

Abstract
Background and purpose:
Tobacco etch virus (TEV) protease is a protease with high sequence specificity which is useful for the cleavage of fusion proteins. A major limitation of this enzyme is its relatively poor solubility. This study aimed to investigate the effects of some suggested mutations by online tools and molecular dynamics simulation to improve the solubility of TEV protease in vitro.
Experimental approach:
We designed a rational multi-stage process to determine the solubilizing mutations of TEV protease. At the first stage, all the possible mutations were predicted using online tools such as PoPMuSiC and Eris servers, in which five mutations include N23F, N23L, Q74L, Q74V, and Q74I were suggested for further studies. In the next step, the three dimensional structure of the wild type (WT) and the best mutations were subjected to molecular dynamic simulations to evaluate the dynamic behaviour of the obtained structures. The selected mutation was introduced into the structure using site-directed mutagenesis and expressed in Escherichia coli BL21DE3. After purification, solubility and activity of the purified mutant and WT-TEV proteases were assayed.
Findings /Results:
By considering the analysis of various factors such as structural and solubility properties, one mutant, N23F, was selected for in vitro studies which led to a 1.5 times increase in the solubility compared to the WT while its activity was decreased somewhat.
Conclusion and implications:
We propose N23F mutation, according to computational and experimental analyses for TEV proteases which resulted in a 150% increase in solubility compared to the WT.
Keywords: Molecular dynamics simulation, Site-directed mutation, Solubility, TEV protease
INTRODUCTION
Tobacco etch virus protease is the 27 kDa catalytic domain of a larger viral protein i.e. NIa. TEV proteases is widely used to remove the N- or C- tags from recombinant fusion proteins (1), an effective tool for in vitro and in vivo biotechnological applications such as in vitro enzymology tests (2), in vivo targeting (3,4,5,6), stoichiometric expression of multiple proteins (7), complementation assays (8), and several other applications (9). It has several outstanding features including strong specificity to recognize its target sequence “ENLYFQG/S” (10,11), ability to create natural N-terminal after cutting in natural proteins (10,12), producibility in various host cells from Escherichia coli (E. coli), to complex organisms such as Drosophila (13), HeLa and other mammalian cell lines (14,15) without showing any toxicity to the host cells, and maintaining activity at different temperatures, buffers and media composition (16).
However, wild-type (WT) TEV proteases suffer from some limitations such as autoproteolysis (self-cutting) between Met 218 and Ser 219 and low solubility (about 1 mg/mL) (17).
Until now, several approaches have been introduced to tackle such issues. For instance, replacing Serine 219 with glutamic acid, valine or proline which resulted in producing variants with higher resistance to autolysis (17). However, addressing the solubility issue is a complex process since the protein solubility depends on various parameters such as amino acid sequences and experimental conditions including pH, temperature and salt concentration (18). Several approaches have so far been introduced to increase the solubility of TEV proteases, which include optimization of environmental conditions, selection of appropriate host strains (19), chaperoneco- expression (20), the use of new tags (21), directed evolution and protein engineering (22,23).
Molecular dynamics (MD) simulation is a powerful complimentary computer-aided approach to study the structure and function of a protein at an atomic level with the possibility of obtaining time-dependent information that otherwise is difficult to achieve from wet-lab experiments (24,25,26). In fact, providing information about protein structure in solution is one of the main advantages of MD (27).
This study aimed to investigate the effects of some suggested mutations by online tools using MD simulation to improve the solubility of TEV protease in vitro.
MATERIALS AND METHODS
The methodology consists of both dry (theoretical) and wet (experimental) lab experiments. The theoretical investigations included a selection of potential mutations using online tools such as PoPMuSiC, Eris, etc. These tools usually predict changes in the folding free energy upon mutations (28). Then, the effects of the applied mutations studied using MD simulations. For the experimental section, screened mutation applied in vitro and its practical effects were assessed.
Theoretical section
Determination of mutations using online tools
PoPMuSiC server (http://dezyme.com/en/ software) was used for rational design. This server introduces single-site mutations into a proteins’ structure and estimates the variations in the ΔΔGs values of such mutations. In the next step, all of the possible single-site mutations (4731 mutations) were sorted by their ΔΔGs in order to exclude the ones with a ΔΔGs ≥ -1 Kcal/mol. Fifteen stabilizing mutations were selected according to these criteria. These favorable replacements (Table 1) are located at three different positions, Q74, N23, and S153. Afterward, the positions of the favorable mutations were screened to remove those interacting with active site residues; hence, the S153 mutation was eliminated (Fig. 1), and the rest of the mutations were submitted to the Eris server (https://dokhlab.med.psu. edu/eris/login.php) in order to validate the corresponding ΔΔGs. Finally, the best mutations were selected for further studies in terms of their scores at both servers.
