

Abstract
Ibrutinib is the first approved therapy for symptomatic patients with Waldenström macroglobulinemia (WM). The approval was based on a single, multicenter, phase II trial in previously treated WM patients. We sought to evaluate whether there were differences in clinical characteristics, response, and survival outcomes to ibrutinib monotherapy between WM patients treated on and off clinical trials. Treatment naïve and previously treated patients who received ibrutinib monotherapy at our institution and participated in two prospective studies (ON trial; n = 72) or a prospective database (OFF trial; n = 157) were included. The median times from WM diagnosis to ibrutinib initiation were 3.1 and 3.5 years for ON and OFF trial patients, respectively (p = 0.38). Similar rates of categorical response at 6, 12, and 24 months and at best response were also observed between ON trial and OFF trial patients. The 4-year PFS and OS rates for ON trial and OFF trial patients were 72% and 63%, respectively (log-rank p = 0.14) and 83% and 81%, respectively (log-rank p = 0.14). CXCR4 mutations impacted response and survival outcomes to ibrutinib monotherapy. The 4-year rates of ibrutinib discontinuation in ON and OFF trial patients were 36% and 44%, respectively (p = 0.11). Ibrutinib is effective in the routine clinical care of both treatment-naïve and previously treated WM patients. The findings of our study validate the efficacy of ibrutinib monotherapy reported in multiple phase II clinical trials.
Introduction
Waldenström macroglobulinemia (WM) is an IgM-secreting lymphoplasmacytic lymphoma.1 Whole genome sequencing has identified highly recurrent activating somatic mutations in MYD88 and CXCR4.2,3 Mutated MYD88 triggers NF-κB activation through Bruton tyrosine kinase (BTK) and IRAK1/IRAK4, and transactivates the SRC family member hematopoietic cell kinase (HCK).4,5CXCR4 mutations promote enhanced AKT and ERK1/2 pro-survival signaling, and confer in vitro and clinical resistance to ibrutinib.6–10
Ibrutinib is an orally administered, small molecule inhibitor of BTK and HCK. In 2015, ibrutinib received approval by the United States Food and Drug Administration for the treatment of symptomatic patients with WM. The regulatory approval of ibrutinib was based on a prospective, multi-center, single-arm phase II study in which 63 patients with relapsed/refractory WM received ibrutinib 420 mg PO QD until disease progression or unacceptable toxicity. In this study, ibrutinib was highly active with an overall response rate (ORR) of 91%, major response rate (MRR) of 73%, and estimated 2-year progression free survival (PFS) and overall survival (OS) of 69% and 95%, respectively.8 Continued durable activity of ibrutinib in these patients was recently reported with an estimated 5-year PFS of 54%.10 An important finding was the identification of MYD88 and CXCR4 mutations as determinants of ibrutinib outcomes. Patients with wild-type (WT) MYD88 had no major responses and a median PFS of 5 months to ibrutinib.11 Among patients with mutated MYD88, the presence of CXCR4 mutations was associated with lower response rates, delayed response attainment, as well as shorter median PFS (42 months vs not reached [NR]) with prolonged follow-up.10 Similar findings for ibrutinib monotherapy have been reported in phase II trials including 31 rituximab-refractory WM patients and 30 treatment-naïve WM patients.9,12
Despite the high efficacy reported in clinical trials, data on outcomes to ibrutinib outside clinical trials are limited in WM patients. It is unclear whether such activity translates to the routine clinical care of WM patients. We therefore designed a comparative study to evaluate the depth of response as well as PFS and OS rates in WM patients treated with ibrutinib monotherapy on and off clinical trials.
Patients and methods
Patient selection
We included WM patients in two prospective studies (ON trial; NCT01614821 and NCT02604511) and WM patients from a prospectively maintained database (OFF trial) who received ibrutinib monotherapy at our institution. Signed informed consent for therapy and medical record review for research purposes was obtained for all patients. All patients were ≥18 years old and met the 2nd International Waldenström Macroglobulinemia Workshop (IWMW-2) criteria for a clinicopathological diagnosis of WM and criteria to treat.1 Patients who received ibrutinib for Bing-Neel syndrome were excluded. This study was approved by the Institutional Review Board at the Dana-Farber Cancer Institute.
