

Abstract
We present eight families with arthrogryposis multiplex congenita and myopathy bearing a TTN intron 213 extended splice-site variant (NM_001267550.1:c.39974-11T>G), inherited in trans with a second pathogenic TTN variant. Muscle-derived RNA studies of three individuals confirmed mis-splicing induced by the c.39974-11T>G variant; in-frame exon 214 skipping or use of a cryptic 3′ splice-site effecting a frameshift. Confounding interpretation of pathogenicity is the absence of exons 213-217 within the described skeletal muscle TTN N2A isoform. However, RNA-sequencing from 365 adult human gastrocnemius samples revealed 56% specimens predominantly include exons 213-217 in TTN transcripts (inclusion rate ≥ 66%). Further, RNA-sequencing of five fetal muscle samples confirms 4/5 specimens predominantly include exons 213-217 (fifth sample inclusion rate 57%). Importantly, contractures improved significantly with age for four individuals, which may be linked to decreased expression of pathogenic fetal transcripts. Our study extends emerging evidence supporting a vital developmental role for TTN isoforms containing metatranscript-only exons.
Keywords: TTN metatranscript-only, congenital titinopathies, arthrogryposis, intronic splice variant, alternative splicing
Titin is the largest known human protein measuring ~1.2μm in length, and is the third most abundant protein in striated muscle (Chauveau, Rowell, & Ferreiro, 2014). Spanning half the length of the sarcomere, titin is a vital structural scaffold for sarcomere formation during development, and underpins the intrinsic elasticity of striated muscles to enable rapid and repeated lengthening and shortening of the sarcomere during muscle contractions (Chauveau et al., 2014). Titin is encoded by TTN, arguably one of the most complex human genes; with 364 exons encoding extensively alternatively-spliced transcripts that are ~100,000 nucleotides in length (Bang et al., 2001; Freiburg et al., 2000; Labeit & Kolmerer, 1995). Further contributing to complexity, TTN bears a triplicated repeat region that encompasses three, near-identical replicated blocks of 9 exons, which are alternatively spliced (Savarese et al., 2018), and technically very challenging to sequence. When a variant is identified within the triplicated repeat region, in many cases it is impossible to be certain in which exon it resides.
Pathogenic variants in TTN are associated with a heterogeneous group of cardiac and muscle disorders with varying ages of onset (Savarese, Sarparanta, Vihola, Udd, & Hackman, 2016). Recently, recessive TTN variants have been linked to a phenotypic spectrum of severe early-onset disorders collectively termed “congenital titinopathies” (Oates et al., 2018), including; centronuclear myopathy, core myopathy with heart disease, early onset myopathy with fatal cardiomyopathy and arthrogryposis multiplex congenita (Chervinsky et al., 2018; Fernández-Marmiesse et al., 2017; Oates et al., 2018).
Due to extensive TTN alternative splicing, variants are often described in reference to the inferred complete TTN metatranscript (NM_001267550.1); a theoretical isoform that includes all putative TTN exons. Although, in the context of a myopathy, typically only variants in exons described in the skeletal muscle isoform N2A (NM_133378.4) are considered. However, six pathogenic variants in TTN exons not included within the described skeletal muscle isoform N2A (NM_133378.4) are recently reported affecting 10 different families with recessive congenital titinopathies (Chervinsky et al., 2018; Fernández-Marmiesse et al., 2017; Oates et al., 2018). These pathogenic variants in TTN metatranscript-only exons support emerging evidence that there are numerous developmental TTN transcripts isoforms yet to be formally described (Savarese et al., 2018).
Herein we describe a recurrent pathogenic TTN haplotype that includes a metatranscript-only intron 213 extended splice site variant (Chr2(GRCh37):g.179514069A>C; NM_001267550.1:c.39974-11T>G) identified in eight unrelated families with autosomal recessive arthrogryposis multiplex congenita and myopathy.
