

Abstract
Aims
A close and bidirectional relationship between alcohol consumption and pain has been previously reported and discussed in influential reviews. The goal of the present narrative review is to provide an update on the developments in this field in order to guide future research objectives.
Methods
We evaluated both epidemiological and neurobiological literature interrogating the relationship between alcohol use and pain for the presence of significant effects. We outlined studies on interactions between alcohol use and pain using both self-reports and objective experimental measures and discussed potential underlying mechanisms of these interactions.
Results
Epidemiological, preclinical and clinical literature point to three major interactions between alcohol use and pain: (a) alcohol use leading to hyperalgesia, (b) alcohol use moderating pain and hyperalgesia and (c) chronic pain as a risk factor predisposing to alcohol relapse. Neurobiological studies using animal models to assess these interactions have transitioned from mostly involuntary modes of experimenter-controlled alcohol administration to self-administration procedures, and increasingly indicate that neuronal circuits implicated in both withdrawal and anticipation stages of alcohol use disorder also have a role in chronic pain. Mechanistically, alterations in GABA, glutamate, the corticotropin-releasing factor system, endogenous opioids and protein kinase C appear to play crucial roles in this maladaptive overlap.
Conclusions
Many of the principles explaining the interactions between alcohol and pain remain on a strong foundation, but continuing progress in modeling these interactions and underlying systems will provide a clearer basis for understanding, and ultimately treating, the damaging aspects of this interaction.
Short summary Alcohol use can either enhance or suppress hyperalgesia. Conversely, chronic pain is associated with increased risk of alcohol relapse. This review focuses on recent progress in understanding the interactions between alcohol use and pain.
INTRODUCTION
Physicians recognized the ability of alcohol to act as an analgesic agent thousands of years ago and offered it to patients for this purpose during medical procedures (Horn-Hofmann et al., 2015). The more general idea that pain and alcohol use are intertwined is substantiated by epidemiological studies showing that alcohol use disorder (AUD) is often comorbid with chronic pain (for reviews, see Egli et al., 2012; Apkarian et al., 2013; Witkiewitz and Vowles, 2018). For example, older problem drinkers report increased disruption in daily activities because of pain and more frequent alcohol use to manage pain compared with non-problem drinkers (Brennan et al., 2005). Over half of individuals seeking treatment for an AUD reportsignificant recurring pain, with a greater prevalence in women (63%) compared with men (54%) (Boissoneault et al., 2018). Chronic pain is also a strong predictor of relapse in problem drinkers (Egli et al., 2012; Witkiewitz and Vowles, 2018). Thus, the relationship between alcohol use and pain appears to be bidirectional: on one hand, alcohol intake modulates pain, and on the other hand, acute and chronic pain influence alcohol-related behaviors.
The comorbidity of AUD and chronic pain is likely a manifestation of common neuronal circuits and neurochemical mechanisms (Egli et al., 2012; Apkarian et al., 2013; Witkiewitz and Vowles, 2018). Given the growing sophistication of neuroscience methodologies and the increased attention to pain management as a consequence of the ongoing opioid epidemic (Rudd et al., 2016; Yeung et al., 2017), the goal of this review is to provide an update on current understanding of the relationship between alcohol use and pain in order to guide future research objectives. We outline the mechanisms regulating excessive alcohol use and hyperalgesia, highlight the overlapping neural circuits involved in the different phases of addiction and chronic pain and touch on major molecular mechanisms. Finally, we outline missing information on the interactions between alcohol and pain, so that readers can draw conclusions on where gaps need to be filled.
OVERLAPPING DYSFUNCTION IN AUD AND PAIN
AUD is defined as a chronic relapsing brain disease characterized by compulsive alcohol use, loss of control over alcohol intake and negative emotional state when not using alcohol (American Psychiatric Association, 2013). In 2017, ~20.2 million adults aged 18 years or older were diagnosed with a substance use disorder. Among all adults diagnosed with a substance use disorder (including AUD and illicit drug use disorder), 4 of 5 are diagnosed with AUD, as compared with 3 out of 10 diagnosed with an illicit drug use disorder (Lipari and Van Horn, 2017). Moreover, AUD has a 29% lifetime prevalence (Grant et al., 2015) and only a 35–40% long-term remission rate (Finney and Moos, 1991). AUD prevalence in men has long been greater than that in women, but the number of women diagnosed with AUD is increasing (Agabio et al., 2017; Grant et al., 2017).
While multiple theories have been proposed to explain the development of addiction, including AUD (Wise and Bozarth, 1987; Robinson and Berridge, 1993; Koob and Le Moal, 2008; Nutt et al., 2015), the opponent process theory of motivation highlights the critical importance of a motivational shift in drinking behaviors from positive to negative reinforcement (Koob, 2003). In brief, the individual becomes tolerant to the initial rewarding, positively reinforcing effects of alcohol, while gradually becoming sensitized to the non-rewarding, negatively reinforcing aspects of drinking, including withdrawal symptoms and preoccupation with alcohol. As the influence of the non-rewarding, negatively reinforcing aspects of alcohol use increases, the balance between the two types of reinforcement is disrupted. Drinking then becomes driven by the motivation to prevent or alleviate negative affective consequences and/or physical withdrawal symptoms experienced upon alcohol intake cessation, thus feeding ‘the dark side’ of addiction (Koob, 2015).
A similar disruption of the typically cautionary function of acute pain is found in patients with chronic pain. Approximately 11% of adults in the USA suffer from daily chronic pain (Nahin, 2015), which is defined as pain that persists for at least 3–6 months (Treede et al., 2015). In healthy individuals, acute pain serves as a salient (meaning important and notable) stimulus that activates sensing, integrating and stabilizing physiological mechanisms to allow the body to respond through homeostatic and allostatic modifications. The power of this ability to re-establish nociceptive balance is illustrated by human and animal studies showing that only a small proportion of individuals who have sustained an injury develop chronic pain (De Felice et al., 2011; Staud, 2012; Heinricher, 2016). Conversely, prolonged use of analgesic drugs—including but not limited to opioids—leads to potentiated pain, as observed by the occurrence of medication-overuse headache and opioid-induced hyperalgesia upon extended use (Compton et al., 2001; Diener et al., 2016; Roeckel et al., 2016). It is now therefore thought that aberrant processing observed in chronic pain states involves both ‘sensitization’ of pain-transmission systems and dysfunction in descending modulatory mechanisms, including those activated by analgesic drugs (Woolf, 2011; Borsook et al., 2013; Bannister and Dickenson, 2016; Arendt-Nielsen et al., 2018).
These parallels in the disruption of balance during the progression to AUD from otherwise non-problematic alcohol use or to chronic pain from acute pain suggest potential common underlying mechanisms and an intersection of relevant nuclei and neural circuits. Possible areas of overlapping circuitry and areas of activation are depicted in Fig. 1 and discussed below.
Fig. 1.
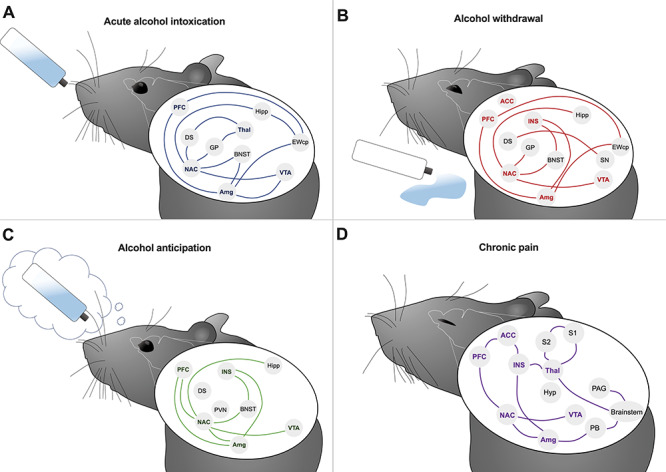
Neurocircuitry activated during alcohol use or pain. Reward, emotional and nociceptive systems contribute to alcohol intoxication (A), alcohol withdrawal (B), alcohol anticipation (C) and pain (D). Anterior cingulate cortex (ACC), amygdala (Amg), bed nucleus of the stria terminalis (BNST), centrally projecting Edinger-Westphal nucleus (EWcp), dorsal striatum (DS), globus pallidus (GP), hippocampus (Hipp), hypothalamus (Hyp), insula (INS), nucleus accumbens (NAC), periaqueductal gray (PAG), parabrachial nucleus (PB), prefrontal cortex (PFC), thalamus (Thal), substantia nigra (SN), primary somatosensory cortex (S1), secondary somatosensory cortex (S2). Bold names indicate overlap between nuclei recruited in both alcohol and pain systems.
NEURONAL ACTIVATION AND CIRCUITRY IN BOTH AUD AND PAIN
Neuronal activation and circuitry implicated in AUD
The escalation of non-problematic alcohol use to AUD can be described in three repeating yet escalating stages: (a) binge/intoxication, (b) withdrawal/negative affect and (c) preoccupation and/or anticipation (or craving) (Koob and Volkow, 2010). Brain regions associated with each of these stages have been identified through brain mapping studies of immediate early gene (IEG) expression at various stages of alcohol exposure (for a full review, see Vilpoux et al. (2009)).
The first stage, binge/intoxication, is best modeled by voluntary alcohol self-administration. Mapping of the IEG c-Fos following voluntary alcohol self-administration has demonstrated inhibition of hippocampus (Hipp) and activation of the centrally projecting Edinger–Westphal nucleus (EWcp) following acute alcohol exposure. The IEG responses in these two regions appear to be specific to the actions of alcohol rather than general novelty, stress, or any other non-specific aspects of the behavioral paradigm (Ryabinin, 1998; Ryabinin and Giardino, 2017). In some (but not all) voluntary alcohol drinking paradigms, the central nucleus of amygdala (CeA) and nucleus accumbens (NAC) have also been reported to be activated (Bachtell et al., 1999; Anacker et al., 2011). The EWcp and CeA have been the only two regions exhibiting c-Fos induction following alcohol exposure in anesthetized animals (Smith et al., 2016b) and thus are linked to alcohol exposure per se rather than to alcohol cues or perception of alcohol intoxication. Additional nuclei are activated during acute, experimenter-administered alcohol exposure and may also relate to intoxication. These include the ventral tegmental area (VTA), prefrontal cortex (PFC), dorsal striatum (DS), globus pallidus, bed nucleus of the stria terminalis (BNST) and subregions of thalamus (Thal) (Hitzemann and Hitzemann, 1997; Ryabinin et al., 1997; Thiele et al., 1997; Bachtell and Ryabinin, 2001). Although it is acknowledged that the non-voluntary exposure paradigms may engage circuits not normally activated during voluntary consumption of alcohol, it has been hypothesized that the regions outlined above comprise an interconnected network activation that is recruited in the first stage of alcohol use, underlying the first stage of addiction (Koob and Volkow, 2010) (Fig. 1A).
