

Abstract
Optically controlled receptor tyrosine kinases (opto-RTKs) allow regulation of RTK signaling using light. Until recently, the majority of opto-RTKs were activated with blue-green light. Fusing a photosensory core module of Deinococcus radiodurans bacterial phytochrome (DrBphP-PCM) to the kinase domains of neurotrophin receptors resulted in opto-RTKs controlled with light above 650 nm. To expand this engineering approach to RTKs of other families, here we combined the DrBpP-PCM with the cytoplasmic domains of EGFR and FGFR1. The resultant Dr-EGFR and Dr-FGFR1 opto-RTKs are rapidly activated with near-infrared and inactivated with far-red light. The opto-RTKs efficiently trigger ERK1/2, PI3K/Akt and PLC-gamma signaling. Absence of spectral crosstalk between the opto-RTKs and GFP-based biosensors enable simultaneous Dr-FGFR1 activation and detection of calcium transients. Action mechanism of the DrBphP-PCM-based opto-RTKs is considered using the available RTK structures. DrBphP-PCM represents a versatile scaffold for engineering of opto-RTKs that are reversibly regulated with far-red and near-infrared light.
Keywords: bacteriophytochrome, DrBphP, BphP1, Dr-RTK, EGFR, FGFR
Introduction
Receptor tyrosine kinases (RTKs) are transmembrane receptors involved in cell proliferation, migration, metabolism and differentiation. Efficient and selective regulation of RTK activity is necessary to study a variety of cell signaling pathways in norm and pathology. Chemical inhibitors are widely used for the studies of RTK signaling, however, they have a limited ability to control RTK signaling reversibly and limited spatiotemporal precision [1]. As opposed to chemical inhibition, regulation of RTK activity with light allows the non-invasive and reversible control over their downstream signaling [1]. Dimerization is necessary but not the only one prerequisite for RTK activation (Fig. 1); many RTKs exist as preformed inactive dimers prior to ligand binding [2]. However, for a long time the opinion that ligand binding activates RTKs by inducing receptor dimerization prevailed [2], therefore, the majority of strategies for chemical or optical RTK regulation exploit an induced dimerization [1].
Figure 1. Luciferase assay of Dr-RTKs.
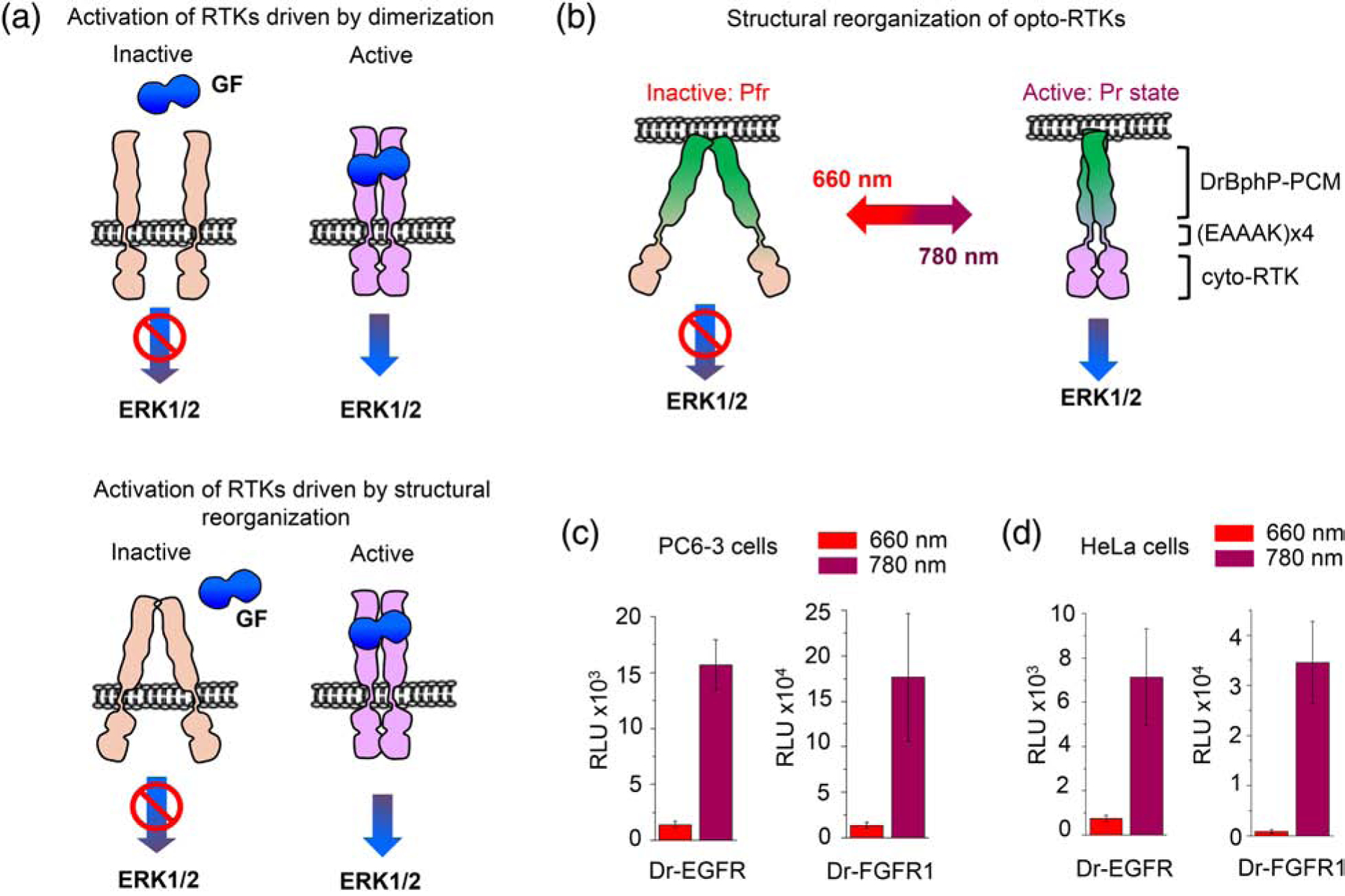
(a). Top: according to a traditional view on RTK activation, binding of a growth factor (GF) causes their dimerization and autophosphorylation, resulting in the activation of the ERK1/2 pathway. Bottom: in alternative view, RTKs exist as pre-formed inactive dimers. Structural reorganization of RTKs caused by a GF binding drives their activation. (b) Light-dependent regulation of a dimeric opto-RTK based on the DrBphP-PCM scaffold. Under FR (660 nm) illumination DrBphP-PCM adopts the Pfr state and the opto-RTKs are inactive. Under NIR (780 nm) light DrBphP-PCM adopts the Pr state in which the cytosolic-RTK domains are autophosphorylated, resulting in the downstream ERK1/2 activation. (c, d) Light-dependent regulation of the Elk-1 transcription by Dr-EGFR1 and Dr-FGFR opto-RTKs in PC6–3 (c) and HeLa (d) cells. 660 nm light inhibits and 780 nm light activates opto-RTK signaling, which causes upregulation of the Elk1-dependent luciferase expression. 25 μM BV was added to culture medium in all experiments. Error bars represent s.d., n=3 experiments.
The first optically regulated RTKs (opto-RTKs) were developed by fusing cytoplasmic RTK domains to CRY2 and LOV photoreceptors that dimerize upon action of blue light [3, 4]. Later, opto-RTKs based on cobalamin-binding domain regulated with green light [5] and on phytochrome regulated with red light [6] were developed. The reversible regulation of these opto-RTKs with light depends on the monomerization-dimerization transitions of photoreceptors fused to the RTK catalytic domains.
Blue- and green-light regulated opto-RTKs enable reversible and non-invasive regulation of RTK signaling, however, because they are activated with visible light they cannot be spectrally multiplexed with common fluorescent proteins and biosensors [7]. For multiplexing far-red (FR, 660 nm) and near-infrared (NIR, 780 nm) opto-RTKs are required. Moreover, because of the low absorbance by hemoglobin and less phototoxicity, FR and NIR light is favorable over shorter wavelengths for use in mammalian tissues [8, 9].