The ten affected individuals presented at delivery with arthrogryposis multiplex congenita and globally reduced muscle bulk (Figure 1A, 1B and Supporting Information Table S1). Contractures were varied, with most occurring distally. Congenital fractures were observed in 3/10 cases. Dysmorphic facial features were observed in all affected individuals; with 7/10 noted to have elongated faces and 5/10 noted with micrognathia. Excluding AII:1 (terminated at 26 weeks gestation), all affected individuals presented at birth with; generalised hypotonia that persisted into early childhood and feeding difficulties in the newborn period. Marked axial weakness was noted for 5/9 cases. 7/9 affected individuals were noted to have facial weakness, 8/9 had high arched palates and 6/9 affected individuals had neonatal respiratory difficulties. GII:1 presented in poor condition at birth, requiring cardiopulmonary resuscitation and epinephrine treatment, and placed on a head cooling protocol for hypoxic ischaemic encephalopathy (see Figure 1Bv). Severe restrictive lung function in HII:1 and HII:2 persisted into infancy, with HII:1 succumbing to respiratory failure aged 2 years.
Figure 1. Eight families presenting with arthrogryposis multiplex congenita and myopathy.
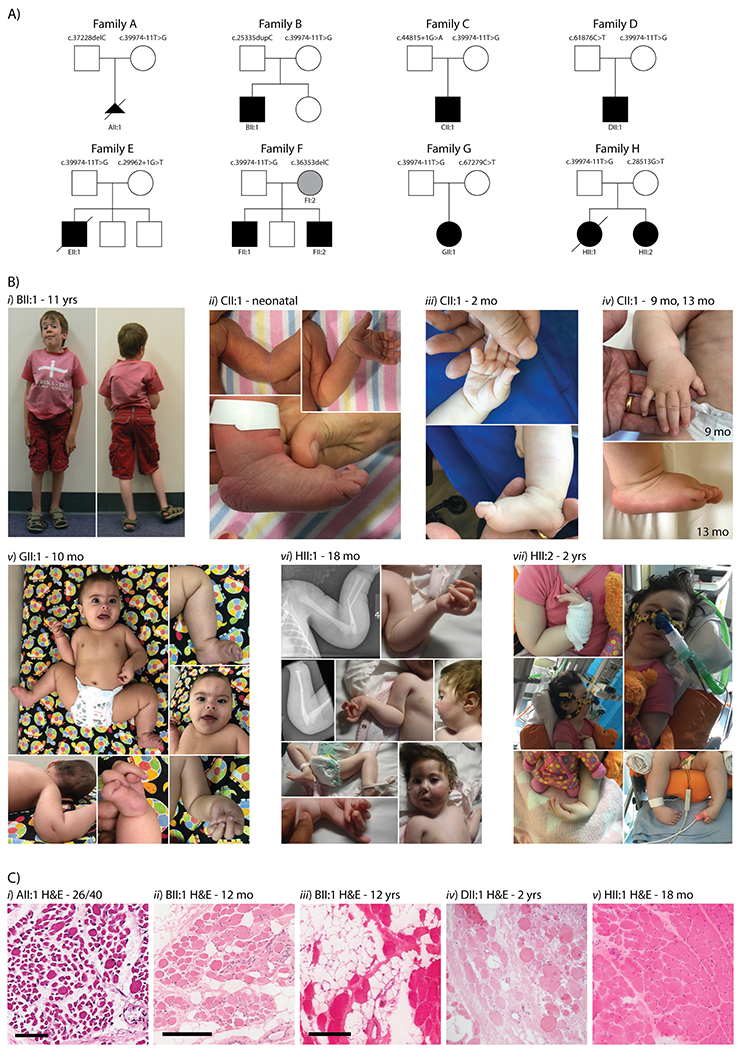
A) Family pedigrees and segregation of the recessive TTN variants, including the common c.39974-11T>G haplotype. NOTE: FI:2 does not carry the c.39974-11T>G variant. B) Clinical photos; i) BII:1 (11 yrs), showing an elongated face, reduced muscle bulk and left talipes valgus; ii) CII:1 (neonatal) showing ulnar deviation and elbow contractures; iii) the ulnar deviation improving in CII:1 at 2 months of age and iv) further resolution of wrist and finger contractures at 13 months of age. v) GII:1 (10 months) presenting with hip dysplasia; finger, wrist, ankle, elbow and knee contractures; and a mild flat nasal bridge; vi) HII:1 (18 months) showing congenital fractures, micrognathia and hip, finger, wrist, ankle and elbow contractures; vii) HII:2 (2 years) presenting with severe restrictive lung disease, micrognathia and wrist, finger and ankle contractures. C) Haematoxylin and eosin (H&E) staining of muscle biopsy cryosections show variation in fibre size, internalised nuclei and areas of fatty/fibrotic replacement of muscle fibres; i) AII:1 psoas from autopsy sample, (scale bar 60 μm); ii) BII:1 quadriceps at 12 months (scale bar 200 μm); iii) BII:1 quadriceps at 12 years of age (scale bar 300 μm); iv) DII:1 vastus lateralis at 2 yrs of age (scale bar unavailable); v) HII:1 vastus lateralis at 18 months of age (scale bar unavailable).
