-Bio CELLSTAR 96 Well Cell Culture Microplate, TC Treated, Black (8 per Pack/ 32 per Case) | Greiner Bio | Cat# 655090 |
| Fetal bovine serum for cell culture (tetracycline-free) | Clontech | Cat# 631367 |
HIGHLIGHTS.
HLTF mediates fork reversal in vivo, associated with DSB formation
HLTF prevents unrestrained replication driven by PRIMPOL or the TLS protein REV1
Unrestrained DNA synthesis promotes S phase progression under replication stress
HLTF loss increases cellular resistance to replication stress and ATR inhibition
ACKNOWLEDGEMENTS
We thank members of the Cimprich laboratory, Gheorge Chistol and Alessandro Vindigni for helpful discussions. We thank Miaw-Sheue Tsai for baculovirus production and insect cell expression of HLTF protein (supported by P01CA092584 from the NIH). We also thank Diana Chavez for help with the ubiquitin ligase assay, and the following individuals for providing reagents: Brian Kelch (RFC), Yuji Masuda (Rad6/18), Mark Glover (Ubc13/Mms2), John Pascal (PCNA), Juan Mendez (PRIMPOL antibody) and Alessandro Vindigni (PRIMPOL guide RNA sequence). This work was supported by grants from the NIH to KAC (ES016486, CA092584), BE (CA092584, GM117299) and MKH (CA233969). HS was supported by a Swiss National Science Foundation Grant (310030_189206), and a Stanford SOM Dean’s Fellowship supported GB and CK. KAC is an ACS Research Professor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Achar YJ, Balogh D, and Haracska L (2011). Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc Natl Acad Sci U S A 108, 14073–14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achar YJ, Balogh D, Neculai D, Juhasz S, Morocz M, Gali H, Dhe-Paganon S, Venclovas C, and Haracska L (2015). Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res 43, 10277–10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansbach CE, Betous R, Lovejoy CA, Glick GG, and Cortez D (2009). The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev 23, 2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, and Cortez D (2012). SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev 26, 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi J, Rudd SG, Jozwiakowski SK, Bailey LJ, Soura V, Taylor E, Stevanovic I, Green AJ, Stracker TH, Lindsay HD, et al. (2013). PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell 52, 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blastyak A, Hajdu I, Unk I, and Haracska L (2010). Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol 30, 684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, and Szakal B (2017). Building up and breaking down: mechanisms controlling recombination during replication. Crit Rev Biochem Mol Biol 52, 381–394. [DOI] [PubMed] [Google Scholar]
- Brzovic PS, Lissounov A, Christensen DE, Hoyt DW, and Klevit RE (2006). A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1-directed ubiquitination. Mol Cell 21, 873–880. [DOI] [PubMed] [Google Scholar]
- Campbell SJ, Edwards RA, Leung CC, Neculai D, Hodge CD, Dhe-Paganon S, and Glover JN (2012). Molecular insights into the function of RING finger (RNF)-containing proteins hRNF8 and hRNF168 in Ubc13/Mms2-dependent ubiquitylation. J Biol Chem 287, 23900–23910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez DA, Greer BH, and Eichman BF (2018). The HIRAN domain of helicase-like transcription factor positions the DNA translocase motor to drive efficient DNA fork regression. J Biol Chem 293, 8484–8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, and Elledge SJ (2009). The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev 23, 2415–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, et al. (2012). Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell 47, 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D (2019). Replication-Coupled DNA Repair. Mol Cell 74, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, and Foiani M (2005). Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell 17, 153–159. [DOI] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhont L, Mascaux C, and Belayew A (2016). The helicase-like transcription factor (HLTF) in cancer: loss of function or oncomorphic conversion of a tumor suppressor? Cell Mol Life Sci 73, 129–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Descheemaeker K, Marynen P, Nelles L, Carvalho T, Carmo-Fonseca M, Collen D, and Belayew A (1996). Characterization of a helicase-like transcription factor involved in the expression of the human plasminogen activator inhibitor-1 gene. DNA and Cell Biology 15, 429–442. [DOI] [PubMed] [Google Scholar]
- Frizzell A, Nguyen JH, Petalcorin MI, Turner KD, Boulton SJ, Freudenreich CH, and Lahue RS (2014). RTEL1 inhibits trinucleotide repeat expansions and fragility. Cell Rep 6, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo D, Kim T, Szakal B, Saayman X, Narula A, Park Y, Branzei D, Zhang Z, and Brown GW (2019). Rad5 Recruits Error-Prone DNA Polymerases for Mutagenic Repair of ssDNA Gaps on Undamaged Templates. Mol Cell 73, 900–914 e909. [DOI] [PubMed] [Google Scholar]
- Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, Powell C, Salido E, Mendez J, Holt IJ, et al. (2013). PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliam TA, Jozwiakowski SK, Ehlinger A, Barnes RP, Rudd SG, Bailey LJ, Skehel JM, Eckert KA, Chazin WJ, and Doherty AJ (2015a). Human PrimPol is a highly error-prone polymerase regulated by single-stranded DNA binding proteins. Nucleic Acids Res 43, 1056–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliam TA, Keen BA, Brissett NC, and Doherty AJ (2015b). Primase-polymerases are a functionally diverse superfamily of replication and repair enzymes. Nucleic Acids Res 43, 6651–6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishiki A, Hara K, Ikegaya Y, Yokoyama H, Shimizu T, Sato M, and Hashimoto H (2015). Structure of a Novel DNA-binding Domain of Helicase-like Transcription Factor (HLTF) and Its Functional Implication in DNA Damage Tolerance. J Biol Chem 290, 13215–13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, and Jentsch S (2002). RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141. [DOI] [PubMed] [Google Scholar]
- Jackson DA, and Pombo A (1998). Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140, 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen BA, Jozwiakowski SK, Bailey LJ, Bianchi J, and Doherty AJ (2014). Molecular dissection of the domain architecture and catalytic activities of human PrimPol. Nucleic Acids Res 42, 5830–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, and Cimprich KA (2015). HLTF’s Ancient HIRAN Domain Binds 3’ DNA Ends to Drive Replication Fork Reversal. Mol Cell 58, 1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Guilliam TA, Tsuda M, Yamamoto J, Bailey LJ, Iwai S, Takeda S, Doherty AJ, and Hirota K (2016). Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides. Cell Cycle 15, 1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881 e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Petermann E, and Boulton SJ (2018). Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov 8, 537–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang L, Kou H, Xie Z, Zhou Y, Feng X, Wang L, and Wang Z (2013). A non-catalytic function of Rev1 in translesion DNA synthesis and mutagenesis is mediated by its stable interaction with Rad5. DNA Repair (Amst) 12, 27–37. [DOI] [PubMed] [Google Scholar]
- Lawrence CW, Gibbs PE, Murante RS, Wang XD, Li Z, McManus TP, McGregor WG, Nelson JR, Hinkle DC, and Maher VM (2000). Roles of DNA polymerase zeta and Rev1 protein in eukaryotic mutagenesis and translesion replication. Cold Spring Harb Symp Quant Biol 65, 61–69. [DOI] [PubMed] [Google Scholar]
- Lecona E, and Fernandez-Capetillo O (2018). Targeting ATR in cancer. Nat Rev Cancer 18, 586–595. [DOI] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, et al. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Zeman MK, Chen JY, Yee MC, and Cimprich KA (2011). SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol Cell 42, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, and Zou L (2015). RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res 25, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda Y, Piao J, and Kamiya K (2010). DNA replication-coupled PCNA monoubiquitination and polymerase switching in a human in vitro system. J Mol Biol 396, 487–500. [DOI] [PubMed] [Google Scholar]
- Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, and Bartek J (2018). High speed of fork progression induces DNA replication stress and genomic instability. Nature 559, 279–284. [DOI] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, et al. (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun 8, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moinova HR, Chen WD, Shen L, Smiraglia D, Olechnowicz J, Ravi L, Kasturi L, Myeroff L, Plass C, Parsons R, et al. (2002). HLTF gene silencing in human colon cancer. Proc Natl Acad Sci U S A 99, 4562–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouron S, Rodriguez-Acebes S, Martinez-Jimenez MI, Garcia-Gomez S, Chocron S, Blanco L, and Mendez J (2013). Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20, 1383–1389. [DOI] [PubMed] [Google Scholar]
- Neelsen KJ, and Lopes M (2015). Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16, 207–220. [DOI] [PubMed] [Google Scholar]
- Neelsen KJ, Zanini IM, Herrador R, and Lopes M (2013). Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol 200, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages V, Bresson A, Acharya N, Prakash S, Fuchs RP, and Prakash L (2008). Requirement of Rad5 for DNA polymerase zeta-dependent translesion synthesis in Saccharomyces cerevisiae. Genetics 180, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero P, and Vindigni A (2017). Nucleases Acting at Stalled Forks: How to Reboot the Replication Program with a Few Shortcuts. Annu Rev Genet 51, 477–499. [DOI] [PubMed] [Google Scholar]
- Peng M, Cong K, Panzarino NJ, Nayak S, Calvo J, Deng B, Zhu LJ, Morocz M, Hegedus L, Haracska L, et al. (2018). Opposing Roles of FANCJ and HLTF Protect Forks and Restrain Replication during Stress. Cell Rep 24, 3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilzecker B, Buoninfante OA, Pritchard C, Blomberg OS, Huijbers IJ, van den Berk PC, and Jacobs H (2016). PrimPol prevents APOBEC/AID family mediated DNA mutagenesis. Nucleic Acids Res 44, 4734–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole LA, and Cortez D (2017). Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit Rev Biochem Mol Biol 52, 696–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porebski B, Wild S, Kummer S, Scaglione S, Gaillard PL, and Gari K (2019). WRNIP1 Protects Reversed DNA Replication Forks from SLX4-Dependent Nucleolytic Cleavage. iScience 21, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A (2017). DNA Fiber Analysis: Mind the Gap! Methods Enzymol 591, 55–82. [DOI] [PubMed] [Google Scholar]