However, the choice of FR and NIR-inducible dimerizers and heterodimerizers is currently limited to optogenetic systems that use phycocyanobilin as a chromophore [8] and optogenetic heterodimerizer systems that use biliverdin IXα (BV) as a chromophore [10–12]. BV-based optogenetic constructs are advantageous over phycocyanobilin-based ones because BV is naturally available in mammalian tissues as a product of heme catabolism [8]. Therefore, an optimal opto-RTK should operate in FR/NIR spectral range and use BV as a chromophore.
Recently we developed two opto-RTKs, called Dr-TrkA and Dr-TrkB, by fusing intracellular domains of RTKs, TrkA and TrkB, to a dimeric photosensory core module (PCM) of Deinococcus radiodurans bacterial phytochrome (DrBphP) [7]. The full-length DrBphP consists of an N-terminal PCM and C-terminal histidine kinase domain. DrBphP-PCM consists of the PAS (Per/Arndt/Sim), GAF (cGMP phosphodiesterase/adenyl cyclase/FhlA) and PHY (phytochrome specific) domains. After absorption of FR light, BV located in the pocket of the GAF domain undergoes Z-E transition that induces structural changes propagating from the PCM to the native histidine kinase of DrBphP. Under NIR light, DrBphP adopts a Pr state in which the PHY and histidine kinase domains of the DrBphP dimer are close to each other. Absorption of FR light converts DrBphP into a Pfr state in which PHY domains are splaying apart [13].
We hypothesized that a “monomerizing” movement of PHY helices of the DrBphP-PCM can be coupled to the regulation of RTK activity in other than Trk RTK families, such as epidermal growth factor receptor (EGFR) and fibroblast growth factor receptor 1 (FGFR1).
As opposed to RTKs of Trk family, which form symmetric dimers upon activation, EGFR and FGFR1 form asymmetric dimers upon activation with relevant growth factors [14]. EGFR is the first member of the human epidermal growth factor receptor family (HER or ErbB family), playing a primary role in skin and liver regeneration [15]. FGFR1 belongs to the family of RTKs, which interact with fibroblast growth factors and regulate earliest stages of embryonic development and organogenesis [16, 17]. Aberrant activity of both EGFR and FGFR1 is associated with pathological cell proliferation including oncogenesis. EGFR and FGFR1 activate several downstream RTK signaling pathways, such as extracellular signal-regulated kinase 1 and 2 (ERK1/2), phosphoinositide-3-kinase (PI3K)/protein kinase B (PKB, also known as Akt) and phospholipase C gamma (PLCγ) pathways [18].
In this paper, by connecting DrBphP-PCM to EGFR and FGFR1 cytoplasmic domains we engineered opto-RTKs for EGFR and FGFR1, which are switched on with NIR and switched off with FR light. We demonstrated that these opto-RTKs activate ERK1/2-dependent immediate early gene transcription, PI3K/Akt pathway, PLCγ signaling and calcium transients. To better understand working mechanism of Dr-RTKs we discussed their structural features. Overall, our results demonstrate that DrBphP-PCM represents a versatile scaffold for engineering of opto-RTKs that are reversibly and non-invasively switchable with FR and NIR light.
Results
Design of Dr-EGFR and Dr-FGFR1 opto-RTKs.
Upon action of FR light DrBphP-PCM undergoes massive structural changes, resulting in the distance increase between the C-termini of the PHY-domain helices [13]. We hypothesized that a prolongation of the PHY-helices with the four-repeat rigid α-helical linkers -EAAAK- ((EAAAK)4), frequently used to achieve efficient separation of proteins [19–21], should allow to manipulate kinase domains fused these linkers. Previously, these linkers have been successfully used by us to design Dr-Trks [7]. We fused the DrBphP-PCM to the EGFR and FGFR1 cytoplasmic domains, consisting of the respective juxtamembrane and catalytic kinase part, via artificial (EAAAK)4 linkers, resulting in the opto-RTKs, termed Dr-EGFR and Dr-FGFR1, respectively. Both Dr-EGFR and Dr-FGFR1 were then anchored to the plasma membrane via an N-terminal myristoylation (Myr) signal (Supplementary data Fig. 1a,b). We hypothesized that because of the similar to Dr-Trk design, kinase activities of Dr-EGFR and Dr-FGFR1 constructs should be efficiently regulated with FR and NIR light.
Optimization of Dr-EGFR and Dr-FGFR1 expression levels.
Primary signaling pathway activated by all RTKs is an ERK1/2 pathway [18] in which the activated ERK1/2 phosphorylates transcription factor Elk-1 (Fig. 1). Analysis of the Elk-1 dependent firefly luciferase expression allows to analyze activation of ERK1/2 signaling in different cell lines [7]. This approach is frequently used for evaluation of opto-RTKs [3, 5, 7]. Overexpression of RTKs may lead to ligand-independent activation [22, 23]; and therefore, determining of expression level of opto-RTKs resulting in a high light-activation contrast should be performed before proceeding to more complex experiments.
To assess optimal expression of opto-RTKs, we used a PathDetect trans-reporting system (Fig. 1c,d). This system consists of the activation domain of Elk-1 fused with the yeast GAL4 DNA binding domain (DBD, 1–147 amino acid residues) in a transactivator pFA-Elk-1 plasmid. Expression of Elk-1-GAL4 DBD fusion was controlled by cytomegalovirus promoter. A pFR-Luc reporter plasmid encodes firefly luciferase under a synthetic promoter, containing the five upstream activating sites (5xUAS) of GAL4. Elk-1 phosphorylation by ERK1/2 leads to the GAL4 DBD dimerization. The dimeric GAL4 DBD binds the 5xUAS sequence in pFR-Luc and activates firefly luciferase expression, reporting the activation of ERK1/2 pathway.
We co-transfected PC6–3 cells with Dr-EGFR or Dr-FGFR1 mixed with pFR-Luc and pFA-Elk1 plasmids in different ratios and kept cells under FR or NIR illumination for 36 h (Supplementary data Fig. 2a,b). Initially, we tested three plasmid ratios, such as 5:100:5, 1:100:5 and 1:200:10, for opto-RTK, pFR-Luc and pFA-Elk1, respectively. These plasmid ratios were chosen because they worked well for the other opto-RTKs, such as Dr-TrkA and TrkB [7]. For Dr-EGFR, the activation contrast of 2-, 7.2- and 11-fold was obtained for the 5:100:5, 1:100:5 and 1:200:10 plasmid ratios. For Dr-FGFR1, the activation contrast of 4-, 5.2- and 13-fold was obtained for the 5:100:5, 1:100:5 and 1:200:10 plasmid ratios. The plasmid ratios 1:100:5 and 1:200:10 proved to be the most efficient for long-term ERK1/2 pathway activation for both opto-RTK.
Because these two ratios worked the best in PC6–3 cells, next, we tested only them in HeLa cells. As a result, with Dr-EGFR we observed the activation contrast of 3.1- and 9.6-fold,and with Dr-FGFR1 we observed the activation contrast of 5.1- and 43-fold for the 1:100:5 and 1:200:10 plasmid ratios, respectively (Supplementary data Fig. 3a,b). These results indicated that the decrease of the opto-RTK concentration led to the increase of the long-term ERK1/2 pathway activation.
We next tested Dr-EGFR and Dr-FGFR1 activity in cells illuminated with white light and cells kept in darkness. Dr-EGFR and Dr-FGFR1 were not activated with white light but stayed active in darkness (Supplementary data Fig. 4a,b). Although addition of exogenous BV improved the photodynamic contrast, Dr-EGFR and Dr-FGFR1 also exhibited light-activation without it (Supplementary data Fig. 5a,b).
Comparison of ERK1/2 activation between Dr-EGFR and Dr-FGFR1.
The analysis of the Elk-1 dependent luciferase expression also reported a substantial difference in the ERK1/2 pathway activation between the Dr-FGFR1 and Dr-EGFR opto-RTKs (Fig. 1c,d). With the plasmid ratio of 1:200:10, Dr-FGFR1 activated the ERK1/2 pathway 11-fold stronger in PC6–3 cells and 4.8-fold stronger in HeLa cells than Dr-EGFR (Fig. 1c,d). Moreover, Dr-FGFR1 demonstrated the higher activation contrast in both cell lines, such as 13-fold versus 11-fold for Dr-EGFR in PC6–3 cells and 43-fold versus 9.6-fold for Dr-EGFR in HeLa cells.