Data gathering
Medical files were manually reviewed to gather pertinent data, including baseline clinical characteristics, MYD88 and CXCR4 mutational status, time to ibrutinib therapy, and response rates as well as PFS and OS to ibrutinib therapy. Time to ibrutinib therapy was defined as the time between diagnosis of WM and ibrutinib initiation. Response to ibrutinib was assessed using modified 6th IWWM criteria,13 in which decrease in extramedullary disease was not required for partial (PR and very good partial response (VGPR) but was required for complete response (CR). PFS was defined as the time from ibrutinib initiation until last follow-up, disease progression, or death. OS was defined as the time from ibrutinib initiation last follow-up or death from any cause. The presence of MYD88 and CXCR4 mutations was assessed using allele-specific polymerase chain reaction (AS-PCR) and Sanger sequencing methods on CD19-sorted cells derived from bone marrow aspirates. AS-PCR was used for MYD88 L265P and nonsense CXCR4 mutations, whereas Sanger sequencing was used for frameshift CXCR4 mutations and non-L265P MYD88 mutations, as previously described.3,14,15
Statistical analysis
Patient characteristics are presented using descriptive statistics. Differences in clinical characteristics and response rates to ibrutinib between groups were assessed using Chi-square and Fisher exact tests. The Kaplan-Meier method for incomplete observations was used to generate time to event estimates, and comparisons were made using the log-rank test. Univariate and multivariate Cox proportional hazard regression models were fitted to evaluate the association between clinical variables and PFS; outcomes are reported as a hazard ratio (HR) with 95% confidence interval. P values <0.05 were considered statistically significant. Graphs and calculations were obtained using STATA (StataCorp, College Station, TX).
Results
Patient characteristics
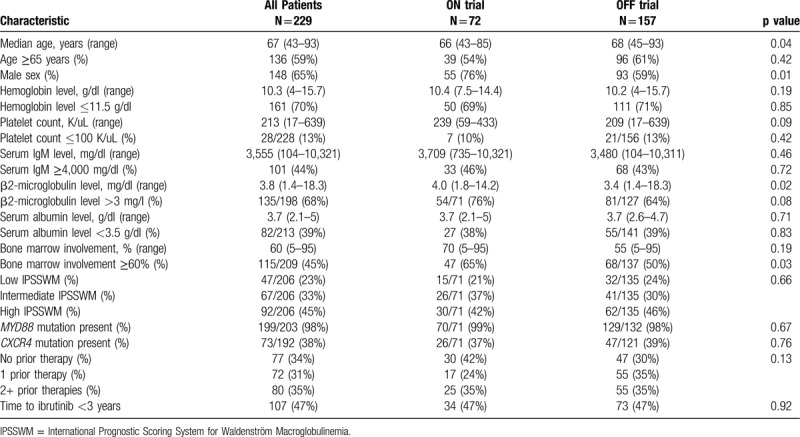
A total of 229 WM patients treated with ibrutinib monotherapy were included in the analysis; 72 patients ON trial and 157 patients OFF trial. The median time to ibrutinib initiation for ON trial and OFF trial patients was 3.1 years (95% CI 2-4.9) and 3.5 years (95% CI 2.3–5.1), respectively (log-rank p = 0.38; Fig. 1). Baseline clinical characteristics at the time of ibrutinib initiation are shown in Table 1. Patients ON trial had a lower median age at ibrutinib initiation (66 vs 68 years; p = 0.04) and had higher median serum β2-microglobulin level (4.0 vs 3.4 mg/dl; p = 0.02) than patients OFF trial. Also, patients ON trial were more likely to have bone marrow involvement ≥60% (65% vs 50%; p = 0.03) and be male (76% vs 59%; p = 0.01) than patients OFF trial. No other statistical differences between ON and OFF trial patients were detected.
Figure 1.
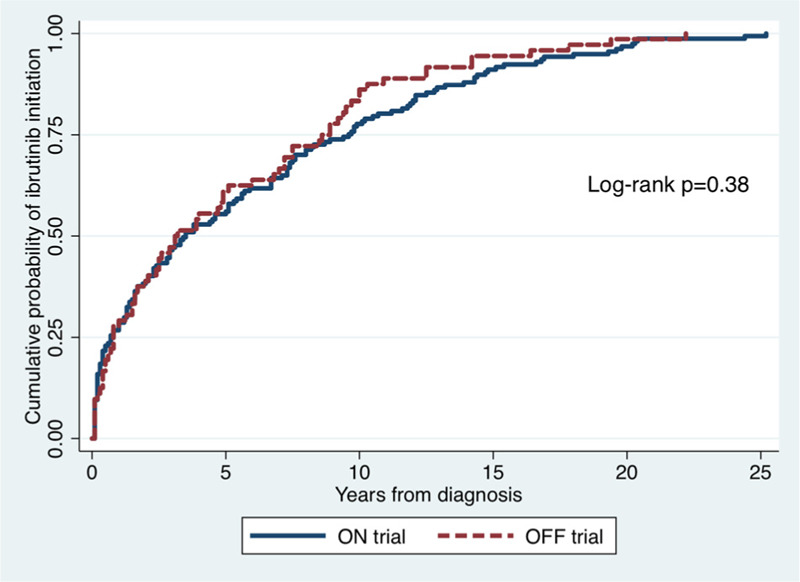
Cumulative incidence of ibrutinib initiation starting at the time of Waldenström macroglobulinemia diagnosis.
Table 1.
