

Abstract
Universal immune receptors represent a rapidly emerging form of adoptive T-cell therapy with the potential to overcome safety and antigen escape challenges faced by conventional chimeric antigen receptor (CAR) T-cell therapy. By decoupling antigen recognition and T-cell signaling domains via bifunctional antigen-specific targeting ligands, universal immune receptors can regulate T-cell effector function and target multiple antigens with a single receptor. Here, we describe the development of the SpyCatcher immune receptor, the first universal immune receptor that allows for the post-translational covalent attachment of targeting ligands at the T-cell surface through the application of SpyCatcher-SpyTag chemistry. The SpyCatcher immune receptor redirected primary human T cells against a variety of tumor antigens via the addition of SpyTag-labeled targeting ligands, both in vitro and in vivo. SpyCatcher T-cell activity relied upon the presence of both target antigen and SpyTag-labeled targeting ligand, allowing for dose-dependent control of function. The mutational disruption of covalent bond formation between the receptor and the targeting ligand still permitted redirected T-cell function but significantly compromised antitumor function. Thus, the SpyCatcher immune receptor allows for rapid antigen-specific receptor assembly, multiantigen targeting, and controllable T-cell activity.
Graphical Abstract
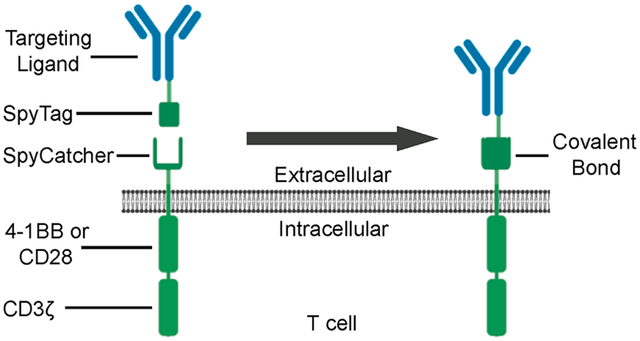
INTRODUCTION
Chimeric antigen receptor (CAR) T cells can mediate dramatic responses in the treatment of certain hematological malignancies, leading to the FDA approval of two CD19-targeting CAR T-cell products, tisagenlecleucel for the treatment of relapse/refractory (r/r) B-cell acute lymphoblastic leukemia (B-ALL) and diffuse large B-cell lymphoma (DLBCL) and axicabtagene ciloleucel for the treatment of (r/ r) large B-cell lymphoma.1–7 Due to high remission rates and prolonged tumor-free survival of CD19 CAR T-cell-treated patients, the field has expanded their use to other malignancies. Clinical trials of CAR T cells targeting other B-cell-specific antigens, such as BCMA, CD20, and CD22,8–10 have produced encouraging results, but several challenges, including those related to unique toxicities and subsequent relapses, need to be addressed before the widespread success of CAR T-cell therapy is achieved in hematologic malignancies and solid tumors.11
CARs are composed of an extracellular antigen targeting domain, such as an scFv, attached to intracellular T-cell signaling and costimulatory domains (e.g., 41BB and/or CD28 in tandem with CD3ζ), allowing for antigen-specific, MHC-independent T-cell targeting.12 This design, though effective for use in single-antigen targeting, presents inherent limitations to broadening the use of CAR T cells across multiple tumor types, as well as the potential for serious adverse events and toxicities.
While most drugs allow dose adjustment and follow predictable pharmacokinetics and pharmacodynamics, conventional CAR T-cell therapies are living drugs that cannot be easily controlled following their infusion. Upon recognition of target antigen, the administered CAR T cells can rapidly proliferate to large numbers in the recipient and release proinflammatory cytokines, in some cases leading to severe and sometimes fatal side effects such as cytokine release syndrome (CRS),3 neurotoxicity, and cerebral edema,13 which require medical management. In some cases, CAR T cells target and destroy nonmalignant tissues that also express the targeted antigen, leading to potentially fatal on-target, off-tumor toxicity.14,15
In addition to these challenges, the rigid CAR architecture also restricts targeting to a single tumor-associated antigen (TAA). Though this approach can be effective when targeting a ubiquitous pan-B cell marker such as CD19, its effectiveness is compromised when targeting tumors with heterogeneous TAA expression or in the setting of a relapsed antigen-negative tumor. About 35% of tisagenlecleucel CD19 CAR T-cell recipients relapse after treatment, and more than half of relapsed disease is associated with a genetic mechanism of CD19 antigen loss due to protein truncation with a nonfunctional or absent transmembrane domain.16 Alternative mechanisms of antigen loss include the emergence of antigen splice variants lacking the targeted antigenic epitope, tumor cell lineage switching, trogocytosis of the CD19 antigen, and, in one rare case, the unintentional introduction of the CAR gene into a leukemic B-cell.17–22 Single antigen targeting is also problematic in the treatment of solid tumors, which are often composed of tumor cells with varying antigen expression patterns. In this case, selective targeting of a single antigen can cause incomplete clearance and adaptive resistance, as has been reported in the targeting of EGFRvIII.23
In order to expand the prototypic CAR architecture to allow for temporal and quantitative control of T-cell effector function, we created the first ever tag-specific receptor capitalizing on the interaction between biotin and avidin.24 This receptor, called the biotin-binding immune receptor, belongs to a rapidly expanding class of orthogonal receptors termed universal immune receptors (UIRs).25–35 UIRs are composed of two split but interactive parts: (i) standard intracellular T-cell signaling domains attached to an extracellular adaptor protein and (ii) targeting ligands that are able to bind the adaptor protein. The targeting ligand acts as an immunologic bridge, binding both the TAA on-target cells and the extracellular adaptor on the receptor, eliciting antigen-specific T-cell effector function.
By decoupling TAA recognition from T-cell signaling, UIRs can address several issues currently faced by CAR T cells. UIRs rely on the presence of antigen-specific targeting ligands to drive T-cell effector function, therefore allowing dose-dependent control of T-cell effector function25 and the mitigation of toxicities seen with CAR T cells due to rapid activation and expansion, such as CRS.33 Additionally, UIRs provide modular platforms capable of targeting multiple TAAs with a single-cell product via the use of multiple targeting ligands, addressing issues of TAA heterogeneity and relapse associated with single antigen loss.
At present, all UIRs rely on noncovalent interactions between the targeting ligand and the UIR. Here, we detail the development of a next-generation UIR system that allows for the post-translational covalent attachment of targeting domains to the receptor via the use of SpyCatcher-SpyTag chemistry. Developed by Zakeri et al., the interaction between the SpyCatcher protein and its cognate peptide SpyTag leads to the rapid and spontaneous formation of a covalent amide bond.36 The reaction between the peptide–protein pair occurs across a range of physiologically relevant temperatures and pH levels, making it a potential candidate for use in vivo.36 SpyCatcher-SpyTag has also been used to label membrane-resident receptors in live mammalian cell culture, as well as in transgenic C. elegans, endogenously expressing proteins containing SpyCatcher and SpyTag domains.37 Taken together, the broad range of robust reactivity conditions, coupled with its use in both mammalian cells and live organisms, makes the SpyCatcher-SpyTag system a favorable choice for the de novo assembly of UIR architectures that mimic CARs.
We have developed a SpyCatcher immune receptor that contains the SpyCatcher protein as its extracellular domain attached to standard second-generation CAR intracellular signaling domains. The addition of TAA-specific targeting ligands labeled with SpyTag allows for the on-demand formation of a CAR-like receptor through spontaneous, autocatalytic isopeptide bond formation. Primary human T cells expressing the SpyCatcher immune receptor can be quantitatively loaded with targeting ligands, allowing for dose-titratable control of redirected T-cell effector function and tumor cell lysis. Even as a single-cell product, SpyCatcher immune receptor T cells can recognize an array of tumor antigens via the addition of clinical-grade antibodies site-specifically labeled with SpyTag, as well as targeting ligands to which SpyTag is genetically fused. The results described here demonstrate the flexibility and efficacy of the SpyCatcher immune receptor system.
EXPERIMENTAL SECTION
Cloning of A24-Protein G-SpyTag (A24-PGST), SpyCatcher, and DARPin-SpyTag Constructs.
The HTB1 domain of protein G containing an amber codon in the 24th amino acid position, fused to a 7xGGS linker and SpyTag, was cloned into the pSTEPL vector via the NdeI and AgeI cloning sites as detailed in previous studies.38,39 myc-DARPin9.26-SpyTag, myc-E01-SpyTag, and myc-Ec1-SpyTag were synthesized (GeneArt) and cloned in the pSTEPL vector via NdeI and AgeI cloning sites.40–42 To generate Flag-E01-SpyTag, polymerase chain reaction (PCR) extension was used to add an NdeI cut site and Flag-tag at the 5′ end of the E01-SpyTag construct. Flag-E01-SpyTag was then cloned into the pSTEPL vector following the methods detailed above. Myc-RFP-SpyTag was generated via the PCR amplification of RFP using primers to add an NdeI cut site and myc-tag at the 5′ end and an XhoI cut site at the 3′ end and was subsequently cloned into the pSTEPL vector upstream of a 7xGGS-SpyTag using the added cut sites. SpyCatcher-Venus was generated via PCR amplification of Venus using primers to add an NheI cut site at the 5′ end and XhoI cut site at the 3′ end and was subsequently cloned into the pSTEPL vector downstream of a SpyCatcher-7xGGs domain using the added cut sites.
To generate SpyTag-DA versions of the above constructs, QuickChange site-directed mutagenesis (Agilent) was used, along with a forward (5′-GCATATCGTTATGGTCGCTGCTTACAA-GCCAACGA-3′) and reverse (5′-TCGTTGGCTTGTAAGCAG-CGACCATAACGATATGC-3′) primer to introduce an A to C point mutation in the SpyTag coding region, mutating Asp117 to Ala117.36
All plasmid sequences were sequence verified.
Expression and Purification of Bacterially Expressed Proteins.
The pSTEPL plasmid containing the A24-protein G-SpyTag (A24-PGST) sequence and pEVOL-pBpF were cotrans-formed into T7 Express Competent E. coli (New England Biolabs). Bacterial starter cultures in lysogeny broth (LB) with 100 mg/mL ampicillin and 25 mg/mL chloramphenicol were grown for 8 h in a shaking incubator (230 rpm, 37 °C). Starter cultures were then diluted 1:1000 in an autoinduction medium (Formedium) containing 100 μg/mL ampicillin, 25 μg/mL chloramphenicol, 500 μM L-benzoylphenylalanine (Bachem), 0.1% w/v L-(+)-arabinose (Sigma-Aldrich), and 0.5% v/v glucose and grown for 16h in a shaking incubator (230 rpm, 37 °C).
All pSTEPL plasmids containing DARPin and RFP constructs were transformed into T7 Express Competent E. coli (New England Biolabs). Cultures were grown as stated above in an autoinduction media (Formedium) containing 100 mg/mL ampicillin and 0.5% v/v glucose.
Cultures were pelleted at 7000 rpm for 10 min. Pellets were resuspended in B-PER lysis buffer supplemented with 0.4 mg/mL lysozyme, 4 μg/mL DNase, and EDTA-free cOmplete protease inhibitor cocktail (Roche) in a ratio of 5 mL of buffer per 0.8–1 g of pellets. Lysate was rotated at room temperature for 1 h, followed by a 30 min freeze at −80 °C. Cell lysate was then thawed at room temperature, followed by centrifugation at 15 000g for 10 min at 4 °C. The aqueous layer of the lysate was collected and incubated for 1 h at room temperature in a 10 mL Poly-Prep chromatography column containing Talon 50/50 metal affinity resin (1 mL of 50/50 resin/100 mL bacterial culture; Clontech). Lysate was allowed to pass through the column, and the resin beads were washed with 5 column volumes of sterile 1×PBS without magnesium and calcium (Corning).
Sortase cleavage purification was run as previously described.38 Briefly, 1×PBS containing 2 mM triglycine and 50 μM calcium chloride was added to each column (500 μL buffer per 1 mL of 50/50 resin) and incubated for 2 h at 37 °C. Elutions were collected, filter sterilized, and stored at 4 °C for later use.
A BCA assay kit (Pierce) was used to determine the protein concentration. To test the functionality of the SpyTag domain, proteins were mixed with excess SpyCatcher-Venus and incubated for 30 min at room temperature. Samples were run on SDS-PAGE reducing gels (Bio-Rad) to confirm isopeptide bond formation. For in vivo studies, endotoxin was removed using a 1% Triton X-114 phase separation method.43 Endotoxin levels were confirmed to be <10 endotoxin units/mL using the Endosafe-PTS system and limulus amebocyte lysate (LAL) test cartridges (Charles River Laboratories).
Light-Activated Site-Specific Conjugation of A24-Protein G-SpyTag to Clinical Grade IgGs.
