

Summary
Cell death pathways regulate various homeostatic processes. Autoimmune lymphoproliferative syndrome (ALPS) in humans and Lymphoproliferative (LPR) disease in mice are a result of abrogated CD95-induced apoptosis. Because Caspase-8 mediates CD95 signaling, we applied genetic approaches to dissect the roles of Caspase-8 in cell death and inflammation. Here we describe oligomerization-deficient Caspase-8F122GL123G/F122GL123G and non-cleavable Caspase-8D387A/D387A mutant mice with defective Caspase-8-mediated apoptosis. Although neither mouse developed LPR disease, removal of the necroptosis effector Mlkl from Caspase-8D387A/D387A mice revealed an inflammatory role for Caspase-8. Ablation of one allele of Fasl, Fadd, or Ripk1 prevented the pathology of Casp8D387A/D387AMlkl−/− animals. Removing both Fadd alleles from these mice resulted in early lethality prior to post-natal day 15 (P15), which was prevented by co-ablation of either Ripk1 or Caspase-1. Our results suggest an in vivo role for the inflammatory RIPK1-Caspase-8-FADD (FADDosome) complex and reveal a FADD-independent inflammatory role for Caspase-8 that involves activation of an inflammasome.
Graphical Abstract
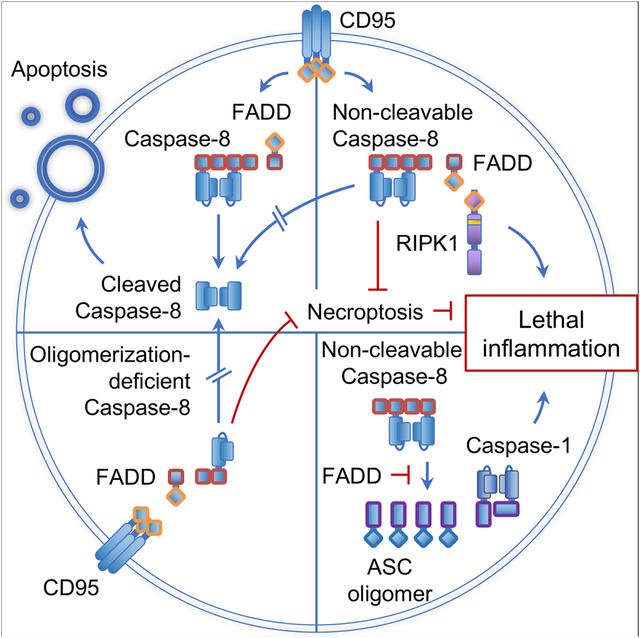
Introduction
Fas (CD95) is a death receptor that belongs to the tumor necrosis factor receptor (TNF-R) family, and its ligation can trigger apoptosis (Strasser et al., 2009). CD95 is expressed on a variety of cells, such as epithelial cells, endothelial cells and immune cells, and is especially important for the regulation of homeostasis of the immune system. Autoimmune lymphoproliferative syndrome (ALPS) is a disorder characterized by the accumulation of TCRαβ+CD3+B220+CD4−CD8− T cells, resulting in lymphadenopathy, splenomegaly, hepatomegaly, autoimmunity and, in some cases, lymphoma (Meynier and Rieux-Laucat, 2019). The equivalent disease in mice is called lymphoproliferative (LPR) syndrome. It is well established that ALPS and LPR disease are a result of abrogated CD95 signaling, either by mutations in the receptor itself, by mutations of its ligand CD95L, or by deleterious mutations in the proteins involved in CD95 signaling (Meynier and Rieux-Laucat, 2019).
The induction of CD95-mediated apoptosis has been well described. Ligation by CD95L activates CD95, leading to oligomerization and the recruitment of the FADD adaptor molecule (Scott et al., 2009). FADD, in turn, recruits the initiator caspase, Caspase-8, thereby forming the death-inducing signaling complex (DISC) on the cytoplasmic tail of CD95. Caspase-8 oligomerizes and subsequently autoproteolytically processes into activated, stabilized dimers that mediate the induction of apoptosis (Pop et al., 2007; Strasser et al., 2009) Caspase-8 binds to FADD via the first (DED1) of its two DED domains. Oligomerization then ensues through the interaction of a pocket in its DED2 domain, which consists of the residues F122L123, with the DED1 domain of a second Caspase-8 molecule, a process that continues with successive interactions (Fu et al., 2016; Hughes et al., 2016). Caspase-8 oligomerization induces its autoproteolytic processing, which initiates at D387, followed by cleavage at D373. The last cleavage at D218 stabilizes and releases the active Caspase-8 dimers into the cytosol, thereby allowing the initiation of apoptosis by processing target proteins (Keller et al., 2009; Pop et al., 2007).
The induction of apoptosis is regulated by the expression of cFLIPL, a Caspase-8-like molecule that does not have catalytic activity and abrogates the ability of Caspase-8 to trigger cell death (Irmler et al., 1997), despite the proteolytic activity of the Caspase-8-FLIP heterodimer (Pop et al., 2011).
Besides mediating death receptor-induced apoptosis, Caspase-8 and FADD have been shown to have other functions. Mice lacking Fadd or Casp8 are embryonically lethal but survive past weaning when either one of the necroptosis-mediating genes, Ripk3 or Mlkl, are co-ablated (Alvarez-Diaz et al., 2016; Dillon et al., 2012; Dillon et al., 2014; Kaiser et al., 2011; Oberst et al., 2011), showing that another function of the FADD-Caspase-8-cFLIPL complex is to block necroptosis via its proteolytic activity.
FADD-Caspase-8 also functions as a scaffold to support cellular signaling. Ligation of TRAIL receptor (TRAIL-R), a death receptor belonging to the same family as CD95, can induce gene expression through the formation of a signaling complex called the FADDosome (Grunert et al., 2012; Hartwig et al., 2017; Henry and Martin, 2017). The FADDosome is a complex of FADD and Caspase-8 that have dissociated from the ligated receptor and have recruited RIPK1 as well as other proteins involved in RIPK1-mediated transcription factor activation (Henry and Martin, 2017). It is currently unknown whether death receptor family members other than TRAIL-R can mediate FADDosome-dependent gene expression upon ligation.
Because mice that are defective for CD95 (Lpr mice) or CD95L (Gld mice), or lacking Fadd or Casp8 in a necroptosis-deficient background, invariably succumb to LPR syndrome, it has been understood for many years that a defect in CD95-induced apoptosis is responsible for this syndrome. Here, we describe two animals in which Caspase-8-mediated apoptosis is defective, but which do not develop LPR disease. These include an oligomerization-deficient Caspase-8 that contains mutations F122G and L123G in the binding pocket in DED2, and a non-cleavable Caspase-8 that contains the D387A cleavage site mutation. Crossing these mice to a necroptosis-deficient background revealed that Caspase-8 plays a role in inflammation that depends on its ability to oligomerize and can only be observed in a setting where neither apoptosis nor necroptosis ensues. Moreover, FADD inhibited an inflammatory function of Caspase-8 that involved RIPK1 and Caspase-1 (and/or Caspase-11). Using these genetic approaches, we have found two ways in which Caspase-8 influences inflammation: it mediates gene expression in a FADDosome-dependent manner, and it activates inflammasome components in epithelial cells in a process that is inhibited by FADD and Caspase-8 auto-cleavage.
Results
Animals expressing oligomerization-deficient or non-cleavable Caspase-8 do not develop LPR disease
Murine Caspase-8 activation requires oligomerization, dependent on the F122L123 pocket in DED2, and subsequent autocleavage at D387 (Fu et al., 2016; Hughes et al., 2016; Shen et al., 2018). We generated two Caspase-8 mutants that were expected to be unable to fully activate Caspase-8. In the first mutant, F122 and L123 were mutated to glycine (Caspase-8 FGLG; (Fu et al., 2016; Hughes et al., 2016), while in the second, D387 was mutated to alanine (Caspase-8 DA). We examined the functions of mutant Caspase-8 in vitro by introducing WT, catalytically inactive Caspase-8 C362A (CA), Caspase-8 DA, or Caspase-8 FGLG into cells that lack Caspase-8 and express MLKL when treated with doxycycline. Cells were then subjected to treatment with CD95L to induce apoptosis (Figure S1A, C), or polyI:C and doxycycline to induce necroptosis (Figure S1B). While neither mutant promoted apoptosis by CD95 ligation (Figure S1A, C), both the Caspase-8 DA and Caspase-8 FGLG mutants, but not the Caspase-8 CA mutant, prevented necroptosis (Figure S1B). Addition of the caspase inhibitor zVAD-fmk permitted polyI:C-induced necroptosis in cells expressing either mutant (Figure S1B), consistent with the idea that the catalytic activity of these mutant caspases, likely in complex with c-FLIPL, is required to prevent necroptosis (Oberst et al., 2011; Pop et al., 2011).
Mice lacking Casp8 die during gestation as a consequence of necroptosis during embryogenesis (Alvarez-Diaz et al., 2016; Dillon et al., 2012; Dillon et al., 2014; Kaiser et al., 2011; Oberst et al., 2011). Because both the Caspase-8 DA and Caspase-8 FGLG mutants blocked necroptosis in vitro, we hypothesized that these mutants should also be able to block necroptosis during mouse embryogenesis, and therefore animals harboring the mutations should be viable. Using CRISPR-Cas9, we generated animals that harbor either the Caspase-8D387A/D387A (Casp8DA/DA) or the Caspase-8F122GL123G/F122GL123G (Casp8FGLG/FGLG) mutations. Both mutant animals were viable and weaned at Mendelian frequencies (Figure S1D, E). Mice with either Caspase-8 mutation were relatively resistant to the lethal effects of administration of the agonistic anti-CD95 antibody, Jo2 (Figure 1A) (Kang et al., 2008). Removal of one DA allele yielded a Casp8DA/− mouse, which showed complete resistance to Jo2 injection (Figure 1A). These observations indicate that Casp8DA/DA, Casp8DA/− and Casp8FGLG/FGLG mutant animals retain the ability to block necroptosis in vivo and are relatively resistant to CD95-mediated apoptosis.
Figure 1: Casp8FGLG/FGLG and Casp8DA/DA mice do not develop LPR syndrome.
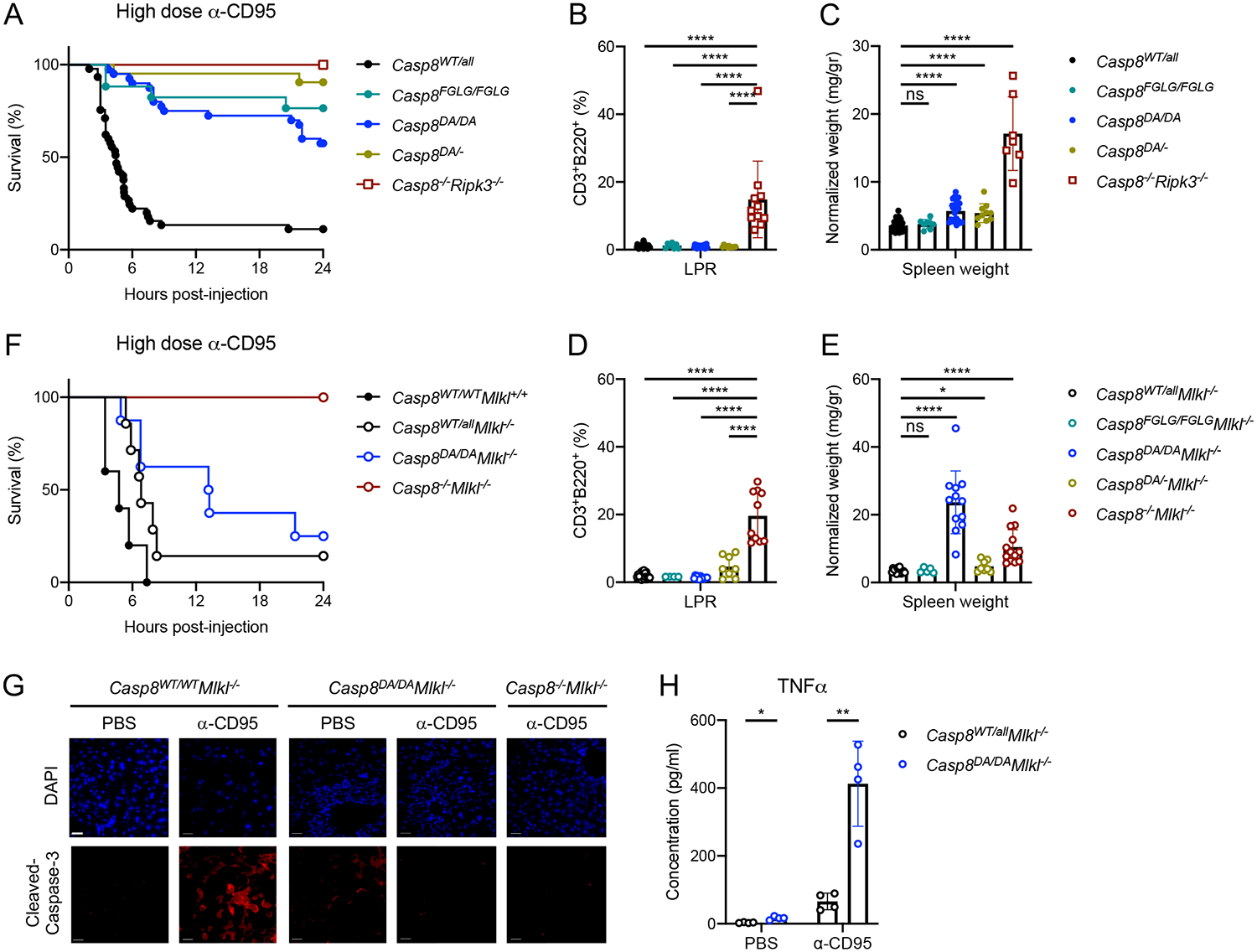
A: Survival of mice of the indicated genotypes after 1.2 mg/kg IP-injected α-CD95 (Jo2 antibody). Casp8WT/all n=45, Casp8FGLG/FGLG n=11, Casp8DA/DA n=40, Casp8DA/− n=21, Casp8−/−Ripk3−/− n=4.
B, D: Splenic CD3+B220+ cell percentages in mice of the indicated genotypes as determined by flow cytometry.
C, E: Normalized spleen weight of mice of the indicated genotypes.
