

SUMMARY
Adenosine-to-inosine RNA editing, catalyzed by adenosine deaminase acting on RNA (ADAR) enzymes, alters RNA sequences from those encoded by DNA. These editing events are dynamically regulated, but few trans regulators of ADARs are known in vivo. Here, we screen RNA-binding proteins for roles in editing regulation with knockdown experiments in the Drosophila brain. We identify zinc-finger protein at 72D (Zn72D) as a regulator of editing levels at a majority of editing sites in the brain. Zn72D both regulates ADAR protein levels and interacts with ADAR in an RNA-dependent fashion, and similar to ADAR, Zn72D is necessary to maintain proper neuromuscular junction architecture and fly mobility. Furthermore, Zn72D’s regulatory role in RNA editing is conserved because the mammalian homolog of Zn72D, Zfr, regulates editing in mouse primary neurons. The broad and conserved regulation of ADAR editing by Zn72D in neurons sustains critically important editing events.
Graphical Abstract

In Brief
Sapiro et al. identify Drosophila Zn72D as an influential regulator of neuronal A-to-I RNA editing and synaptic morphology. Zn72D regulates ADAR levels and editing at a large subset of editing sites, providing insight into the maintenance of critical tissue-specific RNA editing events.
INTRODUCTION
RNA editing expands genetic diversity by altering bases encoded by the genome at the RNA level (Eisenberg and Levanon, 2018; Nishikura, 2016). The deamination of adenosine (A) into inosine (I), a highly prevalent form of mRNA editing, is catalyzed by adenosine deaminase acting on RNA (ADAR) proteins, which are double-stranded RNA-binding proteins that are conserved in metazoans (Bass, 2002). Inosine is recognized by the cellular machinery as guanosine (G); therefore, a single editing event in RNA has the ability to disrupt regulatory mechanisms or to change the protein encoded by the transcript by altering a codon or splice site (Nishikura, 2010). Millions of these RNA editing sites have been identified, necessitating a better understanding of how this process is regulated (Walkley and Li, 2017).
Proper regulation of ADAR proteins and A-to-I RNA editing is essential to organismal health. Humans have two catalytically active ADAR proteins, and functional changes in both proteins are associated with disease. ADAR1 edits endogenous double-stranded RNA, which is critical for proper innate immune function (Liddicoat et al., 2015; Mannion et al., 2014; Pestal et al., 2015), and loss of ADAR1 sensitizes tumors to regression (Gannon et al., 2018; Ishizuka et al., 2019; Liu et al., 2019). ADAR2 edits of a number of ion channels important for regulating neuronal excitability (Rosenthal and Seeburg, 2012), and its dysregulation is associated with a host of neurological diseases including amyotrophic lateral sclerosis, astrocytoma, and transient forebrain ischemia (Slotkin and Nishikura, 2013). In Drosophila, loss of the single Adar homolog, most akin to mammalian Adar2, alters fly locomotion, courtship behaviors, and sleep (Jepson et al., 2011; Robinson et al., 2016); changes synaptic architecture (Bhogal et al., 2011; Maldonado et al., 2013); and leads to neurodegeneration (Palladino et al., 2000). Although maintaining RNA editing levels is critical for proper immune and neuronal function, the regulation of ADAR proteins and editing levels is poorly understood.
Recent studies suggest that regulation of RNA editing levels is highly complex and that critical RNA editing regulators have yet to be identified. RNA editing levels differ across tissues and developmental stages, and these changes do not always correlate with Adar mRNA or protein expression (Sapiro et al., 2019; Tan et al., 2017; Wahlstedt et al., 2009). Trans regulators of ADAR proteins may help explain this variation in editing levels (Li and Church, 2013; Sapiro et al., 2015); however, few ADAR and editing level regulators are known. In mammals, Pin1, WWP2, and AIMP2 regulate ADAR protein levels or localization, leading to changes in editing levels (Behm et al., 2017; Marcucci et al., 2011; Tan et al., 2017). Editing regulators can also be site specific, meaning they regulate ADAR editing at only a subset of editing sites rather than globally regulating ADAR activity. Studies in Drosophila identified FMR1 and Maleless as site-specific regulators of editing (Bhogal et al., 2011; Reenan et al., 2000). Further study has verified that human homologs of both FMR1 (Tran et al., 2019) and Maleless (Hong et al., 2018), along with a number of other RNA-binding proteins and splicing factors, act as site-specific regulators of RNA editing. These factors, including SRSF9, DDX15, TDP-43, DROSHA, and Ro60 (Garncarz et al., 2013; Quinones-Valdez et al., 2019; Shanmugam et al., 2018; Tariq et al., 2013), help to explain some variation in editing levels; however, with thousands of editing sites in flies and millions in humans (Ramaswami and Li, 2014), additional regulators likely remain undiscovered. These previous studies highlight RNA-binding proteins as strong candidates for editing regulators (Washburn and Hundley, 2016). Because of the conserved roles of Drosophila editing regulators as well as the ability to measure nervous system phenotypes, flies serve as an important model for understanding the regulation of editing as it relates to human neurological diseases.
To identify regulators of RNA editing in the brain, we screened 48 RNA-binding proteins for regulation of editing levels using RNA interference (RNAi) in Drosophila neurons. We identified zinc-finger protein at 72D (Zn72D) as a regulator of RNA editing at nearly two-thirds of assayed editing sites. Zn72D knockdown led to a decrease in ADAR protein levels, although that decrease did not fully explain the editing-level changes. We further determined that Zn72D and ADAR physically interact in the brain by binding RNA. In addition to editing changes, loss of Zn72D also led to defects at the neuromuscular junction (NMJ) and impaired locomotion in the fly. Finally, we found that the mouse homolog of Zn72D, Zfr, regulates editing levels in primary cortical neurons, suggesting this mode of editing regulation is highly conserved.
RESULTS
An RNAi Screen Identifies Zn72D as a Dramatic Regulator of RNA Editing
To better understand how ADAR editing is regulated in the brain, we designed an in vivo screen to identify regulators of editing in Drosophila. Because RNA-binding proteins (RBPs) have critical roles in RNA processing and regulate a number of editing events in flies and mammals, we chose to focus on RBPs as candidate regulators of editing. We created a collection of UAS-shRNA fly lines targeting annotated RBPs, as well as GFP as a control, as done previously (Ni et al., 2011). To assay whether loss of these RBPs influenced editing levels, we designed a simple screen in which we crossed UAS-shRNA lines targeting an RBP or GFP to the pan-neuronal driver C155-Gal4 (Figure 1A). We then extracted RNA and produced RNA sequencing (RNA-seq) libraries from two biological replicates of adult knockdown brains. We determined editing levels by calculating the percentage of G-containing reads at adenosines known to be editing sites in control and knockdown brains to identify sites regulated by these RBPs. To validate that approach, we first checked the reproducibility of editing levels between biological replicates of GFP RNAi brains used in the screen as a control and found that editing levels among replicates were highly reproducible (Figure 1B). We then tested the design of the screen by knocking down Adar using two independent short hairpin RNA (shRNA) lines (BDSC28311 and VDRC7763), which reduced Adar mRNA levels by 60% and 72%, respectively. We compared editing levels between two replicates of each Adar knockdown (shAdar) and matched replicates of GFP knockdown (shGFP) at previously identified editing sites. To avoid looking at single nucleotide polymorphisms (SNPs) or false-positive editing sites, we limited the sites queried in our screen to those that were reproducibly edited in controls and altered by Adar knockdowns in these pilot experiments. In total, we identified 1,236 editing sites that were edited reproducibly in the independent sets of GFP RNAi replicates and were reduced significantly by the stronger Adar knockdown as measured by Fisher’s exact tests (Figure 1C).
Figure 1. An RNAi Screen Identifies Zn72D as a Regulator of RNA Editing.
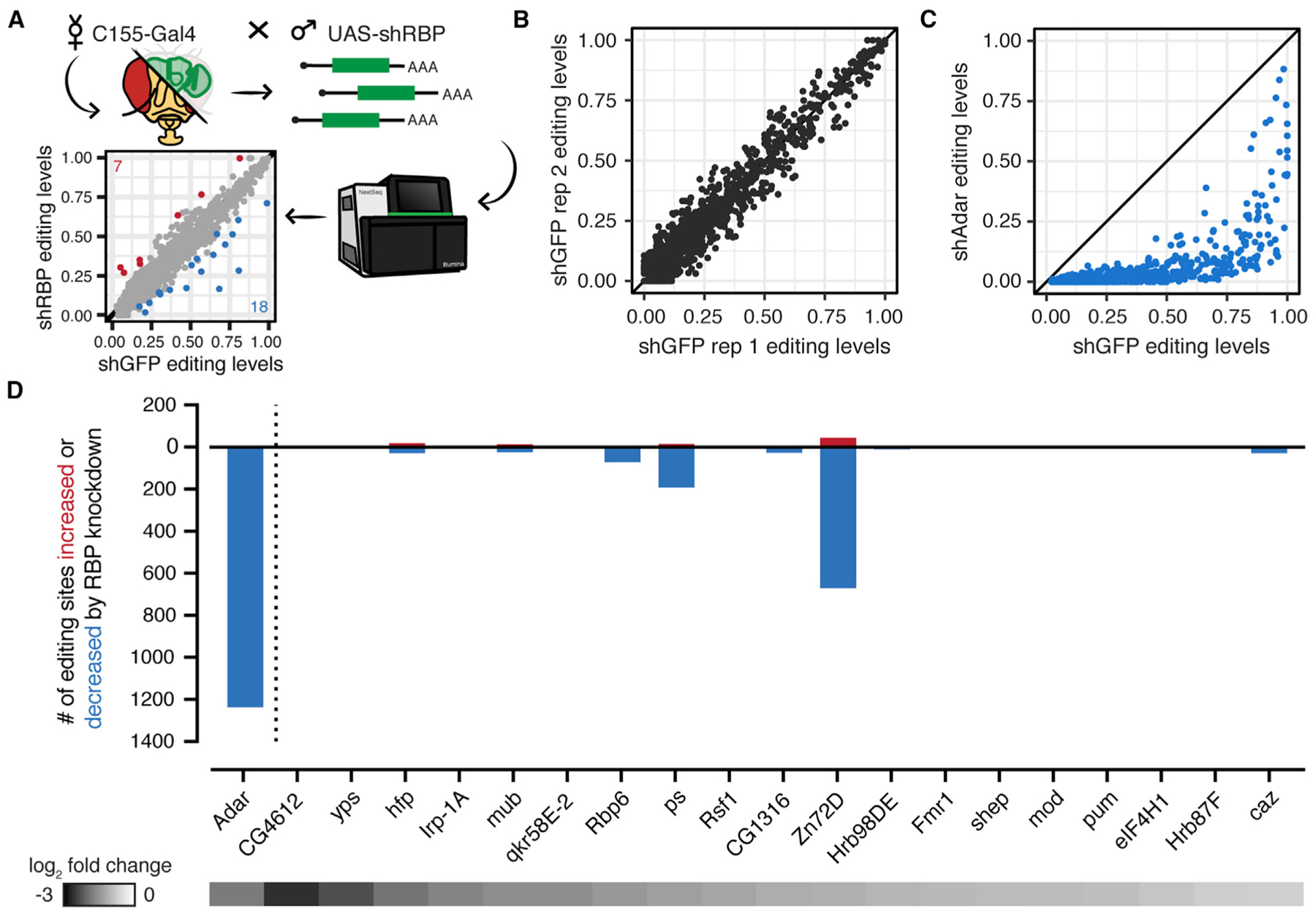
(A) Schematic of RNAi screen. Pan-neuronal driver C155-Gal4 was crossed to UAS-shRNA flies targeting 1 of 20 different RNA-binding proteins. RNA from brains was sequenced to compare editing levels between C155-Gal4; UAS-shGFP controls and C155-Gal4; UAS-shRBP flies.
(B) Comparison of editing levels across two biological replicates of shGFP controls. Biological replicates were highly reproducible.
(C) Comparison of editing levels between C155-Gal4; UAS-shGFP and C155-Gal4; UAS-shAdar (VDRC7763) at sites used in the screen. All sites are reduced by Adar knockdown. Blue dots, p < 0.05, Fisher’s exact tests.
(D) The number of editing sites found to be increased or decreased (p < 0.05, Fisher’s exact tests) upon knockdown of each of 20 RBPs screened. Heatmap shows the log2-fold change of each target RBP between knockdown and control as measured by RNA-seq. shZn72D shows the greatest number of altered editing sites besides shAdar.