Table 1.
The most negative ΔΔGs of suggested mutations using PoPMuSiC and Eris servers.
| Mutations | POPmusic ΔΔG (Kcal/mol) |
Eris energy (Kcal/mol) |
Scorea |
|---|---|---|---|
| Q74I | -2.22 | -9.6 | ++ |
| Q74V | -1.98 | -6.83 | ++ |
| Q74F | -1.82 | 9.01 | + |
| N23I | -1.68 | 2.36 | + |
| Q74L | -1.52 | -5.01 | ++ |
| Q74W | -1.52 | 9.74 | + |
| Q74Y | -1.46 | 4.39 | + |
| N23V | -1.45 | 5.55 | + |
| S153I | -1.43 | 0.8 | + |
| S153V | -1.39 | -2.0 | ++ |
| N23F | -1.32 | -2.26 | ++ |
| N23L | -1.18 | -3.88 | ++ |
| Q74C | -1.1 | 1.28 | + |
| N23Y | -1.06 | 0.72 | + |
| N23C | -1.01 | 3.39 | + |
a, The mutations that had good score energetically in terms of both servers indicated with double plus and the other mutations with one plus approved by Pop music server only.
Fig. 1.
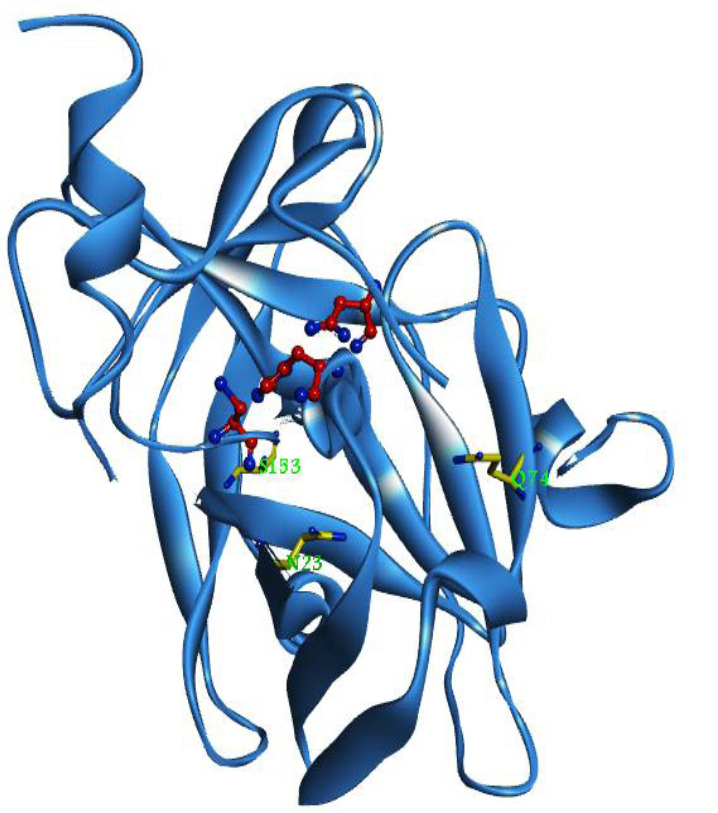
Tobacco etch virus protease protease structure. The catalytic triad H46, D81, and C151 are shown as ball and stick mode in red. The residues prone to mutation (N23, Q74, and S153) represent as stick mode in yellow. The figure was generated by Discovery Studio visualizer 4.0 (http:// www.3dsbiovia.com)
Structure preparation
The initial three dimensional (3D) structure of TEV proteases, a dimer of two similar chains, was obtained from the Protein Data Bank (PDB ID: 1LVM) (11).For this step, just one chain was extracted and used to add a hexa His tag to its N-terminal. Moreover, considering the TEV protease expression plasmid sequence, a moiety containing additional residues of - EEPFQPVKEATQLMNRRRRR- was added to the C-terminal to create the WT structure. Modeller 9.13 software was used to prepare the 3D structures of the WT and mutants. Of all the generated structures the best five models of the WT and mutations were selected in terms of the least dope energy.The selected structures were introduced to the PROCHECK server for quality evaluation (29), and then to the Verify- 3D server to evaluate the model’s environmental profile (30).
MD Simulations
GROMACS 5.1 simulation package with a G43A1 force field (31) was used for MD simulation (32) which was done in four steps:
First, the energy of the initial structures obtained from the homology modeling step was minimized using the steepest descent, and then the conjugate gradient algorithms.
