

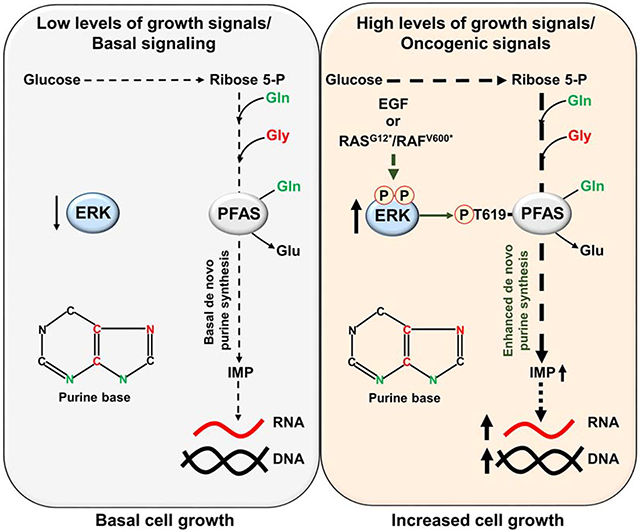
SUMMARY
The RAS-ERK/MAPK (RAS-extracellular signal-regulated kinase/mitogen-activated protein kinase) pathway integrates growth-promoting signals to stimulate cell growth and proliferation, at least in part, through alterations in metabolic gene expression. However, examples of direct and rapid regulation of the metabolic pathways by the RAS-ERK pathway remain elusive. We found that physiological and oncogenic ERK signaling activation leads to acute metabolic flux stimulation through the de novo purine synthesis pathway, thereby increasing building block availability for RNA and DNA synthesis, which is required for cell growth and proliferation. We demonstrate that ERK2, but not ERK1, phosphorylates the purine synthesis enzyme PFAS (phosphoribosylformylglycinamidine synthase) at T619 in cells to stimulate de novo purine synthesis. The expression of nonphosphorylatable PFAS (T619A) decreases purine synthesis, RAS-dependent cancer cell colony formation, and tumor growth. Thus, ERK2-mediated PFAS phosphorylation facilitates the increase in nucleic acid synthesis required for anabolic cell growth and proliferation.
Graphical Abstract
eTOC Blurp:
Ali and Sahu et al. demonstrate that ERK signaling acutely stimulates de novo purine synthesis through direct phosphorylation of the purine enzyme PFAS in response to physiological growth signals or oncogenic activation of RAS or RAF. The ERK-dependent phosphorylation of PFAS is required for cell and tumor growth.
INTRODUCTION
Cells and organisms must synchronize their metabolic activity with changes in their nutrient and hormonal environments. This synchronization is coordinated by the signaling networks that integrate local and systemic nutrient and hormonal signals to convey the metabolic status of the cell (Erez and DeBerardinis, 2015; Ward and Thompson, 2012). The extracellular signal-regulated kinase-mitogen-activated protein kinase (ERK-MAPK) cascade, often hyperactive in cancer cells (Ryan et al., 2015), is a central signaling network that regulates a wide variety of stimulated cellular processes, including proliferation, differentiation, survival, apoptosis and stress responses (Brunet et al., 1999; Cargnello and Roux, 2011; Plotnikov et al., 2011; Shaul and Seger, 2007; Zhang and Liu, 2002). Briefly, the integration of signals by the RAS-ERK cascade involves the binding of an extracellular ligand to a receptor tyrosine kinase (RTK) in the plasma membrane to cause receptor dimerization and tyrosine residue autophosphorylation in the intracellular domains (Lemmon and Schlessinger, 2010). The small G protein RAS is recruited and becomes GTP-loaded, leading to the activation of the serine/threonine protein kinase RAF, which promotes the subsequent activation of MAPK/ERK kinase (MEK) and then ERK. Activated ERK phosphorylates numerous substrates and regulates the activity of different transcription factors, leading to alterations in gene expression (Whitmarsh, 2007). The importance of closely linking ERK signaling to metabolism control is highlighted by the fact that aberrant regulation of this signaling pathway and cellular metabolism have been implicated in the pathophysiology of a diverse set of human diseases, including cancer and metabolic disorders (DeBerardinis and Chandel, 2016; Haigis et al., 2008; Humpton et al., 2019; Shaw and Cantley, 2006). This evidence supports our efforts to identify key metabolic targets downstream of ERK that contribute to cancer cell growth. Previously, in two separate studies, we demonstrated that the mechanistic target of rapamycin complex 1 (mTORC1) stimulates de novo pyrimidine and purine synthesis (Ben-Sahra et al., 2013; Ben-Sahra et al., 2016) to support an increased demand for RNA and DNA, which is required for anabolic growth and proliferation. The synthesis of both purines and pyrimidines requires ribose 5-phosphate from glucose-derived pentose phosphate. Purines and pyrimidines (e.g., ATP, GTP, dATP, dGTP, UTP, CTP, dTTP and dCTP) are essential for growing cells, not only as RNA and DNA building blocks but also for cellular homeostasis maintenance. Cells can acquire purines and pyrimidines through either the salvage pathways, requiring exogenous nucleoside availability and uptake, or de novo synthesis from small molecule precursors such as glutamine, aspartate, glycine, formate, and bicarbonate (Lane and Fan, 2015). Although it is generally accepted that the salvage pathways are sufficient to sustain the demands of quiescent terminally differentiated cells, growing and proliferating cells require activation of de novo synthesis (Villa et al., 2019). With a few remarkable exceptions (Ben-Sahra et al., 2013; Graves et al., 2000; Hoxhaj et al., 2019), most established links between growth factor signaling and cellular metabolism involve the regulation of transcription factors that control the expression of genes encoding metabolic enzymes (Barbie et al., 2009; Stine et al., 2015). ERK signaling is physiologically activated by growth factors in normal settings (Lange-Carter and Johnson, 1994) and pathologically activated by oncogenic RAS and RAF proteins in various cancer types (Downward, 2003; Halilovic and Solit, 2008; Murugan et al., 2019). The ERK signaling network, when activated, promotes anabolic metabolism typically through long-term changes mediated by the transcription factor c-MYC (Davis, 1995; Kerkhoff et al., 1998). The ERK-MYC axis controls various metabolic processes, including glucose and nucleotide metabolism, through transcriptional mechanisms (Dang, 2013; Grassian et al., 2011; Kimmelman, 2015; Liu et al., 2008; Santana-Codina et al., 2018). However, examples of acute and direct regulation of cellular metabolism by ERK signaling remain largely unknown, with one exception of ERK corroborating the immediate regulation of de novo pyrimidine synthesis via the direct phosphorylation of carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, dihydroorotase (CAD), the rate-limiting enzyme in this metabolic pathway (Graves et al., 2000).
In this study, we identified a novel mechanism by which ERK signaling regulates purine synthesis post-translationally to sustain nucleic acid synthesis. Physiological and oncogenic RAS pathway activation results in a rapid increase in metabolic flux through de novo purine synthesis via ERK2-dependent phosphorylation of the purine enzyme phosphoribosylglycinamidine synthase (PFAS) at T619. The expression of nonphosphorylatable PFAS mutant in various normal and cancer cell settings reduced ERK2-dependent flux through purine synthesis. Furthermore, in RAS-driven cancer cells, a PFAS phosphorylation-deficient mutant reduced basal purine synthesis flux, cancer cell growth and proliferation in vitro and in vivo. This study reveals a direct metabolic role for ERK2 signaling in regulating de novo purine synthesis through the acute control of PFAS activity, highlighting purine synthesis as a key anabolic program supporting RAS-ERK-dependent cell growth and proliferation.
RESULTS
Immediate regulation of de novo purine synthesis by ERK signaling
Metabolism is temporally regulated by a variety of cues that can be integrated by signaling pathways, such as the pro-growth RAS-ERK pathway. To determine rapid metabolic processes regulated by the RAS-ERK signaling pathway, we used steady-state metabolite profiling of cancer cell lines exhibiting growth factor-independent ERK signaling due to activating mutations in the RAS or RAF oncogenes (e.g. A549 lung cancer cells (RASG12S) and SK-MEL-28 melanoma cells (BRAFV600E)); we treated these cells for only 1 hour with vehicle or SCH772984, a highly selective ERK inhibitor. The levels of purine intermediates, including IMP, AMP and GMP, were decreased by approximately 20 to 50% following 1 hour of ERK inhibition (Figures 1A and 1B). Moreover, acute HeLa cell stimulation with epidermal growth factor (EGF) increased the steady-state levels of purine intermediates in an ERK-dependent manner (Figure 1C). Notably, we also observed an ERK-dependent increase in intermediates of the pentose phosphate pathway, including glucose 6-phosphate and ribulose 5-phosphate, which can influence upstream steps in nucleotide synthesis (Figure S1A). Because EGF stimulation can activate the mechanistic target of rapamycin complex 1 (mTORC1) signaling (Mendoza et al., 2011), which has been shown to stimulate metabolic pathways supporting nucleotide synthesis (Ben-Sahra et al., 2013; Ben-Sahra et al., 2016), we sought to determine mTORC1 activity in different conditions by evaluating the phosphorylation status of its substrate S6 kinase (S6K). Although S6K phosphorylation was increased in RAS/RAF-driven cancer cell lines or in response to EGF stimulation (HeLa cells), it was not affected by short-term MEK or ERK inhibition (Figures 1A–C, 1E). These results indicate that the decrease in purine synthesis upon MEK or ERK inhibition was independent of mTORC1 signaling. To determine whether the effects of ERK on the relative abundance of purines reflect regulation of the de novo purine synthesis pathway, we measured relative flux in the presence or absence of EGF and the MEK inhibitor U0126 (MEKi), with a 1-hour pulse of either the stable isotope-labeled 15N-glutamine (labeled on the amide nitrogen) or 15N-13C2-glycine which both contribute to purine ring synthesis (Figure 1D). The peak area and fractional abundance of labeled purine intermediates from 15N-glutamine were enhanced in response to EGF stimulation in HeLa cells and reduced by short-term treatment with MEKi U0126, which resulted in ERK inhibition (Figure 1E, Figure S1B, and Table S1). Genetic inhibition of ERK1 and ERK2 with siRNA also led to a decrease in the relative flux through de novo purine synthesis (Figure 1F and Figure S1C). Moreover, acute ERK inhibition in RAS and RAF-driven cancer cells reduced metabolic flux from 5N-13C2-glycine (Figure 1G, Figure S1D, and Table S1) and 15N-glutamine (Figure S1E) through de novo purine synthesis. Similarly, isotopic tracing with 13C4-15N-aspartate, which provides one nitrogen atom to the purine ring (Figure S1F), also revealed rapid ERK-dependent regulation of de novo purine synthesis in HeLa cells (Figure S1G). These results indicate that ERK signaling acutely controls de novo purine synthesis activity in response to physiological and oncogenic signals.
Figure 1. Immediate stimulation of de novo purine synthesis by ERK signaling.