The second stage of escalating alcohol use is withdrawal and negative affect in the absence of alcohol. Again considering IEG as indicators of relevant brain regions, the VTA, Hipp, lateral septum, insula (INS), anterior cingulate cortex (ACC), subregions of the amygdala (Amg), EWcp, substantia nigra (SN), globus pallidus, medial habenula, locus coeruleus, cerebellum and dorsomedial hypothalamus (Hyp) are hypothesized to form a network associated with withdrawal and negative affect that drives further alcohol intake via negative reinforcement (Fig. 1B) (Putzke et al., 1996; Kozell et al., 2005; Chen et al., 2009; Smith et al., 2017). In this stage, the VTA, NAC, habenula and extended Amg are assumed to generate a negative emotional state and stress responses that coincide with an elevated reward threshold, referred to as anhedonia (Garavan et al., 2000). Increased activity of the hypothalamic–pituitary–adrenal axis [including modulations in the corticotropin-released factor (CRF) system], as well as increased dynorphin release (an endogenous kappa-opioid agonist), is observed during this period, resulting in an anxiogenic state (Koob, 2008).
The third stage of AUD is preoccupation, or anticipation. IEG mapping studies have confirmed the PFC, NAC, subregions of Amg and Hipp, BNST, the paraventricular nucleus of Hyp and VTA as regions activated upon exposure to cues predicting alcohol self-administration (Harlan and Garcia, 1998; Zhao et al., 2006; Hill et al., 2007; Vilpoux et al., 2009). Human and rodent studies investigating either cue- or stress-induced reinstatement further implicate three pathways in this response: (a) glutamatergic projections from the PFC, basolateral Amg and Hipp onto the NAC; (b) dopaminergic modulation in the basolateral Amg and DS and (c) increased CRF system activity in the extended Amg and VTA (Valdez et al., 2002; De Witte et al., 2005; Kalivas, 2009) (Fig. 1C).
Circuits implicated in chronic pain
Having both sensory-discriminative and affective dimensions, acute pain serves as a useful warning of impending or actual tissue damage, and its affective aspect motivates escape from the damaging stimulus and supports learning to avoid such stimuli in the future. This adaptive value is not evident in chronic pain. Here, pain is evoked by innocuous sensory inputs that are neither damaging nor normally painful. This latter observation is at least in part due to ‘sensitization’ of pain-processing circuitry, including primary afferent nociceptors, central pain transmission pathways and alterations in descending modulatory systems (Ji and Woolf, 2001; Heinricher, 2016; Thompson and Neugebauer, 2018). The neural circuitry associated with chronic pain has been assessed not using IEG mapping (because expression of these genes tends to habituate with chronic stimulation (Melia et al., 1994)) but instead by a combination of electrophysiological, pharmacological, behavioral, anatomical and, more recently, optogenetic and chemogenetic approaches in a range of animal models. This large body of evidence points to somatosensory pathways with ascending pathways relaying information to reticular formation in the brainstem, Hyp, Amg and cortex (including somatosensory cortex, INS and ACC). These ascending signals engage structures important in motivation and emotion, including mesolimbic dopaminergic systems implicated in salience encoding (Berridge, 2018). Given the increased salience of pain-related information in chronic pain states (Borsook et al., 2013; Kucyi and Davis, 2015), it should not be surprising that alterations in neuronal activation following painful stimuli and alterations in functional connectivity between mesolimbic structures and a number of cortical regions have been observed in functional imaging studies of patients with chronic pain (Apkarian et al., 2005; Baliki and Apkarian, 2015). Increased functional connectivity between the NAC and the PFC predicts pain persistence in patients with chronic lower back pain (Baliki et al., 2012), and painful stimuli evoked increased activity in the PFC and decreased activity in the ACC, primary somatosensory cortex (S1), secondary somatosensory cortex (S2), insular cortex and Thal in patients with clinical pain conditions (Apkarian et al., 2005). Interestingly, in individuals with acute low back pain, activation was concentrated in sensory regions such as the Thal and INS, whereas in patients with chronic back pain, it was focused in emotion-related circuitry (medial PFC, Amg) (Baliki et al., 2006; Hashmi et al., 2013), an observation consistent with an increasing contribution of motivational circuits in chronic pain. The circuitry recruited in both the sensory and affective responses to pain is illustrated in Fig. 1D.
Intersection of circuits implicated in AUD and pain
The idea that the comorbidity between AUD and chronic pain could be the result of a network overlap between regions engaged by excessive alcohol use and pain has been proposed previously (Egli et al., 2012). Further analysis of the neuronal circuits engaged during chronic pain (Fig. 1D) suggests a resemblance to those involved in the withdrawal and preoccupation/anticipation stages of AUD (Fig. 1B and C), reflecting the centrality of incentive salience in both behavioral states and the importance of the emotional components and a potential rationale for the comorbidity of AUD and chronic pain.
MOLECULAR RATIONALE FOR INTERSECTION BETWEEN AUD AND CHRONIC PAIN
The intersection between pain and alcohol in the central nervous system is also apparent due to an overlap in engaged neuronal modulators. Earlier reviews on the interaction between alcohol and pain focused on this overlap (Egli et al., 2012; Apkarian et al., 2013; Witkiewitz and Vowles, 2018). However, with the progress in understanding of these pathways, an update is in order. Here, we emphasize four major molecular mechanisms implicated in both pain and AUD: glutamate and gamma-Aminobutyric acid (GABA), opioids, CRF/urocortins (Ucns) and protein kinase C-epsilon (PKCε).
Glutamate and GABA
Alcohol inhibits the activity of ionotropic glutamate receptors acutely, consequently decreasing excitatory neurotransmission (Lovinger and Roberto, 2013). Alcohol also acts as a potential positive allosteric modulator of the GABAA receptor, thereby increasing GABA-mediated inhibition throughout the central nervous system (Korpi, 1994; Davies, 2003; Olsen et al., 2007; Lovinger and Roberto, 2013). This increased inhibitory effect of GABA is associated with sedation, inhibition of memory formation, altered reward and hypoalgesia following acute alcohol intake (Lobo and Harris, 2008). However, chronic alcohol use is associated with a paradoxical decrease in inhibitory GABA transmission and increase in excitatory glutamatergic transmission, presumably representing a compensatory response to the enhanced inhibitory tone from continued alcohol consumption (Wang et al., 2007; Holmes et al., 2013; Cheng et al., 2017). This disruption in the balance between excitatory and inhibitory transmission is thought to play a role in alcohol tolerance and dependence (defined as adaptive physiological changes where cessation of drug causes withdrawal symptoms) and is accentuated in alcohol withdrawal when the modulatory influence of alcohol on GABA receptors is no longer present (Madamba et al., 1996; Berton et al., 1998).
Altered levels of centrally acting glutamate and GABA are also observed in chronic pain conditions. Increased glutamate and decreased GABA levels have been observed in the anterior INS, ACC and Thal in diabetic neuropathy patients (Petrou et al., 2012), and increased glutamate has also been seen in the INS in patients with fibromyalgia (Kaplan et al., 2019). These observed changes in decreased endogenous GABAergic activity but increased glutamatergic activity in both chronic pain and AUD suggest that modifying the balance between these neurotransmitter levels may reinstate proper pain modulation while decreasing alcohol withdrawal-induced neuronal hyperexcitability. However, studies investigating this claim pharmacologically are limited (Enna and McCarson, 2006; Carter et al., 2014), likely because of the well-known sedative effects of GABA receptor agonism.
Opioids
Opioid alkaloids have long been recognized as potent analgesics, and opioids continue to be the mainstay for treatment of severe acute pain. Endogenous opioids contribute to the rewarding properties of natural reinforcers through their primary actions in the VTA, resulting in disinhibition leading to increased dopamine release onto the NAC (Fields and Margolis, 2015). It should therefore not be surprising that endogenous opioids play a role in the rewarding aspects of other drugs of abuse, including alcohol, and that exogenous opioids themselves come with significant abuse potential.
The opioid receptor family contains three major subtypes—mu (MOR), delta (DOR) and kappa (KOR) opioid receptors. Activity at MOR is required for morphine analgesia, as demonstrated by lack of this analgesia in MOR knockout (KO) animals (Loh et al., 1998). While MOR agonism indeed results in rapid analgesia, tolerance also develops quickly (Ho et al., 1973; Chavkin and Goldstein, 1984), thus leaving MOR-targeted therapeutics a poor choice for chronic pain conditions. Furthermore, direct administration of MOR agonists in reward-associated regions such as the NAC is associated with increased alcohol intake (Richard and Fields, 2016). Conversely, MOR-KO mice do not readily self-administer alcohol (Roberts et al., 2000). Therefore, while MOR agonists provide acute pain relief, they may also increase the risk of AUD, rendering MOR-targeted therapeutics a hazardous choice for those with comorbid chronic pain and AUD (further discussed under ‘AUD and opioid use disorder comorbidity’).
In contrast to the abuse liability concerns of MOR, DOR agonists do not produce potent analgesia in acute pain states, and activation of DOR is not commonly associated with reward (Do Carmo et al., 2009; Pradhan et al., 2014). However, DOR agonists can produce analgesia in persistent pain states (Pradhan et al., 2011; Vicente-Sanchez et al., 2016; Abdallah and Gendron, 2018). Exogenous DOR agonists can also differentially affect voluntary alcohol intake and alcohol reward (van Rijn et al., 2012a; Chiang et al., 2016) and decrease alcohol withdrawal-induced anxiety-like behaviors in mice (van Rijn et al., 2012b). Moreover, DOR-KO mice display greater anxiety-like behavior, increased alcohol intake (Roberts et al., 2001) and increased hyperalgesia following inflammation (Gaveriaux-Ruff et al., 2008), suggesting that endogenous DOR activity is protective in both alcohol-related behaviors and chronic pain.
While MOR and DOR have roles in both pain relief and reward, KORs in the mesolimbic reward system contribute to the aversive affective components of drug withdrawal and chronic pain (Walker et al., 2012; Karkhanis et al., 2017). Increases in the endogenous KOR agonist dynorphin lead to decreased dopamine release in regions such as the NAC, resulting in dysphoria and stress (Goldstein et al., 1979; Anderson and Becker, 2017). Taken as a whole, these finding might suggest that KOR antagonists would be beneficial in treating negative affect in both alcohol withdrawal and chronic pain (Walker et al., 2012; Liu et al., 2019; Massaly et al., 2019). However, KOR antagonists have thus far largely failed in clinical trials for the treatment of drug dependence or depression (Rorick-Kehn et al., 2014; Buda et al., 2015), although investigations are ongoing.
Corticotropin-releasing factor and urocortins
Both injury and AUD engage the CRF system (Vale et al., 1981; Heilig and Koob, 2007). The components of this system in mammals include four ligands (CRF, Ucn1, Ucn2 and Ucn3), two main receptors (CRFR1 and CRFR2) and the CRF-binding protein (Dedic et al., 2018). CRF primarily targets CRFR1, Ucn2 and Ucn3 target CRFR2, while Ucn1 targets both CRFR1 and CRFR2 (Pal et al., 2010). Genetic deletion or pharmacological blockade of CRFR1 can interfere with total fluid consumption, suggesting non-specific regulation of motivated behavior (Giardino and Ryabinin, 2013; Giardino et al., 2017). In contrast, genetic deletion of Ucn1 or short hairpin RNA interference with Ucn1 expression selectively suppresses the escalation of alcohol drinking to excessive drinking in mice (Giardino et al., 2017). Enhanced alcohol consumption in models of severe physiological alcohol dependence selectively depends on CRF acting on CRFR1 receptors (Chu et al., 2007; Heilig and Koob, 2007; Roberto et al., 2010).