Second, the minimized structure was solvated in a cubic box filled with about 10600 SPC/E 216 water molecules with a layer thickness of at least 10 Å in all directions and at neutral pH. The system neutralized by adding 9 Cl[-] ions. Then, the system was evolved using MD simulation at constant volume and temperature (NVT) ensemble for 500 ps at 100 K, and then at a constant pressure and temperature (NPT) ensemble for 1000 ps at 100 K in which the position of the atoms was kept fixed. The thermostat and barostat equilibriums were achieved using the Berendsen algorithm (33).
Third, in order to obtain equilibrium geometry at 300 K and 1 atm, the system was heated at a weak temperature (τ = 0.1 ps) and pressure (τ = 0.5 ps) coupling by adjusting the velocities of the atoms following Maxwell- Boltzmann distribution.
Fourth, 6 separate 20 ns simulations at 300 K with a time step of 2 fs at the NPT ensemble and periodic boundary condition were performed and the final structures were obtained. Nosé-Hoover thermostat and Parrinello-Rahman barostat were used for this step (33,34). The following conditions were considered at the MD simulation; the Van der Waals forces cut-off was set at 10 Å. The electrostatic interactions with a 10 Å cut-off were measured by Particle-Mesh Ewald (35).LINCS algorithm used to constrain the lengths of hydrogen-containing bonds (36). The update frequency of the neighbor list was 10 ps. Finally, the analyses were performed via Gromacs 5 analysis tools.
Experimental section
Production of the TEV protease mutants
The N23F point mutation was introduced into a TEV expression system -pKR793 plasmid (21) as WT-TEV protease sequence- by Biomatik (Canada) and was expressed in E. coli BL21DE3. Briefly, 1 L of the culture was induced by isopropyl b-D- thiogalactopyranoside (0.5 mM) for 3 to 4 h at 30 °C. The cells were then lysed using sonication. Subsequently, the protein was loaded on a 1 mL of Ni-NTA agarose (GE Healthcare, USA), pre-equilibrated with 25 mM sodium phosphate, pH 8.0, 10% (v/v) glycerol, NaCl (0.1 M), and 25 mM imidazole. Then, the protein was eluted with 25 mM sodium phosphate (pH 8.0), 10% (v/v) glycerol, NaCl (0.1 M), 250 mM imidazole and the purity of the enzyme was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). For each protein, all the eluted fractions (which contain TEV protease) were pooled and then stored at -80 °C after a flash-freezing in liquid nitrogen.
Solubility assay
The solubility of TEV protease was assayed by concentrating the purified protein using a centrifugal approach. An initial concentration (0.5 mg/mL) was prepared from purified mutant and WT-TEV protease and pour 5 mL of each to ultra centrifugal filter units (Thermo Fisher Scientific ™, UK) with 10 kDa cut-off. The concentrating process was started with a centrifugal approach at 7000 rpm and at 4 °C. Sampling process performed at regular 1-h intervals without an obvious increase in the initial protein volume. The concentrations were assayed using the Bradford method with bovine serum albumin as the standard. The maximum concentration obtained when equilibrium was reached, which is the plateau in absorbance. The assay was performed in triplicate and reported as a mean ± standard error of mean (SEM).
Activity assay
Enzyme activity was measured using the TEV protease activity assay kit (Biovision, California, USA) following the manufacturer protocol. Briefly, the procedure measures the cleavage of a synthetic fluorescein-based peptide substrate by TEV protease, which is releasing quantified fluorescein, and measuring the absorbance by a fluorescence microplate reader (excitation/emission at 490/560 nm). The fluorescence (excitation/emission at 490/560 nm) was measured for 10 min at 34 °C and at 30 sec intervals. Two time points (T1 and T2) were selected in which their corresponding referenced fluorescence units (RFU) were in a linear range and were used to calculate the ΔRFU and ΔT. Moreover, the standard curve was plot using the 5-FAM standards, and was used to calculate the corresponding RFU of the sampling points and the activity of TEV protease using the equation below:
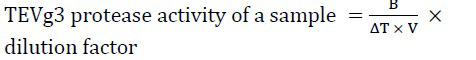
where, B is the calculated TEV protease amount (ng) using the 5-FAM standard curve, V is the initial volume of the sample added into the reaction well (mL), and ΔT is the reaction time (min).
RESULTS
Theoretical results
Homology modeling
The 3D structures of the WT and the mutants were predicted using Modeller 9.13 based on the 1LVM 3D structure as a template. The Ramachandran plots of the suggested models showed a good quality and over 98% residues 9-y angles were in the most favored and additional allowed regions (data not shown). The verified 3D profile revealed that for all models, 89.96% of the residues have an averaged 3D-1D score of ≥ 0.2 (data not shown). The compatibility score is corresponding to acceptable side-chain environments and shows that the models have overall self-consistency in terms of sequence- structure compatibility.