Clinical characteristics of Waldenström macroglobulinemia patients on and off clinical trials at the time of ibrutinib monotherapy initiation.
The most common indications to treat in ON trial patients were anemia (57%), constitutional symptoms (50%), neuropathy (8%), hyperviscosity (7%) and extramedullary involvement (4%). The most common indications to treat in OFF trial patients were anemia (57%), hyperviscosity (25%), constitutional symptoms (24%), extramedullary involvement (11%) and neuropathy (7%). OFF trial patients were less likely to start treatment due to constitutional symptoms (24% vs 50%; p < 0.001) and more likely due to hyperviscosity (25% vs 7%; p = 0.001) than ON trial patients.
Response rates
Data on response at 6, 12, and 24 months and best response were available on 190 (83%), 157 (69%), 125 (55%), and 229 patients (100%), respectively. Categorical responses to ibrutinib in WM patients ON and OFF trials at 6, 12, and 24 months and at best response are shown in Figure 2. No difference in categorical responses were observed at 6 months (p = 0.64), 12 months (p = 0.83), 24 months (p = 0.40), and at best response (p = 0.73) following ibrutinib initiation between ON trial and OFF trial patients.
Figure 2.
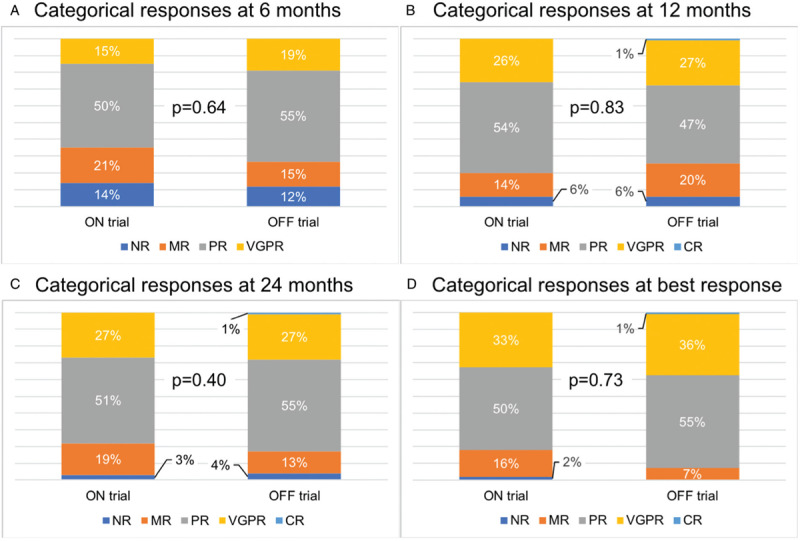
Categorical responses at 6 months, 12 months, 24 months and at best response.
At the best response, the rates of major response for ON trial and OFF trial patients were 81% and 75%, respectively (p = 0.32). In ON trial patients, the rate of major response was higher in patients without than in patients with CXCR4 mutations (93% vs 62%; p = 0.001), and higher in patients with than in patients without MYD88 mutations (0% vs 83%; p = 0.03). In OFF trial patients, the rate of major response was higher in patients without than in patients with CXCR4 mutations (80% vs 62%; p = 0.03), and higher in patients with than in patients without MYD88 mutations (33% vs 74%; p = 0.11). No differences in major response were detected between ON and OFF trial patients among those with CXCR4 mutations (62% vs 62%; p = 0.99), without CXCR4 mutations (93% vs 80%; p = 0.10), with MYD88 mutations (83% vs 74%; p = 0.17) and without MYD88 mutations (33% vs 0%; p = 0.75).
Survival outcomes
The median follow-up was longer for ON trial patients than OFF trial patients at 62 months (32–68 months), and 35 months (95% CI 32–38 months), respectively (log-rank p < 0.001). At the time of this report, 46 patients had progressed (16 ON trial and 30 OFF trial; p = 0.59), and 30 patients had died (10 ON trial and 20 OFF trial; p = 0.81).
There was no difference in PFS on ibrutinib observed between ON trial and OFF trial patients (log-rank p = 0.14; Fig. 3A). The 4-year PFS rates for ON trial and OFF trial patients were 72% (95% CI 56%–83%) and 63% (95% CI 48%–74%), respectively. In ON trial patients, there was a trend towards inferior PFS in CXCR4 mutated compared with CXCR4 WT patients (log-rank p = 0.07). The 4-year PFS rates for CXCR4 mutated and CXCR4 WT patients were 84% (95% CI 68%–93%) and 46% (95% CI 18%–70%), respectively. In OFF trial patients, PFS was inferior in CXCR4 mutated compared with CXCR4 WT patients (log-rank p = 0.02). The 4-year PFS rates for CXCR4 mutated and CXCR4 WT patients were 67% (95% CI 43%–83%) and 47% (95% CI 25%–67%), respectively. No detectable differences in PFS were observed between ON and OFF trial patients among those with mutated CXCR4 (p = 0.42) and CXCR4 WT (p = 0.21). There was an inferior PFS in MYD88 WT versus MYD88 mutated patients in ON trial (p < 0.001) and OFF trial patients (p = 0.03), but there were no differences in PFS between ON and OFF trial patients among those with mutated MYD88 (p = 0.12) and MYD88 WT (p = 0.10). There were, however, too few events in MYD88 WT patients (n = 1 ON trial and n = 1 OFF trial).