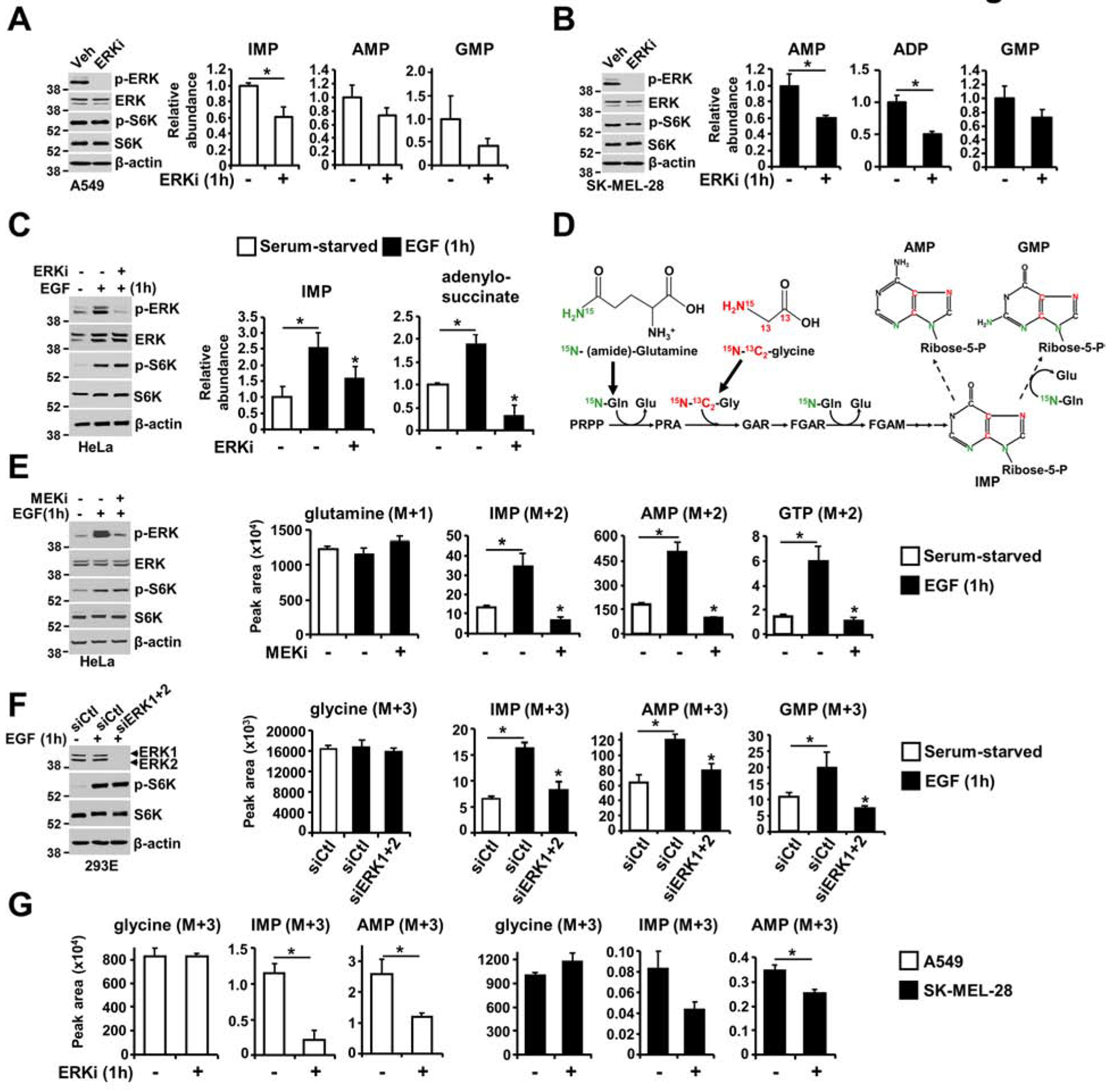
(A, B) The effects of acute ERK inhibition on the steady-state levels of purine intermediates, as measured via LC-MS/MS, in A549 (A) and SK-MEL-28 (B) cells following 15 hours of serum starvation and 1 hour of treatment with SCH772984 (ERKi, 1 μM) or DMSO. (C) Purine intermediates were measured as in (A, B) in HeLa cells serum starved for 15 hours and treated with vehicle or SCH772984 (ERKi, 1 μM) for 30 min prior to 1 hour of stimulation with EGF (50 ng/ml). (D) Schematic of the incorporation of nitrogen and carbon from glutamine and glycine into the purine ring. (E) Normalized peak areas of 15N-labeled purine intermediates, as measured by targeted LC-MS/MS, in HeLa cells serum starved for 15 hours and pretreated with vehicle or U0126 (MEKi, 10 μM) before stimulation with EGF (50 ng/ml) and labeling with 15N-(amide)-glutamine for 1 hour (see also Figure S1B). (F) Normalized peak areas of labeled glycine and purine intermediates HeLa cells transfected with either nontargeting control siRNA (siCtl), or siRNA against ERK1 and ERK2 (siERK1+2) for 48 hours and were treated as in (E) and labeled with 15N-13C2-glycine for the last hour prior to metabolite extraction (see also Figure S1C). (G) A549 and SK-MEL-28 cells serum-starved for 15 hours and treated with vehicle or SCH772984 (ERKi, 1 μM) before labeling with 15N-13C2-glycine for 1 hour (see also Figure S1D). The data are presented as the means ± SDs of biological triplicates and are representative of two independent experiments (A-F). * P<0.05 by two-tailed Student’s t test for pairwise comparisons (A, B, G) and one-way ANOVA with Tukey’s post hoc test for multiple pairwise comparisons (C, E, F).
ERK stimulates newly synthesized purine incorporation into nucleic acids
In addition to incorporating atoms from glutamine, aspartate, and glycine, the de novo purine synthesis pathway combines molecules from other substrates such as, bicarbonate (HCO3−), and formyl-tetrahydrofolate (THF), with the activated ribose derived from the pentose phosphate pathway to form purine nucleotides (Figure 2A) (Lane and Fan, 2015; Villa et al., 2019). To determine whether ERK-dependent regulation of cellular purine levels affects newly assembled nucleic acids synthesis, we used various radiotracers to evaluate the contribution of auxiliary pathways, including glucose metabolism (14C-glucose) and one-carbon metabolism (3-14C-serine), and pathways that directly support purine nucleotide formation, such as de novo purine synthesis (14C-glycine and 14C-formate) and the purine salvage pathway (3H-hypoxanthine). While radiotracer uptake was not altered by ERK activity (Figure S2A), physiological ERK activation increased de novo purine synthesis in a MEK- and ERK-dependent manner in HeLa cells, as evidenced by the alterations in the incorporation of 14C from glycine, formate, serine and glucose into nucleic acids; in contrast, physiological ERK activation did not affect the purine salvage pathway (incorporation of 3H from hypoxanthine into nucleic acids) (Figure 2B and Figure S2B). A similar increase in de novo purine synthesis in response to ERK activation was also observed in human embryonic kidney 293E (HEK293E) cells and wild-type mouse embryonic fibroblasts (MEFs) (Figure 2C).
Figure 2. ERK signaling promotes the integration of newly synthesized purines into nucleic acids.
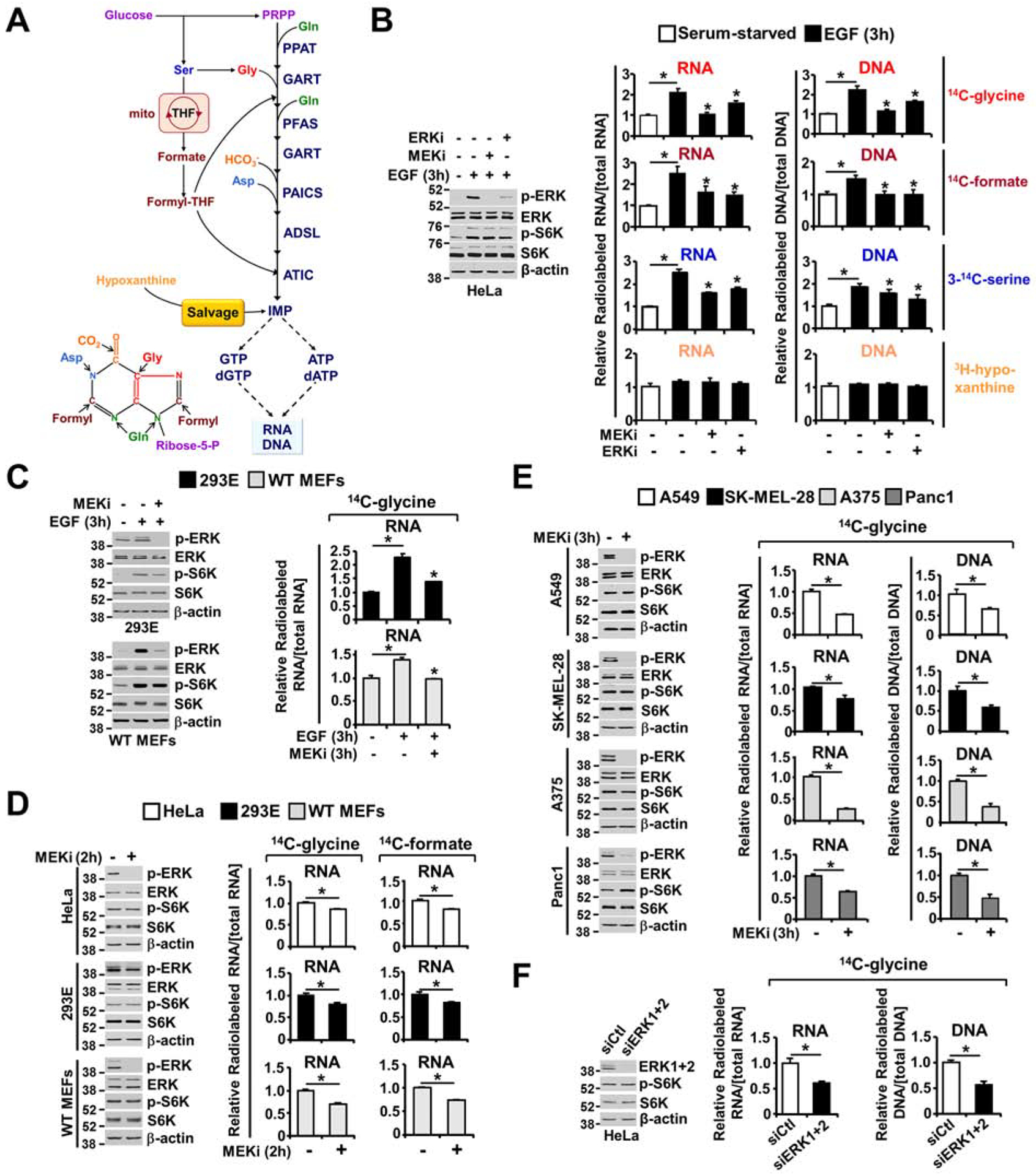
(A) Schematic of the purine synthesis pathways, including the pentose phosphate pathway, serine biosynthesis pathway and the mitochondrial THF cycles providing carbon and nitrogen for de novo purine nucleotide synthesis. Premade nucleobases, such as hypoxanthine, can sustain nucleotide synthesis via the purine salvage pathway. (B) Immunoblots assessing ERK and mTORC1 signaling; the relative incorporation of 14C from glycine, formate, and serine; and the relative incorporation of 3H from hypoxanthine into RNA and DNA. Labeling was performed for 3 hours under serum-free or EGF (50 ng/ml, 3 hours) stimulation conditions in the presence or absence of U0126 (MEKi, 10 μM) or SCH772984 (ERKi, 1 μM), reflecting de novo purine synthesis (14C-glycine, 14C-formate), one-carbon metabolism into purine nucleotides (3-14C-serine) and purine salvage pathway activity (3H-hypoxanthine) (see also Figure S2B and S2C). (C) HEK293E cells and MEFs were treated as in (B) but labeled with only 14C-glycine for 3 hours. The relative levels of incorporation of 14C from glycine into RNA are shown. Immunoblots performed in parallel to the radio-tracing experiments are shown. (D) As in (B, C), but the cells were cultured in 10% dialyzed serum and treated with vehicle or U0126 (MEKi, 10 μM, 2 hours) and labeled with 14C-glycine for 2 hours. (E) Relative incorporation of 14C-glycine into RNA, with labeling for 3 hours under serum-free conditions in the given cancer cell lines with high levels of ERK signaling (A549, SK-MEL-28, A375, and Panc1), treated with vehicle or U0126 (MEKi, 10 μM) for 3 hours (see also Figure S2D and S2E). (F) As in (D), HeLa cells were transfected with ERK1 and ERK2 siRNAs or nontargeting controls (siCtl) for 48 hours. Cells were cultured in 10% dialyzed serum for 15 hours and were then treated with vehicle or U0126 (MEKi, 10 μM, 2 hours) and concurrently labeled with 14C-glycine for 2 hours. The data are graphed as the means ± SDs of biological triplicates and are representative of at least two independent experiments (B-F). * P<0.05 by one-way ANOVA with Tukey’s post hoc test for multiple pairwise comparisons (B, C) and two-tailed Student’s t test for pairwise comparisons (D, E, F).