All liveborn affected individuals had delayed motor milestones, with DII:1 never achieving independent walking. Muscle weakness was observed proximally and distally, with scapular winging in two cases. Neck weakness was pronounced, with all individuals noted to have reduced head control in infancy. Scoliosis developed in 3/9 individuals, with FII:1 presenting with scoliosis at birth. Joint hypermobility was observed in 7/9 cases. Importantly, congenital contractures showed improvement with age for 4/9 affected individuals. For example, multiple contractures present in BII:1 at birth resolved throughout childhood, with resolution of talipes, wrist and knee contractures on examination at 18 years of age, although finger and elbow contractures persisted (see Figure 1Bi–ii). BII:1 showed delayed motor milestones though progressed to walk independently at 6 years; at 18 years BII:1 can walk short distances (20 m) and is dependent on use of a wheelchair for longer distances.
Muscle biopsy performed for six probands (Figure 1C and Supporting Information Table S1) showed variation in fibre size and increased internalised nuclei; adipose replacement and fibre splitting was seen in 3/6 cases, and fibre-type disproportion was seen in 5/6 cases although varied between individuals. Serum creatine kinase was within normal limits for all affected individuals. No cardiac abnormalities have thus far been detected in any of the probands. All affected individuals were recorded to have normal intellect, although BII:1 has autism spectrum disorder.
Parallel sequencing (see Supporting Information Materials and Methods) revealed all affected individuals were heterozygous for the metatranscript-only c.39974-11T>G intron 213 extended splice site variant in TTN; present in the gnomAD population database (Lek et al., 2016) at a frequency of 0.000062 (6/96636 alleles) and not previously reported in ClinVar (Landrum et al., 2018). Each affected individual inherited a second pathogenic or likely pathogenic TTN variant in trans with the c.39974-11T>G TTN variant (see Figure 2Ai and Supporting Information Table S2). In each case, the second TTN variant is a truncating or splicing variant, with low allele frequency or absent from gnomAD and likely to induce severe dysfunction or loss-of-function for the encoded titin protein. Two frameshift variants (Family A c.37228delC and Family F c.36353delC) are also located within metatranscript-only exons (see Figure 2Ai), with c.37228delC lying within the triplicated repeat region. FI:2 is a heterozygous carrier of the TTN c.36353delC variant but does not carry the c.39974-11T>G variant present in her more severely affected children (FII:1, FII:2). FI:2 presented in childhood with mild proximal muscle weakness, slight clinodactyly of toes, and very mild scoliosis. FI:2 has an elongated face with micrognathia and a high arched palate. Scrutiny of parallel sequencing data did not identify an additional likely causative TTN variant in FI:2, and thus remains undiagnosed.
Figure 2. RNA studies of TTN transcripts in muscle.