Figure 3.
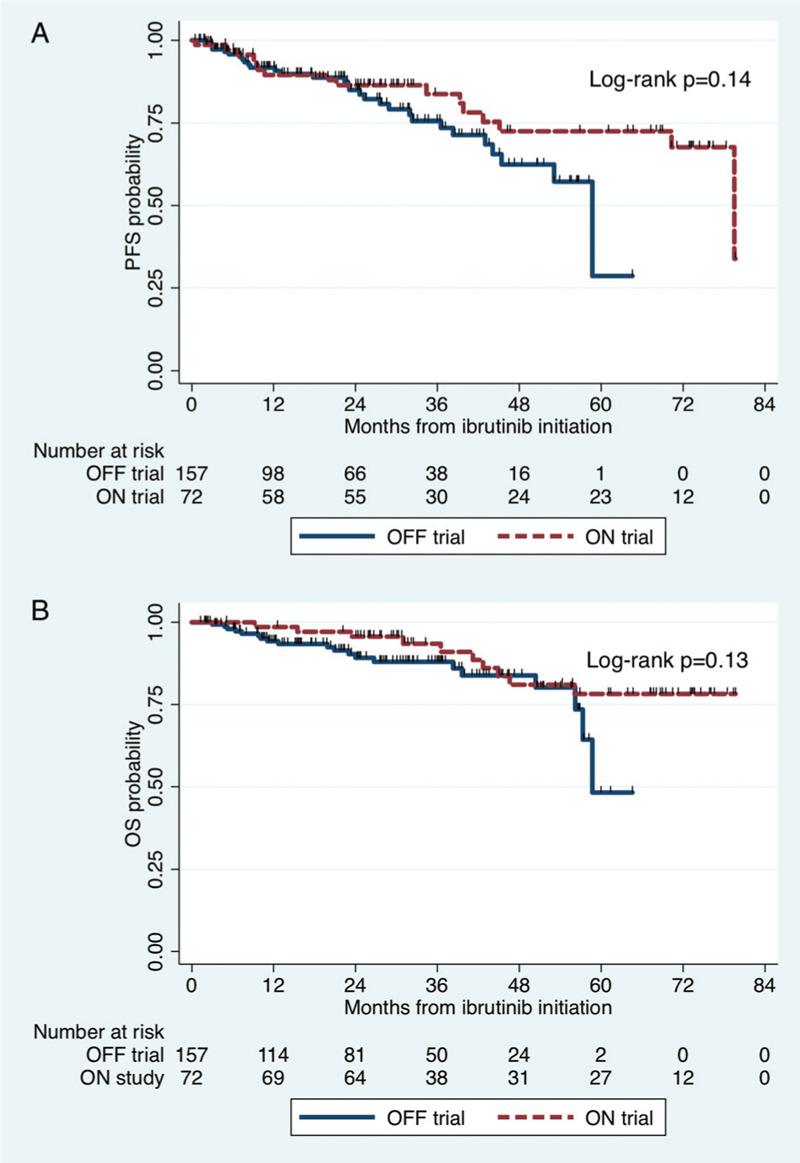
Kaplan–Meier curves for progression-free survival (A) and overall survival (B) on ibrutinib monotherapy in Waldenström macroglobulinemia patients stratified by treatment status on and off a clinical trial.
There were no detectable differences in OS on ibrutinib between ON trial and OFF trial patients (log-rank p = 0.14; Fig. 3B). The 4-year OS rates for ON trial and OFF trial patients were 83% (95% CI 73%–90%) and 81% (95% CI 66%–89%), respectively. No detectable differences in OS were observed between CXCR4 mutated and CXCR4 WT patients in ON trial (p = 0.21) and OFF trial patients (log-rank p = 0.94), and no detectable differences in OS were observed between ON and OFF trial patients among those with mutated CXCR4 (p = 0.12) and CXCR4 WT (p = 0.45). There was an inferior OS in MYD88 WT versus MYD88 mutated patients in ON trial (p < 0.001) and OFF trial patients (p = 0.005), and no detectable differences in OS were observed between ON and OFF trial patients among those with mutated MYD88 (p = 0.13) and MYD88 WT (p = 0.81). There were, however, too few events in MYD88 WT patients (n = 1 ON trial and n = 1 OFF trial).
