

Abstract
Neurodevelopmental disorders (NDDs) affect 7–14% of all children in developed countries and are one of the leading causes of lifelong disability. Epigenetic modifications are poised at the interface between genes and environment and are predicted to reveal insight into NDD etiology. Whole-genome bisulfite sequencing was used to examine DNA cytosine methylation in 49 human cortex samples from 3 different NDDs (autism spectrum disorder, Rett syndrome, and Dup15q syndrome) and matched controls. Integration of methylation changes across NDDs with relevant genomic and genetic datasets revealed differentially methylated regions (DMRs) unique to each type of NDD but with shared regulatory functions in neurons and microglia. NDD DMRs were enriched within promoter regions and for transcription factor binding sites with identified methylation sensitivity. DMRs from all 3 disorders were enriched for ontologies related to nervous system development and genes with disrupted expression in brain from neurodevelopmental or neuropsychiatric disorders. Genes associated with NDD DMRs showed expression patterns indicating an important role for altered microglial function during brain development. These findings demonstrate an NDD epigenomic signature in human cortex that will aid in defining therapeutic targets and early biomarkers at the interface of genetic and environmental NDD risk factors.
Keywords: autism spectrum disorders, DNA methylation, epigenetics, microglia, neurodevelopmental disorders
Introduction
Neurodevelopmental disorders (NDDs) are one of the leading causes of lifelong disability. NDDs, which include autism spectrum disorders (ASDs), intellectual disabilities, attention-deficit/hyperactivity disorder, cerebral palsy, Down, and fetal alcohol syndromes, affect 7–14% of all children in developed countries (Miller et al. 2016). Current treatments consist of labor intensive and expensive behavioral therapies, which are often combined with drug therapies to treat comorbid symptoms such as anxiety. The lifetime cost for a person with ASD, one type of NDD, ranges from $1.2–4.7 million (Buescher et al. 2014) and can cost an additional $5.5 million in caregiver time compared with a neurotypical child (Dudley and Emery 2014). Combined with the rising diagnosis rates of ASD and other NDDs and the limited available treatments, there is an urgent need to accelerate the discovery and development of novel NDD therapeutics and early interventions.
The clinical diversity of idiopathic ASD parallels the genetic complexity of the disorder, which includes hundreds of rare risk variants and potentially thousands of common risk variants (Sanders et al. 2015; De La Torre-Ubieta et al. 2016; The Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium 2017). Monogenic disorders with symptoms that overlap ASD are commonly used to gain insight into the more complex idiopathic ASD cases. For example, Rett syndrome (RTT) is an X-linked, dominant syndromic ASD/NDD caused by mutations in MECP2, affecting females. Duplication 15q11.2-q13.3 (Dup15q) syndrome is an ASD/NDD caused by a maternally inherited copy number variant that results in increased expression of the imprinted gene UBE3A. Recent exome sequencing studies focused on identifying rare, highly penetrant, de novo mutations in patients with ASD identified a number of high-confidence ASD genes with functions in neuronal synapses and transcriptional regulation (Ben-David and Shifman 2012; Sanders et al. 2015). Genetic and environmental risks are hypothesized to interact in the etiology of ASD (Vogel Ciernia and LaSalle 2016), and a multitude of early life environmental perturbations correlate with increased ASD risk, including maternal immune activation and in utero pollutant exposure (Lyall et al. 2014).
The disruption of epigenetic processes regulating brain development is a potential mechanism linking environmental and genetic risk. For example, transcription, DNA methylation, and histone acetylation analyses in postmortem ASD brain have consistently implicated gene pathways involved in synaptic development and immune function (Voineagu et al. 2011; Gupta et al. 2014; Ladd-Acosta et al. 2014; Nardone et al. 2014, 2017; Lin et al. 2016; Parikshak et al. 2016; Sun et al. 2016). However, most analyses of DNA methylation in human ASD brain have been limited to less than 2% of the total Cytosine-p-Guanine (CpG) sites in the human genome (Ladd-Acosta et al. 2014; Nardone et al. 2014, 2017) or a specific syndromic subset of ASD (Dunaway et al. 2016). Consequently, we examined CpG methylation signatures across the human genome using unbiased whole-genome bisulfite sequencing (WGBS) of human cortices from 3 different NDDs and matched controls. By leveraging cortical WGBS data of ASD, RTT, Dup15q, and matched controls, we analyzed regions of differential methylation to identify gene pathways and cell type-specific functions that are convergent and divergent across NDDs. NDD differentially methylated loci were enriched within promoters and for neuronal transcription factor binding sites (TFBSs) with known methylation sensitivity. NDD associated genes were enriched for genes known to be differentially expressed in brain in ASD and in several other neuropsychiatric disorders. Furthermore, we observed functional enrichments related to nervous system development and immune cell types, specifically microglia. Therefore, differential DNA methylation analysis captures gene pathways and cell types converging with both immune and neuronal perturbations in brain development and suggests novel therapeutic pathways for NDDs.
Methods
Sample Acquisition, DNA Isolation, and WGBS Library Preparation ASD BA9
Human cerebral cortex samples from Broadmann Area (BA) 9 were obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland. DNA was isolated using the QIAGEN Puregene kit (Qiagen, 158 667), and WGBS libraries were prepared as described previously (Dunaway et al. 2016). Briefly, 5 μg of DNA was fragmented to ~300 bp using 28 cycles of 15 s on/15 s off on a Diagenode Bioruptor. DNA was end-repaired using 1× T4 DNA ligase buffer, 400 μM dNTPs, 15 U T4 DNA polymerase (NEB), and 50 U PNK (NEB) for 30 min at 20 °C. After Polymerase Chain Reaction (PCR) purification (Qiagen), adenine bases were appended to the ends using 1× NEB 2 buffer, 200 μM dATP, and 15 U Klenow Fragment (3′ to 5′ exo-, NEB) for 30 min at 37 °C. After another DNA purification using the PCR MinElute kit (Qiagen), 3 μL of Illumina’s methylated sequencing adapters were attached using 1× ligase buffer and 5 μL Quick T4 DNA Ligase (NEB) for 30 min at room temperature. After a final PCR purification, 500 ng of library was bisulfite converted using Zymo’s EZ DNA Methylation-Direct kit according to the manufacturer’s instructions. The library was then amplified using 2.5 U PfuTurbo Cx Hotstart DNA Polymerase (Stratagene) for 12 cycles using Illumina’s standard amplification protocol. The library’s quality was assessed on a Bioanalyzer (Agilent) and sequenced (100 bp, single-ended) on an Illumina HiSeq 2000. Each biological sample was sequenced on a single lane.
Sample Acquisition, DNA Isolation, and WGBS Library Preparation RTT BA9
DNA was extracted from BA9 cortex from RTT and Control samples using the Zymo Duet Kit (Zymo). Two hundred nanogram of total genomic DNA was bisulfite converted using the EZ DNA Methylation-Lightning kit (Zymo), and 50 ng of converted DNA was used as input for library construction using the TruSeq DNA Methylation Kit (Illumina). Each sample was given a unique barcode and subjected to 14 cycles of PCR amplification. Final libraries were size selected with 2 rounds of KAPA Pure Bead selection: 0.7X Left and 0.65X Left/0.55X Right for a final library size distribution centered around 370–470 base pairs. Final libraries were assessed with Bioanalyzer (Agilent), quantified, and pooled, then 150 bp paired end sequencing was performed on 2 lanes of the HiSeqX (Illumina).
WGBS Sample Processing
Raw FASTQ files were chastity filtered and then trimmed using trim_galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore) to remove adapters and sequences at the 5′ and 3′ ends with methylation bias. Reads were then aligned to the human genome (hg38), deduplicated, and extracted to a CpG count matrix using Bismark (Krueger and Andrews 2011). QC/QA was performed using Bismark, FastQ screen (Wingett and Andrews 2018), and MultiQC (Ewels et al. 2016). Measures of global methylation were assessed from the Bismark output using a one-way analysis of variance (ANOVA) for diagnosis with adjustments for age for all comparisons and sex for the idiopathic ASD cohort.
DMRs and Blocks
Differentially methylated regions (DMRs) were called for diagnosis (NDD vs. Control) for each NDD separately using the R package dmrseq (Korthauer et al. 2018). This approach utilizes statistical inference, where smoothed methylation values are calculated for CpG sites in a manner that weights them based on their individual coverage. Bismark cytosine reports were processed to collapse strand-symmetric CpGs using bsseq (Hansen et al. 2012). CpGs from unmapped scaffolds and the mitochondrial chromosome were removed from subsequent analysis. For each NDD cohort, the respective cytosine reports were filtered to have at least one read of coverage per CpG across all samples. A generalized least squares regression model with a nested autoregressive correlated error structure for the effect of interest was used to estimate a region statistic for each candidate region. Empirical P-values were calculated by comparing observed region statistics to those generated from a permutation-based pooled null distribution. DMRs were identified using the default parameters, aside from setting the minimum number of CpGs to 4 for a region, a methylation proportion difference coefficient of at least 0.05, and a permutation P-value of P < 0.05 for significance testing without additional corrections. For all analyses, the covariate of age was directly adjusted for, and for the ASD analysis sex was also included as an adjustment.
BG Regions
Background (BG) regions for each NDD cohort were defined as all regions meeting the above criteria for candidate regions (at least one read of coverage per CpG across all samples and groups of at least 4 CpGs), which includes a difference between NDD and control samples for statistical testing. Consequently, the BG regions represent the testable candidate regions subjected to a permutation-based analysis for the identification of significant DMRs. BG regions are regions capable of showing a methylation difference that were subjected to statistical testing to identify DMRs, and thus they are also likely to be similarly affected by any inherent GC bias. For downstream enrichment testing, the 3 testable BG region sets were merged using bedtools to create a set of consensus BG regions, allowing for the use of a consistent BG across NDD cohorts. Individual smoothed methylation values for the DMRs were generated using bsseq (Hansen et al. 2012). Finally, blocks of differential methylation were identified using the recommended block parameters from the dmrseq vignette (Korthauer et al. 2018).