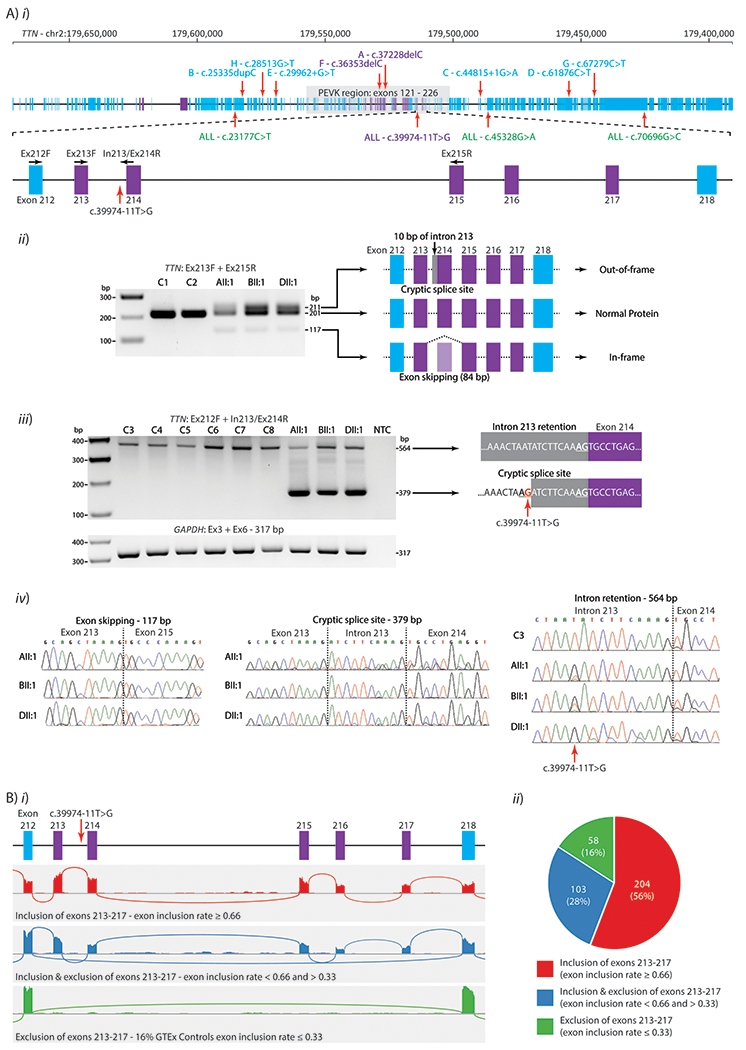
A) i) schematic of TTN genomic locus with exons described in the N2A isoform (NM_133378.4) in blue rectangles and metatranscript-only exons in Purple. Green: Missense variants within the shared haplotype, classified as benign in ClinVar. Zoomed region: Exons 212-218 and the location of the c.39974-11T>G variant and primers used for RT-PCR. ii) RT-PCR of cDNA extracted from skeletal muscle from a fetal control (C1), an adult control (C2), AII:1 (fetal quadriceps), BII:1 (paraspinal muscle, 12 years) and DII:1 (vastus lateralis, 2 years) using primers in TTN exons 213 and 215 (Ex213F + Ex215R, product size 201 bp). Compared to controls, AII:1, BII:1 and DII:1 showed identical additional bands of 117 bp and 211 bp. Sanger sequencing revealed these three bands corresponded to use of a cryptic 3′ splice site (inclusion of 10 bp, product size 211 bp), or exon 214 skipping (loss of 84 bp, product size 117 bp) or normal splicing (product size 201 bp). iii) RT-PCR of cDNA extracted from skeletal muscle from AII:1, BII:1 and DII:1 and six controls (two fetal (C3, C4), 10 months of age (C5), 8 years (C6), 18 years (C7, vastus medialis) and 26 years of age (C8, quadriceps)), using a forward primer in exon 212 and a reverse primer spanning the intron 213 and exon 214 junction (Ex212F + In213/Ex214R). A band corresponding to intron retention was observed in all samples (564 bp product) with cryptic 3′ splice site use only present in AII:1, BII:1 and DII:1 (379 bp product). Primers in exons 3 and 6 of GAPDH (Ex3F + Ex6) were used as a loading control. iv) Sanger sequencing chromatograms of purified gel products. Sanger sequencing of intron retention confirms the c.39974-11T>G variant is present in AII:1, BII:1 and DII:1 (muscle type unknown for C1-C6). B) The inclusion rate of exons 213-217 within TTN transcripts in RNA-seq data from 365 GTEx skeletal muscle samples (mostly gastrocnemius), calculated using the equation (I/6)/[(I/6) + E] (see Supporting Information Materials and Methods). GTEx samples were divided into three groups based on their exon inclusion rate; 1) ≥ 0.66, considered to predominantly include exons 213-217, 2) < 0.66 and > 0.33, considered to have a mix of both events and 3) ≤ 0.33, considered to predominantly skip exons 213-217 i) Examples of sashimi plots of RNA-seq data showing the 3 patterns of exon 213-217 exon inclusion into TTN transcripts. Top: An example of a skeletal muscle biospecimen showing exclusive inclusion of all exons 213-217 (red). Middle: A muscle sample showing a mix of inclusion and skipping of exons 213-217 (blue). Bottom: A muscle specimen showing exclusive skipping of exons 213-217 (green). ii) Pie Chart showing the relative proportion of GTEx muscle samples showing the different patterns of inclusion and skipping of exons 213-217, as defined above.