We then crossed shRNA lines targeting 48 different RBPs, starting with RBPs that are highly expressed in the brain (Table S1). Of the 48 knockdowns, 17 caused lethality before adulthood and were not screened for editing changes. For the 31 knockdowns that were viable as adults, we performed qPCR to determine the level of knockdown of the target: 19 knockdowns showed greater than 40% reduction of the target mRNA, and we made RNA-seq libraries from two replicates of each of those knockdowns and two GFP-targeting controls. We then determined editing levels at the 1,236 sites that were affected by Adar knockdowns. Editing levels among all biological replicates used in the screen were highly reproducible, similar to shGFP replicates (Figure S1A). We determined whether sites differed between control and RBP knockdowns using Fisher’s exact tests comparing the total number of A and G reads from the biological replicates combined. Figure 1D shows the number of editing sites with increased or decreased editing levels in each RBP knockdown compared with the GFP controls, as well as the knockdown efficiency for each target as measured by RNA-seq (Table S1). Most RBP knockdowns showed evidence of positive or negative regulation of editing at fewer than 50 editing sites (Figures 1D and S1B). Two RBP knockdowns had slightly wider-ranging effects on editing levels. Knockdown of Rbp6 decreased editing at 72 sites and increased editing at 2 sites, and knockdown of pasilla decreased editing at 193 sites and increased editing at 15 sites. By far the most robust regulator of RNA editing, however—in terms of both the number of sites altered and the strength of the effect—was Zn72D. Knockdown of Zn72D decreased editing at 670 editing sites and increased editing at 44 sites, affecting 59% of sites measured (Figure 2A). This dramatic regulation of editing exceeded that of all other RBPs screened as well as others previously reported to regulate ADAR editing (Washburn and Hundley, 2016); therefore, we focused on characterizing Zn72D in this work.
Figure 2. Zn72D Knockdown Alters RNA Editing and ADAR Protein Levels.
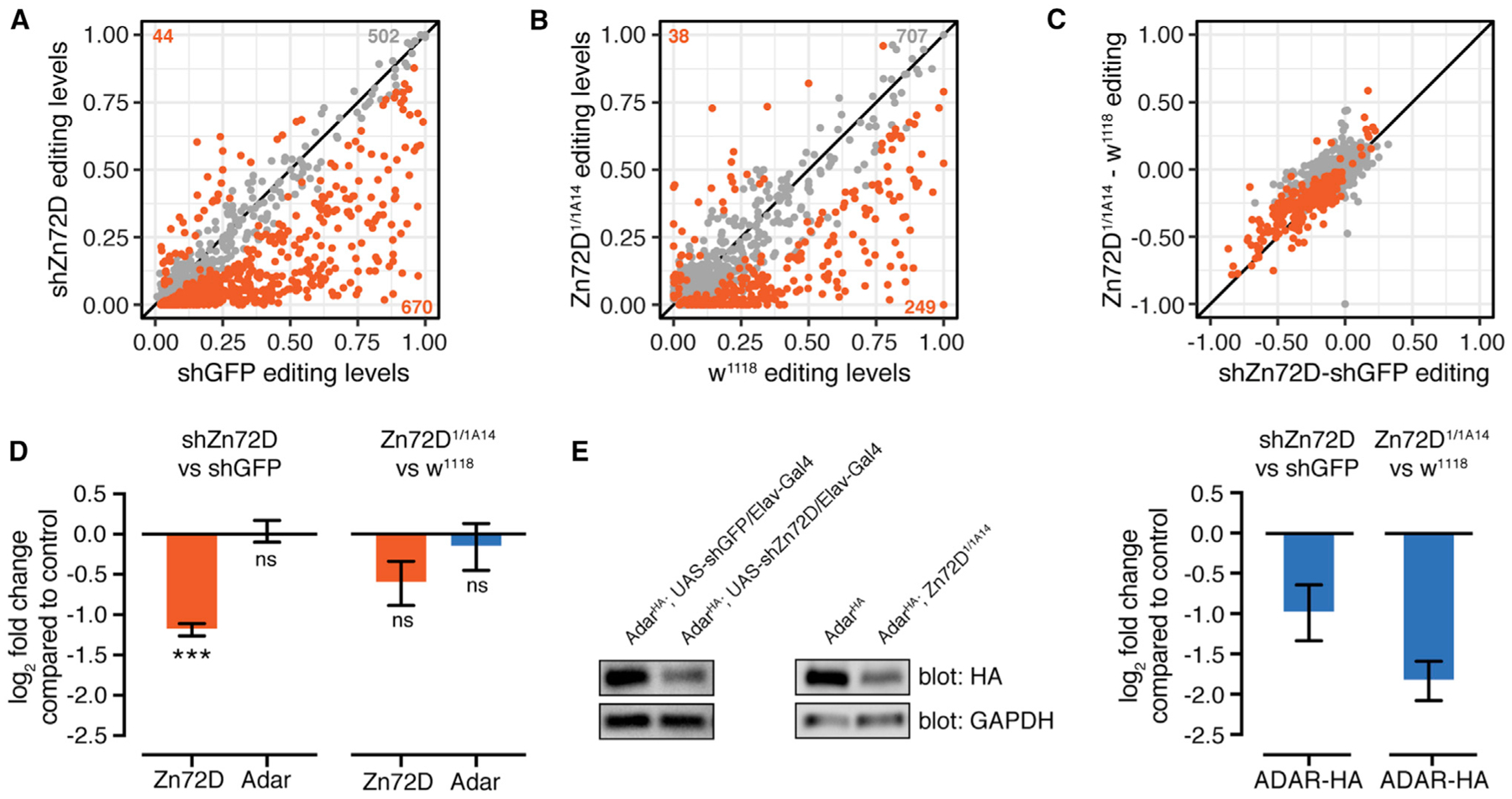
(A) Comparison of editing levels at individual editing sites (dots) between C155-Gal4; UAS-shGFP and C155-Gal4; UAS-shZn72D, from the RNAi screen in Figure 1. Orange dots, p < 0.05, Fisher’s exact tests.
(B) Comparison of editing levels between two replicates of w1118 and Zn72D1/1A14 pupal heads. Orange dots, p < 0.05, Fisher’s exact tests. Many sites are altered in both Zn72D knockdowns and mutants compared with controls.
(C) Comparison of the difference in editing between C155-Gal4; UAS-shZn72D and C155-Gal4; UAS-shGFP and Zn72D1/1A14 and w1118 from (A) and (B). The same sites are significantly altered in both knockdowns and mutants. Orange dots, p < 0.05 in both.
(D) Log2-fold change of Zn72D and Adar mRNA levels in C155-Gal4; UAS-shZn72D compared with C155-Gal4; UAS-shGFP adult brains and Zn72D1/1A14 compared with w1118 pupal heads. Adar mRNA levels are not decreased in Zn72D knockdown and mutants. ***p < 0.0001, ns, p > 0.05, Wald tests. n = 2, error bars indicate SE.
(E) Western blot of ADAR-HA protein in Elav-Gal4 / shGFP and Elav-Gal4 / shZn72D adult brains and w1118 and Zn72D1/1A14 pupal heads. n = 3, a representative result is shown. At right, quantification of HA loss in Zn72D knockdown and mutant compared with controls, normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). ADAR-HA protein levels are decreased in both Zn72D knockdown and mutants. Data are represented as means ± SE.
Zn72D Knockdown Alters RNA Editing and ADAR Protein Levels
Because Zn72D had the strongest effect on editing in our RNAi screen, we wanted to first validate the site-specific nature of that regulation (Figure 2A). Thus, we crossed an independent UAS-shZn72D line (BDSC55635) to C155-Gal4 and sequenced the RNA to confirm the editing-level changes. We observed a similar editing phenotype with that independent shRNA line, with the increased and decreased editing sites showing the same responsiveness to Zn72D knockdown with both shRNAs (Figures S2A and S2B; Table S1). To verify that the editing phenotype was not a consequence of the RNAi system itself or of off-target effects, we measured editing in Zn72D mutants. We crossed two Zn72D mutant alleles, Zn72D1 and Zn72D1A14, which caused premature stop codons at amino acids 38 and 559, respectively. The resulting Zn72D1/1A14 mutant flies died as pupae, as previously reported (Brumby et al., 2004), so we collected heads approximately 72 h after puparium formation and sequenced the RNA to check editing levels. Zn72D1/1A14 pupal heads showed large differences in editing from wild-type pupal heads (Figure 2B; Table S1). We compared the changes in editing observed in the Zn72D knockdowns (shZn72D) to those in the Zn72D mutants. Despite the difference in the developmental stage of the flies, both the increases and decreases in editing levels caused by Zn72D knockdown were similar to those in the mutants (Figure 2C), confirming that the Zn72D editing phenotype was highly reproducible and site specific.
Of the editing sites affected by Zn72D knockdown, 93% showed decreased editing levels, so we wanted to determine whether Zn72D loss reduced Adar mRNA or protein levels. We first checked Adar mRNA levels between shGFP and shZn72D brains and wild-type and mutant heads. Zn72D loss did not lead to a significant decrease in Adar mRNA levels (Figures 2D and S2C). To check ADAR protein levels, we knocked down Zn72D using the pan-neuronal driver Elav-Gal4 in AdarHA flies (Jepson et al., 2011), where the endogenous ADAR protein is tagged with a hem-agglutinin (HA) epitope. We confirmed that the editing phenotype in those flies reproduced that of the Zn72D knockdown driven by the C155-Gal4 driver (Figures S2D–S2F). By western blot, we found that ADAR-HA protein was decreased by 49% in Zn72D-knockdown brains. We also crossed the mutants into the AdarHA background, and we found that ADAR-HA levels were decreased by 72% in AdarHA; Zn72D1/1A14 pupal heads (Figure 2E), verifying an ADAR protein reduction upon loss of Zn72D.
Zn72D and Adar Knockdowns Have Divergent Editing Phenotypes
Although a decrease in the ADAR protein may explain some editing decreases in the Zn72D knockdown and mutant flies, that finding did not appear to fully explain the complex editing phenotype observed. Unlike the Adar knockdown, the Zn72D knockdown did not decrease editing at all sites. To more closely examine the differences in editing between Zn72D and Adar knockdowns, we directly compared the editing levels in Zn72D-knockdown brains to those in Adar-knockdown brains. The stronger Adar knockdown (VDRC7763; see Figure 1C) decreased editing more than the Zn72D knockdown did at 634 sites and less than the Zn72D knockdown at only seven sites (Figure 3A). Concordantly, when crossed into the AdarHA background, this Adar knockdown showed a more dramatic decrease in ADAR protein than Zn72D knockdown did (Figure 3B). The weaker of our two Adar shRNAs (BDSC28311) significantly decreased editing compared with controls at nearly all editing sites but to a lesser extent (Figure 3C). Compared directly to Zn72D knockdown, this Adar knockdown diverged strongly at many sites in both directions, decreasing editing more than Zn72D knockdown did at 279 sites and less than Zn72D knockdown had at 145 sites. It was similar to Zn72D knockdown at 621 sites (Figure 3D). This Adar shRNA did not decrease AdarHA mRNA or protein levels or alter editing in the AdarHA background, so we were unable to compare ADAR protein levels between these knockdowns. Still, the editing differences strongly suggested that Zn72D knockdown altered editing in a manner that did not neatly resemble a decrease in Adar.
Figure 3. Zn72D and Adar Knockdowns Have Divergent Editing Phenotypes.
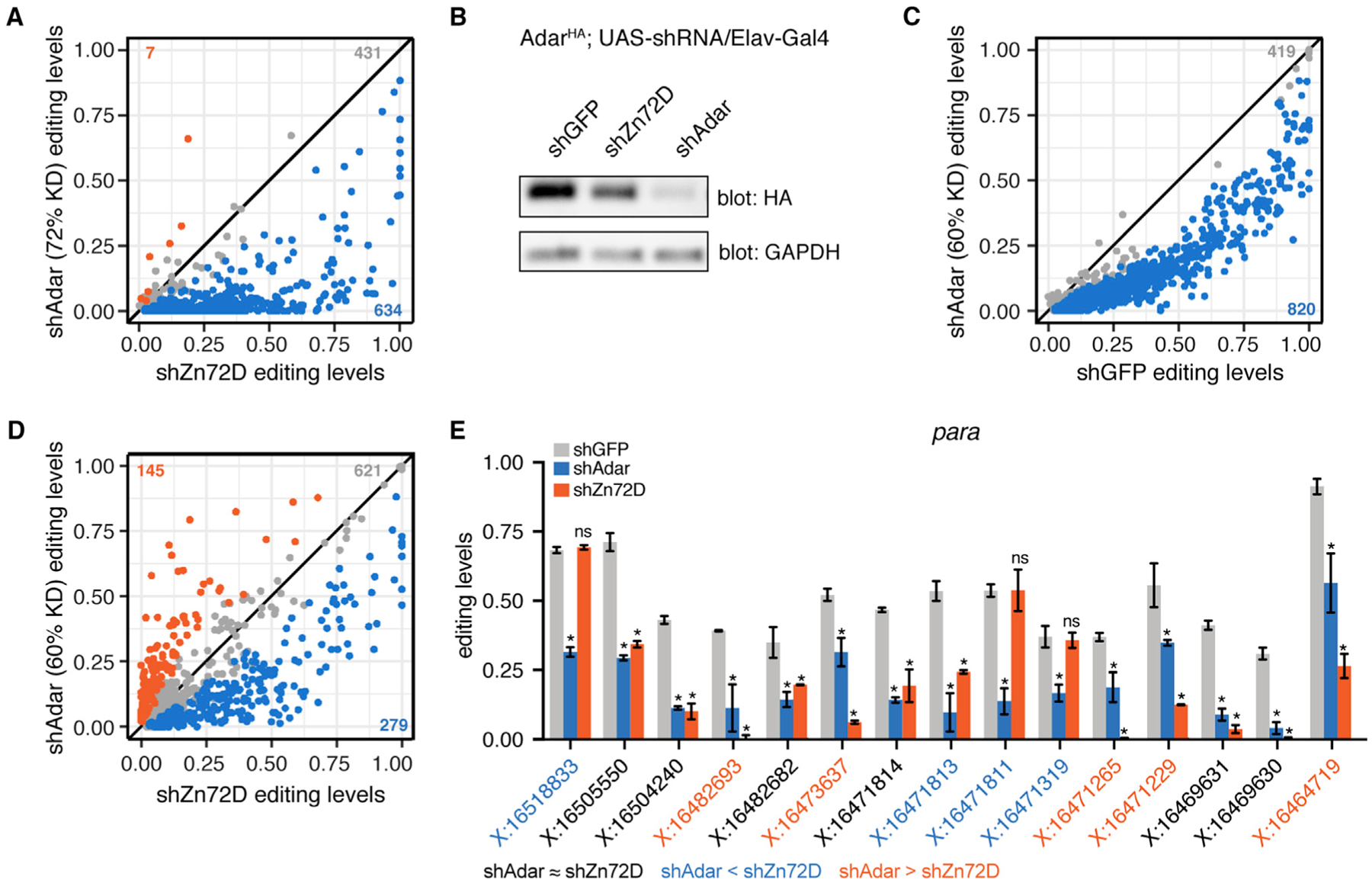
(A) Comparison of RNA editing levels between C155-Gal4; UAS-shZn72D (screen) and C155-Gal4; UAS-shAdar (VDRC7763; editing levels compared with shGFP in Figure 1C). Blue sites, p < 0.05, Fisher’s exact tests, Adar knockdown decreases editing more than Zn72D knockdown does. Gray sites, p > 0.05, shAdar equals shZn72D editing. Orange sites, p < 0.05, Fisher’s exact tests, Zn72D knockdown decreases editing more than Adar knockdown does. The number of sites falling into each category is shown.
