

Abstract
Background and purpose:
The study was launched to use zinc finger nuclease (ZFN) technology to disrupt the cholera toxin gene (ctxA) for inhibiting CT toxin production in Vibrio cholera (V. cholera).
Experimental approach:
An engineered ZFN was designed to target the catalytic site of the ctxA gene. The coding sequence of ZFN was cloned to pKD46, pTZ57R T/A vector, and E2-crimson plasmid and transformed to Escherichia coli (E. coli) Top10 and V. cholera. The efficiency of ZFN was evaluated by colony counting.
Findings/Results:
No expression was observed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blotting in transformed E. coli. The ctxA gene sequencing did not show any mutation. Polymerase chain reaction on pKD46-ZFN plasmid had negative results. Transformation of E. coli Top10 with T/A vectors containing whole ZFN sequence led to 7 colonies all of which contained bacteria with self-ligated vector. Transformation with left array ZFN led to 24 colonies of which 6 contained bacteria with self-ligated vector and 18 of them contained bacteria with vector/left array. Transformation of V. cholera with E2-crimson vectors containing whole ZFN did not produce any colonies. Transformation with left array vectors led to 17 colonies containing bacteria with vector/left array. Left array protein band was captured using western blot assay.
Conclusions and implications:
ZFN might have off target on bacterial genome causing lethal double-strand DNA break due to lack of non-homologous end joining (NHEJ) mechanism. It is recommended to develop ZFNs against bacterial genes, engineered packaging host with NHEJ repair system is essential.
Keywords: ctxA Gene, Gene editing tools, Vibrio cholerae, Zinc finger nuclease
INTRODUCTION
Cholera, caused by Vibrio cholera (V. choerae) O1 and O139 strains, is one of the most prevalent infectious diseases in communities with poor water and sanitation infrastructure (1). The main implication of epidemic cholera is severe dehydration. Annually, 1.4 to 4.3 million individuals worldwide are affected, with an annual mortality of 28,000-143,000 (2). Cholera toxin (CT), an important pathogenesis factor for V.choerae is produced through the integration of CTX$ϕ phage into its genome (3). In 1886, Koch proposed that the symptoms caused by V. cholera could be due to some poisons produced by this organism (4). In 1959, S. N. De showed that cell-free extracts from V. cholerae cultures induced fluid accumulation in rabbits when injected into ligated small intestinal loops. This test confirmed Koch’s postulate (5).
Later, evidences suggested the presence of a toxic protein product in V. cholerae cell-free supernatants (6). After the entrance of V. cholerae into small intestine, the ToxR regulon activates the expression of virulence gene through a regulatory cascade. CT and the toxin- co-regulated pilus (TCP) are the most important virulence genes stimulated by the ToxR regulon. CT enterotoxin is responsible for acute diarrhea and TCP is a type IV pilus essential for colonization (7,8). Disruption of the CTXɸ phage could be an important target to eliminate toxigenesis of V. cholera and ultimately decrease pathogenesis of bacteria (9).
CT is an ADP-ribosylating toxin and belongs to the large family of A-B toxins that contains an AB5 subunit structure. The B subunit (CTB) forms a pentamer that binds to the pentasaccharide part of GM1 gangliosides on the cell surface, and the A subunit (CTA) is cleaved by host proteases into A2 subunit that attaches enzymatically to active A1 subunit using a disulfide bond. A1 subunit releases into cytosol and catalyzes the transfer of an ADP- ribosyl moiety to α subunit of Gs protein, that leads to adenylate cyclase stimulation and triggers cAMP-dependent intestinal fluid hypersecretion via ion channels and transporters (10,11).
Genome editing technologies have become a promising and novel method to develop new therapeutic strategies to fight infectious and monogenic diseases (12,13,14,15,16). These technologies such as zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALENs), clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 9 (Cas9) can target and modify the genome of an organism (17). ZFNs are the first class of these nucleases, which were discovered in 1996 and used for the first time in 2002 for genetic engineering of drosophila and mammalian cells (18,19). ZFNs are composed of two domains: a DNA binding site that is a tandem array of Cys2-His2 zinc- finger and usually contains 3-6 domains that each binds to 3-bp of DNA sequence (19,20) and a cleavage domain of bacterial restriction enzyme FokI which must dimerize in order to make double strand breaks (21) and subsequently stimulate DNA repair pathways including homologous recombination or non- homologous end joining (NHEJ) (22). This research was conducted to investigate the ability of an engineered ZFN to create disruption in ctxA gene and consequently to inhibit CT toxin production in V. cholerae.