There is also evidence that the CRF system contributes to interactions between stress and both pain and AUD. The interactions between stress and pain are complex, with mild stress inducing hyperalgesia and intense stress producing analgesia (Amit and Galina, 1988; Martenson et al., 2009). CRFR1 antagonists interfere with anxiety-like behaviors as well as hyperalgesia in rodents subjected to a chronic inflammatory pain state, and there is emerging evidence that this effect is because of interference with CRFR1-mediated sensitization of nociresponsive neurons in the CeA (Ji et al., 2007; Liang et al., 2007; Fu and Neugebauer, 2008). CRFR1 antagonists can also attenuate hyperalgesia associated with alcohol withdrawal, presumably a form of stress-induced hyperalgesia (Edwards et al., 2012). Stress can also induce alcohol-seeking in rodents with established alcohol dependence. This behavior, which can be considered a model of ‘relapse’ in abstinent individuals with AUD, is reduced by CRFR1 antagonism (Le et al., 2000; Liu and Weiss, 2002). There is thus substantial evidence that peptides acting on the CRFR1 receptor (CRF and Ucn1) could play a role in the chronic pain-AUD comorbidity. Nevertheless, CRFR1 antagonists failed to decrease subjective alcohol-induced cue and stressor craving in human subjects (Kwako et al., 2015; Schwandt et al., 2016). More studies are required to evaluate whether this failure may reflect additional factors brought into play in human populations with AUD.
Protein kinase C-epsilon
Changes in PKCε activity correspond with changes in activity throughout the central nervous system and associated behaviors, including alcohol consumption and pain. For example, inhibition of PKCε kinase activity by small molecule inhibitors decreases alcohol intake and preference in C57Bl/6 mice with no changes in ethanol clearance (Blasio et al., 2018). These effects are in line with genetic studies reporting that PKCε KO mice consume less alcohol than wild-type mice (Hodge et al., 1999) and display aversion to alcohol (Newton and Messing, 2007). PKC inhibitors also decrease alcohol neuropathy-induced hyperalgesia in rats (Dina et al., 2000). Conversely, PKC activators such as 12-myristate 13-acetate increase nociception upon intraplantar injection in mice (Ferreira et al., 2005). As inhibitors of isozymes such as PKCε decrease alcohol-related behaviors and additionally decrease pain responses, inhibitors of PKCε may be central point of molecular convergence between AUD and chronic pain signal transduction, consequently providing a potential therapeutic option for treating comorbid pain and AUDs.
ALCOHOL USE AND POTENTIATED PAIN
Epidemiological studies indicate that AUD is associated with an increased prevalence of chronic pain. In a 2007 study assessing pain persistence in detoxified patients, 73% of patients who identified alcohol as their drug of choice (as compared with opioids or cocaine) reported moderate-to-severe pain in the previous month and 24% reported pain throughout the duration of the 2-year study period (Larson et al., 2007). Another study noted that ~30% of employed, alcoholic patients attending a drug and alcohol treatment program reported severe chronic pain (Sheu et al., 2008). Problem drinking is also associated with increased prevalence of moderate-to-severe pain among older adults (Brennan et al., 2005). Increased complaints of pain in AUD are likely because of multiple factors occurring at distinct time scales. These include not only acutely enhanced pain associated with acute alcohol withdrawal and AUD but also chronic pain due to peripheral neuropathic effects of alcohol (Monforte et al., 1995) (Fig. 2), and perhaps, an increased susceptibility for chronic pain as a result of altered brain circuits associated with AUD progression (as suggested by the overlap in neuronal circuitry in Fig. 1).
Fig. 2.
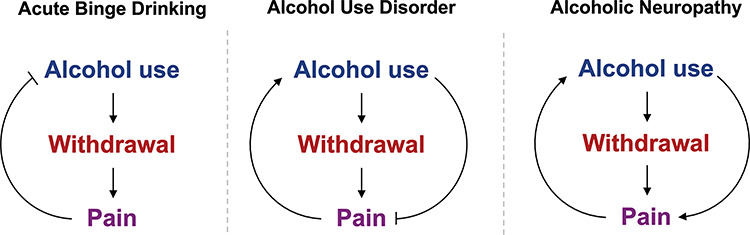
Evolution of pain’s influence on alcohol use during progression from binge drinking to AUD to alcoholic neuropathy. Following cessation of acute binge drinking, hangover causes pain during the acute withdrawal phase, which can increase time between drinking events. In AUD, cessation of alcohol use also causes pain; however, subjects may continue to drink alcohol to alleviate this withdrawal pain or other pain causes. When alcohol use is long-term and chronic, alcoholic neuropathy can develop, where alcohol use no longer provides analgesia and further alcohol intake can increase pain sensation.
Acute hyperalgesia and allodynia associated with withdrawal from alcohol
Cessation of occasional binge alcohol consumption is followed by a hangover (Slutske et al., 2003; Verster, 2008; Penning et al., 2012). During the development of AUD, hangovers increase in frequency and severity. Despite common knowledge linking hangovers with increased pain, surprisingly few studies have investigated the hyperalgesic effect of alcohol withdrawal in humans. In one experimental study, male patients with a history of alcohol dependence undergoing alcohol withdrawal displayed reduced tolerance for noxious thermal stimuli (Jochum et al., 2010), with no change in thermal pain threshold. Potentiated pain during hangover is thus documented primarily by anecdote.
By contrast with limited data in humans, alcohol withdrawal-induced hyperalgesia (increased pain evoked by normally painful stimuli) and allodynia (decrease in threshold so that innocuous stimuli are perceived as painful) have been amply demonstrated in laboratory animals. Different models of alcohol exposure have been used to assess hyperalgesia during withdrawal (Table 1). Experimenter-controlled or no-choice procedures of alcohol administration have allowed researchers to control the amount of administered alcohol and have documented significant hyperalgesia during acute withdrawal. Early studies used liquid diet-based approaches to study alcohol-induced hyperalgesia in rats, where 4–10 days of no-choice exposure to a 6.5% ethanol liquid diet or 72-day exposure to escalating 2–5% ethanol liquid diet resulted in hyperalgesia upon withdrawal (Gatch and Lal, 1999; Dina et al., 2006; Gatch, 2006; Narita et al., 2007b). More recently, mechanical hyperalgesia in rats has also been reported after prolonged alcohol vapor exposure (Edwards et al., 2012; Avegno et al., 2018). Alcohol administered by oral gavage (2 or 3 g/kg over the course of 3 weeks) has been shown to induce mechanical allodynia 24 hours following last alcohol exposure in mice (Alongkronrusmee et al., 2016).
Table 1.
Reported rodent models assessing alcohol-induced hyperalgesia/allodynia
| Rodent models of alcohol-induced hyperalgesia/allodynia | ||||
|---|---|---|---|---|
| Model | Timeline | Species/strain/sex | Analgesic test performed | Reference |
| Self-administration | ||||
| Continuous access two-bottle choice (water vs. increasing concentrations of alcohol, 3–6–10%) | 6 days of alcohol exposure/week Analgesic response measured 24 hours following alcohol removal |
Mouse C57Bl/6J Male only |
Mechanical (von Frey) Thermal (heat, tail-flick) Inflammatory (formalin) |
Smith et al. (2016a) |
| Drinking-in-the-dark two-bottle choice (water vs. 10% alcohol) | 3 weeks of alcohol exposure (4 hours/5 days a week) Analgesic response measured 1, 2, 4, 7 and 14 days following alcohol removal |
Mouse C57Bl/6 Male only |
Mechanical (von Frey) | Alongkronrusmee et al. (2016) |
| Drinking-in-the-dark (water vs. 20% alcohol) | 4 days of alcohol exposure (4 hours/day) Analgesic responses measured 1, 5–11 days following alcohol removal |
Mouse C57Bl/6 Male and female |
Inflammatory (formalin) Mechanical (von Frey) Thermal (cold, acetone evaporation) |
Bergeson et al. (2016) |
| Intermittent access, two-bottle choice (water vs. 20% alcohol) | 12 weeks of alcohol exposure (24 hours/5 days a week) Analgesic responses measured after 4, 8 or 12 weeks of alcohol exposure |
Rat Sprague-Dawley Male only |
Mechanical (von Frey) Thermal (heat, radiant heat) |
Fu et al. (2015) |
| No-choice self-administration | ||||
| Liquid diet containing ethanol (6.5%) | 10 days of liquid diet exposure Analgesic response measured 3, 6, 12 and 36 hours following ethanol removal |
Rat Long–Evans Male only |
Thermal (heat, tail-flick) | Gatch, (2006), Gatch and Lal (1999) |
| Liquid diet containing ethanol (6.5%) | 4 days of liquid diet, 3 days of standard chow (‘4 days on/3 days off’) for 5 weeks Analgesic response measured 3 days following alcohol removal |
Rat Sprague-Dawley Male only |
Mechanical (Randall–Selitto) | Dina et al. (2006) |
| Liquid diet containing ethanol (escalating, 2.5% to 5%) | 72 days of alcohol exposure Analgesic response measured once every 5 days after 7 days following alcohol removal |
Rat Fischer 344 Male |
Mechanical (Randall–Selitto) | Narita et al. (2007b) |
| Experimenter-controlled | ||||
| Chronic intermittent alcohol vapora | 11–12 weeks of alcohol vapor (14 hours/day) Analgesic response measured during withdrawal period (8 hours after vapor off) |
Rat Wistar Male only |
Mechanical (von Frey) | Edwards et al. (2012) |
| Chronic intermittent alcohol vapor | 4–9 weeks of alcohol vapor (14 hours/day) Analgesic response measured during withdrawal period (6–8 hours after vapor off), twice weekly |
Rat Wistar Male only |
Thermal (Hargreaves’) | Avegno et al. (2018), Roltsch Hellard et al. (2017) |
| Oral gavage (2 or 3 g/kg ethanol) | 3 weeks of alcohol exposure (5 consecutive days/week) Analgesic response measured 24 hours after final alcohol exposure |
Mouse C57Bl/6 Male only |
Mechanical (von Frey) | Alongkronrusmee et al. (2016) |
Models described assessed acute withdrawal-induced hyperalgesia or allodynia. Does not include models of alcoholic neuropathy.
aAnimals also exposed to two-bottle choice (10% ethanol vs. water) and ethanol operant self-administration (12 hours/session, 12 sessions) prior to vapor exposure.