The lack of ERK-dependent regulation of hypoxanthine incorporation into nucleic acids demonstrates that the purine salvage pathway and mechanisms controlling RNA and DNA polymerization were not altered by ERK signaling at these short-term treatment times. HEK293E cells, HeLa cells and wild-type MEFs cultured with dialyzed serum and exposed to short-term MEKi treatment also showed a decrease in carbon flux from glycine or formate into nucleic acids (Figure 2D). To determine whether MEK-ERK signaling influences the production of nucleic acids through its specific regulation of de novo purine synthesis in cancer settings with high ERK signaling levels, we measured the incorporation of carbon from glycine into nucleic acids in different RAS- and RAF-driven cancer cell lines. MEK inhibition significantly reduced the incorporation of 14C from glycine into RNA and DNA in these cancer cells (Figure 2E) and in HeLa cells with siRNA-mediated ERK1 and ERK2 depletion cultured in dialyzed serum (Figure 2F). Moreover, two structurally distinct MEKis, U0126 and the clinically approved AZD6244 (Bodoky et al., 2012; Yeh et al., 2007), led to a decrease in de novo purine synthesis in EGF-stimulated HeLa cells (Figure S2C) and RAS-driven A549 cells in serum-free culture (Figure S2D); in addition there were no purine salvage alterations (Figure S2E). Again, mTORC1 activity was not altered by MEK or ERK inhibition, indicating that ERK signaling stimulates the newly synthesized purine incorporation into nucleic acids independent of the metabolic effects mediated by mTORC1 (Figures 2B–F). Collectively, these data suggest that ERK signaling acutely promotes de novo purine synthesis in various settings to sustain nucleic acid synthesis.
ERK signaling regulation does not alter enzyme levels of the de novo purine synthesis pathway
To identify the mechanisms by which ERK signaling stimulates de novo purine synthesis, we first measured transcript levels of purine synthesis pathway components in HeLa cells stimulated with EGF over a time course. Transcript levels did not show discernible changes in HeLa cells exhibiting ERK-dependent purine synthesis flux modulation (Figure 3A and Table S2). We validated these findings by measuring the protein levels of the purine enzymes in HeLa (Figure 3B) and A549 (Figure 3C) cells treated with either EGF or MEKi. Additionally, because MYC activity can be acutely regulated upon ERK activation (Sears et al., 2000), we sought to determine the involvement of MYC in the rapid regulation of purine synthesis. Using the incorporation of radiolabeled carbon from glycine into nucleic acids, we showed that c-MYC knockdown diminished the basal activity of de novo purine synthesis without affecting the ability of EGF or MEKi to further modulate this pathway in HeLa or A549 cells, respectively (Figures 3D and 3E), suggesting that additional mechanisms regulate this pathway. These results demonstrate that the rapid control of de novo purine synthesis downstream of RAS-ERK signaling is independent of the transcript or protein level regulation involved in de novo purine nucleotide synthesis.
Figure 3. ERK signaling regulation does not alter the transcript and protein levels of purine enzymes.
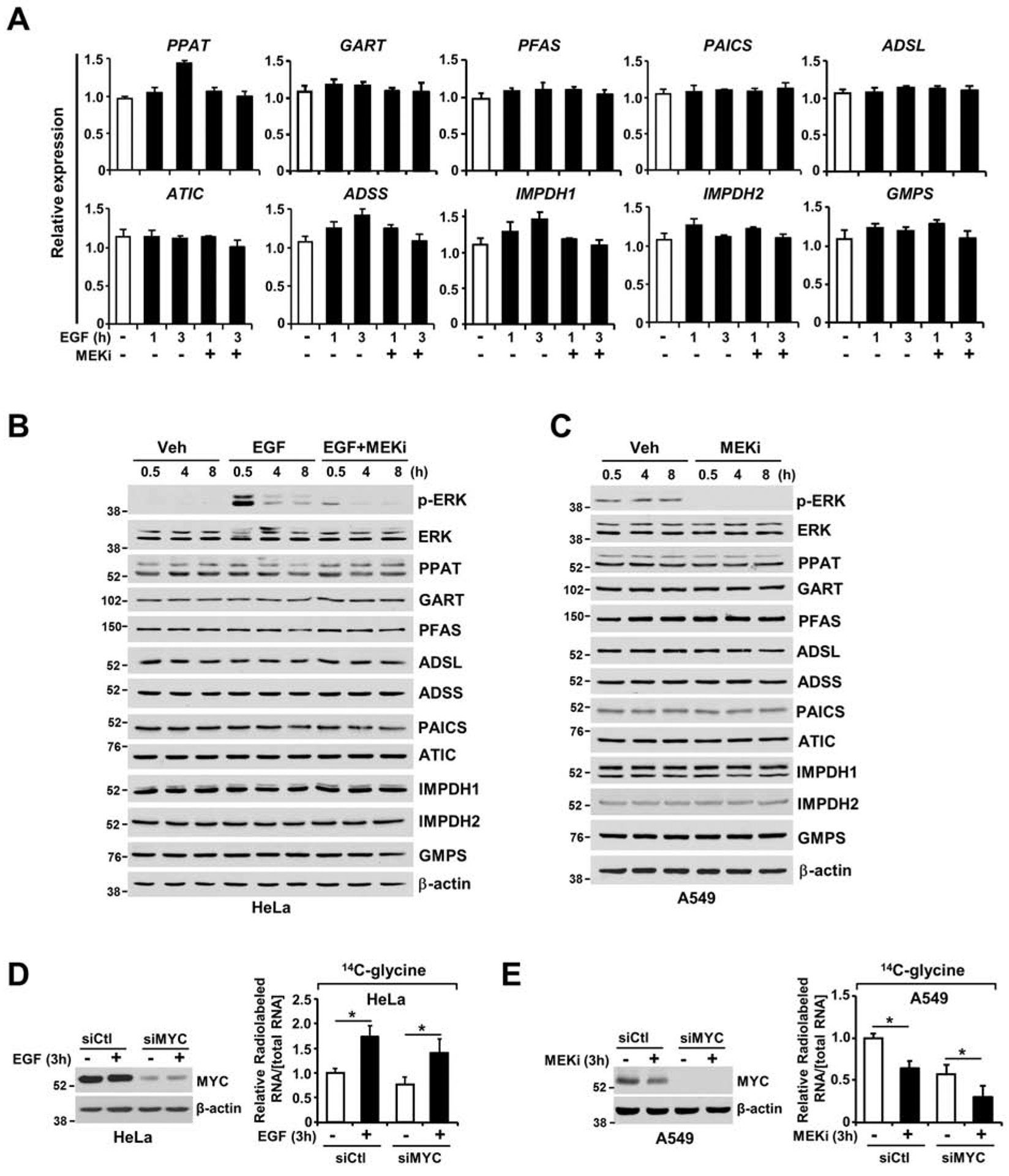
(A) Purine enzyme mRNA levels, as measured by qRT-PCR, in serum-starved HeLa cells (15 hours) and stimulated or not with EGF (50 ng/ml) in the presence or absence of U0126 (MEKi, 10 μM) for 1 or 3 hours. (B) Immunoblot showing all purine enzymes measured in (A). HeLa cells were serum starved for 15 hours and stimulated or not with EGF (50 ng/ml) over a time course (0.5, 4 or 8 hours) in the presence or absence of U0126 (MEKi, 10 μM). (C) Immunoblot showing all the purine enzymes measured in (A) and (B). A549 cells were serum starved for 15 hours and treated with vehicle or U0126 (MEKi, 10 μM) over a time course (0.5, 4 or 8 hours). (D, E) c-MYC knockdown did not abolish EGF- and MEK-dependent regulation of de novo purine synthesis in HeLa (D) and A549 (E) cells. Relative incorporation of radiolabels from 14C-glycine (3 hours of labeling) into RNA from HeLa and A549 cells 48 hours after transfection with c-MYC siRNAs or nontargeting controls (siCtl). Cells were serum starved (15 hours) and stimulated or not with EGF (50 ng/ml, 3 hours) (D) or were serum starved and treated with vehicle or MEKi (U0126, 10 μM) for 3 hours (E). The data are presented as the means±SEMs relative to unstimulated serum-starved HeLa cells (A). The data are plotted as the means ± SDs of biological triplicates (D, E). * P<0.05 by two-tailed Student’s t test for pairwise comparisons. The data are representative of at least three independent experiments (A-E).
The purine enzyme phosphoribosylglycinamidine synthase (PFAS) is a direct substrate of ERK2
Given the acute nature of purine synthesis regulation by ERK signaling, we hypothesized that metabolic enzymes within the de novo purine pathway could be modified posttranslationally in response to ERK activation. A systematic computational analysis using Scansite 4.0 (http://scansite3.mit.edu/) (Obenauer et al., 2003) performed with high stringency for all purine metabolic enzymes revealed phosphoribosylglycinamidine synthase (PFAS) as potentially phosphorylated by ERK1 (MAPK3) at T619, with an ERK docking site (Figure 4A and Table S3). To validate the putative ERK-mediated phosphorylation site in PFAS, we performed tandem mass spectrometry (MS/MS) analyses of PFAS immunopurified from EGF-stimulated cells in the presence or absence of an ERK inhibitor to identify potential ERK-regulated phosphorylation sites. Our analysis, with 82% coverage of the protein, identified three phosphorylation sites: S162, S215 and T619 (Figure 4B and Figures S3A–C). In addition, the phosphorylation sequence motifs are reasonably conserved among vertebrate PFAS orthologs (Figure 4B and Figure S3D). In vitro kinase assays with recombinant purified PFAS over a reaction time course showed that ERK1 and ERK2 directly phosphorylate PFAS (Figures S3E and S3F) and that T619 is the primary residue phosphorylated by ERK1/2 (Figure 4C). Furthermore, the fraction of PFAS molecules phosphorylated by ERK ranges between 19 to 29% depending on the timing of phosphorylation (Figure S3G). A phospho-PFAS antibody recognizing phosphorylated T619 on PFAS (Figure 4D and Figure S4A) established that EGF stimulated ERK-dependent phosphorylation of this PFAS site in HeLa cells (Figure 4E). We also confirmed endogenous ERK-dependent PFAS phosphorylation in response to EGF in HeLa cells (Figure 4F and Figures S4B and S4C). Given that the T619 sequence motif resembles motifs recognized by mTORC1 (Schalm and Blenis, 2002), we treated cells with rapamycin and showed that mTORC1 inhibition did not affect EGF-induced PFAS phosphorylation using immunoblotting (Figure 4G and Figure S4E) and LC-MS/MS-based phosphomapping of PFAS (Figure S4D). Moreover, knockdown of ERK1, ERK2 or both revealed that PFAS is phosphorylated mainly by ERK2 in HeLa and BRAF-driven cancer cells (Figures 4H and S4F). These results indicate that the ERK2 signaling activation induced by growth signals leads to PFAS phosphorylation at the T619 site.
Figure 4. PFAS is a direct substrate of ERK2.