Likely, more efficient upregulation of Elk-1 transcription with Dr-FGFR1 was observed because native FGFR1 causes the sustained long-term ERK1/2 activation, as opposed to native EGFR [18]. Moreover, the upregulation of ERK1/2 signaling by EGFR can be negatively regulated by the larger number of negative feedbacks, causing the faster ERK1/2 activity decay [24].
Light-induced phosphorylation of Dr-EGFR, Dr-FGFR1, ERK1/2 and Akt.
The PathDetect trans-reporting system detects RTK activation in a matter of hours. To characterize activation of Dr-EGFR and Dr-FGFR1 on a minute time scale, we next studied lysates of HeLa cells transfected with Dr-EGFR and Dr-FGFR1 plasmids using Western blot. We directly analyzed the phosphorylation of Dr-EGFR and Dr-FGFR1 and their downstream signaling target ERK1/2 (Fig. 2). NIR illumination of the cells for only 5–10 min led to the phosphorylation of Dr-EGFR (Fig. 2a, Supplementary data Fig. 6,7), Dr-FGFR1 (Fig. 2d, Supplementary data Fig. 6,7) and ERK1/2 proteins (Fig. 2b,e, Supplementary data Fig. 6,7). We also analyzed activation of PI3K/Akt signaling. NIR illumination for 1–5 min led to the Akt phosphorylation (Fig. 2c,f, Supplementary data Fig. 6,7). These results demonstrated that the Dr-EGFR and Dr-FGFR1 chimeric molecules can be non-invasively triggered with NIR light in a matter of minutes.
Figure 2. Phosphorylation of Dr-EGFR, Dr-FGFR1 and downstream Akt and ERK kinases.
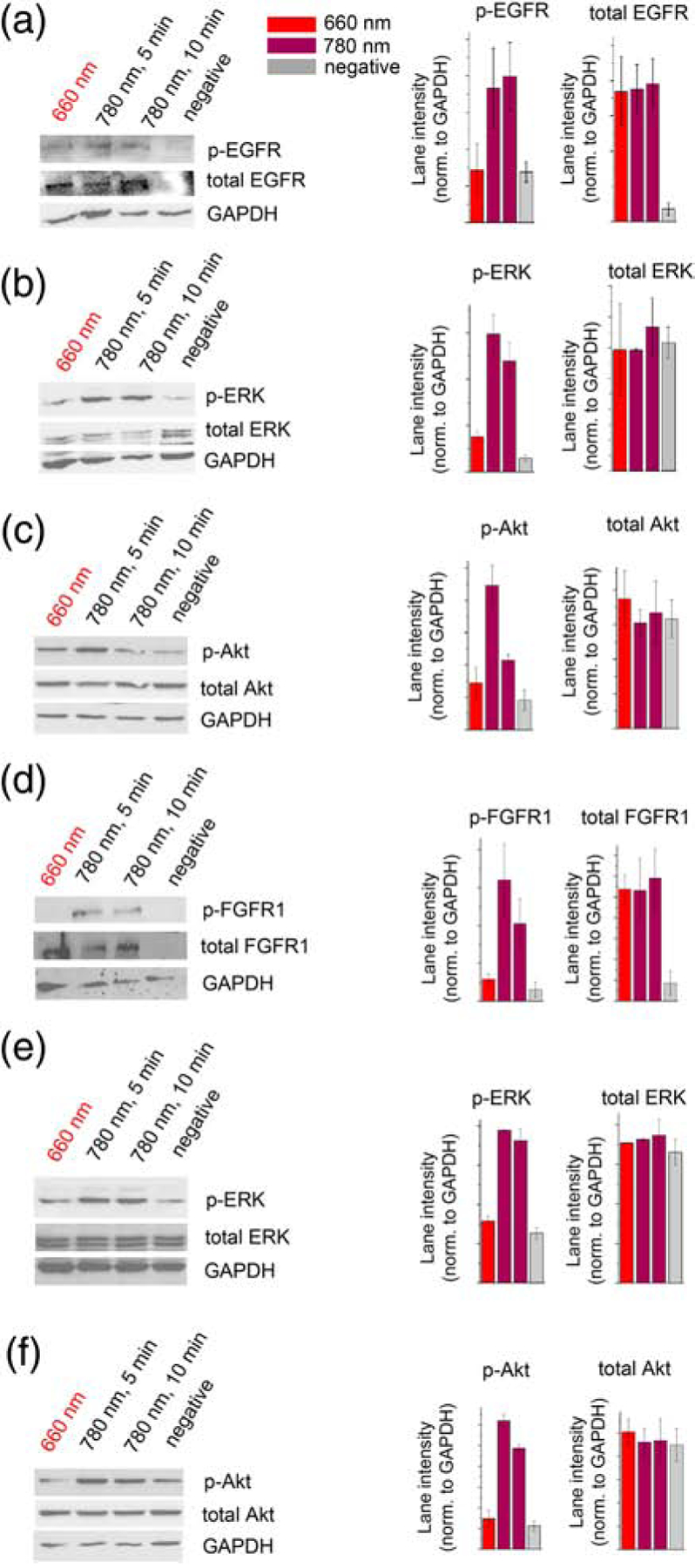
(a) Western blots of phospho-EGFR, total EGFR and GAPDH in lysates of Dr-EGFR-transfected HeLa cells, and quantification of the lane intensities normalized to GAPDH. (b) Western blots of phospho-ERK, total ERK and GAPDH in lysates of Dr-EGFR-transfected HeLa cells, and quantification of lane intensities normalized to GAPDH. (c) Western blots of phospho-Akt, total Akt and GAPDH in lysates of Dr-EGFR-transfected PC-3 cells, and quantification of lane intensities normalized to GAPDH. In (a)-(c) cells transfected with Dr-EGFR were either kept under 660 nm light, activated for 1, 5 or 10 min with 780 nm light, or did not contain Dr-EGFR (negative control). (d) Western blots of phospho-FGFR1, total FGFR and GAPDH in lysates of Dr-FGFR1-transfected HeLa cells, and quantification of lane intensities normalized to GADPH. (e) Western blots of phospho-ERK, total ERK and GAPDH in lysates of Dr-FGFR1-transfected HeLa cells, and quantification of lane intensities normalized to GAPDH. (f) Western blots of phospho-Akt, total Akt and GAPDH in lysates of Dr-FGFR1-transfected PC6–3 cells, and quantification of lane intensities normalized to GAPDH. In (d)-(f) cells transfected with Dr-FGFR1 were either kept under 660 nm light, activated for 1, 5 or 10 min with 780 nm light, or did not contain Dr-FGFR1 (negative control). 25 μM BV was added to culture medium in all experiments. Error bars represent s.d., n=3 experiments.
Reversibility of light activation of ERK1/2 pathway.
To test reversibility of opto-RTKs action, we first analyzed ERK1/2 phosphorylation in HeLa cells expressing Dr-FGFR1. In the first cycle of activation, the cells kept under FR light (Fig. 3a; lane (1)) were triggered for 5 min by NIR light (Fig. 3a; lane (2)). After that, the cells were inactivated with FR light for 5, 10 or 30 min (Fig 3a; lanes (3, 5, 7)). A parallel set of the transfected and initially induced with NIR light for 5 min cells were then kept under NIR light for additional 5, 10 and 30 min, respectively, to serve as a negative control of ERK1/2 inactivation (Fig. 3a; lanes (4, 6, 8)). Although the ERK1/2 activity is tightly regulated by a number of phosphatases and negative feedbacks that facilitate its deactivation [24], FR light substantially accelerated the ERK1/2 inactivation through the switching off Dr-FGFR1 (Fig. 3a,b). The ERK1/2 signaling was attenuated to the initial level already after 5 min of FR illumination (Fig. 3a, lane (3)), whereas in the control set of cells ERK1/2 remained active even after 30 min of NIR illumination.
Figure 3. Reversibility of Dr-FGFR1 and Dr-EGFR activation.