A24-Protein G-SpyTag (A24-PGST) was cross-linked to clinical-grade IgGs (trastuzumab, rituximab, and cetuximab) following methods previously described by Hui et al.39 Briefly, after A24-PGST purification, small pilot conjugation tests were conducted to determine the volume of A24-PGST needed to fully cross-link 1 μg of IgG. Conjugation was run for 2 h at 4 °C under UV light, and samples were run on SDS-PAGE reducing gels (Bio-Rad) to confirm the conjugation of the IgG heavy chains with A24-PGST. The volume/weight ratio determined to achieve maximal conjugation was then used for large batch production following the same steps.
To remove excess A24-PGST, samples were centrifuged in 100 kDa MWCO spin-filtration columns (Millipore Sigma) at 14 000g, followed by a 5× column volume wash with 1×PBS. The protein concentration was determined via a BCA assay kit (Pierce). Samples were stored at 4 °C for later use.
SDS-PAGE.
Sodium dodecyl sulfate polyacrylamide gel electro-phoresis was performed to determine the expression of proteins, the cross-linking of Protein G-SpyTag and IgG heavy chains, and the covalent bond formation between SpyTag-labeled targeting ligands and SpyCatcher reagents. Samples were boiled at 95 °C for 5 min in the presence of 2-mercaptoethanol and loaded onto 4–15% gradient Tris/glycine gels (Bio-Rad). Gels were stained for 1 h with SimplyBlue SafeStain (Invitrogen), followed by overnight destaining with water.
Cloning of Lentiviral Constructs and Lentiviral Packaging.
A gene fragment encoding the truncated version of SpyCatcher (SpyCatcherΔ)44 was synthesized (GeneArt) and digested with restriction enzymes BamHI and NheI. The inset was ligated into a third-generation replication-incompetent lentiviral vector containing either CD28-CD3ζ or 41BB-CD3ζ or lacking functional intracellular T-cell signaling domains (delta zeta, Δζ) intracellular domains. A region encoding GFP and a T2A site were upstream of the receptor, allowing for the detection of transgene expression using GFP as a surrogate marker. The same methodology was used to clone anti-Her2 chimeric antigen receptors using the Herceptin-derived scFv 4D545 containing either CD28-CD3ζ or 41BB-CD3ζ intracellular signaling domains. The expression of all receptors was driven by the EF1α promoter.46
HER2 WT was a gift from Mien-Chie Hung (Addgene plasmid no. 16257; https://www.addgene.org/16257 RRID:Addgene_16257).47 EGFR WT was a gift from Matthew Meyerson (Addgene plasmid no. 11011; https://www.addgene.org/11011 RRID:Addgene_11011).48 The Her2 and EGFR sequences were amplified via PCR and digested with restriction enzymes XbaI and SalI. Gene sequences were ligated into a third-generation replication-incompetent lentiviral packaging vector downstream from an EF1α promoter.
Replication-incompetent lentivirus was produced using HEK293T cells (Invitrogen) as previously described.49 Briefly, pELNS transfer plasmid and lentiviral packaging plasmids pVSV (VSV glycoprotein expression vector), pRSV.REV (Rev expression vector), and pMDLg/ p.RRE (Gag/Pol expression plasmid) were transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen). Supernatants were harvested at 24 and 48 h, and virus was pelleted via ultracentrifugation at 25 000 rpm for 2 h. Concentrated virus was stored at −80 °C until use. The viral titer (IU/mL) was determined using viral transduction of HEK293T cells through measurement of the surrogate marker GFP.
Cell Lines.
Established human tumor cell lines SKOV3, MDA-MB-468, A1847, CRL5803, MDA-MB-361, Ramos, and HEK293T were purchased from the American Type Culture Collection (ATCC). Cells were cultured in complete medium (CM) composed of RPMI 1640 (GIBCO) supplemented with 10% FBS (VWR), 100 U/mL of penicillin, and 100 μg/mL of streptomycin at 37 °C and 5% CO2. All cell lines were routinely tested for mycoplasma.
To generate Ramos-Her2 and Ramos-EGFR lines, Ramos cells were transduced with lentivirus containing the coding sequence of either protein. Cells were then propagated, stained with either anti-Her2 (APC; BioLegend) or anti-EGFR antibody (PE; BioLegend), and sorted for protein expression using fluorescence-activated cell sorting (FACS).
To generate SKOV3-SpyCatcher-BBζ, SKOV3 cells were transduced with lentivirus containing the coding sequence for the GFP-T2A-SpyCatcher-BBζ. Cells were propagated and sorted for GFP expression using FACS.
Healthy donor primary human T cells were purchased from the Human Immunology Core (University of Pennsylvania). CD4+ and CD8+ T cells were combined in equal amounts and stimulated with anti-CD3/CD28 beads (Invitrogen) in a 3:1 ratio. During the expansion process, T cells were maintained in CM at 37 °C and 5% CO2. After 24 h, lentivirus was added at a multiplicity of infection (MOI) of 5–10. Seven days after activation, the anti-CD3/CD28 beads were removed from culture via magnetic separation. T cells were cultured for an additional 7 days at a density of (0.5–1) × 106 cells/mL. CM was supplemented with 50–100 IU/mL IL-2 (Prometheus Therapeutics and Diagnostics) after lentivirus addition and maintained until 2 days before T-cell use in functional assays. The T-cell count and volume were continually tracked during the expansion process using a Coulter Counter (Beckman Coulter). The transduction efficiency was detected by flow cytometry for the expression of GFP.
SpyCatcher T-Cell Arming, Detection of Armed Receptors, and Detection of Surface Proteins.
SpyCatcher-expressing T cells were resuspended in CM containing various concentrations of SpyTag-labeled targeting agent. Cells were incubated for 30–60 min at 37 °C and 5% CO2 and then washed two times with CM. To stain IgG-SpyTag-armed T cells, goat polyclonal antihuman IgG (Sigma-Aldrich) conjugated with LightningLink APC (Expedeon) was used. To stain myc-or Flag-DARPin-SpyTag-armed T cells, either anti-Myc-Alexa647 (Cell Signaling Technologies) or anti-FLAG-BV421 (BioLegend) was used. Stained cells were analyzed by flow cytometry.
For receptor turnover experiments, T cells were expanded and rested to an average volume of <300 fL and then armed with either SpyTag-labeled IgG or SpyTag-labeled DARPin, as described above. For basal turnover, T cells were maintained in CM and stained for loaded receptor every 24 h for a total of 96 h. For antigen-induced turnover, T cells were combined at a 3:1 effector to target ratio and stained after 24 h in culture.
The expression of 4D5-BBζ and 4D5–28ζ receptors was detected through incubation with soluble Her2-his protein (Sino Biological), followed by staining with anti-His-PE antibody (BioLegend). Her2 expression and EGFR expression were detected using anti-Her2-APC (BioLegend) or anti-EGFR-PE (BioLegend), respectively. Stained cells were analyzed by flow cytometry.
Western Blot.
SKOV3-SpyCatcher-BBζ cells were incubated with either 2000 nM myc-RFP-SpyTag or myc-RFP-SpyTag-DA for 1 h at 37 °C. Cells were lysed using RIPA lysis buffer with protease-inhibitor cocktail (Roche, cat. no. 5892970001) and centrifuged for 5 min. Lysate was then collected, and protein concentrations were quantified using a BCA assay (Thermo Scientific). Protein samples (80 μg) were mixed with loading buffer (Lammeli buffer; BioRad) containing 5% β-mercaptoethanol (BioRad) and incubated at 95 °C for 5 min. Samples were loaded in 4–15% Minigels protean TGX (BioRad) and run at 150 V for 1 h. A protein ladder (BioRad) was run along with the samples. Protein samples were transferred to a PVDF membrane (Millipore) at 100 V for 1 h. The membranes were washed with TBST (1% Tween; BioRad) and incubated with primary and secondary antibodies, including purified mouse anti-human CD3ζ (BD Pharmigen; 1:1000), anti-human/mouse/rat GAPDH, (R&D; 1:20 000), and peroxidase AffiniPure goat anti-mouse IgG (Jackson Immunology; 1:10 000). Membranes were washed three times in between the primary and secondary antibody incubation steps. Membranes were developed using the ECL prime Western blotting detection reagent (GE Healthcare no. RPN2236) and imaged using a GE ImageQuant LAS 4000 series imaging system.
Testing Effector Functions of SpyCatcher T Cells.
For cytokine secretion assays, Herceptin-ST proteins were diluted in coating buffer (BioLegend) and incubated in a flat-bottom MaxiSorp plate (Sigma) overnight at 4 °C. Wells were washed twice with PBS, and 70 000 immune receptor-(+) cells were added to each well. Plates were covered with a breathable seal and incubated at 37 °C in 5% CO2 for 16 h. Harvested supernatant was stored at −20 °C for later use. IFNγ secretion levels were assessed using the human IFNγ ELISA kit (BioLegend) following the included protocol.
End-point testing of the lytic function was done using the Luc-Screen Extended-Glow Luciferase Reporter Gene Assay System (Applied Biosystems). For armed SpyCatcher T-cell lysis, SpyCatcher T-cells were incubated with precise concentrations of the appropriate SpyTag-labeled targeting ligand as described above. Armed SpyCatcher T cells and click beetle green luciferase (CBG)-expressing tumor cells were combined in a 7:1 E:T ratio in 200 μL of phenol-free CM and incubated overnight at 37 °C and 5% CO2. Plates were centrifuged at 1200 rpm for 5 min, 100 μL of medium was removed from each well, and luciferase buffer was added according to the manufacturer’s protocol. Luciferase readings were obtained using a microplate reader. For on-demand lysis, SpyCatcher T cells, targeting ligand, and tumor cells were combined simultaneously in phenol-free CM. All other steps were carried out in the same manner as for armed lysis. Cytotoxicity was calculated using the following equation: [1 − (T cells + targeting ligand + target cell)/(target cells alone) − 1 − (T cells + target cell)/(target cells alone)] × 100.
Real-time lysis analysis was carried out using the xCELLigence Real Time Cell Analysis instrument (ACEA Biosciences). Adherent tumor cells were plated and incubated in the instrument overnight at 37 °C with 5% CO2. SpyCatcher T cells were then added after arming as described above. For on-demand lysis, SpyCatcher T cells were added to the tumor cells and incubated for 4 h, followed by targeting ligand addition. Cytotoxicity calculations were made using RTCA software (ACEA Biosciences).
Xenograft Models.
NOD-scid IL2Rγnull mice were purchased from the Stem Cell and Xenograft Core (University of Pennsylvania). Female mice (6–12 weeks old) were kept in a pathogen-free environment following protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee. For intraperitoneal (I.P.) tumor models, mice were injected I.P. with 1 × 106 SKOV3-CBG+GFP tumor cells. After 7 days, 12.5 × 106 SpyCatcher immune receptor T cells were armed with 1000 nM Herceptin-ST and injected I.P. SpyTag-labeled targeting ligand was injected I.P. 1 day after T-cell injection, followed by subsequent injections every 3 days until treatment cessation. Tumor progression was measured via bioluminescence imaging as described previously50 and quantified as the average radiance, gated on the abdomen between the fore and hind limbs. Mice were sacrificed upon gaining 20% body weight due to ascites formation. Mice that formed palpable subcutaneous (S.C.) tumors at the site of injection by day 11 post-tumor injection were excluded from all groups.
Blood samples were collected via retro-orbital bleeds by the Stem Cell and Xenograft Core (University of Pennsylvania). Peripheral blood T cells were quantified using Trucount Tubes following the provided protocol (BD Biosciences). The staining panel consisted of antihuman CD3 (brilliant violet 605; BioLegend), antihuman CD45 (PE; eBiosciences), and antihuman CD8 (APC-H7; BD Biosciences).
For S.C. tumor models, mice were subcutaneously injected in the flank with 1 × 106 SKOV3-CBG+GFP tumor cells. After 6 days, 12.5 × 106 SpyCatcher immune receptor T cells were armed and I.P. injected. SpyTag-labeled targeting ligand was I.P. injected 1 day after T-cell infusion, followed by subsequent injections every 3 days until treatment cessation. Tumor progression was measured via caliper measurement and quantified using the formula volume = 3.14/6(length × (width)2), with length being the longest diameter and width being the shortest diameter.
Statistical Analysis.
Data are reported as mean ± standard deviation (SD) unless otherwise noted. Statistical analysis was performed using an unpaired 2-tailed t test unless otherwise noted. GraphPad Prism 8.0 software was used for statistical analysis. P < 0.05 was considered to be significant.
RESULTS
Generation of SpyTag-Labeled Tumor Antigen Targeting Ligands.