- Quinet A, Martins DJ, Vessoni AT, Biard D, Sarasin A, Stary A, and Menck CF (2016). Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Res 44, 5717–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Jackson J, Svikovic S, Lemacon D, Carvajal-Maldonado D, Gonzalez-Acosta D, Vessoni AT, Cybulla E, Wood M, et al. (2020). PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol Cell 77, 461–474 e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, and Lopes M (2012). Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19, 417–423. [DOI] [PubMed] [Google Scholar]
- Sail V, Rizzo AA, Chatterjee N, Dash RC, Ozen Z, Walker GC, Korzhnev DM, and Hadden MK (2017). Identification of Small Molecule Translesion Synthesis Inhibitors That Target the Rev1-CT/RIR Protein-Protein Interaction. ACS Chem Biol 12, 1903–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Hamperl S, Bocek MJ, Chung M, Bass TE, Cisneros-Soberanis F, Samejima K, Xie L, Paulson JR, Earnshaw WC, et al. (2018). An intrinsic S/G2 checkpoint enforced by ATR. Science 361, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale JE (2013). Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb Perspect Biol 5, a012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu S, Wu X, Nabi Z, Rastegar M, Kung S, Mai S, and Ding H (2012). Loss of HLTF function promotes intestinal carcinogenesis. Mol Cancer 11, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugar I, Ortiz-Bazan MA, and Tercero JA (2014). Tolerating DNA damage during eukaryotic chromosome replication. Exp Cell Res 329, 170–177. [DOI] [PubMed] [Google Scholar]
- Schiavone D, Jozwiakowski SK, Romanello M, Guilbaud G, Guilliam TA, Bailey LJ, Sale JE, and Doherty AJ (2016). PrimPol Is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Mol Cell 61, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelinger M, and Otterlei M (2020). Helicase-Like Transcription Factor HLTF and E3 Ubiquitin Ligase SHPRH Confer DNA Damage Tolerance through Direct Interactions with Proliferating Cell Nuclear Antigen (PCNA). Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, and Foiani M (2002). Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297, 599–602. [DOI] [PubMed] [Google Scholar]
- Soss SE, Yue Y, Dhe-Paganon S, and Chazin WJ (2011). E2 conjugating enzyme selectivity and requirements for function of the E3 ubiquitin ligase CHIP. J Biol Chem 286, 21277–21286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svikovic S, Crisp A, Tan-Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, and Sale JE (2019). R-loop formation during S phase is restricted by PrimPol-mediated repriming. EMBO J 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 68, 414–430 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo L, Neelsen KJ, and Lukas J (2017). Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol Cell 66, 735–749. [DOI] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, and Lukas J (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. [DOI] [PubMed] [Google Scholar]
- Unk I, Hajdu I, Blastyak A, and Haracska L (2010). Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair (Amst) 9, 257–267. [DOI] [PubMed] [Google Scholar]
- Vaisman A, and Woodgate R (2017). Translesion DNA polymerases in eukaryotes: what makes them tick? Crit Rev Biochem Mol Biol 52, 274–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallerga MB, Mansilla SF, Federico MB, Bertolin AP, and Gottifredi V (2015). Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc Natl Acad Sci U S A 112, E6624–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K, et al. (2017). Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol Cell 67, 882–890 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Lou J, Xia Y, Su B, Liu T, Cui J, Sun Y, Lou H, and Huang J (2013). hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep 14, 1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston R, Peeters H, and Ahel D (2012). ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev 26, 1558–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RP, Garcia-Rodriguez N, Zilio N, Hanulova M, and Ulrich HD (2020). Processing of DNA Polymerase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol Cell 77, 3–16 e14. [DOI] [PubMed] [Google Scholar]
- Xu X, Lin A, Zhou C, Blackwell SR, Zhang Y, Wang Z, Feng Q, Guan R, Hanna MD, Chen Z, et al. (2016). Involvement of budding yeast Rad5 in translesion DNA synthesis through physical interaction with Rev1. Nucleic Acids Res 44, 5231–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao N, Coryell L, Zhang D, Georgescu RE, Finkelstein J, Coman MM, Hingorani MM, and O’Donnell M (2003). Replication factor C clamp loader subunit arrangement within the circular pentamer and its attachment points to proliferating cell nuclear antigen. J Biol Chem 278, 50744–50753. [DOI] [PubMed] [Google Scholar]
- Yuan J, Ghosal G, and Chen J (2012). The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol Cell 47, 410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T, Kong X, Yokomori K, and Kadonaga JT (2009). The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev 23, 2400–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, and Lopes M (2018). Dynamic Architecture of Eukaryotic DNA Replication Forks In Vivo, Visualized by Electron Microscopy. Methods Mol Biol 1672, 261–294. [DOI] [PubMed] [Google Scholar]
- Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets or code.