DMR Associated Genes
Genes associated with DMRs or BG regions were found using the bedtools closest function on all hg38 Ensembl annotated genes and a subsequent filter for genes within +/−10 kb from the start or end of each DMR. With this approach, a DMR is first assigned to a gene if it overlaps the gene body. If a DMR does not overlap a gene body, then it is assigned based on the closest gene (or genes in case of a tie) upstream or downstream, within the limit of 10 kb. To test for bias in the association between DMRs and genes of longer size, gene length (gene start to gene end in base pairs) was compared between NDD DMR associated genes, BG regions, and all Ensembl genes in hg38 using permutation testing. Random gene sets of the same number of genes as each DMR list were subsampled from either the BG regions or hg38 genes. The median gene length was then calculated for each sub-sample and used to create a null distribution (100 000 permutations) for comparison against the actual NDD DMR associated genes (see Supplementary Fig. S8). The empirical P-value was calculated as the sum of the number of sub-samples with median gene lengths greater than or equal to the median gene length of the DMR associated genes, divided by the number of permutations.
PCA
Principal components analysis (PCA) was performed using the prcomp and ggbiplot functions in R on the average of smoothed CpG methylation values within 20 kb windows tiled across the genome.
MethylCIBERSORT Cell-Type Deconvolution
Methylation levels from all BA9 cortex samples were extracted for CpGs showing unique cell type-specific methylation levels among glutamatergic neurons, GABAergic neurons, and glial cells (CpG sites used with at least one read/CpG for all samples; see Supplementary Fig. S2) (Kozlenkov et al. 2014). The methylation values from Kozlenkov et al. (2014) were used as a custom signature profile for input to CIBERSORT (Newman et al. 2015) (https://cibersort.stanford.edu) in an approach used previously to deconvolve WGBS data from tumor cells (Chakravarthy et al. 2018). MethylCIBERSORT was utilized to estimate the relative levels of distinct cell types within each WGBS sample by comparing the methylation patterns of the same CpGs between the samples and custom signature profile using machine learning linear support vector regression (Newman et al. 2015).
Average Methylation Analysis over Genomic Features
Average smoothed methylation values were calculated for CpG islands, shores (+/−2 kb from CpG islands), shelves (+/−2 kb from shores), open sea (intergenic CpG IsIands), gene bodies, and promoters. Gene bodies were assigned from transcription start site to transcription end site by the Ensembl hg38 annotation (http://www.ensembl.org/biomart). Promoters were taken from the Ensembl hg38 promoters database (Zerbino et al. 2015), and CpG Islands were taken from the R package annotatr (Cavalcante 2017) for hg38. Average methylation levels and average sequencing coverage were analyzed using a mixed model ANOVA with main effects for diagnosis, genomic feature, sex, the interaction between diagnosis and genomic feature, and the random effect of sample. Post hoc comparisons were made with Benjamini–Hochberg corrected t-tests.
GO Term Enrichment
Gene Ontology (GO) term enrichment on genomic regions was performed for DMRs using the R package GOfuncR (Grote 2018) for GO terms from the current version of the org.Hs.eg.db_3.6.0 package (March 2018). ASD, Dup15q, and RTT DMRs were compared with BG consensus regions using a hypergeometric test to compare the overlap between each DMR and gene extended regions (10 kb up and downstream of each hg38 Ensembl gene start and end site).
Region Overlap Enrichment and Datasets
Testing for the enrichment of DMRs within specific genomic contexts was performed using the Genomic Association Tester (GAT) (Heger et al. 2013) with a workspace defined by the consensus BG regions, isochores for hg38 (controls for G+C Bias during random sampling) and 100 000 permutations. Multiple comparisons were False Discovery Rate (FDR) corrected to P < 0.05. In all figures nonsignificant enrichments are shown in gray. Cell type open chromatin regions datasets were filtered to remove regions in common across cell types for either brain or non-brain samples using bedtools multiIntersectBed.
Cell-type DNase-seq and chromatin states were taken from prefrontal cortex (PFC) from the ENCODE portal (Ernst et al. 2011; Ernst and Kellis 2013; Roadmap Epigenomics Consortium et al. 2015) (http://www.roadmapepigenomics.org/). Human microglial ATAC-seq and PU.1 ChIP-seq datasets were taken from Gosselin et al. (2017). Human data in hg19 or mouse data were transferred to hg38 using LiftOver (Hinrichs et al. 2006).
Machine Learning Model of NDD DMR Diagnosis Prediction
Smoothed methylation values were extracted for a merged list of DMRs from all NDD analyses and corrected for sequencing batch effects using the ComBat function from the sva R package (Johnson et al. 2007). The caret R package was used to train a 5-fold cross-validated random forest model with 4 classes (ASD, Dup15q, RTT, and Control) from the ComBat corrected smoothed methylation values. In 5-fold cross-validation, the dataset is split into 5 groups, where for each of the 5 resampling iterations, the model is trained on 4 of the 5 groups and tested on the remaining fifth group (see diagram in Supplementary Fig. S4). The model was evaluated for overall prediction reliability of classifiers (kappa statistic), overall prediction accuracy, and prediction accuracy within each class. The kappa statistic, which assesses the validity of a model, is a measure of agreement between the actual diagnosis values and predicted diagnosis values and takes into account the expected accuracy by random chance. The kappa statistic can range from −1 to +1, and a value between 0.21 and 0.40 is considered fair, 0.41–0.60 is moderate, 0.61–0.80 is substantial, and 0.81–1.00 is nearly perfect interrater agreement (McHugh 2012). The kappa value was calculated using the following formula: Kappa = (observed accuracy−expected accuracy)/(1−expected accuracy).
For each of the 5 resampling folds, the observed accuracy was the number of correctly classified samples in the fold divided by the total number of samples in the fold. The expected accuracy for each fold was the sum of the marginal frequency of each of the classes in the fold divided by the total number of samples in the fold. The marginal frequency for each class was calculated by multiplying the number of actual samples in a class by the number of samples that were classified as that class and finally dividing by the total number of samples in the fold.
Gene Overlap Enrichment and Datasets
Testing for the enrichment of DMR associated genes within previously published gene lists was performed by permutation testing against a null distribution matched for gene length to account for the bias for expression of longer genes in the brain and in some types of ASD-linked mutations (Shohat and Shifman 2014). For each comparison, a random sub-sample of genes (same number of genes as in the target gene list) was selected from genes annotated to the BG regions so that the sub-sample matched the median gene length of the target gene list (see Supplementary Fig. S8). This median gene length-matched, random sub-sample was then overlapped with the DMR associated genes. This process was repeated for 100 000 permutations to create a null distribution of overlaps between randomly selected groups of BG genes with the same median gene length as the target list. The null distribution was then compared with the actual overlap between the DMR associated gene list and the target gene list and a permutation P-value calculated as the sum of the random sub-sampling of overlaps greater than the actual overlap divided by the number of permutations. Permutation testing for gene enrichment was conducted by randomly sampling a distribution of length-matched genes from among genes assigned to BG regions. Consequently, both length and GC content are similar between the random BG distribution and the target gene list for overlap. All gene lists, overlaps, statistics, and citations are in Supplementary Table S8.
Briefly, ASD and ID genetic risk genes were taken from SFARI (https://s1gene.sfari.org/autdb/GS_Statistics.do), SyS ID database (Kochinke et al. 2016) (https://sysid.cmbi.umcn.nl/table/human-gene-info), and recent exome sequencing studies (Gilissen et al. 2014; Iossifov et al. 2014; Sanders et al. 2015). pLI > 0.9 genes were identified from the Exome Aggregation Consortium (Lek et al. 2016) as genes with a probability of loss of function mutation >0.9, indicating that they are highly intolerant to genetic variation in the human population. Human ASD GWAS hits were taken from The Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium (2017) using a threshold of an association P-value < 0.05 and genes associated with ASD DMRs (Nardone et al. 2017). Lists of differentially expressed genes from postmortem human brain were obtained from published datasets for Dup15q syndrome (Parikshak et al. 2016), ASD (Gupta et al. 2014; Parikshak et al. 2016; Gandal et al. 2018a, 2018b), RTT syndrome (Lin et al. 2016), Alzheimer’s disease (Miller et al. 2013), bipolar disorder, alcoholism, schizophrenia (SCZ), irritable bowel disease, and major depressive disorder (Gandal et al. 2018a, 2018b). Human imprinted genes were obtained from http://www.geneimprint.com. Microglial gene lists were taken from several studies across different microglial isolation approaches and treatments (Hickman et al. 2013; Cronk et al. 2015; Erny et al. 2015; Holtman et al. 2015; Matcovitch-Natan et al. 2016; Hanamsagar et al. 2017; Keren-Shaul et al. 2017; Mattei et al. 2017; Zhao et al. 2017; Vogel Ciernia et al. 2018).
TFBS Analysis
NDD DMRs and BG regions were analyzed for TFBS using the MethMotif Batch Query tool (Xuan Lin et al. 2018) on the MethMotif website (https://bioinfo-csi.nus.edu.sg/methmotif/) for the human SK-N-SH neuronal cell line. This tool combines high-quality ENCODE ChIP-seq data with ENCODE WGBS data for individual cell types, creating a cell type-specific TFBS methylation status database (Xuan Lin et al. 2018). The frequency of TFBS motifs for each set of NDD DMRs was tested for enrichment relative to the frequency in consensus BG regions using a one-tailed Fisher’s Exact test with FDR correction to P < 0.1. Differences in average smoothed methylation levels between Control and NDD subjects within NDD DMRs containing the identified TFBSs were then examined using a 2-way ANOVA with factors for DMR (NDD DMR vs. BG region) and NDD (NDD vs. Ctrl) and sex as a covariate (for the ASD cohort).
Microglial Developmental Time Course Gene Expression
Raw count data per transcript were downloaded from GEO GSE99622 (Hanamsagar et al. 2017). Two samples were excluded due to low total read coverage of <200 000 total reads. The remaining samples were processed in EdgeR to counts per million for differential analysis between time points and sexes with FDR corrected P-values to 0.05. Reads per kilobase per million (RPKM) values were also calculated by normalizing to gene length for weighted gene co-expression network analysis (WGCNA).
