

Graphical abstract
By repeatedly selecting and pooling the most rapidly outgrowing clones during multiple single cell subcloning rounds, the time required for subcloning was reduced from 3 to 2 weeks and the cloning efficiency significantly increased. Transcriptome analyses of the resulting host cell lines revealed a diverse set of pathways differentially enriched in each host cell line treated, with the only shared DE pathway related to changes in extracellular matrix. This indicates that cells struggle with the lack of cell-to-cell communication in isolation during subcloning.
Abbreviations: CHO, Chinese hamster ovary; CoI, clusters of interest; DE, directed evolved; ECM, extracellular matrix; ES, enrichment score; FACS, fluorescent-activated cell sorting; GSEA, gene set analysis; LDC, limiting dilution cloning; POI, product of interest; NES, negative enrichment score; PCA, principal component analysis; PC, principal component; RNA-Seq, RNA sequencing; SCC, single cell cloning; lfcSE, logfoldstandard error
Keywords: Chinese Hamster Ovary Cells, CHO cells, CHO, Cell line development, Single Cell Cloning, Subcloning, Single Cell Subcloning, Directed Evolution, RNA Sequencing, RNA-Seq, Growth enhancement, Growth improvement, FACS, Fluorescent-activated cell sorting, Cell sorting
Abstract
Chinese Hamster Ovary (CHO) cells are the working horse of the pharmaceutical industry. To obtain high producing cell clones and to satisfy regulatory requirements single cell cloning is a necessary step in cell line development. However, it is also a tedious, labor intensive and expensive process. Here we show an easy way to enhance subclonability using subcloning by single cell sorting itself as the selection pressure, resulting in improved subcloning performance of three different host cell lines. These improvements in subclonability also lead to an enhanced cellular growth behavior during standard batch culture. RNA-seq was performed to shed light on the underlying mechanisms, showing that there is little overlap in differentially expressed genes or associated pathways between the cell lines, each finding their individual strategy for optimization. However, in all three cell lines pathways associated with the extracellular matrix were found to be enriched, indicating that cells struggle predominantly with their microenvironment and possibly lack of cell-to-cell contact. The observed small overlap may hint that there are multiple ways for a cell line to achieve a certain phenotype due to numerous genetic and subsequently metabolic redundancies.
1. Introduction
Chinese Hamster Ovary (CHO) cells are one of the most important and commonly used industrial production organisms since the late 1980ies [1]. Advantages of using CHO as a production system are that cells can be grown in chemically defined media and in suspension, that few human viruses propagate in CHO, that several non-antibiotic selection and amplification systems are available and that proteins produced in CHO possess human-like glycosylation [1] Due to these advantages CHO established itself as the predominant production system for biological therapeutics, mainly for monoclonal antibodies (mAb), where 84% of currently approved mAbs are produced in CHO [2].
An important step to achieve commercial production of therapeutic proteins is the generation of a production cell line able to produce the product at high yields and of satisfactory quality. While it is in the interest of the industry and the patient to speed this process up as far as possible, there are also safety and regulatory considerations and rules that require confirmation of monoclonality. Furthermore, subcloning inherently is a limiting factor in promoting rapid cell line development, as both the cloning efficiency, that is the percentage of surviving clones per plate, and the speed of their outgrowth are low [3].
Single Cell Cloning (SCC) is performed during cell line development to ensure that selected cells are producing the POI in a reproducible manner and with the required quality attributes [4]. A typical process of cell line establishment involves random integration of the gene of interest and amplification by a selection system, both steps which cause clonal heterogeneity. In addition, the genome of CHO, as a rapidly growing cell line, exhibits rapid genetic changes, due to random mutations, genetic drifts and chromosomal rearrangements [5], [6], [7], [8], [9]. As high-producing clones occur rarely in heterogenous cell populations, and as they often have a growth disadvantage due to the large amount of cellular resources that is used for recombinant protein production [10], SCC is an essential step to prevent overgrowth of low- or non-producers and to enable stable survival of the high producers. For these reasons, a cell line development campaign can involve the screening of thousands of clones, a process that is labor, time and cost intensive, and may take up to 6 months even in streamlined industrial pipelines that use robotics [1], [3]. These are all strong motivations to improve SCC, where one aim is to increase the percentage of outgrowing clones and the speed at which outgrowth is achieved and in turn to streamline the cell line development.
The simplest way to perform SCC for cell culture is limiting dilution cloning (LDC) where cells are diluted so that statistically less than one cell is seeded per well. Although easy to perform without any dedicated equipment, LDC is a random approach to obtain high producers and thus requires a large number of cells to be screened, to ensure the presence of a sufficient number of high producing clones that can be further screened for product quality and stability. Moreover, using LDC the risk that the obtained cell line originated from more than one single cell is higher compared to the alternative methods. Using fluorescent-activated cell sorting (FACS) in combination with a suitable staining protocol [11] it is possible to focus specifically on the producers, by removing low or non-producers, thus reducing the total number of cells that need to be screened [12]. Deposition of single cells into microtiter plates by FACS has also been used simply for proof of monoclonality [3]. The main disadvantage of cell sorting is that the procedure may further reduce the outgrowth of subclones, due to the shear stress cells are exposed to during sorting and deposition into wells.
To improve clone outgrowth, multiple strategies can be used. One is the use of conditioned media or media additives that promote cell growth [13], [14]. Such additives are commercially available, but may be expensive and not chemically defined. Other options are the use of equipment such as cell printers or microfluidic systems. In addition, robotics and automation may enable the rapid handling of large numbers of clones and plates to help reliable identification of stable high producers [15], [16], [17], [18], [19], [20]. Recent research is focusing on improving the methods for SCC [21], [22] or ensuring the statistical evaluation of clonality [23], [24], [25] to meet regulatory requirements. However, very few if any published studies have systematically addressed the generation of a better host cell line with improved SCC properties.