F: Survival of mice of the indicated genotypes upon 1.2 mg/kg IP-injected α-CD95. Casp8WT/WTMlkl+/+ n=5, Casp8WT/allMlkl−/− n=7, Casp8DA/DAMlkl−/− n=8, Casp8−/−Mlkl−/− n=3.
G: Immunofluorescence (IF) images of liver sections of mice 3 hours post-IP injection with 1.2 mg/kg α-CD95 or PBS. Representative of 2 independent experiments. Scale bar, 50 μm.
H: TNFα concentration in sera of mice 4 hours post-IP injection with 1.2 mg/kg α-CD95 or PBS as measured by ELISA.
**** p<0.0001, ** p<0.005, * p<0.05, ns not significant. Please also see Figure S1.
While LPR disease is believed to be caused by abrogation of CD95-mediated apoptosis, neither mutant mouse developed LPR disease, as assessed by accumulation of splenic CD3+B220+ cells (Figure 1B) and splenomegaly (Figure 1C). Similar to previous reports (Kang et al., 2008; Newton et al., 2019a), thymocytes of the Casp8DA/DA animals were susceptible to CD95L-induced apoptosis (Figure S1F). Similarly, CD95 ligation induced apoptosis of Casp8FGLG/FGLG and Casp8DA/− thymocytes (Figure S1F), albeit to a lesser extent than Caspase-8 WT thymocytes. The susceptibility of the thymocytes of these mutant animals to CD95-mediated death possibly explains the absence of LPR syndrome in the adult animals.
Analysis of the spleens of the mutant mice showed that Casp8FGLG/FGLG animals did not develop splenomegaly (Figure 1C) or alterations in the composition or activation status of hematopoietic cells, as defined by MHC II expression on macrophages and CD44 expression on T lymphocytes (Figure S2A–E). Casp8DA/DA and Casp8DA/− animals developed mild splenomegaly (Figure 1C) with moderately activated splenic myeloid cells (Figure S3D), suggestive of a mild inflammatory syndrome. However, other immune cell populations were unaffected (Figure 2C–E and S3A–G).
Figure 2: Necroptosis controls lethal inflammation in Casp8DA/DA mice.
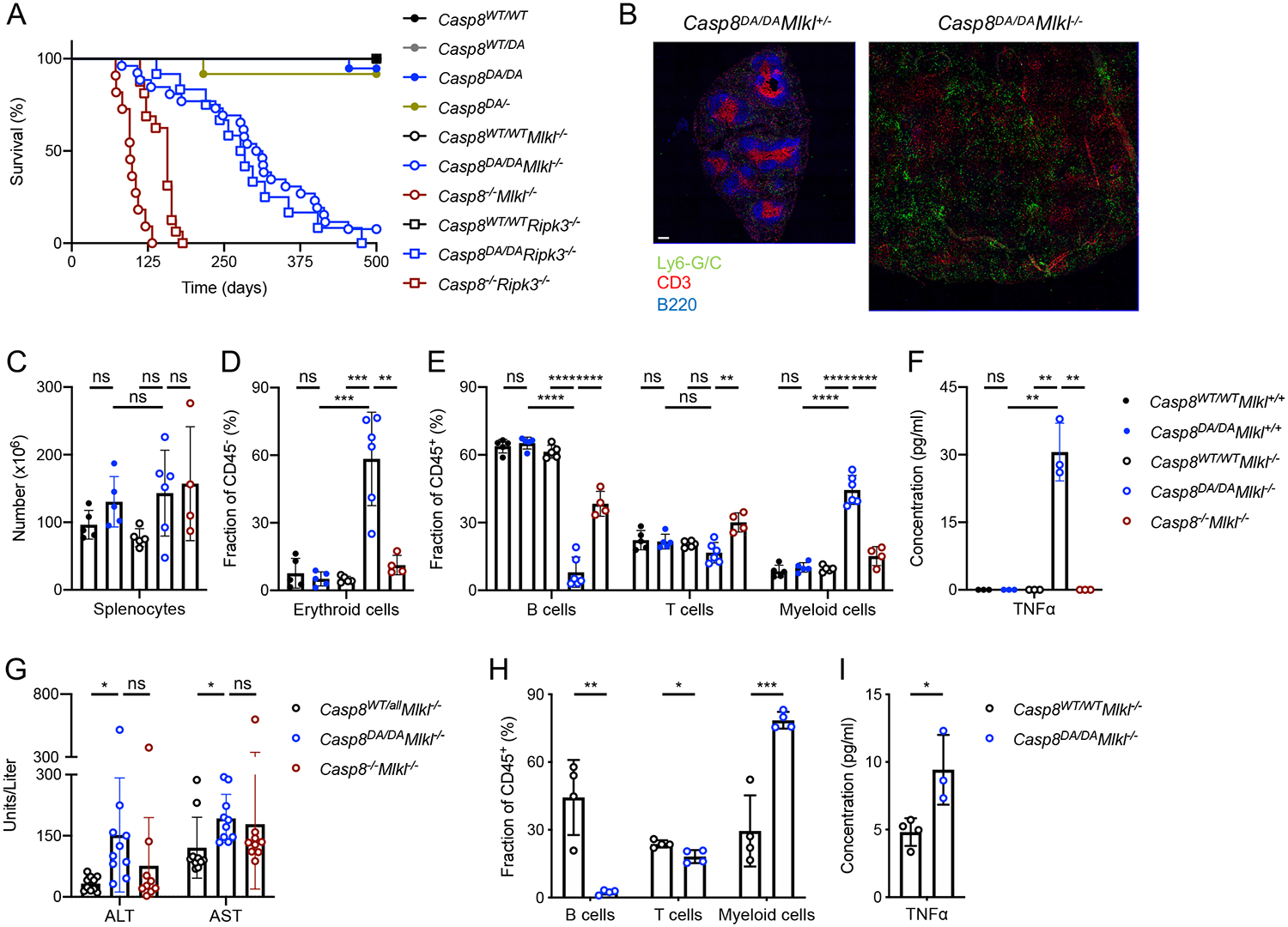
A: Survival of mice of the indicated genotypes. Casp8WT/WT n=16, Casp8WT/DA n=22, Casp8DA/DA n=19, Casp8DA/− n=12, Casp8WT/WTMlkl−/− n=17, Casp8DA/DAMlkl−/− n=25, Casp8−/−Mlkl−/− n=11, Casp8WT/WTRipk3−/− n=7, Casp8DA/DARipk3−/− n=11, Casp8−/−Ripk3−/− n=17.
B: IF images of spleen sections stained for Ly6-G/C (Monocytes/Neutrophils), CD3 (T cells) and B220 (B cells). Representative of 2 independent experiments. Scale bar, 100 μm.
C: Total splenic cell numbers.
D, E: Percentages of splenic erythroid cells (D), B cells, T cells and myeloid cells (E) as determined by flow cytometry.
F: Serum TNFα as measured by Multiplex ELISA.
G: Serum ALT and AST as measured by ELISA.
Irradiated Rag1−/− mice were reconstituted with bone-marrow from Mlkl−/− or Casp8DA/DAMlkl−/− mice.
H: Percentages of B cells, T cells and myeloid cells in blood 6 weeks post-transfer as determined by flow cytometry.
I: Serum TNFα as measured by ELISA.
**** p<0.0001, *** p<0.0005, ** p<0.005, * p<0.05, ns not significant. Please also see Figures S2 and S3.
Necroptosis prevents lethal inflammation in Casp8DA/DA mice
Although the Caspase-8 DA and FGLG mutations inhibited necroptosis, it remained possible that the absence of LPR syndrome was due to necroptotic death of the precursors of the CD3+B220+ cells. Abrogating necroptosis by ablation of Mlkl in Casp8FGLG/FGLG mice yielded viable animals that did not develop LPR disease (Figure 1D), splenomegaly (Figure 1E), alterations in splenic hematopoietic cells (Figures S2A–E) or any other readily detectable phenotype. Likewise, crossing the Casp8DA/DA mouse to a necroptosis-deficient background by removing either Ripk3 or Mlkl did not result in the development of LPR disease (Figures 1D and S4A), suggesting that the necroptosis pathway does not influence the development of LPR syndrome. However, Casp8DA/DAMlkl−/− and Casp8DA/DARipk3−/− mice showed an exacerbated inflammatory phenotype, indicated by pronounced splenomegaly (Figures 1E and S4B). Furthermore, the Casp8DA/DAMlkl−/− mice succumbed to the lethal effects of the in vivo administration of the agonistic anti-CD95 antibody Jo2 (Figure 1F). This lethality was not due to liver failure by apoptosis, as indicated by the healthy appearance of the liver and absence of cleaved Caspase-3 staining, indicative of hepatocytes undergoing anti-CD95-induced apoptosis (Figure 1G). Instead, Jo2 treatment induced a pronounced increase in the pro-inflammatory cytokine TNFα in the serum of these animals (Figure 1H), suggesting that the Casp8DA/DAMlkl−/− mice died as a result of CD95 ligation-induced cytokine production. CD95 ligation-induced serum TNFα in Casp8FGLG/FGLGMlkl−/− mice did not differ from Casp8WT/WTMlkl−/− mice (Figure S2F), therefore, the failure to cleave Caspase-8 in the Casp8DA/DAMlkl−/− mice, rather than the absence of apoptosis (in the Casp8FGLG/FGLGMlkl−/− mice), appears to be responsible for the hyper-inflammatory effect of CD95 ligation. These results indicate that Caspase-8 is able to mediate lethal, pro-inflammatory cytokine production in response to CD95 ligation in vivo.
The Casp8DA/DAMlkl−/− and Casp8DA/DARipk3−/− mice developed pronounced splenomegaly (Figures 1E and S4B) and died prematurely (Figure 2A). Examination of the spleen showed alterations in the splenic architecture (Figures 2B, S4J) and splenic immune cell subsets (Figures 2C–E, S3A, B, S4C–G). Erythroid precursor cells (CD45−TER-119+) and myeloid cells (CD45+CD11b+; predominantly neutrophils, Ly6C+Ly6G+) were increased, whereas T cell (CD45+CD3+) and B cell (CD45+CD19+B220+) populations were decreased or almost absent, respectively. Furthermore, all immune cell populations expressed markers indicative of their activation (Figures S3C–G, S4H, I) and serum pro-inflammatory cytokines were elevated (Figures 2F, S4K). Concomitantly, bone-marrow hematopoiesis was skewed towards a myeloid output (MPP3 (Pietras et al., 2015), Figures S3H, I). Moreover, Casp8DA/DAMlkl−/− animals showed increased serum ALT and AST, indicative of liver damage (Figure 2G). IgG deposition in the kidney glomeruli, indicative of autoimmunity in Casp8−/−Mlkl−/− mice, was not detected in Casp8DA/DAMlkl−/− animals (Figure S3J). The phenotype of Casp8DA/DAMlkl−/− mice was also observed upon transfer of bone marrow from Casp8DA/DAMlkl−/− mice to irradiated Rag1−/− recipient animals (Figure 2H, I), suggesting that it is mediated by the hematopoietic compartment.
Altogether, our results show that ablation of necroptosis in the cleavage-defective Caspase-8 mutant mouse does not support LPR development. Instead, non-cleavable Caspase-8 DA induces an inflammatory environment that is controlled by the ability of cells to undergo necroptosis, suggesting a role for necroptosis in immune homeostasis in vivo.
Casp8DA/DAMlkl−/− mice are hypersensitive to the lethal effects of bacterial LPS
The increase of activated immune cells and myeloid cell populations, and the absence of autoimmunity in the Casp8DA/DAMlkl−/− mice suggested that the animals might be reacting to exogenous stimuli, such as the microbiome, resident in our colony. Therefore, directly upon weaning, Casp8DA/DAMlkl−/− mice were treated with an antibiotic cocktail in the drinking water for 60 days. This treatment reduced spleen weight, the myeloid cell population, and pro-inflammatory cytokines in the sera (Figure 3A–C), suggesting that the microbiome, at least in part, promotes the inflammatory phenotype of the Casp8DA/DAMlkl−/− mice.
Figure 3: Casp8DA/DAMlkl−/− mice are hyper-responsive to microbial ligands.
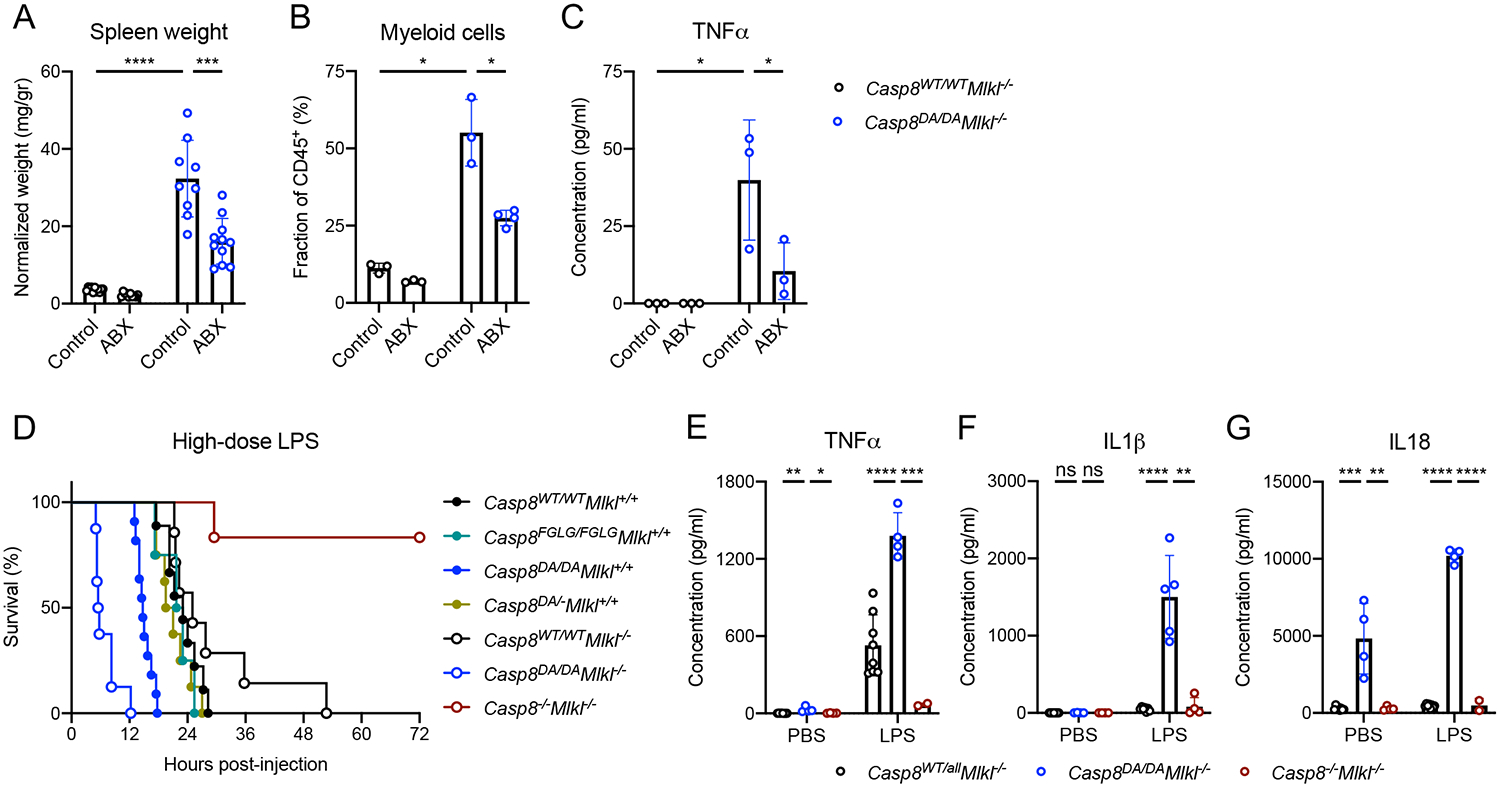
A: Normalized spleen weight.