WGCNA
WGCNA was performed using the WGCNA R package (version 1.61, 5 August 2017) (Langfelder and Horvath 2008, 2012). Average RPKM data from human brain single-cell RNA-seq collected across developmental time (gestational weeks) (GEO GSE104276) (Zhong et al. 2018) were used to construct cell type-specific developmental co-expression networks. Genes with zero variance in expression or a median absolute deviation of zero were removed from the analysis. Genes with a minimum RPKM of 0.001 or higher in at least one sample were kept for analysis for a total of 10 895 genes (unique Ensembl IDs). Values were then transformed to log2(RPKM+1) and clustered to visualize outliers (see Supplementary Fig. S6). A correlation matrix using biweight midcorrelation between all genes was then computed for all samples. An estimated soft thresholding power of 15 was used to derive a signed adjacency matrix with approximately scale-free topology (r2 fit indices >0.80) that was then transformed into a topological overlap matrix (TOM). The matrix 1-TOM was used as input to calculate co-expression modules with hierarchical clustering and a minimum module size of 20 genes. The resulting module eigengenes (MEs) were clustered based on their correlation, and modules were merged at a cutheight of 0.8 to produce co-expression modules and one additional module with genes that did not show module membership (gray). The expression profile of each module was further summarized by the ME, the first principle component of the module. Pearson’s correlation coefficients were used to calculate the correlation between sample traits (cell type and gestational week) and each module’s ME. Overlap between module genes and NDD DMR associated genes was calculated using the EnrichmentAnalysis function in the anRichment R package (version 0.82-1). The BG was set to the intersection between genes in the analysis and the organism database (org.Hs.eg.db_3.4.0) and a Fisher’s Exact test was conducted for each module-gene list pair and corrected to an FDR of P = 0.05. GO term enrichment was similarly calculated using EnrichmentAnalysis with the current hg38 human GO term database and corrected to an FDR of P = 0.05.
WGCNA was performed similarly for microglial samples collected across development in male and female mice. RPKM values were calculated as described above for microglial development data (GEO GSE99622) (Hanamsagar et al. 2017), and mouse Ensembl IDs were converted into human using BioMart. Genes with zero variance in expression or a median absolute deviation of zero were removed from the analysis. Genes with a minimum RPKM of 0.25 or higher in at least one sample were kept for analysis for a total of 2597 for the microglia dataset (Hanamsagar et al. 2017). Values were transformed to log2(RPKM+1) and clustered to visualize outliers, and 2 samples were removed due to poor clustering (see Supplementary Fig. S9). A correlation matrix using biweight midcorrelation between all genes was then computed for all samples. An estimated soft thresholding power of 10 was used to derive a signed adjacency matrix with approximately scale-free topology (r2 fit indices > 0.80) that was then transformed into a TOM. The matrix 1-TOM was used as input to calculate co-expression modules with hierarchical clustering and a minimum module size of 200 genes. The resulting MEs were clustered based on their correlation, and modules were merged at a cutheight of 0.25 (correlation of 0.75) to produce co-expression modules and one additional module with genes that did not show module membership (gray). The expression profile of each module was further summarized by the ME, the first principle component of the module. Pearson’s correlation coefficients were used to calculate the correlation between sample traits and each module’s ME. Intramodule connectivity was calculated as the correlation between every gene in the module with the module ME. Enrichment for NDD DMR associated genes within each module was calculated using the EnrichmentAnalysis function in the anRichment R package (version 0.82-1) by Fisher’s Exact test with correction to an FDR of P = 0.05. The BG was set to the intersection between genes in the analysis and the organism database (org.Hs.eg.db_3.4.0). GO term enrichment was similarly calculated using EnrichmentAnalysis with the current hg38 human GO term database and correction to an FDR of P = 0.05.
Results
DMRs Identified in NDD Cortex
To identify an epigenomic signature for NDDs, we examined genome-wide methylation profiles from postmortem cortices from 3 different NDDs and matched controls (Fig. 1A). WGBS was performed on a total of 49 cortical samples (both previously published and newly generated) and then analyzed through a standardized bioinformatic pipeline (see Supplementary Table S1 and Fig. S1). We compared WGBS methylation levels between brain samples from cortical region BA9 from donors diagnosed with idiopathic ASD (n = 12 male and 5 female ASD vs. n = 5 M and 5 F control), RTT (n = 6 F RTT vs. n = 6 F control), and BA19 from Dup15q syndrome (n = 5 M vs. n = 5 M control) (Dunaway et al. 2016) (see Supplementary Table S1). After filtering for CpG sites with at least 1× sequencing coverage per sample for each NDD cohort (see Supplementary Fig. S1), we assayed a total of 7.2 million CpGs for RTT, 3.5 million CpGs for Dup15q, and 6.5 million CpGs for ASD, making this the most extensive characterization of CpG methylation in NDD brain samples to date. Consistent with previously published findings (Dunaway et al. 2016), global hypomethylation was observed in Dup15q (Dup15q < Ctrl, P = 2.379 × 10−6). There was no significant impact on global levels of methylation at CpG sites (mCpG) and CpH sites in the ASD or RTT cohorts (see Supplementary Table S1). No significant differences in mCpG levels were observed over several types of genomic features (see Supplementary Table S1), and cell-type deconvolution with methylCIBERSORT (Newman et al. 2015) using cell type-specific methylation data from sorted human glutamatergic neurons, GABAergic neurons, and glial cells (Kozlenkov et al. 2014) did not reveal any significant cell-type composition differences (see Supplementary Fig. S2 and Table S2). Dup15q brain exhibited a large block (69 Mb) of hypomethylation (Dup15q < Control) previously identified in chromosome 15 (Dunaway et al. 2016) as well as an additional novel 23 Mb region of hypomethylation on chromosome 16 (see Supplementary Fig. S3). No large megabase block regions were identified in either the ASD or RTT samples. Average smoothed mCpG levels within 20 kb windows revealed strong technical differences between NDD cohorts (disparate brain regions and sequencing platforms), but not between sexes or diagnosis within cohorts (see Supplementary Fig. S3 and Table S1), and consequently further analysis was performed first within each NDD cohort and then compared across cohorts.
Figure 1.
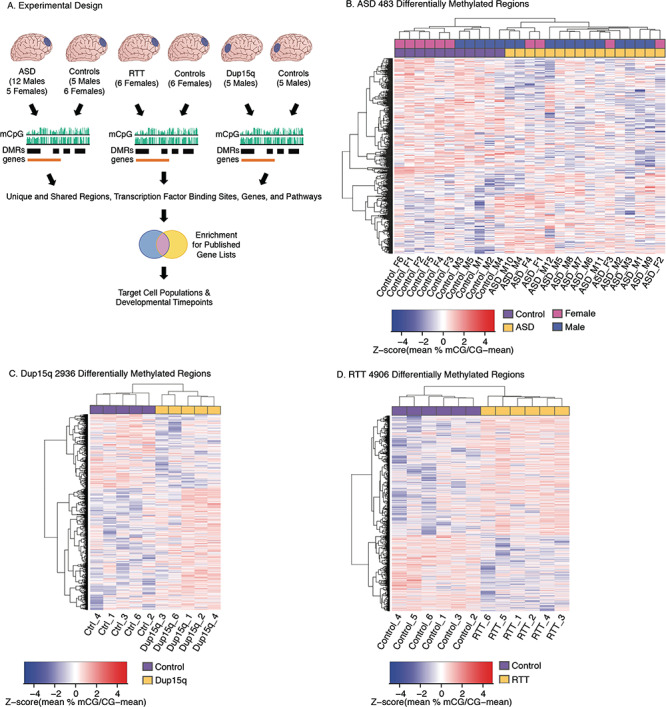
DMRs in ASD, Dup15q, and RTT cortex. (A) Experimental design. (B) Heatmap of 483 significant (empirical P < 0.05) DMRs for ASD compared with control samples. Samples cluster by diagnosis, not sex. (C) Heatmap of 2936 significant (empirical P < 0.05) DMRs for Dup15q compared with control samples. Samples cluster by diagnosis. (D) Heatmap of 4906 significant (empirical P < 0.05) DMRs for RTT compared with control samples. Samples cluster by diagnosis. For A, B, and C each column represents an independent biological replicate, and Z-scores were calculated for each DMR (row) for the percent smoothed methylation values as the mean [mCG divided by (mCG minus the mean mCG)].
We identified DMRs within each of the 3 NDD cohorts when compared with age-matched controls by using an inference and permutation-based statistical approach that is conducive to identifying broad epigenomic signatures of multiple gene loci rather than a few individual high-confidence loci. Using the dmrseq R package (Korthauer et al. 2018) with a direct adjustment for the covariate of age (all cohorts) and an additional direct adjustment for sex in the ASD cohort, we identified 292 significant (permutation P < 0.05) DMRs with lower methylation (hypomethylated) and 191 regions with higher methylation (hypermethylated) in ASD compared with control cortices (Fig. 1B; see Supplementary Table S3). Differences in methylation in ASD DMRs ranged from 5% to 20% with a median difference of 11%. For the Dup15q cohort, 1801 hypermethylated DMRs (Dup15q > Ctrl) and 1135 hypomethylated DMRs (Dupr15q < Ctrl) were identified as significant (permutation P < 0.05) (Fig. 1C; see Supplementary Table S3). Differences in methylation in Dup15q DMRs ranged from 4% to 40% with a median difference of 16%. Comparing the RTT cohort to controls identified 2931 hypermethylated (RTT > Ctrl) and 1975 hypomethylated (Ctrl>RTT) significant DMRs (Fig. 1D; see Supplementary Table S3). Differences in methylation in RTT DMRs ranged from 5% to 43% with a median difference of 15%. The locations of the NDD DMRs were largely nonoverlapping among the 3 different conditions (Fig. 2A; see Supplementary Table S3). However, ASD, Dup15q, and RTT DMRs did impact several common genomic elements including a significant enrichment for all 3 sets of NDD DMRs (Fisher’s Exact test with FDR correction P < 0.05) within promoter regions within 1 kb of the Transcription Start Site (TSS). Dup15q and RTT DMRs were also enriched within exons and the 3′ UTR (Fig. 2B; see Supplementary Table S3). We also trained a 4-class model that was able predict NDD subtype despite technical differences (sequencer, library preparation method, brain region, and sex) with higher accuracy than random chance (see Supplementary Fig. S4), suggesting that the identified DMRs represent biological differences between groups that are largely independent of technical artifacts.