MATERIALS AND METHODS
Design of ZFN construct
The protein sequences coding for the left and right zinc finger protein arms were designed by zinc finger tools (https://www.scripps.edu/barbas/zfdesign/zfdesignhome.php). This ZFN targets the catalytic site of ctxA and its sequence was obtained from uniprot identifier.
The left and right ZFP arrays consist of three fingers that bind to two sites on ctxA gene. The designed protein sequences were then converted to nucleic acid sequence using the EMBOSS Backtranseq (http://www.ebi.ac.uk/Tools/st/emboss_backtranseq/). The heterodimeric variants of Fok I were obtained from EENDB (http://eendb.zfgenetics.org/). The left and right ZFN arrays were linked to the Fok I using linker peptide coding sequences. The order of gene construct is nucleic acid sequences of kanamycin resistance promoter- ATG-6xHIS tag-GLY SER GLY-ZF left array -FOK1 KKR-TGA-chloramphenicol resistance promoter - ATG-HA tag- GLY SER GLY- ZF right array - FOK1 ELD-TGA. GeneCust Company synthesized the pKD-ZFN sequence which was subcloned in pKD46 plasmid between SacI and Ncol restriction sites. It was transformed in chemically competent Escherichia coli (E. coli) Top10F’ with CaCl2 by heat shock protocol (23). The transformed bacteria were cultured on lysogeny broth (LB) agar medium containing 100 μg/mL ampicillin. One colony was cultured overnight and the pKD-ZFN plasmid was extracted by SolGent plasmid extraction kit (SolGent, Korea).
Transformation
V. cholerae ATCC 16961 was purchased from Iranian Research Organization for Science and Technology. The bacteria were cultured at 37 °C in brain heart infusion (BHI) agar medium containing 1% NaCl and then transferred to LB broth for 24 h at 37 °C. The competent V. cholera was prepared using CaCl2 and transformed with pKD-ZFN through heat shock treatment, then spread on LB agar medium containing ampicillin (100 μg/mL) and incubated for 24 h at 37 °C.
Evaluation of ZFN expression using SDS- PAGE and western blot
To evaluate the ZFN expression, a single positive clone of E. coli Top10 containing pKD-ZFN, a clone of E. coli TOP10 containing pKD46 and E. coli Top10 without plasmid were inoculated in LB medium separately and were shaken overnight at 30°C and 200 rpm to achieve OD600 of 0.6-0.8. Cell lysate preparation was performed using lysis buffer (containing 8 M urea, 0.01 M tris-base, 0.1 M NaH2Po4, pH:8, 0.1 g cell/0.4 mL buffer). In order to detect the left and right ZFN array expression, western blot was performed using anti-His antibody (Sigma-Aldrich, MI, USA) and anti-HA antibody (Cell Signaling Technology, USA), respectively (24).
Evaluation of mutation in ctxA gene
The ZFN transformed V. cholera was cultured in AKI medium. After 48 h, DNA was extracted and ctxA gene was amplified using forward primer: 5´ATGGTG- AAATTAATTTTTGATGTAC3´ and reverse primer: 5 ‘ATCACTTTCGTCTACTCCGTTA- C3’. Polymerase chain reaction (PCR) products were sequenced.
Evaluation of ZFN construct
The authenticity of gene construct was checked based on the presence of negative results in western blot. In order to evaluate ZFN construct, a single positive clone was inoculated in LB broth medium containing ampicillin, and plasmid was extracted. The extracted plasmid was amplified using following primers: PKD46 5´ATCACTTT- CGTCTACTCCGTTAC3´ (forward) and PKD46 5´ATGGTGAAATTAATTTTTGAT- GTAC3´ (reverse) and primers that amplified a segment of ampicillin resistant gene including Amp 5 ‘CAGTGCTGCAATGATA-CCG3´ (forward) and Amp 5´AATTAATA-GACTGG- ATGGAGGC3´ (reverse).