(B) Western blot comparing the level of ADAR-HA protein in AdarHA;UAS-shGFP / Elav-Gal4, AdarHA ; UAS-shZn72D / Elav-Gal4, and AdarHA ; UAS-shAdar (VDRC7763) / Elav-Gal4. n = 3, a representative result is shown. This Adar knockdown leads to a greater reduction in ADAR-HA protein than Zn72D knockdown does, consistent with the editing level comparison in (A).
(C) Comparison of RNA editing levels between C155-Gal4; UAS-shGFP and C155-Gal4; UAS-shAdar (BDSC28311). Blue sites, p < 0.05, Fisher’s exact tests.
(D) Comparison of RNA editing levels between C155-Gal4; UAS-shZn72D (screen) and C155-Gal4; UAS-shAdar (BDSC28311). Blue sites, p < 0.05, Fisher’s exact tests, Adar knockdown decreases editing more than Zn72D knockdown does. Gray sites, p > 0.05, shAdar equals shZn72D editing. Orange sites, p < 0.05, Fisher’s exact tests, Zn72D knockdown decreases editing more than Adar knockdown does. The number of sites falling into each category is shown.
(E) Editing levels in C155-Gal4; UAS-shGFP, C155-Gal4; UAS-shAdar, and C155-Gal4; UAS-shZn72D brains in para. Sites within the transcript are differentially affected by Zn72D loss. n = 2, data are represented as means ± SD. *p < 0.001, ns, p > 0.05, Fisher’s exact tests between shGFP and either shAdar (above blue bar) or shZn72D (above orange bar). Orange coordinates, shZn72D decreases editing more than shAdar does. Black coordinates, no difference between shAdar and shZn72D. Blue coordinates, shAdar decreases editing more than shZn72D does. Blue and orange, p < 0.001, Fisher’s exact tests.
The distinct nature of Zn72D editing regulation was also clear across editing sites found in the same transcript. For example, the highly edited transcript paralytic (para) had multiple editing sites that showed different editing changes in response to Zn72D and Adar knockdowns. Figure 3E shows editing levels in para at 15 highly edited sites (>20% in controls) in shGFP, shAdar (BDSC28311), and shZn72D brains. At six sites in para, Zn72D and Adar knockdowns led to similar editing decreases (Figure 3E, coordinates in black), whereas at four sites Adar knockdown decreased editing more than Zn72D knockdown did (Figure 3E, coordinates in blue), and at five sites, Zn72D knockdown decreased editing more than Adar knockdown had (Figure 3E, coordinates in orange). Those sites with different responses to Zn72D knockdown could be found within a few bases of each other, as seen at three editing sites in para located within four bases of each other (chrX:16471811 to chrX:16471814). Another transcript, quiver (qvr), showed similar patterns, including differences between Zn72D and Adar knockdowns and vastly different effects of Zn72D knockdown on four sites that were all more than 70% edited in controls and located within 23 bases of each other (chr2R:11447601 to chr2R:11447623) (Figure S3A). To better understand how Zn72D affected editing differently across sites within the same transcript, we compared phenotypes within all transcripts. Of 187 transcripts in which we measured editing at multiple editing sites, 131 (70%) included at least one site that was affected and at least one site that was not affected by Zn72D knockdown (Figure S3B). Those transcripts in which all sites were either affected or unaffected by Zn72D tended to contain fewer editing sites than those that showed mixed effects (Figure S3C). These results suggested that Zn72D’s site-specific effect on editing was highly localized down to individual editing sites, distinct from a global decrease in ADAR protein.
Zn72D Interacts with ADAR and ADAR-Target mRNAs
Because decreases in ADAR protein levels did not explain the Zn72D knockdown editing phenotype, we hypothesized that site-specific regulation of editing by Zn72D might result from the protein binding the same transcripts as ADAR, as has been previously demonstrated for other known site-specific regulators of editing (Bhogal et al., 2011; Hong et al., 2018; Quinones-Valdez et al., 2019; Rajendren et al., 2018; Shanmugam et al., 2018). We first asked whether Zn72D and ADAR proteins were both found in the nucleus, where most editing in Drosophila occurs (Rodriguez et al., 2012). Using a GFP-exon-trap allele of Zn72D, Zn72DGFP (Zn72DCA07703; BDSC50830) that expresses a GFP-tagged version of Zn72D from the endogenous locus (Morin et al., 2001), we used immunofluorescence microscopy to determine the localization of both ADAR and Zn72D proteins in AdarHA; Zn72DGFP flies. We found that ADAR and Zn72D colocalize in the nuclei in the brain, along with a nuclear marker in the neuron Elav (Figures 4A–4D).
Figure 4. Zn72D Interacts with ADAR in an RNA-Dependent Manner.
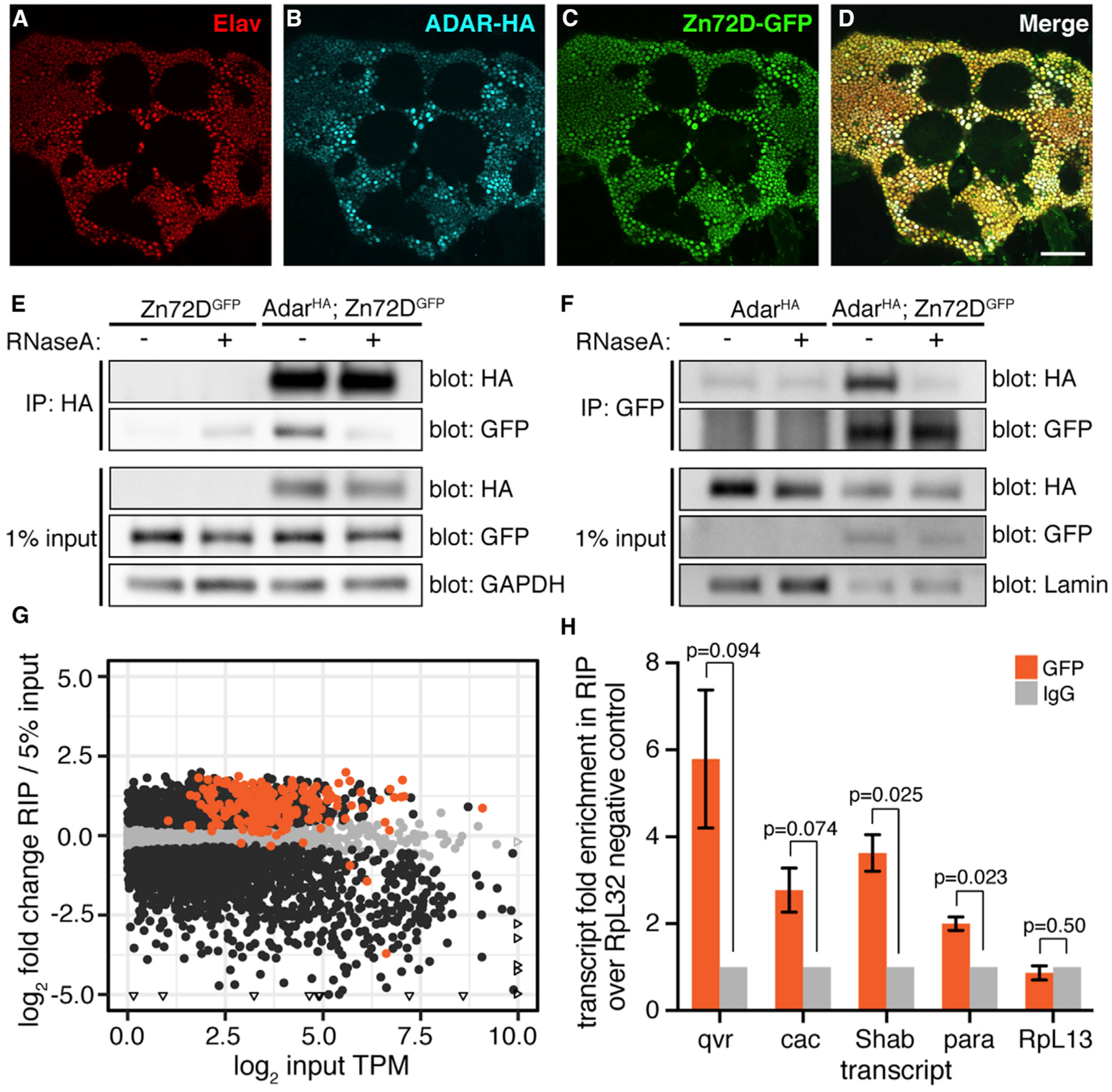
(A–D) Immunofluorescent staining of Elav (A), ADAR-HA (B), Zn72D-GFP (C), and all three merged (D) in the adult fly brain. All proteins are expressed in neuronal nuclei. Images are a single slice, scale bar: 50 μm.
(E) Western blots of HA and GFP after immunoprecipitation of ADAR-HA from Zn72DGFP (control) and AdarHA; Zn72DGFP heads. Half of each IP was treated with RNase A. Blots of HA, GFP, and GAPDH from 1% of input material are shown. n = 3, a representative result is shown.
(F) Western blots of HA and GFP after immunoprecipitation of Zn72D-GFP from AdarHA; Zn72DGFP head nuclei. Half of each IP was treated with RNase A. Blots of HA, GFP, and Lamin from 1% of input material are shown. n = 3, a representative result is shown. ADAR-HA and Zn72D-GFP interact in the presence of RNA.
(G) Scatterplot of transcript enrichment in Zn72D-GFP RIP-seq. Log2-fold change expression in RIP samples compared with 5% input is plotted versus the log2 of the average transcripts per kilobase million (TPM) of each transcript (dot) in the input samples. Orange dots, transcripts have editing sites affected by Zn72D. Black dots, p < 0.05, Wald tests. Triangles represent points falling outside of graph boundaries. n = 3; 185 of 216 transcripts measured with editing sites altered by Zn72D knockdown are enriched in the RIP.
(H) Enrichment of qvr, cac, Shab, para, and negative control RpL13 recovered in the Zn72D-GFP RIP, normalized to IgG control RIP and to enrichment of negative control RpL32, as measured by qPCR. n = 3, error bars represent SE; p values, paired two-tailed t tests. Transcripts enriched in the RIP as measured by RNA-seq(G) are also enriched when measured by qPCR.
We next tested whether ADAR and Zn72D proteins physically interacted. We immunoprecipitated ADAR-HA from AdarHA; Zn72DGFP fly-head lysates and Zn72DGFP fly-head lysates as negative controls. Zn72D-GFP co-immunoprecipitated with ADAR-HA in the anti-HA IP in AdarHA; Zn72DGFP head lysates. However, after treatment with RNase A, the interaction was significantly weakened, suggesting that the two proteins interact in an RNA-dependent manner (Figure 4E). We subsequently performed the reciprocal co-immunoprecipitation (coIP) in nuclear lysates from heads of AdarHA; Zn72DGFP flies, using AdarHA flies as a negative control. We found that ADAR-HA co-immunoprecipitated with Zn72D-GFP in nuclear lysates from flies expressing both tagged proteins but not after RNase A treatment (Figure 4F), which suggested that ADAR and Zn72D interact in an RNA-dependent manner within the nucleus.