Figure 2.
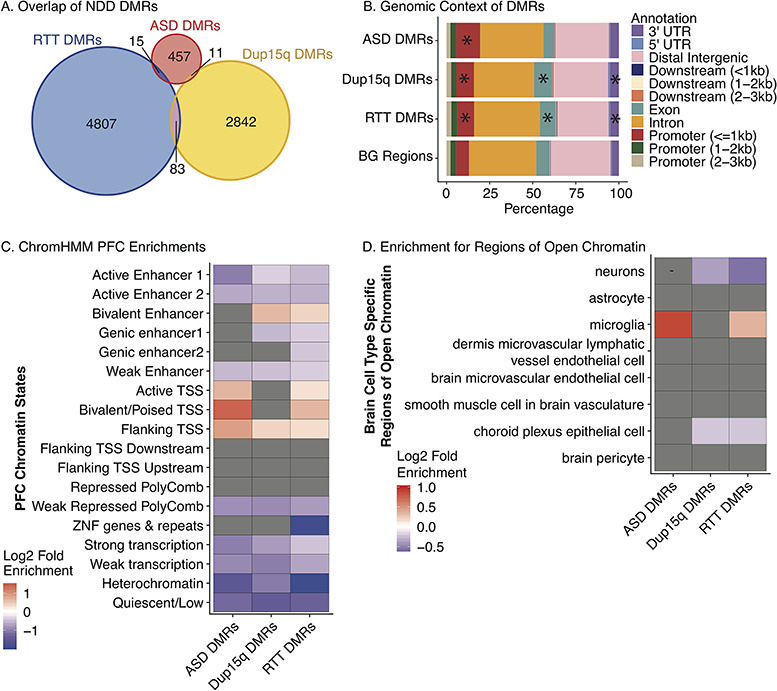
NDD DMR enrichment in epigenomic regulatory regions. (A) Overlap of ASD, Dup15q, and RTT DMRs. No regions were commonly identified across NDDs. (B) DMR enrichment within genomic features relative to BG regions. ASD, Dup15q, and RTT DMRs were enriched within promoter regions within 1 kb of the Transcription Start Site (TSS) (FDR P = 8.83 × 10−46, P = 6.66 × 10−7, and P = 7.06 × 10−8, respectively). Dup15q and RTT DMRs were enriched for exons (FDR P = 6.66 × 10−7 and P = 8.65 × 10−5) and the 3′ UTR (FDR P = 0.014 and P = 0.003). *P < 0.05 FDR corrected two-tailed Fisher’s Exact test. Table S3 includes statistics. UTR, untranslated region. TSS, Transcription Start Site (C) Heatmap of log2(fold enrichment) (FDR P < 0.05) for GAT testing relative to BG regions for overlap between each NDD DMR set and chromatin states identified in human PFC (Ernst et al. 2011; Ernst and Kellis 2013; Roadmap Epigenomics et al. 2015). (D) Heatmap of log2(fold enrichment) (P < 0.05) for GAT testing for overlap between each NDD DMR set and regions of open chromatin unique to different brain cell populations (overlapping regions removed) (Roadmap Epigenomics et al. 2015; Fullard et al. 2017; Gosselin et al. 2017). Statistic details are shown in Supplementary Table S4.
To test the hypothesis that NDD DMRs reflect epigenetic differences in functionally relevant loci, DMRs were overlapped with chromatin state maps from human PFC (Ernst et al. 2011; Ernst and Kellis 2013; Roadmap Epigenomics Consortium et al. 2015). Significant enrichment was observed within promoter regions for all 3 NDD DMRs and bivalent enhancers for Dup15q and RTT DMRs (Fig. 2C; see Supplementary Table S4). ASD, Dup15q, and RTT DMRs were depleted within regions of repressed polycomb, heterochromatin, weak transcription, quiescence, and several subtypes of enhancers (Fig. 2C; see Supplementary Table S4), suggesting that NDD DMRs may commonly impact regions of active chromatin surrounding TSS.
To further examine whether regulatory regions overlapping NDD DMRs were cell type-specific, DMRs were examined for enrichment within differential regions of chromatin accessibility specific to sorted cell populations (Fig. 2D). ASD and RTT DMRs were significantly enriched within regions of open chromatin that were specific to microglia (FDR P = 0.0002 and FDR P = 5.67 × 10−5). Further, RTT DMRs were enriched within binding sites identified in human microglia for the lineage determining immune cell transcription factor PU.1 (see Supplementary Table S4) (P = 2.85 × 10−5). ASD DMRs were also significantly enriched within regions uniquely open in T-helper 2 cells and regulatory T cells (see Supplementary Fig. S5 and Table S4). While T cell populations within the brain are limited, T cells do circulate through the meningeal lymphatic system in the brain and may play an important role in brain function (Louveau et al. 2017). NDD DMRs were not enriched within any other non-brain cell type-specific regions of open chromatin (see Supplementary Fig. S4), highlighting the uniqueness of the immune-related region enrichments.
ASD DMRs Are Enriched for Methylation-Sensitive TFBSs in Neurons
To test the hypothesis that altered methylation at NDD DMRs could affect TF binding, we examined NDD DMRs for enrichment (relative to BG regions composed of all testable candidate regions) of TFBSs with characterized methylation sensitivity in the human neuronal SK-N-SH cell line using MethMotif (Xuan Lin et al. 2018) (Fig. 3A). This tool utilizes highly reproducible ChIP-seq peaks from SK-N-SH cells (ENCODE) to call de novo TF motifs using the MEME-ChIP package. The methylation status within a 200 bp window surrounding each peak motif is then captured from WGBS data from SK-N-SH cells to create a database of TF binding sites and corresponding DNA methylation levels. Examination of NDD DMRs identified 16 TFBS motifs within ASD DMRs, 20 within Dup15q DMRs, and 19 within RTT DMRs, compared with 21 within BG regions (see Supplementary Table S5). Fisher’s Exact testing revealed significant enrichment (compared with BG regions) for 7 TFBSs in ASD DMRs, 10 in Dup15q DMRs, and 4 in RTT DMRs (all FDR P < 0.1) (Fig, 3B; see Supplementary Table S5).
Figure 3.
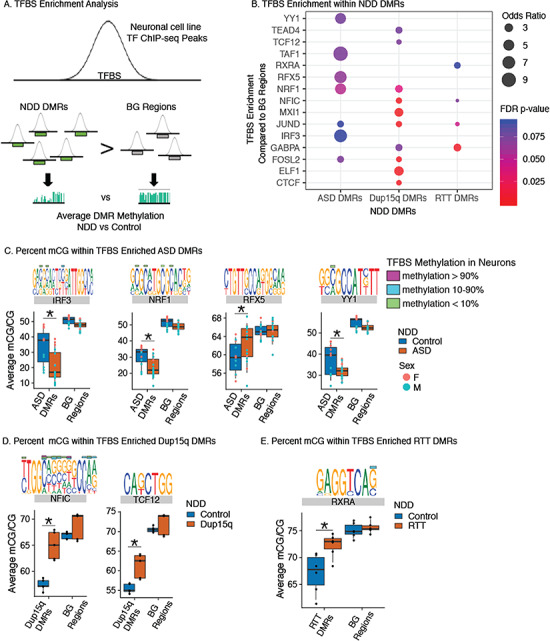
NDD DMRs are enriched for TFBS sensitive to DNA methylation in neurons. (A) TFBS were identified in NDD DMRs and BG regions using ChIP-seq peaks identified in the human SK-N-SH neuronal cell line using MethMotif (Xuan Lin et al. 2018). The frequency of identified TFBS were then compared between NDD and BG regions (B) and the average smoothed methylation values within the NDD DMRs and BG regions were then compared between NDD and Control samples (C and D). (B) TFBS were significantly enriched within NDD DMRs compared with BG regions. Fisher’s Exact test results are shown as dots colored by FDR adjusted P-values (P < 0.1) and scaled by Odds Ratio. (C) Percent smoothed methylation values for ASD DMRs and BG regions containing TFBSs enriched in B. Shown TFs have significant ANOVA and Benjamini Hochberg corrected post hoc comparisons (*P < 0.05) for ASD DMRs but not BG regions (see Supplementary Table S5 for statistics). TFBS MethMotif logos identified using MethMotif (Xuan Lin et al. 2018) are shown above each TF with corresponding color bars for % methylation values at these CpG sites in neuronal SK-N-SH cells. (D) Percent smoothed methylation values for Dup15q DMRs and BG regions containing TFBSs enriched in B. Shown TFs have significant ANOVA and Benjamini Hochberg corrected post hoc comparisons (*P < 0.05) for Dup15q DMRs but not BG regions. TFBS MethMotif logos are shown for each TF as described in C. (E) Percent smoothed methylation values for RTT DMRs and BG regions containing TFBSs enriched in B. Shown TFs have significant ANOVA and Benjamini Hochberg corrected post hoc comparisons (*P < 0.05) for RTT DMRs but not BG regions. TFBS MethMotif logos are shown for each TF as described in C. See Supplementary Table S5 for full statistics.
