

SUMMARY
BET bromodomain inhibitors (BBDI) are candidate therapeutic agents in triple-negative breast cancer (TNBC) and other cancer types, but inherent and acquired resistance to BBDIs limit their potential clinical use. Using CRISPR and small molecule inhibitor screens combined with comprehensive molecular profiling of BBDI response and resistance, we identified synthetic lethal interactions with BBDIs and genes that when deleted confer resistance. We observed synergy with regulators of cell cycle progression, YAP, AXL, and SRC signaling, and chemotherapeutic agents. We also uncovered functional similarities and differences among BRD2, BRD4, and BRD7; while deletion of BRD2 and BRD4 enhances sensitivity to BBDIs, BRD7 loss leads to gain of TEAD/YAP chromatin binding and luminal features associated with BBDI resistance. Single cell RNA-seq and ATAC-seq and cellular barcoding analysis of BBDI responses in sensitive and resistant cell lines highlight significant heterogeneity among samples and demonstrate that BBDI resistance can be either pre-existing or acquired.
Graphical Abstract
eTOC Blurb
Shu et al. conducted integrated comprehensive molecular profiling and functional screens to identify synthetic lethal and resistance interactions with BET bromodomain inhibitors. CDK4 and BRD2 are top synthetic lethal hits, while deletion of BRD7 leads to resistance due to activation of genes suppressed by BET inhibitors in sensitive cells.
INTRODUCTION
TNBC is characterized by the absence of estrogen receptor (ER), progesterone receptor (PR) expression, and the human epidermal growth factor receptor 2 (HER2). Only about 30–40% of TNBC patients respond well to chemotherapy, and even though a subset of TNBC expresses PD-L1 and benefit from immunotherapy (Garrido-Castro et al., 2019), novel therapeutic options are needed for the majority of patients with advanced disease. TNBC genome sequencing studies have failed to identify recurrently mutated therapeutic targets and have revealed extensive inter- and intratumor heterogeneity (Garrido-Castro et al., 2019). This heterogeneity suggests that multiple new therapeutic approaches involving combinations of agents are required for the effective treatment of TNBC patients.
BET bromodomain (BBD) proteins (e.g., BRD2–4) are emerging therapeutic targets in TNBC. Directly acting BET bromodomain inhibitors (BBDIs), including JQ1 (Filippakopoulos et al., 2010), utilize acetyl-lysine competitive binding to displace BBD proteins from chromatin, resulting in selective transcriptional responses and anti-proliferative efficacy (Delmore et al., 2011; Filippakopoulos et al., 2010; Puissant et al., 2013). The selectivity of BBD inhibition arises from the localization of BBDs to super-enhancers (SEs) that regulate cell-specifying and oncogenic transcriptional programs (Chapuy et al., 2013; Hnisz et al., 2013; Loven et al., 2013; Whyte et al., 2013), producing anti-tumor effects in preclinical models and in clinical trials (O’Dwyer et al., 2016; Shapiro et al., 2015; Shu and Polyak, 2016). We have previously identified BBDIs as potential therapeutic agents in TNBC (Shu et al., 2016), but also found that acquired resistance emerges early necessitating the development of rational combinations. Thus, to identify synthetic lethal and resistance interactions with BBDIs and to decipher the underlying mechanisms, we performed unbiased comprehensive functional screens combined with multi-omics profiling using matched pairs of BBDI sensitive and acquired-resistant TNBC cell lines (graphical abstract).
RESULTS
CRISPR screens to uncover synthetic lethal and resistance interactions with JQ1
To identify synthetic lethal targets and mechanisms of resistance to BBDIs, we performed a genome-wide CRISPR/Cas9 knockout screen in the TNBC cell lines SUM159 and SUM149 treated with JQ1 or DMSO, and in their BBDI-resistant derivatives SUM159R and SUM149R, which we previously generated (Shu et al., 2016). Cells with knockout of known essential genes, including ribosomal genes, were strongly depleted in both treatments (Figure S1A), confirming the quality of the data. Across both parental cell lines, sgRNAs that became specifically depleted after JQ1 treatment included those targeting G1-S cell cycle progression (CDK4, SKP2), oncogenic transcriptional regulators (BRD2, EP300, CREBBP, NELFB), and DNA damage repair (PRKDC), indicating that these pathways became more essential under bromodomain inhibition and are synergistic with JQ1 (Figures 1A and 1B, and Table S1). Conversely, sgRNAs that targeted G1-S cell cycle inhibitors (CDKN1A, NF2) and tumor suppressive transcriptional regulators (ARID1A, TCEB3) were enriched, indicating that they contribute to JQ1 resistance. TCEB3 interacts with BRD3 and JQ1 treatment increases its binding (Lambert et al., 2019), which may explain why it was a hit in our screen. Other cell cycle regulators (MYC, CCND1) and transcriptional activators including Mediator complex components (MED23, MED24, MED30) were synergistic hits in SUM159 cells, while cell cycle and transcription-related tumor suppressors (RB1, BRD7) were resistance hits. Many of our CRISPR hits (e.g., CDK4 and EP300) are also dependencies in cell lines that depend on BRD4 in the DepMap data (Tsherniak et al., 2017) (Figures S1B and S1C).
Figure 1. CRISPR screen results and molecular profiles.
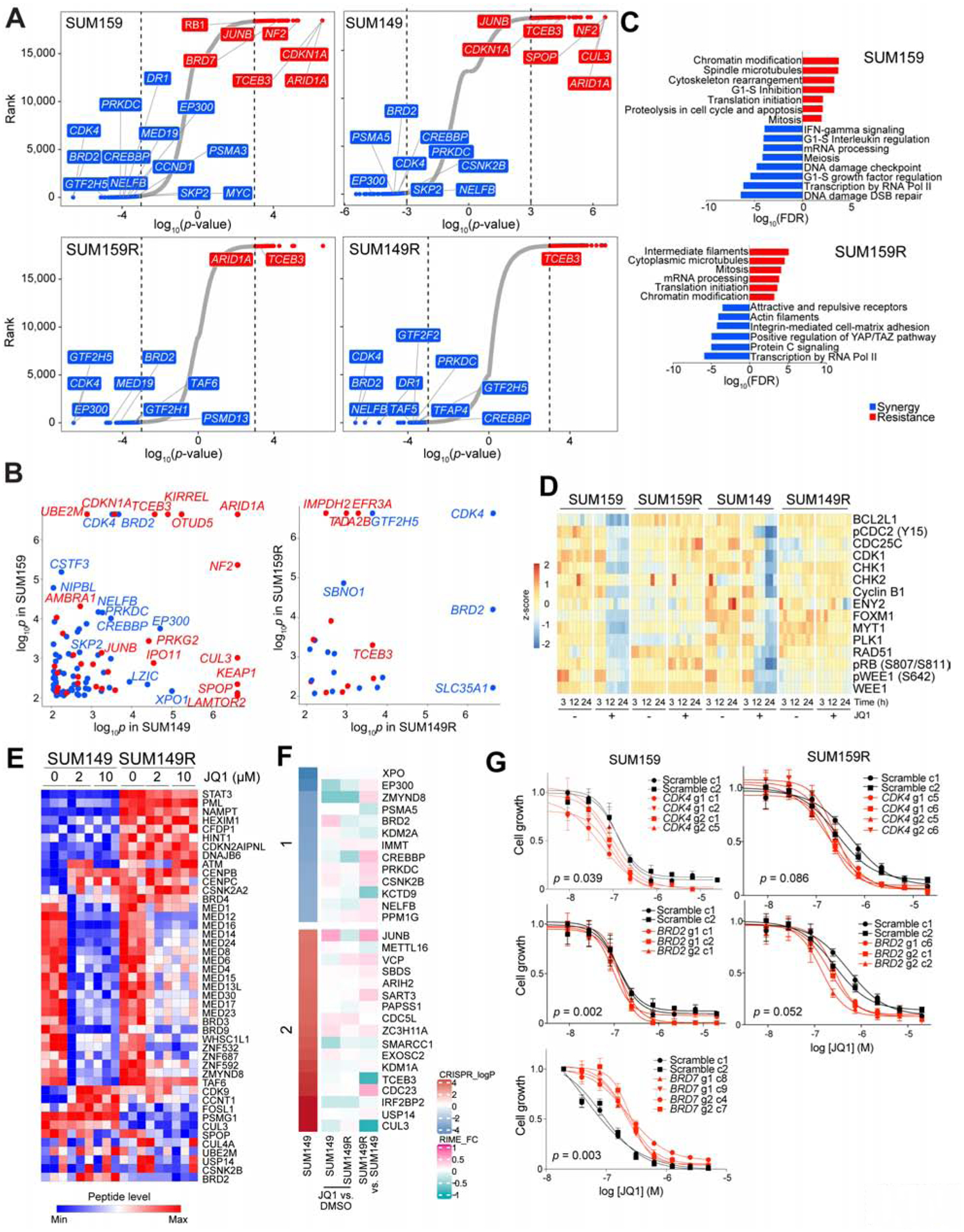
(A) Top hit genes in the CRISPR screen revealed by comparing untreated vs. JQ1-treated cells. Genes are ranked by log10(p-values) defined by MAGeCK. Significant resistance and synthetic lethal hits (p-value < 0.001) are marked in red and blue, respectively. (B) Comparison of CRISPR screen hits between the indicted cell lines. Genes with p-values < 0.001 in both cell lines are shown. (C) Top process networks enriched in CRISPR screen hits. (D) Heatmap of Cluster 8 in RPPA data representing proteins downregulated after JQ1 treatment in parental but not in BBDI-resistant cells. (E) Heatmap of BRD4 RIME data in SUM149 and SUM149R cells −/+ 3h JQ1 treatment at the indicated doses. (F) Heatmap of significant resistant (red) and synthetic lethal (blue) hits CRISPR screen hits and their BRD4 binding changes revealed by RIME in SUM149 and SUM149R cells in JQ1 compared to DMSO and in SUM149R compared to SUM149 cells. (G) Dose response curves of JQ1 of scramble or gene-specific sgRNA-expressing single cell clones derived from the indicated cell lines. See also Figures S1–3, and Tables S1–3.
In SUM149 cells, deletion of CSNK2B, a subunit of CK2 which phosphorylates and stabilizes BRD4 (Wu et al., 2013), was synergistic with JQ1, while deletion of genes known to function in ubiquitination of BRD4 (SPOP, CUL3) and promote proteasomal degradation (Dai et al., 2017; Janouskova et al., 2017; Zhang et al., 2017) made cells more resistant (Figure 1A). Proteasomal subunits were also synergistic hits in both SUM159 and SUM149 cells and ubiquitin-proteasomal proteolysis was the top enriched process network in SUM149 cells (Figure S1D). These data are consistent with our prior finding that CK2-mediated phosphorylation and stabilization of BRD4 on the chromatin is one of the mechanisms of acquired BBDI resistance (Shu et al., 2016).
In the BBDI-resistant SUM159R and SUM149R cells, genes involved in transcription by RNA polymerase II were enriched among the top synergistic hits (Figure 1A). Moreover, RNA polymerase II complex regulation was the most significantly enriched pathway among synergistic genes in both resistant cell lines, implying that BBDI resistance still relies on BRD4/RNA polymerase II complex function (Figures 1C and S1D). This observation is consistent with our prior data demonstrating that BBDI-resistant cells still depend on BRD4 (Shu et al., 2016). Notably, CDK4 and BRD2 were top synergy hits in all four cell lines, suggesting that their deletion not only sensitizes cells to BBDIs but also overcomes acquired BBDI resistance (Figures 1A and 1B).
Integration of CRISPR hits and molecular profiles
To explore mechanisms by which the CRISPR screen hits exert their function, we analyzed genetic alterations, gene expression, DNA methylation, histone and protein modifications, and the BRD4 interactome of parental and BBDI-resistant cells before and after JQ1 treatment (graphical abstract). While we identified several mutations specific to BBDI-resistant SUM149R or SUM159R cells (Figure S1E), none of the mutated genes overlapped with CRISPR screen hits, suggesting that they may not play a role in BBDI response and resistance. BBDI-resistant cell lines displayed a significant increase in global DNA methylation that was not affected by JQ1 (Figure S1F). Although this increase in DNA methylation did not globally correlate with changes in gene expression, a number of genes (e.g., DBC1, PTN) show significant changes in both DNA methylation and expression (Figure S1G). Histone mass spectrometry revealed increased H3K27me2/3 and H3K9me0/1/2/3 levels in both BBDI-resistant cell lines compared to parental cells, but JQ1 treatment did not influence these profiles (Figure S1H).
Functional analysis of differentially expressed genes by Metacore (Nikolsky et al., 2005) revealed upregulation of G1-S growth factor regulation, anti-apoptosis, and epithelial-to-mesenchymal transition (EMT) pathways in resistant compared to parental cells, while G2-M, mitosis, and ubiquitin-proteasomal proteolysis were downregulated (Figure S1I and Table S2). Cell cycle-related pathways were downregulated by JQ1 in parental but upregulated in BBDI-resistant cells. Reverse phase protein array (RPPA) (Tibes et al., 2006) analysis revealed 12 clusters that showed differences due to JQ1 treatment or BBDI resistance (Figure S2A and Table S3), with altered proteins enriched in several oncogenic pathways including NOTCH, WNT, and JAK/STAT signaling (Figure S2B). Specifically, c-MYC, EGFR/pEGFR, JAK2, and STAT3pT705 were elevated in SUM149R cells, YAP had increased expression and phosphorylation (S127) in SUM159R cells, and BCL2, cyclin D1, AXL, BRD4, HER2, STAT5A, and c-MET had higher levels in both BBDI-resistant lines (Figure S2C). Cluster 8 composed of cell cycle-related proteins was downregulated by JQ1 in parental but not in BBDI-resistant cells (Figure 1D). The average expression of all cell cycle proteins based on RPPA showed a similar trend (Figure S2D), indicating that JQ1 has the most pronounced effects on the cell cycle as compared to other cellular programs. These gene expression and protein changes are consistent with our CRISPR screen data showing that deletion of positive regulators of G1-S progression (e.g., CCND1, CDK4) sensitizes cells to JQ1, while deletion of negative regulators (e.g., RB1, CDKN1A) increases JQ1 resistance. The expression levels of several CRISPR screen hits were also differentially regulated in parental and resistant cells with or without JQ1 treatment, including CDK4 and BRD2 (Figures S2A–F). Similarly, the YAP/TAZ pathway, which is specifically upregulated in SUM159R cells based on both RNA-seq and RPPA data was essential only in SUM159R cells (Tables S1–3). These results suggest that JQ1 has the most pronounced effects on signaling pathways and cell cycle proteins regulating G1/S transition.