The SpyCatcher immune receptor system is composed of two main components: targeting ligands containing a SpyTag domain and T cells expressing the SpyCatcher immune receptor (Figure 1A). We used two approaches to generate SpyTag-containing targeting ligands: site-specific conjugation and genetic fusion. Site-specific attachment of SpyTag to clinical-grade IgGs was achieved using a light-activated site-specific conjugation (LASIC) adaptor protein developed by Hui et al.39 This adaptor protein is derived from protein G, an IgG-binding bacterial cell wall protein that interacts with the Fc portion of human IgGs at the CH2–CH3 junction.51 Using an amber-tRNA suppressor aminoacyl-synthase pair, Protein G-SpyTag was expressed with the unnatural amino acid benzoylphenylalanine (BPA) incorporated at the A24 amino acid position. When combined with IgGs, A24-Protein G-SpyTag binds to the Fc domain of the IgG noncovalently. Upon exposure to UV light (365 nm), the BPA molecule is activated and covalently cross-links the Protein G-SpyTag site-specifically to the Fc domain (Figure 1A). The final IgG molecule thus contains two covalently linked Protein G-SpyTag molecules, one on each side of the Fc domain (Figure 1A).
Figure 1.
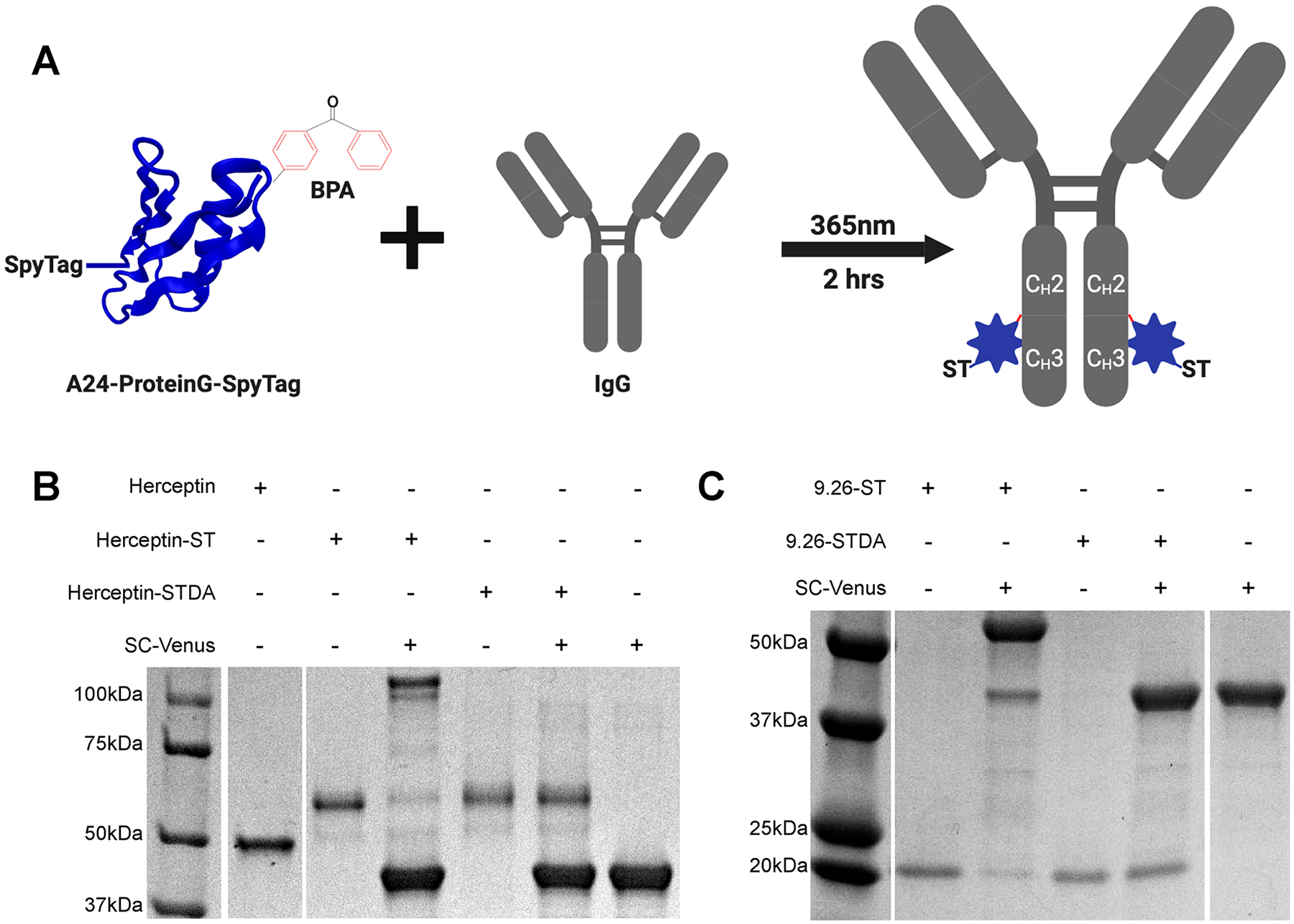
Development of SpyCatcher immune receptor targeting ligands. (A) Schematic representation of Protein G-ST cross-linking to clinical-grade human IgGs. (B) Cross-linking of Herceptin with Protein G-ST or Protein G-STDA, followed by subsequent reaction with SpyCatcher-Venus analyzed by SDS-Page gel under reducing conditions with coomassie staining. (C) DARPin9.26-ST and DARPin9.26-STDA reacted with SpyCatcher-Venus analyzed by SDS-Page gel with Coomassie staining.
An analysis of clinical-grade antibodies cross-linked with Protein G-SpyTag by reducing SDS-page gel demonstrated nearly full cross-linking of the IgG heavy chain for Herceptin (Figure 1B), cetuximab, and rituximab as demonstrated by a band shift equal to the combined masses of the two proteins (~60 kDa combined) (Figure S1A). To demonstrate the maintained functionality of the SpyTag domain after UV exposure, we incubated Herceptin-SpyTag with excess SpyCatcher-Venus (38 kDa). We observed the formation of a shifted band that was maintained under boiling and reducing conditions at the approximate combined mass of both proteins (100 kDa) as well as the loss of the single heavy chain-Protein G band, indicating the formation of a covalent bond between the two proteins (Figure 1B). In order to further broaden the repertoire of targeting ligand types, we expressed Her2 (DARPin9.26; 22.5 kDa)40 (Figure 1C), EGFR (E01; 22.4 kDa),41 or EpCAM (Ec1; 22.8 kDa)42 (Figure S1B) targeting the designed ankyrin repeat proteins (DARPins) containing a C-terminal SpyTag domain using the sortase-tag-expressed protein ligation (STEPL) system.38 SpyTag functionality was again validated via conjugation with excess SpyCatcher-Venus, demonstrating the formation of a shifted band resistant to degradation under boiling and reducing conditions at the approximate combined molecular weights of the two protein components (61 kDa; Figure 1C and Figure S1B).
Previous studies have demonstrated that the reaction of a SpyTag variant containing an aspartic acid to alanine mutation (SpyTag-DA) abolishes covalent bond formation with SpyCatcher while still allowing for the formation of a noncovalent complex with a Kd of 200 nM.36 To serve as a negative control for covalent bond formation, Herceptin-STDA and 9.26-STDA targeting ligand were also produced (Figure 1B,C).
Cloning, Expression, and Detection of the SpyCatcher Immune Receptor.
The truncated, 84 amino acid version of SpyCatcher44 was cloned into previously validated50,52 lentiviral constructs containing either 4–1BB or CD28 costimulatory domains in tandem with CD3ζ to produce SpyCatcher-BBζ and SpyCatcher-28ζ immune receptors. A SpyCatcher receptor lacking intracellular signaling domains was also generated to serve as a negative control (SpyCatcher-Δζ; Figure 2A). All constructs contained eGFP upstream of a T2A self-cleaving peptide, allowing for the coexpression of GFP and the receptor as separate proteins, as well as the use of GFP as a surrogate marker for primary human T-cell transduction.
Figure 2.
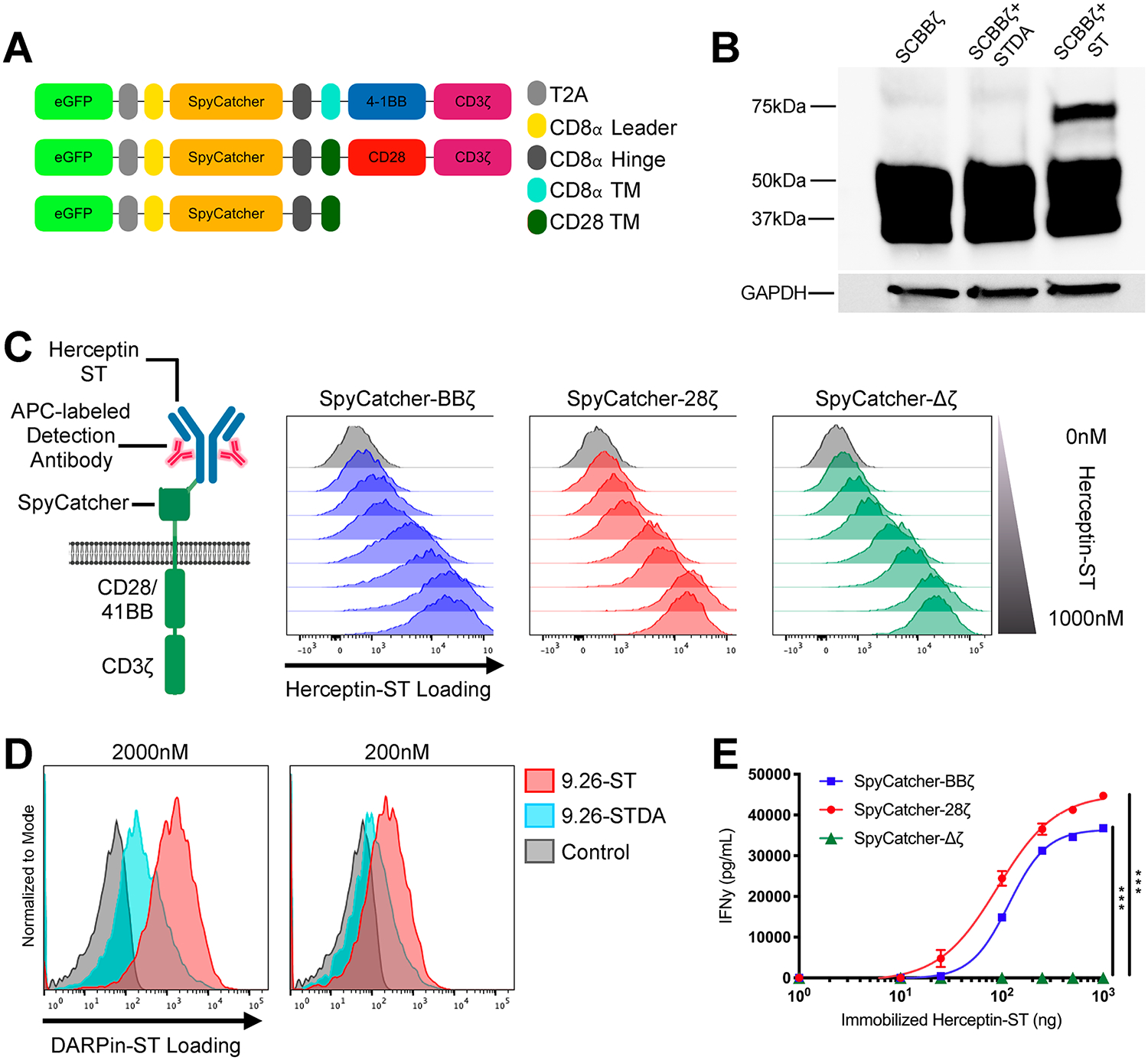
SpyCatcher immune receptor is expressed and capable of covalent loading with SpyTag-labeled ligands. (A) Schematic of the lentiviral SpyCatcher immune receptor constructs. (B) SpyCatcher immune receptor-expressing SKOV3 cells were incubated in culture medium containing 2000 nM RFP-ST or RFP-STDA. Covalent bond formation between the two components was detected via SDS-Page gel under reducing conditions and Western blot staining for total CD3ζ protein. (C) SpyCatcher T cells were armed with varying amounts of Herceptin-ST for 1 h at 37 °C. Herceptin-ST loading onto the SpyCatcher immune receptor was detected by staining with APC polyclonal antihuman IgG and flow cytometric analysis. (D) Comparison of covalent (DARPin9.26-ST) vs noncovalent (DARPin9.26-ST) maximal loading of the SpyCatcher immune receptor at various concentrations. (E) SpyCatcher T cells were incubated in wells containing various amounts of immobilized Herceptin-ST for 16 h. Supernatant was harvested and analyzed for IFNγ by ELISA. Error bars represent the mean ± standard deviation. Data points are the averages of three replicates. A representative T-cell donor is shown. *P < 0.05, **P < 0.01, and ***P < 0.001.