To further test the impact of methylation changes in NDDs on potential TF binding at these sites, smoothed methylation values were extracted for each NDD DMR overlapping the enriched TFBSs and then compared between controls and NDD samples (Fig. 3C; see Supplementary Table S5). ASD samples showed significant differences in methylation levels compared with control samples within ASD DMRs containing binding sites for 4 of the enriched TFs (IRF3, NRF1, RFX5, and YY1), but not within the same TFs within BG regions (Fig. 3C; see Supplementary Fig. S6 and Table S5). Dup15q and RTT samples showed significant differences in methylation when compared with controls over 2 (NFIC and TCF12) or 1 (RXRA) enriched TFBSs, respectively (Fig. 3D,E; see Supplementary Fig. S6). Several of the enriched TFBSs exhibited methyl sensitivity in the SK-N-SH neuronal cell line. For example, both IRF3 and NRF1 show increased binding to sites with lower levels of methylation in SK-N-SH cells, and the ASD DMRs with IRF3 or NRF1 binding sites exhibited lower methylation in ASD compared with control samples. Together, these results are consistent with the enrichment of ASD DMRs for 2 methyl-sensitive TFs, where methylation differences in ASD are in the direction expected for increased binding to an accessible cis-regulatory element in neurons. While neurons were the only brain cell type represented in MethMotif, several of the NDD DMR enriched TFBSs also show preferential cell-type expression patterns, including RFX5 in inhibitory neurons and TCF12 in oligodendrocyte precursor cells (see Supplementary Fig. S7). Together, these analyses provide support for functional consequences related to changes in methylation levels over NDD DMRs.
NDD DMRs Are Enriched for GO terms Critical to Brain Development
To gain deeper insight into the potential functional role of NDD DMRs, we examined enrichment for GO terms. ASD DMRs were significantly enriched for 9 GO terms, Dup15q DMRs for 275 GO terms, and RTT DMRs for 483 GO terms (FDR corrected P < 0.05) (see Supplementary Table S6). The 3 NDDs shared significant enrichment of 8 GO terms (Fig. 4A; see Supplementary Table S6), including terms for nervous system development, cell morphogenesis involved in differentiation, generation of neurons, and anatomical structure development. ASD and RTT DMRs were commonly enriched for Rho GTPase binding, which did not reach significance in Dup15q DMRs (see Supplementary Table S6). In addition, RTT and Dup15q DMRs shared significant enrichments for 197 GO terms, including terms for synapse, neuron part, neurogenesis, and cell projection, indicating a shared disruption of pathways (Fig. 4B). Both RTT and Dup15q DMRs were also significantly enriched for GO terms that were unique to each disorder, including 278 unique terms for RTT and 71 terms for Dup15q. Dup15q unique GO terms included terms related to glial differentiation, morphogenesis, and peripheral nervous system development (Fig. 4C). RTT unique GO terms included terms related to actin and cytoskeleton related processes as well as vasculature development (Fig. 4D). Together, GO term analysis highlights both shared and unique pathways and processes impacted by changes in DNA methylation in NDDs.
Figure 4.
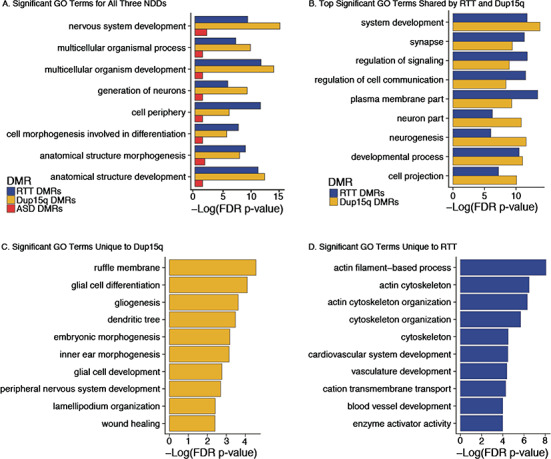
NDD DMRs are enriched for both shared and distinct gene ontologies. NDD DMRs were analyzed for GO term enrichment using gene identifiers for hg38 Ensembl genes with a 10 kb extension up- and downstream of the gene start and end sites, respectively. All comparisons were performed with a hypergeometric test and FDR corrected to P < 0.05 (see Table S6 for statistics). (A) Significantly enriched GO terms shared across all 3 NDD DMRs. (B) Top significantly (by FDR P-value) enriched GO terms shared by RTT and Dup15q DMRs. These terms did not reach significance for ASD DMRs. (C) Top significant GO terms that were unique to Dup15q DMRs. (D) Top GO terms that were unique to RTT DMRs. See Supplementary Table S6 for details and statistics.
ASD DMR Associated Genes Are Enriched for Genes Transcriptionally Impacted by NDDs
To test the hypothesis that previously identified NDD genes identified by mutation, variation, or transcriptional dysregulation may coincide with NDD DMRs, we first assigned NDD DMRs to the closest gene (within the gene body or within +/−10 kb of the gene start or end sites; see Supplementary Fig. S8). ASD DMRs were associated with 431 genes, Dup15q DMRs with 2323 genes, and RTT DMRs with 3647 genes (see Supplementary Table S3). Genes associated with each set of NDD DMRs significantly overlapped all 3 NDDs examined (Fig. 5A; see Supplementary Table S8) with 65 Ensembl genes (57 gene symbols) shared between all 3 DMR lists. Genes shared across NDDs contained many genes previously identified as either ASD or ID genetic risk factors or were differentially expressed in either ASD or SCZ or both disorders (Fig. 5A). To further investigate if NDD DMR associated genes were enriched for genes with differential expression or with increased genetic risk for neuropsychiatric disorders, we performed a permutation-based gene enrichment analysis (see Supplementary Fig. S8) with a series of published datasets of known genetic risk and differentially expressed genes from human brain disorders (see Table S8).
Figure 5.
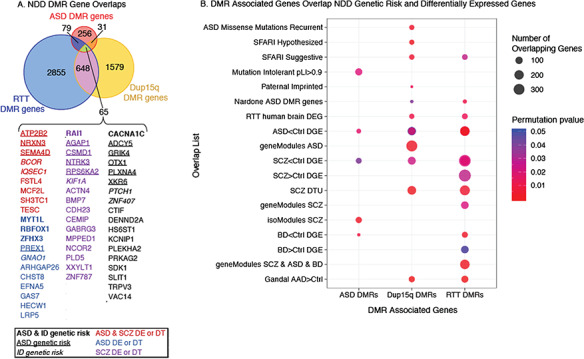
Enrichment of DMR associated genes for NDD genetic risk and differentially expressed genes. (A) ASD, Dup15q, and RTT DMR associated genes significantly overlap with each other (all overlaps are FDR corrected permutation test corrected for gene length P < 1 × 10−5, statistics in Supplementary Table S8). The 65 genes (57 gene symbols) shared across all 3 NDDs are shown below. Genes that are known genetic risk factors for ASD or ID are shown in bold (both ASD and ID), underlined (ASD only), or italics (ID only). Genes differentially expressed (DE) or with differential transcript usage (DT) are shown in red for ASD and SCZ, blue for ASD only, and purple for SCZ only. (B) DMR associated genes are significantly enriched (P-values <0.05 for gene length corrected null distribution permutation testing; see Methods) for genes associated with ASD genetic risk (ASD Missense Mutations Recurrent and SAFARI ASD lists). DMR associated genes are significantly enriched for genes with altered gene expression in numerous neurodevelopmental, neuropsychiatric, and neurodegenerative disorders including ASD, SCZ, ADD, and BD. DEG, differentially expressed genes; SFARI, Simons Foundation Autism Research Initiative; SCZ, schizophrenia; BD, bipolar disorder; AAD, alcohol abuse disorder. Mutation Intolerant pLI > 0.9, probability of being loss-of-function >0.9 (top genes that are intolerant to human mutation) (Lek et al. 2016), paternally imprinted genes for humans from http://www.geneimprint.com, Nardone ASD DMR genes from (Nardone et al. 2017), and RTT human brain DEG: DEG from Lin et al. (2016). ASD < Ctrl DEG are a compilation of overlaps between DMR genes and DEG genes from ASD brain from several papers (Gupta et al. 2014; Parikshak et al. 2016; Gandal et al. 2018a, 2018b). SCZ < Ctrl and SCZ > Ctrl DGE and BD < Ctrl and BD > Ctrl DGE are a compilation of overlaps from Gandal et al. (2018a, 2018b). Individual gene list overlaps from each source as well as all gene lists, citations, and overlap statistics are in Supplementary Table S8.
NDD DMR associated genes were significantly longer than either the BG region associated genes or a random sample of the same number of genes from the human genome (hg38) (see Supplementary Fig. S8). In addition, gene lengths associated with BG regions were also significantly higher than random samples of all hg38 genes, reflecting either the inherent bias of longer genes overlapping a DMR by chance or a bias in defining BG regions as those with sufficient variation in methylation between NDD conditions for testing (see Supplementary Table S7). Given this bias, subsequent gene enrichments were performed by permutation testing against a null distribution matched for median gene length (Methods; see Supplementary Fig. S8).
Overall, DMR associated genes were largely not enriched for genetic risk genes identified across several studies and databases for either ASD or ID risk. In exception to this, Dup15q DMR associated genes were significantly enriched for multiple lists of known risk genes for ASD (Fig. 5B shows significant comparisons from Supplementary Table S8), including SFARI lists and ASD missense recurrent genes identified from exome sequencing studies (Iossifov et al. 2014). RTT DMR genes were also enriched for genes on the “suggestive” SFARI ASD list. ASD DMR genes were not enriched for any of the genetic risk factor lists but were significantly enriched for genes highly intolerant to genetic variation in the human population (probability of loss of function mutation >0.9 [pLI > 0.9]) (Lek et al. 2016), suggesting that ASD DMR associated genes play critical roles in human development.