Ibrutinib discontinuation
At the time of this report, 70 patients had discontinued ibrutinib, 21 ON trial and 49 OFF trial (p = 0.76). In ON trial patients, causes of discontinuation were disease progression (n = 14; 67%), cardiac complications (n = 2; 10%; due to cardiac arrest), death (n = 1; 5%; due to esophageal cancer) and other causes (n = 4; 19%; due to acute myeloid leukemia [n = 1], elevated liver function tests [n = 1], hematoma [n = 1] and thrombocytopenia [n = 1]). Discontinuation was considered ibrutinib-related in 5 ON trial patients (24%). In OFF trial patients, causes of discontinuation were disease progression (n = 29; 59%), cardiac complications (n = 4; 8%; atrial fibrillation [n = 3] and ventricular tachycardia [n = 1]), death (n = 6; 12%; cardiac death [n = 3] and disease progression [n = 3]) and other causes (n = 10; 20%; fatigue [n = 2], musculoskeletal pain [n = 2], acute kidney injury [n = 1], hematuria [n = 1], headache [n = 1], thrombocytopenia [n = 1], elevated liver function tests [n = 1] and mouth sores [n = 1]). Discontinuation was considered ibrutinib-related in 17 ON trial patients (35%). The rate of ibrutinib-related discontinuation was not statistically different between ON and OFF trial patients (p = 0.42). The 4-year rates of ibrutinib discontinuation for ON and OFF trial patients were 36% (95% CI 25%–51%) and 44% (39%–67%), respectively (log-rank p = 0.11; Fig. 4A).
Figure 4.
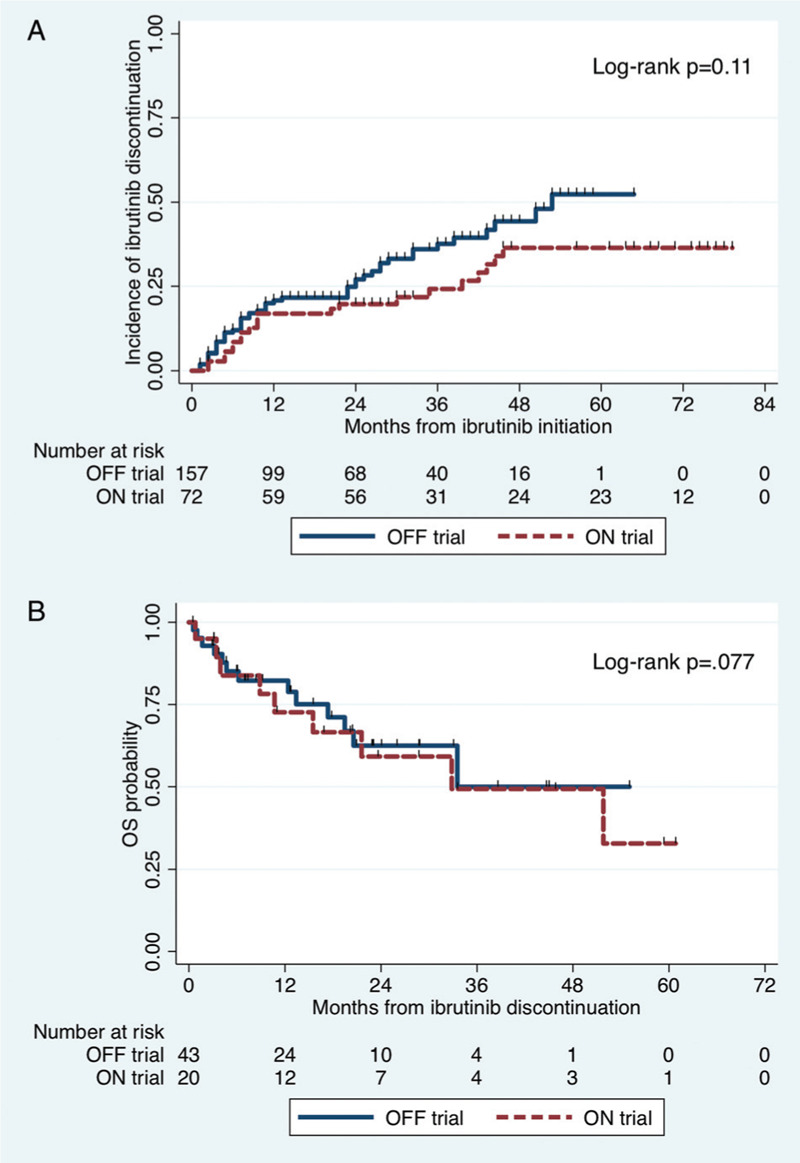
Cumulative incidence of ibrutinib discontinuation starting at the time of ibrutinib initiation (A); and Kaplan-Meier estimated for overall survival starting at the time of ibrutinib discontinuation.