Here we show a simple and straightforward way to improve the ability of CHO to rapidly and efficiently grow from the single cell stage using a directed evolution approach using rapid outgrowth as the evolutionary pressure (Fig. 1). Three different host cell lines were exposed to this treatment, with all three cell lines showing improvement in outgrowth of visible colonies within a 2-week period. Further experiments showed that improvements in growth behavior were also obtained in standard batch cultures. RNA Sequencing (RNA-Seq) demonstrated that each cell line achieved its observed phenotype by regulating individual genes, highlighting the ability of mammalian cells to achieve the same end via multiple routes.
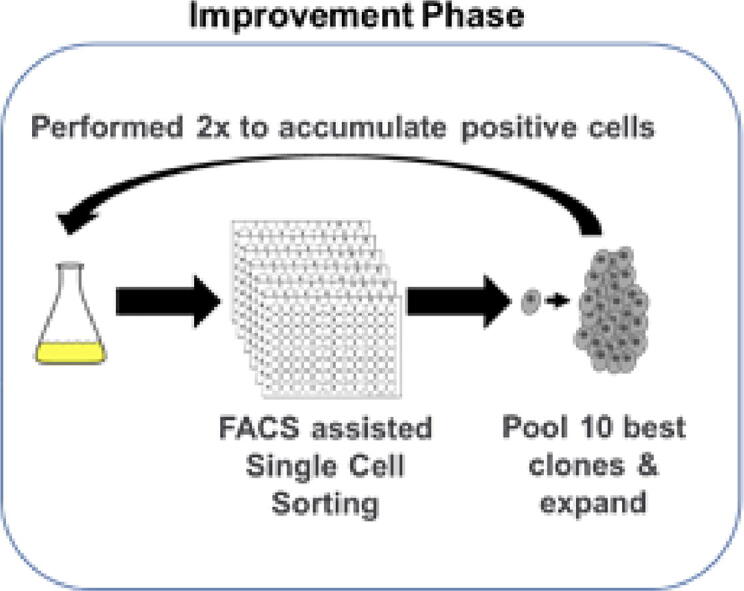
Fig. 1.

Process of directed evolution for SCC enhancement. Cells are single cell sorted by FACS and after outgrowth, the 10 fastest growing clones are pooled to minimize possible clonal effects. The whole process was performed twice to accumulate positive cells. Afterwards the evolved cells were compared to the starting cell line with (1) another round of single cell sorting to compare colony outgrowth, (2) growth characterization in batch cultures and (3) RNA sequencing to determine possible genetic mechanism for future rational cell line design.
2. Material & methods
2.1. Cell culture
All cell lines were routinely cultivated as suspension cultures in TPP TubeSpin® bioreactors (TPP Techno Plastic Products, Switzerland) in CD CHO media (Thermo Fisher Scientific, USA) with supplements as described below at 37 °C, 7% CO2, 85% humidity and 220 rpm shaking with a shaking diameter of 25 mm. Cells were passaged every 3–4 days. CHO-K1 cells (ECACC-CCL61) (“K1 8 mM”) were adapted to serum-free and suspension growth in house [26]. Cultures of these cells were supplemented with 8 mM GlutaMAX (Thermo Fisher Scientific) and 1:500 Anti-clumping agent (Thermo Fisher Scientific). Media for CHO-K1 cells adapted to 0 mM l-Glutamine, isolated from afore-mentioned CHO K1 [27], (“K1 0 mM”) were supplemented only with 1:500 Anti-clumping agent. CHO-K1 Hy (“K1 Hy”) cells were received from Cytiva and grown in medium supplemented with 6 mM GlutaMAX.
2.2. Single cell sorting enhancement
For each cell line, fifteen 96-well-plates (Greiner Bio-One, Austria) were prepared with 200 µL per well of the appropriate medium supplemented with Penicillin-Streptomycin (10.000 U/mL Penicillin, 10 mg/mL Streptomycin, VWR Chemicals, USA). Cell sorting was performed on a MoFlo® Astrios™ (Beckmann Coulter, USA), using a 488 nm laser to determine forward (FSC) and side scatter (SSC). Cells were live gated based on a FSC height to 488 SSC area gate to identify single cells and exclude doublets. The sorted single cells were incubated static at 37 °C, 7% CO2 and 85% humidity for 3 weeks. After three weeks the 10 colonies that had shown fastest outgrowth were pooled, centrifuged at 180×g for 8 min and the supernatant removed. Cells were resuspended in 1 mL of fresh medium and the suspension was transferred to 9 mL TPP TubeSpin® bioreactors and then incubated as described above. After 2 passages, the entire process was repeated.
2.3. Colony counting
Colonies were counted between 13 and 21 days after SCC. Colonies visible by eye were denoted further as “Big” and all colonies visible by microscope were denoted as “All”, where a colony had to consist of at least 5 cells. Moreover, the microscopy data was used to confirm data acquired by visual inspection. Statistical Analysis was performed in the statistical software R version 3.6.2 [28] using a student’s t-test. “*” indicates a p-value <0.05, “**” a p-value <0.01 and “***” a p-value <0.001.
2.4. Batch experiments
Cell lines were seeded at 0.2·106 cells/mL in 20 mL media in TPP TubeSpin® bioreactors and incubated as described above. Viable cell concentration and viability was measured each day by ViCell XR 2.04 (Beckman Coulter) based on the Trypan blue exclusion method. RNA for sequencing was taken on day 2, around 45 h after the start of the batch experiment. 5·106 cells were spun down at 200×g for 8 min, supernatant removed and the cell pellet dissolved in 600 µL TRI Reagent® (Merck KGaA, Germany) and stored at −80 °C for later isolation.
To investigate transient productivity, cells (passage 6) were transfected to express EPO-Fc using the Neon® transfection system with the Neon® transfection system 100 µL kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. In short, 5.5·106 cells were spun down (200×g for 8 min) and resuspended in 100 µL buffer R. After the addition of 15 µg of plasmid (Supplementary Fig. 1), cells were transfected by applying one pulse with 1700 V and 20 ms. A mock transfection was used as control. Cells were allowed to recover for 1.5 h at static 37 °C, 85% humidified air and 7% CO2. Afterwards cells were incubated as described above. Viability and product titer were quantified each day. Cells were spun down at 180×g for 8 min and the supernatant frozen at −20 °C for later quantification. Batches were characterized using an in-house R package vicellR version 0.1.9 [29].