Cloning of whole and left ZFN arrays
The nucleic acid sequences of whole ZFN construct (kanamycin resistance promoter- ATG-HIS tag - GLY SER GLY- ZF left array - FOK1 KKR-TGA - chloramphenicol resistance promoter - ATG-HA tag -GLY SER GLY- ZF right array - FOK1 ELD-TGA) were synthesized by GeneCust Company. It was received as a PCR product and was amplified by Pfu enzyme
The whole ZFN sequence (case group) was amplified by ZFN 5´ATCCATGGT- TTGTACATCAAAAATTAA3´ (forward) and ZFN 5´TAGAGCTCTTTCCGGAA TCAG- AAG3´ (reverse). The left array of ZFN (control group) was amplified by left ZFN 5 ‘TTCCATGGGCAGACAGTTTTATTGTTC ATGATG3´ (forward) and left ZFN 5 ‘TAGAGCTCTTTCCGGAATCAGTAAG3´ (reverse) primers from the main gene construct. PCR products were treated by Taq DNA polymerase to add an A nucleotide in 3´ends and cloned to T/A vector, then E.coli Top10 was transformed with it. In addition, PCR products were cloned to E2-crimson plasmid in NcoI and SacI sites and were used to transform V. cholera. Transformed bacteria were cultured on LB medium containing 100 μg/mL ampicillin and incubated 24 h at 37 °C and resulted colonies were counted.
Evaluation of left array ZFN expression using western blot
A single positive clone of E. coli Top10 containing left array of ZFN was inoculated in LB medium containing ampicillin (100 μg/mL) and was shaken overnight at 35 °C and 200 rpm to achieve OD600 of 0.6-0.8. The cell lysate was prepared as mentioned above and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot was performed using anti-His antibody (Sigma- Aldrich, MI, USA).
RESULTS
Transformation
The transformation of pKD-ZFN into E. coli and V. cholera resulted several colonies.
Evaluation of ZFN expression using SDS- PAGE and western blot
SDS-PAGE protein patterns of V. cholera containing pKD-ZFN, V. cholera containing pKD46, and V. cholera without plasmid did not show detectable changes (Fig. 1) and specific bands (64 kDa) related to left and right ZFN arrays were not captured on western blot (Fig. 2).
Fig. 1.

SDS-PAGE on lysates of Vibrio cholera with and without pKD-ZFN. 1, Protein ladder of Thermo scientific lot No 00433557; 2, lysate of Vibrio cholera without plasmid; 3, lysate of Vibrio cholera contains pKD46; and 4, lysate of Vibrio cholera contains pKD-ZFN. There is not any change in protein bands. The expected band is about 32 and 64 kDa (dimer). SDS-PAGE, Sodium dodecyl sulfate-polyacrylamide gel electrophoresis; ZFN, zinc finger nuclease.
Fig. 2.

Western blot for ZFN protein on Vibria cholera lysate that transormed with pKD-ZFN. 1, CRP protein as a positive control; 2, lysate of Vibria cholera that transormed with pKD-ZFN, there was no band (expected bands were 32 and 64 kDa); 3, pre-stained protein ladder of fermentas. ZFN, zinc finger nuclease; CRP, C-reactive protein.
Evaluation of mutation in ctxA gene
The PCR product for ctxA gene was sequenced and aligned with ctxA gene of a normal bacteria and no mutation was observed (Fig. 3).
Fig. 3.

(A) PCR of ctxA gene on Vibrio cholera genomic DNA. 1, 100 bp DNA ladder of fermentas; 2, negative control (distilled water); 3, PCR of ctxA (365 bp), and (B) sequencing result of ctxA gene. PCR, Polymerase chain reaction; ctxA, cholera toxin A.
Evaluation of ZFN construct
The extracted plasmid was checked by PCR that amplified ZFN and ampicillin resistant sequence (Fig. 4). The PCR of ZFN using PKD46 primer did not result in amplification product, but ampicillin resistant gene showed a 130 bp band as expected (Fig. 4).
Fig. 4.