After excluding the seven patients who discontinued ibrutinib due to death, the median follow times from ibrutinib discontinuation for ON and OFF trial patients were 34 months (95% CI 20-59) and 23 months (95% CI 9–29), respectively (p = 0.001). The 2-year OS rates after ibrutinib discontinuation for ON and OFF trial were 58% (95% CI 31%–78%) and 61% (41%–76%), respectively (p = 0.77; Fig. 4B).
Discussion
Ibrutinib is the first approved therapy for symptomatic patients with WM. The regulatory approval was based on the results of a prospective, multicenter phase II trial in 63 previously treated WM patients.8,10 Similar results were subsequently reported in rituximab-refractory (n = 31) and treatment-naïve (n = 30) WM patients.9,12 We report herein a study comparing outcomes to ibrutinib monotherapy in WM patients treated on and off a clinical trial. The findings validate the high response rates and durable activity of ibrutinib observed in clinical trials by using an independent, real-world cohort of 136 WM patients. Importantly, we provide evidence the efficacy of ibrutinib translates into the routine clinical care of WM patients. To our knowledge, this report is the largest series of ibrutinib-treated WM patients.
In our study, on and off trial patients had similar median times from WM diagnosis to ibrutinib therapy and also had similar distribution of most clinical and laboratory parameters, with exception of on trial patients being slightly younger, with higher median serum β2-microglobulin levels, and higher proportion of bone marrow involvement ≥60% and higher proportion of men than off trial patients. As previously seen in other studies, symptomatic anemia was the most common criterion for initiation of therapy in patients on and off trial.
An important finding was noninferior response and survival outcomes to ibrutinib for WM patients treated outside a clinical trial. This supports the generalizability of ibrutinib clinical trial results in WM, which can be prone to an inherent selection bias from inclusion and exclusion criteria. Among patients treated off trial, we found an estimated 4-year PFS of 69% to ibrutinib. This observation is consistent with cross-trial comparisons between treatment-naïve and rituximab-refractory WM patients, wherein the estimated 18-month PFS to ibrutinib was 86% and 92%, respectively.9,12 Moreover, in the pivotal trial of previously treated WM patients, the median PFS to ibrutinib has not yet been reached and exceeds 5 years; responses in these patients were independent of number of prior lines of therapy and tumor burden.8,10 Differences in sex, serum β2-microglobulin level, and bone marrow involvement between on and off trial patients similarly did not impact ibrutinib outcomes. Taken together, these data highlight the targeted nature of ibrutinib in WM patients, and underscore the need for continued rational drug discovery. Next generation BTK inhibitors, such as zanubrutinib and acalabrutinib, are active and currently in development for WM patients.16,17 A randomized phase III trial comparing ibrutinib and zanubrutinib in WM patients is now fully enrolled and results are awaited (NCT03053440).
Data on a real-world experience with ibrutinib in WM were recently reported in a retrospective study by the Mayo group. Eighty consecutive WM patients treated with ibrutinib monotherapy were included, of whom 54 patients had response data available. After a median ibrutinib duration of 12.5 months, the investigators observed an ORR of 91%, MRR of 78%, and estimated 18-month PFS of 82%.18 Similar results were obtained for both treatment-naïve and relapsed/refractory WM patients, and are in line with the present study as well as outcomes from clinical trials of ibrutinib as frontline and salvage therapy.8–10,12 The investigators also reported the occurrence of an IgM rebound in patients following discontinuation of ibrutinib, including the acute onset of symptomatic hyperviscosity in one patient. We had previously observed an IgM rebound in 73% of WM patients after stopping ibrutinib, of whom 16% developed symptomatic hyperviscosity.19 Nearly half the patients discontinuing ibrutinib had an IgM rebound within 4 weeks. These studies emphasize the importance of closely monitoring serum IgM levels after ibrutinib discontinuation in WM patients.
Outcomes to ibrutinib monotherapy in routine clinical practice were affected by MYD88 and CXCR4 mutational status. Consistent with previous trial data, ibrutinib appears to be less active in WM patients with MYD88 WT disease.11 Recent genomic insights into this subset of WM patients revealed NF-κB activating mutations downstream of BTK, and may explain the lack of activity with ibrutinib.20 We observed CXCR4 mutations were independently associated with an increased risk of progression on ibrutinib. Prior studies have shown CXCR4 mutations confer lower response rates, delayed response attainment, and shorter PFS to ibrutinib.8–10,12 The iNNOVATE study recently evaluated the combination of ibrutinib-rituximab versus rituximab monotherapy in WM patients. Ibrutinib-rituximab resulted in significantly superior response and PFS rates than rituximab for both treatment-naïve and relapsed/refractory patients.21 CXCR4 mutations also adversely impact outcomes to ibrutinib-rituximab with a shorter 36-month PFS for CXCR4 mutated versus CXCR4 WT patients (64% vs 84%, respectively), although response kinetics for CXCR4 mutated patients appeared faster than ibrutinib monotherapy.22 Moreover, recent data suggest the specific CXCR4 mutation as well as its clonal presence may modulate responses to ibrutinib.23,24 Given the importance of CXCR4 mutations, a phase I/II trial evaluating the CXCR4-blocking antibody ulocuplumab and ibrutinib was initiated in CXCR4-mutated WM patients (NCT03225716). These advances may permit a genomic-based treatment approach to WM patients.25
Approximately, 40% of patients had discontinued ibrutinib at 4 years. Rates of ibrutinib cessation due to adverse events appear consistent between clinical trials8,9,12 and retrospective studies18,19 with an estimated 4-year rate of 30%. Disease progression was the most common cause of ibrutinib discontinuation in 60% to 65% of patients, while cardiac complications were the cause in 8% to 10% of patients. Cardiac events included atrial fibrillation, ventricular arrhythmias and cardiac arrest. The current study expands on prior published experience from our group in which the 3-year cumulative incidence of atrial fibrillation in 112 WM patients on ibrutinib was reported at 9%.26 More recently, mounting data suggests also an increased risk of ventricular arrhythmias with ibrutinib therapy.27–29 Based on these emerging data, a thorough discussion on atrial and ventricular events should be undertaken between practitioners and patients prior to ibrutinib initiation.