All 3 groups of NDD DMR associated genes were enriched for genes with differential expression in human brain from either neurodevelopmental or neuropsychiatric disorders. All 3 NDD DMR sets were enriched for genes with lower expression in ASD brain collected from several datasets (ASD < Ctrl DGE) (Gupta et al. 2014; Gandal et al. 2018a, 2018b) (Fig. 5B; see Supplementary Table S8). Both Dup15q and RTT DMR genes were also enriched for genes differentially expressed in RTT brain (Lin et al. 2016). All 3 NDD DMR gene sets were also enriched for genes with decreased expression in SCZ brain (SCZ < Ctrl DGE) (Gandal et al. 2018a, 2018b), and both RTT and ASD DMR genes were enriched for gene co-expression modules with differential gene expression or differential isoform usage in SCZ, respectively. Both RTT and Dup15q DMR genes were also enriched for genes with differential transcript usage in SCZ brain (SCZ DTU) (Gandal et al. 2018b) (Fig. 5B; see Supplementary Table S8). RTT DMR genes also showed significant enrichment for genes differentially expressed in bipolar disorder (Gandal et al. 2018a, 2018b). Together, NDD DMR overlaps with differentially expressed genes across ASD and SCZ indicate that shared gene pathways are disrupted in these disorders. Further examination of the down regulated gene list overlaps between DMR associated genes, and ASD < Ctrl DEG and SCZ < Ctrl DEG gene lists revealed numerous neuronal genes, including NRXN3 and ATP2B2, which are both ASD genetic risk genes (Vaags et al. 2012; Iossifov et al. 2014). Together, this suggests some common pathways and neuronal gene targets with both altered expression and methylation in ASD and SCZ.
Enrichment Analysis of NDD DMR Associated Genes across Cell Types and Human Brain Development
To test the hypothesis that NDD DMR genes are transcriptionally active during brain development, we utilized weighted gene co-expression networks (WGCNA) (Langfelder and Horvath 2008) built from single-cell RNA-seq data collected across early human PFC development by gestational week (GW 8–26) (Zhong et al. 2018). Average expression across GW for cell types identified by Zhong et al. (2018) (stem cells, oligodendrocytes [OPCs], neurons, GABAergic neurons, astrocytes, and microglia) was used to identify modules of genes that significantly co-varied with cell type and developmental time (Fig. 6A). Five co-expression modules (and one uncorrelated gray module) were identified (see Supplementary Fig. S9), including 2 that were significantly correlated with cell type (purple module r2 = 0.69, FDR P = 1.98 × 10−6 and red module r2 = −0.69 and FDR P = 1.98 × 10−6) and 1 that was significantly correlated with developmental time (skyblue module r2 = 0.42, FDR P = 0.02) (Fig. 6A; see Supplementary Table S9). We next looked for enrichment of the NDD DMR associated genes within each module (see Supplementary Table S9). ASD DMR associated genes were not significantly enriched within any of the modules. However, both Dup15q DMR genes and RTT DMR genes were significantly enriched within the purple module (FDR P = 0.027 and FDR P = 7.44 × 10−5). Dup15q DMR genes were also significantly enriched (FDR P = 0.003) within the skyblue module that also associated with gestational age (GW) but not cell type (Fig. 6A). Closer examination of the skyblue module revealed enrichment for GO terms related to the extracellular matrix, membrane function, cellular adhesion, morphogenesis, and other basic process of cellular development (see Supplementary Table S9). The expression levels of skyblue module genes reach a peak at GW 19 across multiple cell types (stem cells, OPCs, and astrocytes) (Fig. 6B), suggesting that Dup15q may impact methylation of regions important for basic cellular process of development occurring around GW 19. In comparison, the purple module revealed a significant enrichment for GO terms related to immune function, cell activation, and immune effector process (see Supplementary Table S9). These enrichments were consistent with microglia showing the highest expression levels across cell types within the purple module (Fig. 6C). Together, this indicates that both RTT and Dup15q DMRs may impact microglial gene expression programs across development.
Figure 6.
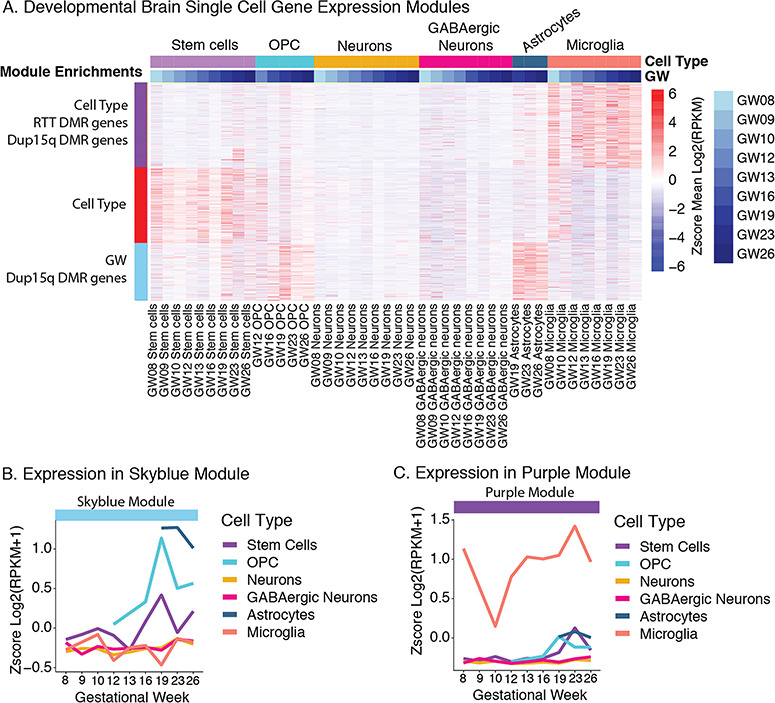
NDD DMR associated genes are enriched in development and cell type-specific gene co-expression modules. (A) Gene expression within modules identified by WGCNA that are significantly associated with human PFC brain development (gestational week) and cell type. Data from human PFC Single-Cell RNA-seq (GEO GSE99622) (Zhong et al. 2018) across development (Gestational Week, GW 8–26). Both the purple and red MEs are significantly correlated with cell type and the skyblue module ME was correlated with cell type (FDR P < 0.05). RTT DMRs were significantly enriched in the purple module. Dup15q DMRs were significantly enriched in the purple and skyblue module (FDR P < 0.05). (B) Z-score of the mean gene expression (log2(RPKM+1)) for each cell type across gestational weeks for the skyblue module. (C) Z-score of the mean gene expression (log2(RPKM+1)) for each cell type across gestational weeks for the purple module. WGCNA network construction, statistics, and enrichments are shown in Supplementary Table S9.
In order to better understand phenotypes associated with the immune signature observed in Figure 6 and previously in ASD brain gene expression datasets (Voineagu et al. 2011; Gupta et al. 2014; Gandal et al. 2016; Lin et al. 2016; Parikshak et al. 2016), we tested the hypothesis that NDD DMR genes are transcriptionally active in microglial development. We constructed weighted gene co-expression modules using a published RNA-seq dataset (Hanamsagar et al. 2017) from microglia isolated from 4 developmental time points from both male and female mice (see Supplementary Fig. S10 and Table S10). Two modules were significantly correlated with both microglial developmental time and enriched for NDD DMRs. Genes in both modules were highly expressed in early microglial development and were enriched for GO terms related to nervous system development and metabolic function, suggesting a role for NDD DMR associated genes within these modules in regulating microglial–neuron interactions during early development (see Supplementary Table S10).
To further explore the role of NDD DMR genes in microglial function, we compared NDD DMR genes to published lists of microglial genes across developmental stages (Hanamsagar et al. 2017) as well as genes dysregulated in microglia isolated from both genetic and maternal immune challenge NDD models (Mecp2 mutation, PolyI:C maternal immune activation, and maternal allergic asthma). All 3 NDD DMR associated gene lists showed significant enrichment for genes regulated during microglial development, including lists from both pre- and postnatal development (see Supplementary Table S11). NDD DMR associated genes were also enriched for microglial differentially expressed genes in several NDD mouse models. Interestingly, all 3 NDD DMR gene sets were significantly enriched for genes that were rescued in the adult animal with minocycline treatment (see Supplementary Table S11). Together, this integration of NDD DMRs with microglial WGCNA and gene enrichments implicates altered microglial regulation during fetal development in NDDs.
Discussion
This is the first study to perform in-depth, genome-wide DNA methylation analysis in brain from NDDs. We greatly extended previous work at the candidate gene loci MECP2 and OXTR (Vogel Ciernia and LaSalle 2016) and the imprinted locus 15q11.2-q13.3 (Nagarajan et al. 2008; Gregory et al. 2009) to identify DMRs within each disorder. We identified significant enrichment for several genetic risk gene sets within NDD DMRs, but the majority of genetic risk loci for ASD and other NDDs were not found within NDD DMRs. This divergence in genetic and epigenetic signatures for NDDs may indicate that epigenetic differences reflect downstream genome-wide impacts of genetic mutations, such as in the case of mutations occurring in chromatin remodelers (i.e., CHD8 or ARID1B) (De Rubeis et al. 2014; Iossifov et al. 2014), DNA methylation regulatory enzymes (DNMT3A) (De Rubeis et al. 2014; Iossifov et al. 2014), or imprinted genes (Lopez et al. 2017). Epigenetic changes may also reflect a convergent signature of molecular events representing abnormal processes at the interface of multiple genetic and environmental risk factors.
While the NDD DMRs did not significantly overlap across disorders, at the gene level there was a significant overlap with 65 genes shared across all 3 NDDs. The higher NDD concordance for genes rather than DMRs suggests a convergence in altered gene functions in NDDs that was not precisely matched at the individual CpG level. The shared sets of enriched GO terms related to nervous system development observed between different NDD DMR associated genes also provide evidence for convergent epigenomic signatures between NDDs, despite a lack of nucleotide precision. This indicates that a subset of regulatory mechanisms is commonly impacted by changes in methylation across NDDs and particularly for genes with decreased expression in ASD and SCZ brains. Prior WGBS analyses have shown both a negative correlation with promoter methylation, as well as a positive correlation with methylation over transcriptionally active gene bodies (LaSalle et al. 2013; Roadmap Epigenomics Consortium et al. 2015), which is consistent with our findings of DMRs and gene expression associations (see Supplementary Table S8). Furthermore, deficiencies in MeCP2 in RTT brain are associated with both increased and decreased transcriptional changes (Chahrour et al. 2008; Lin et al. 2016). Methylation changes may be important at sites of transcription factor binding, particularly neuronal methyl-sensitive TFs, such as IRF3 and NRF1 that we found enriched in ASD DMRs. Future work will be needed to directly test how methylation changes at binding sites for these TFs specifically impact binding and downstream gene regulation in neurons, as well as to identify the methyl sensitivity of TFBSs in non-neuronal cell types such as microglia.