Despite the robust effects of experimenter-administered alcohol, these strategies provide less translation to human studies where subjects consume alcohol voluntarily. Comparison of alcohol withdrawal in mice after alcohol gavage vs. voluntary drinking (via a two-bottle choice procedure) for 3 weeks resulted in a more pronounced (and longer-lasting) mechanical allodynia in gavaged animals, suggesting either that the rate of alcohol administration or the interaction with stress induced by the mode of administration affects the development of allodynia (Alongkronrusmee et al., 2016). Nevertheless, it is reassuring that withdrawal-induced hyperalgesia has been observed in this and other rodent models of self-administration. Mechanical hyperalgesia was observable 24 hours following 4 days of alcohol exposure in a model of exposure using a drinking-in-the-dark protocol between water and 20% alcohol in mice (Bergeson et al., 2016). Additionally, alcohol-induced mechanical, thermal and chemical hyperalgesia has been observed in C57Bl/6J male mice at 24 hours of ethanol withdrawal after continuous access to 10% ethanol and water for 6 days (Smith et al., 2016a). Longer exposure strategies, such as 12 weeks of intermittent access to a two-bottle choice (water vs. 20% alcohol) resulted in very robust hyperalgesia, as assessed through thermal or mechanical hyperalgesia/allodynia, as well as other withdrawal symptoms including tail stiffness, decreased ambulation and lower-limb flexion. Mechanical hypersensitivity was evident for up to 7 days post-withdrawal (Fu et al., 2015). One caveat with prolonged exposures to alcohol is that this strategy might also give rise to peripheral neuropathy, also known as alcoholic neuropathy.
Alcoholic neuropathy
Following years of heavy drinking, AUD patients may be diagnosed with alcoholic neuropathy. Alcoholic neuropathy is estimated to affect 25–66% of AUD patients in the United States and is most common in frequent, heavy drinkers compared with episodic drinkers (Monforte et al., 1995). It is also more common in women than in men (Ammendola et al., 2000). Alcohol-induced neuropathy manifests as pain and abnormalities in sensory, motor and autonomic functions (Chopra and Tiwari, 2012), and these clinical features are associated with axonopathy and reduced nerve fiber density (Koike et al., 2001). The exact cause of alcoholic neuropathy is unknown, and no effective therapeutics are currently available. Patients are nevertheless counseled to decrease and/or cease further alcohol use to prevent disease progression.
One potential mechanism of alcohol-induced neuropathy is thiamine (vitamin B1) deficiency, as ethanol decreases thiamine absorption in the intestine and thus depletes stores of thiamine and thiamine phosphorylation (Singleton and Martin, 2001). This decrease in cellular thiamine levels is proposed to disrupt carbohydrate metabolism, leading to increased oxidative stress or mitochondrial load, resulting in apoptosis or necrosis, respectively (Singleton and Martin, 2001). An additional suggested mechanism for alcoholic neuropathy is increased acetaldehyde accumulation as the result of ethanol metabolism, which can lead to enhanced cytokine production, potentiated oxidative stress and/or increased mitogen-activated protein (MAP) kinase or PKC signaling (Chopra and Tiwari, 2012). Microglial activation and hypertrophy have also been reported (Narita et al., 2007c). These observations raise the possibility that the axonopathy and functional changes with prolonged alcohol consumption reflect a neuroinflammatory insult. Consistent with this, rolipram, a selective phosphodiesterate-4 (PDE4) inhibitor that can reduce levels of proinflammatory cytokines, decreases mechanical allodynia in male ethanol-exposed rats (Pearse et al., 2004; Han et al., 2012).
In addition to neuroimmune mechanisms, alterations in glutamate receptor phosphorylation have been observed in alcoholic neuropathy, and these may contribute to alcoholic neuropathy symptomatology (Bu et al., 2015). Male Fischer 344 rats exposed to an ethanol-containing liquid diet for 70 days displayed mechanical allodynia that persisted for at least 14 weeks after alcohol was removed. There was a significant increase in phosphorylation of the Ser-13030 site of the N-methyl D-aspartate receptor subtype 2B (NR2B) subunit for the N-methyl D-aspartate (NMDA) receptor in the spinal cord of ethanol-fed mice compared with non-ethanol-fed controls (Narita et al., 2007a). It is possible that this phosphorylation is the result of increased PKC activity, since increased phosphorylated-PKC immunoreactivity was observed in the spinal cord of rats fed ethanol chronically (Narita et al., 2007b). These authors also reported dysfunction of MORs. Overall, a host of direct and indirect factors may contribute to the pathogenesis of alcoholic neuropathy and resulting hyperalgesia, although future studies are necessary to assess the causal role of each component.
PAIN GIVING RISE TO INCREASED ALCOHOL USE
As already noted, alcohol has long been used as an analgesic. Alcohol administration has been reported to increase pain tolerance, even at non-intoxicating doses (Woodrow and Eltherington, 1988; Horn-Hofmann et al., 2015). Acute ethanol administration can also relieve hyperalgesia, as described, for example in a study investigating the effect of ethanol on capsaicin-induced hyperalgesia in human volunteers (Arout et al., 2016). These effects of alcohol on acute and potentiated pain raise the possibility that hyperalgesia could drive consumption of alcohol. Consistent with this idea, many drinkers indicate that they consume alcohol to moderate pain (Brennan et al., 2005; Riley and King, 2009).
Acute experimental pain can motivate alcohol consumption. Moderate-to-heavy drinkers report a greater urge and intention to drink when subjected to an experimental hyperalgesia protocol (Moskal et al., 2018). Importantly, however, this study was conducted in individuals exposed to acute pain (apparently otherwise pain-free), and the results do not necessarily reflect alcohol’s effects on chronic pain. A 2015 UK study found a strong correlation between alcohol consumption and reported level of chronic widespread pain. Participants with chronic pain were less likely to report pain symptoms as debilitating if they also reported consuming alcohol regularly (Macfarlane and Beasley, 2015). Similarly, participants who consumed elevated levels of alcohol were also less likely to report chronic widespread pain symptoms in general. In patients with fibromyalgia, low and moderate alcohol consumption was associated with higher quality of life and lower symptom reporting compared with those who did not consume alcohol (Kim et al., 2013). However, a positive relationship between chronic pain and alcohol intake is not a uniform finding. Adolescents with chronic pain were less likely to use alcohol compared with adolescents without chronic pain (Law et al., 2015). Swedish adults diagnosed with long-term musculoskeletal pain reported a lower level of alcohol intake compared with an age-matched control group. Patients with chronic pain drank less often, in small quantities, and became intoxicated less frequently than those without chronic pain (Thelin Bronner et al., 2012). Finally, although alcohol use was associated with a decrease in the frequency of reported pain in individuals with orofacial pain or arthritis, there was no correlation with reduced intensity or chronicity of pain (Riley and King, 2009). These disparate findings may reflect that while alcohol consumption is acutely analgesic, its efficacy, like that of most known analgesic drugs, diminishes with chronic use (Gatch and Lal, 1999; Thompson et al., 2017). A complex interplay between analgesia, pain and other physiological, psychological and social sequelae of alcohol consumption is likely at work in individuals with chronic pain and AUD.
Pain relief is rewarding and produces negative reinforcement in rodents, as shown by increased conditioned place preference to contexts associated lidocaine administration in rats with incisional injury-induced pain (Navratilova et al., 2012). Thus, it is conceivable that rodents consume alcohol for analgesia as a negative reinforcer. However, two questions must be asked regarding alcohol’s analgesic effects: (a) does pain increase alcohol intake? and (b) is potential increased alcohol intake the result of alcohol’s analgesic effects?
A summary of known preclinical investigations addressing these questions is provided in Table 2. With regard to the first question, in a model for osteoarthritis in C57Bl/6 mice, mice in the osteoarthritis group consumed more alcohol and preferred 20% ethanol in a two-bottle choice protocol compared with control mice (Butler et al., 2017). Furthermore, severity of the osteoarthritis correlated with alcohol intake, thus suggesting that the analgesic effects of alcohol led to the increased alcohol consumption. In a different model, male C57Bl/6 mice subjected to localized inflammation of the hind paw exhibited increased alcohol intake in a two-bottle choice (20% alcohol vs. water), continuous access paradigm compared with controls, further supporting the idea that persistent pain increases alcohol intake (Yu et al., 2018). However, it is worth noting that female mice in the same study showed no increase in alcohol consumption in persistent inflammation.
Table 2.
Preclinical reports of changes in drinking in response to pain
| Model | Timeline | Species/strain/sex | Alcohol intake procedure | Changes in alcohol intake | Reference |
|---|---|---|---|---|---|
| Osteoarthritis | Alcohol consumption was assessed 13 weeks after surgical destabilization of the medial meniscus for 24 days | Mice C57Bl/6 Male only |
Two-bottle choice (water vs. gradual increase in 2.5–20% alcohol), continuous access | Osteoarthritis increased alcohol intake and preference at 20% alcohol; severity of osteoarthritis correlated with alcohol intake | Butler et al. (2017) |
| Hind paw inflammation | Alcohol consumption was assessed 3 days after injection of complete Freund’s adjuvant into the hind paw; consumption was monitored for 3 weeks | Mice C57Bl/6 Male and female |
Two-bottle choice (water vs. 20% alcohol), continuous access | Increased alcohol intake in males, no change in alcohol intake in females | Yu et al. (2018) |
| Peripheral nerve injury | Alcohol consumption was assessed 33 days after partial sciatic nerve ligation for 10 days | Mice CD1 Male only |
Single bottle access (20% alcohol), limited access (3 hours) 3 hours into dark/active cycle | Increased alcohol intake | Gonzalez-Sepulveda et al. (2016) |
Models described assessed how pain influences voluntary alcohol intake.
The two studies described above demonstrate that rodents, or at least male rodents, in persistent pain states increase alcohol intake. However, these studies did not determine whether the observed increase in alcohol consumption actually produced antinociception. CD1 mice subjected to a peripheral nerve injury consumed more ethanol in a drinking-in-the-dark procedure compared with sham-operated mice, yet cold hyperalgesia and depressive behaviors were not altered by ethanol consumption (Gonzalez-Sepulveda et al., 2016). Smith and colleagues saw no increase in mechanical withdrawal threshold in mice with persistent inflammation that were given access to alcohol in a two-bottle choice procedure (Smith et al., 2015). The lack of change in thermal and mechanical responding in these studies may be explained by low blood ethanol plasma levels, which may have been inadequate to produce detectable analgesia (Gatch, 2009). Nevertheless, alcohol intake did reverse ethanol withdrawal-induced hyperalgesia, with 4-hour access to 10% alcohol reversing mechanical hypersensitivity induced by withdrawal from a two-bottle choice procedure (Smith et al., 2016a).
Overall, current work in animal models suggests that rodents will consume more alcohol when in pain and that alcohol consumption during pain can result in analgesia, although these findings are not consistent across studies. However, whether or not this increased alcohol intake is motivated by the desire to obtain analgesia or provide potential affective relief has not yet been decisively demonstrated. For affect, an important caveat is that anxiety- and depression-related behaviors typically do not appear until 4–8 weeks following neuropathic pain induction in mice (Yalcin et al., 2011). This delayed appearance suggests that any motivation to consume alcohol following pain induction to alleviate pain-related changes in affect may not be apparent until at least 2–3 months following pain inducement.
ADDITIONAL CONSIDERATIONS
As the above discussion makes apparent, there are strong, bidirectional links between alcohol consumption and chronic pain, and progress has been made in understanding of mechanisms contributing to various aspects of these links (for example better understanding of the relevant neural circuits and molecular mechanisms). Nonetheless, many questions remain, including the influence of sex, social and environmental factors, how pain alters the efficacy of treatments for AUD and whether alcohol and opioid use disorders are related.