The present study is not without limitations. All patients were treated with ibrutinib at a single, specialized referral center. However, our cohort included consecutive WM patients with clinical features consistent with those reported in population-based studies.30 In addition, we were unable to address potential differences in rates of adverse events between patients on and off clinical trials due to non-uniform adverse event assessment. Also, as with most retrospective studies, there were missing data. However, occurrence of missing data was at random, and in most variables with missing data, the proportion was <10%.
In summary, ibrutinib is effective in the routine clinical care of both treatment-naïve and previously treated WM patients. CXCR4 mutational status affects PFS in WM patients on ibrutinib therapy off clinical trials. The findings of our study validate the efficacy of ibrutinib reported in phase II clinical trials.
Acknowledgements
Portions of this research have been presented at the 24th European Hematology Association Congress in Amsterdam, Netherlands in June 2019. Dr. Castillo would like to acknowledge support of the WMR Fund.
Footnotes
Citation: Castillo JJ, Gustine JN, Meid K, Flynn CA, Demos MG, Guerrera ML, Jimenez C, Kofides A, Liu X, Munshi M, Tsakmaklis N, Patterson CJ, Xu L, Yang G, Hunter ZR, Treon SP. Response and Survival Outcomes to Ibrutinib Monotherapy for Patients With Waldenström Macroglobulinemia on and off Clinical Trials. HemaSphere, 2020;4:3:(e363). http://dx.doi.org/10.1097/HS9.0000000000000363
JJC and JNG share first authorship.
JJC has received honoraria and/or research funds from AbbVie, BeiGene, Janssen, Millennium, Pharmacyclics, and TG Therapeutics. SPT has received research funding and consulting fees from Pharmacyclics and Janssen. All the other authors have no conflicts of interest to disclose.
References
- 1.Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom's macroglobulinemia: Consensus Panel Recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol. 2003;30:110–115. [DOI] [PubMed] [Google Scholar]
- 2.Treon SP, Xu L, Yang G, et al. MYD88 L265P Somatic Mutation in Waldenström's Macroglobulinemia. N Engl J Med. 2012;367:826–833. [DOI] [PubMed] [Google Scholar]
- 3.Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123:1637–1646. [DOI] [PubMed] [Google Scholar]
- 4.Yang G, Zhou Y, Liu X, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood. 2013;122:1222–1232. [DOI] [PubMed] [Google Scholar]
- 5.Yang G, Buhrlage SJ, Tan L, et al. HCK is a survival determinant transactivated by mutated MYD88, and a direct target of ibrutinib. Blood. 2016;127:3237–3252. [DOI] [PubMed] [Google Scholar]
- 6.Cao Y, Hunter ZR, Liu X, et al. The WHIM-like CXCR4S338X somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom's Macroglobulinemia. Leukemia. 2015;29:169–176. [DOI] [PubMed] [Google Scholar]
- 7.Cao Y, Hunter ZR, Liu X, et al. CXCR4 WHIM-like frameshift and nonsense mutations promote ibrutinib resistance but do not supplant MYD88L265P-directed survival signalling in Waldenström macroglobulinaemia cells. Br J Haematol. 2015;168:701–707. [DOI] [PubMed] [Google Scholar]
- 8.Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in Previously Treated Waldenström's Macroglobulinemia. N Engl J Med. 2015;372:1430–1440. [DOI] [PubMed] [Google Scholar]
- 9.Treon SP, Gustine J, Meid K, et al. Ibrutinib monotherapy in symptomatic, treatment-naïve patients with waldenström macroglobulinemia. J Clin Oncol. 2018;36:2755–2761. [DOI] [PubMed] [Google Scholar]
- 10.Treon SP, Meid K, Gustine J, et al. Ibrutinib Monotherapy Produces Long-term Disease Control in Previously Treated Waldenstrom's Macroglobulinemia. Final Report of the Pivotal Trial (NCT01614821). Hematol Oncol. 2019;37:184–185. [Google Scholar]
- 11.Treon S P, Xu L, Hunter Z. MYD88 Mutations and Response to Ibrutinib in Waldenström's Macroglobulinemia. N Engl J Med. 2015;373:584–586. [DOI] [PubMed] [Google Scholar]