2.5. EPO-Fc quantification
EPO-Fc concentration was quantified using the Octet® RED96e (FORTÉBIO, USA), equipped with Dip and Read™ Protein A Biosensors (Pall Corp, USA) according to the manufacturer’s recommendations. Samples were diluted 1:2 in non-supplemented CD-CHO media before measurement. Quantification was performed relatively to Trastuzumab (BioVison, USA), as no EPO-Fc standard was commercially available.
2.6. RNA sequencing
Total RNA was isolated using a Direct-zol™ RNA mini prep kit (Zymo Research, USA) according to the manufacturer’s instruction. rRNA depletion and library preparation of 2 replicates per sample was done with the in-house protocol established by the Vienna Biocenter Core Facility NGS Unit. Samples were sequenced as single end 100 bp reads on the HiSeq 2500 system (Ilumina, USA). Data is available under PRJEB37009.
2.7. Analysis of RNA sequencing data and differential gene expression
Raw sequences were trimmed of low quality reads and adapters using Trimmomatic 0.36 [30]. HiSat2, version 2.1.0 [31], was used to map processed reads to the Chinese hamster genome [32]. Reads mapped to coding genes were counted using the HTSeq python package [33]. Read counts were analyzed using the DESeq2 R package, version 1.24.0 [34]. Differential expression analysis was performed using the DESeq function of the package. Differentially expressed genes between samples were analyzed using the log2 fold change threshold 0 and BH adjusted p‐value 0.05 Genes with a foldchange difference of ≥1.5 and BH < 0.05 were deemed significantly differentially expressed. For further analysis, counts were normalized using the DESeq2′s variance stabilizing transformation (vst-normalisation).
Gene Set analysis (GSEA) was performed using GSEA 4.0.3 [35], [36]. GSEAPreranked settings were 1000 permutations, use of c2.cp.v7.0.symbols.gmt as geneset and no collapse. For visualization, Cytoscape 3.7.2 was used [37]. EnrichmentMap v3.2.1 [38] was used to generate the network and AutoAnnotate v1.3.2 [39] was used for grouping of pathways.
2.8. KEGG profiling of gene clusters
Hardclustering of genes according to their z-scores was done using the command heatmap of the R package ComplexHeatmap v2.0.0 [40] via the row_split option. Z-scores were calculated according to:
where x is the count value of a gene of a cell line replicate, µ the mean of all count values of this gene in all cell line replicates of interest and σ the corresponding standard deviation.
Identified genes were mapped to the KEGG pathways [41], [42], using R package KEGGprofile [43], as annotated to identify significantly enriched pathways. Significance was tested using a hypergeometric test with q < 0.05 using a conservative Benjamini-Yekutieli correction. Differentially expressed genes were indicated on the provided pathway maps according to their log2 fold change.
2.9. Metastudy analysis
Genes with a reported correlation to growth were obtained from [44]. Genes with a reported frequency of ≥2 were considered for analysis. The Mouse Genome Information database batch gene lookup tool (http://www.informatics.jax.org/batch) was used to obtain gene names. Upset plot was generated with the R package UpsetR v1.4.0 [45].
3. Results
3.1. Directed evolution improves clone outgrowth during single cell cloning
To improve the SCC ability of the three cell lines, single cells were deposited into microtiter wells by FACS and subsequently allowed to develop into colonies (Fig. 1). To avoid biased clonal effects of the resulting evolved cell line, the 10 biggest colonies of fifteen 96-well plates by visual inspection were pooled roughly 3 weeks after sorting, and again subcloned. After two rounds of this selection for rapid outgrowth, the effect of directed evolution on SCC performance was determined. To monitor SCC improvement, parental cell lines and pools generated by directed evolution (DE) were evaluated by again seeding fifteen 96-well plates per cell line. This approach led to a significant increase in the number of directly visible colonies per plate approximately 2 weeks after sorting in all 3 cell lines (Fig. 2a – “Big”). K1 8 mM showed the best starting SCC, followed by K1 0 mM and K1 Hy. This is inversely represented in the number of colonies, which improved 1.3-fold for K1 8 mM, 2.1-fold for K1 0 mM and 4.8-fold for K1 Hy, respectively. Due to further outgrowth of microscopic colonies, the differences in directly visible colonies are less a week later, however still significant with 1.1-fold more counted colonies for K1 8 mM, 1.4-fold for K1 0 mM and 2.7-fold increase for K1 Hy. Apart from the faster outgrowth of colonies to visible size, the total number of surviving colonies is also significantly improved in K1 8 mM and K1 Hy (Fig. 2a – “All”). For K1 0 mM the difference is not significant due to 4 outlier plates with lower numbers of colonies. Thus, for each DE cell line, a comparable or higher number of directly visible colonies is found 2 weeks after sorting as for the parental cell line after 3 weeks. This corresponds to a significant reduction in the timelines required for the different subcloning steps during a cell line development campaign.
Fig. 2.

Results of SCC improvement (a) Boxplots of colony outgrowth of parental cell lines and DE cell lines. X-axis displays day of analysis after single cell sorting. (b) VCD (blue) and viability (red) of the standard batch experiment with corresponding growth rates (c) and max VCD reached (d). To obtain results in (d) VCD values of K1 8 mM replicate 3 at 190 h was excluded, being an outlier in the measurement. Error bars represent 95% confidence interval. (e) Mean specific productivity (qP) against mean growth rate (µ) DE at the end of the cell line name indicate DE pools. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Improving single cell cloning ability improves overall growth phenotype
Next, we tested whether this enhancement in subcloning ability also translates into faster growth in standard cultivation. Therefore, a batch experiment comparing the parental cell lines to the DE pools was performed. Intriguingly, the different DE cell lines showed diverse phenotypes: 2 cell lines, K1 0 mM DE and K1 Hy DE, showed a significantly faster growth rate (Fig. 2c), whereas K1 8 mM DE reached higher maximum viable cell densities (VCD) after exponential phase (Fig. 2b & d). Growth rates improved 1.3-fold for K1 0 mM DE and 1.2-fold for K1 Hy DE. K1 0 mM DE shows a sudden loss of VCD and viability after 7 days in cultures, possibly due to media exhaustion due to the high cell densities achieved by this cell line and the minimal medium with only glucose as energy substrate.