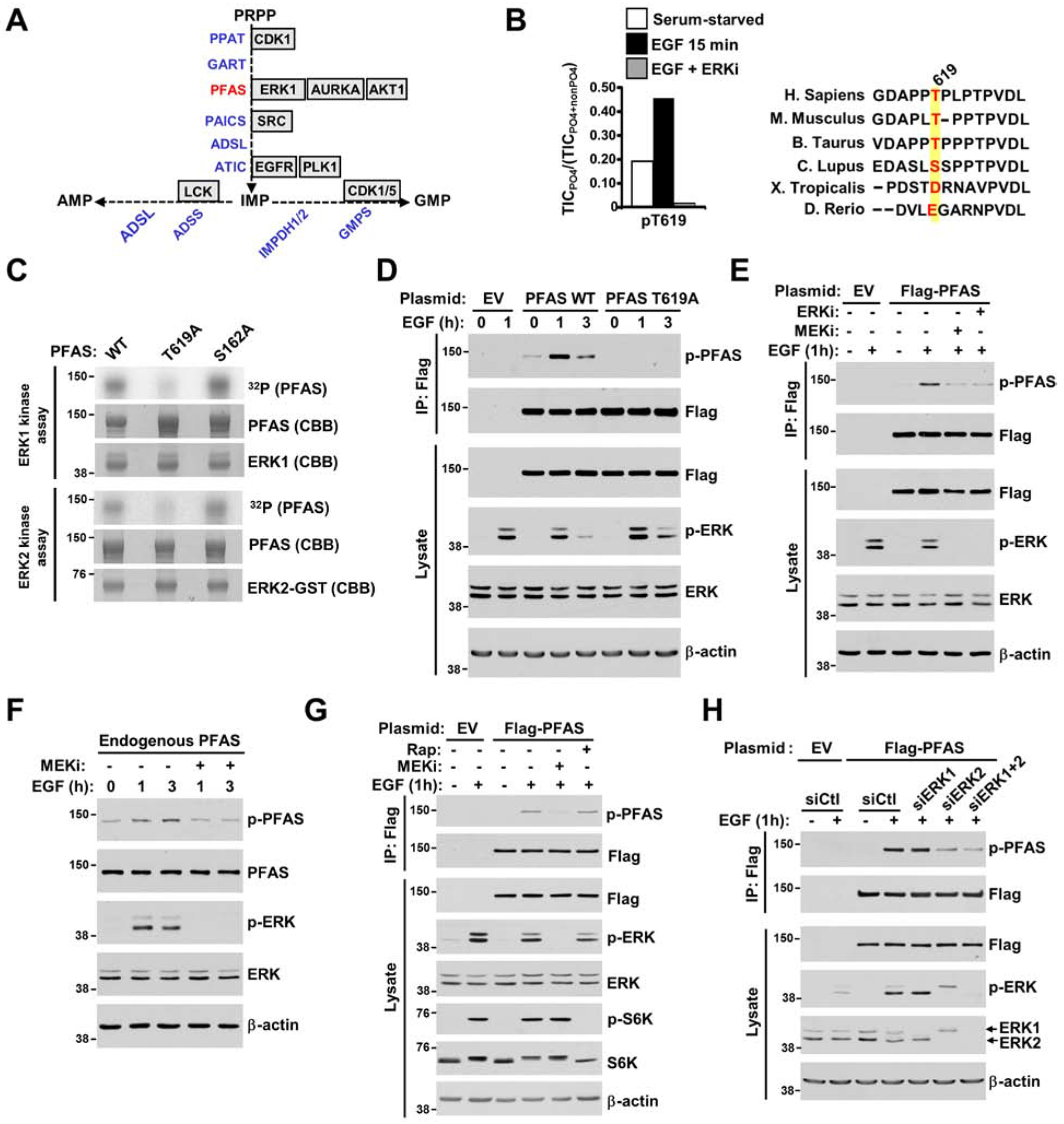
(A) Diagram depicting the purine enzymes predicted to be phosphorylated by canonical kinases based on a computational analysis with Scansite 4.0 software (see also Figure S3A). (B) Effects of EGF and SCH772984 on PFAS phosphorylation. FLAG-PFAS was immunopurified from serum-starved (15 hours) HEK293E cells treated for 30 min with DMSO or SCH772984 (ERKi, 1 μM) prior to stimulation with EGF (15 min, 50 ng/ml). The ratios of phosphorylated T619 peptides on PFAS to the total peptide levels, as measured by the total ion current (TIC) with LC-MS/MS, are plotted. Alignment showing the sequence conservation of T619 among PFAS orthologs (see also Figures S3B, S3C, and S3D). (C) In vitro kinase assays with active ERK1, ERK2 and PFAS variants (wild-type and mutant (T619A, S162A)) were performed with a 10 min reaction time and analyzed by autoradiography (see also Figures S3E, S3F, and S3G). (D) HeLa cells expressing empty vector (EV) or wild-type (WT) or T619A versions of FLAG-PFAS were serum-starved (15 hours) and stimulated with EGF (1 hour, 3 hours, 50 ng/ml). FLAG-immunoprecipitates were immunoblotted with a phospho-PFAS-T619 antibody (see also Figure S4A and S4B). (E) Cells were treated as in (D) and pretreated for 30 min with U0126 (MEKi, 10 μM) or SCH772984 (ERKi, 1 μM) prior to EGF stimulation (1 hour, 50 ng/ml). (F) HeLa cells were serum-starved (15 hours) and pretreated for 30 min with U0126 (MEKi, 10 μM), prior to 1-hour or 3-hour stimulation with EGF (50 ng/mL) (see also Figure S4C). (G) Cells were treated as in (D) and pretreated for 30 min with U0126 (MEKi, 10 μM) or rapamycin (Rap, 20 nM) prior to EGF stimulation (1 hour, 50 ng/ml) (see also Figures S4D and S4E). (H) Cells were treated as in (D), but were transfected with siRNAs targeting ERK1, ERK2, or both, or nontargeting controls (siCtl) (see also Figure S4F).
T619 in PFAS is required to mediate ERK2-dependent purine synthesis stimulation
To determine whether ERK2 signaling activation is sufficient to stimulate PFAS phosphorylation and purine synthesis, HEK293E cells were transfected with either constitutively active ERK2 (ERK2-CA) or catalytically inactive ERK2 (ERK2-KD). Expression of ERK2-CA, but not ERK2-KD, increased PFAS phosphorylation and de novo purine synthesis (Figure 5A and Figure S5A). Then, to determine whether ERK2 could promote de novo purine synthesis through PFAS phosphorylation at T619, we first established HEK293E, HeLa and A549 cell lines with CRISPR-Cas9-mediated PFAS knockout (ΔPFAS) and stably reconstituted these cells with PFAS cDNA encoding wild-type and phosphomutant PFAS [S215A/T619A (2A) or T619A]. Lack of PFAS resulted in defective de novo purine synthesis, as measured via a 14C-glycine assay (Figures S5B and S5C), and rendered the ΔPFAS cells auxotrophic for inosine and/or hypoxanthine and dependent on the purine salvage pathway for cell growth maintenance (Figure S5D). To determine whether these ERK-dependent phosphorylation sites on PFAS contribute to de novo purine synthesis regulation by ERK signaling, we employed 15N-glutamine and 15N-13C2-glycine isotopic tracing techniques to measure de novo purine synthesis activity. Compared with the corresponding cells expressing the wild-type enzyme, both HEK293E and HeLa cells cultured in dialyzed serum (Figures 5B, 5C, and Figure S5E) and A549 cells cultured without serum (Figure 5D) that expressed the phosphomutants (2A and T619A) exhibited diminished basal de novo purine synthesis activity. Moreover, while MEK inhibition decreased the abundance of purine intermediates in wild-type PFAS-expressing cells, it failed to decrease the levels of purine intermediates in cells expressing the phosphodeficient PFAS mutants (Figures 5B–D and Figure S5E). Additionally, EGF stimulated an increase in the amount of the 15N-label from glutamine in purine intermediates in ΔPFAS HeLa cells reconstituted with wild-type PFAS, but this stimulation was prevented in PFAS mutant-expressing cells (Figure S5F). To determine whether ERK signaling influences the nucleic acid production by regulating PFAS-dependent de novo purine synthesis stimulation, we measured the incorporation of carbon from glycine or formate into nucleic acids in ΔPFAS cells reconstituted with either wild-type or phosphomutant (2A or T619A) PFAS. HeLa cells cultured in dialyzed serum (Figure 5E and Figure S5G) and A549 cells cultured without serum (Figure 5D and Figure S5H) that expressed phosphodeficient mutants exhibited lower levels of 14C incorporation into nucleic acids than wild-type cells, and showed minimal response to MEK inhibition (Figures 5E, 5F, and Figures S5G, S5H). These results demonstrate that ERK activation stimulates nucleic acid synthesis by ERK-mediated PFAS phosphorylation.
Figure 5. The T619 site in PFAS is required for ERK-dependent stimulation of the de novo purine synthesis pathway.
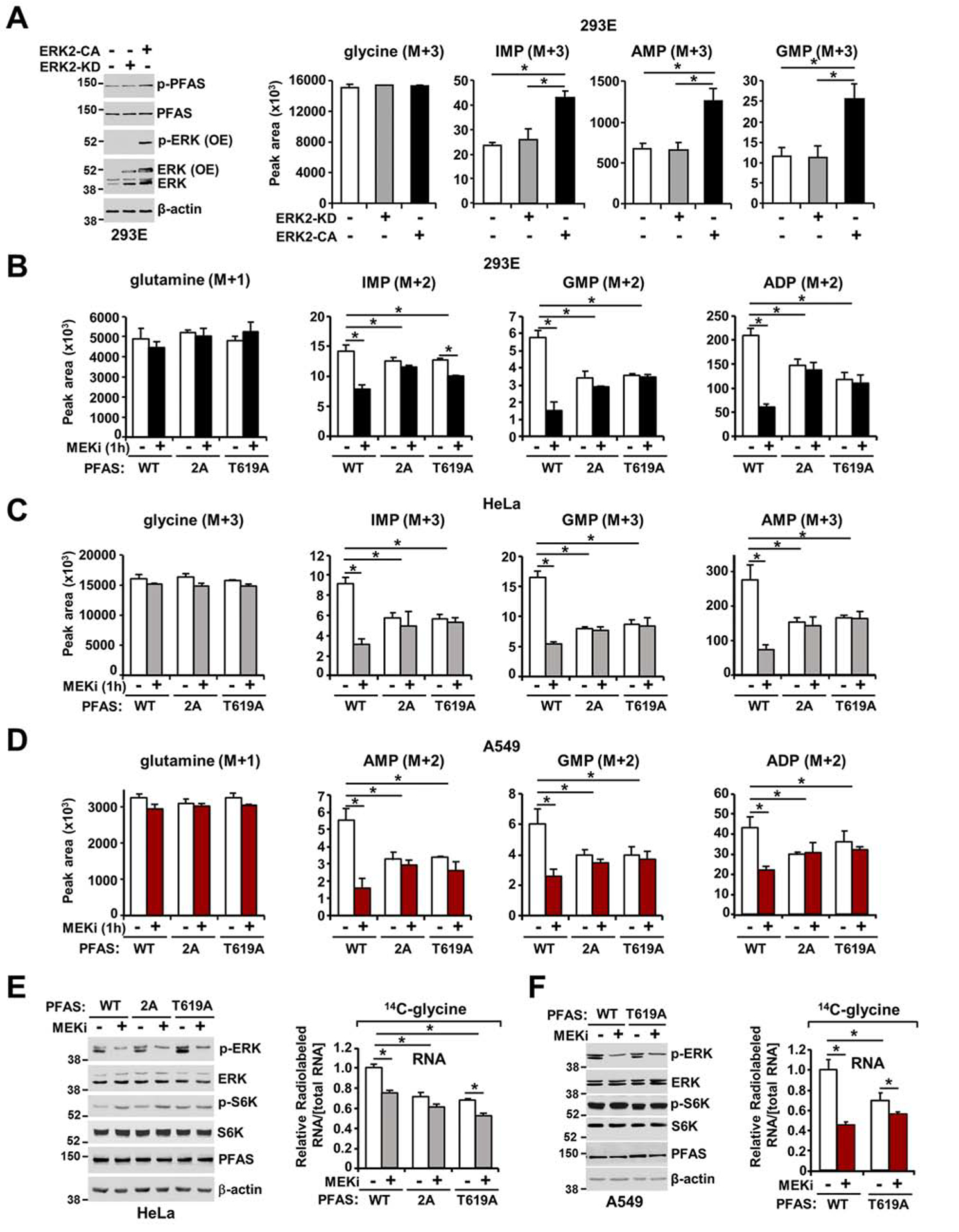
(A) Immunoblots and normalized peak areas of 15N-13C-labeled purine intermediates measured in HEK293E cells transfected with either empty vector, constitutive active ERK2 (ERK2-CA), or catalytically inactive ERK2 (ERK2-KD) (see also Figure S5A). (B) Normalized peak areas of 15N-labeled metabolites measured in HEK293E ΔPFAS cells stably reconstituted with PFAS-WT, PFAS-S215A/T619A (2A) or PFAS-T619A cultured in dialyzed serum for 15 hours, isotopically labeled with 15N-(amide)-glutamine for 1 hour, and treated with vehicle (DMSO) or U0126 (MEKi, 10 μM). (C) Normalized peak areas of 15N-13C-labeled metabolites measured in HeLa ΔPFAS cells stably reconstituted with PFAS-WT, PFAS-2A or PFAS-T619A cultured in dialyzed serum for 15 hours, isotopically labeled with 15N-13C2-glycine for 1 hour and treated as in (B) (see also Figures S5E and S5F). (D) A549 ΔPFAS cells stably reconstituted with PFAS-WT, PFAS-S215A/T619A (2A) or PFAS-T619A cultured without serum for 15 hours, isotopically labeled with 15N-(amide)-glutamine for 1 hour, and treated as in (B). (E) HeLa ΔPFAS cells stably reconstituted with PFAS-WT, PFAS-S215A/T619A (2A) or PFAS-T619A were cultured in dialyzed serum, treated with vehicle or U0126 (MEKi, 10 μM) and concurrently labeled with 14C-glycine for 2 hours. Incorporation of the specific radiolabel into RNA was measured and normalized to the total concentration of RNA (see also Figure S5G). (F) A549 ΔPFAS cells stably reconstituted with PFAS-WT or PFAS-T619A were serum starved for 15 hours and treated as in (E) (see also Figure S5H). (B-D) CRISPR-mediated PFAS knockout validated in Figures S5B, S5C and S5D. The data are plotted as the means ± SDs of biological triplicates. * P<0.05 by one-way ANOVA with Tukey’s post hoc test for multiple pairwise comparisons (A-F). (A-F) The data are representative of at least two independent experiments.