Sex as a biological variable
Throughout this review, the sex of the subjects assessed in each study has been specified to emphasize sex as a potential biological variable. Indeed, sex differences in both pain- and AUD-related behaviors are observed in both human subjects and rodents (Wiesenfeld-Hallin, 2005; Agabio et al., 2017), where both biological and psychosocial mechanisms presumably contribute to these assessed sex differences (Ceylan-Isik et al., 2010; Mogil, 2012; Bartley and Fillingim, 2013; Foster et al., 2014).
For AUD, men have been historically more likely to be diagnosed with AUD (Helzer and Pryzbeck, 1988). However, this gender gap has recently decreased (Foster et al., 2014; Erol and Karpyak, 2015; Agabio et al., 2017). While women are more likely to seek treatment for AUD, both sexes are vulnerable to relapse. Men most commonly relapse because of social pressure, while women relapse because of negative affect (Zywiak et al., 2006), and interestingly women exhibit fewer symptoms of withdrawal (Deshmukh et al., 2003). Sex differences are commonly observed in preclinical research as well. Female rodents voluntarily consume more alcohol than males in both social and isolated housing (Li and Lumeng, 1984; Middaugh and Kelley, 1999; Yoneyama et al., 2008). Notably, no effect of estrous cycle has been observed regardless of strain or drinking paradigm (Priddy et al., 2017). Further investigations are necessary to see if the sex-related differences in withdrawal, negative affect and social support/pressure noted in humans are maintained across species.
As for pain, sex differences in pain threshold and sensitivity remain inadequately understood (Fillingim et al., 2009; Racine et al., 2012a; Bartley and Fillingim, 2013), although it is clear that many of chronic pain conditions (irritable bowel syndrome, fibromyalgia, migraine) are more prevalent in women than in men (Yunus, 2002; Stewart et al., 2008). A variety of biological factors likely contribute to these differences in chronic pain prevalence, including differences in opioid receptor expression, gonadal hormone levels or neuroimmune responses (Bartok and Craft, 1997; Zubieta et al., 2002; Cepeda and Carr, 2003; Loyd et al., 2008; Sorge et al., 2015; Rosen et al., 2017; Sorge and Totsch, 2017). Differences in adaptive vs. innate immune responses observed in rodents in response to pain may explain the increased prevalence of autoimmune and inflammatory chronic pain conditions diagnosed in women compared with men (Mogil, 2012; Sorge et al., 2015; White and Robinson, 2015; Sorge and Totsch, 2017). Further, some of the key psychosocial factors identified above for AUD (negative affect, social environment) may also influence how individuals report and cope with chronic pain (Keogh and Herdenfeldt, 2002; Racine et al., 2012b; El-Shormilisy et al., 2015). One study found that males are more capable of focusing on the sensory-discriminative aspects of pain, while women are more likely to concentrate on the affective dimension (Keogh and Herdenfeldt, 2002). Efforts are ongoing in both animals and humans to better understand how chronic pain expression and coping differs between the sexes and how this relates to treatment outcomes.
Transfer of pain by signals or communication
In addition to alcohol withdrawal causing pain in the individual, the social transfer of pain to ‘bystander’ animal has been described following alcohol withdrawal, opiate withdrawal or inflammatory pain in rodents housed in the same room (Smith et al., 2016a; Walcott et al., 2018). This effect appears to be mediated by olfactory signals in mice, although empathetic pain processing has been reported in humans in response to visual cues (Lamm et al., 2011; Gu et al., 2012). Furthermore, context-dependent pain hypersensitivity has been observed in both male mice and humans (Martin et al., 2019). This implies that the potential effect of empathetic and/or context-dependent pain modulation should be considered in experimental and observational studies of pain in both animals and humans.
Influence of pain on alcohol treatment efficacy
Perceived pain can influence the efficacy of alcohol treatment outcomes. An association between pain and heavy drinking lapses during or after AUD treatment was observed even after controlling for other relapse factors, such as temptation, dependence severity and psychiatric distress (Witkiewitz et al., 2015; Jakubczyk et al., 2016). Similarly, alcohol consumption can influence post-surgical pain outcome. For example, 4 weeks of alcohol exposure prolonged post-surgical pain in C57Bl/6 mice (Liu et al., 2018), and patients with a history of AUD reported greater severity of pain 3 months after traumatic injury than did those without AUD (Holmes et al., 2010). These studies highlight the complexity of managing pain in individuals with AUD and addressing AUD in individuals with chronic pain.
AUD and opioid use disorder comorbidity
In the past few decades, abuse of both prescription and illicit opioids has increased explosively in the USA. This has resulted in increased opioid-related deaths, with ~60% of drug overdose deaths involving opioids in the USA in 2014 (Rudd et al., 2016). Alcohol is consistently a contributing factor to many opioid overdose deaths as both alcohol and opiates act as central nervous system depressants, thus increasing the risk of respiratory depression (White and Irvine, 1999). A 2013 study found that ~23% of adult patients diagnosed with opioid use disorder were also diagnosed with AUD, and the vast majority of these patients also had been diagnosed with a chronic pain condition (Hser et al., 2017), suggesting that chronic pain may be an important ‘third variable’ in the co-abuse of opioids and alcohol (Witkiewitz and Vowles, 2018). Because of the comorbidity of opioid use disorder and the depressant effects of both alcohol and opiates, it is conceivable that those recovering from opioid use disorder with chronic pain may consume alcohol as a means of pain management. However, little is known on the rates of use of alcohol as a substitution therapy, as no studies have formally investigated this following treatment for opioid use disorder. Moreover, opioid use disorder studies frequently exclude individuals with AUD because of the overlapping use of naltrexone for both opioid use disorder and AUD management (Witkiewitz and Vowles, 2018).
CONCLUSIONS
It is clear from preclinical and clinical investigations that alcohol use can lead to enhanced pain either acutely (during alcohol withdrawal) or chronically (via alcoholic neuropathy). Animal models of these effects are improving in sophistication, and progress in deciphering underlying neurocircuitry and neurochemical mechanisms underlying AUD-related hyperalgesia has been made. Nevertheless, this bidirectional relationship between alcohol and pain remains poorly understood. Furthermore, while pain has been demonstrated to increase alcohol intake, the motivation behind increased alcohol intake in those with chronic pain requires deeper analysis, as it could be contributed to either alcohol’s analgesic effects or affective consequences (Koob, 2003; Egli et al., 2012). As a significant overlap in affective responding between chronic pain and alcohol withdrawal and preoccupation is noted (Paulus et al., 2017), studies investigating salience processing and emotional factors in comorbid subjects are warranted. Further studies are required to investigate these overarching questions and to interrogate developed models to help decipher the intricate relationship between pain and alcohol use.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- Abdallah K, Gendron L (2018) The delta opioid receptor in pain control. Handb Exp Pharmacol 247:147–77. [DOI] [PubMed] [Google Scholar]
- Agabio R, Pisanu C, Gessa GL, Franconi F (2017) Sex differences in alcohol use disorder. Curr Med Chem 24:2661–70. [DOI] [PubMed] [Google Scholar]
- Alongkronrusmee D, Chiang T, van Rijn RM (2016) Involvement of delta opioid receptors in alcohol withdrawal-induced mechanical allodynia in male C57BL/6 mice. Drug Alcohol Depen 167:190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association (2013) Diagnostic and Statistical Manual of Mental Disorders, 5th edn. Washington, DC: American Psychiatric Association. [Google Scholar]
- Amit Z, Galina ZH (1988) Stress induced analgesia plays an adaptive role in the organization of behavioral responding. Brain Res Bull 21:955–8. [DOI] [PubMed] [Google Scholar]
- Ammendola A, Gemini D, Iannaccone S, et al. (2000) Gender and peripheral neuropathy in chronic alcoholism: a clinical-electroneurographic study. Alcohol Alcohol 35:368–71. [DOI] [PubMed] [Google Scholar]
- Anacker AM, Loftis JM, Kaur S, Ryabinin AE (2011) Prairie voles as a novel model of socially facilitated excessive drinking. Addict Biol 16:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RI, Becker HC (2017) Role of the dynorphin/kappa opioid receptor system in the motivational effects of ethanol. Alcohol Clin Exp Res 41:1402–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Bushnell MC, Treede RD, Zubieta JK (2005) Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain 9:463–84. [DOI] [PubMed] [Google Scholar]
- Apkarian AV, Neugebauer V, Koob G, et al. (2013) Neural mechanisms of pain and alcohol dependence. Pharmacol Biochem Behav 112:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt-Nielsen L, Morlion B, Perrot S, et al. (2018) Assessment and manifestation of central sensitisation across different chronic pain conditions. Eur J Pain 22:216–41. [DOI] [PubMed] [Google Scholar]
- Arout CA, Perrino AC Jr, Ralevski E, et al. (2016) Effect of intravenous ethanol on capsaicin-induced hyperalgesia in human subjects. Alcohol Clin Exp Res 40:1425–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avegno EM, Lobell TD, Itoga CA, et al. (2018) Central amygdala circuits mediate hyperalgesia in alcohol-dependent rats. J Neurosci 38:7761–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtell RK, Ryabinin AE (2001) Interactive effects of nicotine and alcohol co-administration on expression of inducible transcription factors in mouse brain. Neuroscience 103:941–54. [DOI] [PubMed] [Google Scholar]
- Bachtell RK, Wang YM, Freeman P, et al. (1999) Alcohol drinking produces brain region-selective changes in expression of inducible transcription factors. Brain Res 847:157–65. [DOI] [PubMed] [Google Scholar]
- Baliki MN, Apkarian AV (2015) Nociception, pain, negative moods, and behavior selection. Neuron 87:474–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki MN, Chialvo DR, Geha PY, et al. (2006) Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J Neurosci 26:12165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki MN, Petre B, Torbey S, et al. (2012) Corticostriatal functional connectivity predicts transition to chronic back pain. Nature Neurosci 15:1117–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister K, Dickenson AH (2016) What the brain tells the spinal cord. Pain 157:2148–51. [DOI] [PubMed] [Google Scholar]
- Bartley EJ, Fillingim RB (2013) Sex differences in pain: a brief review of clinical and experimental findings. Br J Anaesth 111:52–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartok RE, Craft RM (1997) Sex differences in opioid antinociception. J Pharmacol Exp Ther 282:769–78. [PubMed] [Google Scholar]
- Bergeson SE, Blanton H, Martinez JM, et al. (2016) Binge ethanol consumption increases inflammatory pain responses and mechanical and cold sensitivity: tigecycline treatment efficacy shows sex differences. Alcohol Clin Exp Res 40:2506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge KC. (2018) Evolving concepts of emotion and motivation. Front Psychol 9:1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton F, Francesconi WG, Madamba SG, et al. (1998) Acamprosate enhances N-methyl-d-apartate receptor-mediated neurotransmission but inhibits presynaptic GABA(B) receptors in nucleus accumbens neurons. Alcohol Clin Exp Res 22:183–91. [PubMed] [Google Scholar]