B: Splenic myeloid cell numbers as determined by flow cytometry.
C: Serum TNFα as measured by ELISA.
D: Survival of mice of the indicated genotypes upon 56 mg/kg IP-injected LPS. Casp8WT/WTMlkl+/+ n=9, Casp8FGLG/FGLGMlkl+/+ n=4, Casp8DA/DAMlkl+/+ n=11, Casp8DA/−Mlkl+/+ n=8, Casp8WT/WTMlkl−/− n=9, Casp8DA/DAMlkl−/− n=8, Casp8−/−Mlkl−/− n=6.
E-G: Serum TNFα (E), IL1β (F), and IL18 (G) in mice 2 hours post-IP injection with 56 mg/kg LPS or PBS as measured by ELISA.
ABX: antibiotics, **** p<0.0001, *** p<0.0005, ** p<0.005, * p<0.05, ns not significant
The absence of Caspase-8-mediated apoptosis is known to protect against the lethal effects of LPS-induced septic shock (Kaiser et al., 2011; Mandal et al., 2018). Since our non-cleavable Caspase-8 mutant is defective in inducing apoptosis, we expected that our mutant animals would also be protected from LPS-induced septic shock. However, both Casp8DA/DA and Casp8DA/DAMlkl−/− animals succumbed more rapidly than WT or Mlkl−/− mice upon high-dose LPS injection, whereas Casp8−/− Mlkl−/− mice were protected (Figure 3D), as previously described for Casp8−/−Ripk3−/− mice (Kaiser et al., 2011; Mandal et al., 2018). Casp8FGLG/FGLG and Casp8DA/− mice were similar to wild-type animals in their susceptibility to LPS-induced death. The cytokines TNFα, IL1β and IL18 were markedly increased in the sera of Casp8DA/DAMlkl−/− mice compared to the other genotypes (Figure 3E–G). These results indicate that it is not the absence of Caspase-8-mediated apoptosis, per se, that protects against the lethal effects of high-dose LPS injection. Rather, Caspase-8 appears to mediate pro-inflammatory cytokine production in response to LPS, a process that is restricted by Caspase-8 cleavage.
The inflammasome components Caspase-1 and Caspase-11 do not mediate the inflammatory phenotype of Casp8DA/DAMlkl−/− mice
LPS resistance in Casp8−/−Mlkl−/− mice suggested an interaction between Caspase-8 and Caspases-1 and -11 (Mandal et al., 2018). Since the Casp8DA/DAMlkl−/− animals were hypersensitive to LPS-induced septic shock and showed increased LPS-induced IL1β and increased constitutive IL18 in sera, we hypothesized that an interaction between Caspase-8 DA and the inflammasome could drive the inflammatory phenotype of the Casp8DA/DAMlkl−/− animals. Therefore, we genetically removed the inflammasome components Caspase-1 (Casp1−/−) from the Casp8DA/DAMlkl−/− mice. It should be noted that this Casp1-null allele is also deficient in functional Casp11 (Kayagaki et al., 2011). Although constitutive serum IL18 was now absent (Figure S5A), the inflammatory phenotype remained present in the Casp1−/−Casp8DA/DAMlkl−/− (Figure S5B–I), suggesting that neither the inflammasome nor IL18 play a role in the pathology of the Casp8DA/DAMlkl−/− animals.
The inflammatory phenotype of Casp8DA/DAMlkl−/− mice is dependent on the FADDosome
Caspase-8 is present in a complex called the FADDosome (Henry and Martin, 2017), which induces gene expression upon activation of TRAIL receptor (TRAIL-R) (Hartwig et al., 2017; Henry and Martin, 2017). In this complex, Caspase-8 binds with FADD and RIPK1 to promote signaling and cytokine production. A role for the FADDosome in CD95-mediated gene expression has not been described. The exacerbated serum cytokine in response to CD95-ligation or LPS in the Casp8DA/DAMlkl−/− mice suggested that these cytokines could be produced in a FADDosome-dependent manner. Previously, it was shown that Caspase-8 is required for CD95 ligation-induced cytokine secretion in macrophages (Bossaller et al., 2012). Similarly, the absence of Caspase-8 abrogated the secretion of TNFα in LPS-pretreated, CD95L-stimulated bone marrow-derived macrophages (BMDMs) (Figure 4A). The inability of Caspase-8 DA to self-cleave enhanced TNFα secretion, whereas the inability to oligomerize decreased TNFα secretion (Figure 4A). CD95L-induced gene expression of TNFα was similar between Caspase-8 DA and Caspase-8 WT cells, whereas in Caspase-8 FGLG cells TNFα expression was blunted (Figure 4B). These results suggested that the formation of the FADDosome requires the ability of Caspase-8 to oligomerize. Indeed, CD95L induced the interaction between RIPK1, FADD and Caspase-8 DA, but not FGLG (Figure 4C). Addition of zVAD-fmk also allowed the interaction between RIPK1 and Caspase-8 WT and FGLG, albeit only slightly. This may be consistent with the idea that complexes of Caspase-8 WT or FGLG are able to cleave RIPK1, perhaps in complex with cFLIPL. The enhanced TNFα secretion by Caspase-8 DA BMDMs was not dependent on the kinase activity of RIPK1, since the addition of the RIPK1 inhibitor, Nec1s, did not reduce secreted TNFα (Figure S6A). CD95 ligation on BMDMs generated from Casp8DA/DAMlkl−/−Casp1−/−, Casp8DA/DA Fadd−/−Mlkl−/−Casp1−/− and Casp8WT/WTFadd−/−Mlkl−/−Casp1−/− animals (discussed in more detail below) showed that FADD was required for the optimal expression of TNFα and IL1β (Figure 4D, E).
Figure 4: Deletion of one allele of Fadd or Ripk1 abrogates the phenotype of Casp8DA/DAMlkl−/−mice.
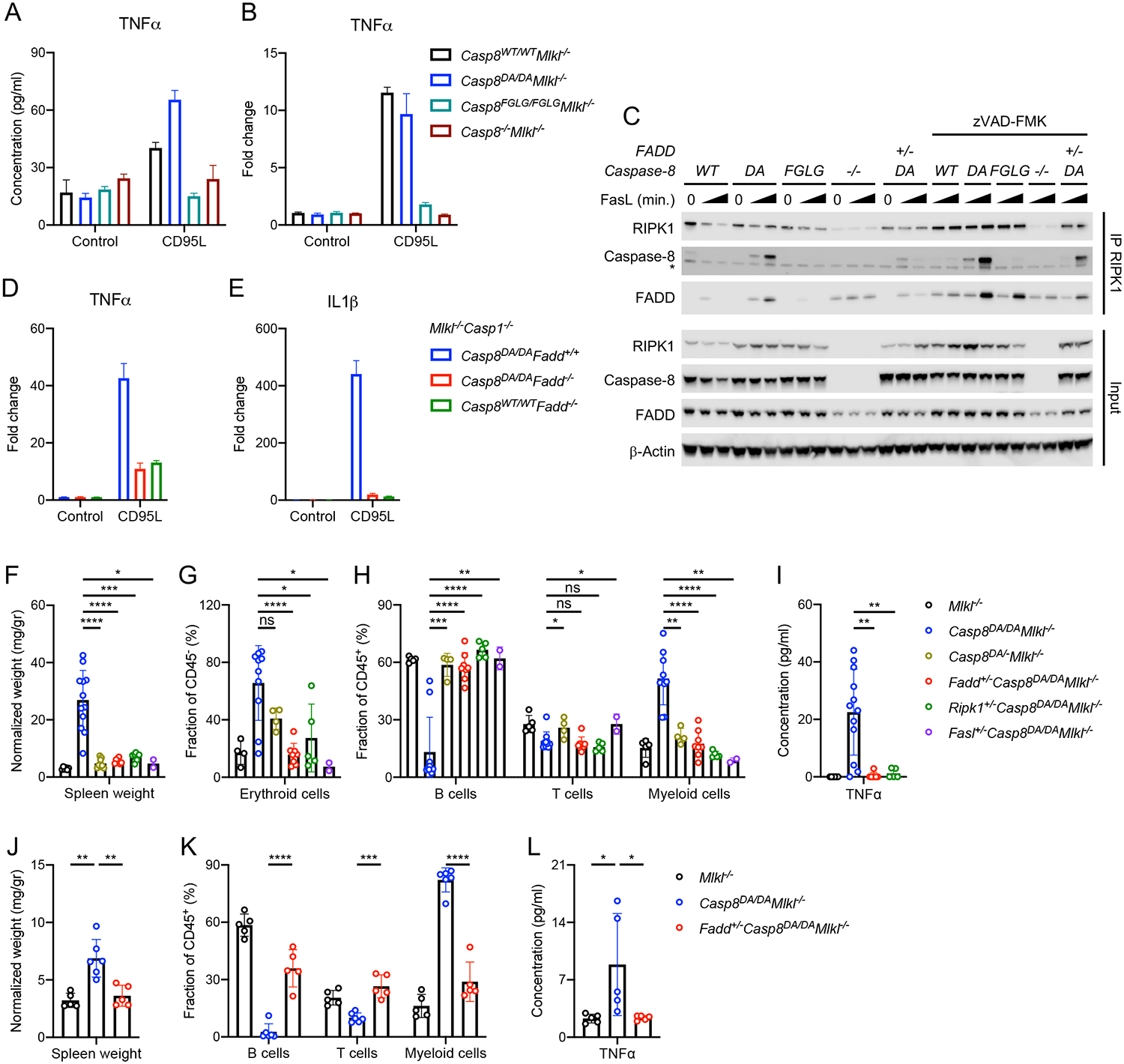
BMDMs generated from mice of the indicated genotypes were pre-treated with LPS and stimulated with CD95L.
A: TNFα in supernatant 24 hours post-CD95L stimulation.
B: Fold change of TNFα gene expression over unstimulated control 2 hours post-CD95L stimulation.
C: Western blots for RIPK1, Caspase-8, FADD and β-Actin. RIPK1 was immunoprecipitated (IP) from BMDMs that were pre-treated with LPS and stimulated with CD95L for 0, 60 or 120 minutes in presence or absence of zVAD-fmk. * IgG heavy chain.
D, E: Fold change of TNFα (D) and IL1β (E) gene expression over unstimulated control 2 hours post-CD95L stimulation.
F: Normalized spleen weight of mice of the indicated genotypes.
G, H: Percentages of splenic erythroid cells (G), B cells, T cells and myeloid cells (H) as determined by flow cytometry.
I: Serum TNFα as measured by ELISA.
Irradiated Rag1−/− mice were reconstituted with bone-marrow from mice of the indicated genotypes.
J: Normalized spleen weight.
K: Percentages of B cells, T cells and myeloid cells in blood 6 weeks post-transfer as determined by flow cytometry.
L: Serum TNFα as measured by ELISA.
**** p<0.0001, *** p<0.0005, ** p<0.005, * p<0.05, ns not significant. Please also see Figure S4.
Together, these results suggest that the increased pro-inflammatory cytokines by Casp8DA/DAMlkl−/− macrophages are a result of both FADDosome formation, defective in Casp8FGLG/FGLGMlkl−/− cells, as well as defective CD95-induced apoptosis, which is intact in the Casp8WT/WTMlkl−/− setting.
Since BMDMs generated from Casp8DA/DAMlkl−/− animals were able to induce TNFα secretion upon CD95 ligation, we surmised that blunting CD95L or the FADDosome by reducing the components (CD95L, FADD, RIPK1, and Caspase-8 DA) would prevent the inflammatory phenotype of the Casp8DA/DAMlkl−/− animals. Indeed, blunting the FADDosome by deletion of one allele of Casp8DA, Fadd, Ripk1 or Fasl abrogated the inflammatory phenotype of Casp8DA/DAMlkl−/− mice (Figure 4F–I). Furthermore, Fadd+/−Casp8DA/DAMlkl−/− animals were less susceptible to the lethal effects of LPS, as indicated by improved survival (Figure S6B) and decreased serum TNFα and IL1β (Figure S6C–E). Basal as well as LPS-induced serum IL18 were higher in the Fadd+/−Casp8DA/DAMlkl−/− animals (Figure S6E). Bone marrow transfer experiments showed that Fadd+/−Caspase-8DA/DAMlkl−/− recipient animals did not develop the inflammatory phenotype observed in animals that received bone marrow from Casp8DA/DAMlkl−/− mice (Figure 4J–L).
Removal of one allele of Fadd blunted FADDosome formation in CD95L-treated BMDMs (Figure 4C), although removal of one allele of Fadd or Ripk1 did not result in reduction of TNFα secretion by CD95L-treated BMDMs (Figure S6A).
Collectively, these data indicate that the inflammatory phenotype of the Casp8DA/DAMlkl−/− mouse is dependent on CD95 signaling mediated by FADD, RIPK1 and Caspase-8 DA and suggest an in vivo function of the FADDosome in hematopoietic cells.