Electrophoresis of PCR products on plasmid which extracted from Escherichia coli containing pKD-ZFN. 1, 100 bp DNA ladder of fermentas; 2, negative control; 3, PCR with forward and reverse primer of PKD46 and PKD46; and 4, PCR on ampicillin resistant gene. PCR, Polymerase chain reaction; ZFN, zinc finger nuclease.
Electrophoresis of PCR product of the whole ZFN and left array showed 2226 and 1300 bp bands, respectively (Fig. 5).
Fig. 5.
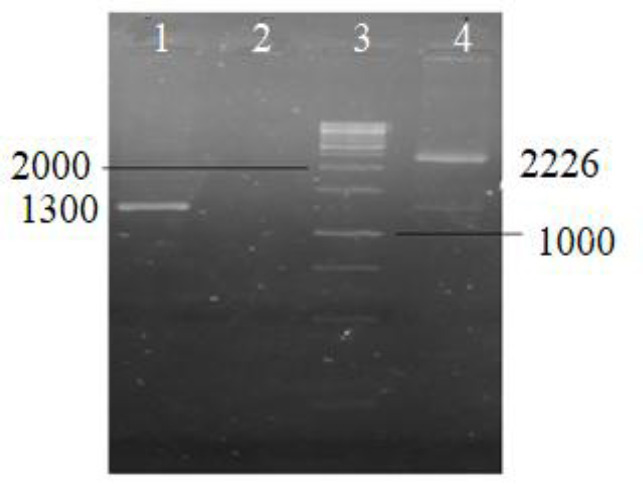
PCR products of whole ZFN construct and left ZFN array. 1, PCR of left ZFN (1300 bp); 2, negative control; 3, 1kb DNA ladder of fermentas; and 4, PCR of whole ZFN construct (2226 bp). PCR, Polymerase chain reaction; ZFN, zinc finger nuclease.
The results of cloning
The number of E. coli Top10 colonies containing whole ZFN in the case group (seven self-ligated colonies) was significantly lower compared to the E. coli Top10 colonies (24 colonies of which 6 were self-ligated and 18 contained left array of the ZFN) in the control group in LB medium containing ampicillin (100 μg/mL). This was also repeated on LB medium without antibiotic selection which showed a considerable decrease in the number of colonies in case group.
The colony count of experiment on V. cholera that was transformed with E2-crimson plasmid containing ZFN was zero and 17 colonies contained the left array of ZFN.
Colony PCR on E. coli Top10 colonies in the case and control group showed that all colonies in the case group contained self-ligated plasmid and had no ZFN (Fig. 6), whereas many colonies in the control group had plasmid containing left array (Fig. 7). For V. cholera, all 17 colonies contained the plasmid with insert.
Fig. 6.

Colony PCR on Escherichia coli transformed by pKD-ZFN (whole construct). Lines 1-3, and 6-9 are self-ligate because PCR bands are about 190 bp; line 4, negative control and line 5, is 100 bp DNA ladder of fermentas. PCR, Polymerase chain reaction; ZFN, zinc finger nuclease.
Fig. 7.

Agarose gel electrophoresis of PCR on colonies resulted from transformation of Escherichia coli with T/A cloning of left arm of ZFN. Line 1, 1 kb DNA ladder of fermentas; lines 2-6, 8, and 9, colonies with insert left arm of ZFN, and lines 3 and 7, self-ligated colonies. PCR, Polymerase chain reaction; ZFN, zinc finger nuclease.
Evaluation of left array ZFN expression using western blot
A detectable band was seen in western blotting of left array of ZFN in E. coli Top10 at about 32 kDa (Fig. 8).
Fig. 8.

Western blot of left arm of ZFN. 1, Prestained protein ladder; 2, left arm of ZFN (about 32 kDa).
DISCUSSION
Cholera, a severe and potentially life- threatening diarrheal disease with high morbidity and mortality, is a major public health concern (25,26) and its prevention and control has always been a challenge. CT, the most important virulence factor of V. cholera (27), induces the secretion of water and salts, main components of stool, from the epithelial cells. Traditional strategies for the prevention and treatment of infectious diseases are vaccination and antibiotic therapy. ZFN, TALEN, and CRISPR are gene editing tools (GETs) that have been widely used to target pathologic island, toxin, and antibiotic resistant genes (28,29,30,31,32). There are several reports about GET application against eukaryotic organism from yeast to human (33,34).