The RNA-dependent interaction between Zn72D and ADAR suggested that the proteins interact by binding the same RNAs or are found within the same ribonucleoprotein (RNP) complexes. We hypothesized that Zn72D interacts with the transcripts containing editing sites affected by Zn72D knockdown. We attempted to perform single-end enhanced crosslinking and immunoprecipitation sequencing (seCLIP-seq) (Van Nostrand et al., 2017) on Zn72D-GFP from fly heads to determine Zn72D’s RNA-binding sites but saw little evidence of RNA binding, likely because of the inefficiency of crosslinking proteins to double-stranded RNA (dsRNA) in vivo using UV light (Wheeler et al., 2018). Instead, we performed RNA immunoprecipitation and sequencing (RIP-seq) on Zn72D by pulling down Zn72D-GFP and its interacting RNAs from fly heads without crosslinking, extracting RNA from inputs and immunoprecipitations (IPs) and making RNA-seq libraries. For negative controls, we split lysates in half and incubated one-half with immunoglobulin G (IgG) antibody rather than GFP antibody; these negative controls did not immunoprecipitate enough RNA to amplify RNA-seq libraries, suggesting our pull-down was specific to RNAs bound by Zn72D-GFP. To determine transcript enrichment in the RIP, we counted the sequencing reads mapping to each gene in both the IP libraries and matched input libraries made from RNA extracted from 5% of the input lysates. We then used those counts as inputs to DESeq2 (Love et al., 2014) to determine genes with increased or decreased expression in the RIP compared with the input. We found that, of the 216 transcripts sequenced in the RIP with at least one editing site affected by Zn72D, 185 (86%) were significantly enriched in the RIP over the input (Figure 4G; Table S2). To validate the results of the RIP-seq, we used qPCR to quantify the relative levels of qvr, cac, para, Shab, and the negative control RpL13 in the three biological replicates of GFP IPs, IgG negative control IPs, and their matched inputs (Figure 4H). From three technical replicates of three biological replicates per IP, we calculated the fold enrichment of each transcript of interest over the enrichment of a negative control transcript RpL32 after normalizing to the IgG-negative control IPs. We found that qvr, cac, para, and Shab, but not RpL13, transcripts showed between 2- and 6-fold enrichment over RpL32 in the Zn72D-GFP IP samples after normalization to the IgG IPs and input amounts. Taken together, these experiments support the hypothesis that Zn72D interacts with the same RNAs as ADAR does, which may help explain its role as a site-specific regulator of editing levels.
Because many edited transcripts immunoprecipitated with Zn72D, and Zn72D and its human homolog ZFR both have reported roles in regulating pre-mRNA splicing (Haque et al., 2018; Worringer and Panning, 2007), we determined whether Zn72D knockdown led to splicing changes in the fly brain. To identify alternative splicing changes in Zn72D-knockdown brains (BDSC55635; see Figure S2A) compared with shGFP controls, we used Mixture of Isoforms (MISO) (Katz et al., 2010), which identifies differentially regulated isoforms across samples. As expected, Zn72D knockdown led to a change in maleless splicing (Worringer and Panning, 2007). We further found that Zn72D knockdown altered splicing in 40 of the 252 transcripts in which we observed editing changes (Table S3). Those 40 transcripts contained 216 of 785 editing sites (28%) altered by Zn72D; 88 of those 216 editing sites (41%) were located within or between the exons bordering the altered splice junction, whereas the others were either upstream or downstream of the differentially spliced region. We found 400 altered splicing events in 257 transcripts; 217 of those transcripts (84%) did not contain editing sites that were altered by Zn72D, suggesting Zn72D regulated both splicing and editing in some transcripts and regulated splicing and editing independently in many transcripts.
Loss of Zn72D Leads to Impaired Locomotion and NMJ Defects
RNA editing is necessary for proper neuronal function in the fly (Jepson et al., 2011; Palladino et al., 2000). Because loss of Zn72D led to such a dramatic change in RNA editing levels, we hypothesized that it might have a similar role to ADAR in regulating neuronal function. First, we tested locomotion in Zn72D-knockdown flies. Although Zn72D mutants died as pupae, C155-Gal4; UAS-shZn72D flies were viable into adulthood, allowing us to test their climbing ability using a negative geotaxis assay. We measured climbing in flies with GFP RNAi and Zn72D RNAi driven by Elav-Gal4 by determining the proportion of flies of each genotype that climbed more than halfway up a 20-cm glass vial over time. We found that an average of 36% of Zn72D-knockdown flies climbed above 10 cm in a glass vial after 2 min compared with 100% of GFP-knockdown flies (Figure S4A). This climbing defect was more severe than what we observed for Adar knockdown, suggesting that Zn72D knockdown led to a locomotion phenotype that was distinct from Adar knockdown. In an independent test of GFP and Zn72D RNAi driven by C155-Gal4, 46% of Zn72D-knockdown flies were found above the 10-cm mark after 5 min compared with 100% of GFP-knockdown flies (Figure S4B).
To more deeply explore the cellular basis for this locomotor defect, we examined how the loss of Zn72D affected the morphology and organization of synapses at the NMJ. ADAR is necessary for proper synaptic architecture and function at the NMJ (Bhogal et al., 2011; Maldonado et al., 2013), and because Zn72D regulates ADAR editing at many sites, we hypothesized that it may similarly be necessary for NMJ organization. In Zn72D mutants, we examined synaptic morphology and observed a 6-fold increase in the number of satellite boutons (Figures 5A–5D and I), a defect typically associated with impaired endocytic cycling and BMP signaling (Dickman et al., 2006; O’Connor-Giles et al., 2008). In vesicle-cycling mutants, such as synaptotagmin I (syt I), endophilin, and synaptojanin, there is a marked increase in satellite bouton number. To determine whether any of those endocytic proteins were affected by the loss of Zn72D, we used immunocytochemistry to examine Syt I levels at the NMJ. At Zn72D-mutant NMJs, Syt I levels were decreased by 31% (Figures 5A–5D and J), suggesting a potential mechanism by which the loss of Zn72D results in excessive satellite boutons. Intriguingly, loss of ADAR increased levels of Syt I (Maldonado et al., 2013), suggesting that Zn72D and ADAR can regulate the levels of synaptic proteins differently. Consistent with that difference, Adar mutants lacked the increased number of satellite boutons (Bhogal et al., 2011), suggesting that both mutants regulate aspects of NMJ architecture differently. However, we also observed similarities between ADAR and Zn72D regulation of protein levels at the NMJ. Loss of ADAR also alters the levels of postsynaptic GluRIIA receptors (Maldonado et al., 2013); this is thought to be in response to changes in presynaptic function. Multiple allelic combinations of Zn72D mutants show a 32% reduction in synaptic GluRIIA staining (Figures 5E–5H and K); this is consistent with the 37% reduction observed in ADAR mutants (Maldonado et al., 2013). Together with the changes in morphology and synaptic Syt I intensity, these results suggest that NMJ phenotypes arising from the loss of Zn72D cannot be completely explained by a loss of ADAR editing. Rather, there are likely to be ADAR-dependent and ADAR-independent roles of Zn72D in regulating NMJ synapse organization.
Figure 5. Loss of Zn72D Regulates NMJ Architecture and Protein Levels.
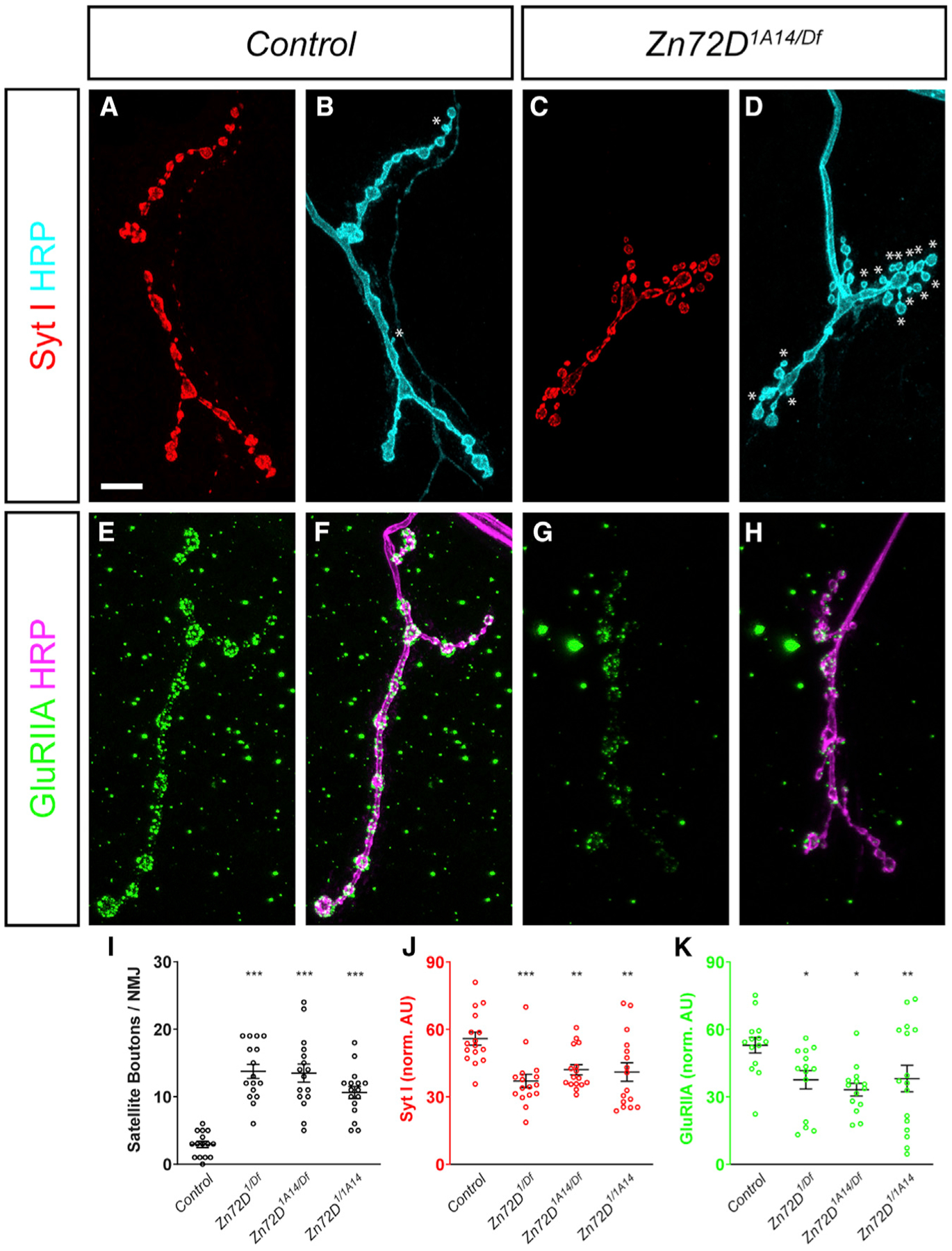
(A–D) Third instar larvae stained with antibodies against Syt I (red) and HRP (cyan) in Control (A and B) and Zn72D1A14/Df mutant larvae (C and D). Df is Df(3L)Exel6127, which lacks the Zn72D locus. Asterisks indicate satellite boutons. Loss of Zn72D markedly increases the incidence of satellite boutons and reduces the apparent fluorescent intensity of Syt I staining.
(E–H) Third instar larvae stained with antibodies against GluRIIA (green) and HRP (magenta) in Control (E and F) and Zn72D1A14/Df mutant larvae (G and H). Loss of Zn72D reduces synaptic GluRIIA staining. Scale bar, 10 mm.
(I–K) Quantification of satellite boutons per NMJ (I), Syt I fluorescence levels (J) and GluRIIA fluorescence levels (K). Multiple allelic combinations of Zn72D mutants show increased satellite bouton numbers and reduced Syt I and GluRIIA staining. Both GluRIIA and Syt I fluorescence levels are normalized to HRP staining at the same NMJs, which is unchanged across all genotypes, suggesting that these deficits are specific. For all graphs, open circles represent each individual value while the mean ± SEM is indicated by the error bars. In all cases, n R 7 animals, 14 NMJs for each genotype. *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA followed by a Dunnett’s multiple comparisons test.
Zn72D Regulation of Editing Is Conserved in Mammalian Neurons
We next sought to determine whether the regulation of ADAR and RNA editing levels by Zn72D was conserved in mammals, as has been demonstrated for other regulators of RNA editing identified in flies (Bhogal et al., 2011; Hong et al., 2018). We designed shRNAs against mouse Zfr, the mammalian homolog of Zn72D, as well as Adar2, which encodes the homolog of Drosophila ADAR, and Adar1, which encodes the other catalytically active mammalian ADAR protein (Figure 6A). We depleted Adar1, Adar2, and Zfr from mouse primary cortical neurons, and performed RNA-seq. We compared editing levels between two combined biological replicates of primary neurons transfected with control shRNAs to those transfected with shRNAs targeting Adar1, Adar2 and Zfr (Figures 6B–6D). In each knockdown, we found more than 100 sites with decreased editing levels, demonstrating that Zfr knockdown alters editing levels in this mammalian neuronal context (Table S4). Among the sites affected by Zfr was the Gria2 Q/R site that is known to have a critical role in neuronal function (Horsch et al., 2011). We compared the sites decreased upon knockdown of Adar1, Adar2, and Zfr and found that all three knockdowns altered a distinct subset of editing sites. Of note, the set of sites decreased by Zfr knockdown more closely overlapped with those decreased by Adar2 knockdown than those decreased by Adar1 knockdown (Figure 6E). This finding is consistent with our findings in Drosophila, because the single ADAR enzyme is a closer homolog of mammalian ADAR2 in sequence and in function (Keegan et al., 2011).