ERK-mediated PFAS phosphorylation is required for cell and tumor growth
To assess the importance of ERK-mediated PFAS phosphorylation in cell proliferation, we assessed the proliferation of ΔPFAS HEK293E and A549 cells stably reconstituted with wild-type PFAS or phosphodeficient PFAS mutants. The T619A PFAS mutants restored cell proliferation; however, cells expressing the mutant variants exhibited a decrease in cell number and cell size relative to wild-type PFAS-expressing cells (Figure 6A and Figures S6A, S6B). Additionally, expression of the T619A PFAS variant in RAS- and BRAF-driven cancer cells (A549 and A375) resulted in a ~30–40% decrease in colony number in a soft agar assay relative to that of the corresponding cells expressing wild-type PFAS (Figure 6B), further indicating that ERK-mediated PFAS phosphorylation contributes to cell growth. Kaplan-Meier curves indicate that high PFAS, KRAS, and BRAF expression is associated with unfavorable overall survival in patients with different cancer types (Figure S6C). To determine the role of PFAS regulation by ERK signaling in tumorigenesis, we injected athymic nude mice subcutaneously with ΔPFAS A549 cells reconstituted with either wild-type PFAS or T619A PFAS mutant. PFAS mutant-expressing cells resulted in significantly reduced tumor growth compared to that of the wild-type counterpart-expressing cells (Figure 6C). These results suggested that ERK-mediated PFAS phosphorylation is required to promote tumor growth in RAS-driven cancer settings. The databases of The Cancer Genome Atlas (TCGA) program c-bioportal revealed hot spot mutations (P618R, P618D, and T619D with a frameshift (FS) mutation) in the AIRS domain of PFAS (Figure S6D). Interestingly, the T619D phosphomimetic mutation was observed in stomach adenocarcinoma and renal clear cell carcinoma patients (Table S4), suggesting that continuous phosphorylation of the T619 residue could provide a selective growth advantage for these cancers by increasing the purine synthesis rate. To test this hypothesis, we reconstituted HEK293E cells ΔPFAS with either wild-type or T619D PFAS and measured cell proliferation and basal purine synthesis flux into nucleotides and nucleic acids. The T619D PFAS-expressing cells had significantly increased de novo purine synthesis and cell proliferation under serum-free or low serum conditions compared to their wild-type counterparts (Figures 6D, S6E, and S6F). Collectively, these data demonstrate that the ERK2-mediated phosphorylation of PFAS at T619 boosts purine synthesis activity to enhance proliferation and tumor growth (Figure 6E).
Figure 6. ERK2-mediated PFAS phosphorylation stimulates RAS-dependent tumor growth.
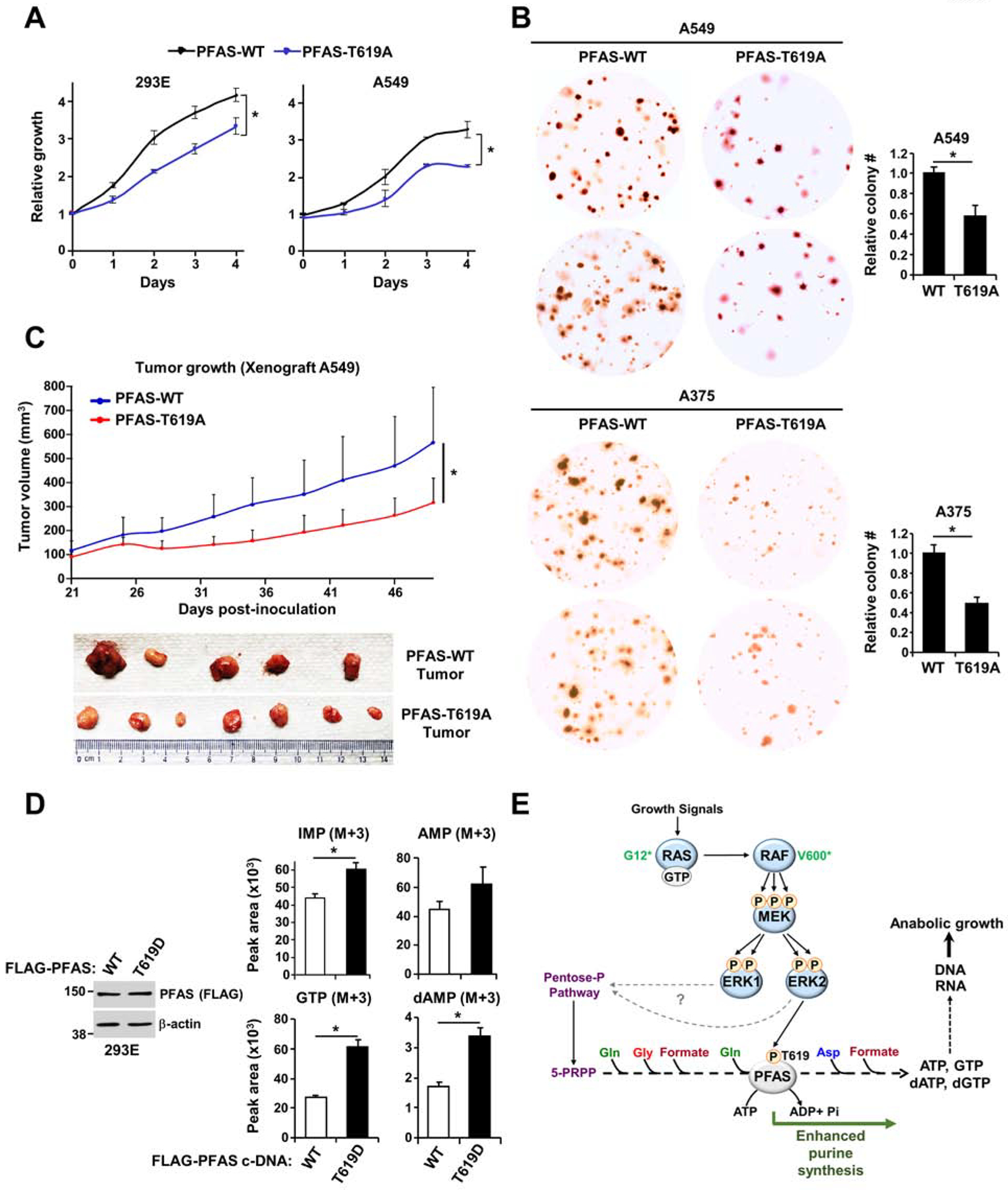
(A) Cell proliferation was measured in HEK293E and A549 ΔPFAS cells stably reconstituted with PFAS-WT or PFAS-T619A (see also Figures S6A and S6B). (B) Soft agar colony formation assay with A549 and A375 ΔPFAS cells stably reconstituted with PFAS-WT or PFAS-T619A. Cell images are at 3x magnification. The relative colony counts are presented as the means ± SDs of biological triplicates. (C) A549 ΔPFAS cells (5 × 106) stably-reconstituted with either WT or T619A-PFAS were injected subcutaneously into athymic nude mice (n = 5 per group). After tumor onset (100 mm3), tumor growth was monitored over time. (D) Normalized peak areas of 15N-13C-labeled purine intermediates measured in HEK293E ΔPFAS cells reconstituted with PFAS-WT or PFAS-T619D cultured in the absence of serum for 15 hours and isotopically labeled with 15N-13C2-glycine for 1 hour (see also Figures S6D, S6E and S6F). (E) Model of purine synthesis stimulation by the RAS-ERK signaling pathway. * P<0.05 by two-tailed Student’s t test for pairwise comparisons (A-D). The data are representative of at least two independent experiments.
DISCUSSION
Aberrant RAS-ERK pathway activation is closely linked to various types of human diseases (Bugaj et al., 2018; Dhillon et al., 2007; Lawrence et al., 2008). The metabolic effects downstream of ERK can be divided broadly into two different categories: rapid short-term and delayed long-term effects. The RAS-ERK pathway has been shown to regulate cancer cell metabolism via long-term mechanisms mediated by the transcription factor c-MYC (Kamphorst et al., 2013; Santana-Codina et al., 2018; Stine et al., 2015; Tsai et al., 2012; White, 2013). In this study, we demonstrate that ERK can function as a direct molecular link between growth-promoting and oncogenic signals and exert rapid control over de novo purine synthesis. Based on our computational analysis, other signaling events may regulate PFAS or other purine enzymes to control nucleotide synthesis. Notably, ERK2 signaling is required to enhance flux through de novo purine synthesis via PFAS phosphorylation, but without a measurement of baseline PFAS activity, we are not able to determine whether the T619 site is required for basal activity of PFAS.
Because of the high level of similarity between ERK1 and ERK2, we cannot eliminate the possibility that ERK1 can redundantly phosphorylate PFAS at T619 site in settings that have not been tested in this study.
Based on the metabolic tracing analysis, our work suggests that specific ERK2-mediated T619 phosphorylation enhances PFAS activity but does not exclude potential additional regulatory nodes from ERK1 and ERK2 signaling that may affect auxiliary pathways to support purine synthesis activity. For example, glucose metabolism has been shown to be one of the major metabolic hubs regulated by the RAS-ERK pathway (Cairns et al., 2011; Stine et al., 2015; Yang et al., 2012). Despite not detecting any significant changes in glucose uptake after short-term activation of ERK signaling, we showed that ERK signaling acutely regulates levels of pentose phosphate pathway intermediates, which can affect substrate availability for de novo nucleotide synthesis. Future studies are needed to dissect the mechanisms by which ERK signaling rapidly modulates glucose metabolism and the pentose phosphate pathway, and how these pathways influence nucleotide synthesis. The control of PFAS by ERK signaling, downstream of glucose metabolism, represents an interesting point of regulation since most of the reported metabolic regulation by signaling pathways occurs at rate-limiting steps (Ben-Sahra et al., 2013; Qian et al., 2018). PFAS and the five other purine biosynthetic enzymes have been shown to compartmentalize into metabolic complexes, namely the purinosomes (Pedley and Benkovic, 2017). The purinosome structure is a naturally occurring example of an enzyme cluster in which reaction rate is boosted by increased enzyme concentration and possibly substrate channeling to enhance flux through de novo purine synthesis (Pareek et al., 2020). PFAS was shown to be an essential component and a marker of the purinosome structure (French et al., 2016; Zhao et al., 2013). A study showed that AMPK activation is associated with the specific removal of PFAS from purinosome structures and a decrease in purine synthesis activity (Schmitt et al., 2016). It is tempting to speculate that the phosphorylation of PFAS by ERK signaling could modulate purinosome assembly and influence substrate channeling, to enhance de novo purine synthesis (Sweetlove and Fernie, 2018; Zhao et al., 2013). This type of regulation could bridge the modulatory nature of signaling pathways with the efficiency of metabolism.
Additionally, our study shows that ERK2-dependent PFAS stimulation is critical for cell proliferation, anchorage-independent growth and tumor growth in RAS-driven cancer. This direct connection between ERK2 and purine synthesis could represent a metabolic liability in cancers exhibiting high ERK activity levels. The sequence containing T619 is well-conserved across mammalian species, and interestingly, it is replaced by a negatively charged residue (T619D) that mimics phosphorylation and possibly increases de novo purine synthesis in a subset of cancers. Co-targeting ERK and purine synthesis with specific metabolic inhibitors could be a new way to more effectively treat tumors arising in patients with RAS or RAF mutations exhibiting ERK signaling hyperactivation.
Taken together, our findings indicate that ERK serves as a molecular link to effectively fine-tune purine metabolism in human cells. This finding supports previous studies demonstrating regulation of cellular metabolism by the RAS signaling network (DeNicola and Tuveson, 2009; Slack, 2017). This direct regulation of purine synthesis by ERK2 complements the ERK1-dependent synthesis of pyrimidine nucleotides (Graves et al., 2000), for the coordinated biosynthesis of nucleic acids. The integral role that the ERK2-PFAS axis plays in controlling cellular metabolism and function, and the conservation of the PFAS-T619 site across vertebrates, suggest that PFAS phosphorylation by ERK2 orthologs may be an ancestral mechanism to modulate the nucleotide pool in response to growth factor availability changes (Li et al., 2011).