All GETs have off target and this can cause side effects in targeted cells (34). Usually the engineered E. coli like Top10 and DH5α are used to amplify GET constructs. Eukaryotic expression cassettes of ZFN, TALEN, and CRISPR/Cas9 are under control of eukaryotic promoter and not expressed in prokaryotic host, although prokaryotic ones can be expressed in E. coli and cause double-strand DNA break (DSB) in host genome. This is lethal for E. coli because it does not have NHEJ repair (35). For this reason, two arms of ZFN, TALEN or Cas9, and sgRNA cassette should be prepared and amplified in separate clones before transformation in targeted host. Co- transformation of bacteria with two arms of ZFN, TALEN or Cas9, and sgRNA is lethal for bacteria and if the aim is homologous recombination, donor DNA should be transformed with them (36). Other studies have tried to disrupt the ampicillin resistant gene in E. coli using ZFN (32). In these investigations, after transformation of bacteria, ZFN proteins were not detected by western blot assay (31,32). The target gene was episomal and the probability of off target presence was ignored in the bacterial genome. In our recent study a ZFN construct was designed against a pseudomonas gene that does not have homolog in E. coli. The ZFN construct could not be synthesized in E. coli after several attempts. It may be due to off target presence in E. coli that ZFN created DSB in E. coli genome and led to bacterial cell death. In this study the ZFN construct was designed against CTX toxin and ordered to the Gene Cust Company for synthesis. This gene does not exist in E. coli genome. ZFN array band was not captured in western blot and ZFN expression was not confirmed, a correct construct was not achieved, and the final construct had some deletions.
It was postulated that ZFN has off target on the genome of the bacterium and killed it and the colonies containing mutated plasmid grew, therefore ZFN could not be expressed. To confirm the result, both ZFN arrays (whole ZFN) and left array were amplified using PCR, separately and cloned in the vector. After transformation of the vector containing left array in E. coli and V. cholera, some colonies grew, but the bacteria that were transformed with vector containing both ZFN arrays did not grow and all of the resulted colonies in case group were self-ligation and did not have insert. It seems that ZFN has off target on bacterial genome that causes DSB and is lethal for them due to lack of NHEJ mechanism.
Inducing DSB in prokaryotes to target pathologic and antibiotic resistant microorganism is an attractive field in GETs technology, but there are some pitfalls (29). In the past decade, there was no report about NHEJ in prokaryotes, although, recently NHEJ repair is known in some bacteria (37). Next generation sequencing showed two proteins that run NHEJ in prokaryotes, KU and ligase D, are not present in E. coli K12 and V. cholera (38). This subject confirms and explains the result of ongoing study.
CONCLUSION
In conclusion, targeting pathologic islands, antibiotic resistance, and toxin genes through engineered armed phage is ideal at least for accessible infections like skin and mucosal infections. Engineered armed phages with GETs could target special pathogenic bacteria and MDR strains (39). Dispersing some special armed phage in environment especially in hospital and urban waste water system could regress population of antibiotic resistant gene, multidrug-resistant, and extensively drug- resistant strain. Packaging of these phages needs some special packaging host that is protected against them. Engineered packaging host with NHEJ repair system is essential and it would be implemented by insertion of KU and ligase D in the genome of packaging host.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest for this study.
AUTHORS´ CONTRIBUTION
N. Hosseini contributed in experimental work, interpretation of data, and drafting the paper, H. Khanahmad, B. Kazemi, M. Bandeh
Pour, and B. Nasr Esfahani contributed in study design and managing the project, L. Shariati contributed in ZFN design, and N. Zahedi contributed in experimental work. All authors read and approved the final manuscript
ACKNOWLEDGMENTS
This study was extracted from Ph.D thesis submitted by Nafiseh Hosseini, which was parcially financially (Grant No. 132) supported by School of Advanced Technologiesin Medicine, Shahid Beheshti University of Medical Sciences, Tehran, I.R. Iran. The research was performed at Microbiology and Virology Department of Isfahan University of Medical Sciences, Isfahan, I.R. Iran.