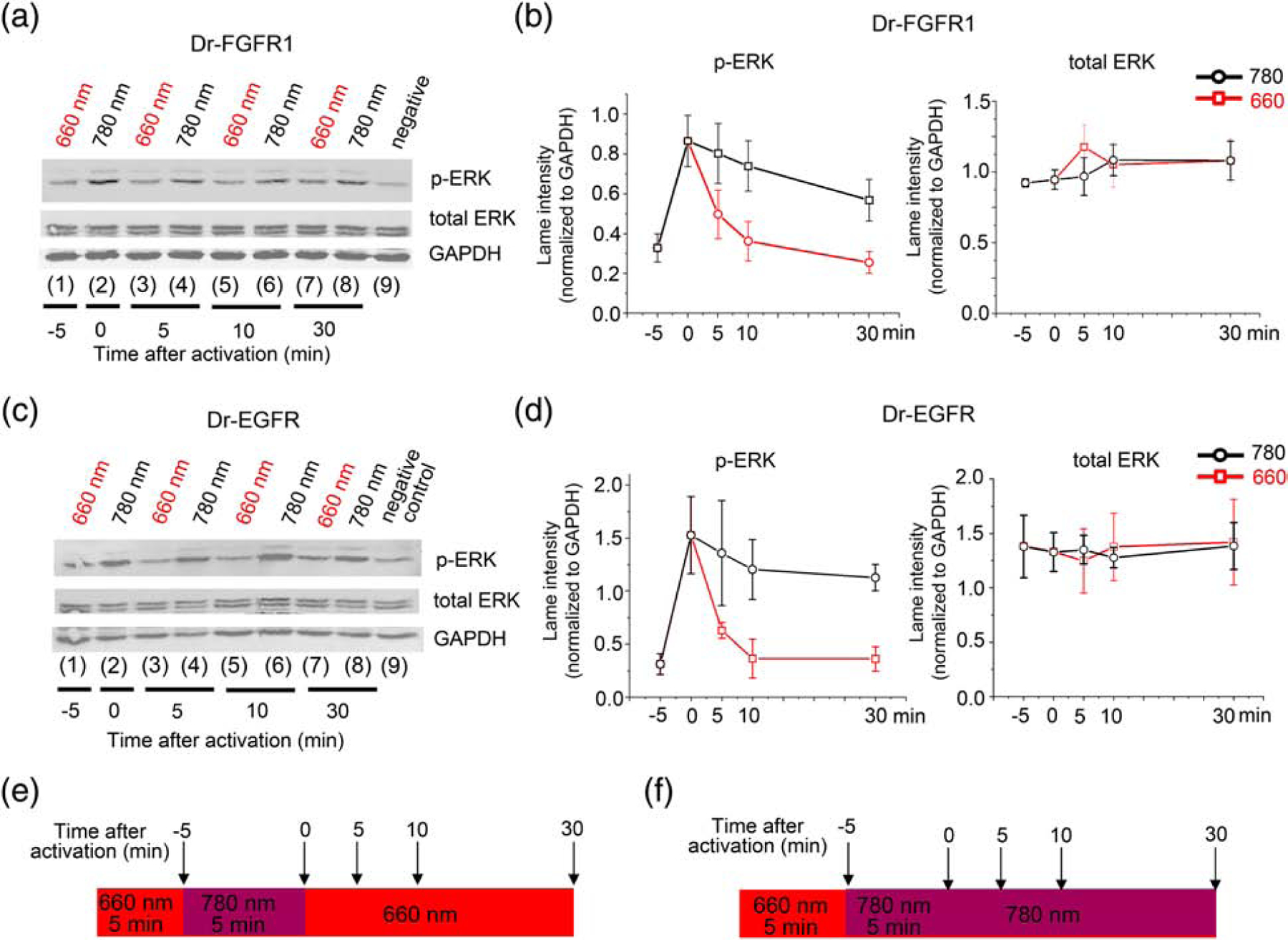
(a) Western blot of phospho-ERK1/2, total ERK1/2 and GAPDH in HeLa cell lysate. From left to right: cells co-transfected with Dr-FGFR1 and carrier DNA (1) kept under 660 nm light before induction; (2) induced for 5 min with 780 nm light; (3), (5) and (7) inactivated with 660 nm light for 5, 10 and 30 min, respectively, after 5 min of 780 nm induction. (4), (6) and (7) are controls of inactivation: cells were kept under 780 nm light for additional 5, 10 and 30 min after step (2). (9) are cells without Dr-FGFR1 as a negative control. (b) Quantification of lane intensities of phospho-ERK and total ERK, normalized to GAPDH represented as graphs. Left: downregulation of phospho-ERK1/2 upon action of 660 nm light (red line) and phospho-ERK1/2 downregulation in constantly activated with 780 nm light cells (black line). Right: total ERK1/2 levels. (c) Western blot of phospho-ERK, total ERK and GAPDH in lysate of HeLa cells co-transfected with Dr-EGFR and reporter plasmids and treated with light as in (a). (d) Quantification of lane intensities of phospho-ERK1/2 and total ERK, normalized to GAPDH represented as graphs. Left: downregulation of phospho-ERK1/2 upon action of 660 nm light (red line) and phospho-ERK1/2 downregulation in constantly activated with 780 nm light cells (black line). Right: total ERK1/2 levels. (e) Illumination pattern: inactivation of ERK signaling with 660 nm FR light was followed by 5 min 780 nm NIR stimulus; after that ERK signaling was inactivated with 660 nm light for 30 min. Samples were collected at 5, 10 and 30 min time points. (f) Illumination pattern for a negative control: inactivation of ERK signaling with 660 nm FR light was followed by 5 min 780 nm NIR light stimulus and by additional 30 min of 780 nm light. 25 μM BV was added to culture medium in all experiments. Error bars represent s.d., n=3 experiments.
The similar results were obtained in HeLa cells expressing Dr-EGFR, with the immediate fast inactivation of ERK1/2 signaling by FR illumination (Fig. 3c,d). The ERK1/2 signaling was attenuated to the initial level already after 5 min of FR illumination (Fig.3a, lane (3)), whereas in the control cells ERK1/2 remained phosphorylated even after 30 min of NIR illumination. Illumination of cells consisted of the overnight FR light, followed by 5 min of activating NIR light, followed by 30 min inactivating FR light (Fig. 3e) or, for a negative reversibility control, of the overnight FR light, followed by 35 min of activating NIR light (Fig. 3f).
Overall, these data revealed that the NIR-light activated opto-RTKs can be efficiently inactivated with FR light on a minute time scale. The observed reversibility of the on-off light-triggering is the important property of the developed Dr-EGFR and Dr-FGFR1.
Light control of PLCγ activity.
Phospholipase Cγ (PLCγ) is activated by FGFR1. PLCγ contains a prototypical Src Homology 2 (SH2) containing substrate, which is recruited to the one phosphorylated FGFR1 molecule and then is phosphorylated by another FGFR1 molecule [26]. Phosphorylation of Y766 residue at the C-terminus of FGFR1 is primarily responsible for recruitment of PLCγ to FGFR1 and subsequent PLCγ phosphorylation at Y783. Phosphorylated PLCγ translocates to the plasma membrane and catalyzes hydrolysis of phosphatidylinositol-4,5-biphosphate (PIP2) to diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3) (Fig. 4a) [27]. To test whether Dr-FGFR1 is able to trigger the PLCγ signaling we studied PLCγ phosphorylation in HeLa cells by Western blot. The Dr-FGFR1-expressing cells were activated with NIR light for 5 or 10 min and its lysate was examined with anti-phospho PLCγ antibodies. The Western blot showed that Dr-FGFR1 activates PLCγ 3-fold after 5 min of NIR illumination, but during additional 5 min of illumination the PLCγ phosphorylation is reduced almost to background level (Fig. 4b,c, Supplementary data Fig. 8), likely because of negative feedbacks regulating PLCγ activity.
Figure 4. Activation of PLCγ and Ca2+ transients with Dr-FGFR1.