To determine whether SpyTag can covalently link to a SpyCatcher immune receptor expressed on the cell surface, an immortalized cancer cell line, SKOV3, was transduced to express the SpyCatcher-BBζ construct (Figure S2A) and then incubated with the soluble RFP-SpyTag protein. Incubation resulted in the formation of a shifted band that was resistant to degradation under reducing conditions when assessed via Western blot staining for CD3ζ, confirming covalent bond formation between the two proteins (Figure 2B). The remaining unshifted band between 50 and 37 kDa likely represents intracellular receptor since the whole cell lysate was loaded onto the gel. Incubation with RFP-SpyTag-DA protein showed that the mutation of the critical aspartic acid residue in SpyTag to an alanine abolished its ability to form a covalent bond with SpyCatcher (Figure 2B). Next, primary human T cells were transduced to express the SpyCatcher immune receptor and incubated with Herceptin-ST at various concentrations, which resulted in dose-dependent loading (“arming”) of the receptor, as detected via staining with APC antihuman IgG polyclonal antibody (Figure 2C). Additionally, covalent bond formation between the SpyTag-conjugated targeting ligand (9.26-ST) and the SpyCatcher receptor was necessary to achieve maximum targeting ligand arming, especially at low concentrations (Figure 2D).
In Vitro Efficacy of SpyCatcher T Cells.
In order to determine if SpyTag engagement of the SpyCatcher immune receptor on primary human T cells would induce specific activation, SpyCatcher T cells were incubated in the presence of various amounts of immobilized Herceptin-ST. Both SpyCatcher-BBζ and SpyCatcher-28ζ T cells, but not SpyCatcher-Δζ T cells, secreted IFNγ in the presence of immobilized Herceptin-ST in a dose-dependent fashion (Figure 2E), with SpyCatcher-28ζ T cells being more immunologically sensitive than SpyCatcher-BBζ T cells (P < 0.001).
Since the SpyCatcher immune receptor is the first universal immune receptor that can be covalently armed with targeting ligands, permanently affixing antigen specificity to the receptor until it is degraded, we evaluated the lytic capabilities of prearmed SpyCatcher T cells in the absence of excess targeting ligand. Here, we sought to determine if a sufficient amount of targeting ligand could be used to covalently arm the receptors at the cell surface to elicit T-cell lysis of cancer cells upon antigen recognition (Figure 3A, left). SpyCatcher-BBζ and SpyCatcher-28ζ T cells armed with Herceptin-ST lysed Her2+ SKOV3 tumor cells, while untransduced and SpyCatcher-Δζ T cells armed with Herceptin-ST showed minimal lytic activity after 20 h of coculture (Figure 3B). Lysis occurred in a dose-dependent manner, with increasing Herceptin-ST arming concentration correlating with increased lytic capacity (Figure 3B), corresponding with the dose-dependent receptor loading observed previously (Figure 2C). The observation of lytic function using live cell imaging confirmed these results while also demonstrating the arming of dose-dependent T-cell clustering (Videos S1–S4). Tumor cell lysis and T-cell clustering were not observed with SpyCatcher-Δζ T cells armed with 1000 nM Herceptin-ST, demonstrating that lysis and clustering are activation-dependent (Video S5).
Figure 3.
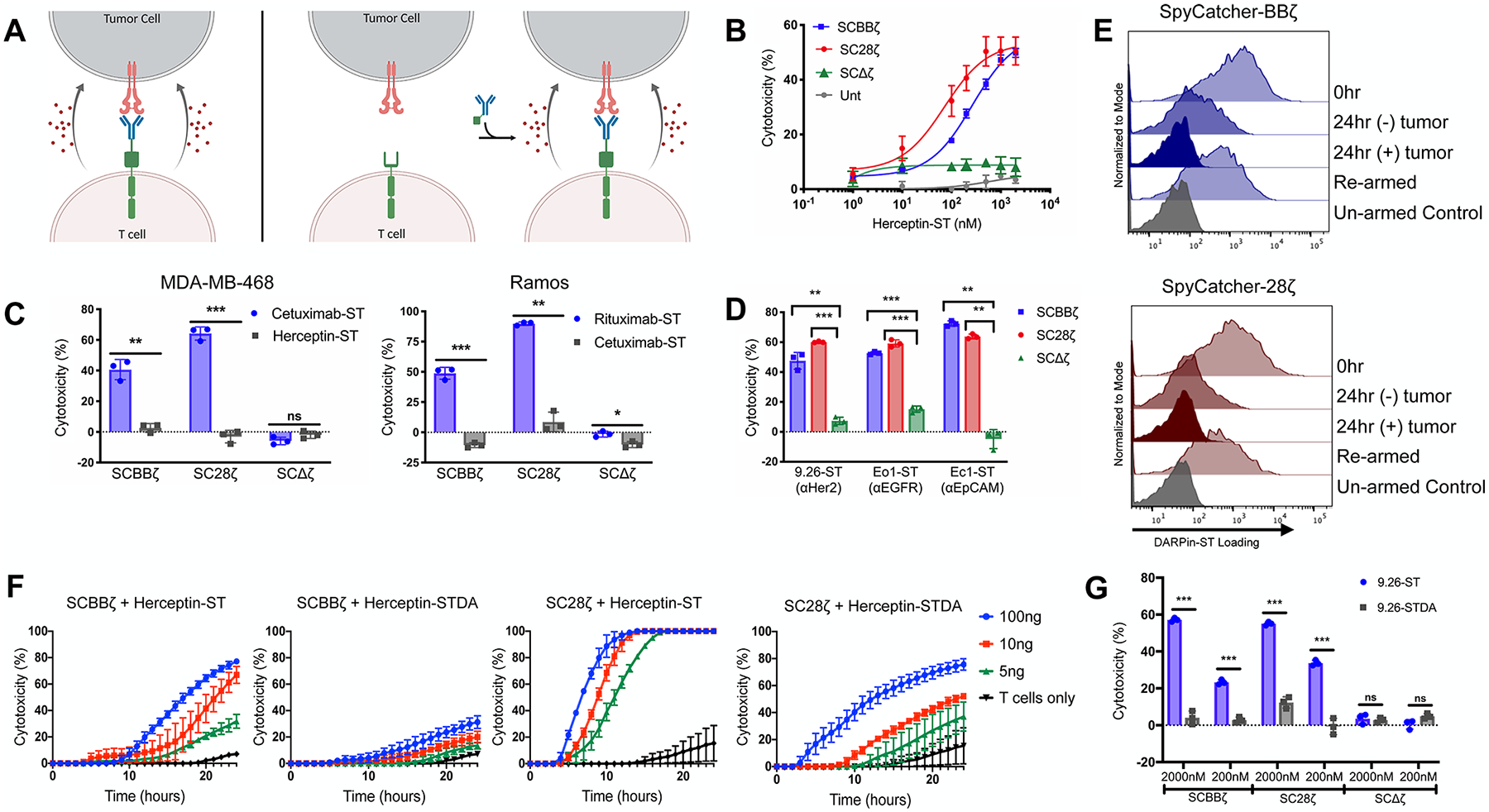
SpyCatcher T cells are capable of in vitro lytic function against a host of different antigen-expressing tumor lines. (A) Schematic representation of armed SpyCatcher immune receptor lysis (left) and on-demand lysis (right). (B) SpyCatcher or untransduced (Unt) T cells were armed with various concentrations of Herceptin-ST and cocultured in the presence of SKOV3 (Her2+) tumor cells. (C) SpyCatcher T cells were armed with antigen-specific and nonspecific IgGs and cocultured with either MDA-MB-468 (EGFR+/Her2-) or Ramos (CD20+/EGFR-) tumor cells. (D) SpyCatcher T cells armed with antigen-specific DARPins were coculture with SKOV3, MDA-MB-468, or A1847 (EpCAM+) tumor cells expressing luciferase. (E) DARPin9.26-ST armed SpyCatcher T cells were incubated with or without SKOV3 tumor cells (E/T = 3:1) for 24 h, removed from culture, and incubated in media ± 2000 nM DARPin9.26-ST. Receptor arming was analyzed via anti-myc staining and flow cytometric analysis. (F) Unarmed SpyCatcher T cells were incubated with SKOV3 tumor cells for 4 h followed by the addition of Herceptin-ST or Herceptin-STDA (time = 0). (G) SpyCatcher T cells were armed with either DARPin9.26-ST or DARPin9.26-STDA at various concentrations and cocultured with SKOV3 tumor cells expressing luciferase. (B, C: MDA-MB-468, F) Lysis was measured using real-time cell analysis. (C: Ramos, D, G) Residual luciferase expression was calculated after 20 h. All cocultures were carried out using a 7:1 E/T ratio. Error bars represent the mean ± standard deviation. Data points are averages of three replicates. A representative T-cell donor is shown. *P < 0.05, **P < 0.01, and ***P < 0.001.
Additionally, dose-dependent arming also impacted the ability of SpyCatcher T cells to recognize tumor cells expressing various levels of target antigen. SpyCatcher-BBζ T cells were capable of lysing tumor cells expressing high levels of antigen in an arming-concentration-dependent manner, but lost functionality against cell lines expressing lower levels of Her2 (Figure S3A,B). In contrast, SpyCatcher-28ζ T cells with high-dose arming were capable of targeting tumor cells with high or low levels of Her2, with lytic capacity correlating with the level of Her2 expression. When the arming concentration was lowered, the SpyCatcher-28ζ T cells lost efficacy against tumor cell lines expressing lower levels of Her2 (Figure S3A,B).
SpyCatcher-BBζ and SpyCatcher-28ζ T cells were also able to lyse EGFR+ or CD20+ tumor cells when armed with EGFR-targeting antibody Cetuximab-ST or CD20-targeting antibody Rituximab-ST, respectively (Figure 3C). The site-specific labeling method used for the off-the-shelf clinical-grade antibodies led to the addition of a SpyTag peptide to both IgG heavy chains, as was previously shown by Hui et al. and further indicated by the nearly complete shift in the heavy-chain band post-conjugation (Figure 1B).39 This multivalent labeling method could potentially cause antigen-independent activation if two SpyCatcher immune receptors were cross-linked by the two SpyTag peptides on the same antibody or if two SpyCatcher T cells were tethered with each other by SpyTag peptides on the same antibody. However, SpyCatcher T cells armed with the nontargeting control antibodies identically conjugated with two SpyTag peptides did not mediate significant lysis, demonstrating that bivalent SpyTag labeling of targeting ligands does not cause appreciable antigen-independent activation of T cells upon arming (Figure 3C). Bivalently labeled antibodies were able to induce T-cell activation when adhered to a plate, indicating the need for the immobilization of the soluble protein to induce activation, as has been seen with other bivalent activating molecules53 (Figure 2E). Covalent loading of SpyTag containing designed ankyrin repeat proteins (DARPins) targeting tumor antigens Her2, EGFR, and EpCAM also led to the specific lysis of antigen-expressing tumor cells, demonstrating the potential to use multiple targeting ligand types with T cells bearing the SpyCatcher receptor (Figure 3D).
Upon antigen recognition and CAR T-cell activation, CARs are internalized, leading to lower detectable levels at the cell surface.54 To evaluate the rate of armed SpyCatcher receptor loss on the T-cell surface in the setting of antigenic stimulation, SpyCatcher T cells were armed, washed, and cocultured with or without antigen-expressing tumor cells. In the absence of antigen-expressing tumor cells, moderate armed receptor loss occurred, with SpyCatcher-28ζ T cells experiencing more rapid loss within 24 h relative to SpyCatcher-BBζ T cells. Similar to CARs,54 when stimulated with antigen-expressing cancer cells, armed receptor expression was not detected on either SpyCatcher-BBζ or SpyCatcher-28ζ T cells at the same time point (Figure 3E). However, both SpyCatcher-BBζ and SpyCatcher-28ζ T cells were amenable to rearming, demonstrating that the expression of SpyCatcher receptors by T cells is maintained and capable of binding newly introduced targeting ligand (Figure 3E). In order to determine the rate of armed receptor loss from the cell surface in nonactivated cells, SpyCatcher T cells were rested, then armed with Herceptin-ST, and subsequently analyzed for a detectable, armed receptor via flow cytometry every 24 h. Results show that armed receptor levels are gradually depleted over time, with full loss occurring approximately 96 h after arming (Figure S4).