REFERENCES
- 1.Chowdhury FR, Nur Z, Hassan N, Von Seidlein L, Dunachie S. Pandemics, pathogenicity and changing molecular epidemiology of cholera in the era of global warming. Ann Clin Microbiol Antimicrob. 2017;16(10):1–6. doi: 10.1186/s12941-017-0185-1. DOI: 10.1186/s12941-017-0185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adagbada AO, Adesida SA, Nwaokorie FO, Niemogha MT, Coker AO. Cholera epidemiology in Nigeria: an overview. Pan Afr Med J. 2012;12(59):1–12. DOI: 10.11604/pamj.02/07/2012.12.59.1627. [PMC free article] [PubMed] [Google Scholar]
- 3.Faruque SM, Rahman MM, Hasan AM, Nair GB, Mekalanos JJ, Sack DA. Diminished diarrheal response to vibrio cholerae strains carrying the replicative form of the CTX(Phi) genome instead of CTX(Phi) lysogens in adult rabbits. Infect Immun. 2001;69(10):6084–6090. doi: 10.1128/IAI.69.10.6084-6090.2001. DOI: 10.1128/IAI.69.10.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howard-Jones N. Robert Koch and the cholera vibrio: a centenary. Br Med J (Clin Res Ed) 1984;288(6414):379–381. doi: 10.1136/bmj.288.6414.379. DOI: 10.1136/bmj.288.6414.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zomer-van Ommen DD, Pukin AV, Fu O, Quarles Van Ufford LH, Janssens HM, Beekman JM, et al. Functional characterization of cholera toxin inhibitors using human intestinal organoids. J Med Chem. 2016;59(14):6968–6972. doi: 10.1021/acs.jmedchem.6b00770. DOI: 10.1021/acs.jmedchem.6b00770. [DOI] [PubMed] [Google Scholar]
- 6.Coelho A, Andrade JR, Vicente AC, Dirita VJ. Cytotoxic cell vacuolating activity from Vibrio cholerae hemolysin. Infect Immun. 2000;68(3):1700–1705. doi: 10.1128/iai.68.3.1700-1705.2000. DOI: 10.1128/iai.68.3.1700-1705.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Lim MS, Li S, Brock M, Pique ME, Woods VL, Jr, et al. Vibrio cholerae toxin-coregulated pilus structure analyzed by hydrogen/deuterium exchange mass spectrometry. Structure. 2008;16(1):137–148. doi: 10.1016/j.str.2007.10.027. DOI: 10.1016/j.str.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craig L, Taylor RK, Pique ME, Adair BD, Arvai AS, Singh M, et al. Type IV pilin structure and assembly: X-ray and EM analyses of Vibrio cholerae toxin- coregulated pilus and Pseudomonas aeruginosa PAK pilin. Mol Cell. 2003;11(5):1139–1150. doi: 10.1016/s1097-2765(03)00170-9. DOI: 10.1016/s1097-2765(03)00170-9. [DOI] [PubMed] [Google Scholar]
- 9.Yu HJ, Cha DSR, Shin DH, Nair GB, Kim EJ, Kim DW. Design and construction of Vibrio cholerae strains that harbor various CTX prophage arrays. Front Microbiol. 2018;9(339):1–8. doi: 10.3389/fmicb.2018.00339. DOI: 10.3389/fmicb.2018.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dittmer JB, Withey JH. Identification and characterization of the functional toxboxes in the Vibrio cholerae cholera toxin promoter. J Bacteriol. 2012;194(19):5255–5263. doi: 10.1128/JB.00952-12. DOI: 10.1128/JB.00952-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stonehouse EA, Hulbert RR, Nye MB, Skorupski K, Taylor RK. H-NS binding and repression of the ctx promoter in Vibrio cholerae. J Bacteriol. 2011;193(4):979–988. doi: 10.1128/JB.01343-09. DOI: 10.1128/JB.01343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trevisan M, Palu G, Barzon L. Genome editing technologies to fight infectious diseases. Expert Rev Anti Infec Ther. 2017;15(11):1001–1013. doi: 10.1080/14787210.2017.1400379. DOI: 10.1080/14787210.2017.1400379. [DOI] [PubMed] [Google Scholar]