Complete ablation of the FADDosome component FADD exacerbates inflammation in Casp8DA/DAMlkl−/− mice
Since removal of one allele of Fadd prevented the inflammatory phenotype of the Casp8DA/DAMlkl−/− mice, we generated Casp8DA/DAMlkl−/− animals that were completely devoid of Fadd. At birth, the pups were indistinguishable from their littermates, were accepted by their parents and suckled normally. However, unexpectedly, the pups displayed runting several days after birth and did not survive beyond two weeks of age (Figure 5A). These animals showed a highly inflammatory phenotype with extensive immune cell infiltrates in the skin, intestine, spleen (Figure 5B) and lungs, which predominantly consisted of myeloid cells (Figure 5C; F4/80+ macrophages).
Figure 5: FADD blocks RIPK1- and Caspase-1-mediated early lethality of Casp8DA/DAMlkl−/− mice.
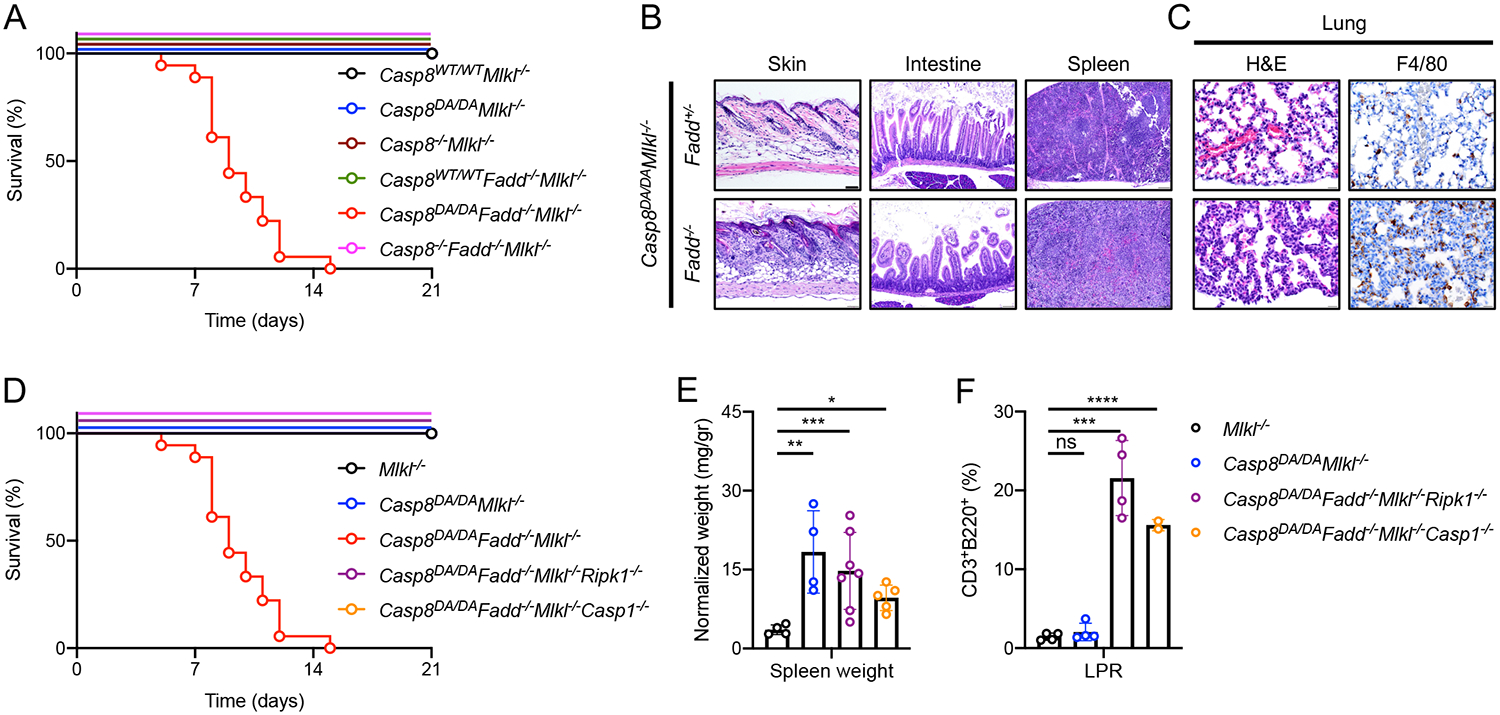
A: Survival of mice of the indicated genotypes. Casp8WT/WTMlkl−/− n>20, Casp8DA/DAMlkl−/− n>20, Casp8−/−Mlkl−/− n>20, Casp8WT/WTFadd−/−Mlkl−/− n>20, Casp8DA/DAFadd−/−Mlkl−/− n=18, Casp8−/−Fadd−/−Mlkl−/− n=7.
B: Images of H&E of skin (left), intestine (middle) and spleen (right). Representative of 2 independent experiments.
C: Images of lung tissue section stained with H&E (left) and IHC for F4/80 (right). Representative of 2 independent experiments. Scale bar, 50 μm.
D: Survival of mice of the indicated genotypes. Casp8DA/DAMlkl−/− n>20, Casp8DA/DAFadd−/−Mlkl−/− n=18, Casp8DA/DAFadd−/−Mlkl−/−Ripk1−/− n=14, Casp8DA/DAFadd−/−Mlkl−/−Casp1−/− n=15. The Fadd−/−Casp8DA/DAMlkl−/− data set is the same set as in A.
E: Normalized spleen weight of mice of the indicated genotypes.
F: CD3+B220+ cell percentages in mice of the indicated genotypes as determined by flow cytometry.
**** p<0.0001, *** p<0.0005, ** p<0.005, * p<0.05, ns not significant.
Fadd−/−Mlkl−/− mice reach adulthood normally and develop LPR disease in later life (Alvarez-Diaz et al., 2016). To evaluate whether the inflammatory phenotype of the Casp8DA/DAFadd−/−Mlkl−/− mice was due to Caspase-8 DA signaling, we generated and evaluated Casp8−/−Fadd−/−Mlkl−/− mice. These animals were born at Mendelian frequencies and survived to adulthood (Figure 5A). This suggests that Caspase-8 has an inflammatory function that is independent of FADD and is inhibited by auto-cleavage of the caspase. Furthermore, these data indicate that FADD suppresses a function of Caspase-8 DA that promotes inflammation.
The pathology of Casp8DA/DAFadd−/−Mlkl−/− mice is mediated by an inflammasome and RIPK1
We had observed that Caspase-1 and −11 do not promote the inflammatory phenotype of Casp8DA/DAMlkl−/− animals (Figure S5). However, ablation of either Ripk1 or Casp1 completely prevented the early lethality of the Casp8DA/DAFadd−/−Mlkl−/− animals (Figure 5D). Moreover, these Casp8DA/DAFadd−/−Mlkl−/−Casp1−/− and Casp8DA/DAFadd−/−Mlkl−/−Ripk1−/− animals developed LPR disease (Figure 5E, F). These data suggest that Caspase-8 DA mediates Caspase-1 (and −11)-dependent inflammation in a process that is inhibited by FADD and self-cleavage.
Caspase-8 influences ASC speck formation and Caspase-1 and/or −11-mediated inflammasome activation in non-immune cells
Our results showed that the Casp8DA/DAFadd−/−Mlkl−/− animals invariably died by a Caspase-1 and/or −11-dependent mechanism. The prevention of the early lethality of Casp8DA/DAFadd−/−Mlkl−/− animals by ablation of Caspase-1 and −11 implied that non-cleavable Caspase-8 may have the ability to interact with components of the inflammasome. Interactions between Caspase-8 and inflammasomes have been suggested (Gurung and Kanneganti, 2015; Lee and Kang, 2019). We therefore asked if our Caspase-8 mutants generally interact with inflammasomes in myeloid cells. We generated primary bone-marrow derived macrophages (BMDMs) from our necroptosis-deficient Caspase-8 mutant animals, primed these cells with LPS and induced classical inflammasome activation by addition of nigericin. Neither the Caspase-8 DA nor the FGLG mutants differed from wild-type Caspase-8; all three induced ASC oligomerization (Figure S7A) and ASC speck formation (Figure S7B). However, ASC oligomerization and speck formation were blunted in the absence of Caspase-8 (Figure S7A, B), as described (Gurung et al., 2014; Lukens et al., 2014). Amounts of the inflammasome components NLRP3, ASC, and Caspase-1 were not different among these cells, suggesting a supporting role for Caspase-8 in inflammasome activation, independent of its oligomerization or self-cleaving abilities.
Our genetic data implied that the absence of FADD exacerbates inflammasome activation in vivo. We therefore generated BMDMs from Fadd proficient and deficient Casp1−/−Casp8DA/DAMlkl−/− animals and asked how the absence of Fadd altered nigericin-mediated ASC oligomerization and speck formation. However, the absence of FADD resulted in blunted ASC oligomerization (Figure S7C) and speck formation (Figure S7D), similar to our observations in Caspase-8-deficient cells.
It remained possible that the Caspase-8 DA mutant could affect bacteria-mediated inflammasome activation. We therefore generated fetal liver-derived macrophages (FLDMs) from Fadd proficient and deficient Casp8DA/DAMlkl−/− embryos and infected these cells with E. coli to activate the inflammasome. Caspase-1, IL1β and Gasdermin D cleavage and release were not affected by the mutants (Figure S7E). However, similar to our observation using nigericin, the absence of Caspase-8 or FADD blunted E. coli-induced inflammasome activation (Figure S7E), again indicating that Caspase-8 and FADD support inflammasome activation in macrophages.
As an alternative to inflammasome activation in macrophages, we examined epithelial cells from the ileum of the small intestine. Cells from Casp1−/−Mlkl−/− mice that were either Fadd-proficient or -deficient and Caspase-8WT/WT or Caspase-8DA/DA were assessed for constitutive ASC oligomerization and speck formation as an indication of inflammasome activation. ASC oligomerization was increased in ileal cells of Fadd−/−Mlkl−/−Casp1−/− animals expressing WT Caspase-8 and this oligomerization was exacerbated when cells expressed Caspase-8 DA (Figure 6A). Ileal sections showed marked ASC speck formation in non-hematopoietic cells (CD45−) of Casp8DA/DAFadd−/−Mlkl−/−Casp1−/−, but not in animals expressing Caspase-8 WT or FADD (Figure 6B). Concomitantly, it has been demonstrated that Caspase-8 can interact with ASC in vitro (Sagulenko et al., 2013; Newton et al., 2019b; Fritsch et al., 2019). Caspase-8 DA from freshly isolated ileal cells co-precipitated with ASC in the absence of FADD (Figure 6A). Moreover, Caspase-8 DA co-localized with ASC in ileum sections of Casp8DA/DAFadd−/−Mlkl−/−Casp1−/− animals (Figure 6C, arrow).
Figure 6: FADD blocks Caspase-8-mediated ASC oligomerization in non-immune cells.
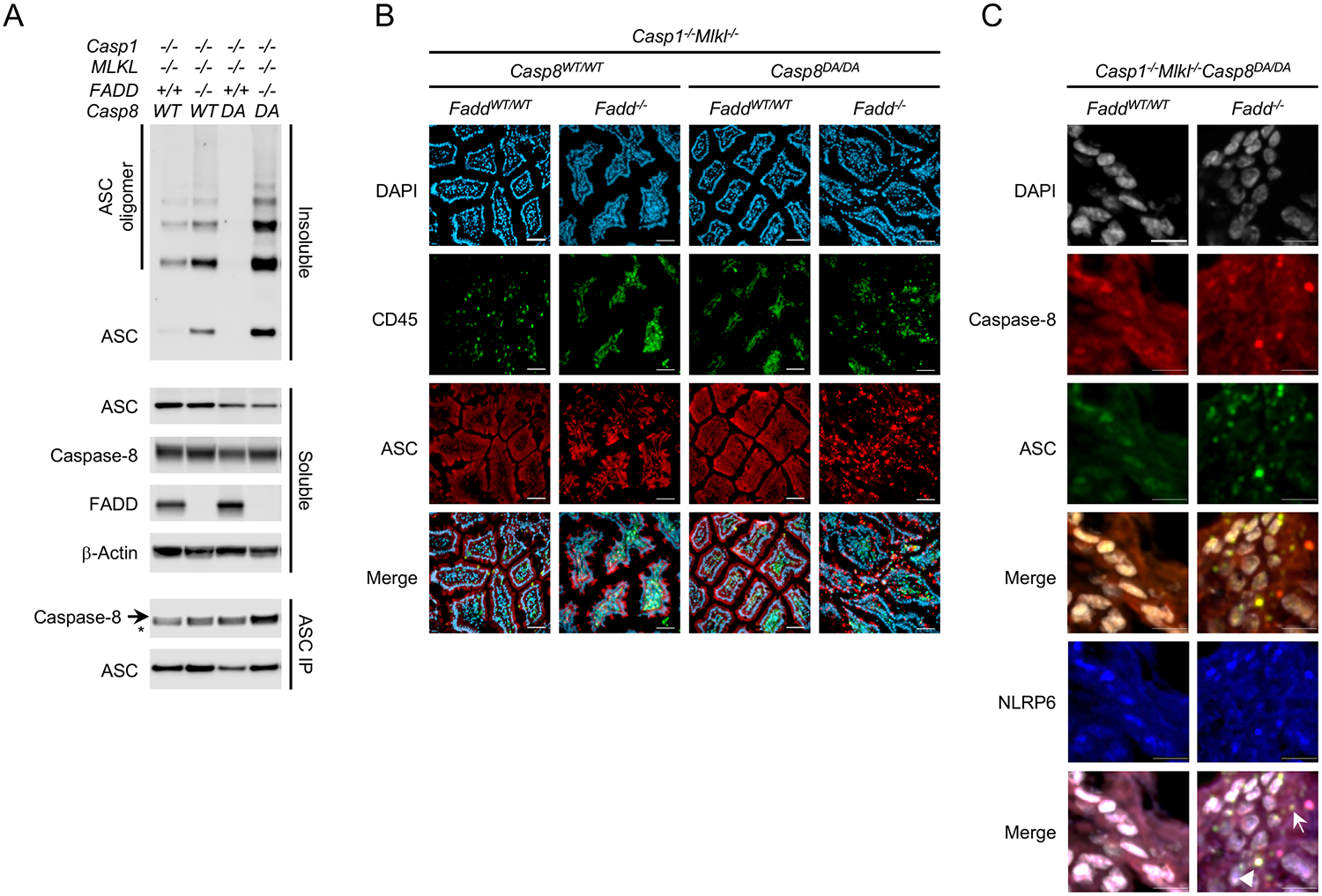
A: Immunoblots for ASC, Caspase-8, FADD and β-Actin from lysed ileal tissue of mice of the indicated genotypes. Soluble and insoluble fractions were generated from total lysates, the insoluble fractions were crosslinked using DSS. ASC was immunoprecipitated (IP) from total lysates. * IgG heavy chain.