To characterize BRD4-associated chromatin complexes, we performed rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) and qPLEX-RIME (Mohammed et al., 2013; Papachristou et al., 2018) in parental and BBDI-resistant cells in the presence and absence of JQ1. We found that JQ1 disrupted BRD4’s interaction with Mediator proteins and epigenetic regulators in both parental cells, while there was less change in BBDI-resistant cells (Figures 1E and S2G, and Table S3). Many of the BRD4-interacting proteins we identified are also part of the BRD4 interactome in other cell types implying a general role in BRD4 function (Lambert et al., 2019). CK2 and STAT3 showed increased binding with BRD4 in SUM149R cells (Figure 1E), which we confirmed for STAT3 by co-immunoprecipitation (Figure S2H). On the other hand, enzymes involved in ubiquitination (CUL3, SPOP) showed the opposite trend (Figure 1E). Analysis of process networks enriched in differentially-bound proteins identified the RNA Pol II complex as most significantly decreased by JQ1 in parental cells, while mRNA processing was the most significantly increased pathway in resistant cells after JQ1 (Figure S2I). The RNA Pol II pathway and chromatin modification complex were the top upregulated process networks in SUM149R compared to SUM149 cells, while cytoskeleton rearrangement and mitosis were the top downregulated ones (Figure S2I). Several BRD4-associated proteins were top CRISPR screen hits, including bromodomain proteins BRD2, ZMYND8, EP300, as well as BRD4 modifiers such as CSNK2B and CUL3 (Figures 1F and S2J.
In sum, our multi-omic profiling of differences between BBDI-sensitive and resistant cells before after JQ1 provide a strong mechanistic explanation for many of the hits we identified in our CRISPR screen.
Validation of CRISPR screen hits
In our CRISPR screen, both positive and negative regulators of G1-S progression were among the top synergistic and resistance hits, respectively, and both RNA-seq and RPPA analyses demonstrated that JQ1 has the most pronounced effects on cell cycle. Since our ultimate goal is to test BBDI combination therapies in the clinic and CDK4/6 inhibitors are already approved for the treatment of ER+ breast cancer (Abraham et al., 2018), we selected CDK4 for further validation. Similarly, we decided to further characterize BRD2 and BRD7, since they were top CRISPR synergy and resistance hits, respectively, and are implicated in JQ1 response based on gene expression and proteomic changes. The read counts of multiple sgRNAs targeting CDK4 and BRD2 displayed specific depletion after JQ1 in both SUM159R and SUM149R cells, while BRD7 sgRNAs showed uniform positive selection in SUM159 cells (Figure S3A).
To further validate the relationships between JQ1 treatment and CDK4, BRD2, and BRD7, we generated single cell clones of SUM159 and SUM159R cells expressing individual sgRNAs targeting CDK4 and BRD2, as well as SUM159 clones expressing BRD7 sgRNAs (Figure S3B). Both SUM159 and SUM159R single cell clones with BRD2 or CDK4 deletion displayed increased sensitivity to JQ1 compared to scrambled sgRNA-expressing clones (Figures 1G and S3C). In contrast, SUM159-derived BRD7 knockout clones were more resistant to JQ1 than clones expressing scrambled controls (Figures 1G and S3C). Overexpression of doxycycline-inducible CDK4 rescued the decreased viability of CDK4 knockout clones after JQ1 treatment validating it as an on-target effect (Figures S3D and S3E).
Small molecule inhibitor (SMI) screen
To identify therapeutic compounds to overcome BBDI resistance we performed a cellular viability screen in both parental and resistant cells, using two SMI libraries targeting a range of pathways (Figure S3F and Table S4). We confirmed the resistance of SUM159R and SUM149R cells to JQ1 and to other BBDIs (Figures 2A–D and S3G). To identify potentially synergistic compounds, we identified drugs that the BBDI-resistant lines were more sensitive than the parental lines (Figure S3H). In SUM149R cells, top hits included DNA damage-inducing agents (topoisomerase, DNA synthesis, microtubule), cell cycle (CDK4/6, PLK, AURK), histone deacetylase (HDAC), and HSP90 inhibitors (Figures 2A–B). Several cell cycle inhibitors targeting CDK4/6, AURK, and PLK especially effectively inhibited the growth of SUM149R compared to SUM149 cells (Figure 2A). For several selected hits we confirmed synergy with JQ1 in SUM149R cells (Figure S3I) and inhibition of their target (Figure S3J). Signaling pathways were most frequent among the top hits in SUM159R cells, including EGFR, MEK, and PI3K, as well as TGF-β and endocrine inhibitors (Figures 2C,D), which is consistent with our RPPA and RNA-seq data demonstrating upregulation of these pathways in resistant cells (Figures S1I and S2A–F). BCL2 inhibitors were hits in both SUM149R and SUM159R cells (Figures 2A and 2C), consistent with our previous findings that upregulation of anti-apoptotic factors contributes to JQ1 resistance (Shu et al., 2016). Topoisomerase inhibitors were hits for both SUM159R and SUM149R cells (Figures 2A and 2C), which may reflect sensitivity to DNA damage and transcription that we also found in our CRISPR screen (Figures 1C and S1D). Furthermore, both BBDI-resistant lines had increased sensitivity to inhibitors of CDK9, a component of P-TEFb complex with BET bromodomain proteins, as well as the non-specific CDK inhibitor AT7519, and SUM149R cells were also more sensitive to a CDK4/6-specific inhibitor palbociclib (Figures 2E and 2F). These findings are in line with our CRISPR and molecular profiling data indicating that G1-S cell cycle progression and transcriptional activators become more essential in cells with acquired BBDI resistance. The identification of the same genes and pathways using multiple independent unbiased methods and the integration of these findings with molecular profiles highlight the robustness of the data and the advantage of using such a combined approach.
Figure 2. Small molecule screen in BBI-resistant TNBC cells.
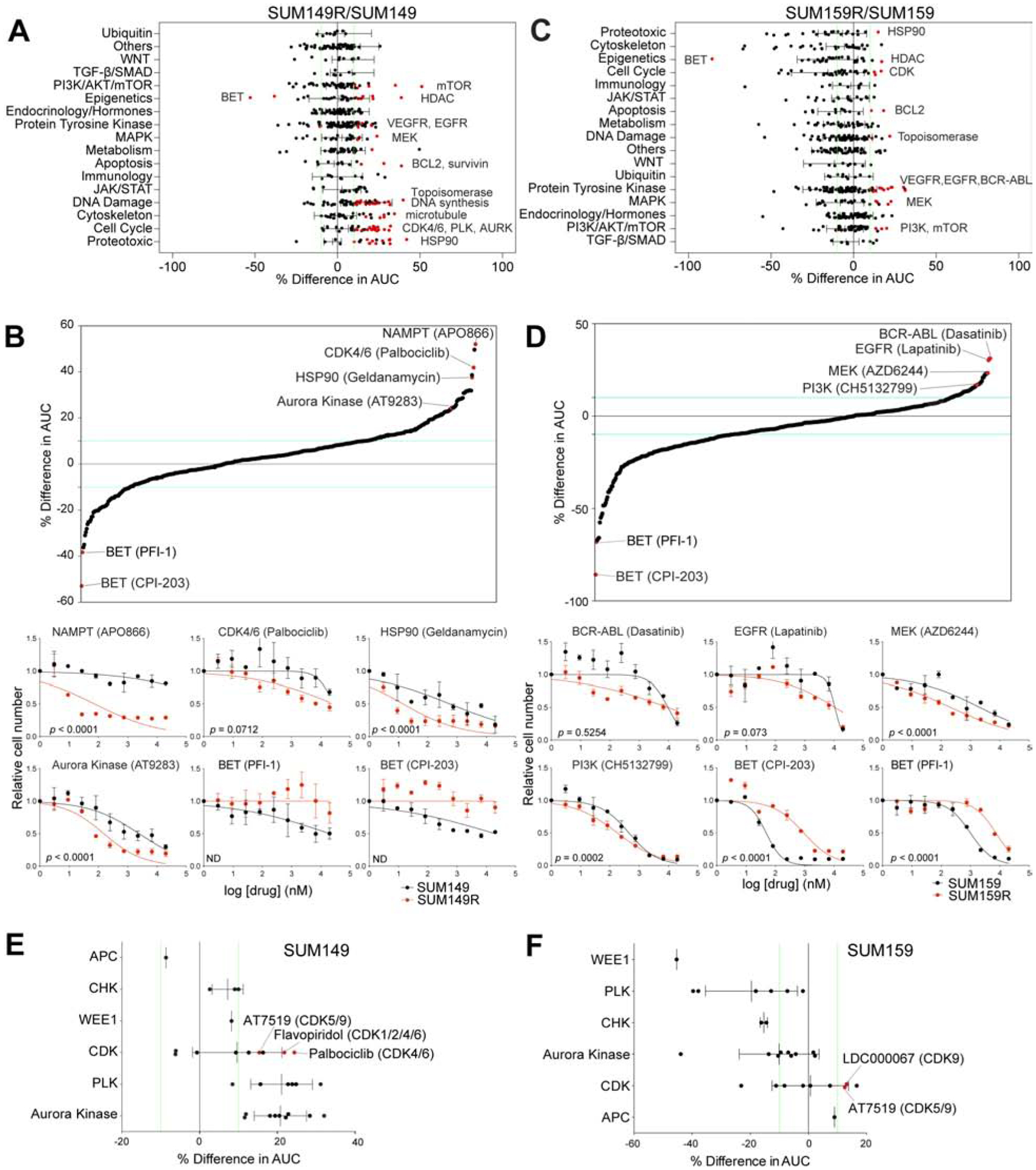
(A-D) Differences in drug sensitivities between SUM149R and SUM149 (A,B) and between SUM159R and SUM159 cells (C,D), grouped by pathways targeted by drugs in the library (A,C), and ordered by percent differences in AUC (B,D). Common targets of top hits are indicated (A,C), and individual dose-response curves of top hits are shown (B,D). (E-F) Differences in sensitivities of drugs targeting the cell cycle, grouped by target, in SUM149 (E), and SUM159 cells (F). See also Figure S3 and Table S4.
Combination treatments in vitro and in vivo
To further validate synthetic lethal targets identified in our CRISPR knockout and SMI screens, we performed synergy screens in SUM149, SUM149R, SUM159, and SUM159R cells. We prioritized FDA-approved drugs to facilitate the clinical translatability of our results. We found that in general, most of these drugs were more synergistic in the resistant compared to the parental lines, in which they were additive or antagonistic. Several DNA-damaging agents (e.g., doxorubicin) and microtubule inhibitors (e.g., vincristine) showed significant synergy with JQ1 in SUM149R cells, while the kinase inhibitor dasatinib and the YAP/TAZ inhibitor verteporfin was synergistic in SUM159R cells (Figure 3A and Table S4). Paclitaxel microtubule inhibitor and TP0903 AXL kinase inhibitor were synergistic in both BBDI-resistant lines (Figure 3A). Notably, palbociclib had the most significant synergy in all parental and resistant cell lines, as well as in several other TNBC cell lines and this was independent of RB1 mutation status (Figures 3A and S4A).
Figure 3. Validation of synthetic lethal interactions with JQ1.
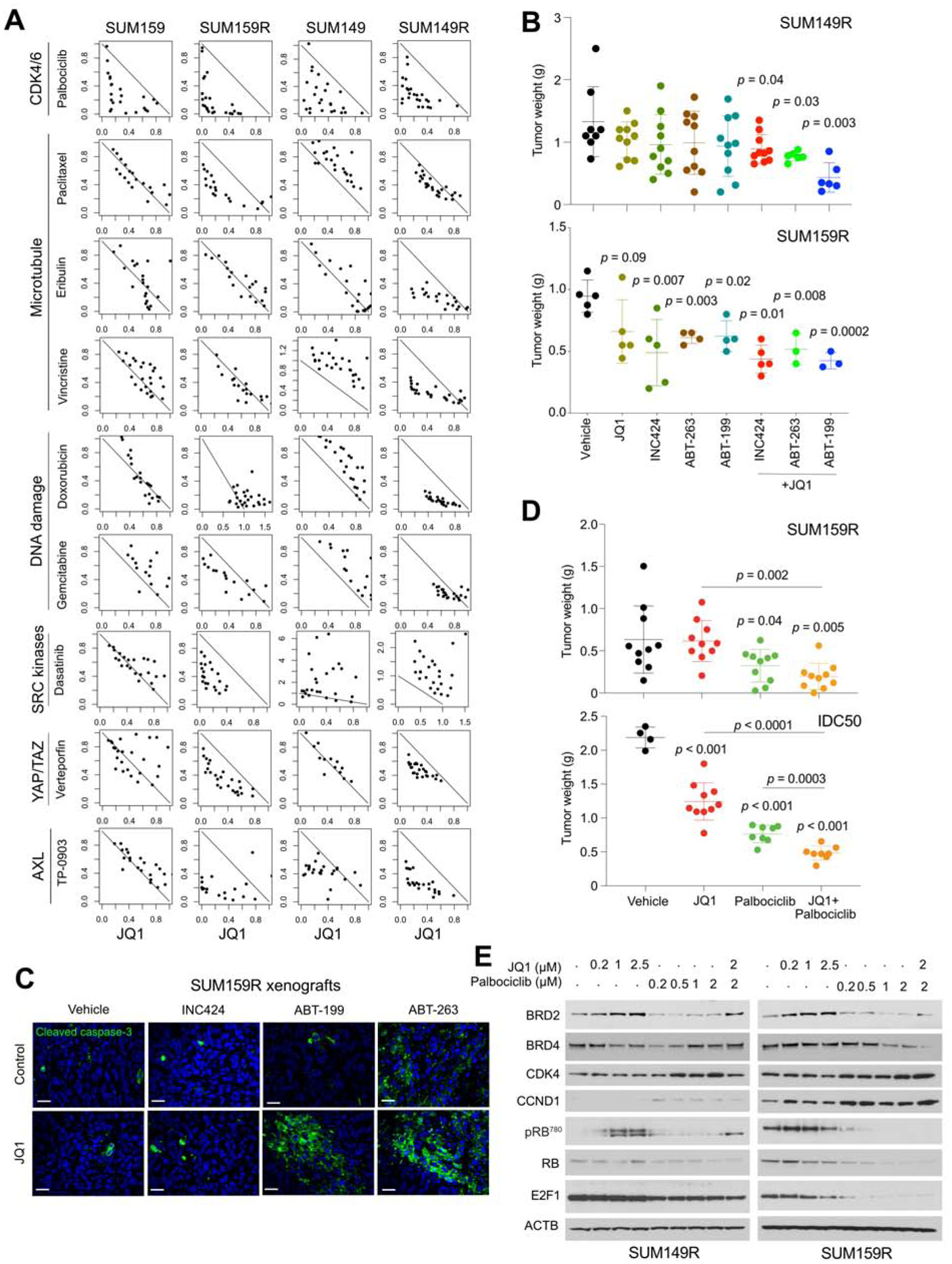
(A) Synergy studies of JQ1 with various inhibitors in cell culture. Points represent paired values of drug concentrations assessed for synergism (see Methods). The diagonal line signifies drug additivity. Points above and below the line represent antagonistic and synergistic drug combinations, respectively. (B) Plots show xenograft weights after treatment with JQ1 and other drugs alone and in combinations. P-values indicate the statistical significance of differences compared to vehicle based on t-tests. (C) Immunofluorescence analysis of cleaved caspase-3 in SUM159R xenografts after single and combination treatments. Scale bars represent 50 μm. (D) Plots show xenograft weights after treatment with JQ1 and palbociclib, alone and in combination. (E) Immunoblot analysis of indicated proteins in SUM149R and SUM159R cells treated with the indicated doses of JQ1, palbociclib, and their combination for 12 hrs. See also Figure S4.