Moreover, we investigated whether this enhanced growth and subclonability had an impact on the protein production capacity of DE cells and whether recombinant protein production affected growth. In all cases, transient EPO-Fc production resulted in non-significant reduction in growth, irrespective of whether the parental cell line or the DE cells were transfected (Supplementary Fig. 2). Overall, while there is a negative correlation between growth rate and specific productivity (Fig. 2e), the difference in qP between the parental cell lines and the DE cell lines is not significant.
3.3. RNA-Seq to identify mechanism of SCC improvement
Next, we analyzed the cellular transcriptome by RNA-Seq to see whether the improvement in SCC observed in the different cell lines has a common underlying mechanism. Using DESeq2 for analysis revealed that each cell line has a different number of genes significantly up or downregulated (Fig. 3a). Interestingly, only little overlap of differentially expressed genes can be observed (Fig. 3b, Supplementary Table S4). There are 44 common genes differentially expressed in all cell lines. However, for genes that are regulated in the same direction the numbers are further reduced (Fig. 3c, Table 1). Surprisingly the found overlaps are smaller than expected compared to the overall number of differentially expressed genes or the similarity in DE phenotype. Additionally, most of the overlapping genes are only identified by an LOC number, representing poorly annotated regions with unknown function.
Fig. 3.

RNA-Seq analysis (a) MA – plots of all 3 cell lines. Significantly upregulated genes are shown in red, significantly downregulated genes are shown in blue. Dotted lines represent the significance limit at 1.5-fold. Detailed information of genes can be found in Supplementary Table S1-S3 (b) Venn diagrams displaying the overlap of differentially expressed genes identified during RNA-Seq analysis. Overlapping genes can be found in Supplementary Table S4. Overlaps of upregulated and downregulated genes are displayed in (c). (d) PCA of the top 500 genes with the highest variance using vst-normalized counts. Black arrows indicate the change of the DE pool. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 1.
Overlap of positively and negatively differentially expressed genes found in all three cell lines.
| K1 8 mM |
K1 0 mM |
K1 Hy |
|||
|---|---|---|---|---|---|
| Gene Symbol | Gene Name | log2Foldchange | |||
| Upregulation | |||||
| Cda | Cytidine Deaminase | 4.00 | 1.38 | 2.88 | |
| LOC113831658 | twist-related protein 2 | 0.87 | 1.30 | 1.61 | |
| Downregulation | |||||
| Bank1 | B Cell Scaffold Protein with Ankyrin Repeats 1 | −1.22 | −0.64 | −1.82 | |
| C3 | Complement C3 | −0.73 | −1.20 | −1.81 | |
| LOC100756987 | NACHT, LRR and PYD domains-containing protein 1a | −0.64 | −0.75 | −2.81 | |
| LOC100763247 | olfactory receptor 1496 | −3.95 | −3.20 | −8.18 | |
| LOC100768488 | uncharacterized (protein coding) | −2.42 | −2.50 | −1.98 | |
| LOC100771648 | cytochrome P450 11B1, mitochondrial | −0.65 | −2.06 | −1.58 | |
| LOC103159507 | lncRNA | −1.30 | −2.77 | −3.16 | |
| LOC113831598 | lncRNA | −2.51 | −1.00 | −3.24 | |
Next a principal component analysis (PCA) of all expressed genes was performed (Fig. 3d). The three parental cell lines were clearly distinguished and their corresponding DE cell lines are clearly separated from the parent. Unexpectedly, the variation of the subclones relative to each other increased (indicated by black arrows in the plot), resulting in a greater distance between the DE cell lines than between the parental cell lines. This increase in distance indicates that each DE pool achieved improved subclonability by its individual way.
3.4. Pathway analysis
As the number of genes regulated in the same direction was unexpectedly low, pathway enrichment was used to see whether changes occurred in similar pathways. GSEA - Preranked was performed, based on the Wald statistic of the differentially expressed analysis. Using GSEA has the advantage that all expressed genes are used in the analysis, not relying on any cutoff as in similar tools [35]. Pathways are seen as significant if they possess a false discovery rate of < 25% and a nominal p-value of <0.01. Using Cytoscape 3.7.2 for enrichment visualization, each cell line has quite a unique pattern of pathway regulation to accomplish the observed phenotype in the DE pools (Supplementary Figs. 3–5). K1 8 mM and K1 0 mM both show a greater number of enriched pathways compared to K1 Hy. However, K1 8 mM seems to undergo more adaptation with an increased number of pathways up- and downregulated, whereas the other cell lines show mainly upregulation of pathways (Fig. 4a & b). As seen before, there is little overlap between the K1 Hy and the other two cell lines, with only 2 pathways enriched in all 3 cell lines (Fig. 4c). Interestingly, both of these are related to the extracellular matrix (ECM), however, with different regulation in each respective cell line. K1 8 mM and K1 Hy share even more pathways affecting the ECM, though again into different directions (Fig. 4d). Moreover, both show changes in pathways affecting DNA repair and synthesis. On the other hand, K1 8 mM and K1 0 mM share 102 enriched pathways (Fig. 4e & Supplementary Fig. 6). Again, ECM affecting pathways are observed, but also pathways dealing with energy production and ER/Golgi transport. K1 0 mM and K1 Hy again mainly share ECM pathways (Fig. 4e).
Fig. 4.

Pathway enrichment analysis (a) Venn diagram of found enriched pathways using GSEA with shared pathways regulated in the same direction displayed in (b). Shared enriched pathways are seen in (b-e). The plot in (c) is showing only a selected number of enriched pathways, the full list can be seen in Supplementary Fig. 6. All enriched pathways are found in Supplementary Table S5-S7.