- Blasio A, Wang J, Wang D, et al. (2018) Novel small-molecule inhibitors of protein kinase C epsilon reduce ethanol consumption in mice. Biolo Psychiatry 84:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissoneault J, Lewis B, Nixon SJ (2018) Characterizing chronic pain and alcohol use trajectory among treatment-seeking alcoholics. Alcohol 75:47–54. [DOI] [PubMed] [Google Scholar]
- Borsook D, Edwards R, Elman I, et al. (2013) Pain and analgesia: the value of salience circuits. Prog Neurobiol 104:93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PL, Schutte KK, Moos RH (2005) Pain and use of alcohol to manage pain: prevalence and 3-year outcomes among older problem and non-problem drinkers. Addiction 100:777–86. [DOI] [PubMed] [Google Scholar]
- Bu F, Tian H, Gong S, et al. (2015) Phosphorylation of NR2B NMDA subunits by protein kinase C in arcuate nucleus contributes to inflammatory pain in rats. Sci Rep 5:15945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buda JJ, Carroll FI, Kosten TR, et al. (2015) A double-blind, placebo-controlled trial to evaluate the safety, tolerability, and pharmacokinetics of single, escalating oral doses of JDTic. Neuropsychopharmacology 40:2059–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler RK, Knapp DJ, Ulici V, et al. (2017) A mouse model for chronic pain-induced increase in ethanol consumption. Pain 158:457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter J, Sharon E, Stern TA (2014) The management of alcohol use disorders: the impact of pharmacologic, affective, behavioral, and cognitive approaches. Prim Care Companion CNS Disord 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda MS, Carr DB (2003) Women experience more pain and require more morphine than men to achieve a similar degree of analgesia. Anesth Analg 97:1464–8. [DOI] [PubMed] [Google Scholar]
- Ceylan-Isik AF, McBride SM, Ren J (2010) Sex difference in alcoholism: who is at a greater risk for development of alcoholic complication? Life Sci 87:133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, Goldstein A (1984) Opioid receptor reserve in normal and morphine-tolerant Guinea pig ileum myenteric plexus. Proc Natl Acad Sci USA 81:7253–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Reilly MT, Kozell LB, et al. (2009) Differential activation of limbic circuitry associated with chronic ethanol withdrawal in DBA/2J and C57BL/6J mice. Alcohol 43:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Huang CCY, Ma T, et al. (2017) Distinct synaptic strengthening of the striatal direct and indirect pathways drives alcohol consumption. Biol Psychiatry 81:918–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang T, Sansuk K, van Rijn RM (2016) Beta-arrestin 2 dependence of delta opioid receptor agonists is correlated with alcohol intake. Br J Pharmacol 173:332–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra K, Tiwari V (2012) Alcoholic neuropathy: possible mechanisms and future treatment possibilities. Br J Clin Pharmacol 73:348–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu K, Koob GF, Cole M, et al. (2007) Dependence-induced increases in ethanol self-administration in mice are blocked by the CRF1 receptor antagonist antalarmin and by CRF1 receptor knockout. Pharmacol Biochem Behav 86:813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton P, Charuvastra VC, Ling W (2001) Pain intolerance in opioid-maintained former opiate addicts: effect of long-acting maintenance agent. Drug Alcohol Depen 63:139–46. [DOI] [PubMed] [Google Scholar]
- Davies M. (2003) The role of GABAA receptors in mediating the effects of alcohol in the central nervous system. J Psychiatry Neurosci 28:263–74. [PMC free article] [PubMed] [Google Scholar]
- De Felice M, Sanoja R, Wang R, et al. (2011) Engagement of descending inhibition from the rostral ventromedial medulla protects against chronic neuropathic pain. Pain 152:2701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Witte P, Littleton J, Parot P, Koob G (2005) Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs 19:517–37. [DOI] [PubMed] [Google Scholar]
- Dedic N, Chen A, Deussing JM (2018) The CRF family of neuropeptides and their receptors—mediators of the central stress response. Curr Mol Pharmacol 11:4–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh A, Rosenbloom MJ, Sassoon S, et al. (2003) Alcoholic men endorse more DSM-IV withdrawal symptoms than alcoholic women matched in drinking history. J Stud Alcohol 64:375–9. [DOI] [PubMed] [Google Scholar]
- Diener HC, Holle D, Solbach K, Gaul C (2016) Medication-overuse headache: risk factors, pathophysiology and management. Nat Rev Neurol 12:575–83. [DOI] [PubMed] [Google Scholar]
- Dina OA, Barletta J, Chen X, et al. (2000) Key role for the epsilon isoform of protein kinase C in painful alcoholic neuropathy in the rat. J Neurosci 20:8614–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina OA, Messing RO, Levine JD (2006) Ethanol withdrawal induces hyperalgesia mediated by PKCepsilon. Eur J Neurosci 24:197–204. [DOI] [PubMed] [Google Scholar]
- Do Carmo GP, Folk JE, Rice KC, et al. (2009) The selective non-peptidic delta opioid agonist SNC80 does not facilitate intracranial self-stimulation in rats. Eur J Pharmacol 604:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Vendruscolo LF, Schlosburg JE, et al. (2012) Development of mechanical hypersensitivity in rats during heroin and ethanol dependence: alleviation by CRF(1) receptor antagonism. Neuropharmacology 62:1142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli M, Koob GF, Edwards S (2012) Alcohol dependence as a chronic pain disorder. Neurosci Biobehav Rev 36:2179–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Shormilisy N, Strong J, Meredith PJ (2015) Associations between gender, coping patterns and functioning for individuals with chronic pain: a systematic review. Pain Res Manag 20:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enna SJ, McCarson KE (2006) The role of GABA in the mediation and perception of pain. Adv Pharmacol 54:1–27. [DOI] [PubMed] [Google Scholar]
- Erol A, Karpyak VM (2015) Sex and gender-related differences in alcohol use and its consequences: contemporary knowledge and future research considerations. Drug Alcohol Depen 156:1–13. [DOI] [PubMed] [Google Scholar]
- Ferreira J, Triches KM, Medeiros R, Calixto JB (2005) Mechanisms involved in the nociception produced by peripheral protein kinase c activation in mice. Pain 117:171–81. [DOI] [PubMed] [Google Scholar]
- Fields HL, Margolis EB (2015) Understanding opioid reward. Trends Neurosci 38:217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillingim RB, King CD, Ribeiro-Dasilva MC, et al. (2009) Sex, gender, and pain: a review of recent clinical and experimental findings. J Pain 10:447–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney JW, Moos RH (1991) The long-term course of treated alcoholism: I. mortality, relapse and remission rates and comparisons with community controls. J Stud Alcohol 52:44–54. [DOI] [PubMed] [Google Scholar]
- Foster KT, Hicks BM, Iacono WG, McGue M (2014) Alcohol use disorder in women: risks and consequences of an adolescent onset and persistent course. Psychol Addict Behav 28:322–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R, Gregor D, Peng Z, et al. (2015) Chronic intermittent voluntary alcohol drinking induces hyperalgesia in Sprague-Dawley rats. Int J Physiol Pathophysiol Pharmacol 7:136–44. [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Neugebauer V (2008) Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J Neurosci 28:3861–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavan H, Pankiewicz J, Bloom A, et al. (2000) Cue-induced cocaine craving: neuroanatomical specificity for drug users and drug stimuli. Am J Psychiatry 157:1789–98. [DOI] [PubMed] [Google Scholar]
- Gatch MB. (2006) Tolerance to the antinociceptive effects of ethanol during ethanol withdrawal. Prog Neuropsychopharmacol Biol Psychiatry 30:946–52. [DOI] [PubMed] [Google Scholar]
- Gatch MB. (2009) Ethanol withdrawal and hyperalgesia. Curr Drug Abuse Rev 2:41–50. [DOI] [PubMed] [Google Scholar]
- Gatch MB, Lal H (1999) Effects of ethanol and ethanol withdrawal on nociception in rats. Alcohol Clin Exp Res 23:328–33. [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Karchewski LA, Hever X, et al. (2008) Inflammatory pain is enhanced in delta opioid receptor-knockout mice. Eur J Neurosci 27:2558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardino WJ, Rodriguez ED, Smith ML, et al. (2017) Control of chronic excessive alcohol drinking by genetic manipulation of the Edinger-Westphal nucleus urocortin-1 neuropeptide system. Trans Psychiatry 7:e1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardino WJ, Ryabinin AE (2013) CRF1 receptor signaling regulates food and fluid intake in the drinking-in-the-dark model of binge alcohol consumption. Alcohol Clin Exp Res 37:1161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A, Tachibana S, Lowney LI, et al. (1979) Dynorphin-(1-13), an extraordinarily potent opioid peptide. Proc Natl Acad Sci USA 76:6666–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Sepulveda M, Pozo OJ, Marcos J, Valverde O (2016) Chronic pain causes a persistent anxiety state leading to increased ethanol intake in CD1 mice. J Psychopharmacol 30:188–203. [DOI] [PubMed] [Google Scholar]
- Grant BF, Chou SP, Saha TD, et al. (2017) Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001–2002 to 2012–2013: results from the National Epidemiologic Survey on alcohol and related conditions. JAMA Psychiatry 74:911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Goldstein RB, Saha TD, et al. (2015) Epidemiology of DSM-5 alcohol use disorder: results from the National Epidemiologic Survey on alcohol and related conditions III. JAMA Psychiatry 72:757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Gao Z, Wang X, et al. (2012) Anterior insular cortex is necessary for empathetic pain perception. Brain 135:2726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KH, Kim SH, Jeong IC, et al. (2012) Electrophysiological and behavioral changes by phosphodiesterase 4 inhibitor in a rat model of alcoholic neuropathy. J Korean Neurosurg Soc 52:32–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlan RE, Garcia MM (1998) Drugs of abuse and immediate-early genes in the forebrain. Mol Neurobiol 16:221–67. [DOI] [PubMed] [Google Scholar]
- Hashmi JA, Baliki MN, Huang L, et al. (2013) Shape shifting pain: chronification of back pain shifts brain representation from nociceptive to emotional circuits. Brain 136:2751–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Koob GF (2007) A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci 30:399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinricher MM. (2016) Pain modulation and the transition from acute to chronic pain. Adv Exp Med Biol 904:105–15. [DOI] [PubMed] [Google Scholar]
- Helzer JE, Pryzbeck TR (1988) The co-occurrence of alcoholism with other psychiatric disorders in the general population and its impact on treatment. J Stud Alcohol 49:219–24. [DOI] [PubMed] [Google Scholar]
- Hill KG, Ryabinin AE, Cunningham CL (2007) FOS expression induced by an ethanol-paired conditioned stimulus. Pharmacol Biochem Behav 87:208–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitzemann B, Hitzemann R (1997) Genetics ethanol and the Fos response: a comparison of the C57BL/6J and DBA/2J inbred mouse strains. Alcohol Clin Exp Res 21:1497–507. [PubMed] [Google Scholar]