MD Simulations
MD simulations were performed to study the dynamic behavior of a protein structure. The backbone root mean square deviation (RMSD) values of the WT and mutant TEV protease during MD simulation at 300 K are shown in Fig. 2A.
Fig. 2.

(A) The backbone RMSD of wild type and mutations at 300 K during 20 ns molecular dynamic simulation. (B) The RMSF of residues of the WT and mutated TEV protease at 300 K during the last 10 ns molecular dynamic simulation. Active sites residues (HIS46, ASP81, and CYS151) are located in 54, 89 and 159 positions duo to exist an additional N-terminal His-tag. RMSD, Root mean square deviation; WT, wild type; TEV, tobacco etch virus.
The structures reached a stable state after 10 ns with a standard deviation of less than 0.4 nm.
Root mean square fluctuations (RMSFs) address the backbone flexibility and mobility of a structure. Figure 2B depicts the RMSF of the Ca of both WT and mutations, indicating that the fluctuations were almost similar for all the systems and the RMSFs of Ca of the residues of the active site (HIS46, ASP81, and CYS151) were small, and in the same range.
Table 2 shows the average value of various properties of the WT and the selected mutants of TEV protease during the last 10 ns of MD simulation.
Table 2.
The average of various properties during the last 10 ns of simulation and differences between the wild type and the mutated tobacco etch virus protease at 300 K.
| Proteins | Radius of gyration (nm) |
|---|---|
| N23F | 1.70 ± 0.008 |
| N23L | 1.69 ± 0.002 |
| Q74L | 1.68 ± 0.007 |
| Q74V | 1.68 ± 0.009 |
| Q74I | 1.67 ± 0.009 |
| WT | 1.66 ± 0.007 |
| Protein-solvent hydrogen bonds | |
| N23F | 452.3 ± 13.3 |
| N23L | 445.2 ± 14.6 |
| Q74V | 443.0 ± 13.2 |
| Q74L | 434.9 ± 12.9 |
| Q74I | 433.4 ± 13.6 |
| WT | 429.2 ± 13.7 |
| Intra-molecular hydrogen bonds | |
| Q74L | 171.0 ± 7.0 |
| N23L | 166.8 ± 7.4 |
| Q74I | 166.7 ± 7.4 |
| Q74V | 163.8 ± 6.8 |
| N23F | 162.8 ± 7.5 |
| WT | 171.6 ± 7.6 |
| Total SASA (nm2) | |
| Q74L | 118.8 ± 1.88 |
| Q74V | 118.5 ± 2.28 |
| N23F | 118.2 ± 2.45 |
| N23L | 118.1 ± 3.64 |
| Q74I | 115.0 ± 2.15 |
| WT | 116.7 ± 2.12 |
| Active site SASA (nm2 ) | |
| Q74V | 4.95 ± 0.10 |
| Q74L | 5.00 ± 0.17 |
| N23L | 5.52 ± 0.13 |
| N23F | 5.55 ± 0.22 |
| Q74I | 5.69 ± 0.15 |
| WT | 4.96 ± 0.17 |
The radius of gyration (Rg) provides a perspective on the size of the protein. The Rg values for all mutants have increased slightly compared to the WT and the TEV protease structures fairly unfold due to the mutations, probably because these mutations are in core positions of the protein. Similar results were obtained for total and active site solvent accessible surface area (SASA) and intramolecular hydrogen bonds in both the WT and the mutants, confirming the observed Rg values. On the other hands, compared to the WT-TEV protease the average number and the pattern of hydrogen bonds in N23F mutant that is formed during MD simulation (Fig. 3) between the proteins and the solvent was significantly increased, indicating a higher solubility of N23F than that of the native protein. The secondary structural elements ofthe protein were calculated by the “do_dssp” function. Secondary structures of all models were almost similar because the mutations were not in a critical position for the secondary structure formation.
Fig. 3.
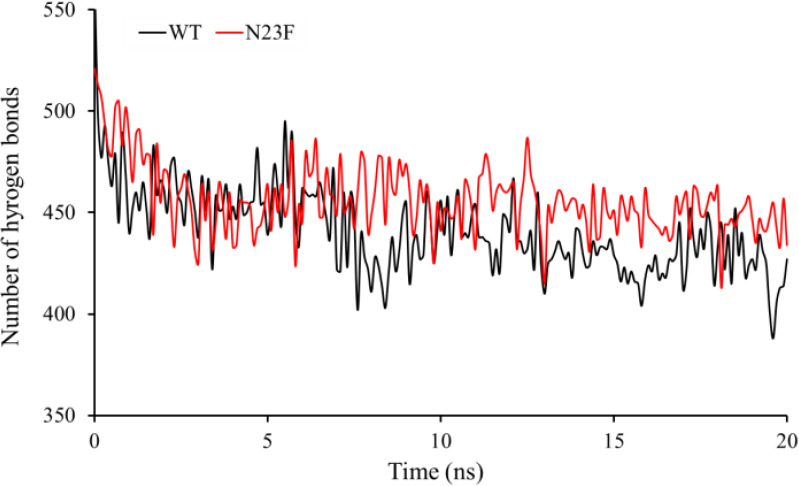
The pattern of the hydrogen bonds during 20 ns molecular dynamics simulation for wild type and N23F mutant.