3.5. Strategies for identifying underlying gene mechanism
The parental K1 8 mM performed best during SCC and had the highest growth rate of all used cell lines. Thus, the observed improvements after DE were much smaller compared to the other two cell lines, indicating that the transcriptome of the parental K1 8 mM was already much better suited for SCC from the beginning. To find possible mechanisms for SCC improvement, the expression level of differentially expressed genes of K1 0 mM and K1 Hy were compared to their corresponding expression level in K1 8 mM by hard-clustering of the z-scores of the vst-normalized counts. Genes that show regulation towards the expression level in K1 8 mM, were deemed Clusters of Interest (CoI) (Fig. 5a & b). These CoI contained 658 genes in K1 Hy and 236 genes in K1 0 mM, with an overlap of only 31 Genes (Fig. 5c), too few for any significant pattern to emerge (Supplementary Table S10). Of note is, however, the high percentage of lncRNAs in this group (26%). KEGG enrichment of the genes in the CoI was performed. Intriguingly, 2 enriched pathways per DE pool were identified (Table 2) with little obvious relevance to growth in K1 0 mM (Supplementary Fig. 9a & b). However, both pathways identified in K1 Hy can be associated with proliferation (Supplementary Fig. 9c & d).
Fig. 5.

Gene clustering (a-b) Heatmaps of Z-scores based on vst-normalised RNA-Seq counts. Z-Scores were hard-clustered and count data was compared to identify clusters where differentially expressed genes were regulated in the direction of constitutively expressed genes in K1 8 mM. Identified clusters are framed in black and seen in more detail in Supplementary Figs. 7 & 8. Gene names and count data can be found in Supplementary Tables S8 and S9. (c) Venn diagram of genes identified in the clusters. Additional information is found in Supplementary Table S10. (d) Upset plot of differentially expressed genes in used cell lines with gene reported to affect growth rate in Metastudy.
Table 2.
Identified Pathways using KEGG enrichment.
| Cell Line | KEGG ID | Name | Genes in Pathway | Total Genes in Pathway | Proportion | p.adjusted |
|---|---|---|---|---|---|---|
| K1 0 mM | 4610 | Complement and coagulation cascades | 3 | 17 | 0.235 | 0.006 |
| 4740 | Olfactory transduction | 5 | 13 | 0.385 | <0.0001 | |
| K1 Hy | ||||||
| 4390 | Hippo signalling pathway | 16 | 83 | 0.193 | 0.002 | |
| 5217 | Basal cell carcinoma | 7 | 27 | 0.259 | 0.043 | |
A second strategy to identify targets was to compare the differentially expressed genes to a recently published Metastudy of publicly available CHO transcriptomic datasets [44]. In this study the authors provide gene lists correlating with either high growth rate or high specific productivity across multiple published datasets. These gene lists were compared to the differentially expressed genes in the DE pools. Analog to the authors of the Metastudy, only genes with a frequency of ≥2 in the growth group were taken for comparison, resulting in 240 possible genes which showed almost no overlap with the genes of the Metastudy (Fig. 5d, Supplementary Table S11).
4. Discussion
A simple directed evolution approach based on repeated selection of cells that grow into visibly sized subclones led to the establishment of cell lines with higher cloning efficiency and faster outgrowth. Their ability to achieve similar clone numbers without the addition of supplements provides additional advantages: (i) the danger of unintentionally selecting cells that are dependent on these substances is avoided; (ii) conditioned media and supplements are not chemically defined and may therefore give unreliable results due to lot to lot variation; and (iii) there is thus significant reduction in cost. Finally, as clones were visible and thus transferable already after 2 weeks, this will translate into a reduction in timelines of cell line development campaigns, where the time required for subclone outgrowth is an unsurpassable limit. For the cell lines used in this study, the improvement in outgrowth was inversely proportional to the starting number of subclones, pointing to the possibility of a systematic genetic, environmental or methodical limit of cloning efficiency. In addition to the higher number, a higher percentage of microscopically visible clones are already macroscopically visible after two weeks (91% for K1 8 mM, 80% for K1 0 mM and 50% for K1 Hy) in the DE cell lines, which in practice would reduce the time required for each subcloning step during cell line development.
Interestingly, the observed improvement in subcloning outgrowth also translated into a better growth phenotype in standard batch experiments. However, two different phenotypes were observed: K1 8 mM, which had the highest growth rate from the beginning, saw no further improvement in growth rate, but reached a higher maximum VCD, whereas the other two cell lines showed increased growth rates with no change in achieved VCD (Fig. 2b-e). These differences in behavior of DE cell lines likely contributed to the observed lack of overlap in differentially expressed genes and affected pathways (Figs. 3 and 4). Again, it may also indicate a fundamental biological limit to growth rate for this cell type and under the given culture conditions.
To obtain a deeper insight into these underlying mechanisms, which would also be useful for rational cell line design or engineering, RNA-Seq was performed (Fig. 3). Surprisingly, identified differentially expressed genes have little overlap. Those cell lines with the initially lower number of clones per plate – K1 0 mM and K1 Hy – show more genes differentially regulated than K1 8 mM. Although K1 0 mM and K1 Hy show the highest number of identical genes regulated, only few of these are regulated in the same direction. The PCA analysis demonstrates that the distance of variance of the subclones actually increases relative to the parental cell lines, while one would have expected that cells that converge in behavior, also approach in transcriptome pattern. This increase in distance is an indication that each cell line was able to transform its transcriptome in its own, very individual way to adapt to the improved subcloning phenotype. Based on the cluster analysis it seems that the majority of differentially expressed genes in K1 Hy were regulated in the direction of similar levels as K1 8 mM, whereas K1 0 mM shows a lower fraction of genes regulated in that way. This may, however, be due to the fact that K1 8 mM and K1 0 mM are more closely related than either of these two is to K1 Hy, as indicated in the PCA (Fig. 3d). However, there are still more than 40% of differentially expressed genes in K1 Hy that show a different, diverging behavior. Thus, it appears that the space of possibilities that cells have at their disposal to achieve the same end is large and highly diverse, consistent with previous research that found very little overlap between differentially expressed genes in cells of similar phenotype [44], [46], [47], [48].