To validate these in vitro synergy results in vivo, we tested the sensitivity of SUM149R and SUM159R, and a patient-derived xenograft (PDX, IDC50) to selected combinations. JAK2 (INC424) and BCL2/BCL-XL (ABT-263 and ABT-199) inhibitors significantly decreased tumor weight even as single agents in SUM159R xenografts, while the combination of all these agents with JQ1 resulted in significantly smaller tumors in both SUM149R and SUM159R xenografts (Figure 3B). The partial JQ1 responsiveness of the JQ1-treated SUM159R xenograft reflects the heterogeneity of this cell line. Hematoxylin-eosin (H&E) stained slides and caspase-3 immunofluorescence demonstrated an increase in apoptosis in combination treatments (Figures 3C, S4B–C). We also detected a more significant decrease in phospho-STAT3 positivity with JQ1+INC424 treatment (Figure S4D). Similarly, palbociclib and JQ1 combination significantly decreased the growth and final tumor weight of SUM159R and IDC50 xenografts with a concomitant decrease in phospho-RB positive cells (Figures 3D and S4E–G). In the IDC50 PDX we even observed slight regression with the combination despite the short treatment due to the extremely fast growth of this tumor.
To explore the mechanism of synergy between palbociclib and JQ1, we analyzed the protein levels and activity of key regulators of G1-S progression as well as BRD2, BRD4, and BRD7 by immunoblots in SUM149R and SUM159R cells. JQ1 treatment increased BRD2 and CCND1 and RB phosphorylation, while palbociclib decreased BRD2, BRD4, and E2F1, and RB phosphorylation in a dose- and time-dependent manner, more markedly in SUM159R than in SUM159 and SUM149R cells (Figures 3E and S4H). These findings suggest that palbociclib can enhance sensitivity to JQ1 via multiple mechanisms including CDK4 inhibition-mediated G1 arrest as well as by enhancing the proteasomal degradation of BRD2 and BRD4, potentially through CDK4-regulated ubiquitination in an RB-independent manner (Jin et al., 2018).
Targeted BET protein degradation overcomes BBDI resistance
Since BBDI-resistant cells are still dependent on BRD2 and BRD4 (Shu et al., 2016) and modulating BBD protein levels is one of the most effective ways to modulate JQ1 sensitivity based on our CRISPR screen and multi-omics data, we tested whether targeted degradation of BBD proteins would overcome BBDI resistance. We recently developed a highly potent and selective small molecule degrader dBET6 with improved cellular permeability and improved efficacy (Winter et al., 2015; Winter et al., 2017). dBET6 is a bifunctional molecule consisting of JQ1 appended to phthalimide-conjugates via a longer linker as compared to dBET1 (Figure S4I). We first tested the effect of these BBD degraders on cellular viability in a panel of TNBC lines and confirmed the highest efficacy of dBET6 (Figure 4A). Both the SUM149R and SUM159R lines were significantly more sensitive to dBET6 as compared to JQ1 (Figure 4B). dBET6 potently degraded BRD2, BRD3, and BRD4 in a dose-dependent manner in both parental and BBDI-resistant cells (Figures 4C and S4J). Consistent with the degradation of BRD4 protein, we observed a global loss of BRD4 from the chromatin in SUM149R cells (Figures 4D and 4E) accompanied with a decrease in mRNA levels across the majority of active genes (Figures 4F and S4K), and a decrease in RNA Pol II phosphorylation (Figure S4L). Thus, dBET6 treatment results in the rapid degradation of BRD4-bound chromatin that in turn causes a global reduction in active gene transcription mediated in part by the loss of initiating and elongated RNA Pol II.
Figure 4. Targeted BET degradation in TNBC.
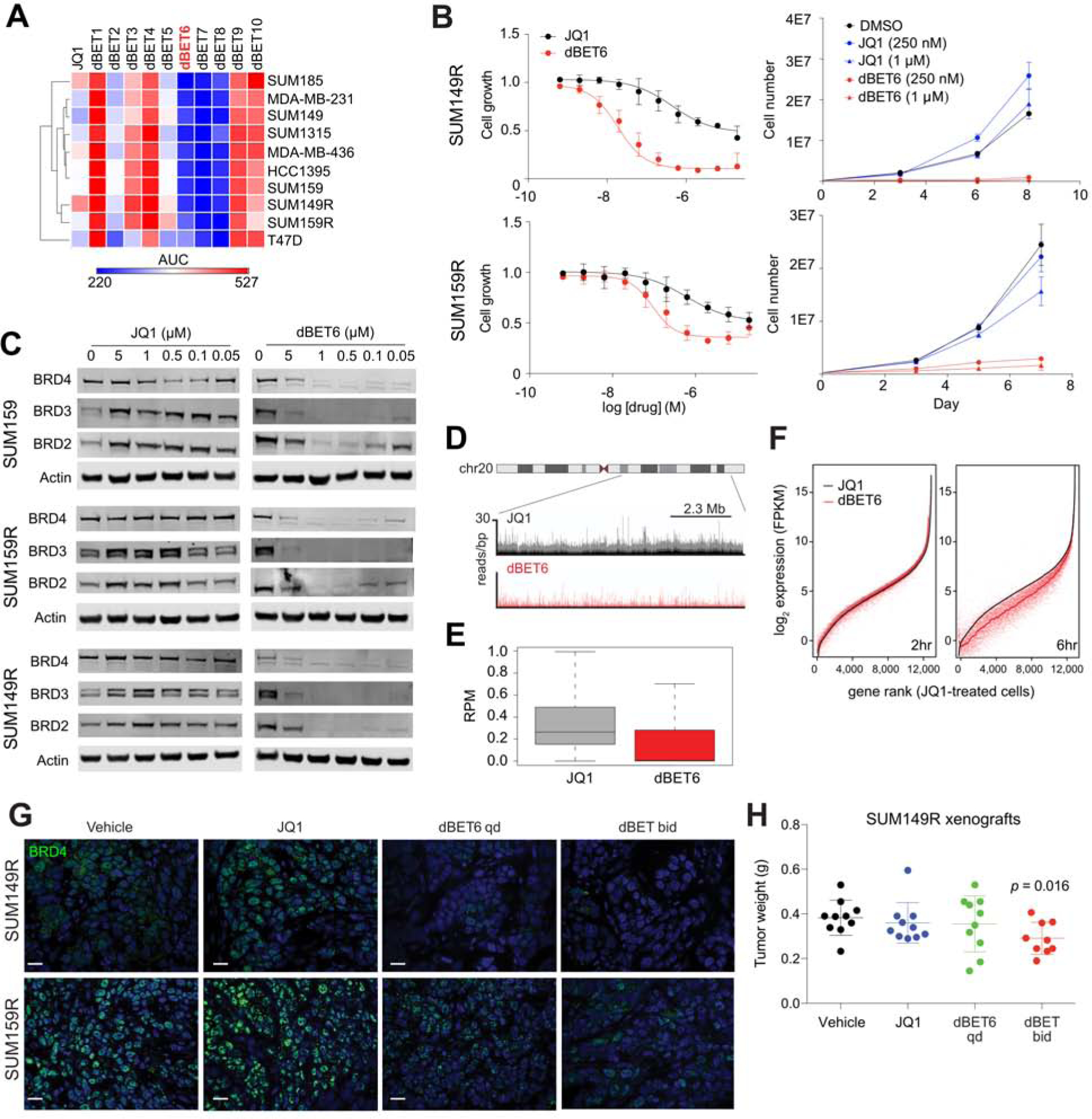
(A) Heatmap of sensitivities to JQ1 and dBET series, by surrogate levels of ATP content. Results of 10-point dose response curves after 72 hours of treatment are represented by area under curve (AUC). (B) Dose response curves (left) and growth curves (right) of JQ1 and dBET6 at indicated concentrations in SUM149R and SUM159R. (C) Immunoblot analysis of BRD2, BRD3, and BRD4 levels after 4 hours of treatment with JQ1 and dBET6 in SUM159, SUM159R, and SUM149R cells. (D) ChIP-seq tracks of BRD4 levels on chromosome 20 in SUM149R. Top track (black) shows the basal resistance state in the presence of 10 μM JQ1, and bottom track (red) shows 2 hours of treatment with 250 nM dBET6. (E) Boxplot of the global levels of chromatin-bound BRD4 after treatment with 10μM of JQ1 and 250 nM dBET6 for 2 hours. RPM, reads per million. (F) Expression levels of all active genes ranked by their expression after treatment with JQ1 for 2 hours (left) and dBET6 for 6 hours (right). (G) Immunofluorescence analysis of BRD4 in SUM149R and SUM159R xenografts treated with JQ1 (50 mg/kg, daily) or dBET6 (7.5 mg/kg, once or twice daily). Scale bars represent 50 μm. (H) Tumor weights of SUM149R xenografts following 2 weeks of treatment with JQ1 or dBET6. P-value indicates the statistical significance of difference compared to vehicle, based on t-test. See also Figure S4.
To further investigate the dBET6 efficacy in vivo, we treated SUM149R and SUM159R xenografts with JQ1 or dBET6. Although dBET6 more efficiently decreased tumor weight compared to JQ1 with a concomitant decrease in BRD4 protein levels (Figures 4G, S4M–N), we had to discontinue the experiment after 10 days of treatment due to significant toxicity. Lower dose (7.5mg/kg) dBET6 still significantly decreased tumor weight in SUM149R xenografts (Figure 4H). These data show that BBD protein degradation overcomes BBDI resistance in both SUM149R and SUM159R cell lines in cell culture and in vivo.
The impact of functional differences between bromodomain proteins on BBDI response and resistance
To investigate whether the identification of BRD2 and BRD7 as top synthetic lethal and resistance hits in our CRISPR screen, respectively, is due to differences in chromatin binding and the sensitivity of this to JQ1, we performed ChIP-seq for BRD2, BRD4, BRD7, and H3K27ac (histone H3 lysine 27 acetyl) as well as ATAC-seq in the presence and absence of JQ1. We found that BRD4, BRD2, and H3K27ac bind to the same promoters and enhancers in both SUM159 and SUM149 cells (Figures 5A and S5A). Although both BRD4 and BRD2 binding was decreased after JQ1 treatment (Figures 5B and S5B) and this was highly correlated in both promoter and enhancer regions (Figures 5C and S5C), the decrease was more significant for BRD4 than BRD2. This finding indicates that BRD2 is less efficiently dissociated from the chromatin by JQ1 than BRD4 potentially explaining why BRD2 was a top synergy hit in our CRISPR screen while BRD4 was not. We observed an increase of BRD4 and BRD2 binding in the same promoter and enhancer regions in SUM159R cells compared to parental cells (Figures 5B–C and S7B–C). Recent reports described that in hematopoietic cells BRD2, but not BRD4, colocalizes with CTCF and enhances its insulator function (Cheung et al., 2017; Hsu et al., 2017). However, in TNBC cells we found that CTCF binding was correlated with all proteins analyzed (BRD2, BRD4, and BRD7) (Figure S5D). We then investigated the effects of these binding changes on transcription and found that alterations in BRD4 binding were associated with gene expression changes especially at SE loci (Figures 5D and S5E) including SEs near BRD2 and CCND1 (Figures 5E and S5F).
Figure 5. BRD4, BRD2 and BRD7 binding changes and their effects upon JQ1 treatment.
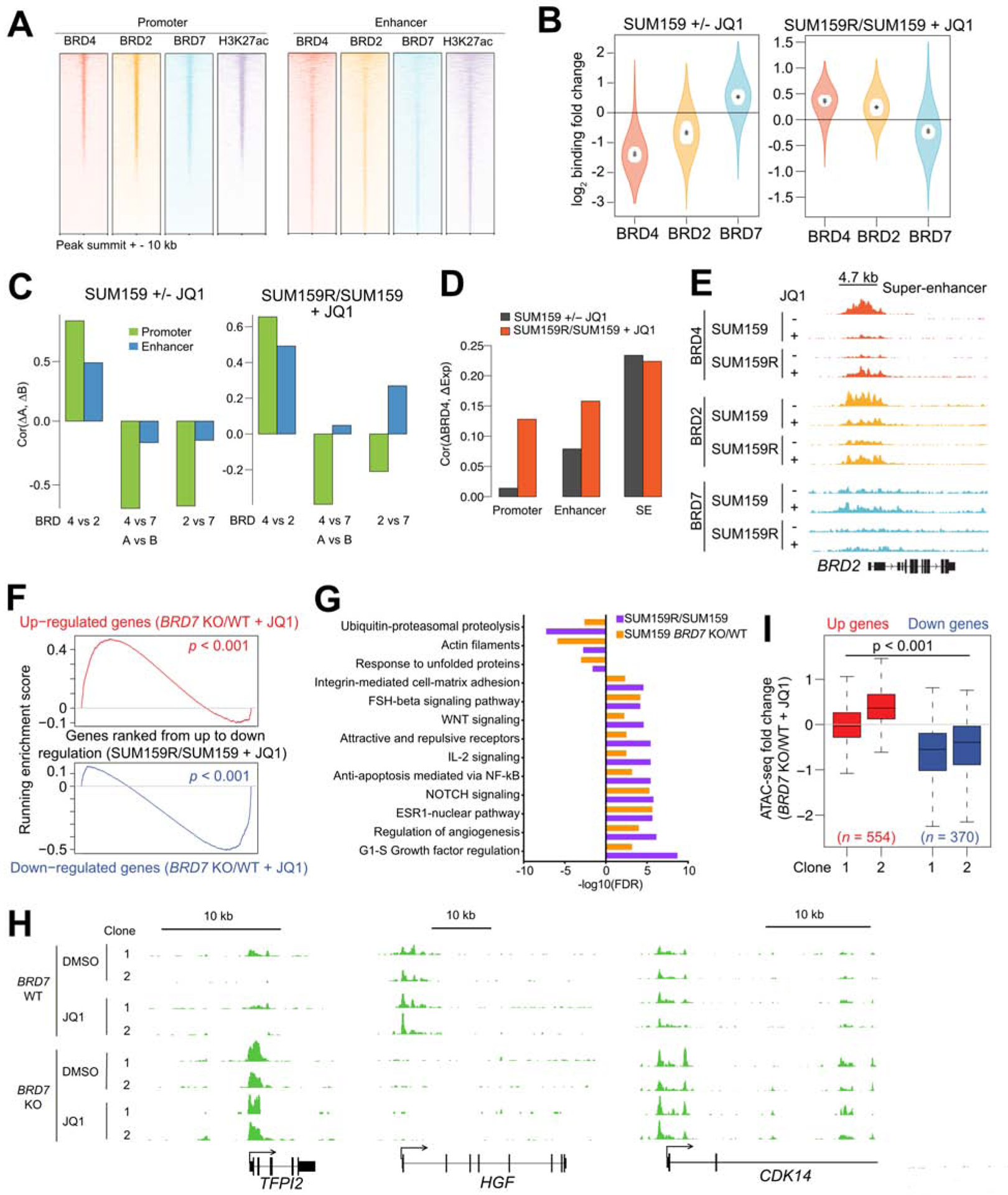
(A) ChIP-seq binding enrichment of BRD4, BRD2, BRD7, and H3K27ac in promoter and enhancer regions in SUM159 cells. (B) BRD4, BRD2, and BRD7 ChIP-seq binding changes between JQ1-treated (+JQ1) and untreated (−JQ1) SUM159 cells and between JQ1-treated resistant (SUM159R) and parental cells. Outer violin indicates the entire distribution, inner violin (white) indicates the IQR, and “.” and “+” indicate the median and mean, respectively. (C) Pairwise correlations of BRD4, BRD2, and BRD7 ChIP-seq binding changes in promoter and enhancer regions. (D) Correlations of BRD4 binding changes in promoter, enhancer, and SE regions and their gene expression changes. (E) Gene tracks depicting BRD4, BRD2, and BRD7 signal at the BRD2 locus. (F) Gene set enrichment analysis (GSEA) depicting the relationship between expression of genes in JQ1-treated BRD7 KO cells and JQ1-treated resistant cells. (G) Top process networks enriched in differentially expressed genes between JQ1-treated BRD7 wild type (WT) and BRD7 knockout (KO) SUM159 cells and between JQ1-treated SUM159R and SUM159 cells. (H) Gene tracks depicting ATAC-seq signal at selected genomic loci in SUM159 BRD7 WT and KO cells. (I) Association between ATAC-seq binding changes and gene expression changes in two JQ1-treated BRD7 KO clones compared with JQ1-treated WT SUM159 cells. See also Figure S5 and Table S5.