B: DAPI staining and IF of CD45 and ASC on ileal sections of mice of the indicated genotypes. Scale bar, 50 μm.
C: DAPI staining and IF of Caspase-8 and ASC on ileal sections of mice of the indicated genotypes. Arrow: Caspase-8, ASC double-positive speck, Arrowhead: Caspase-8, ASC, NLRP6 triple-positive speck. Scale bar, 10 μm.
Please also see Figure S5.
The inflammasome sensor NLRP6 is highly expressed by intestinal cells (Chen et al., 2011; Elinav et al., 2011). NLRP6 was expressed in ileal sections of our animals and was able to co-localize with Caspase-8 DA and ASC when FADD was absent (Figure 6C, arrowhead), suggesting that NLRP6 may be one of the sensors that initiates ASC speck formation in epithelial cells.
Together, these data indicate that Caspase-8 DA can interact with ASC in intestinal epithelial cells. This interaction is blocked by FADD and the ability of Caspase-8 to self-cleave. Furthermore, the suppression of this inflammasome activation in non-immune cells appears to be required for the survival of Casp8DA/DA mice (summarized in Figure S6).
Discussion
Mice defective in CD95, CD95L or the CD95-signaling molecules FADD or Caspase-8 (facilitated by the ablation of Ripk3 or Mlkl) develop LPR disease, characterized by the expansion of TCRαβ+CD3+B220+CD4−CD8− T cells, splenomegaly, and lymphadenopathy. Based on these findings, it is widely concluded that defects in CD95-mediated apoptosis are responsible for LPR disease. Although the Caspase-8 mutant animals described herein were relatively resistant to the lethal effects of agonistic anti-CD95 administration in vivo, we observed no LPR disease in these animals. While it is tempting to suggest that the lack of LPR disease in these mice indicates that apoptosis defects are not responsible, thymocytes from these animals underwent apoptosis in response to CD95 ligation, and therefore the conventional interpretation remains supported.
It has been speculated that necroptosis evolved as a “backup” death mechanism when extrinsic apoptosis fails. However, several studies show that necroptosis is relevant for a variety of pathologies, independent of apoptosis. Necroptosis has roles in the control of inflammation (Pasparakis and Vandenabeele, 2015), cancer (Gong et al., 2019), and ischemic injury (Ruan et al., 2019). We found that the necroptotic cell death pathway is utilized in vivo as a means of immune cell homeostasis, since abrogation of necroptosis, by ablation of Mlkl or Ripk3, from Casp8DA/DA animals resulted in pronounced inflammation. Because necroptosis-deficient Casp8WT/WT and Casp8FGLG/FGLG mice remained disease-free, it is not the ablation of necroptosis per se that induces disease; rather, this indicates that Caspase-8 has an inflammatory role that is dependent on its ability to oligomerize, is blocked by its auto-proteolytic cleavage, and is controlled by necroptosis.
Caspase-8 involvement in inflammasome activation and IL1β cleavage has been suggested previously and appears to depend on the cell type and stimulus (Bossaller et al., 2012; Philip et al., 2016; Philip et al., 2014; Weng et al., 2014; Zhang et al., 2018). Caspase-8 has been shown to be able to process pro-IL1β in Caspase-1-deficient macrophages (Maelfait et al., 2008) when cellular inhibitors of apoptosis (cIAPs) are blocked (Vince et al., 2012), under ER stress (Shenderov et al., 2014), upon HDAC inhibition (Stammler et al., 2015), and in cells undergoing BAX and/or BAK-mediated apoptosis (Chauhan et al., 2018; Vince et al., 2018). Furthermore, Caspase-8 can induce IL1β secretion without pyroptosis in a process called the alternative inflammasome in human, but not murine, monocytes (Gaidt et al., 2016). The Casp8DA/DAMlkl−/− animals showed increased basal serum IL18 and increased anti-CD95- and LPS-induced serum IL18 and IL1β, suggesting that the inflammatory phenotype of the animals was due to enhanced inflammasome activation. However, removal of Casp1 and Casp11 from Casp8DA/DAMlkl−/− animals ablated basal serum IL18, indicating that Caspase-8 DA does not directly mediate IL18 processing. Furthermore, Casp1 and −11 ablation did not prevent the inflammatory phenotype of the Casp8D387A/D387AMlkl−/− mice, indicating that the inflammasome does not mediate the pathology of these animals.
Ablation of one allele of Fasl prevented the inflammatory phenotype of our Casp8DA/DAMlkl−/− animals, indicating that this phenotype may be mediated by CD95 signaling. CD95 is among the death receptors which include TRAIL-R. It has been shown that Caspase-8 can promote inflammatory gene expression upon TRAIL-R ligation in vitro via a complex called the FADDosome, which consists of Caspase-8, FADD and RIPK1 (Hartwig et al., 2017; Henry and Martin, 2017). Our data support a previously unrecognized in vivo role for the CD95L-induced FADDosome, since blunting the FADDosome by removal of one allele of Fasl, Casp8DA, Fadd, or Ripk1 almost completely prevented the inflammatory phenotype of our Casp8DA/DAMlkl−/− animals. Collectively, our data suggest that Caspase-8, FADD, and RIPK1 can form a platform in vivo that results in the induction of inflammation, as has been previously demonstration in vitro (Bossaller et al., 2012; Hartwig et al., 2017; Henry and Martin, 2017), and which is limited by necroptosis.
It is known that Fadd−/−Mlkl−/− mice, which express wild-type Caspase-8, like Casp8−/−Mlkl−/− mice reach adulthood and develop LPR disease in later life (Alvarez-Diaz et al., 2016). We found that Casp8DA/DAMlkl−/− mice reach adulthood and eventually develop a lethal inflammatory disease. However, crossing these animals to produce Casp8DA/DAFadd−/−Mlkl−/− mice resulted in a phenotype that proved lethal within the first two weeks of life. Since removing Casp8 from Fadd−/−Mlkl−/− mice did not alter their phenotype, our genetic models formally show that Caspase-8 has a FADD-independent role in regulating inflammation in a setting where it is unable to self-cleave. Thus, crossing two animal models that exhibit distinct phenotypes in adulthood resulted in death in early life.
Recently, two independent groups reported that mice harboring a mutation in the catalytic site of Caspase-8 (C362S (Fritsch et al., 2019) or C362A (Newton et al., 2019a), Casp8CA allele) die mid-gestation due to exacerbated necroptosis in several tissues, similar to Casp8-deficient animals. However, whereas co-ablation of Mlkl results in viable Casp8−/− animals, Mlkl−/− mice with catalytically inactive Caspase-8 show perinatal lethality, which is dependent on ASC or Caspase-1 (or −11). Similarly, ablation of Fadd resulted in early lethality of Casp8DA/DAMlkl−/− mice, which was dependent on Caspase-1 (or −11). The role for FADD catalytically inactive Caspase-8-mediated ASC oligomerization remains unclear. However, since lethality of both Casp8DA/DAFadd−/−Mlkl−/− and Casp8CA/CAMlkl−/− mice can be prevented by ablation of Ripk1 (Newton et al., 2019b), and RIPK3 seems to have a role in inflammasome activation (Newton et al., 2019b), FADD, speculatively, may inhibit inflammasome activation by mediating RIPK1 cleavage by Caspase-8. Whether Ripk3 ablation can prevent the early lethality of Casp8DA/DAFadd−/−Mlkl−/− mice remains to be determined.
The lethal phenotype of the Casp8CA/CAMlkl−/− mice is not prevented by co-ablation of Nlrp3 (Newton et al., 2019b), and therefore the NLR responsible for inflammasome activation in this setting remains elusive. Epithelial cells are known to express NLRP6, NLRC4 and potentially AIM2 and NLRP3 (Sellin et al, 2015). NLRP6 is highly expressed in the intestine (Chen et al. 2011; Elinav et al., 2011). We observed that NLRP6 partially co-localized with the Caspase-8 DA and ASC specks observed in the ileal sections of our Casp8DA/DAFadd−/−Mlkl−/−Casp1−/− mice, although not all specks showed a presence of NLRP6. This suggests that NLRP6 may be at least one of the receptors responsible for inflammasome activation in intestinal epithelial cells. Whether NLRP6 mediates the pathology of Casp8CA/CAMlkl−/− and/or Casp8DA/DAFadd−/−Mlkl−/− animals, remains to be determined.
In macrophages, Caspase-8 has been shown to be required for NLRP3 inflammasome formation (Antonopoulos et al., 2015; Gurung et al., 2014; Lukens et al., 2014), and for Yersinia-induced pro-inflammatory cytokine expression and pyroptosis (Orning et al., 2018; Philip et al., 2016) in a process that does not require its self-cleavage (Philip et al., 2016). Likewise, FADD is also required for inflammasome activation in macrophages (Antonopoulos et al., 2015; Gurung et al., 2014). Where previous studies focused on Caspase-1 cleavage (Antonopoulos et al., 2015; Gurung et al., 2014; Lukens et al., 2014), we found that in absence of FADD or Caspase-8, ASC failed to oligomerize in these cells. How Caspase-8 and FADD mediate ASC oligomerization remains to be determined, but our findings support the idea that both Caspase-8 and FADD are required for ASC oligomerization in macrophages. Furthermore, this may explain why the Caspase-1 and/or -11-dependent inflammation we observed appeared to be in non-immune cells rather than predominantly in myeloid cells, although the mechanistic differences in inflammasome activation between these cell types remain unclear.
Collectively, our data show that the inflammasome is activated by Caspase-8 DA in the absence of FADD in non-immune cells of barrier tissues, resulting in death within two weeks of life. In the protected Casp1−/−Casp8DA/DAMlkl−/− animals, the absence of FADD enhanced spontaneous ASC oligomerization, indicating that FADD, as well as cleavage of Caspase-8, block ASC activation in non-myeloid cells.
The implications and mechanisms of three cell death pathways of apoptosis, necroptosis and pyroptosis are becoming increasingly clear. However, the interplay between these three pathways remains elusive. Our study supports the idea that perturbation of these pathways can lead to unfavorable outcomes and provides insights into the in vivo functions of necroptosis, the FADDosome, and Caspase-8-mediated inflammation. Furthermore, we found a previously unrecognized FADD-independent role for Caspase-8-induced inflammasome activation in non-immune cells. Given the importance of the three cell death mechanisms, apoptosis, necroptosis and pyroptosis, in the context of inflammation, ALPS, and other pathologies, understanding the interplay between these pathways provides insights into how these modes of cell death are regulated to control disease.
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents may be directed to and will be fulfilled by the Lead Contact, Douglas R. Green (douglas.green@stjude.org). Unique reagents and mouse lines generated in this study may be obtained (pending continued availability) from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The published article includes all datasets analyzed during this study.
