

Abstract
Kinase inhibitors can pose bleeding risks as platelet signaling evolves during clotting. Using microfluidics (200 s−1 wall shear rate) to perfuse Factor XIIa-inhibited or thrombin-inhibited whole blood (WB) over collagen ± tissue factor (TF), we explored the potency of the Src family kinase (SFK) inhibitor dasatinib or the spleen tyrosine kinase (Syk) inhibitor GS-9973 present at clot initiation or added after 90 sec (via rapid switch to inhibitor-pretreated WB). When initially present, dasatinib potently inhibited platelet deposition on collagen (no TF). Furthermore, dasatinib immediately inhibited subsequent platelet deposition when introduced 90 sec after clot initiation. However, when thrombin was generated, dasatinib was markedly less potent against platelet deposition on collagen/TF (but blocked fibrin deposition) and had no effect when added 90 sec after clot initiation. Similarly, dasatinib added at 90 sec had no effect on clotting on collagen/TF when fibrin was also blocked with Gly-Pro-Arg-Pro, indicating that strong thrombin-induced signaling (but not fibrin-induced signaling) can bypass the SFK inhibition at later times. The Syk inhibitor GS-9973 was less potent than dasatinib when present initially, but inhibited clot growth when added at 90 sec, even in the presence of thrombin (± fibrin). Interestingly, the active form (R-406) of fostamatinib inhibits platelet function in only 2 0f 5 healthy blood samples. SFK-inhibitors may have reduced antithrombotic activity and reduced bleeding risks in settings of high TF and local thrombin generation. For oncology patients, SFK-inhibitors like dasatanib may have reduced antithrombotic activity and reduced bleeding risk in settings of local thrombin generation.
Keywords: platelet, thrombosis, kinase, GPVI, collagen
INTRODUCTION
Dasatinib is a commonly used kinase inhibitor for imatinib-resistant Philadelphia chromosome-positive leukemias [1], chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL) [2,3] due to its inhibition of BCR-Abl tyrosine kinase and Src family kinases such as Lyn, Fyn and Src [4]. GS-9973 (entospletinib) is another drug targeting Syk for the treatment of various cancers, including diffuse large B cell lymphoma (DLBCL), mantle cell lymphoma (MCL), and non-Hodgkin lymphoma (NHL) [5]. Fostamatinib [6], the prodrug of R-406 (tamatinib) [7], as inhibits Syk [6,8,9]. However, along with their efficacy in treating various cancers, side effects of Syk or SFK targeting include bleeding [10]. Recent research shows that these coagulation risks are caused by the inhibition effects on Syk/Src within platelets that are linked to platelet inhibition [10], and reduced platelet activation mediated by FcγRIIA [11]. Alternatively, certain kinase inhibitors may have utility as antithrombotic agents [12,13]. For example, a Syk inhibitor could interfere with signaling from GPVI [14], α2β1, αIIbβ3, and potentially GPIb-IX-V to reduce thrombotic risk [15].
Platelets can become activated through diverse receptors, depending on the procoagulant surface, the platelet location in the clot, and the elapsed time since the start of the clotting event. For example, the earliest arriving platelets may immediately encounter collagen and thrombin at the triggering surface of clotting. Platelets arriving later may encounter other clot-adherent platelets, fibrin, and diffusible agonists such as ADP and thromboxane, but have no physical contact with collagen. The core-shell architecture of clots is emblematic of this spatiotemporal heterogeneity of receptor engagement as seen in assays of mouse laser injury [16] or microfluidic human blood clotting [17]. Similarly, pharmacological agents may engage platelet targets at distinct locations or temporal stages of clotting.
Collagen-mediated clustering of platelet GPVI results in phosphorylation of Src family kinases (SFKs) such as Lyn and Fyn [18]. Also, active Lyn is constitutively bound to GPVI to allow for rapid signaling [19]. Lyn phosphorylation of ITAM domains of FcRγ drives spleen tyrosine kinase (Syk) binding and phosphorylation at Tyr352 [18]. Downstream of GPVI activation, phosphorylated Syk (SykpY352) drives activation of phospholipase Cγ2 (PLCγ2) and sustained calcium mobilization [20]. With fibrinogen binding to activated platelets, outside-in signaling through αIIbβ3 results in Src phosphorylation (Src-pY418), ultimately resulting in the generation of phospho-Syk and PLCγ2 activation. Thrombin activation of protease activated receptors, PAR-1 and PAR-4, drives Gαq activation of PLCβ and transient calcium mobilization. During clotting under flow, the generation of fibrin can have diverse influences on platelet signaling by (i) sequestering thrombin [21,22] and potentially (ii) activating GPVI signaling within the clot interior [23–25].