Three heterozygous missense variants were found to co-segregate with the c.39974-11T>G in all families for which full exome sequencing data was available (7/8 families); chr2:179585312G>A, c.23177C>T, p.Ser7726Leu; chr2:79486223C>T, c.45328G>A, p.Asp15110Asn; and chr2:179440163C>G, c.70696G>C, p.Gly23566Arg with gnomAD allele frequencies of 0.007 (19 homozygotes), 0.008 (23 homozygotes) and 0.012 (47 homozygotes) respectively. Further, each TTN missense variant is reported multiple times in ClinVar and LOVD as benign (see Supporting Information Table S2). In addition, we confirmed that the seven carriers of c.39974-11T>G splice variant in gnomAD also carried the c.23177C>T, c.45328G>A and c.70696G>C variants. Thus, collective data infer the c.39974-11T>G splice variant lies within a common TTN haplotype encompassing three missense variants c.23177C>T, c.45328G>A, c.70696G>C (found most commonly in European (Finnish) populations in gnomAD).
RT-PCR performed on mRNA extracted from skeletal muscle biopsies from AII:1, BII:1 and DII:1 showed an identical pattern of abnormal splicing in the three affected individuals, compared to eight controls of different ages, using multiple primer pairs (Figure 2Aii and iii; shows data using two different primer pairs, see Supporting Information Materials and Methods). Sanger sequencing of amplicons confirmed two main abnormal splicing events; 1) Use of a cryptic 3′ splice site that abnormally includes 10 nucleotides of intron 213, leading to a frameshift and premature termination codon (r.39973_39974ins39974-10_39974-1, p.Val13325Aspfs*6) or 2) exon 214 skipping, which is in-frame and results in loss of 28 residues (r.39974_40057del, p.Glu13327_Pro13354del); affecting one of the proline-glutamine-valine-lysine (PEVK) repeat regions (Figure 2Aiv). Multiple primer pairs variably positioned within exons 211 - 218 reproducibly confirmed use of the cryptic 3′ splice site and exon 214 skipping induced by the c.39974-11T>G variant, as well as naturally occurring alternative splicing involving exons 213 - 217 (data not shown). We did not find evidence for increased levels of intron 214 retention resulting from the c.39974-11T>G variant (Figure 2Aiii).
At this point, collective evidence was strongly suggestive of pathogenicity of the TTN c.39974-11T>G variant haplotype. However, formal classification of the c.39974-11T>G as a pathogenic variant remained a challenge, given exons 213-217 are metatranscript-only exons, and are not described within the skeletal muscle TTN N2A isoform (NM_133378.4). We therefore performed detailed analyses of RNA-seq from 365 control human muscle samples (gastrocnemius) within the GTEx database (see Supporting Information Materials and Methods). Our analyses confirm that 56% gastrocnemius specimens predominantly include exons 213-217 in TTN transcripts, 16% specimens predominantly skip exons 213-217, and 28% show a mix of both events (Figure 2B). We further performed RNA-seq of five fetal muscle specimens and showed 4/5 fetal muscle RNA samples showed predominant inclusion of exons 213-217 (inclusion rate ≥ 0.66) with the remaining fetal muscle sample showing an inclusion rate of 0.57. Our data are supported by recent studies, which show that the metatranscript-only exons 213-217 are more highly expressed in fetal muscle than adult muscle (Savarese et al., 2018).