BRD7 binding was detected at the same regions as BRD4 and BRD2; however, BRD7 binding increased after JQ1 treatment at the same loci where BRD4 and BRD2 signal decreased in SUM159 cells, while it decreased in SUM159R cells (Figures 5B and 5C). This anti-correlation between BRD2/4 and BRD7 chromatin binding after JQ1 treatment explains our CRISPR screen data that identified BRD2 and BRD7 as synthetic lethal and resistance hits, respectively, in SUM159 cells.
To investigate the effect of BRD7 deletion on the chromatin landscape and gene expression patterns, we performed ATAC-seq and RNA-seq, respectively, in BRD7 knock out (KO) cells. Intriguingly, the transcriptomic profile of BRD7 KO cells was highly similar to that of SUM159R cells at both the gene and pathway levels and in both DMSO and JQ1-treated conditions (Figures 5F–G and S5G–H, and Table S5). Top enriched process networks included NOTCH, ESR1, and Hh signaling. We also calculated luminal-basal-mesenchymal signature scores of wild-type (WT) and BRD7 KO cells, since our prior data indicated that BBDI-resistance is associated with gain of luminal features (Shu et al., 2016). BRD7 KO cells had an increased luminal and decreased mesenchymal scores (Figure S7I), suggesting that deleting BRD7 may shift the cells to a more luminal phenotype associated with BBDI resistance.
ATAC-seq demonstrated both gain and loss in promoter regions in BRD7 KO cells compared to WT (Figure 5H), and these alterations in open chromatin were correlated with gene expression changes (Figures 5I and S5J). Comparison of ATAC-seq profiles of BRD7 KO and WT clones as well as parental SUM159 and BBDI-resistant SUM159R cells demonstrated higher relatedness of BRD7 KO clones to SUM159R cells, while parental SUM159 cells clustered with scrambled controls (Figure S5K). These results indicate that the deletion of BRD7 changed the chromatin landscape of SUM159 cells making them more similar to BBDI-resistant SUM159R cells. Analysis of ATAC-seq peaks gained in BRD7 KO cells and associated with increased gene expression revealed significant overlap with YAP1 peaks in the CISTROME database and enrichment in TEAD motif suggesting upregulation of YAP1/TEAD transcriptional activity (Figure S5L). Interestingly, our RNA-seq and RPPA analyses as well as our CRISPR screen also identified the YAP/TAZ pathway as upregulated and essential in SUM159R cells highlighting a role for this pathway in BBDI resistance.
The impact of BRD7 loss on BRD4 chromatin binding and histone modification patterns
To further investigate the role of BRD7 in BRD4 chromatin binding and histone modification patterns, we performed ChIP-seq for BRD4, H3K27ac, H3K4me3, and H3K27me3 in SUM159 BRD7 WT and KO cells. Combined analysis of the multi-omics data revealed a positive correlation between changes in BRD4 binding and changes in ATAC-seq, RNA-seq, H3K4me3, and H3K27ac following BRD7 deletion at SEs and less pronouncedly at promoters (Figure S6A). Assessing changes in BRD4 binding at genomic loci also bound by BRD7 in WT cells showed that changes in BRD4 binding were associated with changes in the same direction in H3K27ac, H3K4me3, ATAC-seq signal, and mRNA levels (Figure S6B).
To gauge changes in the SE landscape in BRD7 KO cells and the similarities of this to JQ1-resistant SUM159R cells, we analyzed differences in H3K27ac signal in SEs between BRD7 WT/KO and SUM159/SUM159R cells. Interestingly, many of the SEs gained in the SUM159R cells were also gained in BRD7 KO cells, including BCL2L1 (Figures 6A–B). We further analyzed the changes in H3K27ac signal in BRD7 KO cells in relation to BRD4 genomic binding at both SEs and promoters. In both regions the most significant H3K27ac increase in BRD7 KO cells occurred at loci bound by BRD4 in a JQ1-dependent manner in both SUM159 and SUM159R cells (Figure 6C and S6C). These changes in H3K27ac were also correlated with changes in ATAC-seq signal and the expression of the associated genes. Specifically, the top 1,000 JQ1-sensitive BRD4 binding sites and the top 1,000 downregulated genes by JQ1 showed the most pronounced changes in H3K27ac, H3K4me3 and ATAC-seq signal in BRD7 KO compared to BRD7 WT cells (Figure 6D–E); we found that the former demonstrated significantly larger changes in these markers compared to the latter, suggesting that changes in chromatin modifications are more pronounced than gene expression changes. These data indicate that the loss of BRD7 does not significantly impacts BRD4 chromatin binding but increases H3K27ac and H3K4me3 signal in BRD4 dissociated regions by JQ1 and increases the expression of genes downregulated by JQ1 in SUM159 cells. Thus, these alterations in H3K27ac and gene expression profiles make the BRD7 KO cells resistant to JQ1.
Figure 6. The effects of BRD7 deletion on BRD4 binding and histone modification patterns.
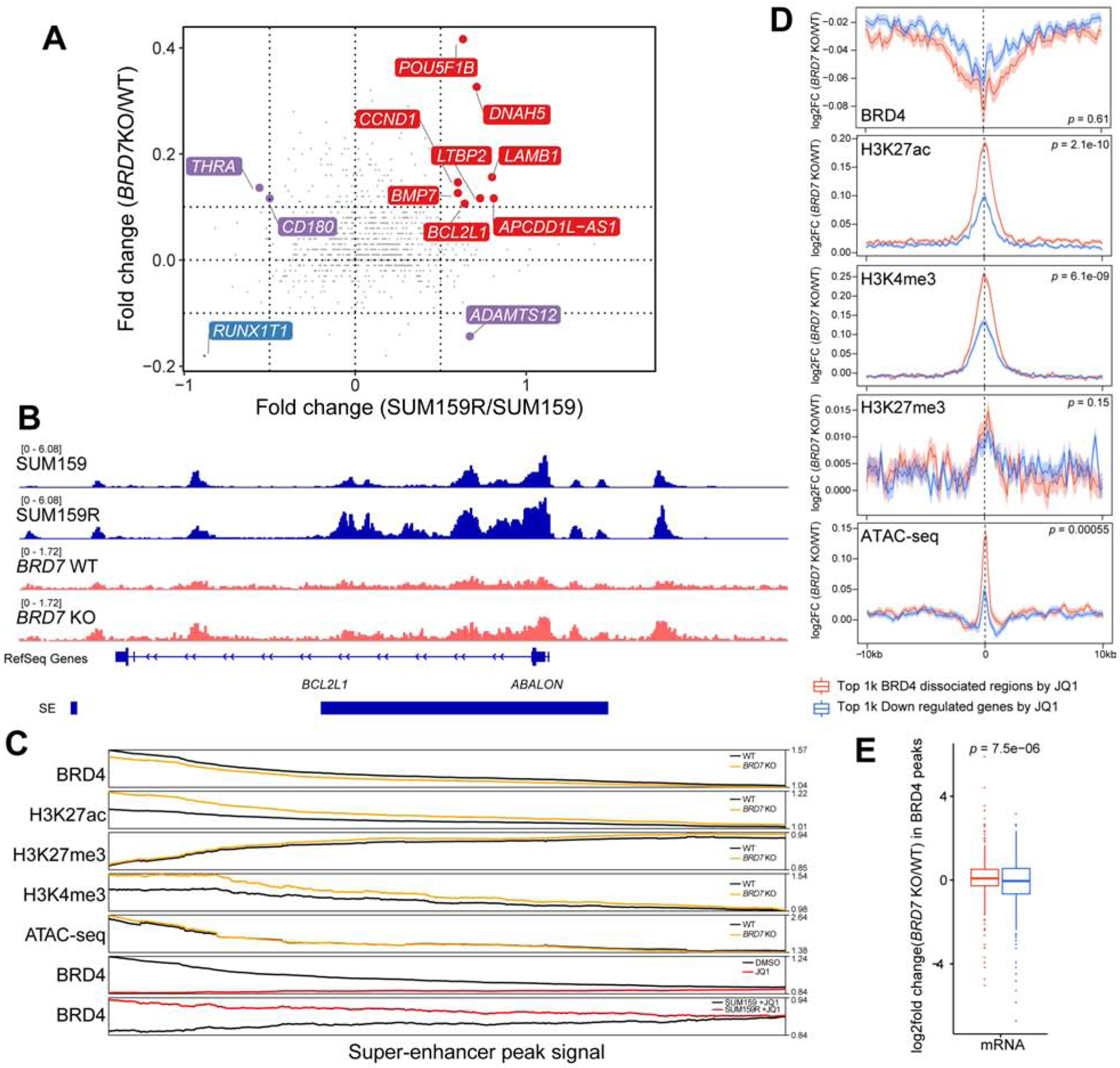
(A) Changes in SEs upon BRD7 knockout and between JQ1 sensitive and resistant SUM159 cells. Red is the co-activated super enhancers; blue is co-repressed super enhancers. Purple is the discrepant regulated super enhancers. (B) Example for a differential H3K27ac signal in a SE region. Normalized H3K27ac ChIP-seq signals (Reads Per Million) are shown as tracks using IGV. (C) Line plot shows smoothed signal of BRD4, H3K27ac, H3K27me3, and H3K4me3 ChIP-seq and ATAC-seq in BRD7 knockout and scramble SUM159 cells at SEs, and comparison to that of BRD4 ChIP-seq signal in DMSO, JQ1-treated and JQ1-resistant SUM159 cells (bottom two tracks). SEs are ranked by the fold change of BRD4 signal between +/− JQ1 from high (left) to low (right). (D) Changes in BRD4, H3K27ac, H3K27me3, H3K4me3, and ATAC-seq signal at top 1,000 BRD4 binding sites sensitive to JQ1 in SUM159 cells (red) and at top 1,000 genes downregulated by JQ1 in SUM159 cells (blue). ChIP-seq signals on peak summit +/− 10kb region. (E) Changes in mRNA levels at BRD4 peaks differential between BRD7 KO and WT cells at top 1,000 BRD4 binding sites sensitive to JQ1 in SUM159 cells (red) and at top 1,000 genes downregulated by JQ1 in SUM159 cells (blue). See also Figures S6.
Clinical significance of BRD4 and BRD7 expression in TNBC
To investigate the clinical relevance of BRD4 and BRD7 interaction in breast cancer, we analyzed their expression patterns in the TCGA cohort (TCGA, 2012). Both BRD4 and BRD7 have significantly higher expression in basal compared to luminal breast tumors, which is in contrast to ARID1A, a subunit of the BAF complex implicated in luminal differentiation (Nagarajan et al., 2020; Xu et al., 2020) (Figure S6D). Even within TNBC, the highest BRD4 and BRD7 expression was observed within basal subtype tumors (Figure S6E) implying that BRD4 and BRD7 function may particularly be relevant in non-luminal breast tumors. Further analysis demonstrated that TNBC patients with lower expression of BRD4 or BRD7 had shorter disease-free survival and this was especially the case when both genes had low expression levels (Figure S6F). BRD7 expression levels were also associated with the expression of JQ1 resistance gene signature derived from our cell line data, low BRD7 expressing tumors have high JQ1 resistance score (Figure S6G), validating the relevance of our data in human breast tumor samples.
Single cell profiling of BBDI-resistant cells
We recently described that higher cellular transcriptomic heterogeneity is associated with therapeutic resistance and that epigenetic regulators modulate this heterogeneity (Hinohara et al., 2018). To assess whether cellular transcriptomic heterogeneity also plays a role in BBDI resistance and if it is modulated by JQ1, we performed single cell RNA-seq (scRNA-seq) profiling of parental and BBDI-resistant cells before and after JQ1 treatment. Untreated and JQ1-treated cells formed distinct clusters in all four lines tested indicating that JQ1 significantly alters cellular transcriptomes (Figures 7A and S7A). Similarly, BBDI-resistant and parental cells separated very clearly and BBDI-resistant cell cluster-specific genes included several CRISPR screen hits such as CDK4, CCND1, and BCL2L1 (Figures 7A and S7A). While both parental cell lines and SUM149R cells formed a single cell cluster, SUM159R cells split up into two groups in both untreated and JQ1-treated conditions (Figure 7B). These two subgroups did not appear to be proliferation-related based on the expression of cell cycle-related genes (Figure S7B), but cluster-specific genes showed enrichment in translation-related functional categories (Figure 7C). Since our prior data indicated that luminal breast cancer cells in general are less sensitive to BBDIs than basal/mesenchymal ones (Shu et al., 2016), we also quantified basal, luminal, and mesenchymal signature scores for each single cell. We observed that SUM159R cells had higher luminal scores compared to SUM159 and that this pattern was further increased by JQ1 (Figure S7C) confirming that gain of luminal features is associated with BBDI resistance. The SUM159R subclusters did not show significant differences in luminal-basal signature scores besides the lower mesenchymal and higher luminal scores observed after JQ1 treatment (Figure S7C). However, analysis of the chromosomal location of cluster-defining genes demonstrated that ~30% of genes specific for clusters 8 and 9 are localized to chromosome 11 (many on 11q) in contrast to 10–17% in clusters 2 and 5 (Table S6) suggesting that cells in clusters 8 and 9 may have a gain of this chromosomal region. CCND1 and YAP1 are located on chromosome 11q and they are cluster 8 and 9 marker genes highlighting their role in BBDI resistance.
Figure 7. Single cell profiling of drug-resistant cells.