Using microfluidics, the bleeding side effects of drugs can be explored under defined hemodynamic flow, defined procoagulant surface triggers, and in human blood. Additionally, the drug may be present in the blood from the start of the clotting event or can be added acutely at a later time by perfusion switching to drug-treated blood. This “perfusion-switch” experimental design allows exploration of platelet signaling at different stages of clotting through the measurement of kinase inhibitor potency to modulate clotting on different procoagulant surface conditions.
METHODS
Materials
Reagents were obtained as follows: anti-human CD61 antibody (BD Biosciences, San Jose, CA. Cat#: 555754), Alexa Fluor 647–conjugated human fibrinogen (Life Technologies, Grand Island, NY. Cat#: F35200), Alexa Fluor 488 phospho-Src (Tyr418) polyclonal antibody (ThermoFisher Scientific, Waltham, MA. Cat#: 44–660A1), Dade Innovin prothrombin time (PT) reagent (Siemens, Malvern, PA. Cat#: B4212–40), collagen (type I; Chrono-Log, Havertown, PA. Cat#: 385), Sigmacote® (Millipore Sigma, Burlington, MA. Cat#: SL2–100ML), H-Gly-Pro-Arg-Pro-OH (GPRP; Millipore Sigma, Burlington, MA. Cat#: 03-34-0001), dasatinib (Selleckchem, Houston, TX. Cat#: S1021), GS-9973 (entospletinib) (Selleckchem, Houston, TX. Cat#: S7523), Alexa Fluor 488-conjugated annexin V (ThermoFisher Scientific, Waltham, MA. Cat#: A13201), GR144053 (Tocris Biosciences, Bristol, UK. Cat#: 1263), Phe-Pro-Arg-chloromethylketone (PPACK, Haematologic Technologies, Essex Junction, VT. Cat#: FPRCK-01) and corn trypsin inhibitor (CTI, Haematologic Technologies, Essex Junction, VT. Cat#: CTI-01).
Preparation and characterization of collagen/TF surface
Glass slides were rinsed with ethanol, then deionized water, and dried with filtered air. Sigmacot® was used to create a hydrophobic surface on the glass. A volume of 5μL of fibrillar collagen was perfused through a patterning channel (250 μm wide × 60 μm high) of a microfluidic device to create a single 250 μm-wide stripe of fibrillar collagen for all experiments, as previously described [26,27]. For experiments without thrombin interaction, collagen was rinsed and blocked with 20 mL 0.5% bovine serum albumin buffer (BSA). For experiments that study the effect of thrombin, lipidated TF was sorbed to the collagen surface by perfusing of 5 μL of Dade Innovin PT reagent (20 nM stock concentration), rinsed and blocked with 20 mL 0.5% BSA, then incubated for 30 min without flow, as previously described [26,27].
Blood collection and preparation
Blood was obtained via venipuncture into a syringe containing either PPACK (1:100 v/v; final concentration of 100 μmol/L) or high concentration of CTI (40 μg/mL) from healthy donors who self-reported as free of alcohol use for at least 72 hr and medication for at least a week prior to blood collection. All donors provided informed consent under approval of the University of Pennsylvania Institutional Review Board. Blood was treated with anti-human CD61 antibody (stock concentration, 1:50 v/v [%] in whole blood) and Alexa Fluor 647-conjugated human fibrinogen (1 mg/mL stock solution, 1:80 v/v [%] in whole blood) immediately after blood collection for platelet labelling and fibrin labeling, respectively. While CD61 can bind both platelets and white blood cells, the platelet deposits made on collagen/TF under flow contain essentially no white blood cells until substantially later times. Even at low resolution, white blood cell staining would be morphologically detected if it were present. Annexin V (stock concentration, 1:80 v/v [%] in whole blood) was added for phosphatidylserine labeling when needed. Dasatinib was dissolved in DMSO and diluted in Millipore water, the final concentration was 10 μM [28] (DMSO < 0.1% v/v) after addition to whole blood. The final concentration of DMSO in perfused blood was < 0.1% by vol. In control experiments, no effect of DMSO at 0.1 % was detected on platelet or fibrin under flow. Additionally, platelet function was essentially unaltered even at 1% DMSO by vol/ (3 donors, 10 clots per condition) (Supplemental Fig. S5). GPRP was dissolved in Millipore water and the final concentration in whole blood was 5 mM. GR144053 (final concentration, 1 μM) was added to block α2bβ3 function for forming monolayer of clots. All experiments were initiated within 5 min after phlebotomy. GS-9973 was dissolved in DMSO and the final concentration was 10 μM after addition to whole blood.