By identifying this temporally-regulated metabolic mechanism, our study provides a better understanding of the dynamic metabolic landscape regulated by RAS-ERK signaling. In addition to the various biological processes stimulated by the ERK pathway, purine synthesis stands as a main anabolic process that responds rapidly to growth factors and oncogenic signals. We anticipate that further investigation of the metabolic effects of the ERK-PFAS axis will provide great insight into the emerging nexus of cancer, diabetes, immune and neurodegenerative disorders in humans (Colucci-D’Amato et al., 2003; Kimmelman, 2015; Ozaki et al., 2016; Richards et al., 2001; Samatar and Poulikakos, 2014).
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Issam Ben-Sahra (issam.ben-sahra@northwestern.edu).
Materials Availability
There are restrictions to the availability of the phospho-PFAS antibody due to the lack of an external centralized repository for its distribution and our need to maintain the stock in our lab. We are glad to share phospho-PFAS antibody with reasonable compensation by requestor for its processing and shipping. All other unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
The dataset supporting the current study have been deposited in a public repository (Mendeley Data: http://dx.doi.org/10.17632/px6zm7ms9r.3).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Details of cell lines used have been provided under section “Cell Lines and Tissue Culture” and details of mice used have been provided under “Xenograft Experiments”.
Cell Lines and Tissue Culture
HeLa, A549, SK-MEL-28, A375 and Panc1 cell lines were obtained from American Type Culture Collection (ATCC). MEFs and HEK293E were kindly provided by Drs. David Kwiatkowski (Brigham and Women’s Hospital) and John Blenis (Weill Cornell Medicine), respectively. MEFs, HeLa, HEK293E, A549, SK-MEL-28, A375 and Panc1 cell lines were cultured in DMEM with 4.5 g.l−1 glucose (CellGro), 10% FBS, 37°C, and 5% CO2. Media of cells knockout for PFAS (ΔPFAS) were supplemented with 50 μM inosine, 100 μM hypoxanthine, and 1mM Penicillin/Streptomycin. ΔPFAS cells stably-transfected with vector or FLAG-PFAS (WT, S215A/T619A or T619A) were maintained in media supplemented with 500 μg/mL G418 (Geneticin). Media were changed to serum free (or 10% dialyzed media according to given experiments) and without inosine/hypoxanthine 15 hours prior to labeling and metabolite or nucleic acid extraction. When needed, a TC-20 Automated Cell Counter (Bio-Rad) was used to count viable cells.
Xenograft Experiments
All animal procedures and studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Northwestern University. All experiments were performed in accordance with relevant guidelines and regulations. A549 cells were deleted for PFAS by CRISPR/Cas9 and stably reconstituted with the constructs expressing either Flag-PFAS WT or Flag-PFAS-T619A. Five female athymic nude mice (5 weeks old) were subcutaneously injected with 5 ×106 A549 cells-expressing either PFAS-WT or PFAS-T619A. When tumors were palpable (~100 mm3), tumor size was monitored twice per week until the end point.
METHOD DETAILS
Constructs, siRNA, and CRISPR/Cas9
Full-length human PFAS cDNA in pCMV6-entry vector containing the FLAG-tag was purchased from Origene (RC217795). QuikChange Site-Directed Mutagenesis method was employed to generate the following mutations: PFAS-S162A, PFAS-T619A, PFAS-T619D, PFAS-S215A/T619A from the PFAS wild-type plasmid, and ERK2-L73P/S151D from ERK2 wild-type plasmid with the KOD Extreme Hot Start DNA Polymerase followed by DpnI digestion. Mutation accuracy was confirmed by Sanger sequencing and the plasmid variants were amplified with Hispeed Qiagen plasmid maxi kit. For transfection, 20 μg of plasmid DNA was used for 10-cm dishes of ~ 60% confluent cells. Lipofectamine 3000 was used to transfect Hela and A549 cells as per manufacturer’s instructions. For transfection of HEK293E cells, polyethylenimine (PEI) was used in a ratio of 2:1 to the plasmids. 48 hours post-transfection, cells were either harvested or selected for generation of stable cell lines with Geneticin. For siRNA experiments, Myc (L-003282-02-0005), ERK1 (L-003592-00-0005) and ERK2 (L-003555-00-0005) siRNAs (On TARGETplus SMARTpool) were purchased from Dharmacon and used at 32 nM (final concentration) with 5 to 6 μL of Lipofectamine RNAimax (Thermo fisher scientific # 13778150) (6-well) for each condition.
To generate PFAS knockout in HEK293E, HeLa and A549 cells, a sgRNA sequence targeting the second exon of PFAS was cloned into the PX458 CRISPR (Addgene, Plasmid # 48138) vector using the following oligos: Forward: GCGAACATAGAAGTGAAGGAC, Reverse: GTCCTTCACTTCTATGTTCGC. After two days transfection, GFP positive cells were sorted into 96-well plates as single cell per well. Clonal cells were screened by immunoblotting with anti-PFAS antibody.
Generation of the phospho-PFAS-T619 antibody by Cell Signaling Technology
Rabbit polyclonal antibody was raised against phosphorylated PFAS (Thr619) by injecting rabbits with a synthetic, KLH-conjugated phosphopeptide corresponding to residues surrounding Thr619 of human PFAS protein. Rabbits were immunized subcutaneously with 0.5 mg of antigen in Complete Freund’s Adjuvant, followed by five boost injections of antigen (0.25 mg) in Incomplete Freund’s Adjuvant every three weeks thereafter. Crude bleeds were collected 10 days following the last boost injection, and antibody was purified from crude bleeds using peptide affinity chromatography.
Immunoblotting and immunoprecipitation
For protein extract preparation, cells were lysed in ice-cold Triton lysis buffer (40 mM HEPES, pH 7.4, 120 mM NaCl, 1 mM EDTA, 1% Triton X-100, 10 mM glycerol 2-phosphate, 10 mM sodium pyrophosphate, 0.5 mM sodium orthovanadate and 50 mM NaF; 1 μM Microcystin-LR and protease inhibitor cocktail was added just before lysis of cells) and incubated on ice for 30 min. Lysates were centrifuged at 20,000 × g for 15 min at 4°C. Protein concentrations were measured by Bradford assay and protein lysates were normalized for each experiment.
For western blots, equal amounts of proteins (10 to 15 μg) from each samples were separated by standard SDS-PAGE and transferred onto nitrocellulose blotting membranes. Membranes were blocked in 5% milk in TBST (50 mM Tris pH 7.4, 150 mM NaCl, 0.1% Tween 20) and incubated overnight at 4°C with the indicated antibodies followed by incubation with HRP-tagged anti-mouse or anti-rabbit antibodies for one hour. Bands were revealed with chemiluminescent substrates (Thermo fisher scientific # 34577, #34095). In all immunoblotting experiments, β-actin was used as a loading control. For immunoprecipitations, FLAG-tagged PFAS was immunoprecipitated from cell lysates by incubation with anti-FLAG antibodies for overnight with prior clearance with protein A/G beads. Protein A/G agarose was added to the incubation mixture for an additional 1–2 hours to pull down antibody-protein complexes. Immunoprecipitates were washed three times with lysis buffer, resuspended in 2x Laemmli buffer, incubated at 95°C on heat block for 5 min and separated on SDS-PAGE.
ERK kinase assays on immunopurified PFAS and measure of stoichiometry
To perform ERK kinase assays on PFAS, recombinant proteins MAPK3 (ERK1) and MAPK1 (ERK2, GST tagged on N-terminus) were purchased from MRC-PPU, University of Dundee, UK. To purify PFAS, 5×106 HEK293E ΔPFAS cells were seeded in 15-cm dishes and 24 hours later, transfected with either FLAG-PFAS wild-type, FLAG-PFAS-S162A or FLAG-PFAS-T619A. 48 hours of post-transfection, cells were treated with the MEK inhibitor (U0126) with a concentration of 10 μM for 1 hour. Cells were lysed according to the Immunoblot section, PFAS and its variants were immunoprecipitated with anti-FLAG antibodies and protein A/G beads. The immunoprecipitated PFAS and its variants were washed twice with the ERK reaction buffer (50 mM Tris-HCl pH 7.5, 1 mM DTT, 0.1 mM EGTA, 10 mM magnesium acetate) and incubated with 2 μg or indicated quantities of ERK1 or ERK2. This assay was carried out in 30 μl reaction buffer with 100 μM [γ−32P]-ATP at 30°C for the indicated times (Figure S3E, S3G) or 10 min (Figure 4C and Figure S3F). The PFAS bands on gel were excised, and stoichiometry of incorporation of 32P-radioactivity into protein was quantified as previously described (Hastie et al., 2006). Reactions were stopped by addition of SDS sample buffer followed by a 5-min incubation at 95°C. Samples were separated on standard SDS-PAGE and developed by autoradiography.
Phospho-peptide analysis of PFAS by mass spectrometry
Phospho-peptide analysis by mass spectrometry was performed as described previously28. For protein phosphorylation site mapping of PFAS, FLAG-PFAS immunoprecipitates were mixed with Laemmli Sample buffer and separated by SDS-PAGE. The gel was stained with Coomassie blue followed by destaining (Coomassie destain: 15% methanol, 10% glacial acetic acid) the gel overnight at 4°C with at least five solvent changes in the first 2 hours, and subsequently the PFAS band was excised. Gel slices were reduced with 55 mM DTT, alkylated with 10 mM iodoacetamide, and digested overnight with TPCK modified trypsin (Promega) at pH 8.3. Peptides were extracted, dried (by SpeedVac), and resuspended in 10 μL of 50% ACN, 6% TFA. Peptide mixture was loaded on TiO2 tips to enrich the phosphopeptides and LC-MS/MS was performed using an Easy-nLC nanoflow HPLC (Thermo Fisher Scientific) with a self-packed 75 mm id × 15 cm C18 column connected to a hybrid linear ion trap LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) in the data-dependent acquisition and positive ion mode at 300 nL/min.
Measurements of glucose, glycine, hypoxanthine, serine, and formate uptakes
Glucose uptake was measured as the incorporation of radiolabeled 1,2-3H-2-deoxy-D-glucose. Cells were grown in 6-well plates and serum starved for 15 hours and treated with vehicle (DMSO), EGF, MEKi or ERKi for 3 hours. Cells were then washed twice with glucose free media, and incubated for 10 min with 900 μl glucose free media with the effector concentrations, subsequently added 0.1 mM cold 2-deoxy-D-glucose and 0.5 μCi 1,2-3H-2-deoxy-D-glucose for 5 min. Cells were then placed on ice to stop the reactions, washed twice with ice-cold PBS, and lysed in 0.5 ml of 1% Triton X-100 lysis buffer. 0.4 ml lysate was counted in 3 ml of scintillation liquid using a Beckman LS6500 scintillation counter.
In a similar procedure (mentioned above), uptake of glycine, hypoxanthine, serine and formate were measured where concentration of cold glycine, hypoxanthine, serine, and formate were 0.3, 0.1, 0.3 and 1mM, respectively, and for each case, 1 μCi of specific activity (14C-glycine, 3H-hypoxanthine, 14C-serine or 14C-formate) was added for 5 min. Cells were then placed on ice to stop the reactions, washed twice with ice-cold PBS, and lysed in 0.5 ml of 1% Triton X-100 lysis buffer. 0.4 ml lysate was counted in 5 ml of scintillation fluid using a Beckman LS6500 scintillation counter. All conditions were performed in biological triplicates, normalized to protein concentration, and reproduced at least twice.
Metabolite profiling for steady state and targeted flux analyses
To determine the relative abundances of intracellular metabolites, extracts were prepared and analyzed by LC-MS/MS. Briefly, for targeted steady-state samples, metabolites were extracted on dry ice with 4-mL 80% methanol (−80°C), as described previously (Weinberg et al., 2019; Yuan et al., 2019). Insoluble material was pelleted by centrifugation at 3000 g for 5 min, followed by two subsequent extractions of the insoluble pellet with 0.5-ml 80% methanol, with centrifugation at 20,000g for 5 min. The 5-ml metabolite extract from the pooled supernatants was dried down under nitrogen gas using the N-EVAP (Organomation, Inc, Associates).
For 13C4-15N-aspartic acid tracing studies, cells seeded in biological triplicate (~80% confluent), were washed once with serum-free DMEM and then incubated in the same medium containing 4-mM 13C4-15N-aspartic acid for 1 h and metabolites were extracted as described in the steady-state studies (Weinberg et al., 2019).