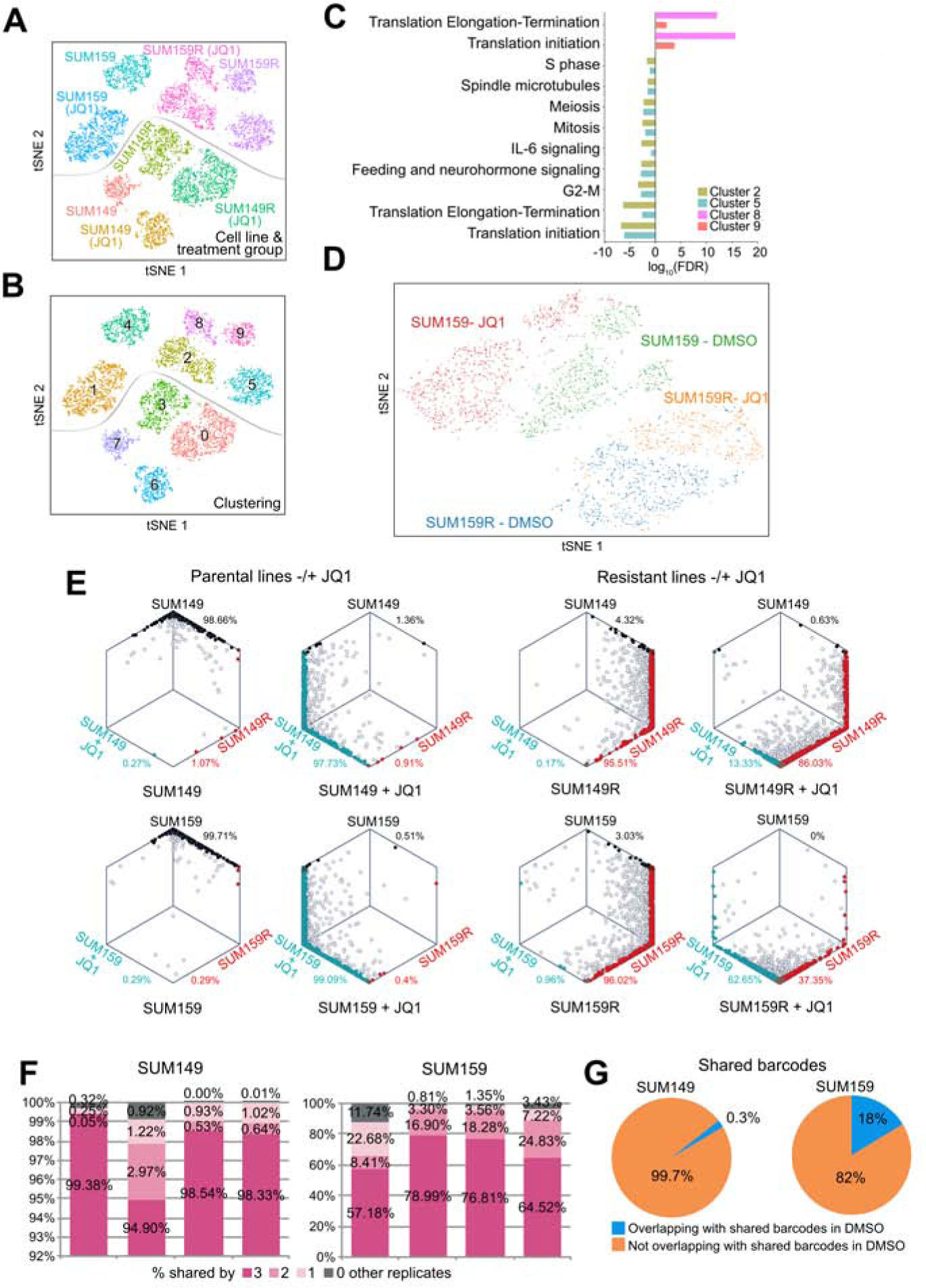
A-B, t-SNE plots depicting single cells by single cell RNA-seq in populations of JQ1-treated and untreated SUM149 and SUM159 parental and BBDI-resistant SUM149R and SUM159R cells, colored by cell line and treatment group (A), and by gene expression cluster (B). (C) Top process networks enriched in differentially expressed genes between clusters 6 and 9 (DMSO) and between clusters 2 and 8 (JQ1) within SUM159R cells. (D) t-SNE plots depicting single cells by single cell ATAC-seq in populations of JQ1-treated and untreated SUM159 parental and BBDI-resistant SUM159R cells. (E) Hexagonal plots depicting the bootstrap classification of single cells in populations of parental, JQ1-treated, BBDI-resistant, and JQ1-treated BBDI-resistant cells. Each point is a single cell and is positioned along axes according to its bootstrapping classification score for the indicated cell identity. Black, blue, and red cells are classified as parental, JQ1-treated, and BBDI-resistant cells, respectively, and gray cells are unclassified. (F) Bar graphs show percentages of total barcodes shared among all replicates in JQ1-treated SUM149 and SUM159 cells. (G) Pie charts show percentages of shared barcodes overlapping between untreated and JQ1-treated SUM149 and SUM159 cells. See also Figure S7 and Table S6.
We also performed single cell ATAC-seq (scATAC-seq) to assess potential cellular epigenetic heterogeneity that could underlie the clustering of the cells based on scRNA-seq. Pooled scATAC-seq correlated very well with ATAC-seq on bulk cell populations confirming the quality of the data (Figure S7D). Interestingly, the cell lines clustered differently based on scATAC-seq compared to scRNA-seq. Control and JQ1-treated SUM159R cells formed one large cluster without clear separation of the two treatment conditions (Figure 7D). In contrast, JQ1-treated SUM159 cells were clearly distinct from DMSO-treated controls. These data demonstrate that SUM159R cells are epigenetically distinct from SUM159 cells and that their epigenetic state is less responsive to JQ1 treatment than that of SUM159 cells.
To further investigate differences in BBDI resistance mechanisms between the two cell lines and the heterogeneity of SUM159R cells, we explored our scRNA-seq data to determine whether we could detect rare cells with gene expression signatures of BBDI-resistant cells prior to treatment and whether the gene expression profiles of BBDI-resistant and BBDI-treated cells are related. Thus, we selected genes differentially expressed between parental and BBDI-resistant or JQ1-treated parental cells based on bulk RNA-seq data (Figure S1I, Table S2) and investigated if single cells could be classified into one of these three transcriptionally distinct groups (i.e., parental, resistant, and JQ1-treated). While most single cells in SUM149R and SUM159R populations were classified as resistant, very few such cells were present in parental and in JQ1-treated populations (Figure 7E), suggesting that JQ1-resistant clones were selected from a mixed population during treatment. Almost all JQ1-treated parental cells had the JQ1-treatment signature in both cell lines. In contrast, JQ1-treated BBDI-resistant cells showed striking differences in the distribution of cellular identities between SUM149R and SUM159R cells. While only 13% of the cells changed their identity to the JQ1-treated type in the SUM149R cell line, nearly 50% of SUM159R cells were classified as the JQ1-treated type after JQ1 treatment (Figure 7E). Although this shift was not directly associated with the two distinct clusters identified in SUM159R cells, these observations suggest that SUM159R cells are more heterogeneous than the SUM149R line and almost 50% of the cells still demonstrate a transcriptional response to JQ1 even though the cells continue proliferating in the presence of the drug.
To further interrogate mechanisms of BBDI resistance in the two cell lines, we carried out a cellular barcoding experiment using the ClonTracer barcode library (Bhang et al., 2015). We cultured four replicates of barcoded SUM149 and SUM159 cells with comparable starting barcode representations in the presence of JQ1 until resistance was achieved to distinguish pre-existing clones selected during treatment from acquired alterations. We observed very different patterns of barcode selection between SUM149 and SUM159 cells. JQ1 treatment significantly reduced barcode complexity in SUM149 cells and more than 94% of barcodes were shared by all four replicates (Figures 7F–G and S7E). These findings strongly suggest that the vast majority of JQ1-resistant clones were pre-existing in the parental SUM149 cell population and were highly selected for during treatment. In contrast, in SUM159 cells there was minimal selection during JQ1 treatment since the barcode pool of the JQ1-resistant population was not significantly different from DMSO-treated parental cells at the same passage (Figures 7F–G and S7E), suggesting that resistance to JQ1 in SUM159 cells is not likely due to selection for pre-existing resistant cells but acquired during treatment. These results suggest distinct patterns of JQ1 response and resistance mechanisms in TNBC.
DISCUSSION
We previously described BBDIs as candidate therapeutic agents in TNBC and also showed that acquired resistance develops quickly necessitating combination therapies (Shu et al., 2016). Here we describe a multi-omic profiling and functional screening approach in TNBC for the identification of synthetic lethal and resistance interactions with JQ1. The advantage of this combined approach is the high confidence in the data when the same genes and pathways are identified using multiple different unbiased approaches. We identified several genes and pathways that make TNBCs more responsive or resistant to JQ1, which can be grouped into five general functional categories.
The first category encompasses chromatin modifiers and transcriptional regulators including BBD proteins that are the direct targets of JQ1 itself. The deletion of BRD2 sensitized both parental and BBDI-resistant cell lines to JQ1. Based on our data, BRD2 and BRD4 play largely overlapping roles in TNBC, but they differ in the JQ1 sensitivity of their chromatin binding. Because BRD2 is less efficiently removed from the chromatin by JQ1 than BRD4, and JQ1 induces BRD2 expression, deletion of BRD2 significantly synergized with JQ1 treatment. Deletion of genes encoding multiple different Mediator proteins (MED1, MED19, MED24) and EP300 also increased sensitivity to JQ1 likely due to their direct effects on the assembly and recruitment of transcriptional complexes to active promoters. In contrast, loss of components of the BAF chromatin remodeling complex (BRD7, ARID1A and PBRM1) significantly increased resistance to JQ1 of parental and BBDI-resistant cells, while deletion of BRD9 increased JQ1 sensitivity of all four cell lines. Somatic mutations of ARID1A, CTCF, SMARCA4, and PBRM1 are detected at low (1–5%) frequencies in treatment naïve breast cancers, while ARID1A mutations are more common in treatment-resistant distant metastases (Zehir et al., 2017) and in metaplastic breast cancer (Ng et al., 2017). These results suggest that alterations of the BAF complex may in general be associated with therapeutic resistance and perturbed differentiation.
The second category comprises regulators of protein stability and degradation, some of which may regulate the stability of BRD4 and other BBD proteins. Deletion of ubiquitination-related genes (e.g., SPOP, UBE2M, CUL3, and USP14) resulted in resistance to JQ1. SPOP is a regulator of ubiquitination-mediated degradation of BRD4 and other BBD proteins and SPOP mutations in prostate cancer have been associated with BBDI resistance (Dai et al., 2017; Janouskova et al., 2017; Zhang et al., 2017). Our data are consistent with these observations and further expand the role of these proteins as regulators of BBDI sensitivity in TNBC.
The third category involves kinase signaling pathways. Deletion of TGFBR1, TGFBR2, PIK3CA, EGFR, and TEAD1 increases JQ1 responsiveness. Small molecule inhibitors blocking AXL, BCRABL, EGFR, JAK/STAT, IGFR-1, SRC, VEGFR, MEK, PIK3/AKT/mTOR, and YAP/TAZ also sensitized cells to JQ1. The effect of these signaling pathways was not always observed in all cell lines. However, AXL and TEAD1 were CRISPR synergy hits in all four cell lines and they were also validated by SMIs. Consistent with our findings, a recent study reported that BRD4 is a mediator of transcriptional addiction to YAP/TAZ in colorectal cancer and described synthetic lethal interactions between BRD4 and YAP (Zanconato et al., 2018). Similarly, BBDIs were shown to be able to overcome cetuximab resistance in head and neck squamous cell carcinoma with activated AXL (Leonard et al., 2018).
The fourth group of genes and compounds targeting them comprises regulators of apoptosis including BCL2 and BCL2L1 (BCLxl), which is consistent with our prior data (Shu et al., 2016). While we validated the synergy between JQ1 and BCL2 and BCLxl inhibitors both in vitro and in vivo, the toxicity of some of these compounds (especially dual BCL2/BCLxl inhibitors) limits their potential for clinical development.
Lastly, the fifth and the largest category includes cell cycle regulators and genes involved in DNA repair. Genetic deletion or pharmacologic inhibition of promoters of G1/S transition including CDK4, CDK6, CCND1, SKP2, and CDK2 synergized with JQ1, while inactivation of genes encoding negative regulators CDKN1A, CDKN1B, and RB1 had the opposite effect leading to JQ1 resistance. Multiple mechanisms could explain the synergy between JQ1 and CDK4/6 inhibitors. The mRNA and protein levels of CDK4, CCND1, and SKP2 are increased after JQ1 treatment in BBDI-resistant cells, leading to higher CDK4 kinase activity. CDK4 also regulates BRD4 protein levels via phosphorylating SPOP and DUB3 that play a role in BRD4 ubiquitination and degradation (Dai et al., 2017; Janouskova et al., 2017; Jin et al., 2018; Zhang et al., 2017). Our data showing decreased BRD4 after palbociclib treatment implies this regulatory loop in TNBC and could explain the JQ1 CDK4 inhibition synergy in RB1null cells.
Whereas the interaction of regulators of G1 phase progression with JQ1 was largely expected, since JQ1 causes G1 arrest and most transcription occurs in G1, we also identified synthetic lethal interactions between JQ1 and proteins involved in G2/M phase. Deletion of BUB3, inhibitors of PLK1 and AURKA kinases, and chemotherapeutic agents targeting microtubules such as paclitaxel, and vincristine all demonstrated potent synergy with JQ1 in both parental and BBDI-resistant cell lines. Lastly, the deletion of DNA repair genes (e.g., BRCA1 and BRCA2) and topoisomerase inhibitors sensitize TNBC cells to JQ1. BBDIs suppress DNA repair and BET inhibition sensitizes homologous recombination-proficient tumors to PARP inhibitors (Karakashev et al., 2017; Yang et al., 2017), suggesting that breast tumors with BRCA1 or BRCA2 mutations may particularly be sensitive to BBDIs.
In summary, our data highlights the heterogeneity of cellular responses and resistance to JQ1 in TNBC and identifies several promising combination therapies with BBDIs that could be used for the more effective treatment of chemotherapy-resistant TNBC.
STAR*METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Kornelia Polyak, Dana-Farber Cancer Institute, 450 Brookline Ave., SM1070B, Boston, MA 02215, USA. kornelia_polyak@dfci.harvard.edu; tel: 617-632-2106; fax: 617-582-8490.
Materials availability
SUM159 cell line derivatives generated by CRISPR-Cas9 (BRD7 KO, CDK4 KO, and BRD2 KO) will be made available upon request and following the execution of an MTA.
DATA AND CODE AVAILABILITY
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplemental Information. All raw genomic data was deposited to GEO under accession number: GSE131102. This study did not generate custom code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Breast cancer cell lines
Breast cancer cell lines were obtained from ATCC, DSMZ, or generously provided by Steve Ethier (SUM cell lines, University of Michigan) and cultured following the provider’s recommendations except for SUM cell lines we used SUM medium (1:1 mix of DMEME/F12 10% FBS with complete HMEC medium). The identity of the cell lines was confirmed based on STR and exome-seq analyses. Cells were regularly tested for mycoplasma.
Animal model
For xenograft assays female NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) and NOG (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) mice at 5–6 weeks of age were purchased from the Jackson Laboratory and Taconic, respectively. Animal experiments were performed by S.Sh. and by research technicians in the Lurie Family Imaging Center. Animal studies were performed according to protocol 11–023 or by the Lurie Family Imaging Center according to protocol 04–111, approved by the Dana-Farber Cancer Institute Animal Care and Use Committee. Mice were housed 5 to a cage with ad libitum access to food and water in 20°C ambient temperature, 40–50% humidity, and 12-hour light/12-hour dark cycle.