In addition, RNA-seq data was available for BII:1 (paraspinal muscle biopsy taken at 12 yrs) and HII:A (quadriceps biopsy at age 4 years). RNA-seq for BII:1 showed predominant skipping of exons 213-217 (inclusion rate of 0.1 at 12 yrs). Due to the low number of reads for exons 213-217, abnormal splicing events arising from the c.39974-11T>G variant were unable to be determined with confidence using RNA-seq data (see Supporting Information Figure S1). RNA-seq for HII:A confirmed the same mis-splicing events detected by RT-PCR for AII:1, BII:1 and DII:1, showing abnormal use of the upstream 3′ cryptic splice site (490 junctional reads) and exon 214 skipping (517 junctional reads bridging exons 213-215) (see Figure S1).
In conclusion, we identify eight families with arthrogryposis multiplex congenita and myopathy with a novel TTN c.39974-11T>G variant inherited in trans with a second pathogenic TTN variant. RT-PCR of muscle RNA confirms the c.39974-11T>G variant induces abnormal use of a cryptic 3′ splice site resulting in a frameshift, or exon 214 skipping, which removes 28 amino-acids from the encoded titin protein. While use of the cryptic 3′ splice-site inducing a frameshift may readily be interpreted as exerting damaging consequences for the encoded titin protein; it remains difficult to interpret the functional implications attributable to the loss of 28 residues within the differentially-spliced PEVK region of titin. Exons within the PEVK region are extensively alternatively spliced, regulating passive tension and muscle elasticity (Freiburg et al., 2000; Ottenheijm et al., 2009; Savarese et al., 2018). Importantly, exon 214 skipping is not observed in control muscle (Savarese et al., 2018), is a very rare/absent event among our muscle RNA-seq data from ~ 50 disease controls (data not shown) and absent from 8 control samples by RT-PCR (results for C1 and C2 are shown in figure 2Aii).
Weighting collective evidence from eight families presenting with an overlapping clinical and histopathological phenotype consistent with congenital titinopathy (Oates et al., 2018), plausible combined deleterious effects evoked by splicing abnormalities (frameshift or deletion of 28 amino-acids), and experimental evidence confirming the affected alternatively-spliced exons 213-217 are expressed highly in both fetal and adult skeletal muscle specimens; the c.39974-11T>G variant has been classified as a pathogenic variant, when inherited in trans with a second, loss-of-function likely/pathogenic TTN variant. While evidence from gnomAD and ClinVar infer the three TTN missense variants within the haplotype are benign due to frequent homozygosity; we cannot exclude potential additive pathogenic contributions of these TTN missense variants to the manifesting phenotype. However, RNA studies support abnormal splicing induced by the TTN c.39974-11T>G variant as the primary pathogenic element within the haplotype.
Genetic diagnosis of a recessive congenital titinopathy was further complicated for Families A and F whose second TTN frameshift variants involved metatranscript-only exons (exons 181 and 170, respectively). In support of pathogenicity, there are several recent reports of autosomal recessive congenital titinopathies associated with variants within metatranscript-only exons 163, 172, 181, 201 (Oates et al., 2018), 197 (Fernández-Marmiesse et al., 2017) and 167 (Chervinsky et al., 2018). Junctional reads bridging exons 181 and 170 are detected in adult skeletal muscle (Savarese et al., 2018), inferring the novel frameshift variants found in families A and F affect transcripts expressed in skeletal muscle. Importantly, exons 213-217 are not expressed at significant levels in cardiac tissue (Savarese et al., 2018) and reported individuals with a metatranscript-only pathogenic TTN variant did not present with a cardiac phenotype (herein and in Chervinsky et al., 2018; Fernández-Marmiesse et al., 2017; Oates et al., 2018).