Experimental results
Solubility assay
According to the MD simulations results, the N23F mutation was a good choice to enhance the solubility, therefore it was introduced into a soluble TEV expression system. To calculate the solubility plateau, both purified WT and N23F proteins were subjected to the concentration process starting from an initial concentration of 0.5 mg/mL. For both proteins, the sampling was performed at the same interval times in order to determine their concentration due time. As shown in Fig. 4, the maximum concentration (a plateau in absorbance readings) for WT and N23F were 1.5 and 2.2 mg/mL, respectively; in other words, the solubility of the N23F mutant was increased 1.5 folds.
Fig. 4.
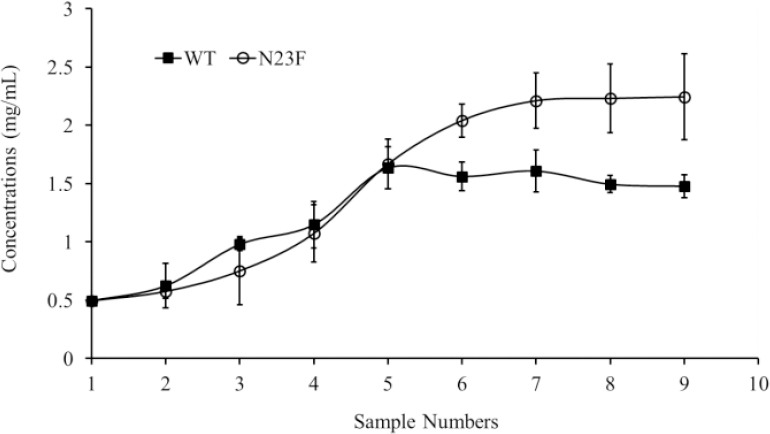
The Solubility curves for wild type and N23F mutant. The maximum concentration was obtained from the observed plateau in a concentration test.
Activity Assay
The activity assays were performed following the kit protocol. The kinetics curves were obtained by measuring the enzyme activity for up to 10 min for the same concentrations (0.5 mg/mL) of the samples (Fig. 5). As shown in the previous step, although the solubility of the N23F mutant was increased, the activity of the mutant was decreased compared to the WT.
Fig. 5.
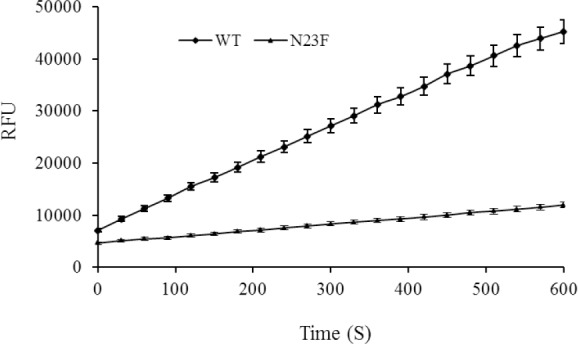
The kinetics progress curves for wild type and mutant N23F. Calculated RFUs (for 10 min with 30 seconds intervals) for the same amounts of samples were presented. RFU, Referenced fluorescence units.
DISCUSSION
TEV protease is an enzyme with stringent specificity that is very useful for the cleavage of fusion proteins; however, its low solubility is a major limitation. To overcome this drawback, we followed a rational multi-stage process to improve the solubility of the WT-TEV protease. At the first stage, all the possible mutations were predicted by PoPMuSiC and Eris server, in which five mutations were screened for further studies in terms of the obtained scores from both servers. However, there are some limitations to such servers. Being unable to change the protein structure in a dynamics mode is one of the most important issues, in other words, the applied mutations are evaluated in a rigid mode (22), but it cannot be described in a live molecules situation. Therefore, such servers may only provide a clue. Therefore, we only used the obtained data from these servers as a clue and subjected the obtained structures into MD simulations.
In the next step, 6 separate 20 ns MD simulations executed at 300 K to compare the dynamic behavior of the WT and the mutant structures. The backbone RMSD values of the structures during the MD simulation showed that the structures reach a stable state. RMSFs of the Ca WT and mutations in 300 K were almost similar for all systems and the RMSFs of the Ca of active sites residues (HIS46, ASP81, and CYS151) were small, indicating that probably their activity was very similar to the WT.