Figure 6. Zfr Affects Editing Levels and Adar2 mRNA Levels in Mouse Primary Neurons.
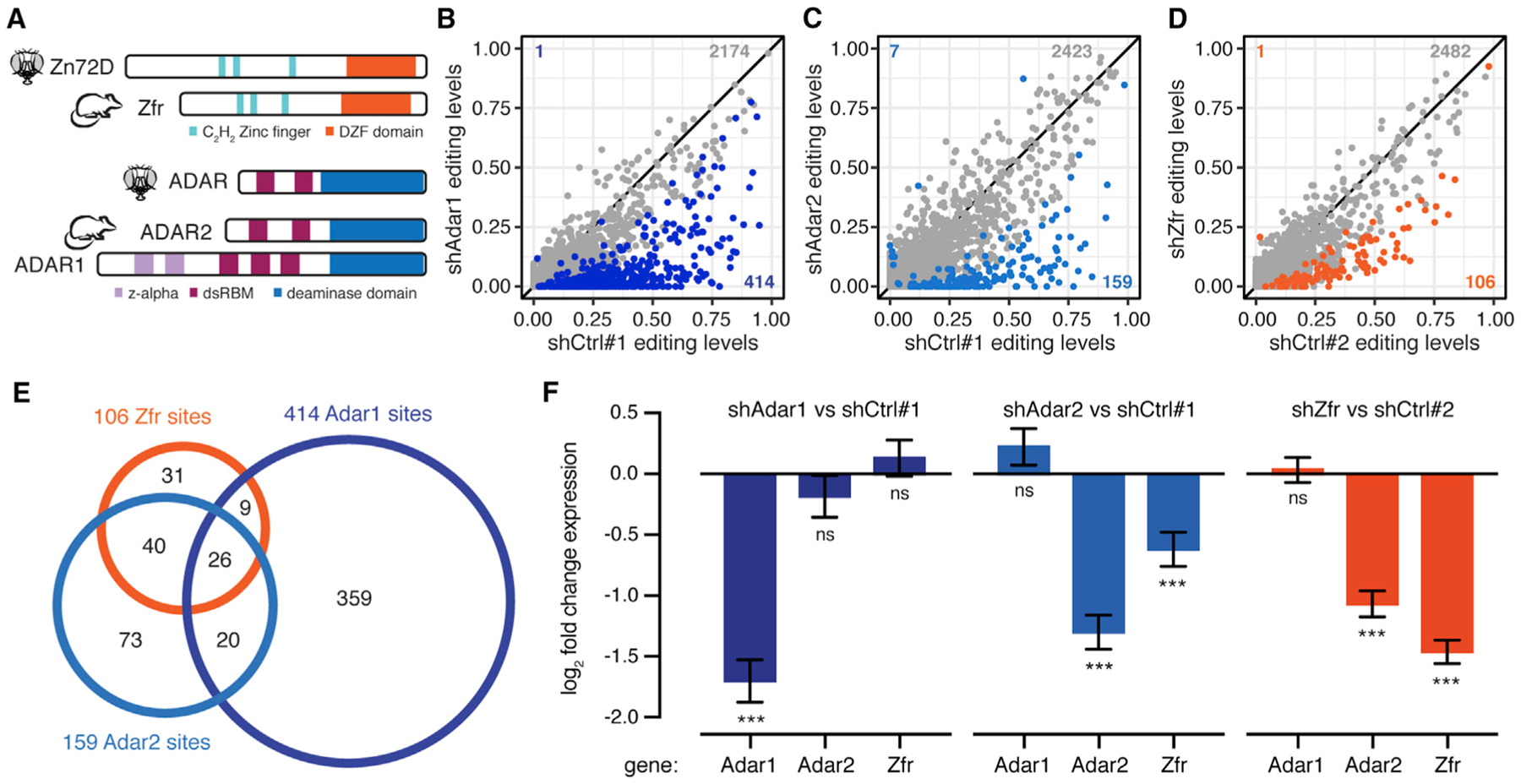
(A) Schematic of protein domains of Zn72D and its mouse homolog, Zfr (top). Schematic of protein domains of Drosophila ADAR and its mouse homolog ADAR2 along with the other catalytically active mouse ADAR, ADAR1 (bottom).
(B) Comparison of editing levels between mouse primary neurons transfected with a control shRNA versus shAdar1. Blue dots, p < 0.05, Fisher’s exact tests. n = 2.
(C) Comparison of editing levels between mouse primary neurons transfected with a control shRNA versus shAdar2. Blue dots, p < 0.05, Fisher’s exact tests. n = 2.
(D) Comparison of editing levels between mouse primary neurons transfected with control shRNA versus shZfr. Orange dots, p < 0.05, Fisher’s exact tests. n = 2. The number of changed sites is indicated. Many editing sites show decreased editing upon Adar1, Adar2, and Zfr knockdown.
(E) Venn diagram showing the overlap of affected sites between shAdar1, shAdar2, and shZfr. shZfr sites share a larger overlap with ADAR2-affected sites, although the three sets are distinct.
(F) Log2-fold changes of mRNA levels of Adar1, Adar2, and Zfr in shAdar1, shAdar2, and shZfr neurons compared with shControl neurons. n = 2, error bars represent SE. ***p < 0.001, ns, p > 0.05, Wald tests. Adar1 knockdown does not affect Adar2 or Zfr levels, whereas Adar2 knockdown decreases Zfr levels, and Zfr knockdown decreases Adar2 levels.
Because Zfr affected mostly ADAR2-regulated sites, we used the RNA-seq data to measure Adar1, Adar2, and Zfr mRNA expression levels in all three knockdowns. We found that Zfr knockdown led to a decrease in Adar2 mRNA expression (Figure 6F), suggesting that Zfr regulated Adar2 levels in mouse primary neurons. Further supporting an ADAR2-centric role for editing level regulation by Zfr, we found that knocking down ZFR in human HEK293T cells (Haque et al., 2018) led to no change in editing (Figures S5A–S5C; Table S4). Unlike mouse primary neurons, Adar2 is scantily expressed in HEK293T cells (Figure S5D), and therefore, ADAR1 is likely responsible for most editing events in these cells. Taken together, these data suggest that the broad mechanisms of Zn72D regulation of editing—regulating both ADAR levels and editing at specific sites—are conserved in the mouse brain between Zfr and ADAR2.
DISCUSSION
RNA editing is dynamically regulated during development and across tissue and cell types (Graveley et al., 2011; Sapiro et al., 2019; Tan et al., 2017; Wahlstedt et al., 2009), but few factors responsible for that regulation are known. Because RBPs such as ADAR form extensive cross-regulatory networks (Dassi, 2017), they are top-candidate editing regulators, and many known regulators of editing are proteins that interact with ADARs by binding the same RNAs (Quinones-Valdez et al., 2019; Washburn and Hundley, 2016). Our previous work suggested a role for trans regulators of editing in the fly brain (Sapiro et al., 2015, 2019), so we perturbed RBP levels in that context. Most of the RBPs we screened had only a small influence on editing, suggesting that editing levels are fairly stable, even as the RBP landscape changes. This result is consistent with our previous finding that many editing sites have stable editing levels across different neuronal populations in the fly brain (Sapiro et al., 2019) as well as a study of the role of RBPs in regulating editing levels in human cells (Quinones-Valdez et al., 2019). However, our screen results may include false-negatives because of incomplete knockdown. Future experiments testing additional double-stranded RBPs for roles in RNA editing may help identify more critical trans regulators because ADARs interact with double-stranded RNA species (Bass, 2002).
We identified Zn72D as a broadly influential regulator of RNA editing. This zinc finger RBP was first identified as a suppressor of a mutation in the cell-cycle regulator cyclin E (Brumby et al., 2004). Zn72D has three C2H2 zinc finger domains and a DZF domain that facilitates protein dimerization and contributes to RNA binding (Castello et al., 2016; Wolkowicz and Cook, 2012). The mouse homolog of Zn72D, Zfr, is predicted to bind A-form dsRNA helices because of similarities between its zinc finger domains and those of other dsRNA-binding proteins: long linkers between zinc fingers, an interhistidine distance of five amino acids, and a reversal of characteristic aromatic and hydrophobic residues (Meagher et al., 1999). Zfr alters splicing in human macrophages, regulating innate immunity (Haque et al., 2018), suggesting it has a broad role in RNA processing. Zn72D also regulates the male-specific lethal (MSL) dosage compensation complex in flies by altering the splicing of maleless, which encodes a critical member of the complex (Worringer and Panning, 2007). Interestingly, a gain-of-function mutation in maleless regulates RNA editing levels in para (Reenan et al., 2000), although loss-of-function mutations did not have the same effect. Because the human homolog of Maleless, DHX9, is also known to regulate editing (Hong et al., 2018), some of the editing phenotype upon loss of Zn72D may be caused indirectly through regulation of maleless.
Zn72D knockdown regulated editing at a large subset of sites, whereas many were unaffected. We hypothesize that Zn72D facilitates ADAR editing at some sites by binding the same dsRNAs as ADAR. Zn72D regulation of editing differs within transcripts and even among sites found within a few bases of each other. Although Zn72D loss leads to an overall decrease in the ADAR protein, the effects of this loss are distributed asymmetrically across edited adenosines, in a manner that is distinct from the effect of knocking down Adar itself. Although some of the observed editing decreases may be a consequence of lower ADAR levels, we hypothesize that for at least a subset of RNA species, the presence of Zn72D nearby alters the efficiency at which particular adenosines are edited. For sites with increased editing upon Zn72D knockdown, Zn72D binding may inhibit ADAR binding when present.
Zn72D may affect editing in a number of ways. For example, Zn72D may alter the structure of ADAR-bound dsRNAs by modulating splicing kinetics; however, although we found 40 transcripts with both splicing and editing changes, there were 212 transcripts with editing changes in which we did not find evidence of splicing changes. Although splicing efficiency can alter editing levels (Licht et al., 2016), editing can also affect splicing (Hsiao et al., 2018), complicating this question. Our observation that Zn72D affects some, but not all, adenosines in clusters of linked editing sites suggests that Zn72D could change which adenosines are edited in certain dsRNA structures. It could also modify ADAR’s ability to move along a substrate to edit multiple adenosines within a few bases of each other. Future studies are needed to clarify the precise mechanisms by which Zn72D affects ADAR function.
In addition to molecular phenotypes, we found that loss of Zn72D leads to cellular and organismal changes; however, these phenotypes differ somewhat from those that we or others have found in Adar mutants and knockdown flies. For instance, ADAR and Zn72D reduction lead to opposite effects on Syt I levels at the NMJ, which may stem from the fact that Zn72D inhibits ADAR editing at only some editing sites. It is also likely that Zn72D has ADAR-independent functions. Overall, these results demonstrate that Zn72D has a distinct and critical role in neurons and fly physiology.
We found that the neuronal role for Zn72D in RNA editing is conserved in mammals. Knockdown of Zfr affected mainly editing sites that were regulated by ADAR2, although at a subset distinct from those affected by Adar2 knockdown. Knockdown of Zfr also led to a decrease in Adar2 mRNA, suggesting that some portion of the editing phenotype may be due to decreased ADAR2 levels in mouse neurons. In a biochemical screen for proteins that interact with human ADARs, we identified human ZFR as a top ADAR1-and ADAR2-interacting protein and demonstrated an RNA-dependent interaction between ZFR and ADAR1 and ADAR2 (Freund et al., 2020 [this issue of Cell Reports]). Together, these results suggest that ZFR regulates editing in mammals by interacting with ADARs on RNA. The regulation of RNA editing by ZFR may have implications relevant to human disease. ADAR1 mutations can lead to Aicardi-Goutières syndrome (AGS) (Rice et al., 2012) and spastic paraplegia (Crow et al., 2014). These auto-immune diseases can have neurological symptoms and are caused by an increase in interferon expression after loss of ADAR1 editing of endogenous dsRNAs. ZFR has also been implicated, through one missense mutation, in spastic paraplegia (Novarino et al., 2014). Although our data suggest ZFR’s effect on editing is mainly exerted through ADAR2, rather than ADAR1, future studies should explore the consequences of this ZFR mutation on editing in more human contexts. Furthermore, because targeting ADAR1 has been shown to be an effective strategy to enhance cancer treatment (Ishizuka et al., 2019; Liu et al., 2019), ZFR—either through its regulation of editing or independent mechanisms of innate immune activation (Haque et al., 2018)—may prove to be a candidate drug target. As a broadly influential trans regulator of ADAR, a detailed understanding of how Zn72D and ZFR regulate editing will provide needed insight into the RNA editing process.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jin Billy Li (jin.billy.li@stanford.edu).
Materials Availability
All unique reagents generated in this study are available without restriction from the lead contact.