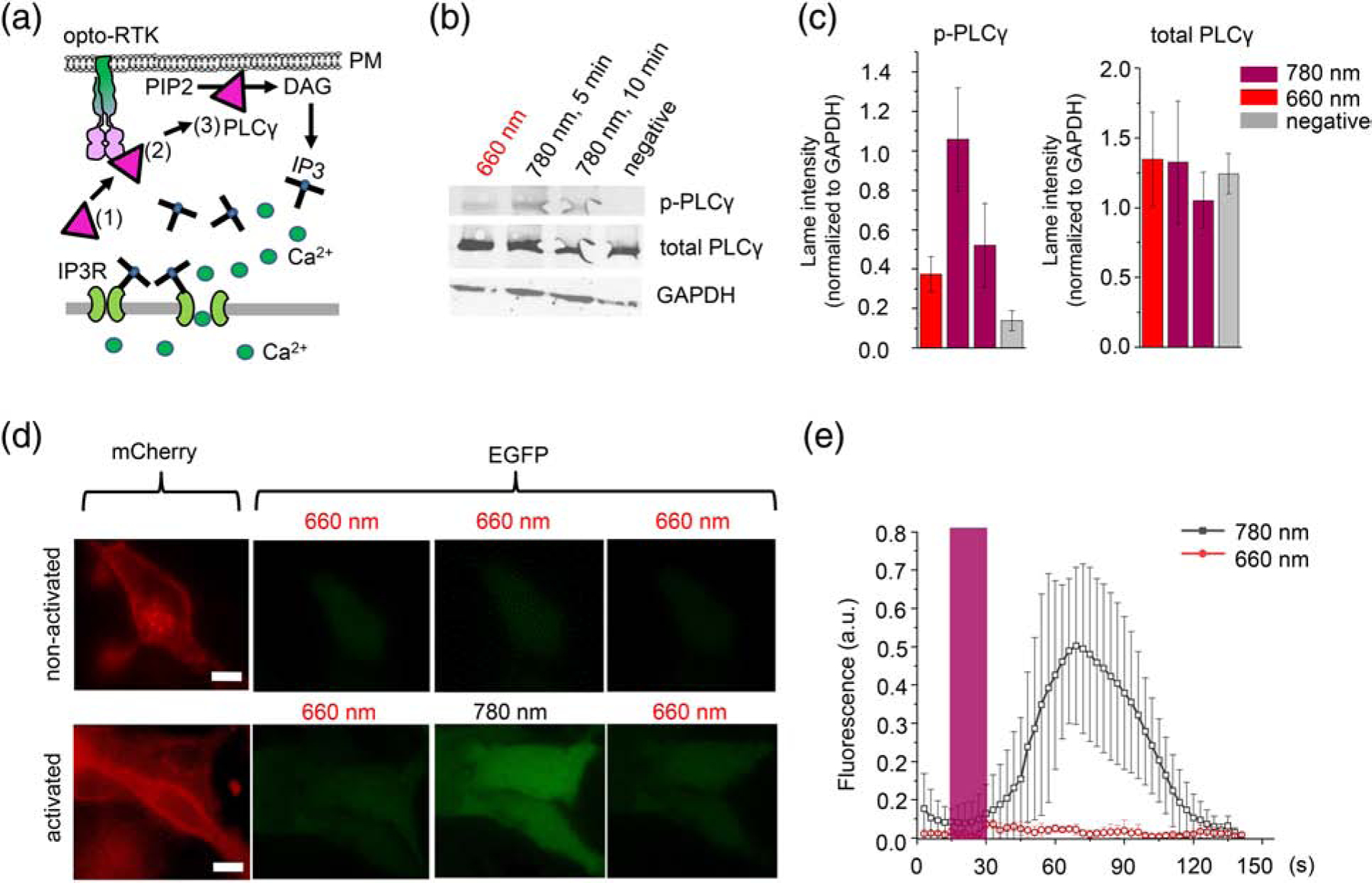
(a) Activation of PLCγ and Ca2+ transients by opto-RTK: (1) inactive PLCγ in the cytoplasm, (2) PLCγ is phosphorylated by opto-RTK, (3) PLCγ translocases to the plasma membrane and cleaves PIP2 to DAG and IP3, which activates IP3R channels in endoplasmic reticulum and induces Ca2+ entry into the cytoplasm. (b) Western blot of phospho-PLCγ, total PLCγ and GAPDH in HeLa cell lysate. Cells co-transfected with Dr-FGFR1 and carrier DNA were either kept under 660 nm light, activated for 5 or 10 min with 780 nm light, or did not contain Dr-FGFR1 (negative control). (c) Quantification of lane intensities of phospho-PLCγ and total PLCγ, normalized to GAPDH. (d) HeLa cells co-expressing Dr-FGFR1 with mCherry tag and Ca2+ biosensor GCaMP6m. Upper row: non-activated cells kept under constant 660 nm light. Bottom row: cells initially kept under 660 nm light were activated with 15 s pulse of 780 nm light. Cells in both rows are imaged in mCherry (left) and EGFP (right) channels. 780 nm light caused increase of GCaMP6m fluorescence (cells in the bottom row). Scale bar, 10 μm. (e) Changes of GCaMP6m fluorescence in Dr-FGFR1-expressing cells either kept under 660 nm light (red line) or stimulated with 780 nm light (black line) for 15 s (dark-grey bar). GCaMP6m fluorescence was normalized to baseline fluorescence. 25 μM BV was added to culture medium in all experiments. Error bars represent s.d., n=10 cells.
Light activation of cellular calcium transients.
Because PLCγ signaling is related to induction of cellular Ca2+, we next evaluated the possibility of multiplexing Dr-RTKs with green fluorescent protein (GFP)-based Ca2+ biosensor GCaMP6m [28]. Triggering of HeLa cells co-expressing Dr-FGFR1 and GCaMP6m with 15 s NIR light pulse caused an ~10-fold increase of GCaMP6m fluorescence, which then decreased after the light was changed to FR (Fig. 4d,e). From the beginning of the NIR pulse, the maximal Ca2+ level was achieved at ~70 s, with a rise half-time of ~30 s. In contrast, in cells constantly illuminated with FR light GCaMP6m fluorescence did not change (Fig. 4d,e). Ca2+ transients were reversed after application of FR light for 60 s, with a decrease half-time of ~20 s (Fig. 4d,e). Notably, imaging of GCaMP6m was spectrally compatible with activation and inactivation light of Dr-FGFR1, indicating that DrBphP-PCM-based opto-RTKs allow their crosstalk-free combination with the visible-light fluorescent proteins and biosensors.
Discussion
Organization of opto-RTKs strongly resembles active dimer of RTKs with dimeric PCM playing the role of extracellular receptor part and (EAAAK)4 playing the role of transmembrane (TM) domain. The resemblance is particularly striking due to a close correspondence of (EAAAK) −4 linker length to the length of TM regions of many RTKs. Note that earlier, variation of the linkers in Dr-Trks has shown that linkers with both 3 and 5 α-helical repeats demonstrated substantially lower kinase activity than (EAAAK)4 [7]. The structural rearrangements occurring in DrBphP-PCM upon FR-NIR illumination are well known (Fig. 5a), and the separation of the PHY domain C-termini accompanying Pr/Pfr transition changing from 14 to 40 Å is sufficient to separate catalytic kinase domains attached to the PHY helices of DrBphP-PCM.
Figure 5. Structural modules of DrBphP and RTKs used for opto-RTK modeling.