- 13.Modares Sadeghi M, Shariati L, Hejazi Z, Shahbazi M, Tabatabaiefar MA, Khanahmad H. Inducing indel mutation in the SOX6 gene by zinc finger nuclease for gamma reactivation: an approach towards gene therapy of beta thalassemia. J Cell Biochem. 2018;119(3):2512–2519. doi: 10.1002/jcb.26412. DOI: 10.1002/jcb.26412. [DOI] [PubMed] [Google Scholar]
- 14.Shariati L, Khanahmad H, Salehi M, Hejazi Z, Rahimmanesh I, Tabatabaiefar MA, et al. Genetic disruption of the KLF1 geneto overexpress the y- globin gene using the CRISPR/Cas9 system. J Gene Med. 2016;18(10):294–301. doi: 10.1002/jgm.2928. DOI: 10.1002/jgm.2928. [DOI] [PubMed] [Google Scholar]
- 15.Shariati L, Modarressi MH, Tabatabaiefar MA, Kouhpayeh S, Hejazi Z, Shahbazi M, et al. Engineered zinc-finger nuclease to generate site- directed modification in the KLF1 gene for fetal hemoglobin induction. J Cell Biochem. 2019;120:8438–8446. doi: 10.1002/jcb.28130. DOI: 10.1002/jcb.28130. [DOI] [PubMed] [Google Scholar]
- 16.Shariati L, Rohani F, Heidari Hafshejani N, Kouhpayeh S, Boshtam M, Mirian M, et al. Disruption of SOX6 gene using CRISPR/Cas9 technology for gamma-globin reactivation: an approach towards gene therapy of p-thalassemia. J Cell Biochem. 2018;119(11):9357–9363. doi: 10.1002/jcb.27253. DOI: 10.1002/jcb.27253. [DOI] [PubMed] [Google Scholar]
- 17.Ma D, Liu F. Genome editing and its applications in model organisms. Genomics Proteomics Bioinformatics. 2015;13(6):336–344. doi: 10.1016/j.gpb.2015.12.001. DOI: 10.1016/j.gpb.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandrasegaran S, Carroll D. Origins of programmable nucleases for genome engineering. J Mol Biol. 2016;428(5):963–989. doi: 10.1016/j.jmb.2015.10.014. DOI: 10.1016/j.jmb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bibikova M, Golic M, Golic KG, Carroll D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161(3):1169–1175. doi: 10.1093/genetics/161.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29(1):183–212. doi: 10.1146/annurev.biophys.29.1.183. DOI:10.1146/annurev.biophys.29.1.183. [DOI] [PubMed] [Google Scholar]
- 21.Vanamee ES, Santagata S, Aggarwal AK. FokI requires two specific DNA sites for cleavage. J Mol Biol. 2001;309(1):69–78. doi: 10.1006/jmbi.2001.4635. DOI: 10.1006/jmbi.2001.4635. [DOI] [PubMed] [Google Scholar]
- 22.Mashimo T. Gene targeting technologies in rats: zinc finger nucleases, transcription activator-like effector nucleases, and clustered regularly interspaced short palindromic repeats. Dev Growth Differ. 2014;56(1):46–52. doi: 10.1111/dgd.12110. DOI: 10.1111/dgd.12110. [DOI] [PubMed] [Google Scholar]
- 23.Chang AY, Chau V, Vivian WY, Landas JA, Pang Y. Preparation of calcium competent Escherichia coli and heat-shock transformation. JEMI Methods. 2017;1:22–25. [Google Scholar]
- 24.Boshtam M, Khanahmad Shahreza H, Feizollahzadeh S, Rahimmanesh I, Asgary S. Expression and purification of biologically active recombinant rabbit monocytechemoattractant protein1 in Escherichia coli. FEMS Microbiol Lett. 2018;365(9):1–7. doi: 10.1093/femsle/fny070. DOI: 10.1093/femsle/fny070. [DOI] [PubMed] [Google Scholar]
- 25.Shikanga OT, Mutonga D, Abade M, Amwayi S, Ope M, Limo H, et al. High mortality in a cholera outbreak in western Kenya after post-election violence in 2008. Am J Trop Med Hyg. 2009;81(6):1085–1890. doi: 10.4269/ajtmh.2009.09-0400. DOI: 10.4269/ajtmh.2009.09-0400. [DOI] [PubMed] [Google Scholar]