CRISPR libraries
We used CRISPR Knock-out H1 and H2 libraries essentially as described (Jeselsohn et al., 2018).
Small molecule inhibitor libraries
The two drug libraries used for the SMI screen were the Target Selective Inhibitor Library (Catalog No.L3500) and the Anti-cancer Compound Library (Catalog No.L3000). These drug libraries were purchased from Selleck Chemicals and were compiled as previously described (Harris et al., 2019).
Barcoding and selection for resistant cells
The high-complexity barcode library, ClonTracer, was a kind gift from Frank Stegmeier (Novartis). Barcoding experiments were performed as previously described (Bhang et al., 2015). Briefly, cells were barcoded by lentiviral infection using 8 μg/ml polybrene. After a 24 h incubation with virus, infected cells were selected with 2 μg/ml puromycin. To ensure that the majority of cells were labeled with a single bar- code per cell, for lentiviral infection we used a target m.o.i. of approximately 0.1, corresponding to 10% infectivity after puromycin selection. Infected cell populations were expanded in culture for the minimal time period to obtain a sufficient number of cells to set up replicate experiments. Barcoded SUM149 and SUM159 cells were treated with increasing dose of JQ1(200nm, 500nm, 1um, 2um, 5um, and 10uM). The control groups were treated with 0.1% DMSO. Each group was cultured in quadruplicate. Cells were cultured in SUM medium. To keep the baseline control population as close as possible to that of the treatment group, each treatment group was cultured at the same passage as their corresponding control group, because random barcode loss during passaging has been reported previously. Genomic DNA was extracted from the frozen cell populations with a QIAamp DNA Mini Kit (Qiagen). We used PCR to amplify the barcode sequence for NGS by introducing Illumina adaptors and 5-bp-long index sequences. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on an Illumina NextSeq500 with single-end 75 bp reads by the Dana-Farber Cancer Institute Molecular Biology Core Facilities.
METHOD DETAILS
Xenograft assays
Animal experiments were performed by the Lurie Family Imaging Center or by our lab following protocols approved by the Dana-Farber Cancer Institute Animal Care and Use Committee. For xenograft assays 5–6-weeks old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ and NOG (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) mice were purchased from Jackson Laboratory and Taconic, respectively. Tumors were induced by bilateral orthotopic mammary fat pad injection of 2 × 106 cells suspended in 50 μl of culture medium/Matrigel Growth Factor Reduced Basement Membrane Matrix, Phenol Red-Free (Corning) in a 1:1 ratio. After 14 days, mice were randomized to treatment groups based on tumor size. Mice were administered JQ1 (30 mg/kg, weekly), INC424 (75 mg/kg, daily), ABT-263 (75mg/kg, every other day per dose), ABT-199 (75mg/kg, every other day per dose), dBET6 (7.5mg/kg, daily or twice per day), palbociclib (75 mg/kg, daily), combination of JQ1 with IN424, ABT-263, ABT-199, palbociclib, or vehicle only (control) for 14 days. Tumors implanted in mice were imaged using magnetic resonance imaging (MRI) and by caliper measurement. Mice were euthanized and tumors collected, fixed overnight in 4% formalin, stored in 70% ethanol, followed by paraffin embedding, sectioning, and hematoxylin and eosin staining by the Pathology Core of the Brigham and Women’s Hospital.
Immunofluorescence staining
After deparaffinization and rehydration, slides were subjected to antigen retrieval in citrate buffer pH 6 (Dako, cleaved caspase 3, BRD4, and phospho-RB) and in EDTA pH 9 buffer (Dako, pSTAT3) for 20 min in a steamer. Blocking solution (100% goat serum) was applied for 10–20 min. Incubation with primary antibody in PBS with 5% goat serum was held overnight at 4 °C in a moist chamber. Secondary antibody was applied for 1 h at room temperature. Samples were mounted with VectaShield HardSet Antifade Mounting Medium with DAPI (Vector Laboratories).
Antibodies and Inhibitors
Antibodies used for ChIP-seq were BRD4 (Bethyl, A301–985A), BRD2 (Cell signaling, 5848), BRD7 (Cell Signaling, 14910), histone H3K27ac (Abcam, ab4729), histone H3K4me3 (Abcam, ab1012), and histone H3K27me3 (Cell Signaling, 9733). Antibodies used for immunoblotting were RB (Cell Signaling, 9313), CCND1 (Cell Signaling, 2978), CDK4 (Cell Signaling, 12790), E2F1 (Cell Signaling, 3742), phospho-Rb-Ser780 (Cell Signaling, 3590), phospho-Rb-Ser807/811 (Cell Signaling, 9308). Antibodies used for Immunofluorescence were BRD4 (Bethyl, A301–985A), cleaved Caspas-3 (9664), phospho-Rb-Ser807/811 (Cell Signaling, 8516), and pSTAT3 (Cell Signaling, 9131).
The JAK2 (INC424), Bcl-2 (ABT-199, S8048), and Bcl-xl/Bcl-2 (ABT263, S1001) inhibitors were from Selleckchem. The CDK4/6 inhibitor (palbociclib, HY-50767A) was from MedChemExpress. The dBET series (dBET1 - dBET10) was synthesized by Dr. Dennis Buckley (Dana Farber Cancer Institute). JQ1 was synthesized by Dr. Jun Qi (Dana Farber Cancer Institute).
Small molecule inhibitor (SMI) screens
The two drug libraries used for the SMI screen were the Target Selective Inhibitor Library (Catalog No.L3500) and the Anti-cancer Compound Library (Catalog No.L3000). These drug libraries were purchased from Selleck Chemicals and were compiled as previously described (Harris et al., 2019). Parental and BBDI-resistant (R) SUM149 and SUM159 cells were cultured DMEM/F-12 media (Life Technologies #11330057) with mammary epithelial cell growth medium supplement, 5% FBS and 1% penicillin/streptomycin until cells were seeded for screening, where RPMI 1640 media (Life Technologies #11875119) with 5% FBS and 1% penicillin/streptomycin was used. Additionally, SUM149R and SUM159R cells were cultured in the presence of 10 μM and 20 μM JQ-1, respectively, until the cells were seeded for screening, after which JQ-1 was not included in media. For the SMI screen, 500 cells per well for SUM149/SUM149R cells and 250 cells per well for SUM159/SUM159R were seeded. A Z’ factor for each cell line was determined using vincristine (1 μM) as a cytotoxic agent (Zhang et al., 1999). The Z’ factor for cell lines were as follows: SUM149 = 0.35; SUM149R = 0.56; SUM159 = 0.60; SUM159R = 0.38. Cells were seeded in 30 μl and after one day, 100 nl of molecules from the drug libraries were delivered by pin-transfer and 20 μl of fresh media was added. After 72 hours, plates were harvested and data was collected as previously described (Harris et al., 2019). All dose-response curves were completed in duplicate. Statistical analysis of dose-curves was completed in GraphPad Prism 8 (nonlinear regression, variable slope, four parameters, bottom constant equal to 0, top constant equal to 1). LogIC50 values were compared with an extra sum-of-squares F Test.
CRISPR Screen and data analyses
We used CRISPR Knock-out H1 and H2 libraries essentially as described (Jeselsohn et al., 2018). For the pooled genome-wide CRISPR screen, 200 million SUM149 and SUM159 parental and BBDI-resistant cells were infected with the pooled lentiviral CRISPR knock-out H1 and H2 library at a multiplicity of infection of 0.3 to ensure that most cells receive only one viral construct with high probability. After 5 days of puromycin selection, the surviving cells were divided into day 0 control cells and cells cultured for five passages treated with DMSO or JQ1 (500nM and 1uM for SUM159; 100nM, 200nM, 400nM, 800nM for SUM149; 20uM for SUM159R; 10uM for SUM149R cells) before genomic DNA extraction and library preparation. The CRISPR screens were also repeated in SUM159 cells with DMSO or JQ1 treatment (500nM, 1000nM). PCR was performed on genomic DNA to construct the sequencing library. Each library was sequenced at 30~40 million reads to achieve ~300× average coverage over the CRISPR library. The day 0 sample library of each screen served as controls to identify positively or negatively selected genes or pathways.
Generation of Single CRISPR/Cas9 Knock-Out cells
Construction of lenti-CRISPR/Cas9 vectors targeting BRD2, BRD7, and CDK4 was performed following the protocol associated with the backbone vector lentiCRISPR V2 (Sanjana et al., 2014) (49535, Addgene). The sgRNA sequences used are listed in the Key resources table. SUM159 and SUM159R cells were infected with lentivirus expressing sgRNAs targeting CDK4, BRD2, or BRD7. After puromycin selection, single cell clones were picked and expanded, and knockout was verified by western blot analyses.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-BRD4 | Bethyl laboratories | Cat# A301–985A; RRID:AB_1576498 |
| Rabbit monoclonal anti- BRD2 | Cell Signaling Technology | Cat# 5848; RRID:AB_10835146 |
| Rabbit monoclonal anti- BRD7 | Cell Signaling Technology | Cat# 14910; |
| Mouse monoclonal anti-H3K4me3 | Abcam | Cat# ab1012; RRID: AB_442796 |
| Rabbit Anti-Histone H3, Trimethyl (Lys27) Monoclonal Antibody, | Cell Signaling Technology | Cat# 9733, RRID:AB_2616029 |
| Rabbit monoclonal anti- RB | Cell Signaling Technology | Cat# 9313; RRID:AB_1904119 |
| Rabbit monoclonal anti- CCND1 | Cell Signaling Technology | Cat# 2978; RRID:AB_2259616 |
| Rabbit monoclonal anti- CDK4 | Cell Signaling Technology | Cat# 12790; RRID:AB_2631166 |
| Mouse monoclonal anti-beta-Actin | Sigma-Aldrich | Cat# A2228; RRID: AB_476697 |
| Rabbit polyclonal anti-H3K27Ac | Abcam | Cat# ab4729; RRID: AB_2118291 |
| Mouse monoclonal anti- E2F1 | Cell Signaling Technology | Cat# 3742; RRID:AB_2096936 |
| Rabbit monoclonal anti- Phospho-Rb-Ser780 | Cell Signaling Technology | Cat# 3590; AB_2177182 |
| Rabbit monoclonal anti- Phospho-Rb-Ser807/811 | Cell Signaling Technology | Cat# 9308; RRID:AB_331472 |
| Rabbit monoclonal anti- Phospho-Rb-Ser807/811 | Cell Signaling Technology | Cat# 8516; RRID:AB_11178658 |
| Rabbit monoclonal anti- cleaved Caspase-3 | Cell Signaling Technology | Cat# 9664; RRID:AB_2070042 |
| Rabbit polyclonal Phospho-Stat-3 (tyr705) antibody | Cell Signaling Technology | Cat# 9131, RRID:AB_331586 |
| Mouse monoclonal anti- BrdU | Roche | Cat# 11170376001; RRID:AB_514483 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| JQ1 | Jun Qi (Dana-Farber Cancer Institute) | N/A |
| Palbociclib | MedChem Express | HY-50767A |
| ABT-199 | Selleckchem | S8048 |
| ABT263 | Selleckchem | S1001 |
| INC424 | InCyte | N/A |
| Dasatinib | Selleckchem | S1021 |
| TP-0903 | Selleckchem | S7846 |
| Paclitaxel | Dana-Farber Cancer Institute Research Pharmacy | N/A |
| Doxorubicin | Dana-Farber Cancer Institute Research Pharmacy | N/A |
| Vincristine | Sigma-Aldrich | V8879 |
| Gemcitabine | Sigma-Aldrich | G6423 |
| dBET series (dBET1 - dBET6) | Dennis Buckley (Dana-Farber Cancer Institute) | N/A |
| Flavopiridol | Selleckchem | S1230 |
| Barasertib | Selleckchem | S1147 |
| Target Selective Inhibitor Library | Selleckchem | L3500 |
| Anti-cancer Compound Library | Selleckchem | L3000 |
| Critical Commercial Assays | ||
| ThruPLEX DNA-seq 48S Kit | RUBICON | R400427 |
| Experimental Models: Cell Lines | ||
| SUM159 cell line | Steve Ethier (University of Michigan) | N/A |
| SUM149 cell line | Steve Ethier (University of Michigan) | N/A |
| SUM159R cell line | Shu et al., 2016 | N/A |
| SUM149R cell line | Shu et al., 2016 | N/A |
| SUM159 cell line, BRD2 KO | This paper | N/A |
| SUM159 cell line, BRD7 KO | This paper | N/A |
| SUM159 cell line, CDK4 KO | This paper | N/A |
| CAL-51 cell line | DSMZ | ACC 302 |
| CAL-120 cell line | DSMZ | ACC 459 |
| HCC38 cell line | ATCC | CRL-2314 |
| HCC3153 cell line | Adi Gazdar/LBL Joe Gray | N/A |
| PMC42 cell line | R.Whitehead | N/A |
| MDA-MB-436 cell line | ATCC | HTB-130 |
| MDA-MB-231 cell line | ATCC | HTB-26 |
| Hs578T cell line | ATCC | HTB-126 |
| Oligonucleotides - sgRNA targeting sequences | ||
| BRD2–1: CTTCTCATCGTAACTCATG | This paper | N/A |
| BRD2–2: CCACGCAAAGACTTGCCTG | This paper | N/A |
| BRD7–1: CGGGCAGCTCGGGGCACGA | This paper | N/A |
| BRD7–2: CCTACAATGGGATCCACAG | This paper | N/A |
| CDK4–1: CCAGAGGATGACTGGCCTC | This paper | N/A |
| CDK4–2: CCTCACGAACTGTGCTGAT | This paper | N/A |
| Recombinant DNA | ||
| CRISPR-V2 | Sanjana et al, 2014 | Addgene Cat# 52961 |
| Plv(Exp)-Bsd-TRE-CDK4 | This paper | Vector builder |
| Deposited Data | ||
| All raw genomic data | GEO | GSE131102 |
Generation of TET-inducible exogenous CDK4-expressing CDK4 knockout cells
SUM159 cells were infected with TET-inducible GFP-CDK4 resistant to CDK4 sgRNA-1 virus. After blasticidin selection, three days of doxycycline treatment induces the GFP-CDK4 expression. GFP positive cells were sorted by FACS. The SUM159 inducible CDK4 positive cells were infected with CDK4 knockout sgRNA virus for endogenous CDK4 knockout. After puromycin selection, the cells from single cell cloning were grown and treated with or without doxycycline. CDK4 expression was confirmed by western blot.