On the basis of the finding that armed, but not unarmed, SpyCatcher T cells are capable of targeting and killing antigen-expressing tumor cells, we hypothesized that the unarmed SpyCatcher T-cell effector function could be temporally triggered upon later addition of targeting ligand to unarmed SpyCatcher T cells in coculture with antigen-expressing tumor cells (Figure 3A, right). To test for “on-demand” cancer cell lysis, unarmed SpyCatcher T cells were incubated with SKOV3 (Her2+) tumor cells for 4 h in the absence of targeting ligand. Herceptin-ST was added after 4 h of induced rapid tumor cell lysis in cultures containing either SpyCatcher-BBζ or SpyCatcher-28ζ T cells (Figure 3F). Compared to SpyCatch-er-BBζ T cells, SpyCatcher-28ζ T cells reacted more rapidly upon addition of Herceptin-ST, lysed target cells at lower targeting ligand concentrations, and reached a higher level of maximum lysis. This observation is consistent with prior findings showing that the use of the CD28 costimulatory domain in CARs leads to more potent T-cell effector function than 4–1BB in vitro.55 Though equivalent levels of maximum lysis were achieved with SpyCatcher-28ζ T cells at different doses of Herceptin-ST, the initial lysis kinetics occurred in a dose-dependent fashion (Figure 3F). SpyCatcher-BBζ T cells displayed a more titratable response, with lower maximal lysis at lower Herceptin-ST doses. The induction of “on-demand” lysis was also observed using live cell imaging, where unarmed SpyCatcher-BBζ T cells reamined inactive until addition of exogenous Herceptin-ST (Video S6).
Testing the Effects of Covalent Bond Formation on Lytic Function.
To test the effect of covalent bond formation on receptor arming and T-cell activation, both “arming” and “on-demand” lysis experiments were performed comparing Herceptin-ST and Herceptin-STDA, or the anti-Her2 DARPins 9.26-ST and 9.26-STDA, respectively (Figure 3F,G). SpyCatcher-BBζ and SpyCatcher-28ζ T cells armed with 9.26-ST lysed Her2+ SKOV3 tumor cells, while those armed with 9.26-STDA exhibited reduced lysis or no lysis (Figure 3G). This result corresponds to previous data demonstrating that covalent bond formation is necessary for maximal loading of the SpyCatcher receptor, particularly at lower arming concentrations (Figure 2D). Covalent bond formation also impacted on-demand lysis. Herceptin-STDA addition led to slower lysis kinetics and lower maximal lysis compared to Herceptin-ST at equivalent doses (Figure 3F). In all cases, SpyCatcher-28ζ T cells exhibited increased effector function, compared to SpyCatcher-BBζ T cells, when armed or cocultured with excess Herceptin-STDA (Figure 3F,G).
Simultaneous Receptor Arming and Dual Antigen Targeting.
One advantage of covalent universal immune receptor loading is the ability to affix multiple targeting ligands with different specificities onto receptors at the T-cell surface, creating a single-cell product with the capability of targeting multiple antigens simultaneously (Figure 4A). To test this, Her2-targeting (9.26-ST) and EGFR-targeting (E01-ST) DARPins containing unique tags for detection by flow cytometry were loaded onto SpyCatcher T cells either alone or in combination in a 1:1 molar ratio (Figure 4B). Tag staining and flow cytometric analysis revealed that single DARPin loaded SpyCatcher T cells were stained for only a single tag, while SpyCatcher T cells coincubated with both DARPins displayed equivalent staining for each tag and thus equivalent arming (Figure 4B). These dual armed T cells were capable of lysing both Her2+/EGFR-and EGFR+/Her2-Ramos cells (Figure S2B), while SpyCatcher T cells armed with a single DARPin targeting ligand were capable only of lysing cells expressing the prescribed target antigen. Levels of specific cell lysis mediated by dual-armed T cells were similar to those of their single-armed counterparts, demonstrating the capacity of this single-cell product to simultaneously target multiple antigens (Figure 4C). SpyCatcher-Δζ T cells remained inactive regardless of the arming agent (Figure 4C).
Figure 4.
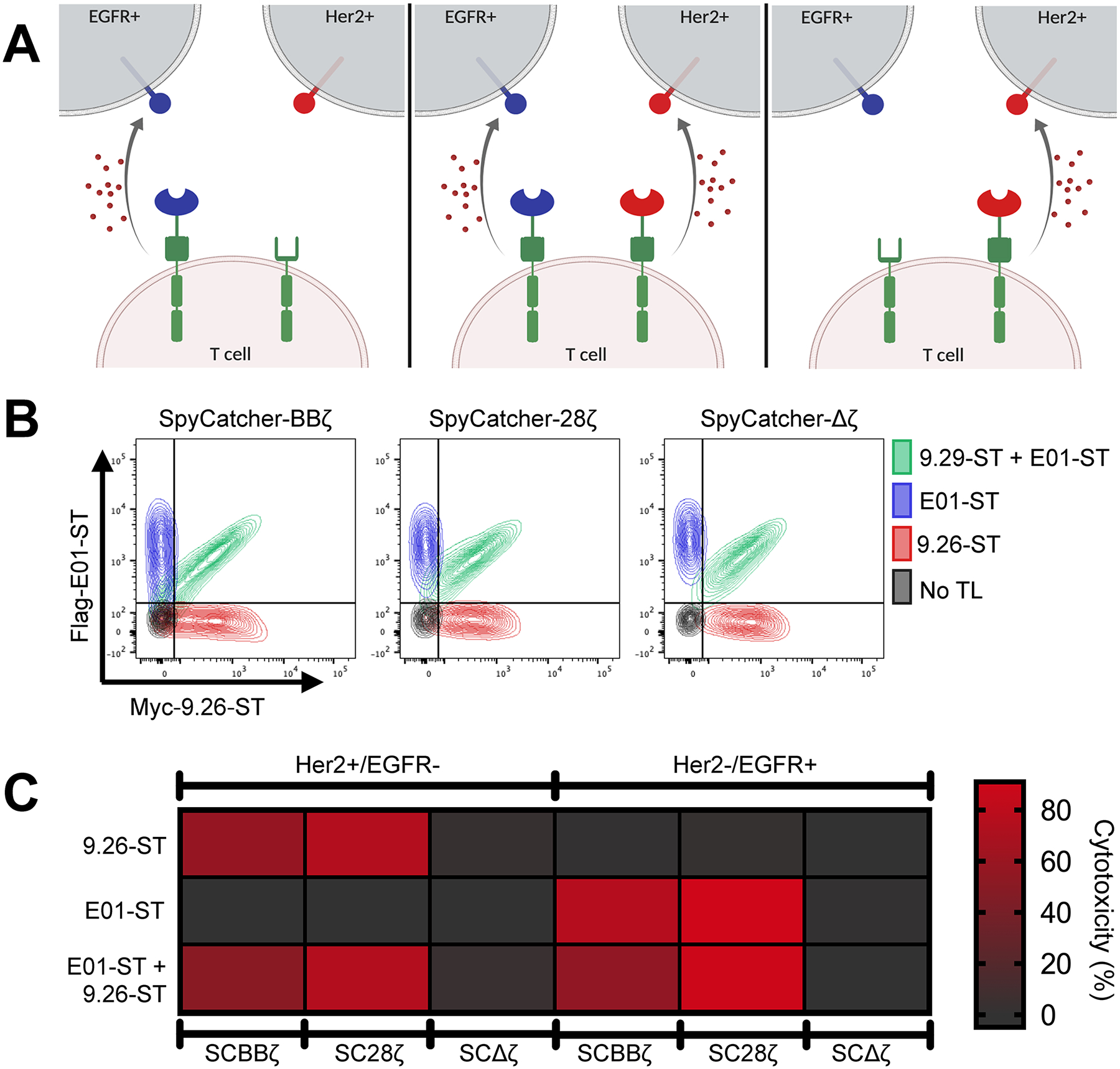
Simultaneous arming of SpyCatcher T cells with two targeting ligands generates a single cell product capable of dual antigen targeting. (A) Schematic representation of single vs dual targeting ligand loading. (B) SpyCatcher T cells were armed with either 1000 nM myc-9.26-ST (αHer2; red), 1000 nM Flag-E01-ST (αEGFR; blue), or both simultaneously at 1000 nM each (green). Receptor loading was detected with a combination of fluorescently conjugated anti-myc and anti-flag antibodies and assessed via flow cytometry. (C) Single- or dual-armed SpyCatcher T cells were cocultured with Ramos cells expressing either Her2 or EGFR and luciferase. Residual luciferase expression was calculated after 20 h. All cocultures were carried out using a 7:1 E/T ratio. Data points are averages of three replicates. A representative T-cell donor is shown.
To assess the capability of combined the targeting ligand arming enhancing SpyCatcher T-cell function against cancer cells that coexpress two distinct antigens, SpyCatcher-BBζ T cells were armed with low doses of either 9.26-ST or E01-ST, or both in combination, and then cocultured with the Her2+/ EGFR+ SKOV3 tumor line (Figure S3C). Dual-armed SpyCatcher-BBζ T cells achieved increased tumor lysis relative to single-armed cells alone (Figure S3D).
In Vivo Efficacy in Xenograft Tumor Models.
We next tested the effectiveness of the SpyCatcher T-cell system in xenograft tumor models using nonobese diabetic (NOD)-scid gamma (NSG) mice. Though our in vitro results showed that SpyCatcher-28ζ T cells displayed more potent effector function than SpyCatcher-BBζ T cells, we chose to move forward with preclinical models using the SpyCatcher-BBζ receptor, as previous studies have demonstrated the enhanced prolonged survival capabilities of T-cell-bearing CARs containing the 4–1BB costimulatory domain in vivo.55,56
We tested the in vivo efficacy of SpyCatcher-BBζ T cells using an intraperitoneal model of ovarian cancer. Her2+ SKOV3 ovarian cancer tumor cells were injected into the peritoneal cavity (I.P.) and allowed to establish for 7 days. On day 7, mice received an I.P. injection of SpyCatcher-BBζ T cells armed with Herceptin-ST at a concentration of 1000 nM. One group was administered armed SpyCatcher-Δζ T cells to control for any tumor reduction caused by T-cell infusion or targeting ligand administration independent of antigen-dependent T-cell stimulation. Beginning on day 8, additional targeting ligand was administered, followed by continual dosing every 3 days during the dosing window (Figure 5A, orange box).
Figure 5.
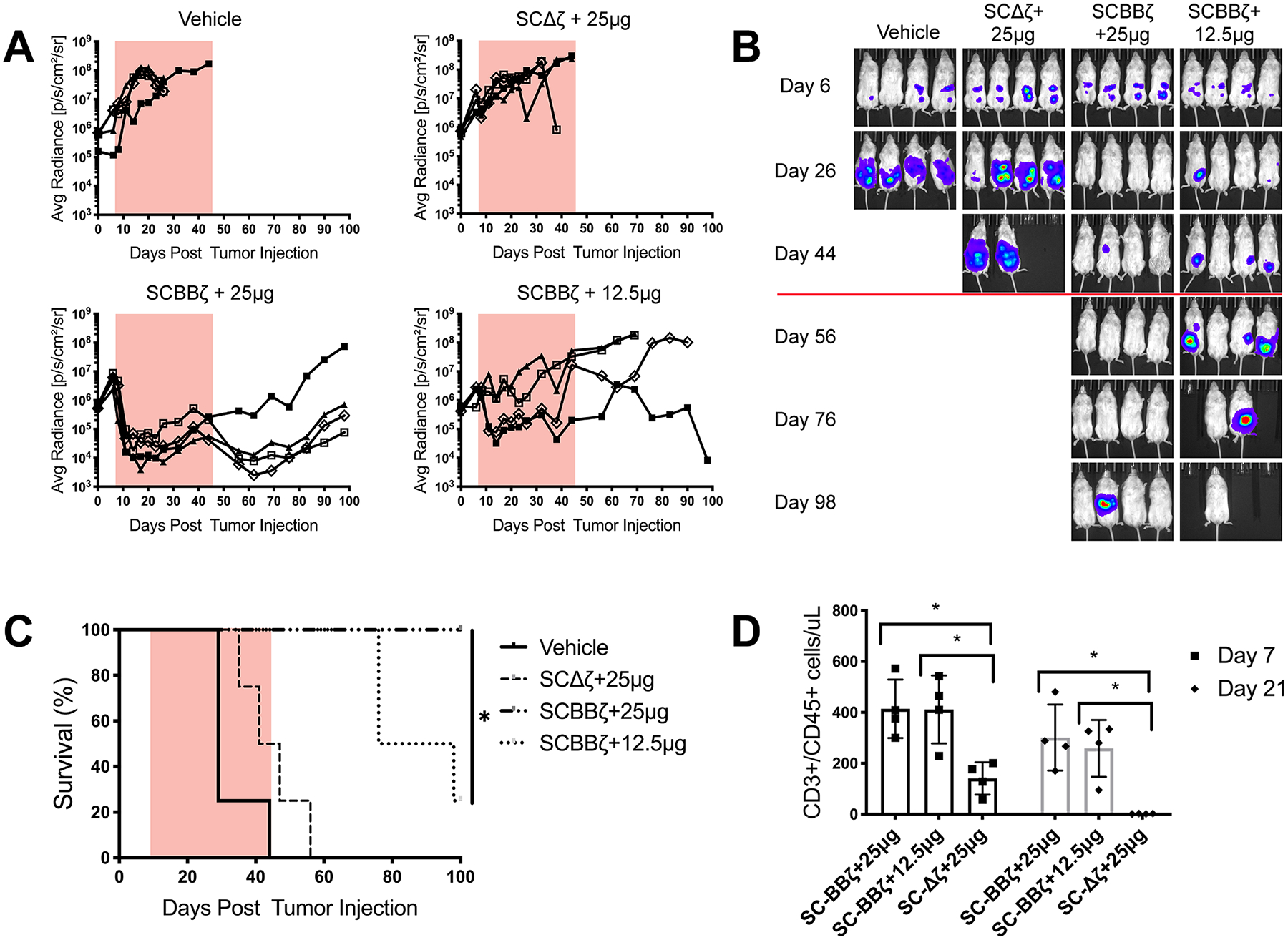
SpyCatcher-BBζ T cells prevent tumor growth in vivo. NSG mice (n = 4 per group) were injected intraperitoneally with 1 × 106 SKOV3 tumor cells expressing luciferase on day 0, followed by 12.5 × 106 Herceptin-ST armed SpyCatcher-BBζ T cells on day 7. Herceptin-ST was administered on day 8, followed by injections every 3 days during the dosing window, indicated by either an orange box (A) or red line (B). Injection amounts indicate the dose per mouse. (A) Tumor growth was monitored by luminescence and plotted as the average radiance for each individual mouse. (B) Luminescence images of treated mice. (C) Survival curve for mice treated in (A). (D) TruCount analysis of total human T cells (CD3+/CD45+) on days 7 and 21 post-T-cell injection. Error bars represent the mean ± standard deviation (D). *P < 0.05, **P < 0.01, and ***P < 0.001.