For targeted 15N-glutamine and 13C2-15N-glycine isotopic tracing experiments, cells seeded in biological triplicate (~80% confluent) were incubated in serum-free medium or dialyzed serum containing medium for 15 hours, with a medium change 2 h prior to extraction. For 15N-glutamine (amide-labeled) flux studies, cells were washed once with glutamine-free DMEM and then incubated in the same medium containing 4 mM 15N-(amide)-glutamine and associated treatments specific for the given experiments for 1 hour21. For 13C2-15N- glycine flux studies, cells were washed once with serine and glycine free DMEM and then incubated in DMEM containing 1 mM 13C2 15N-glycine for 1 hour. Metabolites were extracted as described in this protocol (Yuan et al., 2019).
U-14C-glycine, L-3-14C-serine, D-14C-glucose, 3H-hypoxanthine and 14C-formate incorporation into RNA and DNA
Cells (~80% confluent) were serum starved (or in dFBS medium) for 15 hours, then treated and labeled as indicated in the figures. Cells were labeled with 2-μCi of either U-14C-glycine, 3-14C-serine, 14C-glucose, 3H-hypoxanthine and 14C-formate. Cells were harvested and RNA or DNA was isolated using Allprep DNA/RNA kits according to the manufacturer’s instructions and quantified using a spectrophotometer. 70 μl of eluted DNA or 30 μl of eluted RNA were added to scintillation vials and radioactivity was measured by liquid scintillation counting and normalized to the total DNA or RNA concentrations, respectively. All conditions were analyzed with biological triplicates and representative of at least two independent experiments.
Bioinformatics analysis and sequence alignments
The search for putative sites on the purine synthesis enzymes to be phosphorylated by specific protein kinases was performed with SCANSITE 4.0 (https://scansite4.mit.edu/4.0/#home), with “Scan a Protein” as a feature with high stringency. The data are presented in Table S3. Putative protein phosphorylation sites can be further investigated by evaluating evolutionary conservation of the site sequence. PFAS amino acid sequences and alignments for different species were obtained from Uniprot (http://www.uniprot.org/).
mRNA expression analysis
Total RNA was extracted (RNeasy kit, #74104, Qiagen) from HeLa cells at the indicated times and 1 μg of RNA was used for reverse-transcription (SuperScript III kit, #1808005, Invitrogen). The resulting cDNAs were then diluted (1:20) in nuclease free water and quantified by real-time PCR with Bio-Rad Advanced universal SYBR green supermix (#1725271) using a Biorad CFX Connect Real-Time PCR Detection System. The human primers used for the qPCR are described in Table S2. The expression levels of each transcript were normalized with the RPLP0 transcript level and data were represented as fold change relative to the average of control samples. Data are representative of least from two independent experiments.
Cell size and proliferation
Cells were plated in 96-well plates at density of 3000 cells per well. For proliferation of wild-type or ΔPFAS cells (HEK293E, HeLa, and A549) cells were seeded in media containing 10% (HEK293E and HeLa) or 1% dFBS (A549) and were grown for 5 days in the presence or absence of inosine (50 μM) and hypoxanthine (100 μM). Relative cell proliferation was measured using CellTiter-Glo® every 24 h. Cell size was measured in solution, following trypsinization, using a Countess II Automated Cell Counter (Thermo Fisher Scientific) and data points were extracted with the WebPlotDigitizer software. For cell proliferation, the data were normalized to day 0. For proliferation of cells stably-reconstituted with wild-type or mutant PFAS (T619A), cells were seeded in media containing 10% (HEK293E) or 1% dFBS (A549) in 96-well plates. Cell proliferation was measured using 0.1% crystal violet (or CellTiter-Glo®) every 24 hours for 5 days. All proliferation assays were performed in biological duplicates seeded in technical quadruplicates for each condition, and are representative of at least two independent experiments.
Soft agar colony formation assay
ΔPFAS A549 cells stably-reconstituted with PFAS-wild type and PFAS-T619A were employed for soft agar assay. 5000 cells in 1.5 ml of growth media (DMEM with 1% dFBS) containing 0.3% agarose were seeded in 12-well plates with a bottom layer of 0.6% agarose (1ml, prepared in same media as top layer). An additional 0.5 ml growth media was poured over top layer, once a week. After 7 weeks, colonies were stained with 0.2 ml of 1 mg/ml of thiazolyl Blue Tetrazolium Bromide solution for 5 hours. Number of Colonies was quantified using ImageJ software (Schneider et al., 2012) and represented as average of technical triplicates. Images shown in the manuscript (Figure 6B) are representative of biological triplicates from three independent experiments.
Survival analysis of cancer patients with differentially expressed PFAS
Kaplan-Meier Plotter (KM plotter, http://kmplot.com/analysis/) compiles publicly available data from repositories such as Gene Expression Omnibus (GEO), European Genome-Phenome Archive (EGA), and The Cancer Genome Atlas’ (TCGA). With a total of 64 datasets from 21 cancer types. The largest datasets include breast (n=6,234), ovarian (n=2,190), lung (n=3,452), and gastric (n=1,440) cancer. PostgreSQL server handles the gene expression and clinical data concomitantly. To examine the prognostic value of KRAS, BRAF and PFAS mRNA expression simultaneously, Pan-cancer RNA-seq database was used to evaluate the overall survival of cancer patients (n=7462). The two patient cohorts showing differential gene expression were compared by a Kaplan-Meier survival plot, and the hazard ratio with 95% confidence intervals and logrank P values were calculated.
Publicly available PFAS mutation data
The PFAS mutation dataset from multiple cancer types from 73,103 patients (75,955 samples in 253 studies) was downloaded from c-BioPortal for cancer genomics. (https://www.cbioportal.org/).
QUANTIFICATION AND STATISTICAL ANALYSIS
For two pairwise comparisons, two-tailed Student’s t tests were performed in Microsoft Excel 2016. One-way ANOVA followed by Tukey’s post hoc tests were performed in GraphPad Prism 7.0 to determine differences between each group when more than two conditions were present. All error bars represent standard deviation (SD), except for Figure 3A in which error bars represent standard error of the mean (SEM). A value of P < 0.05 was considered significant. The number of independent experiments done is described in each figure legend.
Supplementary Material
Table S3. Computational scan for protein motifs with high stringency in the purine synthesis pathway (Scansite 4.0). Related to Figure 4.
Table S4. 548 PFAS Mutations: Including 176 duplicate mutations in patients with multiple samples. Related to Figure 6.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (E10) Mouse mAb antibody | Cell Signaling Technology | Cat# 9106S; RRID: AB_331768 |
| p44/42 MAPK (Erk1/2) Antibody | Cell Signaling Technology | Cat# 9102S; RRID: AB_330744 |
| Phospho-p70 S6 Kinase (Thr389) (108D2) Rabbit mAb | Cell Signaling Technology | Cat# 9234S; RRID: AB_2269803 |
| Rabbit Anti-p70 S6 Kinase Monoclonal Antibody | Cell Signaling Technology | Cat# 2708S; RRID: AB_390722 |
| c-MYC antibody | Cell Signaling Technology | Cat#9402; RRID: AB_2151827 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat#7076; RRID: AB_330924 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat#7074; RRID: AB_2099233 |
| Phospho-PFAS T619 Rabbit antibody | Cell Signaling Technology | In development |
| PFAS Antibody | Bethyl Laboratories | A304–218A; RRID: AB_2620417 |
| ATIC Antibody | Bethyl Laboratories | A304–272A; RRID: AB_2620468 |
| PPAT Antibody | Proteintech Group Inc | 15401–1-AP; RRID: AB_2166532 |
| GART Antibody | Proteintech Group Inc | 13659–1-AP; RRID: AB_2107937 |
| ADSL Antibody | Proteintech Group Inc | 15264–1-AP; RRID: AB_2225668 |
| ADSS Antibody | Proteintech Group Inc | 16373–1-AP; RRID: AB_2225676 |
| PAICS Antibody | Proteintech Group Inc | 12967–1-AP; RRID: AB_10638449 |
| IMPDH1 Antibody | Proteintech Group Inc | Cat#22092–1-AP |
| IMDPH2 Antibody | Proteintech Group Inc | 12948–1-AP; RRID: AB_2127351 |
| GMPS Antibody | Proteintech Group Inc | 16376–1-AP; RRID: AB_2232412 |
| β-actin Antibody | Sigma-Aldrich | A5316; RRID: AB_476743 |
| Anti-FLAG M2 | Sigma-Aldrich | F1804; RRID: AB_262044 |
| Bacterial and Virus Strains | ||
| NEB Stable Competent E. coli | NEB | C3040 |
| DH5 alpha | Fisher Scientific | 18258012 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| U0126 MEK inhibitor | Tocris Biosciences | 1144 |
| AZD6244 | Selleckchem | S1008 |
| SCH772984 | Selleckchem | S7101 |
| EGF (human recombinant) | Life Technologies (Gibco) | PHG0311 |
| L-glutamine | Corning | MT25005Cl |
| 13C2-15N-glycine | Sigma-Aldrich | 489522 |
| 13C4-15N-aspartic acid | Sigma-Aldrich | 607835–250MG |
| L-glutamine-(amide-15N) | Sigma-Aldrich | 490024 |
| thiazolyl Blue Tetrazolium Bromide | Sigma-Aldrich | M5655 |
| DMSO | Sigma-Aldrich | D2650–100ML |
| 2-mercaptoethanol | Sigma-Aldrich | M6250–100ML |
| Hypoxanthine | Sigma-Aldrich | H9636–1G |
| Inosine | Sigma-Aldrich | I4125–1G |
| 1,2-3H-2-deoxy-D-Glucose | Perkin-Elmer | NET328A250UC |
| U-14C-glycine | Perkin-Elmer | NEC276E050UC |
| U-14C-glucose | Perkin-Elmer | NEC042V250UC |
| 3-14C-serine | Perkin-Elmer | NEC853050UC |
| 3H-hypoxanthine | Perkin-Elmer | NET177001MC |
| [γ−32P]-ATP | Perkin-Elmer | BLU002A100UC |
| 14C-formate | American Radiolabeled Chemicals | ARC 0163A |
| Polyethylenimine (PEI) | Polysciences | 23966–2 |
| Opti-MEM media | Thermo Fisher | 31985062 |
| Glutamine-free DMEM | Gibco | 11960–044 |
| Geneticin Selective Antibiotic | Fisher Scientific | 10131035 |
| Lipofectamine 3000 Transfection reagent | Invitrogen | L3000008 |
| Glucose-free DMEM | Invitrogen | 11966025 |
| Lipofectamine RNAi max | Invitrogen | 13778150 |
| DpnI | ||
| SuperSignal™ West Femto Chemiluminescent | Thermo Fisher scientific | PI34096 |
| SuperSignal™ West Pico PLUS | Thermo Fisher scientific | PI34580 |
| Protein A/G AGAROSE 3ML/PK | Thermo Scientific | 20421 |
| Recombinant Protein - MAPK3 (ERK1) | MRC-PPU | 2 – 379 |
| Recombinant Protein - MAPK1 (ERK2) | MRC-PPU | 2 – 360 |
| DMEM | Corning CellGro | 10–017-CV |
| Glycine (and serine)-free DMEM | US Biologicals | D9800–16 |
| Fetal bovine serum (FBS) | Sigma-Aldrich | F2442 |
| Dialyzed FBS (dFBS) | Sigma-Aldrich | F0392 |
| Microcystin-LR | Fisher Scientific | ALX350012C500 |
| Protease inhibitor cocktail | Sigma-Aldrich | P8340 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAZEN | 74104 |
| HiSpeed Plasmid Maxi Kit (25) | QIAZEN | 12663 |
| AllPrep DNA/RNA Mini Kit | QIAZEN | 80204 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | G7573 |
| KOD Extreme Hot Start DNA polymerase kit | Merck Millipore | 71975–3 |
| Deposited Data | ||
| A study of Ali, Sahu et al_Unprocessed file | Mendeley Data | http://dx.doi.org/10.17632/px6zm7ms9r.3 |
| Experimental Models: Cell Lines | ||
| HEK293E cells | John Blenis lab (Weill Cornell Medicine) | N/A |
| HeLa cells | ATCC | CCL-2 |
| A549 cells | ATCC | CCL-185 |
| SKMEL-28 cells | ATCC | HTB-72 |
| Panc1 cells | ATCC | CRL-1469 |
| Mouse Embryonic Fibroblasts (MEFs) | David Kwiatkowski (Brigham and Women’s Hospital) | N/A |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| See Table S3 for list of primers used in this study | Integrated DNA Technologies | |
| siRNA targeting siMYC | Dharmacon | L-003282-02-0005 |
| Non-targeting siRNA pool | Dharmacon | D-001810-10-20 |
| Recombinant DNA | ||
| PFAS Human Tagged ORF Clone | Origene | RC217795 |
| pCMV6-Entry Tagged Cloning Vector | Origene | PS100001 |
| pLenti-III-PGK vector | Applied Biological Materials | G305 |
| pCMV6-PFAS-S162A | This study | NA |
| pCMV6-PFAS-S215A | This study | NA |
| pCMV6-PFAS-T619A | This study | NA |
| pCMV6-PFAS-T619D | This study | NA |
| pCMV6-PFAS-S215A-T619A | This study | NA |
| pLenti-III-PGK-PFAS | This study | NA |
| pLenti-III-PGK-PFAS-S215A-T619A | This study | NA |
| p3xFlag-CMV7-Erk2-L73P-S151D (ERK2-CA) | This study | NA |
| p3xFlag-CMV7-Erk2 KR (ERK2-KD) | Addgene | 39224 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | RRID: SCR_003070 |
| SCANSITE 4.0 | Obenauer et al., 2003 | https://scansite4.mit.edu/4.0/#cite |
| Prism 7 | Prism | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Multipurpose Scintillation Counter | Beckman | Model: LS 6500 |
| Microplate Reader | Tecan | Infinite M1000 Pro |
| Nitrocellulose membrane 0.2 μm | GE Healthcare | 10600001 |
Highlights.