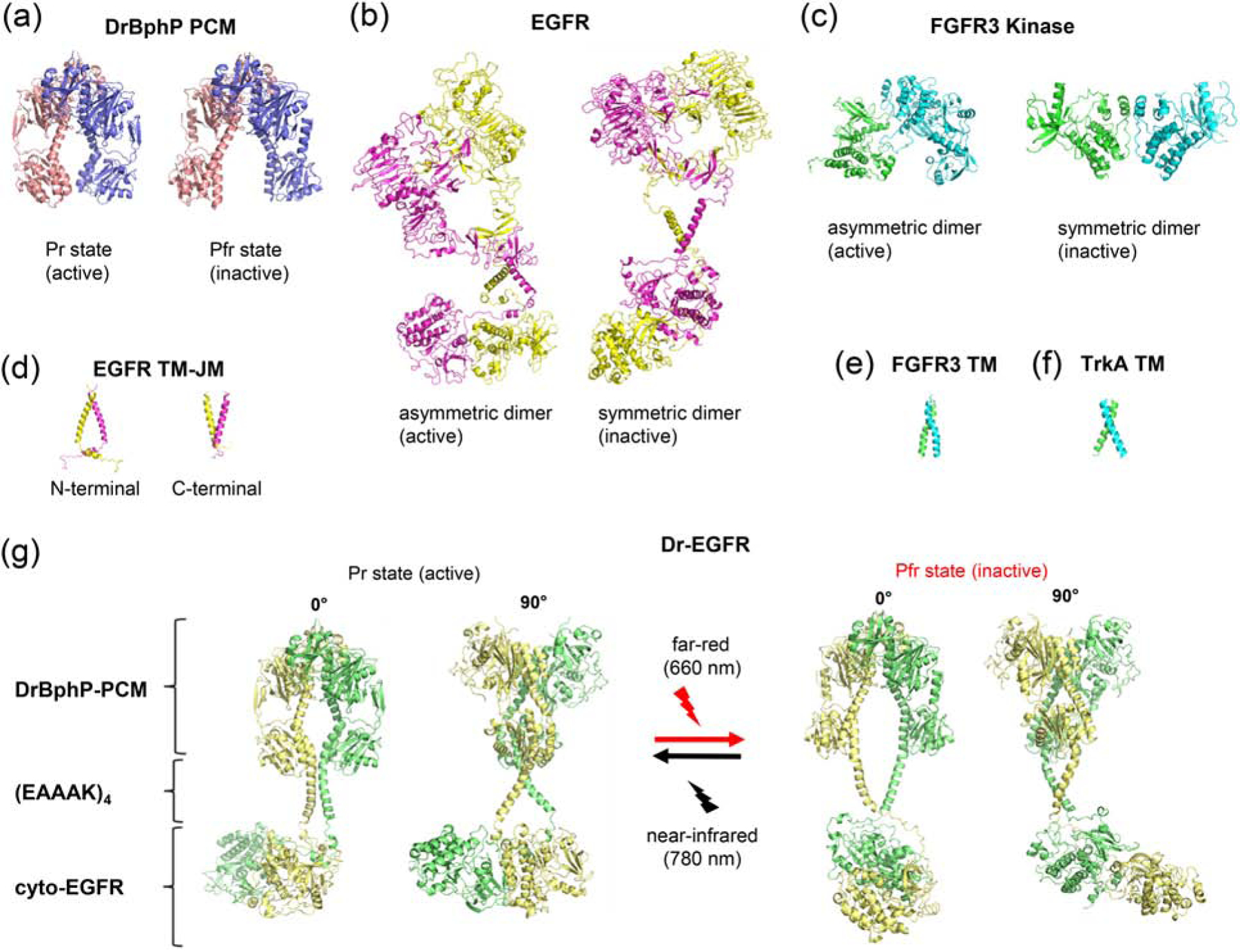
(a) X-ray structures of DrBphP-PCM in the active (Pr) (PDB 4O01) and inactive (Pfr) (PDB 4O0P) states [13]. (b) ab initio model of full-length EGFR [32] (c) X-ray structures of asymmetric (active) (PDB 3GQI [37]) and symmetric (inactive) (PDB 1FGK [35]) (d) NMR structures of transmembrane (TM) and juxtamembrane (JM) domains of EGFR in the active (PDB 2M20 [30]) and inactive (PDB 2M0B [30]) states. (e) NMR structure of TM of FGFR3 (PDB 2LZL [30]). (f) NMR structure of TM of TrkA (PDB 2N90 [36]). (g) Structural model of Dr-EGFR opto-RTK in its active (Pr) and inactive (Pfr) states composed of DrBphP-PCM, (EAAAK)4 and cytoplasmic EGFR JM and kinase domains.
The model of EGFR was proposed based on CryoEM data [29], NMR data for TM and JM domains [30, 31], and ab initio calculations for TM and JM regions [32] (Fig. 5b–g). Molecular dynamic simulations of TM regions of active and inactive states of EGFR suggested that they form distinctly different dimers. In active state, the extracellular domains of EGFR favor dimerization of the TM helices near their N-termini, followed by dimerization of the JM segments and formation of asymmetric (active) kinase dimer. In ligand-free inactive state, the extracellular domains of EGFR favor formation of C-termini dimer by TM domain helices. This results in dissociation and membrane burial of JM segments, and formation of symmetric (inactive) kinase dimer [18, 22, 33]. Both N- and C-terminal helices are stabilized by -GxxxG-motifs located at the respective terminus of TM domains. The distance between C-terminus of extracellular domains of EGFR dimer in active and inactive states have been calculated to be 29 and 47 Å, respectively, and the distance between C-terminus of TM domains to be 9 and 34 Å.
In the absence of experimental or ab initio data for the whole FGFR1, we had to rely on the results of structural studies of isolated receptor parts. Taking into account inherently dimeric nature of opto-RTKs, we excluded concentration-dependent and activation-based dimerization scenarios. Based on NMR data, it was proposed that unliganded FGFR3 dimer (structurally similar to FGFR1) is stabilized by the heptad motif located in the upper region of TM domain [34]. In the resulting TM dimer individual helixes are crossed at 23° angle and the distance between their C-terminus is ~10 Å (Fig. 5e). On ligand binding, the full-length FGFR3 dimer undergoes a further conformational transition, in which the TM domain dimer switches into the alternative structure stabilized by the N-terminal GG4-like motifs; individual helixes of TM dimer are crossed at 40° angle and the distance between their C-terminus is ~20 Å. The kinase domains of FGFR form an asymmetric dimer during receptor activation, whereas the symmetric kinase dimer is attributed to an auto-inhibited conformation (Fig. 5c) [14, 35]. Asymmetric kinase domain dimer of FGFR1 is different from asymmetric kinase domain dimer of EGFR [14]. This is not surprising, since FGFR kinases are activated via cross-phosphorylation of tyrosines on the activation loop, and not allosterically like EGFR [14].
By linking kinase and JM domains of EGFR and FGFR1 to DrBphP-PCM via (EAAAK)4 the following considerations must be kept in mind. First, the nature of (EAAAK)4-linkers, even though their length matches 20–21 amino acids of TM domains of EFGR and FGFR1, is different from the native TM domains of RTKs. The (EAAAK)4-linkers contain 4 positively and 4 negatively charged side groups that could potentially form salt bridges. Second, to provide for sufficient difference between opto-RTK basal and active states, DrBphP-PCM-(EAAAK)4 construct should be able to afford for adequate transition between active and inactive kinase dimers. Dr-FGFR1 exhibited higher than Dr-EGFR activation level, suggesting that behavior of the (EAAAK)4-linkers is closer to that of TM domains of FGFR1 where both the stabilizing GxxxG- and the heptad motifs are located in the upper region of TM domain (Fig. 5e), rather than of TM-domains of EGFR undergoing rearrangement between N- and C-terminal dimers (Fig. 5d). This suggestion is in agreement with the earlier observed high activation (35-fold) for the similar Dr-Trk constructs [7]. Indeed, NMR data revealed that dimers of TM domains of TrkA are very similar to those of FGFR with N-terminal dimer corresponding to active and GxxxG-motif dimer to inactive states of TrkA (Fig. 5f) [36].
Initially, we hypothesized that the modular engineering approach used to develop Dr-Trks opto-RTKs could be applied to kinase domains of the other RTKs to regulate their signaling with light. Indeed, this approach proved to be successful for engineering of FR-NIR opto-RTKs with kinase domains from other RTK families, such as EGFR and FGFR1. The developed Dr-EGFR and Dr-FGFR1 are fast activated by NIR light in tens of seconds. This activation is fully and fast reversed by FR illumination. The DrBphP-based opto-RTKs enabled efficient spectral multiplexing with EGFP-based biosensor. Transduction of the light-induced conformational changes of the developed opto-RTKs into the activation/inactivation of the C-terminal kinase domains can be explained on the basis of the RTK structures.
We anticipate that the opto-RTK engineering approach based on DrBphP-PCM will be further applied to develop other light-controllable enzymes and transcription factors whose natural activity depends on their dimeric states.
Materials and Methods
Molecular cloning.