Looking at the small number of genes regulated the same way in all three cell lines, the two upregulated genes, cytidine deaminase (Cda) and twist-related protein 2 (Twist2) could be potential targets for rational cell line development. Cda is involved in maintaining the cellular pyrimidine pool [49], a benefit for a better growth phenotype thus is likely. Twist2 is a transcription factor important for embryogenesis, but has also been shown to play a role in tumorigenesis and during epithelial- to-mesenchymal transition and as an inhibitor of senescence [50], [51]. Cda may be upregulated to use cellular resources more efficiently and support the necessary DNA synthesis during enhanced growth, while Twist2 may help cells to adapt to different conditions, in particular to those in isolation during subcloning. Downregulated genes are difficult to interpret, with six out of eight genes, including two lncRNAs, lacking proper functional annotation. Most of the identified proteins do not have any apparent function in CHO, However, Cytochrome P450 11B1, is needed for the production of steroid hormones, specifically cortisol and was reported to be responsible for the generation of this major glucocorticoid in rat, which is, amongst other mechanisms, also involved in glucose metabolism [52], [53]. The precise connection to the observed phenotype is unclear, though.
Pathway enrichment via GSEA shows a similar pattern as observed on the gene level. The majority of identified pathways are unique to each cell line, and again some of those that are shared are regulated in opposite directions. It is striking, however, that throughout the majority of differentially expressed genes is found in pathways related to the ECM, which may indicate that cells struggle with the situation of being in isolation during subcloning and try to counteract by modifying their immediate matrix environment. One of the few pathways identified by cluster analysis that potentially has a direct functional connection to growth, is the “Hippo signaling pathway”, which is reported to be of importance for animal development and the regulation of cellular proliferation to ensure proper differentiation [54]. Looking closely at this pathway reveals that four anti-apoptotic and pro-proliferation genes are upregulated (Supplementary Fig. 9c). Serpine1, Axin2 and Ccnd1 are controlled by the transcriptional transducers Smad3 with its partner Smad4 [55] and Tcf7l1 respectively, both of which are downregulated in K1 Hy to reach similar expression levels as in K1 8 mM. Smad3 downregulation was already shown to increase cell proliferation [56], [57], [58] whereas Tcf7l is a transcriptional repressor of the Wnt signaling pathway [59], [60], which controls cell proliferation [61]. The second pathway “Basal Cell Carcinoma” shows also changes in proliferation associated transcription factors. Both pathways share genes in the Gli transcription factor family, which were shown to be important for tumor growth [57], [58].
In conclusion, we here present a simple and inexpensive way to improve the outgrowth of clones during subcloning and to reduce the time required for cell line development in the biopharmaceutical industry, independent of the used cell line. While all cell lines achieve the same aim – to grow faster into visible colonies and to have a higher cloning efficiency – there was no common pattern found in the transcriptomes of the cell lines studied, indicating that each strengthened pathways and regulated genes according to their individual needs. The only broader overlap is associated with genes related to the extracellular matrix, which indicates that a major limitation of subcloning is due to the fact that cells struggle with isolation.
Taken together these results indicate that cells have high genetic redundancies that provide them with multiple possibilities to adapt. The presence of such redundancies may have implications for cell line development and engineering strategies.
Conflict of interest
Authors hereby confirm that they have no conflict of interest.
CRediT authorship contribution statement
Marcus Weinguny: Methodology, Investigation, Software, Validation, Formal analysis, Data curation, Writing - original draft, Visualization, Project administration. Gerald Klanert: Methodology, Supervision, Formal analysis, Writing - review & editing, Project administration. Peter Eisenhut: Methodology, Writing - review & editing, Visualization. Andreas Johnson: Conceptualization, Methodology. Daniel Ivansson: Conceptualization, Methodology. Ann Lövgren: Conceptualization, Methodology, Supervision, Writing - review & editing, Funding acquisition. Nicole Borth: Conceptualization, Methodology, Resources, Writing - review & editing, Supervision, Funding acquisition.
Acknowledgments
Acknowledgments
The authors acknowledge Thomas Hackl for technical help. Next Generation Sequencing was performed by the NGS Facility at the Vienna Biocenter Core Facilities (VBCF), member of the Vienna Biocenter (VBC), Austria.
Funding
This work was supported by the COMET center: acib: Austrian Center of Industrial Biotechnology, of the Austrian Reseacht Promotion Agency FFG and funded by BMVIT, BMDW, SFG, Standortagentur Tirol, Government of Lower Austria und Vienna Business Agency. PE and MW were also supported by the PhD program BioToP (Biomolecular Technology of Proteins) funded by the Austrian Science Fund [FWF Project W1224].