- Ho IK, Loh HH, Way EL (1973) Effects of cyclic 3′,5′-adenosine monophosphate on morphine tolerance and physical dependence. J Pharmacol Exp Ther 185:347–57. [PubMed] [Google Scholar]
- Hodge CW, Mehmert KK, Kelley SP, et al. (1999) Supersensitivity to allosteric GABA(A) receptor modulators and alcohol in mice lacking PKCepsilon. Nature Neurosci 2:997–1002. [DOI] [PubMed] [Google Scholar]
- Holmes A, Spanagel R, Krystal JH (2013) Glutamatergic targets for new alcohol medications. Psychopharmacology 229:539–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Williamson O, Hogg M, et al. (2010) Predictors of pain severity 3 months after serious injury. Pain Med 11:990–1000. [DOI] [PubMed] [Google Scholar]
- Horn-Hofmann C, Buscher P, Lautenbacher S, Wolstein J (2015) The effect of nonrecurring alcohol administration on pain perception in humans: a systematic review. J Pain Res 8:175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hser YI, Mooney LJ, Saxon AJ, et al. (2017) Chronic pain among patients with opioid use disorder: results from electronic health records data. J Subst Abuse Treat 77:26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubczyk A, Ilgen MA, Kopera M, et al. (2016) Reductions in physical pain predict lower risk of relapse following alcohol treatment. Drug Alcohol Depen 158:167–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Fu Y, Ruppert KA, Neugebauer V (2007) Pain-related anxiety-like behavior requires CRF1 receptors in the amygdala. Mol Pain 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Woolf CJ (2001) Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis 8:1–10. [DOI] [PubMed] [Google Scholar]
- Jochum T, Boettger MK, Burkhardt C, et al. (2010) Increased pain sensitivity in alcohol withdrawal syndrome. Eur J Pain 14:713–8. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. (2009) The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10:561–72. [DOI] [PubMed] [Google Scholar]
- Kaplan CM, Schrepf A, Vatansever D, et al. (2019) Functional and neurochemical disruptions of brain hub topology in chronic pain. Pain 160:973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkhanis A, Holleran KM, Jones SR (2017) Dynorphin/kappa opioid receptor signaling in preclinical models of alcohol, drug, and food addiction. Int Rev Neurobiol 136:53–88. [DOI] [PubMed] [Google Scholar]
- Keogh E, Herdenfeldt M (2002) Gender, coping and the perception of pain. Pain 97:195–201. [DOI] [PubMed] [Google Scholar]
- Kim CH, Vincent A, Clauw DJ, et al. (2013) Association between alcohol consumption and symptom severity and quality of life in patients with fibromyalgia. Arthritis Res Ther 15:R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike H, Mori K, Misu K, et al. (2001) Painful alcoholic polyneuropathy with predominant small-fiber loss and normal thiamine status. Neurology 56:1727–32. [DOI] [PubMed] [Google Scholar]
- Koob GF. (2003) Alcoholism: allostasis and beyond. Alcohol Clin Exp Res 27:232–43. [DOI] [PubMed] [Google Scholar]
- Koob GF. (2008) A role for brain stress systems in addiction. Neuron 59:11–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. (2015) The dark side of emotion: the addiction perspective. Eur J Pharmacol 753:73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M (2008) Review. Neurobiological mechanisms for opponent motivational processes in addiction. Philos Trans R Soc Lond B Biol Sci 363:3113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology 35:217–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpi ER. (1994) Role of GABAA receptors in the actions of alcohol and in alcoholism: recent advances. Alcohol Alcohol 29:115–29. [PubMed] [Google Scholar]
- Kozell LB, Hitzemann R, Buck KJ (2005) Acute alcohol withdrawal is associated with c-Fos expression in the basal ganglia and associated circuitry: C57BL/6J and DBA/2J inbred mouse strain analyses. Alcohol Clin Exp Res 29:1939–48. [DOI] [PubMed] [Google Scholar]
- Kucyi A, Davis KD (2015) The dynamic pain connectome. Trends Neurosci 38:86–95. [DOI] [PubMed] [Google Scholar]
- Kwako LE, Spagnolo PA, Schwandt ML, et al. (2015) The corticotropin releasing hormone-1 (CRH1) receptor antagonist pexacerfont in alcohol dependence: a randomized controlled experimental medicine study. Neuropsychopharmacology 40:1053–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamm C, Decety J, Singer T (2011) Meta-analytic evidence for common and distinct neural networks associated with directly experienced pain and empathy for pain. Neuroimage 54:2492–502. [DOI] [PubMed] [Google Scholar]
- Larson MJ, Paasche-Orlow M, Cheng DM, et al. (2007) Persistent pain is associated with substance use after detoxification: a prospective cohort analysis. Addiction 102:752–60. [DOI] [PubMed] [Google Scholar]
- Law EF, Bromberg MH, Noel M, et al. (2015) Alcohol and tobacco use in youth with and without chronic pain. J Pediatr Psychol 40:509–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le AD, Harding S, Juzytsch W, et al. (2000) The role of corticotrophin-releasing factor in stress-induced relapse to alcohol-seeking behavior in rats. Psychopharmacology 150:317–24. [DOI] [PubMed] [Google Scholar]
- Li TK, Lumeng L (1984) Alcohol preference and voluntary alcohol intakes of inbred rat strains and the National Institutes of Health heterogeneous stock of rats. Alcohol Clin Exp Res 8:485–6. [DOI] [PubMed] [Google Scholar]
- Liang Y-J, Wu D-F, Yang L-Q, et al. (2007) Interaction of the {micro}-opioid receptor with synaptophysin influences receptor trafficking and Signaling. Mol Pharmacol 71:123–31. [DOI] [PubMed] [Google Scholar]
- Lipari RN, Van Horn SL (2017) Trends in substance use disorders among adults aged 18 or older . The CBHSQ Report Rockville, MD; pp. 1–10 [PubMed] [Google Scholar]
- Liu S, Zhao Z, Guo Y, et al. (2018) Spinal AMPA receptor GluA1 Ser831 phosphorylation controls chronic alcohol consumption-produced prolongation of postsurgical pain. Mol Neurobiol 55:4090–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SS, Pickens S, Burma NE, et al. (2019) Kappa opioid receptors drive a tonic aversive component of chronic pain. J Neurosci 39:4162–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weiss F (2002) Additive effect of stress and drug cues on reinstatement of ethanol seeking: exacerbation by history of dependence and role of concurrent activation of corticotropin-releasing factor and opioid mechanisms. J Neurosci 22:7856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo IA, Harris RA (2008) GABA(A) receptors and alcohol. Pharmacol Biochem Behav 90:90–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh HH, Liu HC, Cavalli A, et al. (1998) Mu opioid receptor knockout in mice: effects on ligand-induced analgesia and morphine lethality. Brain Res Mol Brain Res 54:321–6. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, Roberto M (2013) Synaptic effects induced by alcohol. Curr Top Behav Neurosci 13:31–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyd DR, Wang X, Murphy AZ (2008) Sex differences in micro-opioid receptor expression in the rat midbrain periaqueductal gray are essential for eliciting sex differences in morphine analgesia. J Neurosci 28:14007–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane GJ, Beasley M (2015) Alcohol consumption in relation to risk and severity of chronic widespread pain: results from a UK population-based study. Arthritis Care Res (Hoboken) 67:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madamba SG, Schweitzer P, Zieglgansberger W, Siggins GR (1996) Acamprosate (calcium acetylhomotaurinate) enhances the N-methyl-d-aspartate component of excitatory neurotransmission in rat hippocampal CA1 neurons in vitro. Alcohol Clin Exp Res 20:651–8. [DOI] [PubMed] [Google Scholar]
- Martenson ME, Cetas JS, Heinricher MM (2009) A possible neural basis for stress-induced hyperalgesia. Pain 142:236–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Acland EL, Cho C, et al. (2019) Male-specific conditioned pain hypersensitivity in mice and humans. Curr Biol 29:192–201e4. [DOI] [PubMed] [Google Scholar]
- Massaly N, Copits BA, Wilson-Poe AR, et al. (2019) Pain-induced negative affect is mediated via recruitment of the nucleus Accumbens kappa opioid system. Neuron 102:564–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melia KR, Ryabinin AE, Schroeder R, et al. (1994) Induction and habituation of immediate early gene expression in rat brain by acute and repeated restraint stress. J Neurosci 14:5929–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middaugh LD, Kelley BM (1999) Operant ethanol reward in C57BL/6 mice: influence of gender and procedural variables. Alcohol 17:185–94. [DOI] [PubMed] [Google Scholar]
- Mogil JS. (2012) Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nat Rev Neurosci 13:859–66. [DOI] [PubMed] [Google Scholar]
- Monforte R, Estruch R, Valls-Sole J, et al. (1995) Autonomic and peripheral neuropathies in patients with chronic alcoholism. A dose-related toxic effect of alcohol. Arch Neurol 52:45–51. [DOI] [PubMed] [Google Scholar]
- Moskal D, Maisto SA, De Vita M, Ditre JW (2018) Effects of experimental pain induction on alcohol urge, intention to consume alcohol, and alcohol demand. Exp Clin Psychopharmacol 26:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahin RL. (2015) Estimates of pain prevalence and severity in adults: United States, 2012. J Pain 16:769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Miyoshi K, Narita M, Suzuki T (2007a) Changes in function of NMDA receptor NR2B subunit in spinal cord of rats with neuropathy following chronic ethanol consumption. Life Sci 80:852–9. [DOI] [PubMed] [Google Scholar]
- Narita M, Miyoshi K, Narita M, Suzuki T (2007b) Functional reduction in mu-opioidergic system in the spinal cord under a neuropathic pain-like state following chronic ethanol consumption in the rat. Neuroscience 144:777–82. [DOI] [PubMed] [Google Scholar]
- Narita M, Miyoshi K, Narita M, Suzuki T (2007c) Involvement of microglia in the ethanol-induced neuropathic pain-like state in the rat. Neurosci Lett 414:21–5. [DOI] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, Okun A, et al. (2012) Pain relief produces negative reinforcement through activation of mesolimbic reward-valuation circuitry. Proc Natl Acad Sci USA 109:20709–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton PM, Messing RO (2007) Increased sensitivity to the aversive effects of ethanol in PKCepsilon null mice revealed by place conditioning. Behav Neurosci 121:439–42. [DOI] [PubMed] [Google Scholar]
- Nutt DJ, Lingford-Hughes A, Erritzoe D, Stokes PR (2015) The dopamine theory of addiction: 40 years of highs and lows. Nat Rev Neurosci 16:305–12. [DOI] [PubMed] [Google Scholar]
- Olsen RW, Hanchar HJ, Meera P, Wallner M (2007) GABAA receptor subtypes: the “one glass of wine” receptors. Alcohol 41:201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal K, Swaminathan K, Xu HE, Pioszak AA (2010) Structural basis for hormone recognition by the human CRFR2{alpha} G protein-coupled receptor. J Biol Chem 285:40351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus DJ, Bakhshaie J, Ditre JW, et al. (2017) Emotion dysregulation in the context of pain and alcohol use among Latinos in primary care. J Stud Alcohol Drugs 78:938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Marcillo AE, et al. (2004) cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med 10:610–6. [DOI] [PubMed] [Google Scholar]
- Penning R, McKinney A, Verster JC (2012) Alcohol hangover symptoms and their contribution to the overall hangover severity. Alcohol Alcohol 47:248–52. [DOI] [PubMed] [Google Scholar]