Treatment with SpyCatcher-Δζ T cells coadministered with a 25 μg dose of Herceptin-ST was similar to the vehicle control, demonstrating that the tumor was not sensitive to targeting ligand alone or the infusion of signaling-deficient T cells (Figure 5A,B). SpyCatcher-BBζ T cells coadministered with a 25 μg dose of Herceptin-ST were able to clear detectable tumor in three of the four mice, which resulted in prolonged survival relative to all other treatment groups (Figure 5A–C). Tumor relapse was seen in one mouse beginning 30 days after the cessation of targeting ligand treatment. SpyCatcher-BBζ T cells coadministered with a 12.5 μg dose of Herceptin-ST showed a transient reduction of tumor burden in some mice, but did not maintain efficacy over the course of treatment, demonstrating that sufficient levels of targeting ligand must be provided to drive tumor clearance. Peripheral blood T-cell levels were detected on day 7 after T-cell infusion in all groups, with signaling-enabled SpyCatcher-BBζ T-cell counts exceeding those in the SpyCatcher-Δζ group (Figure 5D). Human T-cell counts in SpyCatcher-BBζ T-cell groups were similar between the two targeting ligand dose groups, implicating targeting ligand availability as one limitation to effective treatment. Additionally, weight loss due to toxicity was not seen in treatment groups, while weight gain was seen in control group mice due to ascites formation during tumor progression (Figure S5). To assess the systemic delivery of the SpyCatcher system, preclinical testing was performed in a rapidly growing, highly aggressive subcutaneous Her2+ SKOV3 tumor model. Results showed that I.P.-injected SpyCatcher-BBζ T cells coadministered with Herceptin-ST lead to a reduction in tumor volume out to day 22 post-tumor inoculation, compared to SpyCatcher-BBζ T cells alone (Figure S6).
DISCUSSION
Universal immune receptors (UIRs) are an emerging technology aimed at improving standard CAR T-cell therapies and addressing limitations in therapeutic design.25 Through the use of a targeting ligand to redirect T cells against antigen expressing tumor cells, UIRs allow for the dose-dependent control of the T-cell effector function while also enabling the use of a single-receptor, single-cell product to target multiple tumor antigens. Current UIR platforms rely on noncovalent interactions between their extracellular adapter protein and targeting ligand tag, rendering them unable to be covalently loaded with antigen specificity prior to infusion.
In this study, we developed a novel UIR platform that allows for the post-translational covalent attachment of targeting ligands to an immune receptor via SpyCatcher/SpyTag chemistry, mediating the redirection of T cells against multiple tumor-associated antigens. We generated two functional SpyCatcher immune receptor constructs containing extracellular SpyCatcher domains and either CD28-CD3ζ or 4–1BB-CD3ζ intracellular domains and demonstrated their expression in primary human T cells. To redirect the SpyCatcher immune receptor expressing T cells against target tumor antigens, we used two methods of targeting ligand production. Using the LASIC adaptor protein method developed by Hui et al., we were able to site-specifically label off-the-shelf clinical-grade antibodies with SpyTag moieties.39 This methodology allows for a potentially advantageous pairing of SpyCatcher T cells and clinical-grade antibodies in patients who do not benefit from or become resistant to antibody monotherapy alone, if the resistance in not due to antigen loss.57,58 We further generated monovalent targeting ligands through the genetic fusion of the SpyTag to various designed ankyrin repeat proteins (DARPins). We demonstrated the capability of the SpyCatcher immune receptor to target multiple tumor antigens in vitro. The transfer of SpyCatcher T cells, along with SpyTag targeting ligands, led to the control of outgrowth in aggressive subcutaneous tumors and the clearance of established intraperitoneal disease in immunodeficient mouse xenograft models.
Notably, the formation of a covalent bond between SpyCatcher and SpyTag was critical for optimal T-cell effector function. The transient bond formed between the SpyTag-DA mutant and SpyCatcher receptor (Kd = 200 nM) led to a substantial loss of receptor loading (Figure 2D), and this in turn resulted in decreased lytic capacity (Figure 3G). In instances where the targeting ligand was added directly to the culture, the inability to form a covalent bond greatly diminished or completely abolished the effector function at low concentrations (Figure 3F). This indicates that covalent bond formation between the targeting ligand and the receptor may be important in instances where targeting ligand concentrations are low.
When compared to standard CAR T cells, armed SpyCatcher T cells achieved similar activity at high E/T ratios but were less effective at low E/T ratios (Figure S7A,B). While CAR T cells displayed enhanced potency, we have shown that the SpyCatcher immune receptor system is capable of addressing some of the limitations currently facing CAR T-cell therapy. One of these limitations is the inability to control CAR T cells post-infusion. Upon binding to target antigen, CAR T-cell activation and proliferation can occur at a rapid rate and can lead to issues such as CRS or the on-target, off-tumor lysis of normal tissue.3,14,15 We have shown that the effector function of SpyCatcher T cells is titratable based on the amount of targeting ligand either covalently loaded onto the receptor prior to tumor exposure or injected post-infusion, and that SpyCatcher T cells without targeting ligand are unable to target tumor cells (Figure 3B,F). We saw that suboptimal doses of targeting ligand led to tumor outgrowth in our I.P. mouse model, indicating that the continued administration of targeting ligand at sufficient levels is necessary for prolonged T-cell function (Figure 5A–C). The ability to escalate, decrease, or withdraw the targeting ligand dose as a means of attenuating the T-cell effector function with the SpyCatcher system could allow for the mitigation of on-target, off-tumor toxicity. SpyCatcher T cells function with the use of antibodies, as well as DARPins, and have the potential to be expanded further for the use of scFvs, Fabs, and small-molecule conjugates. This could allow for another potential layer of control, as targeting ligands with short half-lives would lead to the more rapid cessation of SpyCatcher T-cell effector function. Patients who receive CD19 targeted CAR T-cell therapy sustain long-term B-cell aplasia due to the continual elimination of CD19+ normal B cells. SpyCatcher T cells are unreactive in the absence of targeting ligand and therefore could allow for the re-establishment of a normal B-cell population after tumor clearance via discontinuation of the targeting ligand.
The outgrowth of an antigen-negative tumor population is another issue facing single-antigen-targeting T-cell therapy. This can occur for a multitude of reasons, which include heterogeneous tumor antigen expression, the down regulation of tumor antigens, trogocytosis, or the expression of splice variants.17–22 To overcome tumor escape via antigen loss, other groups have proposed the expression of multiple CARs in a single T-cell product,59 the expression of CARs with multiple epitope binding domains,60–62 or making a combined T-cell product containing two distinct CAR populations.63 While these approaches may help mitigate antigen loss, they do not address safety and toxicity concerns and still limit the total number of targetable antigens. SpyCatcher T cells are capable of lysing a broad range of tumor antigens, with a proof of principle established here for Her2, EGFR, EpCAM, and CD20. We have further demonstrated the ability to arm SpyCatcher immune receptor T cells with multiple targeting ligands simultaneously, allowing for the creation of a single vector and single-cell product capable of targeting multiple tumor antigens simultaneously (Figure 4A–C). The turnover of armed receptors coupled with the continual expression of new SpyCatcher receptor by the T cells enables rearming with targeting ligand (Figure 3E) and the potential for sequential antigen targeting, as we have demonstrated with a biotin-binding immune receptor.24
Though the SpyCatcher immune receptor system has shown promise both in vitro and in vivo, it is important to acknowledge the potential limitations of its expanded use. The SpyCatcher/SpyTag system itself was derived from the second immunoglobulin-like collagen adhesin domain (CnaB2) of Streptococcus pyogenes fibronectin-binding protein FbaB. The bacterial origin of these proteins means that they are potentially immunogenic and therefore may be suppressed by the patient’s immune system. A similar issue has plagued CARs with murine-derived scFv domains, against which patients develop human antimouse antibodies (HAMA).64 However, previous studies have tested the SpyCatcher immunogenicity in immune-competent mice and have reported that truncated versions of the SpyCatcher protein, similar to that used here, induced significantly lower antibody levels.65
The SpyCatcher immune receptor and UIRs in general present an exciting new avenue for therapeutic investigation. However, much like in the field of CAR T-cell therapy, optimal clinical translation will rely on facing unique challenges related to UIRs. For instance, the ability to target multiple antigens with a single receptor relies upon the discovery, validation, and safety profiling of tumor-associated antigens and clinical-grade targeting ligands against those antigens. To target some antigens, there may also be challenges in designing an optimal universal immune receptor and their targeting ligand pairs since the optimal targeting ligand size and receptor hinge domain spacing can vary on the basis of the targeted antigenic epitope.33,34 The shedding of target antigen by tumor cells is another potential issue facing both UIR and CAR T cells. We previously showed that conventional CAR T cells can mediate highly efficient cancer cell attack even in the presence of high concentrations of soluble targeted antigen secreted by solid tumors.50 To what degree UIR systems are impacted by shed antigen is not fully understood; however, the SKOV3 cell line used in our studies sheds HER2 antigen,66 and HER2-redirected SpyCatcher T cells mediate the efficient killing of SKOV3 ovarian cancer cells in vitro and in vivo, suggesting that UIR T cells can function in the presence of soluble antigen with proper targeting ligand dosing. In addition, the clinical production of UIR T cells will rely upon patient T cells as the starting source cell material, akin to CAR T-cell therapy. Accordingly, UIR therapies will also need to address the shared issues of pre-existing T-cell exhaustion and senescence, poor proliferation, persistence after infusion, and the lack of T-cell infiltration into solid tumors4,67–69 that can limit otherwise effective CAR T-cell therapy in certain indications.
In conclusion, the SpyCatcher immune receptor described here is the first universal immune receptor to allow for post-translational covalent loading of targeting ligands for subsequent redirection of T cells against an array of tumor antigens in vitro and in vivo. This platform technology provides a method for single-vector, single-receptor targeting of multiple antigens simultaneously while also allowing for continual rearming to generate, regulate, and diversify a sustained T-cell response over time.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health National Cancer Institute (NCI) grant RO1-CA168900 and the ACC Support Grant (P30-CA016520). The authors thank the University of Pennsylvania (UPenn) Stem Cell and Xenograft Core, UPenn Small Animal Imaging Facility, UPenn Human Immunology Core, and UPenn Flow Cytometry Core for their technical support. The authors thank Saar Gill, Nicholas Petty, and Michael Klichinsky for the provision of tumor cell lines CRL5803, MDA-MB-361, SKOV3, and MDA-MB-468. All graphics were created with BioRender.com.
Footnotes
Supporting Information
Supporting Information consists of Figures S1–S7 and videos SV1–SV6. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b11622.