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.05.020.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Lai T., Yang Y., Ng S. Advances in mammalian cell line development technologies for recombinant protein production. Pharmaceuticals. 2013;6:579–603. doi: 10.3390/ph6050579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh G. Biopharmaceutical benchmarks 2018. Nat Biotechnol. 2018;36:1136–1145. doi: 10.1038/nbt.4305. [DOI] [PubMed] [Google Scholar]
- 3.Frye C., Deshpande R., Estes S., Francissen K., Joly J., Lubiniecki A. Industry view on the relative importance of “clonality” of biopharmaceutical-producing cell lines. Biologicals. 2016;44:117–122. doi: 10.1016/j.biologicals.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Welch J.T., Arden N.S. Considering “clonality”: a regulatory perspective on the importance of the clonal derivation of mammalian cell banks in biopharmaceutical development. Biologicals. 2019;62:16–21. doi: 10.1016/j.biologicals.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Feichtinger J., Hernández I., Fischer C., Hanscho M., Auer N., Hackl M. Comprehensive genome and epigenome characterization of CHO cells in response to evolutionary pressures and over time. Biotechnol Bioeng. 2016;113:2241–2253. doi: 10.1002/bit.25990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelly PS, Clarke C, Costello A, Monger C, Meiller J, Dhiman H, Borth N, Betenbaugh MJ, Clynes M, Barron, N. Ultra-deep next generation mitochondrial genome sequencing reveals widespread heteroplasmy in Chinese hamster ovary cells. Metab. Eng., 10.1016/j.ymben.2017.02.001. [DOI] [PubMed]
- 7.Cao Y., Kimura S., Itoi T., Honda K., Ohtake H., Omasa T. Construction of BAC-based physical map and analysis of chromosome rearrangement in Chinese hamster ovary cell lines. Biotechnol Bioeng. 2012;109:1357–1367. doi: 10.1002/bit.24347. [DOI] [PubMed] [Google Scholar]
- 8.Derouazi M., Martinet D., Besuchet Schmutz N., Flaction R., Wicht M., Bertschinger M. Genetic characterization of CHO production host DG44 and derivative recombinant cell lines. Biochem Biophys Res Commun. 2006;340:1069–1077. doi: 10.1016/j.bbrc.2005.12.111. [DOI] [PubMed] [Google Scholar]
- 9.Vcelar S., Jadhav V., Melcher M., Auer N., Hrdina A., Sagmeister R. Karyotype variation of CHO host cell lines over time in culture characterized by chromosome counting and chromosome painting. Biotechnol Bioeng. 2018;115:165–173. doi: 10.1002/bit.26453. [DOI] [PubMed] [Google Scholar]
- 10.Kallehauge T.B., Li S., Pedersen L.E., Ha T.K., Ley D., Andersen M.R. Ribosome profiling-guided depletion of an mRNA increases cell growth rate and protein secretion. Sci Rep. 2017;7:40388. doi: 10.1038/srep40388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sleiman R.J., Gray P.P., McCall M.N., Codamo J., Sunstrom N.-A.S. Accelerated cell line development using two-color fluorescence activated cell sorting to select highly expressing antibody-producing clones. Biotechnol Bioeng. 2008;99:578–587. doi: 10.1002/bit.21612. [DOI] [PubMed] [Google Scholar]
- 12.Mattanovich D., Borth N. Applications of cell sorting in biotechnology. Microb Cell Factories. 2006;5:12. doi: 10.1186/1475-2859-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim U.M., Yap M.G.S., Lim Y.P., Goh L.-T., Ng S.K. Identification of autocrine growth factors secreted by CHO cells for applications in single-cell cloning media. J Proteome Res. 2013;12:3496–3510. doi: 10.1021/pr400352n. [DOI] [PubMed] [Google Scholar]
- 14.Xu W., Yu X., Zhang J., Bhushan S., Prasad S., Prasad K. Soy hydrolysate mimic autocrine growth factors effect of conditioned media to promote single CHO-K1 cell proliferation. Tissue Cell. 2019;58:130–133. doi: 10.1016/j.tice.2019.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Riba J., Schoendube J., Zimmermann S., Koltay P., Zengerle R. Single-cell dispensing and ‘real-time’ cell classification using convolutional neural networks for higher efficiency in single-cell cloning. Sci Rep. 2020;10:1–9. doi: 10.1038/s41598-020-57900-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Priola J.J., Calzadilla N., Baumann M., Borth N., Tate C.G., Betenbaugh M.J. High-throughput screening and selection of mammalian cells for enhanced protein production. Biotechnol J. 2016;11:853–865. doi: 10.1002/biot.201500579. [DOI] [PubMed] [Google Scholar]
- 17.Le K., Tan C., Gupta S., Guhan T., Barkhordarian H., Lull J. A novel mammalian cell line development platform utilizing nanofluidics and optoelectro positioning technology. Biotechnol Prog. 2018;34:1438–1446. doi: 10.1002/btpr.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gross A., Schoendube J., Zimmermann S., Steeb M., Zengerle R., Koltay P. Technologies for single-cell isolation. Int J Mol Sci. 2015;16:16897–16919. doi: 10.3390/ijms160816897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yim M., Shaw D. Achieving greater efficiency and higher confidence in single-cell cloning by combining cell printing and plate imaging technologies. Biotechnol Prog. 2018;34:1454–1459. doi: 10.1002/btpr.2698. [DOI] [PubMed] [Google Scholar]
- 20.Le K., Tan C., Le H., Tat J., Zasadzinska E., Diep J. Assuring clonality on the beacon digital cell line development platform. Biotechnol J. 2020;15:1900247. doi: 10.1002/biot.201900247. [DOI] [PubMed] [Google Scholar]
- 21.Beketova E.V., Ibneeva L.R., Abdulina Y.A., Dergousova E.A., Filatov V.L., Kozlovsky S.V. Optimized dual assay for the transgenes selection and screening in CHO cell line development for recombinant protein production. Biotechnol Lett. 2019;41:929–939. doi: 10.1007/s10529-019-02711-4. [DOI] [PubMed] [Google Scholar]
- 22.Klottrup K.J., Miro-Quesada G., Flack L., Pereda I., Hawley-Nelson P. Measuring the aggregation of CHO cells prior to single cell cloning allows a more accurate determination of the probability of clonality. Biotechnol Prog. 2018;34:593–601. doi: 10.1002/btpr.2500. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Y., Shaw D., Lam C., Tsukuda J., Yim M., Tang D. Beating the odds: the poisson distribution of all input cells during limiting dilution grossly underestimates whether a cell line is clonally-derived or not. Biotechnol Prog. 2018;34:559–569. doi: 10.1002/btpr.2560. [DOI] [PubMed] [Google Scholar]
- 24.Chen C., Le K., Le H., Daris K., Soice N., Stevens J. Methods for estimating the probability of clonality in cell line development. Biotechnol J. 2020;15:1900289. doi: 10.1002/biot.201900289. [DOI] [PubMed] [Google Scholar]