Experimental Model and Subject Details
Mice
Ripk3−/− (Newton et al., 2004), Casp8−/−Ripk3−/− (Oberst et al., 2011), Mlkl−/− (Murphy et al., 2013), Casp8−/−Mlkl−/− (Dillon et al., 2014), Casp1−/− (Kayagaki et al., 2011), Ripk1−/− (Kelliher et al., 1998), Fadd−/− (Yeh et al., 1998), and Rag1−/− (Mombaerts et al., 1992) mice have been previously described. Casp8D387A/D387A and Casp8F122GL123G/F122GL123G mice were generated using CRISPR/Cas9 technology as detailed previously (Pelletier et al., 2015). Briefly, pronuclear stage C57BL/6J zygotes were injected with Cas9 mRNA transcript (100 ng/μL), a single guide RNA transcript (50 ng/μL) and a homology-directed repair template in the from of a single stranded DNA molecule (1.2 pmol/μL). To prevent further cleavage by Cas9 upon repair of the double strand break and to facilitate genotyping, the homology directed repair templates were designed to introduce, in addition to the desired mutations, silent mutations altering the guide target region and encoding a restriction enzyme site (Key Resource Table). Zygotes were then transferred to pseudo pregnant CD1 females. Pups obtained from zygote injections were analyzed at the genomic level using PCR, restriction enzyme digest and Sanger sequencing as detailed previously (Pelletier et al., 2015). Pups carrying the desired mutation were selected and used to establish colonies. Mutant mice were backcrossed to the C57BL/6J background for more than 5 generations. Mice bearing WT and/or Casp8D387A alleles were routinely genotyped by PCR amplification and restriction digest using Casp8–78-F01 and Casp8–78-R01 primers and SacI restriction enzyme. DNA fragments of 610 bp were obtained for both alleles of which only the mutant allele is cleaved by SacI, generating 2 fragments of 345 and 265 base pairs. Similarly, mice bearing WT and/or Casp8F122GL123G alleles were genotyped by PCR and restriction digest using Casp8_FL-GG_F01 and Casp8_FL-GG_R01 primers and DraI restriction enzyme. DNA fragments of 430 bp were obtained for both alleles of which only the mutant allele can be cleaved by DraI, resulting in 2 fragments of 313 and 117 base pairs. For guide RNA and homology-directed repair (HDR) and primer oligonucleotide sequences, please see the Key Resource table. Both male and female mice were used in this study. For all studies mice were age- and sex-matched. Two- to sixteen-week old mice were used for in vivo studies and six- to twelve-week old mice were used for in vitro studies. All mice were bred and housed in specific pathogen-free facilities, in a 12-hour light/dark cycle in ventilated cages, with chow and water supply ad libitum, at the Animal Resources Center at St. Jude Children’s Research Hospital. Necropsy was performed by the St. Jude Veterinary Pathology Core facility. The St. Jude Institutional Animal Care and Use Committee approved all procedures in accordance with the Guide for the Care and Use of Animals.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD95 (Jo2) | BD Pharmingen | Cat# 554254; RRID:AB_395326 |
| anti-ASC | Santa Cruz Technologies | Cat# sc-22514; RRID:AB_2174874 |
| anti-ASC | Adipogen | Cat# AG-25B-0006; RRID:AB_2490440 |
| anti-β-Actin | Cell signaling Technology | Cat# 3700; RRID:AB_2242334 |
| anti-β-Actin-HRP | Santa Cruz Technologies | Cat# sc-47778; RRID:AB_2714189 |
| anti-B220 AF488 | BioLegend | Cat# 103228; RRID:AB_492874 |
| anti-B220 APC | BD Pharmingen | Cat# 553092; RRID:AB_398531 |
| anti-B220 APC-eFluor780 | eBioscience | Cat# 47-0452-82; RRID:AB_1518810 |
| anti-B220 FITC | eBioscience | Cat# 11-0452-82; RRID:AB_465054 |
| anti-Caspase-1 | Cell signaling Technology | Cat# 3866; RRID:AB_2069051 |
| anti-Caspase-1 p20 | eBioscience | Cat# 14-9832-82; RRID:AB_2016691 |
| anti-caspase-3 | Cell signaling Technology | Cat# 9662; RRID:AB_331439 |
| anti-Caspase-8 | Cell signaling Technology | Cat# 4790; RRID:AB_10545768 |
| anti-CD117 APC-e780 | eBioscience | Cat# 47-1171-80; RRID:AB_1272213 |
| anti-CD11b Brilliant Violet 421 | Biolegend | Cat# 101235; RRID:AB_10897942 |
| anti-CD135 APC | BD Pharmingen | Cat# 560718; RRID:AB_1727425 |
| anti-CD150 PE-Cy7 | BioLegend | Cat# 115914; RRID:AB_439797 |
| anti-CD19 APC-Cy7 | BD Pharmingen | Cat# 557655; RRID:AB_396770 |
| anti-CD19 FITC | BD Pharmingen | Cat# 553785; RRID:AB_395049 |
| anti-CD3 PE | BioLegend | Cat# 100205; RRID:AB_312662 |
| anti-CD3e FITC | Invitrogen | Cat# 11-0031-85; RRID:AB_464883 |
| anti-CD4 Brilliant Violet 605 | Biolegend | Cat# 100547; RRID:AB_11125962 |
| anti-CD4 eFluor450 | Invitrogen | Cat# 48-0042-82; RRID:AB_1272194 |
| anti-CD4 FITC | eBioscience | Cat# 11-0042-82; RRID:AB_464896 |
| anti-CD44 PE | BD Pharmingen | Cat# 553134; RRID:AB_394649 |
| anti-CD45 AF647 | Biolegend | Cat# 103123; RRID:AB_493534 |
| anti-CD45.2 PerCP-Cy5.5 | Invitrogen | Cat# 45-0454-82; RRID:AB_953590 |
| anti-CD48 Alexa Fluor700 | BioLegend | Cat# 103425; RRID:AB_10612754 |
| anti-CD62L APC | BD Pharmingen | Cat# 553152; RRID:AB_398533 |
| anti-CD8 eFluor450 | Invitrogen | Cat# 48-0081-82; RRID:AB_1272198 |
| anti-CD8a Brilliant Violet 605 | Biolegend | Cat# 100743; RRID:AB_2561352 |
| anti-CD8a FITC | BD Pharmingen | Cat# 553031; RRID:AB_394569 |
| anti-CD95 PE | BD Pharmingen | Cat# 554258; RRID:AB_395330 |
| anti-cleaved caspase-3 | Cell signaling Technology | Cat# 9661; RRID:AB_2341188 |
| anti-cleaved Caspase-8 | Novus | Cat# NB100-56116; RRID:AB_837874 |
| anti-F4/80 | Invitrogen | Cat# MF48000; RRID:AB_10376289 |
| anti-FADD | Abcam | Cat# ab124812; RRID:AB_10976310 |
| anti-GasderminD | Sigma-Aldrich | Cat# G7422; RRID:AB_1850381 |
| anti-I-A/I-E PE | BD Pharmingen | Cat# 557000; RRID:AB_396546 |
| anti-IL-1β | R&D systems | Cat# AF-401; RRID:AB_416684 |
| anti-Ly-6A/E APC-Cy7 | BD Pharmingen | Cat# 560654; RRID:AB_1727552 |
| anti-Ly-6A/E PerCp-Cy5.5 | BioLegend | Cat# 122523; RRID:AB_893621 |
| anti-Ly-6C Brilliant Violet 711 | Biolegend | Cat# 128037; RRID:AB_2562630 |
| anti-Ly-6G PE-Cy7 | BD Pharmingen | Cat# 560601; RRID:AB_1727562 |
| anti-Ly-6G/Ly-6C AF647 | Biolegend | Cat# 108420; RRID:AB_493481 |
| anti-Ly-6G/Ly-6C FITC | Invitrogen | Cat# 11-5931-82; RRID:AB_465314 |
| anti-mouse CD16/CD32 | BD Pharmingen | Cat# 553142; RRID:AB_394657 |
| anti-mouse HRP | GE Lifesciences | Cat # NA931; RRID:AB_772210 |
| anti-myeloperoxidase (MPO) | DAKO | Cat# A0398; RRID:AB_2335676 |
| anti-NLRP3 | Cell signaling Technology | Cat# 15101; RRID:AB_2722591 |
| anti-NLRP3 | Adipogen | Cat# AG-20b-0014; RRID:AB_2490202 |
| anti-NLRP6 | Sigma-Aldrich | Cat# SAB1302240; RRID:AB_2750643 |
| anti-rabbit AF555 | Thermo Fischer Scientific | Cat# A-21429; RRID:AB_2535850 |
| anti-rabbit HRP | GE Lifesciences | Cat# NA934; RRID:AB_772206 |
| anti-rat HRP | Vector Laboratories | Cat# BA-4001; RRID:AB_10015300 |
| anti-RIPK1 | Sigma-Aldrich | Cat# SAB3500420; RRID:AB_10643987 |
| anti-RIPK1 | BD Biosciences | Cat# 610459; RRID:AB_397832 |
| anti-TER-119 FITC | Invitrogen | Cat# 11-5921-82; RRID:AB_465311 |
| Bacterial and Virus Strains | ||
| E. coli K12 DH5α thyA− | J.M. Blander | Sander et al., 2011 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Amersham ECL Prime Western Blotting Detection Reagent | GE Lifesciences | Cat# RPN2232 |
| Background Sniper | BioCare Medical | Cat# BS966H |
| Bacitracin | St. Jude pharmacy | N/A |
| Blasticidin | A.G. Scientific | Cat# B-1247 |
| Biocare TBS wash buffer | BioCare Medical | Cat# TWB954M |
| ChromoMap DAB | Roche | Cat# 760-159 |
| Ciprofloxacin | St. Jude pharmacy | N/A |
| Clarity Western ECL substrate | Bio-Rad | Cat# 1705060 |
| Cycloheximide | Sigma-Aldrich | Cat# 239763 |
| DAB Quanto Chromogen and Substrate | ThermoShandon | Cat# TA-125-HDX |
| DMEM | Thermo Fischer Scientific | Cat# 11995-073 |
| Doxycycline (by Clontech) | Thermo Fischer Scientific | Cat# NC0424034 |
| Etoposide | Sigma-Aldrich | Cat# E1383 |
| Equal sweetener | Equal | Cat# 20019141 |
| FasL, soluble (human) (recombinant) set | Enzo Lifesciences | Cat# ALX-850-014-KI02 |
| Gentamycin | Sigma Aldrich | Cat# G4918 |
| L-Glutamine | Thermo Fischer Scientific | Cat# 25030081 |
| Lipofectamine 2000 | Thermo Fischer Scientific | Cat# 11668019 |
| LPS from E. coli (055:B5) | Sigma-Aldrich | Cat# L2880 |
| Lycopersicon Esculentum (Tomato) Lectin (LEL, TL), DyLight® 488 | Vector Laboratories | Cat# DL-1174 |
| Metronidazole | Sigma Aldrich | Cat# M1547 |
| Nec1s | Calbiochem | Cat# 5.04297.0001 |
| Nigericin | Invivogen | Cat# tlrl-nig |
| Non-essential amino acids | Thermo Fischer Scientific | Cat# 11140-050 |
| NucBlue Live ReadyProbes Reagent (DAPI) | Thermo Fischer Scientific | Cat# R37606 |
| OmniMap Rabbit Detection kit | Roche | Cat# 760-4311 |
| Penicillin and streptomycin | Thermo Fischer Scientific | Cat# 15070-063 |
| Phosphatase inhibitor | Roche | Cat# 04 906 837 001 |
| PolyI:C | Invivogen | Cat# tlrl-pic |
| Protease inhibitor | Roche | Cat# 04 693 159 001 |
| Puromycin | Sigma-Aldrich | Cat# P-8833 |
| Q-VD(OMe)-Oph | APExBIO | Cat# A8165 |
| Restriction enzyme DraI | New England BioLabs | Cat# R0129 |
| Restriction enzyme SacI | New England BioLabs | Cat# R0156 |
| Sterile PBS | Gibco | Cat# 14190-144 |
| Streptavidin-HRP | ThermoShandon | Cat# TS-125-HR |
| SYBR Green | Thermo Fischer Scientific | Cat# 4309155 |
| SYTOX Green | Invitrogen | Cat# S7020 |
| Target Retrieval | DAKO | Cat# S2367 |
| Thermo Scientific™ Pierce™ ECL 2 Western Blotting Substrate | Thermo Fischer Scientific | Cat# PI80196 |
| Ultrapure LPS from E. coli (0111:B4) | Sigma-Aldrich | Cat# L4391 |
| Ventana Reaction Buffer | Ventana | Cat# 950-300 |
| XT sample buffer 4x | Bio-Rad | Cat# 161-0791 |
| zVAD-FMK | APExBIO | Cat# A1902 |
| Critical Commercial Assays | ||
| Dynabeads Co-Immunoprecipitation Kit | Invitrogen | Cat# 14321D |
| IL1 beta mouse uncoated ELISA kit | Thermo Fischer Scientific | Cat# 88-7013-88; RRID:AB_2574946 |
| M-MLV Kit | Invitrogen | Cat# 28025013 |
| Mouse IL18 ELISA kit | MBL | Cat# 7625 |
| Multiplex ELISA | Millipore | Cat# MCYTOMAG-70K |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| TNF alpha mouse uncoated ELISA kit | Thermo Fischer Scientific | Cat# 88-7324-88; RRID:AB_2575080 |
| UltraComp eBeads Compensation Beads | Invitrogen | Cat# 01-2222-42 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Immortalized Casp8−/−Mlkl−/−MEFs | Dillon et al., 2014 | N/A |
| NCTC clone 929 (L-929) | ATCC | CCL-1 |
| Phoenix-AMPHO | ATCC | CRL-3213 |
| Primary murine BMDMs | This paper | N/A |
| Primary murine FLDMs | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129S7-Rag1tm1Mom/J | The Jackson Laboratory | Cat# 002216 |
| Casp1−/− | Kayagaki et al., 2011 | N/A |
| Casp8D387A/D387A | This paper | N/A |
| Casp8F122GL123G/F122GL123G | This paper | N/A |
| Casp8−/−Mlkl−/− | Dillon et al., 2014 | N/A |
| Casp8−/−Ripk3−/− | Oberst et al., 2011 | N/A |
| Fadd−/− | Yeh et al., 1998 | N/A |
| Ripk1−/− | Kelliher et al., 1998 | N/A |
| Mlkl−/− | Murphy et al., 2013 | N/A |
| Ripk3−/− | Newton et al., 2004 | N/A |
| Oligonucleotides | ||
|
Casp8
D387A guide 5’-TAATACGACTCACTATAGGACAGAACCACACTTTAGAAGGTTTTAGAGCTAGAAATAGCA-3’ |
This paper | N/A |
|
Casp8
D387A HDR oligo 5’-CCAAGATCTTTTTCATTCAGGCTTGCCAAGGAAGTAACTTCCAGAAAGGAGTGCCTGATGAGGCAGGCTTCGAGCAACAGAACCACACTTTGGAGGTTGCGAGCTCATCTCACAAGAACTATATTCCGGATGAGGCAGACTTTCTGCTGGGAATGGCTACGGTGAAGAACTGCGTTTCCTACCGAGATCCTGTGAATGGA-3’ (underscored is the SacI site) |
This paper | N/A |
|
Casp8
D387A PCR Casp8-78-F01: 5’-TACCAAAGCGCAGACCACAA Casp8-78-R01: 5’-CCATCTCTCCCACTTTGCCA |
This paper | N/A |
|
Casp8
F122GL123G guide 5’- TAATACGACTCACTATAGGTGGGGATCTCATTGTTCAAAGTTTTAGAGCTAGAAATAGCA-3’ |
This paper | N/A |
|
Casp8
F122GL123G HDR oligo 5’- GTTTCCTGCCACAGGGTCATGCTCTTTAAGCTCTCAGAAGAAGTGAGCGAGTTGGAATTGAGATCTTTTAAAGGTGGTTTGAACAATGAGATCCCCAAATGTAAGCTGGAAGATGACTTGGTAAGACCTAATCTCCTGAAGATGGGTCACCTCTGG-3’ (underscored is the DraI site) |
This paper | N/A |
|
Casp8
F122GL123G PCR Casp8_FG-GG_F01: 5’-TTCCCCCAAATCCTCGCATC Casp8_FG-GG_R01: 5’-TTAGCAGGGCTCTCTGGTCT |
This paper | N/A |
| Actin qPCR F: 5’-ATGGAGGGGAATACAGCCC R: 5’-TTCTTTGCAGCTCCTTCGTT |
This paper | N/A |
| IL1β qPCR F: 5’-CACAGCAGCACATCAACAAG R: 5’-GTGCTCATGTCCTCATCCTG |
This paper | N/A |
| TNFα qPCR F: 5’-CCTGTAGCCCACGTCGTAGC R: 5’-AGCAATGACTCCAAAGTAGACC |
This paper | N/A |
| Recombinant DNA | ||
| pBABE-puro plasmid | Addgene | Cat# 1764 |
| pRetroX-TRE3G plasmid | Clontech | Cat# 631188 |
| psPAX2 plasmid | Addgene | Cat# 12260 |
| pVSVg plasmid | Addgene | Cat# 8454 |
| Software and Algorithms | ||
| FlowJo v10.4 | Tree Star | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism 7.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| IncuCyte Base Software | Essen Biosciences | https://www.essenbioscience.com/en/products/software/incucyte-base-software/ |
| LiCOR Image Studio | LiCOR | https://www.licor.com/bio/image-studio/ |
| Nikon NIS-elements Advanced Research | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| Slidebook 6 | 3i | https://www.intelligent-imaging.com/slidebook |
| SP6800 software | Sony | https://www.sonybiotechnology.com/us/instruments/sp6800/ |
| Other | ||
Bone marrow- and fetal liver-derived macrophages
Both male and female mice were used to generate BMDMs. For preparation of bone marrow-derived macrophages (BMDMs), bone-marrow cells were harvested from femur and tibia. For preparation of fetal liver-derived macrophages (FLDMs), dames were euthanized at 11–15 days of pregnancy and fetal livers were harvested from the embryos. The sex of embryos was not determined before generation of FLDMs. BMDMs and FLDMs were differentiated in DMEM medium (Gibco) containing 30% L929-conditioned medium, 20% FBS, 200 mM L-glutamine, 1X MEM non-essential amino acids (NEAA), 1 mM sodium pyruvate, 55 μM 2-Mercaptoethanol and 100 units/ml penicillin-streptomycin. For the preparation of L-929-conditioned medium, NCTC clone 929 (L-929) cells (ATCC) were grown until confluent in DMEM medium containing 10% fetal bovine serum (FBS), 200 mM L-glutamine, 1X MEM non-essential amino acids (NEAA), 1 mM sodium pyruvate, 55 μM 2-Mercaptoethanol and 100 units/ml penicillin-streptomycin. The medium was refreshed, and the cells were incubated for one week after which supernatants were harvested and filtered. Cells were cultivated at 37°C with 5% CO2. NCTC clone 929 is a mouse cell line of male origin.