- 26.Ali M, Lopez AL, You YA, Kim YE, Sah B, Maskery B, et al. The global burden of cholera. Bull World Health Organ. 2012;90:209–218. doi: 10.2471/BLT.11.093427. DOI: 10.2471/BLT.11.093427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broeck DV, Horvath C, De Wolf MJ. Vibrio cholerae: cholera toxin. Int J Biochem Cell Biol. 2007;39(10):1771–1775. doi: 10.1016/j.biocel.2007.07.005. DOI: 10.1016/j.biocel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol. 2015;81(7):2506–2514. doi: 10.1128/AEM.04023-14. DOI: 10.1128/AEM.04023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pursey E, Sunderhauf D, Gaze WH, Westra ER, Van Houte S. CRISPR-Cas antimicrobials: challenges and future prospects. PLoS Pathog. 2018;14(6):e1006990,1–8. doi: 10.1371/journal.ppat.1006990. DOI: 10.1371/journal.ppat.1006990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo ML, Leenay RT, Beisel CL. Current and future prospects for CRISPR-based tools in bacteria. Biotechnol Bioeng. 2016;113(5):930–943. doi: 10.1002/bit.25851. DOI: 10.1002/bit.25851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shahbazi Dastjerdeh M, Kouhpayeh S, Sabzehei F, Khanahmad H, Salehi M, Mohammadi Z, et al. Zinc finger nuclease: a new approach to overcome beta- lactam antibiotic resistance. Jundishapur J Microbiol. 2016;9(1):e29384,1–11. doi: 10.5812/jjm.29384. DOI: 10.5812/jjm.29384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabzehei F, Kouhpayeh S, Dastjerdeh MS, Khanahmad H, Salehi R, Naderi S, et al. A novel prokaryotic green fluorescent protein expression system for testing gene editing tools activity like zinc finger nuclease. Adv Biomed Res. 2017;6:155. doi: 10.4103/2277-9175.219420. DOI: 10.4103/2277-9175.219420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta RM, Musunuru K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J Clin Invest. 2014;124(10):4154–4161. doi: 10.1172/JCI72992. DOI: 10.1172/JCI72992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez Rodriguez DR, Ramirez Solis R, Garza Elizondo MA, Garza Rodriguez ML, Barrera Saldana HA. Genome editing: a perspective on the application of CRISPR/Cas9 to study human diseases (Review) Int J Mol Med. 2019;43(4):1559–1574. doi: 10.3892/ijmm.2019.4112. DOI: 10.3892/ijmm.2019.4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bowater R, Doherty AJ. Making ends meet: repairing breaks in bacterial DNA by non-homologous end- joining. PLoS Genet. 2006;2(2):e8,0093–0099. doi: 10.1371/journal.pgen.0020008. DOI:10.1371/journal.pgen.0020008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Wang S, Chen W, Song L, Zhang Y, Shen Z, et al. CRISPR-CasP and CRISPR-assisted cytidine deaminase enable precise and efficient genome editing in Klebsiella pneumoniae. Appl Environ Microbiol. 2018;84(23):e01834. doi: 10.1128/AEM.01834-18. DOI: 10.1128/AEM.01834-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitcher RS, Wilson TE, Doherty AJ. New insights into NHEJ repair processes in prokaryotes. Cell Cycle. 2005;4(5):675–680. doi: 10.4161/cc.4.5.1676. DOI: 10.4161/cc.4.5.1676. [DOI] [PubMed] [Google Scholar]
- 38.Malyarchuk S, Wright D, Castore R, Klepper E, Weiss B, Doherty AJ, et al. Expression of Mycobacterium tuberculosis Ku and Ligase D in Escherichia coli results in RecA and RecB- independent DNA end-joining at regions of microhomology. DNA Repair (Amst) 2007;6(10):1413–1424. doi: 10.1016/j.dnarep.2007.04.004. DOI: 10.1016/j.dnarep.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pires DP, Cleto S, Sillankorva S, Azeredo J, Lu TK. Genetically engineered phages: a review of advances over the last decade. Microbiol Mol Biol Rev. 2016;80(3):523–543. doi: 10.1128/MMBR.00069-15. DOI: 10.1128/MMBR. 00069-15. [DOI] [PMC free article] [PubMed] [Google Scholar]