Data and Code Availability
The accession number for the high-throughput sequencing data reported in this work, including the RNA binding protein RNAi screen, Zn72D-GFP RIP-seq, and the mouse primary neuron RNA-seq, is GEO: GSE126631. This SuperSeries includes the Drosophila RNAi screen RNA-seq data (GEO: GSE126628), the Zn72D RIP-seq data (GEO: GSE126630), and the mouse primary neuron knockdown data (GEO: GSE126629). R and python scripts used for data analysis are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly stocks and husbandry
RNA binding protein shRNA lines for the screen were created as in Ni et al. (2011); see Table S1 for shRNA sequences and vectors used. C155-GAL4 (BDSC458) flies were obtained from Bloomington Drosophila Stock Center (BDSC), along with one UAS-shAdar line (BDSC28311) and the independent UAS-shZn72D line (BDSC55635) which were created by the Transgenic Drosophila RNAi project (TRiP) (Perkins et al., 2015). The stronger shAdar line was obtained from the Vienna Drosophila Resource Center (v7763) (Dietzl et al., 2007). For the RNAi screen, C155-Gal4 virgin females were crossed to males containing UAS-driven shRNAs against individual RNA binding proteins. If viable, F1 females were collected at 0–2 days old and aged for three days. Approximately 15 brains were dissected from females for each replicate, with two replicates per shRNA line. Zn72DGFP (BDSC50830), Zn72D1 (BDSC5061), Zn72D1A14 (BDSC32668) (Brumby et al., 2004), and Df(3L)Exel6127 (BDSC7606) (Parks et al., 2004), which deletes chromosomal region 72D1–72D9 including Zn72D and surrounding genes, were obtained from BDSC. AdarHA (Jepson et al., 2011) flies were a generous gift from the R. Reenan lab and Adar5G1 mutants (Palladino et al., 2000) a generous gift from L. Keegan. Flies were raised at 25°C on molasses-based food on a 12 hr light/dark cycle.
Mouse primary neuron culture
Primary mouse cortical neurons of E16.5 Mus musculus (strain: C57BL/6J) from whole mixed-sexed litters were dissociated and mixed into single cell suspensions using a papain dissociation system (Worthington Biochemical Corporation). Neurons were seeded onto poly-L-lysine coated plates (0.1% w/v) and grown in Neurobasal media (GIBCO) supplemented with B-27 serum-free supplement (GIBCO), GlutaMAX, and Penicillin-Streptomycin (GIBCO) in a humidified incubator at 37°C, with 5% CO2. Half media changes were performed every 4–5 days, or as required. For gene silencing experiments, neurons were infected the day after seeding with a 6-well pellet worth of concentrated frozen virus. The media was changed 12–16 hours later and every 4 days following (neurobasal + B-27 + glutamine). Neurons were harvested on day 7 for RNA extractions. All mouse experiments were approved by the Stanford Administrative Panel on Animal Care (APLAC).
METHOD DETAILS
RNA extraction and cDNA synthesis
RNA was extracted from dissected brains or heads using Agencourt RNAdvanced Tissue Kit (Beckman Coulter: A32645) following the standard protocol using one fourth of all volumes. To bind RNA to beads, final Bind Buffer was prepared by adding 10 μL of Bind Buffer beads to 90 μL of isopropanol. Following RNA extraction, 1 μL of TURBO DNase (Invitrogen: AM1907) was used to remove DNA by incubating for 20–30 minutes at 37°C. cDNA was synthesized from half of each RNA sample using SuperScript III (Invitrogen: 18080093) following the standard protocol using random hexamers as primers. The other half of the RNA was used as input for RNA-seq libraries.
qPCR determination of RNAi efficiency
qPCR was performed using KAPA SYBR Fast (Kapa Biosystems: KK4600) to determine whether knockdown of the target exceeded 40% before proceeding to RNA-seq. qPCR primers were designed by FlyPrimerBank (Hu et al., 2013), and primer efficiency was tested to ensure 90%–105% efficiency. qPCR was performed on a Bio-Rad CFX96 Real-Time System. Averaging three technical replicates, fold changes were calculated using the △△Ct method for the change between the gene of interest and reference gene GAPDH. Knockdown levels reported in Figure 1 were calculated using DESeq2 (Love et al., 2014) after RNA-sequencing.
RNA-seq library preparation
rRNA was depleted from total RNA following RNase H-based protocols adopted from Adiconis et al. (2013) and Morlan et al. (2012). We mixed approximately 150 ng of RNA with 150 ng of pooled DNA oligos designed antisense to Drosophila rRNA in 50 base pair sections (Table S5) in an 8 μL reaction with μL of 5X Hybridization buffer (500 mM Tris-HCl pH 7.4, 1 M NaCl). We annealed rRNA antisense oligos to total RNA samples for 2 minutes at 95°C, slowly reduced the temperature to 45°C and then added 2U of Hybridase Thermostable RNase H (Epicenter, Lucigen: H39500) and 1 μL of 10X Digestion buffer (500 mM Tris-HCl, 1 M NaCl, 200mM MgCl2) and incubated for 30 minutes at 45°C. rRNA-depleted RNA was then purified using 2.2X reaction volume of Agencourt RNAClean XP beads (Beckman Coulter: A63987), treated with TURBO DNase (Invitrogen: AM1907), and then purified with RNAClean XP beads again. rRNA-depleted RNA was used as input to the KAPA Stranded RNA-seq Kit (Kapa Biosystems: KK8400) to make RNA-sequencing libraries for fly knockdowns. For mouse primary neuron RNA-seq libraries, the KAPA HyperPrep RNA-seq Kit (Kapa Biosystems: KK8540) was used to create libraries after rRNA depletion using oligos antisense to human rRNA sequences (Adiconis et al., 2013). All libraries were sequenced with 76 base pair paired-end reads using an Illumina NextSeq.
Brain immunofluorescence microscopy
Fly brains were dissected from 3-to-5-day-old adult females and stained exactly as in Wu and Luo (2006). The following primary antibodies were used: mouse anti-HA antibody (HA.11, Covance, BioLegend: 901514) and rabbit anti-GFP antibody (Abcam: ab290) were used at 1:500, and rat anti-Elav antibody (Developmental Studies Hybridoma Bank, Iowa City, IA, deposited by G. M. Ruben: 7E8A10) was used at 1:25. Cross absorbed secondary antibodies used were: goat anti-mouse IgG Alexa Fluor Plus 555 (Invitrogen: A32727), goat anti-rabbit IgG Alexa Fluor 488 (Invitrogen: A11034), and goat anti-rat IgG Alexa Fluor 647 (Invitrogen: A21247). Brains were imaged on an Inverted Zeiss LSM 780 Multiphoton Laser Scanning Confocal Microscope with a 20X objective.
NMJ immunofluorescence microscopy
Zn72D mutant alleles were maintained over GFP-tagged balancer chromosomes or the larval-selectable Tb marker to enable selection as third instar larvae. Third instar larvae were dissected and stained as previously described (Mosca et al., 2012) in 0 mM Ca2+ modified Drosophila saline (Mosca et al., 2005). Larvae were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 20 minutes (for all antibodies except GluRIIA) or in Bouin’s Fixative (Electron Microscopy Sciences) for 5 minutes (for GluRIIA staining). The following primary antibodies were used: rabbit anti-Syt I at 1:4000 (Loewen et al., 2001), mouse anti-GluRIIA at 1:100 (Parnas et al., 2001), Cy5-conjugated goat anti-HRP at 1:100 (Jackson ImmunoResearch). The following secondary antibodies were used: Alexa 488 conjugated goat anti-mouse (Jackson ImmunoResearch) and Alexa568-conjugated goat anti-rabbit (Invitrogen), both at 1:250. Larvae were imaged on a Zeiss LSM 880 confocal microscope with a 40X, NA 1.3 or a 63X, NA 1.4 lens. NMJs on muscle 4 in segment A3 on both the right and left sides were imaged and quantified. All images were scored with the experimenter blind to genotype and processed using ImageJ (NIH) and Adobe Photoshop. Immunofluorescence was quantified using ImageJ (NIH) and each channel (Syt I or GluRIIA) normalized to the HRP fluorescence of the corresponding image. Data was analyzed and statistical analysis completed using GraphPad Prism 8.4.0.
Co-immunoprecipitation
Immunoprecipitation of ADAR-HA was performed as described in Bhogal et al. (2011) with slight modifications as follows. Flies were flash frozen in liquid N2, their heads were removed by vortexing and then collected using a liquid N2 cooled sieve. Approximately 500 μL of fly heads were homogenized in lysis buffer (150 mM NaCl, 0.1% NP40, 20 mM HEPES (pH 7.4), 2 mM MgCl2, 1 mM DTT, cOmplete protease inhibitor (Sigma-Aldrich: 4693159001)) for input protein. Homogenates were centrifuged at 600 xg, supernatants were collected, and then additional lysis buffer was added, pellets were homogenized and centrifuged again, and supernatants combined. Half of each lysate was treated with 100 μg RNase A (Thermo Scientific: EN0531) per mg of lysate for 30 minutes on ice. Equal amounts of lysate (approximately 1 mg) were rotated at 4°C overnight with 20 μL of mouse anti-HA agarose (Sigma-Aldrich: A2095) and washed 5X for 10 minutes each with 1 mL of lysis buffer. Protein was eluted in 2X Laemmli Sample Buffer (Bio-Rad: 161–0747) at 95°C for 10 minutes. Samples were run on 4%–15% SDS-PAGE gels (Bio-Rad: 456–1086) and transferred to nitrocellulose membranes (Bio-Rad) for western blots. For immunoprecipitation of Zn72D-GFP, nuclei were collected from fly heads and immunoprecipitation was performed following the protocols described in Lo Piccolo et al. (2015), with slight modifications as follows. 20 μL of Protein G Dynabeads (Invitrogen: 10003D) were incubated with 5 mg of rabbit anti-GFP antibody (Abcam: ab290). Following overnight incubation at 4C, IPs were washed 5 times with 1 mL of IP Wash Buffer, and Protein was eluted in 2X Laemmli Sample Buffer (Bio-Rad) at 95μC for 10 minutes. Samples were run on 4%–15% SDS-PAGE gels (Bio-Rad) and transferred to nitrocellulose membranes (Bio-Rad) for western blots.
Western Blotting
Antibodies used in western blots were: mouse anti-HA antibody (Covance: HA.11) at1:500, rabbit anti-GFP (Abcam: ab290) at 1:10000, mouse anti-GAPDH (Invitrogen: GA1R) at 1:2000, and mouse anti-Lamin (Developmental Studies Hybridoma Bank, deposited by P. A. Fisher: ADL67.10 s) at 1:50 in 5% milk. Horseradish Peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoResearch) were used 1:5000. Western blots were imaged after exposing to Pierce ECL Plus Western Blotting Substrate (Thermo Scientific: 32132) using a BioRad ChemiDoc imaging system running Image Lab Touch Software (v1.1.04). Quantification of western blots was performed using BioRad Image Lab 5.2. Bands were manually traced, and adjusted volumes of HA were normalized to GAPDH controls before comparisons between genotypes.
RNA-immunoprecipitation and sequencing
RNA immunoprecipitation was performed after homogenizing three biological replicates of ~500 μL of fly heads in IP Buffer (150 mM NaCl, 20 mM HEPES pH 7.5, 2 mM MgCl2, 0.1% NP40, cOmplete protease inhibitor, RNaseOUT RNase inhibitor 1U/ μL (Invitrogen)). Lysates were split in half, and 5% of input was removed for input control libraries. IP lysates were incubated overnight at 4°C with Dynabeads Protein G (Invitrogen: 10003D), plus 5 μg of anti-GFP antibody (Abcam: ab290) or IgG (Sigma-Aldrich: I8765). IPs were washed 8 times in IP buffer. Beads and saved inputs were added to 1 mL of TRIzol (Thermo Fisher: 15596026). 200 μL of chloroform was added, and samples were centrifuged at 14000 xg at 4°C for 15 minutes. Aqueous phases were collected, mixed with 1 volume of 70% ethanol and then transferred to a RNeasy MinElute column (QIAGEN, Hilden, Germany: 74204) for purification following the standard protocol. RIP-seq libraries were made using KAPA HyperPrep RNA-seq Kits (Kapa Biosystems: KK8540) after rRNA depletion as described above for RNA-seq library preparation. Libraries were sequenced with 76 base pair paired-end reads on an Illumina NextSeq. RNA-seq reads were mapped using STAR v2.54b (-outFilterMultimapNmax 10-outFilterMultimapScoreRange 1-outFilterScoreMin 10-alignEndsType EndToEnd) (Dobin et al., 2013) to the dm6 genome (Aug 2014, BDGP Release 6 + ISO1 MT/dm6) (Hoskins et al., 2015). Reads hitting annotated genes in the transcriptome were counted using RSEM v1.2.30 (Li and Dewey, 2011). RSEM TPMs were used for plotting and expected counts were rounded to the nearest integer and then used as input to DESeq2 v1.22.0 (Love et al., 2014). Log2 fold changes were calculated using the DESeq() function followed by lfcShrink(type = “apeglm”) (Zhu et al., 2019). For qPCR, cDNA was made with iScript Advanced (Bio-Rad: 1708842), and qPCR was performed using KAPA SYBR Fast (Kapa Biosystems: KK4600) with 1 μL of input cDNA in three technical replicates for each biological replicate. All primers were designed by FlyPrimerBank (Hu et al., 2013), and their efficiency was determined by amplification of serial dilutions. Primer sequences and efficiencies were: RpL32 Forward 5′-GCCCAAGGGTATCGACAACA, Reverse 5′-GCGCTTGTTCGATCCG TAAC, 97% efficiency; RpL13 Forward 5′-GTGGTCGAGTTCCGTGAGG, Reverse 5′-CCTTCTTGGGGTCTCCCTT, 98% efficiency; qvr Forward 5′-CCTTTCAACTATACAGCCCTGC, Reverse 5′-TGTAACTGTGACGTACACATGC, 98% efficiency; cac Forward 5′-GCGATGGCACCTTTACTGC, Reverse 5′-GTGCGCCCGAATAAAACTCG, 102% efficiency; Shab Forward 5′-CATCAGTCACGG GATCAGGAT, Reverse 5′-AAAGGGGCGTAGCGAACTTC, 105% efficiency; para Forward 5′-ACGAGGATGAAGGTCCACAAC, Reverse 5′- ACGACGTATCGGATTGAATGG, 100% efficiency. GFP and IgG RIP Cts were normalized to inputs: △Ct [RIP] = (Ct [RIP] - Ct [Input]). Fold changes for each replicate of each transcript of interest were calculated for the Zn72D-GFP RIPs normalized to the IgG RIPs after input subtraction, and then further normalized to the fold change of negative control transcript RpL32, so that the fold changes represented in Figure 4 are: 2−(△Ct[GFP-input] - △Ct[IgG-input]) for transcript of interest / 2−(△Ct[GFP-input] - △Ct[IgG-input]) for RpL32.