One of the convergent findings across analyses was the enrichment for NDD DMRs in regulatory regions, genes, and developmental expression modules for immune genes. All 3 sets of NDD DMRs were significantly enriched within promoter regions and near transcription start sites with active chromatin signatures in PFC. Specifically, both ASD and RTT DMRs were significantly enriched for regions of open chromatin unique to microglia, RTT DMRs were enriched for microglial PU.1 binding sites, and both RTT and Dup15q DMR associated genes were enriched within developmental gene expression modules with the highest cell-type expression in microglia. In addition, ASD DMRs with TFBSs for IRF3, a critical TF for immune gene signaling, showed significantly lower levels of methylation in ASD compared with control samples. These patterns led us to further explore a potential role for NDD DMR associated genes in microglial development. Genes associated with all 3 types of NDD DMRs were specifically enriched within mouse microglial gene expression modules characterized by a high level of gene expression in early development. These 2 microglial developmental modules were characterized by GO terms critical for neuron–microglia interactions and energy regulation, indicating that NDD DMR associated genes may be shaping early microglial maturation and nervous system interactions. This is also consistent with alterations in microglia morphology and density previously observed in a subset of postmortem ASD brains (Yuen et al. 2016), as well as previous transcriptional and epigenomic profiling in human ASD brain that has consistently revealed both synaptic and immune dysfunction (Voineagu et al. 2011; Gupta et al. 2014; Gandal et al. 2016; Lin et al. 2016; Parikshak et al. 2016). Together with the immune gene expression signature observed across multiple ASD human brain cohorts (Gupta et al. 2014; Parikshak et al. 2016; Gandal et al. 2018a, 2018b), our data support a convergent role for altered immune and nervous system function in NDDs. As the resident immune cells in the brain, microglia may be a common cell type impacted by diverse NDD etiologies because they serve as important sentinels that respond to both genetic and environmental disruptions. Microglia not only constantly monitor the brain for signs of infection but also respond to genetic abnormalities that impair neuronal function (Derecki et al. 2012; Cronk et al. 2015; Horiuchi et al. 2016), suggesting the immune signatures observed across transcriptomic and epigenomic studies in human ASD brain (Gupta et al. 2014; Parikshak et al. 2016; Gandal et al. 2018a, 2018b) may be partially driven by an immune response to abnormal neuronal processes that arise from genetic and epigenetic etiologies. Regardless of the initial cause of the NDD, microglia and other immune cells may be an appropriate target for therapy, since altered immune function may have profound impacts on neuronal development and ongoing brain function. Our analysis provides initial support for this premise as all 3 NDD DMR associated gene lists significantly overlapped genes normalized by minocycline treatment in adult mice that had received PolyI:C MIA in utero (Mattei et al. 2017). Together, these results suggest that therapeutics that alter inflammation are candidates for further study in relation to resetting the microglial transcriptome and function in NDDs.
In addition to convergent immune signatures across NDDs, we also identified regulatory regions, TFBSs, and pathways that were unique to each disorder. For example, both Dup15q and RTT DMRs were significantly enriched for sets of unique GO terms. Dup15q was uniquely enriched for several GO terms involved in glial cell development, and RTT DMRs were uniquely enriched for terms involved in actin cytoskeleton and blood vessels. Dup15q DMRs were also uniquely enriched within the single-cell gene expression module related to developmental timing of gene expression. This module peaked in expression at GW 19 in several cell types including stem cells, OPCs, and astrocytes. These results suggest that changes in DNA methylation in Dup15q may be particularly important for cell type transitions occurring near week 19 of gestation.
One limitation of this work is the relatively small sample size due to limited region-matched availability of human brain samples and relatively high cost of WGBS. However, this work is similar or larger in sample size than previous DNA methylation microarray studies when considering the evaluation of a single brain region. While our analysis is not sufficiently powered to directly examine sex differences, we did identify epigenomic signatures relevant to both male and female ASD cases. Future work will be needed to more fully explore differences in males and females across NDDs. The average WGBS genome coverage is not sufficient to identify single CpG methylation differences, but does allow for the assessment of DMRs (McGill et al. 2017), which represent biologically relevant regional methylation differences (Korthauer et al. 2018).
There are several potential explanations for the different numbers of DMRs obtained among the different disorders. The most likely explanation is biological; individuals with idiopathic ASD are expected to be more heterogeneous in their etiology and pathology than the 2 genetic syndromic NDDs and therefore have fewer DMRs that reach significance. The ASD analysis also included both males and females, and by including sex as a covariate we exclude DMRs that have sex-specific methylation differences in ASD.
The relatively limited sample size in this study also precluded FDR correction of permutation P-values. However, within the environmental epigenetics field, it is common to identify reproducible and biological relevant epigenetic changes with small magnitude effect sizes (Breton et al. 2017), and our machine learning model was able to predict NDD diagnosis from the DMRs with a moderate level of accuracy despite these limitations. Our study does not focus on a single gene or region, but instead leverages multiple epigenomic and genetic datasets to examine the epigenomic signature of cell types and gene functions altered in NDDs. With the continued decrease in cost of sequencing technologies and increased brain bank advocacy, future work can more fully characterize additional NDD brain samples by WGBS on a single sequencing platform with additional brain regions, disorders, and cell type-specific sorting.
In conclusion, findings from this study reveal a critical epigenomic signature in NDD cortex that overlaps with known neuronal and immune dysfunction in NDDs. It remains unclear if alterations in DNA methylation in NDDs are a contributing cause or indirect consequence of the disorder; however, regardless of the direction of causality, the identified DMRs may serve as a unique read-out of the intersection between multiple genetic and environmental perturbations to the developing brain. Integration with multiple data sources identified both neuronal and microglial cell types and pathways as potentially relevant therapeutic avenues that may be commonly dysregulated mediators at the interface of genetic and environmental risk factors.
Supplementary Material
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Previously published datasets for human ASD and Control BA9 as well as Dup15q and Control BA19 samples are available at GEO GSE81541. Single-Cell RNAseq data from human brain across gestational weeks are from GEO GSE104276 (Breton et al. 2017). Microglial developmental RNAseq data are available at GEO GSE99622 (Zhong et al. 2018). Previously unpublished human ASD, Rett and Control BA9 samples are available at GEO GSE119981. All analysis code is available on Github at https://github.com/aciernia, https://github.com/ben-laufer/CpG_Me, https://github.com/ben-laufer/DMRichR, and https://github.com/hyeyeon-hwang.
Funding
National Institutes of Health (R01ES021707, R01NS081913, and R01AA027075 to J.M.L.); Brain & Behavior Research Foundation (NARSAD Young Investigator Award to A.V.C.); National Institutes of Mental Health (1K01MH116389-01A1 K01 Mentored Research Scientist Development award to A.V.C); the Canadian Institutes of Health Research (MFE-146824 postdoctoral fellowship award to B.I.L.); Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley (NIH S10 OD018174 Instrumentation Grant); University of California, Davis Intellectual and Developmental Disabilities Research Center (NIH U54 HD079125).
Notes
The authors would like to thank Dr Shreejoy Tripathy at the University of British Columbia for helpful discussion on the human single-cell RNAseq data. We would like to thank Dr Matt Settles, Dr Ian Korf, and Dr Blythe Durbin-Johnson at the University of California, Davis Genome Center for helpful discussions about bioinformatic and statistical analyses. We would also like to thank Dr Keegan Korthauer at Harvard University for helpful discussions and troubleshooting of the implementation of dmrseq for this project. Conflict of Interest: None declared
Author Contributions
A.V.C. designed the research approach, performed bioinformatic and statistical analysis, and wrote the manuscript. B.I.L. developed the WGBS alignment and DMR calling pipeline, contributed to the bioinformatic analysis, and helped prepare the manuscript. K.W.D. prepared sequencing libraries and initial bioinformatics processing. H.H., C.E.M., and R.L.C. provided additional bioinformatics analysis. D.H.Y. assisted with experimental design and manuscript preparation. J.M.L. assisted with research design and manuscript preparation. All authors read, edited, and approved of the final manuscript.