Supplemental Figures - Quantitative Control of Gene- Engineered T cell Activity Through Covalent Attachment of Targeting Ligands to a Universal Immune Receptor (PDF)
SV1: GFP-SpyCatcher-BBζ T cells armed with 1000 nM Herceptin-ST targeting Her2+ SKOV3 cells expressing nuclear RFP (AVI)
SV2: GFP-SpyCatcher-BBζ T cells armed with 100 nM Herceptin-ST targeting Her2+ SKOV3 cells expressing nuclear RFP (AVI)
SV3: GFP-SpyCatcher-BBζ T cells armed with 10 nM Herceptin-ST targeting Her2+ SKOV3 cells expressing nuclear RFP (AVI)
SV4: GFP-SpyCatcher-BBζ T cells armed with 0 nM Herceptin-ST targeting Her2+ SKOV3 cells expressing nuclear RFP (AVI)
SV5: GFP-SpyCatcher-Δζ T cells armed with 1000 nM Herceptin-ST targeting Her2+ SKOV3 cells expressing nuclear RFP (AVI)
SV6: Addition of 100 ng of Herceptin-ST to unarmed GFP-SpyCatcher-BBζ T cells co-cultured with Her2+ SKOV3 cells induces lytic function (Herceptin-ST added at 48 h) (AVI)
The authors declare the following competing financial interest(s): Daniel Powell and Andrew Tsourkas have a patent filed on this technology.
Contributor Information
Nicholas G. Minutolo, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, Center for Cellular Immunotherapies, Perelman School of Medicine, and Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States;.
Prannda Sharma, Department of Pathology and Laboratory Medicine, Perelman School of Medicine and Center for Cellular Immunotherapies, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Mathilde Poussin, Department of Pathology and Laboratory Medicine, Perelman School of Medicine and Center for Cellular Immunotherapies, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Lauren C. Shaw, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, Center for Cellular Immunotherapies, Perelman School of Medicine, and Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Daniel P. Brown, Center for Cellular Immunotherapies, Perelman School of Medicine and Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Erin E. Hollander, Department of Cancer Biology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Anzě Smole, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, Center for Cellular Immunotherapies, Perelman School of Medicine, and Parker Institute for Cancer Immunotherapy, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Alba Rodriguez-Garcia, Department of Pathology and Laboratory Medicine, Perelman School of Medicine and Center for Cellular Immunotherapies, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
James Z. Hui, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States
Fabiana Zappala, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States;.
Andrew Tsourkas, Department of Bioengineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States;.
Daniel J. Powell, Jr., Department of Pathology and Laboratory Medicine, Perelman School of Medicine and Center for Cellular Immunotherapies, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States;
REFERENCES
- (1).Porter DL; Levine BL; Kalos M; Bagg A; June CH Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med 2011, 365 (8), 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kalos M; Levine BL; Porter DL; Katz S; Grupp SA; Bagg A; June CH T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med 2011, 3 (95), 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Grupp SA; Kalos M; Barrett D; Aplenc R; Porter DL; Rheingold SR; Teachey DT; Chew A; Hauck B; Wright FJ; Milone MC; Levine BL; June CH Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med 2013, 368 (16), 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Maude SL; Frey N; Shaw PA; Aplenc R; Barrett DM; Bunin NJ; Chew A; Gonzalez VE; Zheng Z; Lacey SF; Mahnke YD; Melenhorst JJ; Rheingold SR; Shen A; Teachey DT; Levine BL; June CH; Porter DL; Grupp SA Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med 2014, 371 (16), 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Schuster SJ; Svoboda J; Chong EA; Nasta SD; Mato AR; Anak Ö; Brogdon JL; Iulian P-M; Bhoj V; Landsburg D; Wasik M; Levine BL; Lacey SF; Melenhorst JJ; Porter DL; June CH Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med 2017, 377 (26), 2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Locke FL; Neelapu SS; Bartlett NL; Siddiqi T; Chavez JC; Hosing CM; Ghobadi A; Budde LE; Bot A; Rossi JM; Jiang Y; Xue AX; Elias M; Aycock J; Wiezorek J; Go WY Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol. Ther 2017, 25 (1), 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pehlivan KC; Duncan BB; Lee DW CAR-T Cell Therapy for Acute Lymphoblastic Leukemia: Transforming the Treatment of Relapsed and Refractory Disease. Current Hematologic Malignancy Reports 2018, 13 (5), 396–406. [DOI] [PubMed] [Google Scholar]
- (8).Ali S; Shi V; Maric I; Wang M; Stroncek D; Rose J; Brudno J; Stetler-Stevenson M; Feldman S; Hansen B; Fellowes V; Hakim F; Gress R; Kochenderfer J T Cells Expressing an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Multiple Myeloma. Blood 2016, 128 (13), 1688–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Fry TJ; Shah NN; Orentas RJ; Maryalice S-S; Yuan CM; Ramakrishna S; Wolters P; Martin S; Delbrook C; Yates B; Shalabi H; Fountaine TJ; Shern JF; Majzner RG; Stroncek DF; Sabatino M; Feng Y; Dimitrov DS; Zhang L; Nguyen S; Qin H; Dropulic B; Lee DW; Mackall CL CD22-Targeted CAR T Cells Induce Remission in B-ALL That Is Naive or Resistant to CD19-Targeted CAR Immunotherapy. Nat. Med 2018, 24 (1), 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang W; Wang Y; Guo Y; Dai H; Yang Q; Zhang Y; Zhang Y; Chen M; Wang C; Feng K; Li S; Liu Y; Shi F; Luo C; Han W Treatment of CD20-Directed Chimeric Antigen Receptor-Modified T Cells in Patients with Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma: An Early Phase IIa Trial Report. Signal Transduction and Targeted Therapy 2016, 1 (1), sigtrans20162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mirzaei HR; Rodriguez A; Shepphird J; Brown CE; Badie B Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front. Immunol 2017, 8, 1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gill S; June CH Going Viral: Chimeric Antigen Receptor T-Cell Therapy for Hematological Malignancies. Immunological reviews 2015, 263 (1), 68. [DOI] [PubMed] [Google Scholar]
- (13).Santomasso B; Park J; Riviere I; Mead E; Li D; Senechal B; Purdon T; Halton E; Diamonte C; Sadelain M Neurotoxicity Associated with CD19-Specific Chimeric Antigen Receptor T Cell Therapy for Adult Acute Lymphoblastic Leukemia (B-ALL). Neurology 2018, 90, S23 008. [Google Scholar]
- (14).Morgan RA; Yang JC; Kitano M; Dudley ME; Laurencot CM; Rosenberg SA Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther 2010, 18 (4), 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lamers CH; Sleijfer S; van Steenbergen S; van Elzakker P; van Krimpen B; Groot C; Vulto A; den Bakker M; Oosterwijk E; Debets R; Gratama JW Treatment of Metastatic Renal Cell Carcinoma With CAIX CAR-Engineered T Cells: Clinical Evaluation and Management of On-Target Toxicity. Mol. Ther 2013, 21 (4), 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Orlando EJ; Han X; Tribouley C; Wood PA; Leary RJ; Riester M; Levine JE; Qayed M; Grupp SA; Boyer M; Moerloose B; Nemecek ER; Bittencourt H; Hiramatsu H; Buechner J; Davies SM; Verneris MR; Nguyen K; Brogdon JL; Bitter H; Morrissey M; Pierog P; Pantano S; Engelman JA; Winckler W Genetic Mechanisms of Target Antigen Loss in CAR19 Therapy of Acute Lymphoblastic Leukemia. Nat. Med 2018, 24 (10), 1504–1506. [DOI] [PubMed] [Google Scholar]
- (17).Bagashev A; Sotillo E; Wu G; Andrei T-T The Importance of CD19 Exon 2 for Surface Localization: Closing the Ig-like Loop. Blood 2015, 126, 3433. [Google Scholar]
- (18).Sotillo E; Barrett DM; Black KL; Bagashev A; Oldridge D; Wu G; Sussman R; Lanauze C; Ruella M; Gazzara MR; Martinez NM; Harrington CT; Chung EY; Perazzelli J; Hofmann TJ; Maude SL; Raman P; Barrera A; Gill S; Lacey SF; Melenhorst JJ; Allman D; Jacoby E; Fry T; Mackall C; Barash Y; Lynch KW; Maris JM; Grupp SA; Thomas-Tikhonenko A Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discovery 2015, 5 (12), 1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jacoby E; Nguyen SM; Fountaine TJ; Welp K; Gryder B; Qin H; Yang Y; Chien CD; Seif AE; Lei H; Song YK; Khan J; Lee DW; Mackall CL; Gardner RA; Jensen MC; Shern JF; Fry TJ CD19 CAR Immune Pressure Induces B-Precursor Acute Lymphoblastic Leukaemia Lineage Switch Exposing Inherent Leukaemic Plasticity. Nat. Commun 2016, 7 (1), 12320 DOI: 10.1038/ncomms12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Gardner R; Wu D; Cherian S; Fang M; Hanafi L-A; Finney O; Smithers H; Jensen MC; Riddell SR; Maloney DG; Turtle CJ Acquisition of a CD19-Negative Myeloid Phenotype Allows Immune Escape of MLL-Rearranged B-ALL from CD19 CAR-T-Cell Therapy. Blood 2016, 127 (20), 2406–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hamieh M; Dobrin A; Cabriolu A; van der Stegen SJ; Giavridis T; Mansilla-Soto J; Eyquem J; Zhao Z; Whitlock BM; Miele MM; Li Z; Cunanan KM; Huse M; Hendrickson RC; Wang X; Rivière I; Sadelain M CAR T Cell Trogocytosis and Cooperative Killing Regulate Tumour Antigen Escape. Nature 2019, 568, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ruella M; Xu J; Barrett DM; Fraietta JA; Reich TJ; Ambrose DE; Klichinsky M; Shestova O; Patel PR; Kulikovskaya I; Nazimuddin F; Bhoj VG; Orlando EJ; Fry TJ; Bitter H; Maude SL; Levine BL; Nobles CL; Bushman FD; Young RM; Scholler J; Gill SI; June CH; Grupp SA; Lacey SF; Melenhorst JJ Induction of Resistance to Chimeric Antigen Receptor T Cell Therapy by Transduction of a Single Leukemic B Cell. Nature 2018, 24 (10), 1499–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).O’Rourke DM; Nasrallah MP; Desai A; Melenhorst JJ; Mansfield K; Morrissette JJ; Martinez-Lage M; Brem S; Maloney E; Shen A; Isaacs R; Mohan S; Plesa G; Lacey SF; Navenot J-M; Zheng Z; Levine BL; Okada H; June CH; Brogdon JL; Maus MV A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med 2017, 9 (399), eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Urbanska K; Lanitis E; Poussin M; Lynn RC; Gavin BP; Kelderman S; Yu J; Scholler N; Powell DJ A Universal Strategy for Adoptive Immunotherapy of Cancer through Use of a Novel T-Cell Antigen Receptor. Cancer Res 2012, 72 (7), 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Minutolo NG; Hollander EE; Powell DJ The Emergence of Universal Immune Receptor T Cell Therapy for Cancer. Front. Oncol 2019, 9, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Clémenceau B; Congy-Jolivet N; Gallot G; Vivien R; Gaschet J; Thibault G; Vié H Antibody-Dependent Cellular Cytotoxicity (ADCC) Is Mediated by Genetically Modified Antigen-Specific Human T Lymphocytes. Blood 2006, 107 (12), 4669. [DOI] [PubMed] [Google Scholar]
- (27).Tamada K; Geng D; Sakoda Y; Bansal N; Srivastava R; Li Z; Davila E Redirecting Gene-Modified T Cells toward Various Cancer Types Using Tagged Antibodies. Clin. Cancer Res 2012, 18 (23), 6436. [DOI] [PubMed] [Google Scholar]
- (28).Kudo K; Imai C; Lorenzini P; Kamiya T; Kono K; Davidoff AM; Chng W; Campana D T Lymphocytes Expressing a CD16 Signaling Receptor Exert Antibody-Dependent Cancer Cell Killing. Cancer Res 2014, 74 (1), 93. [DOI] [PubMed] [Google Scholar]