ERK signaling rapidly stimulates de novo purine synthesis Phosphoribosylglycinamidine synthase (PFAS) is phosphorylated by ERK2 at T619 Mutation of PFAS-T619 abolishes ERK2-dependent purine synthesis stimulation ERK2-mediated PFAS phosphorylation is required for cell and tumor growth
ACKNOWLEDGMENTS
The authors thank B. D. Manning and G. Hoxhaj for helpful discussion and comments and Z. Djabari and M. Yuan, for technical assistance. This works was supported by the Lynn Sage (I.B.-S.) and LAM Foundations LAM0127C01-18 (I.B.-S.), grants from the National Institutes of Health (NIH) R00CA194192 (I.B.-S.), R01GM135587 (I.B.-S.), 5P01CA120964 (J.M.A.) and 5P30CA006516 (J.M.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
All authors reviewed the manuscript and declare no competing financial interests.
REFERENCES
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Howell JJ, Asara JM, and Manning BD (2013). Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science (New York, N.Y.) 339, 1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, and Manning BD (2016). mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, and Tebbutt NC (2012). A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investigational new drugs 30, 1216–1223. [DOI] [PubMed] [Google Scholar]
- Brunet A, Roux D, Lenormand P, Dowd S, Keyse S, and Pouyssegur J (1999). Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. The EMBO journal 18, 664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugaj LJ, Sabnis AJ, Mitchell A, Garbarino JE, Toettcher JE, Bivona TG, and Lim WA (2018). Cancer mutations and targeted drugs can disrupt dynamic signal encoding by the Ras-Erk pathway. Science (New York, N.Y.) 361, eaao3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, and Mak TW (2011). Regulation of cancer cell metabolism. Nature reviews. Cancer 11, 85–95. [DOI] [PubMed] [Google Scholar]
- Cargnello M, and Roux PP (2011). Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiology and molecular biology reviews : MMBR 75, 50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci-D’Amato L, Perrone-Capano C, and di Porzio U (2003). Chronic activation of ERK and neurodegenerative diseases. BioEssays : news and reviews in molecular, cellular and developmental biology 25, 1085–1095. [DOI] [PubMed] [Google Scholar]
- Dang CV (2013). MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harbor perspectives in medicine 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ (1995). Transcriptional regulation by MAP kinases. Molecular reproduction and development 42, 459–467. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, and Tuveson DA (2009). RAS in cellular transformation and senescence. European journal of cancer (Oxford, England : 1990) 45 Suppl 1, 211–216. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, and Kolch W (2007). MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290. [DOI] [PubMed] [Google Scholar]
- Downward J (2003). Targeting RAS signalling pathways in cancer therapy. Nature reviews. Cancer 3, 11–22. [DOI] [PubMed] [Google Scholar]
- Erez A, and DeBerardinis RJ (2015). Metabolic dysregulation in monogenic disorders and cancer - finding method in madness. Nature reviews. Cancer 15, 440–448. [DOI] [PubMed] [Google Scholar]
- French JB, Jones SA, Deng H, Pedley AM, Kim D, Chan CY, Hu H, Pugh RJ, Zhao H, Zhang Y, et al. (2016). Spatial colocalization and functional link of purinosomes with mitochondria. Science (New York, N.Y.) 351, 733–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassian AR, Metallo CM, Coloff JL, Stephanopoulos G, and Brugge JS (2011). Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Genes Dev 25, 1716–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves LM, Guy HI, Kozlowski P, Huang M, Lazarowski E, Pope RM, Collins MA, Dahlstrand EN, Earp HS 3rd, and Evans DR (2000). Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature 403, 328–332. [DOI] [PubMed] [Google Scholar]
- Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, et al. (2008). Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nature genetics 40, 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halilovic E, and Solit DB (2008). Therapeutic strategies for inhibiting oncogenic BRAF signaling. Current opinion in pharmacology 8, 419–426. [DOI] [PubMed] [Google Scholar]
- Hastie CJ, McLauchlan HJ, and Cohen P (2006). Assay of protein kinases using radiolabeled ATP: a protocol. Nature protocols 1, 968–971. [DOI] [PubMed] [Google Scholar]
- Hoxhaj G, Ben-Sahra I, Lockwood SE, Timson RC, Byles V, Henning GT, Gao P, Selfors LM, Asara JM, and Manning BD (2019). Direct stimulation of NADP(+) synthesis through Akt-mediated phosphorylation of NAD kinase. Science (New York, N.Y.) 363, 1088–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humpton TJ, Alagesan B, DeNicola GM, Lu D, Yordanov GN, Leonhardt CS, Yao MA, Alagesan P, Zaatari MN, Park Y, et al. (2019). Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer discovery 9, 1268–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, and Rabinowitz JD (2013). Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proceedings of the National Academy of Sciences of the United States of America 110, 8882–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkhoff E, Houben R, Loffler S, Troppmair J, Lee JE, and Rapp UR (1998). Regulation of c-myc expression by Ras/Raf signalling. Oncogene 16, 211–216. [DOI] [PubMed] [Google Scholar]
- Kimmelman AC (2015). Metabolic Dependencies in RAS-Driven Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research 21, 1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AN, and Fan TW (2015). Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43, 2466–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange-Carter CA, and Johnson GL (1994). Ras-dependent growth factor regulation of MEK kinase in PC12 cells. Science (New York, N.Y.) 265, 1458–1461. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Jivan A, Shao C, Duan L, Goad D, Zaganjor E, Osborne J, McGlynn K, Stippec S, Earnest S, et al. (2008). The roles of MAPKs in disease. Cell research 18, 436–442. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, and Schlessinger J (2010). Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Liu J, and Zhang C (2011). Evolutionary history of the vertebrate mitogen activated protein kinases family. PloS one 6, e26999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI, and Dang CV (2008). Global regulation of nucleotide biosynthetic genes by c-Myc. PloS one 3, e2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza MC, Er EE, and Blenis J (2011). The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 36, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugan AK, Grieco M, and Tsuchida N (2019). RAS Mutations in Human Cancers: Roles in Precision Medicine. Seminars in cancer biology. [DOI] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, and Yaffe MB (2003). Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 31, 3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki KI, Awazu M, Tamiya M, Iwasaki Y, Harada A, Kugisaki S, Tanimura S, and Kohno M (2016). Targeting the ERK signaling pathway as a potential treatment for insulin resistance and type 2 diabetes. Am J Physiol Endocrinol Metab 310, E643–E651. [DOI] [PubMed] [Google Scholar]
- Pareek V, Tian H, Winograd N, and Benkovic SJ (2020). Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science (New York, N.Y.) 368, 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedley AM, and Benkovic SJ (2017). A New View into the Regulation of Purine Metabolism: The Purinosome. Trends in biochemical sciences 42, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov A, Zehorai E, rocaccia S, and Seger R (2011). The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochimica et biophysica acta 1813, 1619–1633. [DOI] [PubMed] [Google Scholar]
- Qian X, Li X, Tan L, Lee JH, Xia Y, Cai Q, Zheng Y, Wang H, Lorenzi PL, and Lu Z (2018). Conversion of PRPS Hexamer to Monomer by AMPK-Mediated Phosphorylation Inhibits Nucleotide Synthesis in Response to Energy Stress. Cancer discovery 8, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JD, Dave SH, Chou CH, Mamchak AA, and DeFranco AL (2001). Inhibition of the MEK/ERK signaling pathway blocks a subset of B cell responses to antigen. Journal of immunology (Baltimore, Md. : 1950) 166, 3855–3864. [DOI] [PubMed] [Google Scholar]
- Ryan MB, Der CJ, Wang-Gillam A, and Cox AD (2015). Targeting RAS-mutant cancers: is ERK the key? Trends in cancer 1, 183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samatar AA, and Poulikakos PI (2014). Targeting RAS-ERK signalling in cancer: promises and challenges. Nature reviews. Drug discovery 13, 928–942. [DOI] [PubMed] [Google Scholar]
- Santana-Codina N, Roeth AA, Zhang Y, Yang A, Mashadova O, Asara JM, Wang X, Bronson RT, Lyssiotis CA, Ying H, et al. (2018). Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nature communications 9, 4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalm SS, and Blenis J (2002). Identification of a conserved motif required for mTOR signaling. Current biology : CB 12, 632–639. [DOI] [PubMed] [Google Scholar]
- Schmitt DL, Cheng YJ, Park J, and An S (2016). Sequestration-Mediated Downregulation of de Novo Purine Biosynthesis by AMPK. ACS chemical biology 11, 1917–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, and Nevins JR (2000). Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 14, 2501–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul YD, and Seger R (2007). The MEK/ERK cascade: from signaling specificity to diverse functions. Biochimica et biophysica acta 1773, 1213–1226. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, and Cantley LC (2006). Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441, 424–430. [DOI] [PubMed] [Google Scholar]
- Slack C (2017). Ras signaling in aging and metabolic regulation. Nutr Healthy Aging 4, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, and Dang CV (2015). MYC, Metabolism, and Cancer. Cancer discovery 5, 1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetlove LJ, and Fernie AR (2018). The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nature communications 9, 2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, and Kuo MT (2012). Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res 72, 2622–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa E, Ali ES, Sahu U, and Ben-Sahra I (2019). Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, and Thompson CB (2012). Signaling in control of cell growth and metabolism. Cold Spring Harbor perspectives in biology 4, a006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, Gao P, Helmin KA, Abdala-Valencia H, Sena LA, et al. (2019). Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E (2013). Exploiting the bad eating habits of Ras-driven cancers. Genes & development 27, 2065–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ (2007). Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochimica et biophysica acta 1773, 1285–1298. [DOI] [PubMed] [Google Scholar]
- Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, and Lu Z (2012). ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nature cell biology 14, 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, et al. (2007). Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clinical cancer research : an official journal of the American Association for Cancer Research 13, 1576–1583. [DOI] [PubMed] [Google Scholar]
- Yuan M, Kremer DM, Huang H, Breitkopf SB, Ben-Sahra I, Manning BD, Lyssiotis CA, and Asara JM (2019). Ex vivo and in vivo stable isotope labelling of central carbon metabolism and related pathways with analysis by LC-MS/MS. Nature protocols 14, 313–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, and Liu HT (2002). MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell research 12, 9–18. [DOI] [PubMed] [Google Scholar]
- Zhao H, French JB, Fang Y, and Benkovic SJ (2013). The purinosome, a multi-protein complex involved in the de novo biosynthesis of purines in humans. Chemical communications (Cambridge, England) 49, 4444–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. Computational scan for protein motifs with high stringency in the purine synthesis pathway (Scansite 4.0). Related to Figure 4.
Table S4. 548 PFAS Mutations: Including 176 duplicate mutations in patients with multiple samples. Related to Figure 6.
Data Availability Statement
The dataset supporting the current study have been deposited in a public repository (Mendeley Data: http://dx.doi.org/10.17632/px6zm7ms9r.3).