The DrBphP-PCM prolonged with the C-terminal rigid α-helices (Dr-hel-4) was amplified from the previously described Myr-Dr-hel4-TrkA plasmid [7] and inserted via HindIII/XhoI sites into the pcDNA3.1+ plasmid. cDNA encoding human EGFR and FGFR1 cytoplasmic domains was PCR-amplified from the total HeLa cDNA prepared using a ProtoScriptII First Strand cDNA Synthesis Kit (NEB). Human RNA was purified from HeLa cells using a NucleoSpin RNA purification kit (Macherey-Nagel). Encoding EGFR and FGFR1 cytoplasmic domains PCR products were inserted via XhoI and XbaI sites downstream the DrBphP-PCM prolonged with the C-terminal rigid α-helices (Dr-hel-4). To generate mCherry-tagged Dr-FGFR1, a Myr-Dr-Cherry sequence was amplified from the Myr-mCherry-Dr-hel4-TrkA plasmid and inserted via HindIII/XhoI sites into the Dr-FGFR1 encoding plasmid. The protein sequences of the Dr-EGFR and Dr-FGFR1 constructs are shown in Supplementary data Fig. 1.
Mammalian cell culture and transfection.
PC6–3 cells were obtained from ATCC and cultured in RPMI-1640 medium supplemented with 10% horse serum (HS) and 5% fetal bovine serum (FBS) (both from Biowest). HeLa cells were obtained from ATCC and cultured in DMEM medium supplemented with 10% FBS and penicillin-streptomycin mixture (Gibco) at 37°C. For luciferase assay, 20,000 of PC6–3 cells were seeded in 0.5 ml medium per well in 24-well plates and transfected with 1 μg of opto-RTK and pFr-Luc and pFA-Elk-1 plasmids from the PathDetect trans-reporting system (Agilent) in mass ratios of 5:100:5, 1:100:5 and 1:200:10. Prior transfection, a Turbofect reagent (ThermoFisher Scientific) was added to plasmid DNA at a volume-to-mass ratio of 2:1. After 6 h of incubation, the transfection medium was changed to a serum-starving medium (RPMI with 2.5% HS and 25 μM BV). For induction of Elk-1 dependent luciferase transcription in HeLa cells without BV, culture medium was changed to DMEM with 10% FBS 6 h after transfection. Cells then were kept for 30 h under either 660 nm (0.5 mW cm-2) or 780 nm (0.5 mW cm−2) light. For Western blot, HeLa cells were seeded in 6-well plates and co-transfected with 4 μg of total DNA per well with opto-RTK and pcDNA3.1+ plasmids in a mass ratio of 1:5. 6 h after transfection, the medium was changed to DMEM with 1% FBS. For Ca2+ measurements, HeLa cells plated on Nunc glass-bottom dishes were co-transfected with 2.5 μg of total DNA plasmids encoding mCherry-Dr-FGFR1 and GCAMP6m in a mass ratio of 1:1 using Lipofectamine 2000 reagent.
Bioluminescence assay.
PC6–3 and HeLa cells transfected and kept under 660 nm (0.5 mW cm−2) or 780 nm (0.5 mW cm−2) light as described above were then lysed in 100 μl of lysis buffer (20 mM Tris-HCl, 10% glycerol, 0.1% Triton X-100, 1 mM PMSF, 0.1% β-mercaptoethanol, pH 8.0) for 30 min at room temperature on swinging platform shaker. Luciferase assay was performed in 96-well half-area white plates (Costar) by mixing 10 μl of cell lysate with 20 μl of firefly luciferase substrate (Nanolight Technology). Bioluminescence was measured immediately with a Victor X3 multilabel plate reader (Perkin Elmer), and data were analyzed with an OriginPro v. 8.6 software.
Immunoblotting and antibodies.
For immunoblotting of EGFR, FGFR1, ERK, Akt and PLCγ, HeLa cells were plated in 6-well plates and transfected at 90% confluence with Turbofect reagent (ThermoFisher Scientific). Transfection mixtures were prepared by dilution of 4 μg DNA with 400 μl DMEM medium and 6 μl of Turbofect reagent. For detection of ERK1/2 phosphorylation, a pcDNA-DrEGFR or Dr-FGFR1 plasmids was mixed with a pcDNA3.1+ at a mass ratio of 1:5. Turbofect/DNA complexes were formed for 15 min and added to the wells drop-wise. After 6 h the medium was changed to DMEM medium with 1% FBS and 25 μM BV. Then cells were kept under 660 nm light (0.5 mW cm−2) for additional 20 h. Dr-EGFR and Dr-FGFR1 were activated for relevant time intervals by transferring plates under 780 nm light (0.5 mW cm−2), whereas non-induced cells were kept under 660 nm light (0.5 mW cm−2). Induced and non-induced cells were put on ice, washed with ice-cold PBS, and lysed in 300 μl of ice-cold RIPA buffer (Thermo-Scientific) supplemented with phosphatase and protease inhibitors (ThermoFisher Scientific). Cell lysis was performed for 5 min. Lysates were centrifuged at 12,000 rpm for 20 min in an Eppendorf centrifuge at 4°C. For phospho-ERK detection, 20 μl of lysate per lane was loaded to 10% gel. For phospho-EGFR, phospho-FGFR1 and PLCγ detection, 30 μl of lysate per lane was loaded to 10% gel. Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked in 5% solution of non-fat dry milk for 1 h. Then the membranes were incubated overnight at +4°C with antibodies against phosphorylated ERK (1:2000, #2234, Cell Signaling Technology), phosphorylated EGFR (1:1000, #2234, Cell Signaling Technology), phosphorylated FGFR1 (1:1000, #9740, Cell Signaling Technology), Akt phosphorylated at Thr308 (1:1000, #9275, Cell Signaling Technology), or phosphorylated PLCγ (1:1000, #2821, Cell Signaling Technology) diluted in 5% solution of non-fat dry milk. Membranes were washed 4 times by TBS with 0.5% Tween and incubated with goat anti-rabbit HRP conjugate (1:2000) for 2 h at room temperature and then washed by TBS with 0.5% Tween. Bioluminescence was detected using a Clarity Western ECL Substrate (BioRad). Images were taken with a ChemiDoc imaging system. After detection of phosphorylated ERK, Akt and PLCγ, the membranes were stripped as described before [7] and re-stained with antibodies against total ERK, Akt or PLCγ (all from Cell Signaling Technology). For detection of EGFR and FGFR1, similar samples were transferred to two different membranes and stained separately with antibodies against phosphorylated and non-phosphorylated EGFR (#2232, Cell Signaling Technology) and FGFR-1(#3471, Cell Signaling Technology). GAPDH was used as loading control. Antibodies against GAPDH (sc-47724, Santa-Cruz) were used in 1:1000 dilution.
Phosphorylation of Akt.
PC6–3 cells were co-transfected with Dr-FGFR1 or Dr-EGFR encoding plasmids and empty pcDNA3.1+ plasmid in a 1:10 mass ratio. 6 h after cell transfection culture medium was changed to RPMI medium containing 10% HS, 5% FBS and 25 μM BV. Cells were kept under 660 nm light (0.5 mW cm−2) overnight and then serum-starved in RPMI medium supplemented with 1% HS and 25 μM BV for 4 h before induction with 780 nm light (0.5 mW cm−2). Cell lysates were analyzed using Western blot as described above.
Reversibility of Dr-FGFR1 and Dr-EGFR signaling.
HeLa cells were co-transfected with Dr-FGFR1 or Dr-EGFR and pcDNA3.1+ plasmids with a 1:5 mass ratio. Transfected cells were kept under 660 nm light (0.5 mW cm−2) and after that they were induced for 5 min with 780 nm light (0.5 mW cm−2). Then the cells were transferred to 660 nm light (0.5 mW cm−2) and were kept under for 5, 10 or 30 min. The parallel control set of cells was kept under 780 nm light (0.5 mW cm−2) for the same periods of time. Cell lysates then were analyzed with Western blot as described above.
Activation of calcium signaling by Dr-FGFR1.