- (29).Urbanska K; Lynn RC; Stashwick C; Thakur A; Lum LG; Powell DJ Targeted Cancer Immunotherapy via Combination of Designer Bispecific Antibody and Novel Gene-Engineered T Cells. J. Transl. Med 2014, 12 (1), 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ochi F; Fujiwara H; Tanimoto K; Asai H; Miyazaki Y; Okamoto S; Mineno J; Kuzushima K; Shiku H; Barrett J; Ishii E; Yasukawa M Gene-Modified Human α/β-T Cells Expressing a Chimeric CD16-CD3ζ Receptor as Adoptively Transferable Effector Cells for Anticancer Monoclonal Antibody Therapy. Cancer Immunol. Res 2014, 2 (3), 249–262. [DOI] [PubMed] [Google Scholar]
- (31).Feldmann A; Arndt C; Bergmann R; Loff S; Cartellieri M; Bachmann D; Aliperta R; Hetzenecker M; Ludwig F; Albert S; Ziller-Walter P; Kegler A; Koristka S; Gärtner S; Schmitz M; Ehninger A; Ehninger G; Pietzsch J; Steinbach J; Bachmann M Retargeting of T Lymphocytes to PSCA- or PSMA Positive Prostate Cancer Cells Using the Novel Modular Chimeric Antigen Receptor Platform Technology “UniCAR. Oncotarget 2017, 8, 31368–31385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kim M; Ma JS; Yun H; Cao Y; Kim J; Chi V; Wang D; Woods A; Sherwood L; Caballero D; Gonzalez J; Schultz PG; Young TS; Kim C Redirection of Genetically Engineered CAR-T Cells Using Bifunctional Small Molecules. J. Am. Chem. Soc 2015, 137 (8), 2832. [DOI] [PubMed] [Google Scholar]
- (33).Rodgers DT; Mazagova M; Hampton EN; Cao Y; Ramadoss NS; Hardy IR; Schulman A; Du J; Wang F; Singer O; Ma J; Nunez V; Shen J; Woods AK; Wright TM; Schultz PG; Kim CH; Young TS Switch-Mediated Activation and Retargeting of CAR-T Cells for B-Cell Malignancies. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (4), E459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ma JS; Kim JY; Kazane SA; Choi S-HH; Yun HY; Kim MS; Rodgers DT; Pugh HM; Singer O; Sun SB; Fonslow BR; Kochenderfer JN; Wright TM; Schultz PG; Young TS; Kim CH; Cao Y Versatile Strategy for Controlling the Specificity and Activity of Engineered T Cells. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (4), E450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cho JH; Collins JJ; Wong WW Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173 (6), 1426–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Zakeri B; Fierer JO; Celik E; Chittock EC; Schwarz-Linek U; Moy VT; Howarth M Peptide Tag Forming a Rapid Covalent Bond to a Protein, through Engineering a Bacterial Adhesin. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (12), E690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bedbrook CN; Kato M; Kumar SR; Lakshmanan A; Nath RD; Sun F; Sternberg PW; Arnold FH; Gradinaru V Genetically Encoded Spy Peptide Fusion System to Detect Plasma Membrane-Localized Proteins In Vivo. Chem. Biol 2015, 22 (8), 1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Warden-Rothman R; Caturegli I; Popik V; Tsourkas A Sortase-Tag Expressed Protein Ligation: Combining Protein Purification and Site-Specific Bioconjugation into a Single Step. Anal. Chem 2013, 85 (22), 11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hui JZ; Tamsen S; Song Y; Tsourkas A LASIC: Light Activated Site-Specific Conjugation of Native IgGs. Bioconjugate Chem 2015, 26 (8), 1456–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kasaraneni N; Chamoun-Emanuelli AM; Wright G; Chen Z Retargeting Lentiviruses via SpyCatcher-SpyTag Chemistry for Gene Delivery into Specific Cell Types. mBio 2017, 8 (6), No. e01860–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Steiner D; Forrer P; Plückthun A Efficient Selection of DARPins with Sub-Nanomolar Affinities Using SRP Phage Display. J. Mol. Biol 2008, 382 (5), 1211–1227. [DOI] [PubMed] [Google Scholar]
- (42).Stefan N; Martin-Killias P; Wyss-Stoeckle S; Honegger A; Zangemeister-Wittke U; Plückthun A DARPins Recognizing the Tumor-Associated Antigen EpCAM Selected by Phage and Ribosome Display and Engineered for Multivalency. J. Mol. Biol 2011, 413 (4), 826–843. [DOI] [PubMed] [Google Scholar]
- (43).Liu S; Tobias R; Shannon M; Styba G; Shi Q; Jackowski G Removal of Endotoxin from Recombinant Protein Preparations. Clin. Biochem 1997, 30 (6), 455–463. [DOI] [PubMed] [Google Scholar]
- (44).Li L; Fierer JO; Rapoport TA; Howarth M Structural Analysis and Optimization of the Covalent Association between SpyCatcher and a Peptide Tag. J. Mol. Biol 2014, 426 (2), 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhao Y; Wang QJ; Yang S; Kochenderfer JN; Zheng Z; Zhong X; Sadelain M; Eshhar Z; Rosenberg SA; Morgan RA A Herceptin-Based Chimeric Antigen Receptor with Modified Signaling Domains Leads to Enhanced Survival of Transduced T Lymphocytes and Antitumor Activity. J. Immunol 2009, 183 (9), 5563–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Milone MC; Fish JD; Carpenito C; Carroll RG; Binder GK; Teachey D; Samanta M; Lakhal M; Gloss B; Danet-Desnoyers G; Campana D; Riley JL; Grupp SA; June CH Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy in Vivo. Mol. Ther 2009, 17 (8), 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Li YM; Pan Y; Wei Y; Cheng X; Zhou BP; Tan M; Zhou X; Xia W; Hortobagyi GN; Yu D; Hung M-C Upregulation of CXCR4 Is Essential for HER2-Mediated Tumor Metastasis. Cancer Cell 2004, 6 (5), 459–469. [DOI] [PubMed] [Google Scholar]
- (48).Greulich H; Chen T-H; Feng W; Jänne PA; Alvarez JV; Zappaterra M; Bulmer SE; Frank DA; Hahn WC; Sellers WR; Meyerson M Oncogenic Transformation by Inhibitor-Sensitive and -Resistant EGFR Mutants. Plos Med 2005, 2 (11), No. e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Smith JB; Lanitis E; Dangaj D; Buza E; Poussin M; Stashwick C; Scholler N; Powell DJ Tumor Regression and Delayed Onset Toxicity Following B7-H4 CAR T Cell Therapy. Mol. Ther 2016, 24 (11), 1987–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lanitis E; Poussin M; Hagemann IS; Coukos G; Sandaltzopoulos R; Scholler N; Powell DJ Redirected Antitumor Activity of Primary Human Lymphocytes Transduced with a Fully Human Anti-Mesothelin Chimeric Receptor. Mol. Ther 2012, 20 (3), 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Sauer-Eriksson A; Kleywegt G; Uhlén M; Jones T Crystal Structure of the C2 Fragment of Streptococcal Protein G in Complex with the Fc Domain of Human IgG. Structure (Oxford, U. K.) 1995, 3 (3), 265. [DOI] [PubMed] [Google Scholar]
- (52).Song D-G; Ye Q; Santoro S; Fang C; Best A; Powell DJ Chimeric NKG2D CAR-Expressing T Cell-Mediated Attack of Human Ovarian Cancer Is Enhanced by Histone Deacetylase Inhibition. Hum. Gene Ther 2013, 24 (3), 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Geppert TD; Lipsky PE Activation of T Lymphocytes by Immobilized Monoclonal Antibodies to CD3. Regulatory Influences of Monoclonal Antibodies to Additional T Cell Surface Determinants. J. Clin. Invest 1988, 81 (5), 1497–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Walker AJ; Majzner RG; Zhang L; Wanhainen K; Long AH; Nguyen SM; Lopomo P; Vigny M; Fry TJ; Orentas RJ; Mackall CL Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther 2017, 25 (9), 2189–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Salter AI; Ivey RG; Kennedy JJ; Voillet V; Rajan A; Alderman EJ; Voytovich UJ; Lin C; Sommermeyer D; Liu L; Whiteaker JR; Gottardo R; Paulovich AG; Riddell SR Phosphoproteomic Analysis of Chimeric Antigen Receptor Signaling Reveals Kinetic and Quantitative Differences That Affect Cell Function. Sci. Signaling 2018, 11 (544), No. eaat6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Imai C; Mihara K; Andreansky M; Nicholson I; Pui C-H; Geiger T; Campana D Chimeric Receptors with 4–1BB Signaling Capacity Provoke Potent Cytotoxicity against Acute Lymphoblastic Leukemia. Leukemia 2004, 18 (4), 676–684. [DOI] [PubMed] [Google Scholar]
- (57).Rezvani AR; Maloney DG Rituximab Resistance. Best Practice & Research Clinical Haematology 2011, 24 (2), 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Karapetis CS; Shirin K-F; Jonker DJ; Chris JO; Tu D; Tebbutt NC; Simes JR; Chalchal H; Shapiro JD; Robitaille S; Price TJ; Shepherd L; Au H-J; Langer C; Moore MJ; Zalcberg JR K-Ras Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer. N. Engl. J. Med 2008, 359 (17), 1757–1765. [DOI] [PubMed] [Google Scholar]
- (59).Duong CP; Westwood JA; Berry LJ; Darcy PK; Kershaw MH Enhancing the Specificity of T-Cell Cultures for Adoptive Immunotherapy of Cancer. Immunotherapy 2011, 3 (1), 33–48. [DOI] [PubMed] [Google Scholar]
- (60).Grada Z; Hegde M; Byrd T; Shaffer DR; Ghazi A; Brawley VS; Corder A; Schönfeld K; Koch J; Dotti G; Heslop HE; Gottschalk S; Wels WS; Baker ML; Ahmed N TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther.–Nucleic Acids 2013, 2 (7), No. e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Hegde M; Mukherjee M; Grada Z; Pignata A; Landi D; Navai SA; Wakefield A; Fousek K; Bielamowicz K; Chow K; Brawley VS; Byrd TT; Krebs S; Gottschalk S; Wels WS; Baker ML; Dotti G; Mamonkin M; Brenner MK; Orange JS; Ahmed N Tandem CAR T Cells Targeting HER2 and IL13Rα2 Mitigate Tumor Antigen Escape. J. Clin. Invest 2016, 126 (8), 3036–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Schneider D; Xiong Y; Wu D; Nölle V; Schmitz S; Haso W; Kaiser A; Dropulic B; Orentas RJ A Tandem CD19/CD20 CAR Lentiviral Vector Drives on-Target and off-Target Antigen Modulation in Leukemia Cell Lines. J. Immunother Cancer 2017, 5 (1), 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ruella M; Barrett DM; Kenderian SS; Shestova O; Hofmann TJ; Perazzelli J; Klichinsky M; Aikawa V; Nazimuddin F; Kozlowski M; Scholler J; Lacey SF; Melenhorst JJ; Morrissette J; Christian DA; Hunter CA; Kalos M; Porter DL; June CH; Grupp SA; Gill S Dual CD19 and CD123 Targeting Prevents Antigen-Loss Relapses after CD19-Directed Immunotherapies. J. Clin. Invest 2016, 126 (10), 3814–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Beatty GL; Haas AR; Maus MV; Torigian DA; Soulen MC; Plesa G; Chew A; Zhao Y; Levine BL; Albelda SM; Kalos M; June CH Mesothelin-Specific Chimeric Antigen Receptor MRNA-Engineered T Cells Induce Antitumor Activity in Solid Malignancies. Cancer Immunol. Res 2014, 2 (2), 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Liu Z; Zhou H; Wang W; Tan W; Fu Y-XX; Zhu M A Novel Method for Synthetic Vaccine Construction Based on Protein Assembly. Sci. Rep 2015, 4, 7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Gall VA; Philips AV; Qiao N; Clise-Dwyer K; Perakis AA; Zhang M; Clifton GT; Sukhumalchandra P; Ma Q; Reddy SM; Yu D; Molldrem JJ; Peoples GE; Alatrash G; Mittendorf EA Trastuzumab Increases HER2 Uptake and Cross-Presentation by Dendritic Cells. Cancer Res 2017, 77 (19), 5374–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Fraietta JA; Lacey SF; Orlando EJ; Pruteanu-Malinici I; Gohil M; Lundh S; Boesteanu AC; Wang Y; O’Connor RS; Hwang W-T; Pequignot E; Ambrose DE; Zhang C; Wilcox N; Bedoya F; Dorfmeier C; Chen F; Tian L; Parakandi H; Gupta M; Young RM; Johnson BF; Kulikovskaya I; Liu L; Xu J; Kassim SH; Davis MM; Levine BL; Frey NV; Siegel DL; Huang AC; Wherry JE; Bitter H; Brogdon JL; Porter DL; June CH; Melenhorst JJ Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat. Med 2018, 24, 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Lanitis E; Dangaj D; Irving M; Coukos G Mechanisms Regulating T-Cell Infiltration and Activity in Solid Tumors. Ann. Oncol 2017, 28 (suppl_12), xii18–xii32. [DOI] [PubMed] [Google Scholar]
- (69).Mueller KT; Waldron E; Grupp SA; Levine JE; Laetsch TW; Pulsipher MA; Boyer MW; August KJ; Hamilton J; Awasthi R; Stein AM; Sickert D; Chakraborty A; Levine BL; June CH; Tomassian L; Shah SS; Leung M; Taran T; Wood PA; Maude SL Clinical Pharmacology of Tisagenlecleucel in B-Cell Acute Lymphoblastic Leukemia. Clin. Cancer Res 2018, 24 (24), 6175–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.