Since samples were normalized to IgG RIP within the same replicate, IgG fold changes are 1 with no error. Paired two-tailed t tests were performed using GraphPad Prism 8.
Climbing assay
The negative geotaxis assay was performed with groups of 10 flies at a time counting the number of flies above the 10 cm mark on a glass vial every 30 s or 1 minute. Flies were given 24 hours to recover from CO2 exposure before tests.
Lentivirus production
shRNAs targeted against mouse Adar1 (5′-CTCACTGAGGACAGGCTGGCGAGATGGTG), Adar2 (5′-AGCAATGGTCACTCCAAGTACCGCCTGAA), and Zfr (5′-GAGTATACTGTGTTGCACCTTGGC), and non-targeting controls (control #1, matched with Adar1 and Adar2 knockdowns: 5′-ATCGCACTTAGTAATGATTGAA; control #2, matched with Zfr knockdown: 5′-AACCGATGTACTTCCCGTTAAT) were cloned into the pGreenPuro backbone from System Biosciences. This construct was used to produce lentivirus in a 6-well according to standard protocols in HEK293T cells using the third-generation system and concentrated 1:100 with lenti-X (Clontec). The virus pellet was stored at −80°C.
Gene Knockdown in Mouse Primary Neurons
Primary mouse cortical neurons were dissociated into single cell suspensions from E16.5 mouse (strain: C57BL/6J) cortices using a papain dissociation system (Worthington Biochemical Corporation, Lakewood, NJ). Neurons were seeded onto poly-L-lysine coated plates (0.1% w/v) and grown in Neurobasal media (GIBCO) supplemented with B-27 serum-free supplement (GIBCO), GlutaMAX, and Penicillin-Streptomycin (GIBCO) in a humidified incubator at 37°C, with 5% CO2. Half media changes were performed every 4–5 days, or as required. For gene silencing experiments, neurons were infected the day after seeding with a 6-well pellet worth of concentrated frozen virus (see above). The media was changed 12–16 hours later and every 4 days following (neurobasal + B-27 + glutamine). Neurons were harvested on day 7 and RNA was extracted using the PARIS kit from Ambion followed by TURBO DNase. Adar1, Adar2, and control shRNA#1 knockdowns were matched from the same mouse, while Zfr and control shRNA#2 knockdowns were matched from the same mouse. 250 ng of RNA was used to make RNA-seq libraries using the KAPA HyperPrep Kit from two biological replicates of each genotype. For Adar2 knockdowns, we sequenced three technical replicates of the first biological replicate and combined all A and G counts to increase coverage. Editing levels were determined after requiring 20X coverage total. Adar and Zfr mRNA expression changes between groups were determined using DESeq2.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA Editing Level Quantification
RNA-seq reads were mapped using STAR v2.54b (-outFilterMultimapNmax 10-outFilterMultimapScoreRange 1-outFilterScoreMin 10-alignEndsType EndToEnd) (Dobin et al., 2013) to the dm6 genome (Aug 2014, BDGP Release 6 + ISO1 MT/dm6) (Hoskins et al., 2015). Mapped reads were filtered for primary hits only. Editing levels were determined using the Samtools v1.9 (Li et al., 2009) mpileup command to count A and G reads at known editing sites from Duan et al. (2017), Graveley et al. (2011), Mazloomian and Meyer (2015), Ramaswami et al. (2015, 2013), Rodriguez et al. (2012), Sapiro et al. (2015), St Laurent et al. (2013), Yu et al. (2016), and Zhang et al. (2017). We required 20X coverage in each replicate, except for in Zn72D mutant versus wild-type pupal head and mouse primary neuron comparisons, where we required 20X coverage total between the two replicates. A and G counts from two replicates of each shRNA or mutant combined were compared to A and G counts from two control replicates combined using Fisher’s exact test with a Benjamini-Hochberg multiple hypothesis testing correction in R v3.5.1 (Benjamini and Hochberg, 1995).
Gene Expression Quantification
Gene expression levels were determined by counting reads hitting annotated genes in the transcriptome using RSEM v1.2.30 (Li and Dewey, 2011). RSEM outputted expected counts were rounded to the nearest integer and then used as input to DESeq2 v1.22.0 (Love et al., 2014). The DESeq() and results() functions were used to calculate gene expression differences between pairs of cell types, using Wald tests with Benjamini-Hochberg multiple hypothesis testing corrections.
Splicing Event Quantification
To analyze splicing changes in Zn72D knockdown flies, we trimmed all reads to 75 bp and then mapped reads using STAR v2.54b (-twoPassMode Basic), filtering for uniquely mapped reads. We ran MISO (Katz et al., 2010) after merging reads from two biological replicates of shGFP and shZn72D (BDSC55635). We used the modENCODE Drosophila splice junctions available through MISO (https://miso.readthedocs.io/en/fastmiso/annotation.html), lifted over from dm3 to dm6 using the UCSC Genome Browser LiftOver function (http://genome.ucsc.edu). After comparing events, we filtered for significant changes using-num-inc 1-num-exc 1-numsum-inc-exc 10-delta-psi 0.12-bayes-factor 20.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-HA | BioLegend (Covance) | Cat#901514; RRID: AB_2565336 |
| Rabbit anti-GFP | Abcam | Cat#ab290; RRID: AB_303395 |
| Rat anti-Elav | Developmental Studies Hybridoma Bank | DSHB#7E8A10; RRID: AB_528218 |
| Mouse anti-lamin (Dm0) | Developmental Studies Hybridoma Bank | DSHB#ADL67.10 s; RRID: AB_528336 |
| Mouse anti-GAPDH | Invitrogen | Cat#MA5–15738; RRID: AB_10977387 |
| Rabbit anti-Syt I | Loewen et al., 2001; Noreen Reist Lab | N/A |
| Mouse anti-GluRIIA | Parnas et al., 2001 | RRID:AB_2568752 |
| Critical Commercial Assays | ||
| Agencourt RNAdvanced Tissue Kit | Beckman Coulter | Cat#A32649 |
| KAPA SYBR Fast | KAPA Biosystems | Cat#KK4601 |
| KAPA Stranded RNA-seq Kit | KAPA Biosystems | Cat#KK8400 |
| KAPA HyperPrep RNA-seq Kit | KAPA Biosystems | Cat#KK8540 |
| Hybridase Thermostable RNase H | Lucigen (Epicenter) | Cat#H39500 |
| Deposited Data | ||
| All data generated herein as a SuperSeries | This paper | GEO: GSE126631 |
| Drosophila RNAi RNA-seq Screen | This Paper | GEO: GSE126628 |
| Zn72D RIP-seq | This Paper | GEO: GSE126630 |
| Mouse primary neuron RNA-seq | This Paper | GEO: GSE126629 |
| ZFR knockdown HEK293T RNA-seq | Haque et al., 2018 | GEO: GSE99231 |
| Experimental Models: Cell Lines | ||
| Mouse primary cortical neurons; C57BL/6J; E16.5 | This Paper | N/A |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: AdarHA | Jepson et al., 2011 | FlyBaseID: FBal0298344 |
| D. melanogaster: Zn72DGFP (y[1] w[*]; P{w [+mC] = PTT-GA}Zn72D[CA07703]) | Bloomington Drosophila Stock Center | BDSC50830 |
| D. melanogaster: RNAi lines, see Table S1 | This Paper; Bloomington Drosophila Stock Center; Vienna Drosophila Resource Center | N/A |
| D. melanogaster: Adar5G1 | Palladino et al., 2000 | FlyBaseID: FBal0118605 |
| D. melanogaster. Zn72D[1]/TM2 | Bloomington Drosophila Stock Center | BDSC5061 |
| D. melanogaster. w[*]; Zn72D[1A14]/TM6B, Tb[+] | Bloomington Drosophila Stock Center | BDSC32668 |
| D. melanogaster: Zn72DDf (w[1118]; Df(3L) Exel6127, P{w[+mC] = XP-U}Exel6127/TM6B, Tb[1]) | Bloomington Drosophila Stock Center | BDSC7606 |
| Oligonucleotides | ||
| qPCR primers, see STAR METHODS | Hu et al., 2013 | https://www.flyrnai.org/FlyPrimerBank |
| rRNA oligos, see Table S5 | This Paper | N/A |
| Recombinant DNA | ||
| pGreenPuro (CMV) shRNA Expression Lentivector | System Biosciences | Cat#SI505A-1 |
| Mouse shRNAs, see STAR METHODS | This Paper | N/A |
| Software and Algorithms | ||
| STAR v2.54b | Dobin et al., 2013 | RRID:SCR_015899; https://github.com/alexdobin/STAR |
| Samtools v1.9 | Li et al., 2009 | RRID:SCR_002105; http://samtools.sourceforge.net/ |
| RSEM v1.2.30 | Li and Dewey 2011 | RRID:SCR_013027; http://deweylab.biostat.wisc.edu/rsem/ |
| DESeq2 v1.22.0 | Love et al., 2014 | RRID:SCR_015687; https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MISO v0.5.4 | Katz et al., 2010 | RRID:SCR_003124; https://miso.readthedocs.io/en/fastmiso/ |
Highlights.