References
- Ben-David E, Shifman S. 2012. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol Psychiatry. 18(10):1054–1056. [DOI] [PubMed] [Google Scholar]
- Breton CV, Marsit CJ, Faustman E, Nadeau K, Goodrich JM, Dolinoy DC, Herbstman J, Holland N, LaSalle JM, Schmidt R et al. 2017. Small-magnitude effect sizes in epigenetic end points are important in children’s environmental health studies: the children’s environmental health and disease prevention research center’s epigenetics working group. Environ Health Perspect. 125(4):511–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher AVS, Cidav Z, Knapp M, Mandell DS. 2014. Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatr. 168:721–728. [DOI] [PubMed] [Google Scholar]
- Cavalcante RG, Sartor MA. 2017. annotatr: genomic regions in context. Bioinformatics. R package version 1.10.0 https://bioconductor.org/packages/release/bioc/html/annotatr.htmldate accessed: April 2019. [DOI] [PMC free article] [PubMed]
- Chahrour M, Sung YJ, Shaw C, Zhou X, Wong STC, Qin J, Zoghbi HY. 2008. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 320:1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy A, Furness A, Joshi K, Ghorani E, Ford K, Ward MJ, King EV, Lechner M, Marafioti T, Quezada SA et al. 2018. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun. 9:3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronk JC, Derecki NC, Ji E, Xu Y, Lampano AE, Smirnov I, Baker W, Norris GT, Marin I, Coddington N et al. 2015. Methyl-CpG binding protein 2 regulates microglia and macrophage gene expression in response to inflammatory stimuli. Immunity. 42:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Torre-Ubieta L, Won H, Stein JL, Geschwind DH. 2016. Advancing the understanding of autism disease mechanisms through genetics. Nat Med. 22:345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek EA, Kou Y, Liu L, Fromer M, Walker S et al. 2014. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 515:209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SBG, Guyenet PG, Kipnis J. 2012. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 484:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley C, Emery JCH. 2014. The value of caregiver time: costs of support and care for individuals living with autism spectrum disorder. SPP Res Pap. 7:1–49. [Google Scholar]
- Dunaway KW, Islam MS, Coulson RL, Lopez SJ, Vogel Ciernia A, Chu RG, Yasui DH, Pessah IN, Lott P, Mordaunt C et al. 2016. Cumulative impact of polychlorinated biphenyl and large chromosomal duplications on DNA methylation, chromatin, and expression of autism candidate genes. Cell Rep. 17:3035–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2013. ChromHMM: automating chromatin state discovery and characterization. Nat Methods. 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T, Jakobshagen K, Buch T et al. 2015. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 18:965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, Käller M. 2016. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32:3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullard JF, Giambartolomei C, Hauberg ME, Xu K, Voloudakis G, Shao Z, Bare C, Dudley JT, Mattheisen M, Robakis NK et al. 2017. Open chromatin profiling of human postmortem brain infers functional roles for non-coding schizophrenia loci. Hum Mol Genet. 26:1942–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Haney J, Parikshak N, Leppa V, Horvath S, Geschwind DH. 2018a. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science. 693:693–697. [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van H, Varghese M, Wang Y et al. 2018b. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 362:eaat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen C, Hehir-Kwa JY, Thung D, van de M, van B, Willemsen M, Kwint M, Janssen I, Hoischen A, Schenck A et al. 2014. Genome sequencing identifies major causes of severe intellectual disability. Nature. 511:344–347. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP et al. 2017. An environment-dependent transcriptional network specifies human microglia identity. Science. 23:6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory SG, Connelly JJ, Towers AJ, Johnson J, Biscocho D, C a M, Lintas C, Abramson RK, Wright HH, Ellis P et al. 2009. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 7:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote S. 2018. GOfuncR: Gene Ontology Enrichment Using FUNC.
- Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, West AB, Arking DE. 2014. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun. 5:5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamsagar R, Alter MD, Block CS, Sullivan H, Bolton JL, Bilbo SD. 2017. Generation of a microglial developmental index in mice and in humans reveals a sex difference in maturation and immune reactivity. Glia. 65(9):1504–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Langmead B, Irizarry RA, Hansen K, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald O, Wen B et al. 2012. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 13:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger A, Webber C, Goodson M, Ponting CP, Lunter G. 2013. GAT: a simulation framework for testing the association of genomic intervals. Bioinformatics. 29:2046–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L, Means TK, El Khoury J. 2013. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 16:1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F et al. 2006. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 34:D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Möller T, Orre M, Kamphuis W et al. 2015. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun. 3:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Smith L, Maezawa I, Jin L-W. 2016. CX3CR1 ablation ameliorates motor and respiratory dysfunctions and improves survival of a Rett syndrome mouse model. Brain Behav Immun. 60:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE et al. 2014. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 515:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 8:118–127. [DOI] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B et al. 2017. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 169:1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, Honti F, Keerthikumar S, Oortveld MAW, Kleefstra T, Kramer JM et al. 2016. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet. 98:149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korthauer K, Chakraborty S, Benjamini Y, Irizarry RA. 2018. Detection and accurate false discovery rate control of differentially methylated regions from whole genome bisulfite sequencing. Biostatistics 00:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlenkov A, Roussos P, Timashpolsky A, Barbu M, Rudchenko S, Bibikova M, Klotzle B, Byne W, Lyddon R, Di Narzo AF et al. 2014. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 42:109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd-Acosta C, Hansen KD, Briem E, Fallin MD, Kaufmann WE, Feinberg AP. 2014. Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry. 19:862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. 2012. Fast R functions for robust correlations and hierarchical clustering. J Stat Softw. 46:i11. [PMC free article] [PubMed] [Google Scholar]
- LaSalle JM, Powell WT, Yasui DH. 2013. Epigenetic layers and players underlying neurodevelopment. Trends Neurosci. 36:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Nicholls L, Assareh H, Fang Z, Amos TG, Edwards RJ, Assareh AA, Voineagu I. 2016. Transcriptome analysis of human brain tissue identifies reduced expression of complement complex C1Q genes in Rett syndrome. BMC Genomics. 17:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez SJ, Dunaway K, Islam MS, Mordaunt C, Vogel Ciernia A, Meguro-Horike M, Horike SI, Segal DJ, LaSalle JM. 2017. UBE3A-mediated regulation of imprinted genes and epigenome-wide marks in human neurons. Epigenetics. 12:982–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A, Plog BA, Antila S, Alitalo K, Nedergaard M, Kipnis J. 2017. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest. 127(9):3210–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall K, Schmidt RJ, Hertz-Picciotto I. 2014. Maternal lifestyle and environmental risk factors for autism spectrum disorders. Int J Epidemiol. 43:443–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matcovitch-Natan O, Winter DR, Giladi A, Aguilar SV, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Gonzalez FZ, Perrin P et al. 2016. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 353(6301):aad8670. [DOI] [PubMed] [Google Scholar]
- Mattei D, Ivanov A, Ferrai C, Jordan P, Guneykaya D, Buonfiglioli A, Schaafsma W, Przanowski P, Deuther-Conrad W, Brust P et al. 2017. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry. 7(5):e1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill BE, Barve RA, Maloney SE, Strickland A, Rensing N, Wang P, Wong M, Head R, Wozniak DF, Milbrandt J. 2017. Abnormal microglia and enhanced inflammation-related gene transcription in mice with conditional deletion of Ctcf in Camk2a-Cre-expressing neurons. J Neurosci. 3(38):200–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh ML. 2012. Interrater reliability: the kappa statistic. Biochem Med. 22:276–282. [PMC free article] [PubMed] [Google Scholar]
- Miller A, Shen J, Mïesse LC. 2016. Child functional characteristics explain child and family outcomes better than diagnosis: population-based study of children with autism or other neurodevelopmental disorders/disabilities. Health Rep. 27:9–18. [PubMed] [Google Scholar]
- Miller JA, Woltjer RL, Goodenbour JM, Horvath S, Geschwind DH. 2013. Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Med. 5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Patzel KA, Martin M, Yasui DH, Susan E, Hertz-Picciotto I, Hansen RL, Van de Water J, Isaac N, Jiang R et al. 2008. MECP2 promoter methylation and X chromosome inactivation in autism. Autism Res. 1:169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardone S, Sams DS, Reuveni E, Getselter D, Oron O, Karpuj M, Elliott E. 2014. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl Psychiatry. 4:e433–e439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardone S, Sams DS, Zito A, Reuveni E, Elliott E. 2017. Dysregulation of cortical neuron DNA methylation profile in autism spectrum disorder. Cereb Cortex. 27(12):5739–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. 2015. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 12:453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta LT, Huang J et al. 2016. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. 540:423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature. 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Devlin B, Roeder K, State MW, Sanders SJ, He X, Willsey AJ, Ercan-sencicek AG et al. 2015. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 87:1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohat S, Shifman S. 2014. Bias towards large genes in autism. Nature. 512:E1–E2. [DOI] [PubMed] [Google Scholar]
- Sun W, Poschmann J, Cruz-Herrera del Rosario R, Parikshak NN, Hajan HS, Kumar V, Ramasamy R, Belgard TG, Elanggovan B, CCY W et al. 2016. Histone acetylome-wide association study of autism spectrum disorder. Cell. 167:1385–1397. [DOI] [PubMed] [Google Scholar]
- The Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium 2017. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism. 8(21):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaags AK, Lionel AC, Sato D, Goodenberger M, Stein QP, Curran S, Ogilvie C, Ahn JW, Drmic I, Senman L et al. 2012. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am J Hum Genet. 90:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel Ciernia A, Careaga M, LaSalle JM, Ashwood P. 2018. Microglia from offspring of dams with allergic asthma exhibit epigenomic alterations in genes dysregulated in autism. Glia. 66:505–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel Ciernia A, LaSalle J. 2016. The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nat Rev Neurosci. 17:411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. 2011. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 474:380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingett SW, Andrews S. 2018. FastQ Screen: a tool for multi-genome mapping and quality control. F1000Res. 7:1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Lin QX, Sian S, An O, Thieffry D, Jha S, Benoukraf T, et al. 2019. MethMotif: an integrative cell specific database of transcription factor binding motifs coupled with DNA methylation profiles. Nucleic Acids Res. 8(47):D145–154.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RK, Merico D, Cao H, Pellecchia G, Alipanahi B, Thiruvahindrapuram B, Tong X, Sun Y, Cao D, Zhang T et al. 2016. Genome-wide characteristics of de novo mutations in autism. NPJ Genomic Med. 1:16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR. 2015. The Ensembl regulatory build. Genome Biol. 16:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Mokhtari R, Pedrosa E, Birnbaum R, Zheng D, Lachman HM. 2017. Transcriptome analysis of microglia in a mouse model of Rett syndrome: differential expression of genes associated with microglia/macrophage activation and cellular stress. Mol Autism. 8(17):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Zhang S, Fan X, Wu Q, Yan L, Dong J, Zhang H, Li L, Sun L, Pan N et al. 2018. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature. 555:524–528. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Previously published datasets for human ASD and Control BA9 as well as Dup15q and Control BA19 samples are available at GEO GSE81541. Single-Cell RNAseq data from human brain across gestational weeks are from GEO GSE104276 (Breton et al. 2017). Microglial developmental RNAseq data are available at GEO GSE99622 (Zhong et al. 2018). Previously unpublished human ASD, Rett and Control BA9 samples are available at GEO GSE119981. All analysis code is available on Github at https://github.com/aciernia, https://github.com/ben-laufer/CpG_Me, https://github.com/ben-laufer/DMRichR, and https://github.com/hyeyeon-hwang.