Careful analyses of developmentally-regulated Ttn expression in mice and rabbits reveal that titin protein is observably larger in fetal muscle, than adult muscle; with clear, age-related decrement in titin size (Ottenheijm et al., 2009). Accompanying transcriptomics infer that the increased molecular weight of titin relates primarily to alternative-splicing of the complex PEVK region - and more common inclusion of these exons during development (Ottenheijm et al., 2009). Increased inclusion of the metatranscript-only exons 213-217 in fetal muscle, compared with adult muscle, has been independently confirmed in our study, and in detailed transcriptomic analyses reported in Savarese et al., 2018. Therefore, it is plausible that the improvement of severe contractures present at delivery for 4/9 individuals may be due to decreased reliance on PEVK repeats in mature muscle transcripts; an important finding for prognostic counselling. This hypothesis is supported by individual BII:1, whose contractures had mostly resolved at 18 yrs, and for whom RNA-seq shows skipping of exons 213-217 in 90% of TTN transcripts in a muscle biopsy taken at 12 years of age.
TTN is an extraordinarily complex gene, with the full extent of TTN alternative splicing only beginning to be elucidated (Guo, Bharmal, Esbona, & Greaser, 2010; Ottenheijm et al., 2009; Savarese et al., 2018). Interpretation of the functional impact of putative pathogenic variants in TTN metatranscript-only exons will benefit greatly from emerging technologies in long read RNA-seq, potentially from isolated fibers, to better define TTN isoforms expressed in developing and adult muscles of different fiber-types.
Titin is the cornerstone for sarcomere assembly and is largely responsible for the passive tension and elasticity in muscle (Chauveau et al., 2014; Ottenheijm et al., 2009). It is conceivable that TTN variants leading to abnormal muscle development may yet be associated with a range of developmental phenotypes. Muscles with unique tensile or contractile properties may uniquely depend on a sub-group of TTN isoforms, which may be dispensable in other muscles.
In conclusion, we identify a recurrent TTN c.39974-11T>G splice variant haplotype as the likely causal basis for arthrogryposis multiplex congenita and myopathy in eight families, when co-inherited with a second, loss-of-function, likely/pathogenic TTN variant. The TTN c.39974-11T>G variant may be missed by genomics platforms that do not assess or capture all TTN metatranscript exons. We advocate screening for this variant in any individual presenting with arthrogryposis who bears one TTN likely/pathogenic variant, and is shown to also carry missense variants within the common haplotype (c.23177C>T, c.45328G>A, and c.70696G>C). Our results extend emerging evidence linking recessive metatranscript-only TTN variants with severe, arthrogryposis multiplex congenita and myopathy; due to a crucial role for TTN transcripts bearing metatranscript-only exons during development.
Supplementary Material
Acknowledgements:
We thank the families for their invaluable contributions to this research, and the clinicians and health care workers involved in their assessment and management.
Funding Information:
This study was supported by the National Health and Medical Research Council of Australia (APP1048816 and APP1136197 S.T.C, 1080587 S.T.C., D.G.M.). SB is supported by a Muscular Dystrophy New South Wales PhD scholarship. WES and RNA-seq was provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 to D.G.M and Heidi Rehm.
Footnotes
Competing Interests:
Professor Sandra Cooper is director of Frontier Genomics Pty Ltd (Australia). Frontier Genomics has not traded (as of October, 2019). Frontier Genomics Pty Ltd (Australia) has no existing financial relationships that will benefit from publication of these data. The remaining co-authors do not have any relationships, financial or otherwise, that may result in a perceived conflict of interest.