- Petrou M, Pop-Busui R, Foerster BR, et al. (2012) Altered excitation-inhibition balance in the brain of patients with diabetic neuropathy. Acad Radiol 19:607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Befort K, Nozaki C, et al. (2011) The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci 32:581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Smith ML, Zyuzin J, Charles A (2014) Delta-opioid receptor agonists inhibit migraine-related hyperalgesia, aversive state and cortical spreading depression in mice. Br J Pharmacol 171:2375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priddy BM, Carmack SA, Thomas LC, et al. (2017) Sex, strain, and estrous cycle influences on alcohol drinking in rats. Pharmacol Biochem Behav 152:61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzke J, Spanagel R, Tolle TR, Zieglgansberger W (1996) The anti-craving drug acamprosate reduces c-fos expression in rats undergoing ethanol withdrawal. Eur J Pharmacol 317:39–48. [DOI] [PubMed] [Google Scholar]
- Racine M, Tousignant-Laflamme Y, Kloda LA, et al. (2012a) A systematic literature review of 10 years of research on sex/gender and experimental pain perception—part 1: are there really differences between women and men? Pain 153:602–18. [DOI] [PubMed] [Google Scholar]
- Racine M, Tousignant-Laflamme Y, Kloda LA, et al. (2012b) A systematic literature review of 10 years of research on sex/gender and pain perception—part 2: do biopsychosocial factors alter pain sensitivity differently in women and men? Pain 153:619–35. [DOI] [PubMed] [Google Scholar]
- Richard JM, Fields HL (2016) Mu-opioid receptor activation in the medial shell of nucleus accumbens promotes alcohol consumption, self-administration and cue-induced reinstatement. Neuropharmacology 108:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley JL 3rd, King C (2009) Self-report of alcohol use for pain in a multi-ethnic community sample. J Pain 10:944–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Cruz MT, Gilpin NW, et al. (2010) Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry 67:831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Gold LH, Polis I, et al. (2001) Increased ethanol self-administration in delta-opioid receptor knockout mice. Alcohol Clin Exp Res 25:1249–56. [PubMed] [Google Scholar]
- Roberts AJ, McDonald JS, Heyser CJ, et al. (2000) Mu-opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther 293:1002–8. [PubMed] [Google Scholar]
- Robinson TE, Berridge KC (1993) The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev 18:247–91. [DOI] [PubMed] [Google Scholar]
- Roeckel LA, Le Coz GM, Gaveriaux-Ruff C, Simonin F (2016) Opioid-induced hyperalgesia: cellular and molecular mechanisms. Neuroscience 338:160–82. [DOI] [PubMed] [Google Scholar]
- Roltsch Hellard EA, Impastato RA, Gilpin NW (2017) Intra-cerebral and intra-nasal melanocortin-4 receptor antagonist blocks withdrawal hyperalgesia in alcohol-dependent rats. Addict Biol 22:692–701. [DOI] [PubMed] [Google Scholar]
- Rorick-Kehn LM, Witkin JM, Statnick MA, et al. (2014) LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology 77:131–44. [DOI] [PubMed] [Google Scholar]
- Rosen S, Ham B, Mogil JS (2017) Sex differences in neuroimmunity and pain. J Neurosci Res 95:500–8. [DOI] [PubMed] [Google Scholar]
- Rudd RA, Seth P, David F, Scholl L (2016) Increases in drug and opioid-involved overdose deaths—United States, 2010–2015. MMWR Morb Mortal Wkly Rep 65:1445–52. [DOI] [PubMed] [Google Scholar]
- Ryabinin AE. (1998) Role of hippocampus in alcohol-induced memory impairment: implications from behavioral and immediate early gene studies. Psychopharmacology 139:34–43. [DOI] [PubMed] [Google Scholar]
- Ryabinin AE, Criado JR, Henriksen SJ, et al. (1997) Differential sensitivity of c-Fos expression in hippocampus and other brain regions to moderate and low doses of alcohol. Mol Psychiatry 2:32–43. [DOI] [PubMed] [Google Scholar]
- Ryabinin AE, Giardino WJ (2017) Contribution of Urocortin to the development of excessive drinking. Int Rev Neurobiol 136:275–91. [DOI] [PubMed] [Google Scholar]
- Schwandt ML, Cortes CR, Kwako LE, et al. (2016) The CRF1 antagonist verucerfont in anxious alcohol-dependent women: translation of neuroendocrine, but not of anti-craving effects. Neuropsychopharmacology 41:2818–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu R, Lussier D, Rosenblum A, et al. (2008) Prevalence and characteristics of chronic pain in patients admitted to an outpatient drug and alcohol treatment program. Pain Med 9:911–7. [DOI] [PubMed] [Google Scholar]
- Singleton CK, Martin PR (2001) Molecular mechanisms of thiamine utilization. Curr Mol Med 1:197–207. [DOI] [PubMed] [Google Scholar]
- Slutske WS, Piasecki TM, Hunt-Carter EE (2003) Development and initial validation of the hangover symptoms scale: prevalence and correlates of hangover symptoms in college students. Alcohol Clin Exp Res 27:1442–50. [DOI] [PubMed] [Google Scholar]
- Smith ML, Hostetler CM, Heinricher MM, Ryabinin AE (2016a) Social transfer of pain in mice. Sci Adv 2:e1600855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ML, Li J, Cote DM, Ryabinin AE (2016b) Effects of isoflurane and ethanol administration on c-Fos immunoreactivity in mice. Neuroscience 316:337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ML, Li J, Ryabinin AE (2015) Increased alcohol consumption in urocortin 3 knockout mice is unaffected by chronic inflammatory pain. Alcohol Alcohol 50:132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ML, Walcott AT, Heinricher MM, Ryabinin AE (2017) Anterior cingulate cortex contributes to alcohol withdrawal- induced and socially transferred hyperalgesia. eNeuro 13:710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Mapplebeck JC, Rosen S, et al. (2015) Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nature Neurosci 18:1081–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Totsch SK (2017) Sex differences in pain. J Neurosci Res 95:1271–81. [DOI] [PubMed] [Google Scholar]
- Staud R. (2012) Abnormal endogenous pain modulation is a shared characteristic of many chronic pain conditions. Expert Rev Neurother 12:577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Wood C, Reed ML, et al. (2008) Cumulative lifetime migraine incidence in women and men. Cephalalgia 28:1170–8. [DOI] [PubMed] [Google Scholar]
- Thelin Bronner KB, Wennberg P, Kallmen H, Schult ML (2012) Alcohol habits in patients with long-term musculoskeletal pain: comparison with a matched control group from the general population. Int J Rehabil Res 35:130–7. [DOI] [PubMed] [Google Scholar]
- Thiele TE, van Dijk G, Bernstein IL (1997) Ethanol-induced c-Fos expression in rat lines selected for low and high alcohol consumption. Brain Res 756:278–82. [DOI] [PubMed] [Google Scholar]
- Thompson JM, Neugebauer V (2018) Cortico-limbic pain mechanisms. Neurosci Lett 702:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson T, Oram C, Correll CU, et al. (2017) Analgesic effects of alcohol: a systematic review and meta-analysis of controlled experimental studies in healthy participants. J Pain 18:499–510. [DOI] [PubMed] [Google Scholar]
- Treede RD, Rief W, Barke A, et al. (2015) A classification of chronic pain for ICD-11. Pain 156:1003–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez GR, Roberts AJ, Chan K, et al. (2002) Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res 26:1494–501. [DOI] [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J (1981) Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213:1394–7. [DOI] [PubMed] [Google Scholar]
- van Rijn RM, Brissett DI, Whistler JL (2012a) Distinctive modulation of ethanol place preference by delta opioid receptor-selective agonists. Drug Alcohol Depen 122:156–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn RM, Brissett DI, Whistler JL (2012b) Emergence of functional spinal delta opioid receptors after chronic ethanol exposure. Biol Psychiatry 71:232–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verster JC. (2008) The alcohol hangover—a puzzling phenomenon. Alcohol Alcohol 43:124–6. [DOI] [PubMed] [Google Scholar]
- Vicente-Sanchez A, Segura L, Pradhan AA (2016) The delta opioid receptor tool box. Neuroscience 338:145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilpoux C, Warnault V, Pierrefiche O, et al. (2009) Ethanol-sensitive brain regions in rat and mouse: a cartographic review, using immediate early gene expression. Alcohol Clin Exp Res 33:945–69. [DOI] [PubMed] [Google Scholar]
- Walcott AT, Smith ML, Loftis JM, Ryabinin AE (2018) Social transfer of alcohol withdrawal-induced hyperalgesia in female prairie voles. Soc Neurosci 13:710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BM, Valdez GR, McLaughlin JP, Bakalkin G (2012) Targeting dynorphin/kappa opioid receptor systems to treat alcohol abuse and dependence. Alcohol 46:359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Carnicella S, Phamluong K, et al. (2007) Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: implications for alcohol drinking behavior. J Neurosci 27:3593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White HD, Robinson TD (2015) A novel use for testosterone to treat central sensitization of chronic pain in fibromyalgia patients. Int Immunopharmacol 27:244–8. [DOI] [PubMed] [Google Scholar]
- White JM, Irvine RJ (1999) Mechanisms of fatal opioid overdose. Addiction 94:961–72. [PubMed] [Google Scholar]
- Wiesenfeld-Hallin Z. (2005) Sex differences in pain perception. Gend Med 2:137–45. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA (1987) A psychomotor stimulant theory of addiction. Psychol Rev 94:469–92. [PubMed] [Google Scholar]
- Witkiewitz K, Vowles KE (2018) Alcohol and opioid use, co-use, and chronic pain in the context of the opioid epidemic: a critical review. Alcohol Clin Exp Res 42:478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewitz K, Vowles KE, McCallion E, et al. (2015) Pain as a predictor of heavy drinking and any drinking lapses in the COMBINE study and the UK alcohol treatment trial. Addiction 110:1262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodrow KM, Eltherington LG (1988) Feeling no pain: alcohol as an analgesic. Pain 32:159–63. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. (2011) Central sensitization: implications for the diagnosis and treatment of pain. Pain 152:S2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin I, Bohren Y, Waltisperger E, et al. (2011) A time-dependent history of mood disorders in a murine model of neuropathic pain. Biol Psychiatry 70:946–53. [DOI] [PubMed] [Google Scholar]
- Yeung EW, Craggs JG, Gizer IR (2017) Comorbidity of alcohol use disorder and chronic pain: genetic influences on brain reward and stress systems. Alcohol Clin Exp Res 41:1831–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama N, Crabbe JC, Ford MM, et al. (2008) Voluntary ethanol consumption in 22 inbred mouse strains. Alcohol 42:149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Hwa LS, Makhijani VH, et al. (2018) Chronic inflammatory pain drives alcohol drinking in a sex-dependent manner for C57BL/6J mice. Alcohol 77:135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunus MB. (2002) Gender differences in fibromyalgia and other related syndromes. J Gend Specif Med 5:42–7. [PubMed] [Google Scholar]
- Zhao Y, Dayas CV, Aujla H, et al. (2006) Activation of group II metabotropic glutamate receptors attenuates both stress and cue-induced ethanol-seeking and modulates c-fos expression in the hippocampus and amygdala. J Neurosci 26:9967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, et al. (2002) Mu-opioid receptor-mediated antinociceptive responses differ in men and women. J Neurosci 22:5100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zywiak WH, Stout RL, Trefry WB, et al. (2006) Alcohol relapse repetition, gender, and predictive validity. J Subst Abuse Treat 30:349–53. [DOI] [PubMed] [Google Scholar]