Thymocytes
Both male and female mice were used to harvest thymocytes. Thymocytes were harvested from the thymus of 4- to 6-week old mice. Thymocytes were cultured in DMEM medium containing 10% charcoal-stripped FBS, 200 mM L-glutamine, 1X MEM non-essential amino acids (NEAA), 1 mM sodium pyruvate, 55 μM 2-Mercaptoethanol and 100 units/ml penicillin-streptomycin. Cells were cultivated at 37°C with 5% CO2.
Mouse Embryonic Fibroblasts (MEFs)
Dames were euthanized at 11–15 days of pregnancy and MEFs were harvested from the embryos. Immortalized Casp8−/−Mlkl−/− MEFs (iMEFs) were generated previously (Dillon et al., 2014). All MEFs were cultured in DMEM medium containing 10% fetal bovine serum (FBS), 200 mM L-glutamine, 1X MEM non-essential amino acids (NEAA), 1 mM sodium pyruvate, 55 μM 2-Mercaptoethanol and 100 units/ml penicillin-streptomycin. Cells were cultivated at 37°C with 5% CO2. The sex of embryo’s was not determined before generation of primary MEF cultures.
Phoenix-AMPHO cells
Phoenix-AMPHO cells (ATCC) were maintained in complete DMEM media (10% fetal bovine serum (FBS), 200 mM L-glutamine, 1X MEM non-essential amino acids (NEAA), 1 mM sodium pyruvate, 55 μM 2-Mercaptoethanol and 100 units/ml penicillin-streptomycin). Cells were cultivated at 37°C with 5% CO2. Phoenix-AMPHO cells are human cells derived from HEK293T/17 cells.
Bacterial strains
Escherichia coli K12, strain DH5α (innocuous) was purchased from Invitrogen. Naturally occurring thymidine auxotrophs (thyA−) were selected as previously described (Sander et al., 2011).
Method Details
Reconstitution of Casp8−/−Mlkl−/− iMEFs
The mouse Mlkl gene was cloned into the Dox-inducible vector pRetroX-TRE3G (Clontech). Caspase-8 WT, or C362A, D387A or F122GL123G mutant constructs were cloned in frame with a T2A-GFP sequence into the pBABE-puro retroviral expression vector. Retroviruses were produced using Phoenix-AMPHO cells co-expressing psPAX2 and pVSVg plasmids (Addgene) and our pBABE-puro or pRetroX-TRE3G vectors using Lipofectamine 2000 (Thermo Fisher Scientific). Retrovirus containing supernatants were harvested and filtered 48 hours post-transfection. Casp8−/−Mlkl−/− iMEFs were transduced to stably express DOX-inducible MLKL and were selected using puromycin (2 μg/ml). Selected cells were re-transduced to stably express GFP and Caspase-8 WT or mutants and were selected three times by FACS sort based on GFP.
Cell stimulations
BMDM or FLDM were harvested and re-plated 12–16 hours before stimulation at a concentration of 125,000 cells per cm2. For CD95 ligation experiments, cells were pre-treated with 20 ng/ml LPS (E. coli O55:B5, Sigma) for 16–20 hours. Where indicated, cells were treated with 30 μM Nec1s (Calbiochem) 2 hours prior to CD95 ligation. Cells were stimulated with 10 – 100 ng/ml CD95L (Enzo) together with 1 μg/ml enhancer solution (Enzo), 25 μM zVAD-FMK (APExBIO), and/or 30 μM Nec1s (Millipore). For LPS and nigericin stimulation, cells were treated with 100 ng/ml LPS for 6 hours, after which nigericin was added to reach a final concentration of 6 μM for 45 minutes. For bacterial stimulation of phagocytes, E. coli were grown to mid-log phase (optical density at 600 nm of 0.6) in the appropriate medium, washed three times in PBS, and counted before addition to cells. Bacteria were added at a ratio of 20 to 1 cell (MOI 20). After addition of bacteria, cells were briefly centrifuged (1250 rpm, 2 minutes) to help synchronizing infection. One-hour post stimulation with bacteria, antibiotics (cocktail of Penicillin, Streptomycin and Gentamycin) were added to the medium and kept until the end of the experiments.
Thymocytes were treated as indicated with 100 ng/ml CD95L (Enzo), 1 μg/ml enhancer solution (Enzo), 20 μM Q-VD-Oph (APExBIO) or 10 μg/ml Etoposide (Sigma) for 24 hours. Stimulated thymocytes were harvested and propidium iodide (PI) uptake was determined by Flow cytometry.
Reconstituted immortalized Casp8−/−Mlkl−/− MEFs were re-plated 12–16 hours before stimulation at a concentration of 30,000 cells per cm2. For necroptosis assessment assays, MLKL expression was induced by pre-treating the cells for 6 hours with 1 μg/ml doxycycline. Cells were treated as indicated with complete DMEM containing 1 μM SYTOX Green (Invitrogen), 100 ng/ml CD95L (Enzo), 1 μg/ml enhancer solution (Enzo), 0.5 μM Cycloheximide (Sigma), 20 μM Q-VD-Oph (APExBIO), 100 μg/ml PolyI:C (Invivogen), 25 μM zVAD-FMK (APExBIO) and/or 30 μM Nec1s (Millipore). SYTOX Green uptake was determined using an IncuCyte Kinetic Live Cell Imager and base software (Essen Biosciences).
Antibiotic treatment
Antibiotic cocktail water consisted of sterilized water containing 5% w/v Equal sweetener (Equal), 125 mg/L Ciprofloxacin (St. Jude pharmacy), 50.000 U/L Bacitracin (St. Jude pharmacy), 1.5 g/L Flagyl (Metronidazole, Sigma Aldrich) and 172 mg/L Gentamycin (Sigma Aldrich). Placebo water consisted of sterilized water containing 5% w/v Equal sweetener (Equal). Directly upon weaning, mice were separated to treatment cages and received freshly prepared respective water two to three times a week for a total of 8 weeks.
Bone marrow reconstitution
Recipient Rag1−/− mice were irradiated with 900 rad and received donor cells within 4 hours after radiation. Donor mice were euthanized, and bone-marrow cells were harvested from femur and tibia. Cells were resuspended at 10×106 cells/200 μl in sterile PBS and injected into the tail vein of irradiated recipient mice.
Cytokine analysis
For serum cytokine analysis, mice were euthanized, and blood was collected via cardiac puncture. Blood was centrifuged and serum was harvested. Cell supernatants were pre-cleared by centrifugation. Sera and supernatants were analyzed for indicated cytokines by Multiplex ELISA (Millipore) or mouse TNFα, IL1β (both Thermo Fischer Scientific) or IL18 (MBL) ELISA according to the manufacturer’s instructions.
Flow cytometry
Splenocytes were isolated from spleens of animals of the indicated genotypes. Single cell suspensions were blocked using anti-mouse CD16/CD32 (Fc Block, clone 2.4G2; BD Pharmingen) at 4°C for 15 minutes prior to staining. Surface antigens were stained with indicated conjugated primary antibodies at 4°C for 20 minutes. Antibodies used were: PerCP-Cy5.5 anti-CD45.2 (clone 104; Invitrogen), FITC anti-CD3 (clone 145–2C11; Invitrogen), eFluor450 anti-CD4 (clone RM4–5; Invitrogen), Brilliant Violet 605 anti-CD8a (clone 53–6.7; Biolegend), eFluor450 anti-CD8 (clone 53–6.7; Invitrogen), Brilliant Violet 605 anti-CD4 (clone RM4–5; Biolegend), PE anti-CD44 (clone IM7; BD Pharmingen), APC anti-CD62L (clone MEL-14; BD Pharmingen), APC-eFluor780 anti-B220 (clone RA3–6B2; eBioscience), APC anti-B220 (clone RA3–6B2; BD Pharmingen), APC-Cy7 anti-CD19 (clone 1D3; BD Pharmingen), PE anti-CD95 (clone Jo2; BD Pharmingen), Brilliant Violet 421 anti-CD11b (clone M1/70; Biolegend), Brilliant Violet 711 anti-Ly-6C (clone HK1.4; Biolegend), PE-Cy7 anti-Ly-6G (clone 1A8; BD Pharmingen), PE anti-I-A/I-E (clone M5/114.15.2; BD Pharmingen), PE anti-TER-119 (clone TER-119; Invitrogen), APC-Cy7 anti-Ly-6A/E (clone D7; BD Pharmingen), FITC anti-CD4 (clone RM4–5; eBioscience), FITC anti-CD8a (clone 53–6.7; BD Pharmingen), FITC anti-Ly-6G/Ly-6C (clone RB6–8C5; Invitrogen), FITC anti-CD19 (clone 1D3; BD Pharmingen), FITC anti-B220 (clone RA3–6B2; eBioscience), APC anti-CD135 (clone A2F10; BioLegend), Alexa Fluor700 anti-CD48 (clone HM48–1; BioLegend), PE-Cy7 anti-CD150 (clone TC15–12F12.2; BioLegend), PerCp-Cy5.5 anti-Ly-6A/E (clone D7; BioLegend), APC-e780 anti-CD117 (clone 2B8; eBioscience). For all experiments, cells were analyzed using a Sone SP6800 Spectral Analyzer (Sony). Panel setup and fluorescent compensation were performed using BD compensation beads (BD Bioscience). All analyses were performed using FlowJo software (FlowJo, LLC).
Immunoprecipitation and Immunoblot
Dynabeads were coated with indicated antibody according to the recommendations of the manufacturer (Invitrogen). Cells were lysed in buffer provided by the manufacturer with additional protease inhibitors (Roche) for 30–45 minutes on ice. Lysates were centrifuged and supernatants were transferred to fresh tubes containing antibody-coated Dynabeads. IPs were performed following the recommendations of the manufacturer (Invitrogen). Elution products were diluted in 4x XT sample buffer (Biorad) and boiled at 95°C for 5–10 minutes.
Cells were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris (pH 7.4) 1% NP-40, 0.5% deoxycholate, 0.1% SDS, protease inhibitors (Roche) and phosphatase inhibitors (Roche)) for 30–45 minutes on ice. Lysates were centrifuged, 15 minutes, max speed in a refrigerated microcentrifuge. Supernatants (soluble fraction) were transferred to a fresh tube and diluted in 4x XT sample buffer (BioRad). For ASC oligomerization assays of LPS and LPS+nigericin-treated cells, pellets (insoluble fraction) were washed with PBS and treated with 4 mM DSS in PBS for 15 minutes at RT. Samples were centrifuged and pellets were resuspended in 1x XT sample buffer (Biorad). All samples were boiled at 95°C for 5–10 minutes.
After SDS-PAGE resolution (BioRad), proteins were transferred onto nitrocellulose (BioRad) or PVDF membranes (Millipore). Membranes were blocked with 7% evaporated milk in PBS 0.2% Tween and were incubated with primary antibodies and peroxidase-conjugated secondary antibodies (all diluted in PBS 0.2% Tween). Bound antibodies were visualized using the Amersham™ ECL, Pierce® ECL2 or Clarity Western ECL substrate (BioRad) detection reagents and imaged using Amersham™ Imager 600 (GE Healthcare) or LiCOR Odyssey Fx imaging system (LiCOR). Antibodies used were: anti-β-Actin (Santa Cruz Technologies and CST), anti-Caspase-1 p20 (eBiosciences), anti-Caspase-1 (CST), anti-ASC (Santa Cruz Technologies and Adipogen), anti-GasderminD (Sigma), anti-NLRP3 (Adipogen and CST), anti-IL-1β (R&D), anti-FADD (Abcam), anti-RIPK1 (Sigma), anti-Caspase-8 (CST), anti-cleaved Caspase-8 (Novus), anti-caspase-3 (CST) and anti-cleaved caspase-3 (CST).