- 12.Dimopoulos MA, Trotman J, Tedeschi A, et al. Ibrutinib for patients with rituximab-refractory Waldenstrom's macroglobulinaemia (iNNOVATE): an open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol. 2017;18:241–250. [DOI] [PubMed] [Google Scholar]
- 13.Owen RG, Kyle RA, Stone MJ, et al. Response assessment in Waldenström macroglobulinaemia: update from the VIth International Workshop. Br J Haematol. 2013;160:171–176. [DOI] [PubMed] [Google Scholar]
- 14.Xu L, Hunter ZR, Yang G, et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013;121:2051–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu L, Hunter ZR, Tsakmaklis N, et al. Clonal architecture of CXCR4 WHIM-like mutations in Waldenström Macroglobulinaemia. Br J Haematol. 2016;172:735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Owen RG, McCarthy H, Rule S, et al. Acalabrutinib monotherapy in patients with Waldenstrom macroglobulinemia: a single-arm, multicentre, phase 2 study. Lancet Haematol. 2019;7:e112–e121. [DOI] [PubMed] [Google Scholar]
- 17.Trotman J, Opat S, Marlton P, et al. Updated safety and efficacy data in a phase 1/2 trial of patients with waldenström macroglobulinaemia (WM) treated with the bruton tyrosine kinase (BTK) inhibitor zanubrutinib (BGB-3111). HemaSphere. 2019;3 (S1):192–193. [Google Scholar]
- 18.Abeykoon JP, Zanwar S, Ansell SM, et al. Ibrutinib monotherapy outside of clinical trial setting in Waldenström macroglobulinaemia: practice patterns, toxicities, and outcomes. Br J Haematol. 2019;188:394–403. [DOI] [PubMed] [Google Scholar]
- 19.Gustine JN, Meid K, Dubeau T, et al. Ibrutinib discontinuation in Waldenström macroglobulinemia: etiologies, outcomes, and IgM rebound. Am J Hematol. 2018;93:511–517. [DOI] [PubMed] [Google Scholar]
- 20.Hunter ZR, Xu L, Tsakmaklis N, et al. Insights into the genomic landscape of MYD88 wild-type Waldenström macroglobulinemia. Blood Adv. 2018;2:2937–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimopoulos MA, Tedeschi A, Trotman J, et al. Phase 3 Trial of Ibrutinib plus Rituximab in Waldenström's Macroglobulinemia. N Engl J Med. 2018;378:2399–2410. [DOI] [PubMed] [Google Scholar]
- 22.Buske C, Tedeschi A, Trotman J, et al. Ibrutinib treatment in Waldenström's macroglobulinemia: follow-up efficacy and safety from the iNNOVATE study [abstract]. Blood. 2018;132:149. [Google Scholar]
- 23.Castillo JJ, Xu L, Gustine JN, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br J Haematol. 2019;187:356–363. [DOI] [PubMed] [Google Scholar]
- 24.Gustine JN, Xu L, Tsakmaklis N, et al. CXCR4S338X clonality is an important determinant of ibrutinib outcomes in patients with Waldenström macroglobulinemia. Blood Adv. 2019;3:2800–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunter ZR, Yang G, Xu L, et al. Genomics, signaling, and treatment of Waldenström macroglobulinemia. J Clin Oncol. 2017;35:994–1001. [DOI] [PubMed] [Google Scholar]
- 26.Gustine JN, Meid K, Dubeau TE, et al. Atrial fibrillation associated with ibrutinib in Waldenstrom macroglobulinemia. Am J Hematol. 2016;91:E312–E313. [DOI] [PubMed] [Google Scholar]
- 27.Cheng C, Woronow D, Nayernama A, et al. Ibrutinib-associated ventricular arrhythmia in the FDA adverse event reporting system. Leuk Lymphoma. 2018;59:3016–3017. [DOI] [PubMed] [Google Scholar]
- 28.Guha A, Derbala MH, Zhao Q, et al. Ventricular arrhythmias following ibrutinib initiation for lymphoid malignancies. J Am Coll Cardiol. 2018;72:697–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lampson BL, Yu L, Glynn RJ, et al. Ventricular arrhythmias and sudden death in patients taking ibrutinib. Blood. 2017;129:2581–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olszewski AJ, Treon SP, Castillo JJ. Evolution of management and outcomes in Waldenström macroglobulinemia: a population-based analysis. Oncologist. 2016;21:1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]