Drosophila Zn72D is a broadly influential regulator of neuronal A-to-I RNA editing
Zn72D regulates ADAR protein levels and editing levels at hundreds of editing sites
Loss of Zn72D causes morphological defects at the neuromuscular junction
Zn72D regulation of editing is conserved in mammalian neurons
ACKNOWLEDGMENTS
We thank all members of the Li lab for their input, TRiP at Harvard Medical School (NIH/NIGMS R01-GM084947), the Bloomington Drosophila Stock Center (NIH P40OD018537), and the Vienna Drosophila Resource Center for fly stocks and FlyBase for curation. The Developmental Studies Hybridoma Bank, created by the NICHD of the NIH at The University of Iowa, Department of Biology, provided antibodies. We thank N. Kramer and A. Gitler for mouse primary neurons. This work is funded by NIH grants R01-GM102484, R01-GM124215, and R01-MH115080 (to J.B.L.); the National Science Foundation graduate research fellowship grant DGE-114747 (to A.L.S.); an NIH-NIGMS training grant T32 GM007790 (to A.L.S.); an American Heart Association postdoctoral grant 16POST27700036 (to E.C.F.); NIH grants R00-DC013059 and R01-NS110907 (to T.J.M.); and the Alfred P. Sloan Foundation and the Whitehall Foundation (to T.J.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107654.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adiconis X, Borges-Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, Sivachenko A, Thompson DA, Wysoker A, Fennell T, et al. (2013). Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 10, 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL (2002). RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem 71, 817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm M, Wahlstedt H, Widmark A, Eriksson M, and Öhman M (2017). Accumulation of nuclear ADAR2 regulates adenosine-to-inosine RNA editing during neuronal development. J. Cell Sci 130, 745–753. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 57, 289–300. [Google Scholar]
- Bhogal B, Jepson JE, Savva YA, Pepper AS-R, Reenan RA, and Jongens TA (2011). Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat. Neurosci 14, 1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumby A, Secombe J, Horsfield J, Coombe M, Amin N, Coates D, Saint R, and Richardson H (2004). A genetic screen for dominant modifiers of a cyclin E hypomorphic mutation identifies novel regulators of S-phase entry in Drosophila. Genetics 168, 227–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Frese CK, Horos R, Alleaume A-M, Foehr S, Curk T, Krijgsveld J, and Hentze MW (2016). Comprehensive identification of RNA-binding domains in human cells. Mol. Cell 63, 696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Zaki MS, Abdel-Hamid MS, Abdel-Salam G, Boespflug-Tanguy O, Cordeiro NJV, Gleeson JG, Gowrinathan NR, Laugel V, Renaldo F, et al. (2014). Mutations in ADAR1, IFIH1, and RNASEH2B presenting as spastic paraplegia. Neuropediatrics 45, 386–393. [DOI] [PubMed] [Google Scholar]
- Dassi E (2017). Handshakes and fights: the regulatory interplay of RNA-binding proteins. Front. Mol. Biosci 4, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman DK, Lu Z, Meinertzhagen IA, and Schwarz TL (2006). Altered synaptic development and active zone spacing in endocytosis mutants. Curr. Biol 16, 591–598. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su K-C, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Dou S, Luo S, Zhang H, and Lu J (2017). Adaptation of A-to-I RNA editing in Drosophila. PLoS Genet. 13, e1006648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, and Levanon EY (2018). A-to-I RNA editing—immune protector and transcriptome diversifier. Nat. Rev. Genet 19, 473–490. [DOI] [PubMed] [Google Scholar]
- Freund EC, Sapiro AL, Li Q, Linder S, Moresco JJ, Yates JR III, and Li JB (2020). Unbiased identification of trans regulators of ADAR and A-to-I RNA editing. Cell Reports 31, Published online May 19, 2020. 10.1016/j.celrep.2020.107656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, Ivan AP, Buchumenski I, Berger AC, Goldstein JT, et al. (2018). Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun 9, 5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garncarz W, Tariq A, Handl C, Pusch O, and Jantsch MF (2013). A high-throughput screen to identify enhancers of ADAR-mediated RNA-editing. RNA Biol. 10, 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. (2011). The developmental transcriptome of Drosophila melanogaster. Nature 471, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque N, Ouda R, Chen C, Ozato K, and Hogg JR (2018). ZFR coordinates crosstalk between RNA decay and transcription in innate immunity. Nat. Commun 9, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, An O, Chan THM, Ng VHE, Kwok HS, Lin JS, Qi L, Han J, Tay DJT, Tang SJ, et al. (2018). Bidirectional regulation of adenosineto-inosine (A-to-I) RNA editing by DEAH box helicase 9 (DHX9) in cancer. Nucleic Acids Res. 46, 7953–7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsch M, Seeburg PH, Adler T, Aguilar-Pimentel JA, Becker L, Calzada-Wack J, Garrett L, Götz A, Hans W, Higuchi M, et al. (2011). Requirement of the RNA-editing enzyme ADAR2 for normal physiology in mice. J. Biol. Chem 286, 18614–18622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, Carlson JW, Wan KH, Park S, Mendez I, Galle SE, Booth BW, Pfeiffer BD, George RA, Svirskas R, et al. (2015). The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 25, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao YE, Bahn JH, Yang Y, Lin X, Tran S, Yang E-W, Quinones-Valdez G, and Xiao X (2018). RNA editing in nascent RNA affects pre-mRNA splicing. Genome Res. 28, 812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Sopko R, Foos M, Kelley C, Flockhart I, Ammeux N, Wang X, Perkins L, Perrimon N, and Mohr SE (2013). FlyPrimerBank: an online database for Drosophila melanogaster gene expression analysis and knockdown evaluation of RNAi reagents. G3 (Bethesda) 3, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. (2019). Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson JEC, Savva YA, Yokose C, Sugden AU, Sahin A, and Reenan RA (2011). Engineered alterations in RNA editing modulate complex behavior in Drosophila: regulatory diversity of adenosine deaminase acting on RNA (ADAR) targets. J. Biol. Chem 286, 8325–8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, and Burge CB (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan LP, McGurk L, Palavicini JP, Brindle J, Paro S, Li X, Rosenthal JJC, and O’Connell MA (2011). Functional conservation in human and Drosophila of Metazoan ADAR2 involved in RNA editing: loss of ADAR1 in insects. Nucleic Acids Res. 39, 7249–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, and Church GM (2013). Deciphering the functions and regulation of brain-enriched A-to-I RNA editing. Nat. Neurosci 16, 1518–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht K, Kapoor U, Mayrhofer E, and Jantsch MF (2016). Adenosine to Inosine editing frequency controlled by splicing efficiency. Nucleic Acids Res. 44, 6398–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, and Walkley CR (2015). RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, Bullock CP, Jones MD, Kerr G, Li L, et al. (2019). Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med 25, 95–102. [DOI] [PubMed] [Google Scholar]
- Loewen CA, Mackler JM, and Reist NE (2001). Drosophila synaptotagmin I null mutants survive to early adulthood. Genesis 31, 30–36. [DOI] [PubMed] [Google Scholar]
- Lo Piccolo L, Bonaccorso R, and Onorati MC (2015). Nuclear and cytoplasmic soluble proteins extraction from a small quantity of Drosophila’s whole larvae and tissues. Int. J. Mol. Sci 16, 12360–12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado C, Alicea D, Gonzalez M, Bykhovskaia M, and Marie B (2013). ADAR is essential for optimal presynaptic function. Mol. Cell. Neurosci 52, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. (2014). The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 9, 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci R, Brindle J, Paro S, Casadio A, Hempel S, Morrice N, Bisso A, Keegan LP, Del Sal G, and O’Connell MA (2011). Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 30, 4211–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazloomian A, and Meyer IM (2015). Genome-wide identification and characterization of tissue-specific RNA editing events in D. melanogaster and their potential role in regulating alternative splicing. RNA Biol. 12, 1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher MJ, Schumacher JM, Lee K, Holdcraft RW, Edelhoff S, Disteche C, and Braun RE (1999). Identification of ZFR, an ancient and highly conserved murine chromosome-associated zinc finger protein. Gene 228, 197–211. [DOI] [PubMed] [Google Scholar]
- Morin X, Daneman R, Zavortink M, and Chia W (2001). A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc. Natl. Acad. Sci. USA 98, 15050–15055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlan JD, Qu K, and Sinicropi DV (2012). Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS ONE 7, e42882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca TJ, Carrillo RA, White BH, and Keshishian H (2005). Dissection of synaptic excitability phenotypes by using a dominant-negative Shaker K+ channel subunit. Proc. Natl. Acad. Sci. USA 102, 3477–3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca TJ, Hong W, Dani VS, Favaloro V, and Luo L (2012). Trans-synaptic Teneurin signalling in neuromuscular synapse organization and target choice. Nature 484, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J-Q, Zhou R, Czech B, Liu L-P, Holderbaum L, Yang-Zhou D, Shim H-S, Tao R, Handler D, Karpowicz P, et al. (2011). A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 8, 405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K (2010). Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem 79, 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K (2016). A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol 17, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD, Abdellateef M, Rosti B, Scott E, Mansour L, et al. (2014). Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor-Giles KM, Ho LL, and Ganetzky B (2008). Nervous wreck interacts with thickveins and the endocytic machinery to attenuate retrograde BMP signaling during synaptic growth. Neuron 58, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O’Connell MA, and Reenan RA (2000). A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 102, 437–449. [DOI] [PubMed] [Google Scholar]
- Parks AL, Cook KR, Belvin M, Dompe NA, Fawcett R, Huppert K, Tan LR, Winter CG, Bogart KP, Deal JE, et al. (2004). Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet 36, 288–292. [DOI] [PubMed] [Google Scholar]
- Parnas D, Haghighi AP, Fetter RD, Kim SW, and Goodman CS (2001). Regulation of postsynaptic structure and protein localization by the Rho-type guanine nucleotide exchange factor dPix. Neuron 32, 415–424. [DOI] [PubMed] [Google Scholar]
- Perkins LA, Holderbaum L, Tao R, Hu Y, Sopko R, McCall K, Yang-Zhou D, Flockhart I, Binari R, Shim H-S, et al. (2015). The Transgenic RNAi Project at Harvard Medical School: resources and validation. Genetics 201, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, and Stetson DB (2015). Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones-Valdez G, Tran SS, Jun H-I, Bahn JH, Yang E-W, Zhan L, Brümmer A, Wei X, Van Nostrand EL, Pratt GA, et al. (2019). Regulation of RNA editing by RNA-binding proteins in human cells. Commun. Biol 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendren S, Manning AC, Al-Awadi H, Yamada K, Takagi Y, and Hundley HA (2018). A protein-protein interaction underlies the molecular basis for substrate recognition by an adenosine-to-inosine RNA-editing enzyme. Nucleic Acids Res. 46, 9647–9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, and Li JB (2014). RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 42, D109–D113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O’Connell MA, and Li JB (2013). Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods 10, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Deng P, Zhang R, Anna Carbone M, Mackay TFC, and Billy Li J (2015). Genetic mapping uncovers cis-regulatory landscape of RNA editing. Nat. Commun 6, 8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reenan RA, Hanrahan CJ, and Ganetzky B (2000). The mlenapts RNA helicase mutation in Drosophila results in a splicing catastrophe of the para Na+ channel transcript in a region of RNA editing. Neuron 25, 139–149. [DOI] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. (2012). Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet 44, 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JE, Paluch J, Dickman DK, and Joiner WJ (2016). ADAR-mediated RNA editing suppresses sleep by acting as a brake on glutamatergic synaptic plasticity. Nat. Commun 7, 10512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Menet JS, and Rosbash M (2012). Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol. Cell 47, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal JJC, and Seeburg PH (2012). A-to-I RNA editing: effects on proteins key to neural excitability. Neuron 74, 432–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapiro AL, Deng P, Zhang R, and Li JB (2015). Cis regulatory effects on A-to-I RNA editing in related Drosophila species. Cell Rep. 11, 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapiro AL, Shmueli A, Henry GL, Li Q, Shalit T, Yaron O, Paas Y, Billy Li J, and Shohat-Ophir G (2019). Illuminating spatial A-to-I RNA editing signatures within the Drosophila brain. Proc. Natl. Acad. Sci. USA 116, 2318–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugam R, Zhang F, Srinivasan H, Charles Richard JL, Liu KI, Zhang X, Woo CWA, Chua ZHM, Buschdorf JP, Meaney MJ, and Tan MH (2018). SRSF9 selectively represses ADAR2-mediated editing of brain-specific sites in primates. Nucleic Acids Res. 46, 7379–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin W, and Nishikura K (2013). Adenosine-to-inosine RNA editing and human disease. Genome Med. 5, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Laurent G, Tackett MR, Nechkin S, Shtokalo D, Antonets D, Savva YA, Maloney R, Kapranov P, Lawrence CE, and Reenan RA (2013). Genome-wide analysis of A-to-I RNA editing by single-molecule sequencing in Drosophila. Nat. Struct. Mol. Biol 20, 1333–1339. [DOI] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. ; GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; Biospecimen Collection Source Site—RPCI; Biospecimen Core Resource—VARI; Brain Bank Repository—University of Miami Brain Endowment Bank; Leidos Biomedical—Project Management; ELSI Study; Genome Browser Data Integration &Visualization—EBI; Genome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa Cruz (2017). Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariq A, Garncarz W, Handl C, Balik A, Pusch O, and Jantsch MF (2013). RNA-interacting proteins act as site-specific repressors of ADAR2-mediated RNA editing and fluctuate upon neuronal stimulation. Nucleic Acids Res. 41, 2581–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran SS, Jun H-I, Bahn JH, Azghadi A, Ramaswami G, Van Nostrand EL, Nguyen TB, Hsiao YE, Lee C, Pratt GA, et al. (2019). Widespread RNA editing dysregulation in brains from autistic individuals. Nat. Neurosci 22, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Nguyen TB, Gelboin-Burkhart C, Wang R, Blue SM, Pratt GA, Louie AL, and Yeo GW (2017). Robust, cost-effective profiling of rna binding protein targets with single-end enhanced crosslinking and immunoprecipitation (seCLIP) In mRNA Processing, Methods in Molecular Biology, Shi Y, ed. (Humana Press; ), pp. 177–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstedt H, Daniel C, Ensterö M, and Öhman M (2009). Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 19, 978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, and Li JB (2017). Rewriting the transcriptome: adenosine-toinosine RNA editing by ADARs. Genome Biol. 18, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MC, and Hundley HA (2016). Controlling the editor: the many roles of RNA-binding proteins in regulating A-to-I RNA editing. Adv. Exp. Med. Biol 907, 189–213. [DOI] [PubMed] [Google Scholar]
- Wheeler EC, Van Nostrand EL, and Yeo GW (2018). Advances and challenges in the detection of transcriptome-wide protein-RNA interactions. Wiley Interdiscip. Rev. RNA 9, e1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolkowicz UM, and Cook AG (2012). NF45 dimerizes with NF90, Zfr and SPNR via a conserved domain that has a nucleotidyltransferase fold. Nucleic Acids Res. 40, 9356–9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worringer KA, and Panning B (2007). Zinc finger protein Zn72D promotes productive splicing of the maleless transcript. Mol. Cell. Biol 27, 8760–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JS, and Luo L (2006). A protocol for dissecting Drosophila melanogaster brains for live imaging or immunostaining. Nat. Protoc 1, 2110–2115. [DOI] [PubMed] [Google Scholar]
- Yu Y, Zhou H, Kong Y, Pan B, Chen L, Wang H, Hao P, and Li X (2016). The landscape of A-to-I RNA editome is shaped by both positive and purifying selection. PLoS Genet. 12, e1006191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Deng P, Jacobson D, and Li JB (2017). Evolutionary analysis reveals regulatory and functional landscape of coding and non-coding RNA editing. PLoS Genet. 13, e1006563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Ibrahim JG, and Love MI (2019). Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2048–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the high-throughput sequencing data reported in this work, including the RNA binding protein RNAi screen, Zn72D-GFP RIP-seq, and the mouse primary neuron RNA-seq, is GEO: GSE126631. This SuperSeries includes the Drosophila RNAi screen RNA-seq data (GEO: GSE126628), the Zn72D RIP-seq data (GEO: GSE126630), and the mouse primary neuron knockdown data (GEO: GSE126629). R and python scripts used for data analysis are available upon request.