Immunoblotting and immunoprecipitation experiments
Cells were lysed in RIPA buffer. Proteins were resolved in SDS-polyacrylamide gels (4%–12%) and transferred to PVDF membranes by using a Tris-glycine buffer system. Membranes were blocked with 5%milk powder in 0.1% Tween20 in PBS (PBS-T) for 1 hr at room temperature followed by incubation with primary antibodies at 1:1000 dilution in 2.5% milk PBS-T. For immunoprecipitation, nuclear extracts were prepared as follow: 10× 106cells were resuspended in 5ml Buffer A: 10mM Tris pH 7.9, 1.5mM MgCl2, 10mM KCl, 0.05% NP-40, 1mM DTT, and protease and phosphatase inhibitors. Cells were incubated on ice for 15 min and gently vortexed every 5 min. After centrifugation at 2,000g for 5 min, pellets were suspended in 0.3 ml buffer B (20mM Tris pH 7.9, 25% glycerol, 0.42 M NaCl, 1.5mM MgCl2, 1mM KCl, 0.5% NP40, 0.2mM EDTA, 1mM DTT, and protease and phosphatase inhibitors) and incubated for 5 min on ice. After centrifugation of the lysates at 14g for 10 min at 4°C, supernatant was diluted with 0.6 ml buffer A, and added NP-40 to final 0.5% and treated with DNase I. The samples were then incubated at 4°C overnight with BRD4 antibody at 1:100 dilution and immunoprecipitates were collected on Dynabeads Protein G for 2 hr. Beads were washed with buffer B containing 150mM NaCl and 0.5% NP-40 three times and then resuspended in gel loading buffer.
ChIP-seq
For BRD2 and BRD4 ChIP-Seq, SUM149, SUM159, SUM149R, and SUM159R cells (1×107) were grown in SUM Medium. The media were then removed and replaced with PBS containing 1% formaldehyde (EM grade; Tebu-bio) and crosslinked for 8 min at 37°C. Crosslinking was quenched by adding glycine to a final concentration of 0.2 M. For BRD7 ChIP-Seq, SUM159 and SUM159R cells (8×107) were grown in SUM medium. For BRD4, H3K27ac, H3K4me3, and H3K27me3 ChIP-Seq SUM159 WT and BRD7 KO cells (1×107) were grown in SUM Medium. The medium was removed and replaced with PBS containing 2mM DSG (disuccinimidyl glutarate, ThermoFisher) and incubated for 30 min at room temperature, then removed and replaced with PBS containing 1% formaldehyde (EM grade; Tebu-bio) and crosslinked for 8 minutes at 37°C. Crosslinking was quenched by adding glycine to a final concentration of 0.2 M. The cells were washed with ice-cold PBS, harvested in PBS, and the cell pellet was washed with PBS. The nuclear fraction was extracted by first resuspending the pellet in 10 ml of LB1 buffer (50mM HEPES-KOH [pH 7.5], 140mMNaCl, 1mMEDTA, 10% glycerol, 0.5% NP-40 or Igepal CA-630, and 0.25% Triton X-100) for 10 min at 4°C. Cells were pelleted, resuspended in 10 ml of LB2 buffer (10 mM Tris-HCL [pH 8.0], 200 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA), and mixed for 5 min. Cells were pelleted and resuspended in 1ml LB3 buffer (10 mM Tris-HCl [pH 7.4], 1 mM EDTA, 0.1%SDS, 1%Triton X-100, 0.1% Na-deoxycholate, 1Mm DTT, 0.25% N-lauroylsarcosine, protease inhibitors and phosphatase inhibitors) and sonicated in a Covaris sonicator for 10 min. A total of 30 μl of 5M NaCl was added, and lysate was centrifuged for 10 min at 20,000 rcf to purify the debris. The supernatant was then incubated with 50 μl of magnetic beads (Life Technologies) prebound with 20 μg BRD4 antibody (Bethyl, A301–985A), 20 μl BRD2 antibody (Cell signaling, 5848), 40 μl BRD7 antibody (Cell Signaling, 14910) and immunoprecipitation (IP) was conducted overnight in the cold room. The beads were washed ten times in 1 ml of RIPA buffer and twice in 100mM ammonium hydrogen carbonate (AMBIC) solution. DNA was eluted in elution buffer (50 mM Tris-HCl pH 8, 10 mM EDTA, and 1% SDS). Cross-links were reversed overnight at 65°C. RNA and protein were digested with 0.2 mg/mL RNase A for 2 hr followed by 0.2 mg/mL Proteinase K for 1 hr. DNA was purified with phenol chloroform extraction and ethanol precipitation. Libraries for Illumina sequencing were prepared following the Rubicon ThruPLEX-FD kit for 10–12 cycles.
RNA-seq
SUM149 and SUM149R were incubated in biological duplicates for 12 hr with 500 nM of JQ1 or DMSO treatment. Total RNA was extracted using the standard QIAGEN RNeasy kit (74106). RNA concentrations were measured and quality controlled on a Bioanalyzer, RNA-Seq libraries were made using Illumina True-Seq RNA kits using the Sciclone NGSx workstation.
qPLEX-RIME
Samples were digested and purified with the Ultra-Micro C18 Spin Columns (Harvard Apparatus) as previously described (Papachristou et al., 2018). After purification, each sample was dried and reconstituted in 100ul 0.1M TEAB (triethylammonium bicarbonate) and labelled with the TMT-10plex reagents (Thermo Fisher). The peptide mixture was fractionated with Reversed-Phase spin columns at high pH (Pierce, #84868) and each fraction was analyzed on a Dionex Ultimate 3000 UHPLC system coupled with the LTQ Orbitrap Velos mass spectrometer (Thermo Scientific). Mobile phase A was composed of 2% acetonitrile, 0.1% formic acid, 5% dimethyl sulfoxide (DMSO) and mobile phase B was composed of 80% acetonitrile, 0.1% formic acid, 5% DMSO. The precursor scans were performed in the Orbitrap in the range of 380–1500 m/z at 60K resolution. The MS2 scans were performed in the ion trap with CID collision energy 30% and in the Orbitrap with HCD collision energy 40% back-to-back for each precursor. The raw data were processed on Proteome Discoverer 2.1 using the SequestHT search engine. The node for SequestHT included the following settings: Precursor Mass Tolerance 20ppm, Fragment Mass Tolerance 0.5Da for the CID spectra and 0.05Da for the HCD spectra, Dynamic Modifications were Oxidation of M (+15.995Da), Deamidation of N/Q (+0.984Da) and Static Modifications were TMT6plex at any N-Terminus/K (+229.163Da).
Mass spectrometry analysis of histone modifications
Histones were isolated from cell nuclei using acid extraction, biochemically prepared, and analyzed by mass spectrometry against a reference of stable isotope-labeled synthetic peptide standards exactly as described (Creech et al., 2015).
DNA methylation
DNA methylation profiling was carried out on Infinium HumanMethylation450K BeadChip arrays (Illumina, discontinued) at the Harvard Medical School-Partners HealthCare Center for Genetics and Genomics.
Single cell RNA-Seq
Cells were collected by trypsinization and processed according to the 10xGenomics scRNA-seq sample preparation protocol (Chromium Single Cell 3’ v2 Reagent Kit, 10xGenomics). 2,000 cells were targeted for each sample.
ATAC-Seq
50,000 cells were resuspended in 1 ml of cold ATAC-seq resuspension buffer (RSB; 10 mM Tris-HCl pH 7.4, 10 mM NaCl, and 3 mM MgCl2 in water). Cells were centrifuged at max speed for 5 min in a pre-chilled (4 C) fixed-angle centrifuge. After centrifugation supernatant was carefully aspirated. Cell pellets were then resuspended in 50 μl of ATAC-seq RSB containing 0.1% NP40, 0.1% Tween-20, and 0.01% digitonin by pipetting up and down three times. This cell lysis reaction was incubated on ice for 3 min. After lysis, 1 ml of ATAC-seq RSB containing 0.1% Tween-20 (without NP40 or digitonin) was added, and the tubes were inverted to mix. Nuclei were then centrifuged for 5 min at max speed in a pre-chilled (4 C) fixed-angle centrifuge. Supernatant was removed and nuclei were resuspended in 50 μl of transposition mix (reference to OMNI paper) 2.5 μl transposase (100 nM final), 16.5 μl PBS, 0.5 μl 1% digitonin, 0.5 μl 10% Tween-20, and 5 μl water) by pipetting up and down six times. Transposition reactions were incubated at 37 C for 30 min in a thermomixer with shaking at 1,000 r.p.m. Reactions were cleaned up with Qiagen columns. Libraries were amplified as described previously (Buenrostro et al., 2015).
Single cell ATAC-seq
Cells were collected by trypsinization and cryopreserved in 10% DMSO. Subsequently cells were thawed and nuclei prepared and transposed according to the OMNI-ATAC protocol (Corces et al., 2017). From that point ~2,000 cells were targeted for each sample and processed according to the 10xGenomics scATAC-seq sample preparation protocol (Chromium Single Cell ATAC Library & Gel Bead Kit, V1.0 10xGenomics).
Public data analysis
RNA-seq gene expression profile (RSEM counts) and the major breast tumor subtypes (Luminal A, Luminal B, Normal, HER2, and Basal) in the TCGA cohort were obtained from Broad GDAC Firehose database (https://gdac.broadinstitute.org/). The three TNBC subtypes were defined by using GSVA algorithm and gene expression signatures identified in our lab (Jovanovic et al. unpublished). JQ1 resistance score was defined by using GSVA algorithm on the differentially expressed genes (DEGs) between untreated and JQ1-treated cells. The intersected DEGs identified in SUM149 and SUM159 were used. Survival analyses were performed by using survminer R package (https://cran.r-project.org/web/packages/survminer/index.html). Gene co-dependency data were obtained from DepMap portal (https://depmap.org/portal/). The relationship between CRISPR screen results and gene co-dependency was studied.
QUANTIFICATION AND STATISTICAL ANALYSIS
Software used in this study
cutadapt v1.8.1, bowtie2 v2.3.3, samtools v1.9, picard v1.123, MACS2 v2.1.0.20150731, Tophat2 v2.0.11, HT-seq v0.6.1p1, DEseq2 1.22.2, BWA, GATK, MuTect v1.1.4, ROSE v0.1, Cell Ranger v2.0.2, Seurat v2, MAGeCK v0.5.7
ChIP-seq data analysis
Adapter sequences of ChIP-seq raw reads are removed by using cutadapt (DOI:10.14806/ej.17.1.200). Trimmed reads are aligned by bowtie2 using default parameters to version hg19 of human genome. The samtools (Li et al., 2009) and picard (http://broadinstitute.github.io/picard) are used to sort and remove duplicated reads to avoid PCR bias from the sequencing process. Peak calling (identification of regions of ChIP-seq enrichment over background) is performed by using MACS2 (Zhang et al., 2008) with parameters of “--extsize=146 – nomodel”. Enhancers are identified from H3K27ac data in SUM149 and SUM159 by using ROSE pipeline (Hnisz et al., 2013).
RNA-seq data analysis
Raw RNA-seq reads are aligned to version hg19 of human genome by using Tophat2 (Kim et al., 2013) with the default parameters. Gene counts are quantified by using HT-seq (Anders et al., 2015) with REFSEQ annotation. Differentially expressed genes are identified by using DESeq2 (Love et al., 2014) with cutoff of q value < 0.001, ranked by the statistics.
Barcoding data analysis
We followed the method used in Bhang et al. 2015 with small modifications. In details, all sequencing reads are trimmed by using 3′ adaptor sequence: AGCAGAGCTACGCACTCTATGCTAGTGCTAGAGATCGGAAGAGCACACGTCTGAACTCCAGTCACTCACGATCGTATCTCGTATGCCGTCTTCTGCTTG with minimum alignment length of 40-nt. The trimmed reads with Ns or less than 30-nt or without the WS x 15 pattern are removed. Then the 30-nt barcode sequences are extracted from the 3′ prime end of the trimmed sequences. Barcodes with an estimated Phred quality score of at least 10 for all nucleotides and with an average Phred quality score greater than 30 are kept as qualified barcodes. The barcodes with only one count are excluded from the analyses to avoid the noise derived from the sequencing error.
Drug synergy analysis
Synergy studies were performed in 384-well plates (Corning). SUM159 cells were seeded at a density of 200 cells/well, SUM159R and SUM149 at a density of 500 cells/well, and SUM149R, CAL-120, CAL-51, HCC38, HCC3153, Hs578-T, MDA-MB-231, MDA-MB-436, and PMC42 at a density of 1000 cells/well, in 50 μL of media. The following day, the cells were imaged and counted using the Celigo Imaging Cytometer (Nexcelom). Drugs were then pin-transferred into the wells from a 500X concentrated drug plate made in a 384-well plate (Greiner Bio-One) using the JANUS Automated Workstation (Perkin Elmer). Five concentrations for each drug were chosen between 20% and 80% inhibition, and half doses were used for combinations (see Table S5 for list of concentration of each compound used), with 4 replicate wells for each concentration of each drug alone and 8 replicates of each concentration pair. After approximately 3 cell doublings (3 days for SUM series, CAL-51, and MDA-MB-231; 4 days for PMC42; 5 days for CAL-120, HCC3153, Hs578-T, and MDA-MB-436; and 6 days for HCC38), cells were stained with 5 μg/mL Hoechst 33342 (Sigma-Aldrich) and 2 μg/mL propidium iodide (Sigma-Aldrich) in PBS, and nuclei were imaged and counted using the Celigo Imaging Cytometer. Propidium iodide-positive dead cells were subtracted from the total Hoechst-positive nuclei to determine the number of live cells, which were then normalized to the baseline number of cells. Combination indices were determined using the median-effect principle of Chou & Talalay (Chou and Talalay, 1984). Isobologram plots were generated using R.
Single cell RNA-seq data analysis
Single cell RNA-seq data generated by 10x genomics were preprocessed using the Cell Ranger (https://www.10xgenomics.com/) to obtain the UMI (unique molecular identifier) counts for each gene. To get a reliable single cell transcriptome dataset, we excluded the cells with less than 200 genes expressed (UMI > 0) or the cells with more than 80% UMIs from mitochondrial genes. The filtered data were then normalized and scaled by using Seurat to remove unwanted source of variations (Lun et al., 2016). T-SNE was performed on the normalized data to visualize the single cells in two-dimensional space by using the top 10 dimensions of principal component analysis (PCA). Unsupervised clustering was performed by using “FindClusters” function in Seurat package with parameter of resolution = 0.8. Cell cycle phases of all single cells were assigned by using cyclone function in scran package (Lun et al., 2016). Genes with differential expression between clusters were obtained by using Wilcoxon rank-sum test. FDR was then calculated to correct for multiple testing.
Identification of pre-existing resistant cells from single cell transcriptome
Cell identity signatures of SUM149, JQ1-treated SUM149 and SUM149R cells: For each of the three cell types, we compare the bulk gene expression of it with the other two cells together. We choose the top most 100 up-regulated and down-regulated genes as the (up and down) signatures of the cell type. Cell identity signatures of SUM159, JQ1-treated SUM159 and SUM159R cells are obtained in the same way. Calculation of cell identity score: For each single cell, we calculated the average expression of each set of up signature genes minus the average expression of each set of down signature genes as the cell identity score. We carried out a bootstrap procedure to estimate the significance of the cell identity score. We randomly select 1,000 sets of up and down signatures with the same size of the original true signatures, generated the bootstrap distribution of the cell identity score, and calculated the bootstrap p value based on the distribution. We classified the single cells based on the bootstrap p value cutoff of 5%. If a cell did not pass the test of any signature, it is annotated as unclassified. Hexagonal plots were used to show the bootstrap classification of single cells in indicated cell populations, in which cells showed clear identity (passed the 5% threshold) are positioned on the edge of the plot.