Immunofluorescence
Spleen and liver tissues were fixed in 2% paraformaldehyde, 0.1% Triton-100 and 1% DMSO for 24h prior to cryoprotection with 30% sucrose in PBS for an additional 24h. Analyses of inflammasome pathway components in small intestines were performed on tissues fixed for 15 min with 2% paraformaldehyde, 0.05% glutaraldehyde, 0.1% Triton-100 and 1% DMSO prior to embedding in tissue freezing medium. Tissues were cryo-sectioned at 10 μm thickness and blocked in buffer comprised of PBS containing 2% bovine serum albumin and 5% donkey serum. Tissues were stained overnight in blocking buffer containing the following antibodies, all used at 1:200 dilution: AF488-conjugated B220 (clone RA3–6B2, Biolegend), PE-conjugated CD3 (clone 17A2, Biolegend), AF647-labeled Gr1 (clone RB6–8C5, Biolegend), AF647-conjugated CD45 (clone 30-F11, Biolegend), rabbit anti-ASC (Adipogen), rabbit anti-caspase 8 (CST), rabbit anti-NLRP6 (Sigma), and rabbit anti-cleaved caspase 3 (CST). Rabbit antibodies were subsequently detected with AF555-conjugated goat anti-rabbit (Thermo Fisher Scientific), while some experiments included fluorescent tomato lectin (Vector labs, catalog DL-1174) as a counterstain, and were mounted with prolong glass antifade mounting medium with NucBlue (Thermo Fisher Scientific). High resolution images were acquired using a Marianis spinning disk confocal microscope (Intelligent Imaging Innovations) equipped with a 20x 1.0 NA or 40X 1.3NA objective, 405nm, 488nm, 561nm and 647nm laser lines and Prime 95B CMOS camera (Photometrics) and analyzed using Slidebook software (Intelligent Imaging Innovations).
Immunohistochemistry
All assay steps for myeloperoxidase (MPO), including deparaffinization, rehydration, and epitope retrieval, were performed on the Ventana Discovery Ultra autostainer with Ventana Reaction Buffer (cat # 950–300, Ventana) rinses between steps. Epitope retrieval was performed with CC1 buffer for 32 minutes. The primary antibody for MPO (DAKO, #A0398) was incubated at 1:1200 for 32 minutes. Antibody binding was detected by the OmniMap Rabbit Detection kit (Roche, #760–4311) for 16 minutes, followed by ChromoMap DAB (Roche, #760–159) for 10 minutes. For F4/80 and CD68, heat-induced epitope retrieval was performed by heating slides in a BioCare Medical Decloaking Chamber at 110°C for 30 minutes in Target Retrieval (pH 9.0, DAKO #S2367) followed by a 30-minute cool-down period. Non-specific binding was blocked by incubating slides in Background Sniper (cat #BS966H, BioCare Medical) for 30 minutes. The following steps were performed on the Biocare intelliPATH with Biocare TBS wash buffer (cat # TWB954M, Biocare) rinses between steps. Slides were incubated with the primary antibody for F4/80 (Invitrogen #MF48000) at 1:500 for 30 minutes and for CD68 (BioRad, #MCA1957) at 1:1000 for 120 minutes. Both were followed by the secondary antibody (rabbit anti-rat, Vector Labs #BA-4001) at 1:200 for 30 minutes. For detection of F4/80, slides were additionally incubated with streptavidin conjugated to horse radish peroxidase (ThermoShandon, #TS-125-HR, 10 minutes). Detection of both was completed by incubation in substrate containing the chromagen DAB (ThermoShandon, #TA-125-HDX, 5 minutes).
RT-qPCR
For quantitative PCR, total RNA was isolated from cells using the RNeasy Kit (Qiagen) according to the instructions of the manufacturer. First-strand synthesis was performed using M-MLV reverse transcriptase (Invitrogen). Realtime PCR was performed using SYBR GREEN PCR master mix (Applied Biosystems) in an Applied Biosystems 7900HT thermocycler using SyBr Green detection protocol as outlined by the manufacturer using the following PCR conditions: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. mRNA was normalized to actin allowing for comparison of mRNA expression. Please see key reagents table for qPCR primer sequences.
Statistics
Please refer to the legend of the figures for description of sample size (n) and statistical significance. Data were analyzed with GraphPad Prism 7.0 software using the two-tailed unpaired Student t test. Differences were considered statistically significant when the p value was less than 0.05, where **** p<0.0001, *** p<0.0005, ** p<0.005, * p<0.05, ns not significant.
Supplementary Material
Acknowledgements
The authors thank Dr. David Boyd and Dr. Marie Wehenkel for thoughtful discussion, suggestions and technical assistance. This work was supported by grants from the US National Institutes of Health, AI44828 and CA231620 to D.R.G, AI127658 to J.M.B. and the Paul Barrett Endowed Fellowship to B.T.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
D.R.G. consults for Ventus Therapeutics and Inzen Therapeutics.
References
- Alvarez-Diaz S, Dillon CP, Lalaoui N, Tanzer MC, Rodriguez DA, Lin A, Lebois M, Hakem R, Josefsson EC, O’Reilly LA, et al. (2016). The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 45, 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, Kaiser WJ, Mocarski ES, and Dubyak GR (2015). Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J Biol Chem 290, 20167–20184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossaller L, Chiang PI, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VA, Mocarski ES, Subramanian D, Green DR, Silverman N, et al. (2012). Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via Caspase-8 in an RIP3-independent manner. J Immunol 189, 5508–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan D, Bartok E, Gaidt MM, Bock FJ, Herrmann J, Seeger JM, Broz P, Beckmann R, Kashkar H, Tait SWG, et al. (2018). BAX/BAK-Induced Apoptosis Results in Caspase-8-Dependent IL-1beta Maturation in Macrophages. Cell Rep 25, 2354–2368 e2355. [DOI] [PubMed] [Google Scholar]
- Chen GY, Liu M, Wang F, Bertin J, Núñez G. (2011). A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol 186, 7187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, and Green DR (2012). Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep 1, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, et al. (2014). RIPK1 blocks early postnatal lethality mediated by Caspase-8 and RIPK3. Cell 157, 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch M, Günther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, Schiffmann LM, Stair N, Stocks H, Seeger JM, Lamkanfi M, Krönke M, Pasparakis M, Kashkar H. (2019). Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 575, 683–687. [DOI] [PubMed] [Google Scholar]
- Fu TM, Li Y, Lu A, Li Z, Vajjhala PR, Cruz AC, Srivastava DB, DiMaio F, Penczek PA, Siegel RM, et al. (2016). Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex. Mol Cell 64, 236–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, Robertson AA, Cooper MA, Graf T, and Hornung V (2016). Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 44, 833–846. [DOI] [PubMed] [Google Scholar]
- Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, Cheng H, Jin K, Ni Q, Yu X, et al. (2019). The role of necroptosis in cancer biology and therapy. Mol Cancer 18, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunert M, Gottschalk K, Kapahnke J, Gundisch S, Kieser A, and Jeremias I (2012). The adaptor protein FADD and the initiator Caspase-8 mediate activation of NF-kappaB by TRAIL. Cell Death Dis 3, e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, and Kanneganti TD (2014). FADD and Caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192, 1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung P, and Kanneganti TD (2015). Novel roles for Caspase-8 in IL-1beta and inflammasome regulation. Am J Pathol 185, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig T, Montinaro A, von Karstedt S, Sevko A, Surinova S, Chakravarthy A, Taraborrelli L, Draber P, Lafont E, Arce Vargas F, et al. (2017). The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol Cell 65, 730–742 e735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zeng MY, Yang D, Motro B, and Nunez G (2016). NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CM, and Martin SJ (2017). Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol Cell 65, 715–729 e715. [DOI] [PubMed] [Google Scholar]
- Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, Schwabe JW, Leverkus M, Cain K, and MacFarlane M (2016). Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol Cell 61, 834–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, et al. (1997). Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, and Mocarski ES (2011). RIP3 mediates the embryonic lethality of Caspase-8-deficient mice. Nature 471, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TB, Oh GS, Scandella E, Bolinger B, Ludewig B, Kovalenko A, and Wallach D (2008). Mutation of a self-processing site in Caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J Immunol 181, 2522–2532. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. (2011). Non-canonical inflammasome activation targets Caspase-11. Nature 479, 117–121. [DOI] [PubMed] [Google Scholar]
- Keller N, Mares J, Zerbe O, and Grutter MG (2009). Structural and biochemical studies on proCaspase-8: new insights on initiator caspase activation. Structure 17, 438–448. [DOI] [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, and Leder P (1998). The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8, 297–303. [DOI] [PubMed] [Google Scholar]
- Lee KH, and Kang TB (2019). The Molecular Links between Cell Death and Inflammasome. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens JR, Gurung P, Vogel P, Johnson GR, Carter RA, McGoldrick DJ, Bandi SR, Calabrese CR, Vande Walle L, Lamkanfi M, Kanneganti TD. (2014). Dietary modulation of the microbiome affects autoinflammatory disease. Nature. 516(7530):246–249. 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, and Beyaert R (2008). Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by Caspase-8. J Exp Med 205, 1967–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Feng Y, Lyons JD, Berger SB, Otani S, DeLaney A, Tharp GK, Maner-Smith K, Burd EM, Schaeffer M, et al. (2018). Caspase-8 Collaborates with Caspase-11 to Drive Tissue Damage and Execution of Endotoxic Shock. Immunity 49, 42–55 e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meynier S, and Rieux-Laucat F (2019). FAS and RAS related Apoptosis defects: From autoimmunity to leukemia. Immunol Rev 287, 50–61. [DOI] [PubMed] [Google Scholar]
- Mombaerts P; Iacomini J; Johnson RS; Herrup K; Tonegawa S; Papaioannou VE. (1992). RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68(5):869–77. [DOI] [PubMed] [Google Scholar]
- Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, Lamming DW, Chen ZJ, Horng T, Yeretssian G, et al. (2017). STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell 171, 809–823 e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti J, Vabret N, and Blander JM (2018). Measuring Innate Immune Responses to Bacterial Viability. Methods Mol Biol 1714, 167–190. [DOI] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. [DOI] [PubMed] [Google Scholar]
- Newton K, Sun X, and Dixit VM (2004). Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 24, 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose-Girma M, Dohse M, Komuves L, Webster JD, and Dixit VM (2019a). Cleavage of RIPK1 by Caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 475, 428–431. [DOI] [PubMed] [Google Scholar]
- Newton K, Wickliffe KE, Maltzman A, Dugger DL, Reja R, Zhang Y, Roose-Girma M, Modrusan Z, Sagolla MS, Webster JD, Dixit VM. (2019b). Activity of Caspase-8 determines plasticity between cell death pathways. Nature 575, 679–682. [DOI] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, and Green DR (2011). Catalytic activity of the Caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al. (2018). Pathogen blockade of TAK1 triggers Caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, and Vandenabeele P (2015). Necroptosis and its role in inflammation. Nature 517, 311–320. [DOI] [PubMed] [Google Scholar]
- Pelletier S, Gingras S, Green DR. (2015). Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity 42(1):18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip NH, DeLaney A, Peterson LW, Santos-Marrero M, Grier JT, Sun Y, Wynosky-Dolfi MA, Zwack EE, Hu B, Olsen TM, et al. (2016). Activity of Uncleaved Caspase-8 Controls Anti-bacterial Immune Defense and TLR-Induced Cytokine Production Independent of Cell Death. PLoS Pathog 12, e1005910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip NH, Dillon CP, Snyder AG, Fitzgerald P, Wynosky-Dolfi MA, Zwack EE, Hu B, Fitzgerald L, Mauldin EA, Copenhaver AM, et al. (2014). Caspase-8 mediates Caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kappaB and MAPK signaling. Proc Natl Acad Sci U S A 111, 7385–7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Reynaud D, Kang YA, Carlin D, Calero-Nieto FJ, Leavitt AD, Stuart JM, Gottgens B, and Passegue E (2015). Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 17, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop C, Fitzgerald P, Green DR, and Salvesen GS (2007). Role of proteolysis in Caspase-8 activation and stabilization. Biochemistry 46, 4398–4407. [DOI] [PubMed] [Google Scholar]
- Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, and Salvesen GS (2011). FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J 433, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan ZH, Xu ZX, Zhou XY, Zhang X, and Shang L (2019). Implications of Necroptosis for Cardiovascular Diseases. Curr Med Sci 39, 513–522. [DOI] [PubMed] [Google Scholar]
- Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, Silke J, Stacey KJ. (2013). AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ 20, 1149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, Guy CS, Briard B, Place DE, Bhattacharya A, et al. (2019). DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573, 590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, Swanson JA, Muller M, and Blander JM (2011). Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature 474, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, and Hornung V (2016). A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem 291, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott FL, Stec B, Pop C, Dobaczewska MK, Lee JJ, Monosov E, Robinson H, Salvesen GS, Schwarzenbacher R, and Riedl SJ (2009). The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 457, 1019–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin ME, Maslowski KM, Maloy KJ, Hardt WD. (2015). Inflammasomes of the intestinal epithelium. Trends Immunol 36, 442–50. [DOI] [PubMed] [Google Scholar]
- Shen C, Pei J, Guo X, Zhou L, Li Q, and Quan J (2018). Structural basis for dimerization of the death effector domain of the F122A mutant of Caspase-8. Sci Rep 8, 16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenderov K, Riteau N, Yip R, Mayer-Barber KD, Oland S, Hieny S, Fitzgerald P, Oberst A, Dillon CP, Green DR, et al. (2014). Cutting edge: Endoplasmic reticulum stress licenses macrophages to produce mature IL-1beta in response to TLR4 stimulation through a Caspase-8- and TRIF-dependent pathway. J Immunol 192, 2029–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, et al. (2016). NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17, 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stammler D, Eigenbrod T, Menz S, Frick JS, Sweet MJ, Shakespear MR, Jantsch J, Siegert I, Wolfle S, Langer JD, et al. (2015). Inhibition of Histone Deacetylases Permits Lipopolysaccharide-Mediated Secretion of Bioactive IL-1beta via a Caspase-1-Independent Mechanism. J Immunol 195, 5421–5431. [DOI] [PubMed] [Google Scholar]
- Strasser A, Jost PJ, and Nagata S (2009). The many roles of FAS receptor signaling in the immune system. Immunity 30, 180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince JE, De Nardo D, Gao W, Vince AJ, Hall C, McArthur K, Simpson D, Vijayaraj S, Lindqvist LM, Bouillet P, et al. (2018). The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and -7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1beta Activation. Cell Rep 25, 2339–2353 e2334. [DOI] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, Mason K, Gross O, Ma S, Guarda G, et al. (2012). Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36, 215–227. [DOI] [PubMed] [Google Scholar]
- Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK, Kelliher MA, et al. (2014). Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U S A 111, 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh WC, de la Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, et al. (1998). FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279, 1954–1958. [DOI] [PubMed] [Google Scholar]
- Zhang CJ, Jiang M, Zhou H, Liu W, Wang C, Kang Z, Han B, Zhang Q, Chen X, Xiao J, et al. (2018). TLR-stimulated IRAKM activates Caspase-8 inflammasome in microglia and promotes neuroinflammation. J Clin Invest 128, 5399–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets analyzed during this study.