Data Availability Statement:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References:
- Bang M-L, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, … Labeit S (2001). The Complete Gene Sequence of Titin, Expression of an Unusual ≈700-kDa Titin Isoform, and Its Interaction With Obscurin Identify a Novel Z-Line to I-Band Linking System. Circulation Research, 89(11), 1065–1072. 10.1161/hh2301.100981 [DOI] [PubMed] [Google Scholar]
- Chauveau C, Rowell J, & Ferreiro A (2014). A Rising Titan: TTN Review and Mutation Update. Human Mutation, 35(9), 1046–1059. 10.1002/humu.22611 [DOI] [PubMed] [Google Scholar]
- Chervinsky E, Khayat M, Soltsman S, Habiballa H, Elpeleg O, & Shalev S (2018). A homozygous TTN gene variant associated with lethal congenital contracture syndrome. American Journal of Medical Genetics Part A, 176(4), 1001–1005. 10.1002/ajmg.a.38639 [DOI] [PubMed] [Google Scholar]
- Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, … MacArthur DG (2017). Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Science Translational Medicine, 9(386), eaa15209 10.1126/scitranslmed.aal5209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, … Taschner PEM (2016). HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Human Mutation, 37(6), 564–569. 10.1002/humu.22981 [DOI] [PubMed] [Google Scholar]
- Fernández-Marmiesse A, Carrascosa-Romero MC, Alfaro Ponce B, Nascimento A, Ortez C, Romero N, … Couce ML (2017). Homozygous truncating mutation in prenatally expressed skeletal isoform of TTN gene results in arthrogryposis multiplex congenita and myopathy without cardiac involvement. Neuromuscular Disorders, 27(2), 188–192. 10.1016/J.NMD.2016.11.002 [DOI] [PubMed] [Google Scholar]
- Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, & den Dunnen JT (2011). LOVD v.2.0: the next generation in gene variant databases. Human Mutation, 32(5), 557–563. 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, … Labeit S (2000). Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circulation Research, 86(11), 1114–1121. 10.1161/01.RES.86.11.1114 [DOI] [PubMed] [Google Scholar]
- Ghaoui R, Cooper ST, Lek M, Jones K, Corbett A, Reddel SW, … Clarke NF (2015). Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy. JAMA Neurology, 72(12), 1424 10.1001/jamaneurol.2015.2274 [DOI] [PubMed] [Google Scholar]
- GTEx Consortium T Gte. (2013). The Genotype-Tissue Expression (GTEx) project. Nature Genetics, 45(6), 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Bharmal SJ, Esbona K, & Greaser ML (2010). Titin diversity-alternative splicing gone wild. Journal of Biomedicine and Biotechnology. 10.1155/2010/753675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labeit S, & Kolmerer B (1995). Titins: Giant Proteins in Charge of Muscle Ultrastructure and Elasticity. Science, 270(5234), 293–296. Retrieved from http://www.jstor.org.ezproxy1.library.usyd.edu.au/stable/2888540 [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, … Maglott DR (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–D1067. 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … Consortium EA (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Grady GL, Lek M, Lamande SR, Waddell L, Oates EC, Punetha J, … North K (2016). Diagnosis and etiology of congenital muscular dystrophy: We are halfway there. Annals of Neurology, 80(1), 101–111. 10.1002/ana.24687 [DOI] [PubMed] [Google Scholar]
- Oates EC, Jones KJ, Donkervoort S, Charlton A, Brammah S, Smith JE, … Laing NG (2018). Congenital Titinopathy: Comprehensive characterization and pathogenic insights. Annals of Neurology, 83(6), 1105–1124. 10.1002/ana.25241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottenheijm CAC, Knottnerus AM, Buck D, Luo X, Greer K, Hoying A, … Granzier H (2009). Tuning Passive Mechanics through Differential Splicing of Titin during Skeletal Muscle Development. Biophysical Journal, 97(8), 2277–2286. 10.1016/J.BPJ.2009.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, & Mesirov JP (2011). Integrative genomics viewer. Nature Biotechnology, 29(1), 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarese M, Jonson PH, Huovinen S, Paulin L, Auvinen P, Udd B, & Hackman P (2018). The complexity of titin splicing pattern in human adult skeletal muscles. Skeletal Muscle, 8(1), 11 10.1186/s13395-018-0156-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarese M, Sarparanta J, Vihola A, Udd B, & Hackman P (2016). Increasing Role of Titin Mutations in Neuromuscular Disorders. Journal of Neuromuscular Diseases, 3(3), 293–308. 10.3233/JND-160158 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.