For analysis of Ca2+ signaling, HeLa cells were co-transfected with the GCaMP6m and Myr-mCherry-Dr-FGFR1 plasmids in a mass ratio of 1:1. 24 h after transfection cells were serum-starved under the constant 660 nm illumination (0.5 mW cm−2) for 4 h. After that starving medium was changed to Hank’s balanced salt solution with Ca2+. Induction of Ca2+ transients was performed with the 15 s pulse of 780 nm light (0.5 mW cm−2). Imaging of GCaMP6m fluorescence changes was performed as previously described [7].
Supplementary Material
Research Highlights.
Optically-controlled receptor tyrosine kinases (opto-RTKs) allow regulation of cell signaling non-invasively using light.
Fusion of photosensory core module of DrBphP bacterial phytochrome to cytoplasmic domains of EGFR and FGFR1 results in opto-RTKs, termed Dr-EGFR and Dr-FGFR1.
Dr-EGFR and Dr-FGFR1 are reversibly switchable with far-red and near-infrared light that makes them spectrally compatible with probes operating in visible spectral range.
Photosensory core module of DrBphP represents a versatile molecular template for engineering of opto-RTKs of different families.
Acknowledgements
We thank J. Ihalainen (University of Jyväskylä, Finland) for the DrBphP gene, D. Lindholm (all from University of Helsinki, Finland) for the cell lines, and all members of the lab for the useful discussions. This work was supported by grants GM122567 and NS103573 from the US National Institutes of Health (NIH) and 322226 from the Academy of Finland. This project was also funded in part with US federal funds from the Frederick National Laboratory for Cancer Research, NIH contract HHSN261200800001E, and the Intramural Research Program of the NIH, Frederick National Laboratory, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2020.04.005
Declarations of Interest: none
References
- [1].Leopold AV, Chernov KG, Verkhusha VV. Optogenetically controlled protein kinases for regulation of cellular signaling. Chem Soc Rev. 2018;47:2454–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Maruyama IN. Mechanisms of activation of receptor tyrosine kinases: monomers or dimers. Cells. 2014;3:304–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Grusch M, Schelch K, Riedler R, Reichhart E, Differ C, Berger W, et al. Spatio-temporally precise activation of engineered receptor tyrosine kinases by light. EMBO J. 2014;33:1713–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chang KY, Woo D, Jung H, Lee S, Kim S, Won J, et al. Light-inducible receptor tyrosine kinases that regulate neurotrophin signalling. Nat Commun. 2014;5:4057. [DOI] [PubMed] [Google Scholar]
- [5].Kainrath S, Stadler M, Reichhart E, Distel M, Janovjak H. Green-Light-Induced Inactivation of Receptor Signaling Using Cobalamin-Binding Domains. Angew Chem Int Ed Engl. 2017;56:4608–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Reichhart E, Ingles-Prieto A, Tichy AM, McKenzie C, Janovjak H. A Phytochrome Sensory Domain Permits Receptor Activation by Red Light. Angew Chem Int Ed Engl. 2016;55:6339–42. [DOI] [PubMed] [Google Scholar]
- [7].Leopold AV, Chernov KG, Shemetov AA, Verkhusha VV. Neurotrophin receptor tyrosine kinases regulated with near-infrared light. Nat Commun. 2019;10:1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chernov KG, Redchuk TA, Omelina ES, Verkhusha VV. Near-Infrared Fluorescent Proteins, Biosensors, and Optogenetic Tools Engineered from Phytochromes. Chem Rev. 2017;117:6423–46. [DOI] [PubMed] [Google Scholar]
- [9].Shcherbakova DM, Shemetov AA, Kaberniuk AA, Verkhusha VV. Natural photoreceptors as a source of fluorescent proteins, biosensors, and optogenetic tools. Annu Rev Biochem. 2015;84:519–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kaberniuk AA, Shemetov AA, Verkhusha VV. A bacterial phytochrome-based optogenetic system controllable with near-infrared light. Nat Methods. 2016;13:591–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Redchuk TA, Kaberniuk AA, Verkhusha VV. Near-infrared light-controlled systems for gene transcription regulation, protein targeting and spectral multiplexing. Nat Protoc. 2018;13:1121–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Redchuk TA, Omelina ES, Chernov KG, Verkhusha VV. Near-infrared optogenetic pair for protein regulation and spectral multiplexing. Nat Chem Biol. 2017;13:633–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Takala H, Lehtivuori H, Hammaren H, Hytonen VP, Ihalainen JA. Connection between absorption properties and conformational changes in Deinococcus radiodurans phytochrome. Biochemistry. 2014;53:7076–85. [DOI] [PubMed] [Google Scholar]
- [14].Bae JH, Schlessinger J. Asymmetric tyrosine kinase arrangements in activation or autophosphorylation of receptor tyrosine kinases. Mol Cells. 2010;29:443–8. [DOI] [PubMed] [Google Scholar]
- [15].Komposch K, Sibilia M. EGFR Signaling in Liver Diseases. Int J Mol Sci. 2015;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ornitz DM, Itoh N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015;4:215–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16:105–22. [DOI] [PubMed] [Google Scholar]
- [18].Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001;14:529–32. [DOI] [PubMed] [Google Scholar]
- [20].Leopold AV, Shcherbakova DM, Verkhusha VV. Fluorescent Biosensors for Neurotransmission and Neuromodulation: Engineering and Applications. 2019;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang WH, Herde MK, Mitchell JA, Whitfield JH, Wulff AB, Vongsouthi V, et al. Monitoring hippocampal glycine with the computationally designed optical sensor GlyFS. Nat Chem Biol. 2018;14:861–9. [DOI] [PubMed] [Google Scholar]
- [22].Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, et al. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 2009;137:1293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tsai CJ, Nussinov R. Emerging Allosteric Mechanism of EGFR Activation in Physiological and Pathological Contexts. Biophys J. 2019;117:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lake D, Correa SA, Muller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci. 2016;73:4397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Csanaky K, Hess MW, Klimaschewski L. Membrane-Associated, Not Cytoplasmic or Nuclear, FGFR1 Induces Neuronal Differentiation. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Huang Z, Marsiglia WM, Basu Roy U, Rahimi N, Ilghari D, Wang H, et al. Two FGF Receptor Kinase Molecules Act in Concert to Recruit and Transphosphorylate Phospholipase Cgamma. Mol Cell. 2016;61:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cocco L, Follo MY, Manzoli L, Suh PG. Phosphoinositide-specific phospholipase C in health and disease. J Lipid Res. 2015;56:1853–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mi LZ, Lu C, Li Z, Nishida N, Walz T, Springer TA. Simultaneous visualization of the extracellular and cytoplasmic domains of the epidermal growth factor receptor. Nat Struct Mol Biol. 2011;18:984–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, et al. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell. 2013;152:543–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bocharov EV, Lesovoy DM, Pavlov KV, Pustovalova YE, Bocharova OV, Arseniev AS. Alternative packing of EGFR transmembrane domain suggests that protein-lipid interactions underlie signal conduction across membrane. Biochim Biophys Acta. 2016;1858:1254–61. [DOI] [PubMed] [Google Scholar]
- [32].Arkhipov A, Shan Y, Das R, Endres NF, Eastwood MP, Wemmer DE, et al. Architecture and membrane interactions of the EGF receptor. Cell. 2013;152:557–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Landau M, Ben-Tal N. Dynamic equilibrium between multiple active and inactive conformations explains regulation and oncogenic mutations in ErbB receptors. Biochim Biophys Acta. 2008;1785:12–31. [DOI] [PubMed] [Google Scholar]
- [34].Bocharov EV, Lesovoy DM, Goncharuk SA, Goncharuk MV, Hristova K, Arseniev AS. Structure of FGFR3 transmembrane domain dimer: implications for signaling and human pathologies. Structure. 2013;21:2087–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mohammadi M, Schlessinger J, Hubbard SR. Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell. 1996;86:577–87. [DOI] [PubMed] [Google Scholar]
- [36].Franco ML, Nadezhdin KD, Goncharuk SA, Mineev KS, Arseniev AS, Vilar M. Structural Basis of the Transmembrane Domain Dimerization in the Activation Mechanism of TrkA by NGF. 2019:721233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bae JH, Lew ED, Yuzawa S, Tome F, Lax I, Schlessinger J. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell. 2009;138:514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.