On the other hand, in the case of N23F mutation, the average number and the pattern of hydrogen bonds during MD simulation between protein and the solvent increased significantly (compared to the WT-TEV protease), indicating that the solubility of N23F is higher than that of the native protein.
The average values of other properties namely Rg, SASA, intramolecular hydrogen bonds changed slightly. Although the changes did not significantly influence the solubility, they were likely to change the activity. Therefore, by considering the analysis of various factors such as structural and solubility properties, the N23F mutant was selected for in vitro studies.
Generally, protein solubility is a complex phenomenon that depends on various parameters such as amino acid sequences and environmental conditions namely pH, temperature, and salt concentration. In constant environmental conditions, there are two general approaches to alter the stability and solubility of a protein, changing the surface residues in order to change the surface interaction, or changing the core residues in order to change the hydrophobic core of the protein. The positions of the suggested mutations (N23 and Q74) are in the core region of the TEV protease in which the polar residues replaced with nonpolar residues. Therefore, it is expected that such mutations might improve the hydrophobic core packing of the protein and the stability of TEV protease (37). On the other hand, according to the MD results, in N23F mutation, the number of hydrogen bonds between the protein and the solvent significantly increased compared to the WT-TEV protease. Subsequently, the experimental solubility assay of both purified WT and N23F TEV protease proteins confirmed that, compared to the WT, the solubility of the N23F mutant increased by 1.5 times while the activity was decreased somewhat. It can be argued that the used servers were not designed to predict the changes in the activity. Also, It is due to the fact that the activity of an enzyme cannot be addressed via MD simulation since enzyme activity is a chemical process and MD simulation does not directly consider chemical reactions because we omit electrons transition in enzyme’s active site in MD simulation. Thus, MD simulation can only provide indirect insight into the enzyme activity for example by calculation of RMSF or SASA of active site residues of both mutants and WT-TEV protease. According to the MD simulation results, a slight change in activity was expectable for all of the mutants.
Cabrita et al. reported a double mutant L56V/S135G resulting in an increase in the solubility to almost 40 times compared to the WT. The in vitro catalytic activity was also improved compared to the WT-TEV protease (22). Moreover, Berg et al. reported the T17S/N68D/I77V mutant generated by a directed evolution with a 5 folds increase in its production level, but they did not reported the in vitro solubility and activity of the purified protein. In fact, in this work, a fluorescence- based in vivo solubility screening was carried out by cloning of the libraries into a plasmid encoding a C-terminal GFP fusion (23). Fang et al. investigated the solubility of two previous TEV protease mutants: a double mutant TEV protease 2M (L56V/S135G), a triple mutant TEV protease 3M (T17S/N68D/I77V), and a combination of the above five mutations as a quintuple mutant TEVp5M (T17S/L56V/N68D/I77V/S135G). The results showed that the maximum achievable concentration for the WT, TEVp2M, TEVp3M, and TEVp5M was 17, 22, 50, and 80 mg/mL, respectively. They measured the level of soluble TEV protease in the bacterial lysate supernatants using an indirect approach in terms of the S-tag fusion system. The observed contradiction in the results of the studies mentioned above might be due to differences in measurement methods and sample types. In our study, we used the purified enzyme sample for this test and the in vitro solubility assayed directly by a simple concentration method. Wang et al. studied the mutations S219V,L56V/S135G, and T17S/N68D/I77V from a molecular perspective with molecular dynamics simulations. The reported SASA of the three mutants during MD simulation was less than that of the WT while it was expected to be higher because the mutations had a higher solubility than the WT. They also concluded that the stability of the mutants is higher than the WT due to the compacting of the hydrophobic cores in mutants (38).
CONCLUSION
The major aim of this study was to investigate the effects of some suggested mutations by online tools, and using molecular dynamics simulation to improve the solubility of TEV protease in vitro. N23F mutation was the best according to computational and experimental analyzes which resulted in a 150% increase in the solubility compared to that of the WT. Although our main objective, i.e. increasing the solubility of the TEV enzyme was achieved, the in vitro studies revealed that the enzyme activity was reduced due to limitation of the servers and MD studies to predict changes in the activity. For future studies, we propose further in silico studies (for example using MD-docking investigations) to predict any changes in the enzyme activity as well.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest for this study.
AUTHORS’ CONTRIBUTION
All authors contributed equally in this work.