- 25.Evans K., Albanetti T., Venkat R., Schoner R., Savery J., Miro-Quesada G. Assurance of monoclonality in one round of cloning through cell sorting for single cell deposition coupled with high resolution cell imaging. Biotechnol Prog. 2015;31:1172–1178. doi: 10.1002/btpr.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hackl M., Jakobi T., Blom J., Doppmeier D., Brinkrolf K., Szczepanowski R. Next-generation sequencing of the Chinese hamster ovary microRNA transcriptome: identification, annotation and profiling of microRNAs as targets for cellular engineering. J Biotechnol. 2011;153:62–75. doi: 10.1016/j.jbiotec.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bort J.A.H., Stern B., Borth N. CHO-K1 host cells adapted to growth in glutamine-free medium by FACS-assisted evolution. Biotechnol J. 2010;5:1090–1097. doi: 10.1002/biot.201000095. [DOI] [PubMed] [Google Scholar]
- 28.R Core Team . R Foundation for Statistical Computing Vienna; Austria: 2019. R: A language and environment for statistical computing. [Google Scholar]
- 29.Klanert G., Fernandez D.J., Weinguny M., Eisenhut P., Bühler E., Melcher M. A cross-species whole genome siRNA screen in suspension-cultured Chinese hamster ovary cells identifies novel engineering targets. Sci Rep. 2019;9:1–11. doi: 10.1038/s41598-019-45159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rupp O., MacDonald M.L., Li S., Dhiman H., Polson S., Griep S. A reference genome of the Chinese hamster based on a hybrid assembly strategy. Biotechnol Bioeng. 2018 doi: 10.1002/bit.26722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mootha V.K., Lindgren C.M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 37.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE. 2010;5 doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kucera M., Isserlin R., Arkhangorodsky A., Bader G.D. AutoAnnotate: a cytoscape app for summarizing networks with semantic annotations. F1000Research. 2016;5 doi: 10.12688/f1000research.9090.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinforma Oxf Engl. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 41.Kanehisa M., Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao S, Guo Y, Shyr Y. (2019) KEGGprofile: An annotation and visualization package for multi-types and multi-groups expression data in KEGG pathway.
- 44.Tamošaitis L., Smales C.M. Meta-analysis of publicly available Chinese hamster ovary (CHO) cell transcriptomic datasets for identifying engineering targets to enhance recombinant protein yields. Biotechnol J. 2018;13:1800066. doi: 10.1002/biot.201800066. [DOI] [PubMed] [Google Scholar]
- 45.Conway J.R., Lex A., Gehlenborg N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics. 2017;33:2938–2940. doi: 10.1093/bioinformatics/btx364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vishwanathan N., Le H., Le T., Hu W.-S. Advancing biopharmaceutical process science through transcriptome analysis. Curr Opin Biotechnol. 2014;30:113–119. doi: 10.1016/j.copbio.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 47.Vishwanathan N., Yongky A., Johnson K.C., Fu H.-Y., Jacob N.M., Le H. Global insights into the Chinese hamster and CHO cell transcriptomes: Chinese hamster and CHO cell transcriptomes. Biotechnol Bioeng. 2015;112:965–976. doi: 10.1002/bit.25513. [DOI] [PubMed] [Google Scholar]
- 48.Doolan P., Clarke C., Kinsella P., Breen L., Meleady P., Leonard M. Transcriptomic analysis of clonal growth rate variation during CHO cell line development. J Biotechnol. 2013;166:105–113. doi: 10.1016/j.jbiotec.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 49.Frances A., Cordelier P. The emerging role of cytidine deaminase in human diseases: a new opportunity for therapy? Mol Ther. 2020;28:357–366. doi: 10.1016/j.ymthe.2019.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ansieau S., Bastid J., Doreau A., Morel A.-P., Bouchet B.P., Thomas C. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14:79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Yu H., Jin G.-Z., Liu K., Dong H., Yu H., Duan J.-C. Twist2 is a valuable prognostic biomarker for colorectal cancer. World J Gastroenterol WJG. 2013;19:2404–2411. doi: 10.3748/wjg.v19.i15.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vinson G.P. Glomerulosa function and aldosterone synthesis in the rat. Mol Cell Endocrinol. 2004;217:59–65. doi: 10.1016/j.mce.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 53.Schiffer L., Anderko S., Hannemann F., Eiden-Plach A., Bernhardt R. The CYP11B subfamily. J Steroid Biochem Mol Biol. 2015;151:38–51. doi: 10.1016/j.jsbmb.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 54.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hill C.S. Transcriptional control by the SMADs. Cold Spring Harb Perspect Biol. 2016;8 doi: 10.1101/cshperspect.a022079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou Y.-Z., Li C.-Z., Gao H., Zhang Y.-Z. The effects of Smad3 on adrenocorticotropic hormone-secreting pituitary adenoma development, cell proliferation, apoptosis, and hormone secretion. World Neurosurg. 2018;114:e329–e337. doi: 10.1016/j.wneu.2018.02.181. [DOI] [PubMed] [Google Scholar]
- 57.Sato F., Otsuka T., Kohsaka A., Le H.T., Bhawal U.K., Muragaki Y. Smad3 suppresses epithelial cell migration and proliferation via the clock gene Dec1, which negatively regulates the expression of clock genes Dec2 and Per1. Am J Pathol. 2019;189:773–783. doi: 10.1016/j.ajpath.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 58.Bailey K.L., Agarwal E., Chowdhury S., Luo J., Brattain M.G., Black J.D. TGFβ/Smad3 regulates proliferation and apoptosis through IRS-1 inhibition in colon cancer cells. PLoS ONE. 2017;12 doi: 10.1371/journal.pone.0176096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shy B.R., Wu C.-I., Khramtsova G.F., Zhang J.Y., Olopade O.I., Goss K.H. Regulation of Tcf7l1 DNA binding and protein stability as principal mechanisms of Wnt/β-catenin signaling. Cell Rep. 2013;4:1–9. doi: 10.1016/j.celrep.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hrckulak D., Kolar M., Strnad H., Korinek V. TCF/LEF transcription factors: an update from the internet resources. Cancers. 2016;8 doi: 10.3390/cancers8070070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Willert K. Wnt signaling: is the party in the nucleus? Genes Dev. 2006;20:1394–1404. doi: 10.1101/gad.1424006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.