CRISPR screen data analysis
CRISPR data were analyzed by MAGeCK and MAGeCK-VISPR essentially as described (Chen et al., 2018; Jeselsohn et al., 2018; Li et al., 2015). Briefly, raw sequencing data are pre-processed by using MAGeCK (Li et al., 2014) to obtain the read counts for each sgRNA. Control sgRNAs are used to normalize the data. MAGeCK TEST algorithm is used to compare treatment with control samples to obtain the significant enriched and depleted sgRNAs and genes. Genes with p value less than 0.001 are candidate hits. Different doses and replicates of JQ1 treatment libraries in the same cell line are tested separately and merged together by choosing the lowest p value. If one gene is enriched in one library and depleted in the other, or vice versa, the gene is set as not changed. MageckFlute package (Li et al., 2014) was used to visualize the data.
DNA methylation
DNA methylation profiling was carried out on Infinium HumanMethylation450K BeadChip arrays (Illumina, discontinued) at the Harvard Medical School-Partners HealthCare Center for Genetics and Genomics. The methylation status of the cell lines was obtained using the Infinium HumanMethylation450 BeadChip Kit from Illumina. The unnormalized probe-level beta-values (Du et al., 2010) of each sample were obtained from the raw data using the Illumina GenomeStudio software (GSGX v1.1.0). The normalization of the beta-values was performed using the R Bioconductor package lumi. The normalized M-value thus obtained were converted back to normalized beta-values using the following formula:
The normalized beta-values of each sample were then collapsed to promoters and enhancers of genes. Briefly, each probe is attempted to be assigned to either a promoter, enhancer, or gene-body of a gene using the annotation provided in the output of the GenomeStudio software mentioned above. The methylation status of the enhancer of a gene is determined by taking the average normalized beta-values of all the probes assigned to the enhancer of that. Depending on the position of the probes relative to the TSS of a gene, the probes are also assigned to either promoters (TSS200, TSS1500, 1stExon, and 5′UTR) or gene-body (Body, 3′UTR) of genes. Following that, single gene-level methylation values of each gene’s promoter and gene-body are obtained using the average normalized beta-values as explained for enhancers. The gene-level enhancer, promoter, and gene-body methylation values of each sample have been used to determine the correlation between the samples and their tissue of origin (lineage).
ATAC-seq
ChiLin pipeline 2.0.0 (Qin et al., 2016) is used for QC and preprocess of the ATAC-seq. We use Burrows-Wheeler Aligner (BWA) (Li and Durbin, 2009) as a read mapping tool, and Model-based Analysis of ChIP-Seq (MACS2) (Zhang et al., 2008) as a peak caller, with a q-value (FDR) threshold of 0.01.Based on a dynamic Poisson distribution MACS2 can effectively capture local biases in the genome sequence, allowing for more sensitive and robust prediction of binding sites. Unique read for a position for peak calling is used to reduce false positive peaks, statically significant peaks are finally selected by calculated false discovery rate of reported peaks. Deeptools (Ramirez et al., 2016) is used for the heatmap plots. ATAC-seq peaks from all study samples were merged to create a union set of sites. Read densities were calculated for each peak for each sample, differential peaks between WT and KO were identified by DEseq2 (Love et al., 2014) with adjusted P ≤0.05, |log2fold change| ≥ 1.
Single cell ATAC-seq
Single-cell ATAC-seq data were processed using the Cell Ranger ATAC pipeline v1.1.0, which provides QC and clustering. Any cell that had Fraction of reads in peaks (FRiP) < 0.2 and total fragments < 1,000 was removed from the analysis, total 2,701 cells passed the QC. Median fragments per cell is 27,599, fraction of transposition events in peaks is 32.8%. The t-SNE analysis was performed using the implementation from the Loupe Cell Browser 3.1.0.
Exome Sequencing
Exome sequencing was performed in the Dana-Farber Cancer Institute Center for Cancer Genome Discovery as previously described (Shu et al., 2016).
RIME
Peptide intensities were normalized using median scaling and protein level quantification was obtained by the summation of the normalized peptide intensities. A statistical analysis of differentially-regulated proteins was carried out using qPLEXanalyzer a Bioconductor R-package (Papachristou et al., 2018), which internally uses limma R-package from Bioconductor (Ritchie et al., 2015). Multiple testing correction of p-values was applied using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995) to control the false discovery rate.
Supplementary Material
Table S1. Related to Figure 1. CRISPR Screen Data. Log(P-value) of enrcihment/drop out are listed. Provided as an Excel file.
Table S2. Related to Figure 1. List of differentially expressed genes in SUM149/SUM149R and SUM159/SUM159R cells −/+ JQ1. Provided as an Excel file.
Table S3. Related to Figure 1. RPPA Data and list of BRD4-interacting proteins identified by RIME. Provided as an Excel file.
Table S4. Related to Figures 2 and 3. List of SMI drugs, screen results, and drug concentrations used in drug synergy experiments. Provided as an Excel file.
Table S5. Related to Figure 5. Genes differentially expressed between scramble or BRD7 KO and WT SUM159 cells in the presence or absence of JQ1. Provided as an Excel file.
Table S6. Related to Figure 7. Marker genes specific for each of the cell clusters identified based on scRNA-seq. Provided as an Excel file.
HIGHLIGHTS.
Synthetic lethal and resistance interactions with BET inhibitors in TNBC
Multiple independent unbiased screens identify the same genes/pathways
CDK4/6 inhibitors and paclitaxel are top synergies with BET bromodomain inhibitors
Deletion of SNF/SWI complex components leads to BET inhibitor resistance
ACKNOWLEDGMENTS
We thank members of our laboratories for their critical reading of this manuscript and useful discussions. We thank the Dana-Farber Cancer Institute Molecular Biology Core Facility for their outstanding sequencing service. This research was supported by the National Cancer Institute PSOC U54 CA193461 (F.M. and K.P.), F30 CA228208 (J.Y.G.), R35 CA197623 (K.P.), P01 CA080111 (K.P., M.B.), SPORE P50 CA168504 (K.P.), the Susan G. Komen Foundation (S.Sh.), and the Ludwig Center at Harvard (K.P., F.M., M.B., J.S.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
M.B. and K.P. received research support and were consultants to Novartis Institutes for BioMedical Research during the execution of this study. K.P. serves on the scientific advisory board of Farcast Biosciences and Acrivon Therapeutics. M.B. receives sponsored research support from Novartis and serves on the SAB of Kronos Bio, and is a consultant to GTx, Inc., Aleta Biotherapeutics, and H3 Biomedicine. R.Z. and J.B. is a current employee of C4 Therapeutics Inc. and Novartis, respectively. C.K. is a Scientific Founder, fiduciary Board of Directors member, Scientific Advisory Board member, consultant, and shareholder of Foghorn Therapeutics, Inc. (Cambridge, MA). S.Sh, K.P., and J.B. are inventors of a patent on BET inhibitor resistance that DFCI licensed to Roche.
REFERENCES
- Abraham J, Coleman R, Elias A, Holmes FA, Kalinsky K, Kittaneh M, Lower E, Mahtani R, Terry Mamounas E, Pegram M, et al. (2018). Use of cyclin-dependent kinase (CDK) 4/6 inhibitors for hormone receptor-positive, human epidermal growth factor receptor 2-negative, metastatic breast cancer: a roundtable discussion by The Breast Cancer Therapy Expert Group (BCTEG). Breast Cancer Res Treat 171, 11–20. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing J. Royal Statistical Society 57, 289–300. [Google Scholar]
- Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V, Caushi JX, Zhao R, Hims MM, Singh AP, Kao I, Rakiec D, Shaw P, et al. (2015). Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med 21, 440–448. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. (2013). Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Xiao T, Xu H, Jiang P, Meyer CA, Li W, Brown M, and Liu XS (2018). Improved design and analysis of CRISPR knockout screens. Bioinformatics 34, 4095–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KL, Zhang F, Jaganathan A, Sharma R, Zhang Q, Konuma T, Shen T, Lee JY, Ren C, Chen CH, et al. (2017). Distinct Roles of Brd2 and Brd4 in Potentiating the Transcriptional Program for Th17 Cell Differentiation. Mol Cell 65, 1068–1080 e1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, and Talalay P (1984). Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22, 27–55. [DOI] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creech AL, Taylor JE, Maier VK, Wu X, Feeney CM, Udeshi ND, Peach SE, Boehm JS, Lee JT, Carr SA, et al. (2015). Building the Connectivity Map of epigenetics: chromatin profiling by quantitative targeted mass spectrometry. Methods 72, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, Liu S, Zhong Q, Guo J, Zhang J, et al. (2017). Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med 23, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, and Lin SM (2010). Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Castro AC, Lin NU, and Polyak K (2019). Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov 9, 176–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris IS, Endress JE, Coloff JL, Selfors LM, McBrayer SK, Rosenbluth JM, Takahashi N, Dhakal S, Koduri V, Oser MG, et al. (2019). Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab 29, 1166–1181 e1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinohara K, Wu HJ, Vigneau S, McDonald TO, Igarashi KJ, Yamamoto KN, Madsen T, Fassl A, Egri SB, Papanastasiou M, et al. (2018). KDM5 Histone Demethylase Activity Links Cellular Transcriptomic Heterogeneity to Therapeutic Resistance. Cancer Cell 34, 939–953 e939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SC, Gilgenast TG, Bartman CR, Edwards CR, Stonestrom AJ, Huang P, Emerson DJ, Evans P, Werner MT, Keller CA, et al. (2017). The BET Protein BRD2 Cooperates with CTCF to Enforce Transcriptional and Architectural Boundaries. Mol Cell 66, 102–116 e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A, Bernasocchi T, Civenni G, Losa M, Svinkina T, et al. (2017). Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med 23, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, Xiao T, Li W, Qiu X, Buchwalter G, et al. (2018). Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 33, 173–186 e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Yan Y, Wang D, Ding D, Ma T, Ye Z, Jimenez R, Wang L, Wu H, and Huang H (2018). DUB3 Promotes BET Inhibitor Resistance and Cancer Progression by Deubiquitinating BRD4. Mol Cell 71, 592–605 e594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakashev S, Zhu H, Yokoyama Y, Zhao B, Fatkhutdinov N, Kossenkov AV, Wilson AJ, Simpkins F, Speicher D, Khabele D, et al. (2017). BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep 21, 3398–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JP, Picaud S, Fujisawa T, Hou H, Savitsky P, Uuskula-Reimand L, Gupta GD, Abdouni H, Lin ZY, Tucholska M, et al. (2019). Interactome Rewiring Following Pharmacological Targeting of BET Bromodomains. Mol Cell 73, 621–638 e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard B, Brand TM, O’Keefe RA, Lee ED, Zeng Y, Kemmer JD, Li H, Grandis JR, and Bhola NE (2018). BET Inhibition Overcomes Receptor Tyrosine Kinase-Mediated Cetuximab Resistance in HNSCC. Cancer Res 78, 4331–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Koster J, Xu H, Chen CH, Xiao T, Liu JS, Brown M, and Liu XS (2015). Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol 16, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, Bach K, and Marioni JC (2016). Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol 17, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H, D’Santos C, Serandour AA, Ali HR, Brown GD, Atkins A, Rueda OM, Holmes KA, Theodorou V, Robinson JL, et al. (2013). Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep 3, 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan S, Rao SV, Sutton J, Cheeseman D, Dunn S, Papachristou EK, Prada JG, Couturier DL, Kumar S, Kishore K, et al. (2020). ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat Genet 52, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CKY, Piscuoglio S, Geyer FC, Burke KA, Pareja F, Eberle CA, Lim RS, Natrajan R, Riaz N, Mariani O, et al. (2017). The Landscape of Somatic Genetic Alterations in Metaplastic Breast Carcinomas. Clin Cancer Res 23, 3859–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolsky Y, Ekins S, Nikolskaya T, and Bugrim A (2005). A novel method for generation of signature networks as biomarkers from complex high throughput data. Toxicol Lett 158, 20–29. [DOI] [PubMed] [Google Scholar]
- O’Dwyer PJ, Piha-Paul S, French C, Harward S, Ferron-Brady G, Wu Y, Barbash O, Wyce A, Annan M, Horner T, et al. (2016). GSK525762, a selective bromodomain (BRD) and extra termina protein (BET) inhibitor: results from part 1 of a phase I/II open-label single-agent study in patients with NUT midline carcinoma (NMC) and other cancers. Proc Am Assoc Cancer Res, CT014 [abstract]. [Google Scholar]
- Papachristou EK, Kishore K, Holding AN, Harvey K, Roumeliotis TI, Chilamakuri CSR, Omarjee S, Chia KM, Swarbrick A, Lim E, et al. (2018). A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes. Nat Commun 9, 2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, et al. (2013). Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov 3, 308–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Mei S, Wu Q, Sun H, Li L, Taing L, Chen S, Li F, Liu T, Zang C, et al. (2016). ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC Bioinformatics 17, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nature Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro GI, Dowlati A, LoRusso PM, Eder JP, Anderson A, Do KT, Kagey MH, Sirard C, Bradner JE, and Landau SB (2015). Clinical efficacy of the BET bromodomain inhibitor TEN-010 in an open-label substudy with patients with documented NUT-midline carcinoma. Proc AACR-NCI-EORTC, A49 [abstract]. [Google Scholar]
- Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, et al. (2016). Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu S, and Polyak K (2016). BET Bromodomain Proteins as Cancer Therapeutic Targets. Cold Spring Harb Symp Quant Biol 81, 123–129. [DOI] [PubMed] [Google Scholar]
- TCGA (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, and Kornblau SM (2006). Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther 5, 2512–2521. [DOI] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015). DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. (2017). BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell 67, 5–18 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Lai HT, Zhang H, and Chiang CM (2013). Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell 49, 843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Chhangawala S, Cocco E, Razavi P, Cai Y, Otto JE, Ferrando L, Selenica P, Ladewig E, Chan C, et al. (2020). ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat Genet 52, 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Zhang Y, Shan W, Hu Z, Yuan J, Pi J, Wang Y, Fan L, Tang Z, Li C, et al. (2017). Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Battilana G, Forcato M, Filippi L, Azzolin L, Manfrin A, Quaranta E, Di Biagio D, Sigismondo G, Guzzardo V, et al. (2018). Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med 24, 1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. (2017). Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature Medicine 23, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, and Oldenburg KR (1999). A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
- Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z, Wang S, Pan CW, Zhu Y, Yan Y, et al. (2017). Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med 23, 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 1. CRISPR Screen Data. Log(P-value) of enrcihment/drop out are listed. Provided as an Excel file.
Table S2. Related to Figure 1. List of differentially expressed genes in SUM149/SUM149R and SUM159/SUM159R cells −/+ JQ1. Provided as an Excel file.
Table S3. Related to Figure 1. RPPA Data and list of BRD4-interacting proteins identified by RIME. Provided as an Excel file.
Table S4. Related to Figures 2 and 3. List of SMI drugs, screen results, and drug concentrations used in drug synergy experiments. Provided as an Excel file.
Table S5. Related to Figure 5. Genes differentially expressed between scramble or BRD7 KO and WT SUM159 cells in the presence or absence of JQ1. Provided as an Excel file.
Table S6. Related to Figure 7. Marker genes specific for each of the cell clusters identified based on scRNA-seq. Provided as an Excel file.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplemental Information. All raw genomic data was deposited to GEO under accession number: GSE131102. This study did not generate custom code.