ACKNOWLEDGMENTS
This work was financially supported by Isfahan University of Medical Sciences, Isfahan, I.R. Iran under the Grant No. 394944. We thank Fatemeh Moazen, the technician of Molecular Biology Laboratory, Department of Pharmaceutical Sciences, Isfahan University of Medical Sciences, Iran, for her technical assistance in this study
REFERENCES
- 1.Nunn CM, Jeeves M, Cliff MJ, Urquhart GT, George RR, Chao LH, et al. Crystal structure of tobacco etch virus protease shows the protein C terminus bound within the active site. J Mol Biol. 2005;350(1):145–155. doi: 10.1016/j.jmb.2005.04.013. DOI:10.1016/j.jmb.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Miladi B, Bouallagui H, Dridi C, El Marjou A, Boeuf G, Di Martino P, et al. A new tagged-TEV protease: construction, optimisation of production, purification and test activity. Protein Expr Purif. 2011;75(1):75–82. doi: 10.1016/j.pep.2010.08.012. DOI: 10.1016/j.pep.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 3.Yi L, Gebhard MC, Li Q, Taft JM, Georgiou G, Iverson BL. Engineering of TEV protease variants by yeast ER sequestration screening (YESS) of combinatorial libraries. Proc Natl Acad Sci U S A. 2013;110(18):7229–7234. doi: 10.1073/pnas.1215994110. DOI: 10.1073/pnas.1215994110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uhlmann F, Wernic D, Poupart MA, Koonin EV, Nasmyth K. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell. 2000;103(3):375–386. doi: 10.1016/s0092-8674(00)00130-6. DOI: 10.1016/s0092-8674(00)00130-6. [DOI] [PubMed] [Google Scholar]
- 5.Xiao F, Widlak P, Garrard WT. Engineered apoptotic nucleases for chromatin research. Nucleic Acids Res. 2007;35(13):1–7. doi: 10.1093/nar/gkm486. DOI: 10.1093/nar/gkm486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yogev O, Karniely S, Pines O. Translation-coupled translocation of yeast fumarase into mitochondria in vivo. J Biol Chem. 2007;282(40):29222–29229. doi: 10.1074/jbc.M704201200. DOI: 10.1074/jbc.M704201200. [DOI] [PubMed] [Google Scholar]
- 7.Ghiaci P, Norbeck J, Larsson C. 2-Butanol and butanone production in Saccharomyces cerevisiae through combination of a B12 dependent dehydratase and a secondary alcohol dehydrogenase using a TEV- based expression system. PLoS One. 2014;9(7):1–7. doi: 10.1371/journal.pone.0102774. DOI:10.1371/journal.pone.0102774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wehr MC, Rossner MJ. Split protein biosensor assays in molecular pharmacological studies. Drug Discov Today. 2016;21(3):415–429. doi: 10.1016/j.drudis.2015.11.004. DOI: 10.1016/j.drudis.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Cesaratto F, Burrone OR, Petris G. Tobacco etch virus protease: a shortcut across biotechnologies. J Biotechnol. 2016;231:239–249. doi: 10.1016/j.jbiotec.2016.06.012. DOI: 10.1016/j.jbiotec.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Kapust RB, Tozser J, Copeland TD, Waugh DS. The P1’ specificity of tobacco etch virus protease. Biochem Biophys Res Commun. 2002;294(5):949–955. doi: 10.1016/S0006-291X(02)00574-0. DOI: 10.1016/S0006-291X(02)00574-0. [DOI] [PubMed] [Google Scholar]
- 11.Phan J, Zdanov A, Evdokimov AG, Tropea JE, Peters HK, Kapust RB, et al. Structural basis for the substrate specificity of tobacco etch virus protease. J Biol Chem. 2002;277(52):50564–50572. doi: 10.1074/jbc.M207224200. DOI: 10.1074/jbc.M207224200. [DOI] [PubMed] [Google Scholar]
- 12.Shih YP, Wu HC, Hu SM, Wang TF, Wang AH. Self- cleavage of fusion protein in vivo using TEV protease to yield native protein. Protein Sci. 2005;14(4):936–941. doi: 10.1110/ps.041129605. DOI: 10.1110/ps.041129605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pauli A, Althoff F, Oliveira RA, Heidmann S, Schuldiner O, Lehner CF, et al. Cell-type-specific TEV protease cleavage reveals cohesin functions in Drosophila neurons. Dev Cell. 2008;14(2):239–251. doi: 10.1016/j.devcel.2007.12.009. DOI: 10.1016/j.devcel.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cesaratto F, Lopez-Requena A, Burrone OR, Petris G. Engineered tobacco etch virus (TEV) protease active in the secretory pathway of mammalian cells. J Biotechnol. 2015;212:159–166. doi: 10.1016/j.jbiotec.2015.08.026. DOI: 10.1016/j.jbiotec.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 15.Wehr MC, Laage R, Bolz U, Fischer TM, Grunewald S, Scheek S, et al. Monitoring regulated protein- protein interactions using split TEV. Nat Methods. 2006;3(12):985–993. doi: 10.1038/nmeth967. DOI: 10. 1038/nmeth967. [DOI] [PubMed] [Google Scholar]
- 16.Sun C, Liang J, Shi R, Gao X, Zhang R, Hong F, et al. Tobacco etch virus protease retains its activity in various buffers and in the presence of diverse additives. Protein Expr Purif. 2012;82(1):226–231. doi: 10.1016/j.pep.2012.01.005. DOI: 10.1016/j.pep.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, et al. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng Des Sel. 2001;14(12):993–1000. doi: 10.1093/protein/14.12.993. DOI: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 18.Schein CH. Solubility as a function of protein structure and solvent components. Biotechnology (N Y) 1990;8(4):308–317. doi: 10.1038/nbt0490-308. DOI: 10.1038/nbt0490-308. [DOI] [PubMed] [Google Scholar]
- 19.Blommel PG, Fox BG. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr Purif. 2007;55(1):53–68. doi: 10.1016/j.pep.2007.04.013. DOI: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang L, Jia KZ, Tang YL, Ma DY, Yu M, Hua ZC. An improved strategy for high-level production of TEV protease in Escherichia coli and its purification and characterization. Protein Expr Purif. 2007;51(1):102–109. doi: 10.1016/j.pep.2006.07.003. DOI: 10.1016/j.pep.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 21.Kapust RB, Waugh DS. Escherichia coli maltose- binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8(8):1668–1674. doi: 10.1110/ps.8.8.1668. DOI: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabrita LD, Gilis D, Robertson AL, Dehouck Y, Rooman M, Bottomley SP. Enhancing the stability and solubility of TEV protease using in silico design. Protein Sci. 2007;16(11):2360–2367. doi: 10.1110/ps.072822507. DOI: 10.1110/ps.072822507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Den Berg S, Lofdahl PA, Hard T, Berglund H. Improved solubility of TEV protease by directed evolution. J Biotechnol. 2006;121(3):291–298. doi: 10.1016/j.jbiotec.2005.08.006. DOI: 10.1016/j.jbiotec.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;9(9):646–652. doi: 10.1038/nsb0902-646. DOI: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 25.Sotomayor M, Schulten K. Single-molecule experiments in vitro and in silico. Science. 2007;316(5828):1144–1148. doi: 10.1126/science.1137591. DOI: 10.1126/science.1137591. [DOI] [PubMed] [Google Scholar]
- 26.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. DOI: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 27.Banisharif-Dehkordi F, Mobini-Dehkordi M, Shakhsi-Niaei M, Mahnam K. Design and molecular dynamic simulation of a new double-epitope tolerogenic protein as a potential vaccine for multiple sclerosis disease. Res Pharm Sci. 2019;14(1):20–26. doi: 10.4103/1735-5362.251849. DOI: 10.4103/1735-5362.251849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saunders HM, Gilis D, Rooman M, Dehouck Y, Robertson AL, Bottomley SP. Flanking domain stability modulates the aggregation kinetics of a polyglutamine disease protein. Protein Sci. 2011;20(10):1675–1681. doi: 10.1002/pro.698. DOI: 10.1002/pro.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laskowski R, MacArthur M, Thornton J. Procheck-a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. DOI: 10.1107/S0021889892009944. [Google Scholar]
- 30.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356(6364):83–85. doi: 10.1038/356083a0. DOI: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 31.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. DOI: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 32.Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. DOI: 10.1016/j.softx.2015.06.001. [Google Scholar]
- 33.Evans DJ, Holian L. The Nose-Hoover thermostat. J Chem Phys. 1985;83(8):4069–4074. DOI: 10.1063/1.449071. [Google Scholar]
- 34.Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 1981;52(12):7182–7190. DOI: 10.1063/1.328693. [Google Scholar]
- 35.Darden T, York D, Pedersen L. Particle mesh Ewald: An N-log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. DOI: 10.1063/1.464397. [Google Scholar]
- 36.Hess B, Bekker H, Berendsen HJ, Fraaije JG. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. DOI: 10.1002/(SICI)1096-987X(199709)18. [Google Scholar]
- 37.Munson M, Balasubramanian S, Fleming KG, Nagi AD, O’Brien R, Sturtevant JM, et al. What makes a protein a protein hydrophobic core designs that specify stability and structural properties. Protein Sci. 1996;5(8):1584–1593. doi: 10.1002/pro.5560050813. DOI: 101002/pro5560050813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Zhu GF, Ren SY, Han YG, Luo Y, Du LF. Insight into the structural stability of WT and mutants o f the tobacco etch virus protease with molecular dynamics simulations. J Mol Model. 2013;19(11):4865–4875. doi: 10.1007/s00894-013-1930-9. DOI: 10.1007/s00894-013-1930-9. [DOI] [PubMed] [Google Scholar]