Microfluidic clotting assay on collagen surfaces with or without TF
An 8-channel polydimethylsiloxane (PDMS) flow device was vacuum-mounted perpendicularly to collagen/TF surfaces forming 8 parallel-spaced prothrombotic patches (250 × 250 μm), as previously described [29]. Treated blood was perfused across the 8 channels by withdrawal through a single outlet. Drug-treated blood was added to the inlet reservoir without stopping flow, thus providing a rapid change in perfusion pharmacology within < 15 sec without hemodynamics crosstalk between channels during the perfusion switch. All clotting events were initiated simultaneously on the chips. Initial wall shear rate was controlled by a syringe pump (Harvard PHD ULTRA; Harvard Apparatus, Holliston, MA) connected to the outlet on the flow device. For experiments using whole blood with CTI, thrombi were formed under constant flow rate (constant Q) condition [30]. Platelet, fibrin and/or phosphatidylserine activities were monitored simultaneously by epifluorescence microscopy (IX81; Olympus America Inc., Center Valley, PA) at 10× and/or 40× magnification. Each experiment contains three 8-channel devices, so in total 6 independent channels/image streams were used for control, 6 channels/images were used for dasatinib or GS-9973 at t=0s, and 12 channels/images were used for dasatinib or GS-9973 switch at t=90s. For each set of experiments (dasatinib/GS-9973 ± GPRP with PPACK/HCTI blood), blood samples from N=3 donors were taken, so in total 18 samples have been analyzed for control and dasatinib/GS-9973 at t=0s and 36 samples analyzed for dasatinib/GS-9973 at t=90s. Additionally, each clot is extremely well localized on the 250 μm × 250 μm collagen feature and contains tens of thousands of platelets, ideal for obtaining whole clot fluorescence intensities (i.e. total clot mass) with time. However, since we image 24 clots simultaneously, high magnification single-cell imaging or morphological analysis was not possible. Since we image in real time and under flow conditions, bright field imaging is also more difficult to quantify to obtain clot mass since the thickness of the flowing blood above the clot changes with time and alters the prevailing background signal for brightfield imaging. Images were captured with a charged coupled device camera (Hamamatsu, Bridgewater, NJ) and were analyzed with ImageJ software (National Institutes of Health). To avoid side-wall effects, fluorescence values were taken only from the central 75% of the channel.
Post-staining using phospho-Src (Tyr418) polyclonal antibody
In some experiments, clots formed under flow conditions were prepared for fixation and staining to detect Src in cells. After clotting under flow, a solution of 0.5% BSA was perfused in the same manner as whole blood to rinse out blood and then 4% paraformaldehyde was perfused and incubated in the channels. Blocking buffer (3% BSA/TBST/0.1% Triton X-100) was then perfused and incubated for 30 min before clot staining using phospho-Src (pTyr418) polyclonal antibody (1:50 v/v [%] in blocking buffer). Clots were then incubated with phospho-Src antibody for 2 hr under room temperature or overnight under 4C. After rinsing excessive antibody with HBS, images of clots were then taken by the same microscope and camera at 10× and/or 40× magnification and analyzed with ImageJ software.
RESULTS
Dasatinib inhibits platelet deposition on collagen, but is modulated by thrombin
Using an 8-channel microfluidic device, PPACK-treated whole blood (± dasatinib) was perfused over collagen at 200 s−1 wall shear rate. In some channels of the device, perfusion was switched after 90 sec of clotting to PPACK-treated blood with dasatinib (10 μM). Dasatinib present at the start of perfusion potently limited platelet deposition to a sparse monolayer (Figs. 1A, 2A). Switching to dasatinib-treated blood at 90 sec of flow resulted in an immediate ablation of subsequent platelet deposition, demonstrating the role of SFKs in both the initial platelet activation on collagen as well as later stages of platelet deposition. In this experiment with PPACK-treated whole blood and collagen (no TF) that was designed to prevent thrombin generation, no fibrin was detected (Fig. 2B). See Fig. S1 for dasatinib inhibition of phospho-Src staining in a platelet monolayer under thrombin free condition. Clearly, the potency of dasatinib to block platelet accumulation when added at 90 sec was independent of antagonism of any signaling driven by either thrombin or fibrin, neither of which were present in the experiment.
Figure 1. The montage image of microfluidic assay at 0.75, 3.75 and 6.75 minute for platelet and fibrin under 10×.

Whole blood with (A) PPACK, (B) high CTI and (C) high CTI with GPRP were perfused over (A) collagen or (B, C) collagen incubated with TF under venous shear rate (200 s−1). The left two channels are control conditions, the next two channels are control conditions with dasatinib at t = 0 sec, and the right four channels are control conditions switched to blood with dasatinib at t = 90 sec. Direction of blood flow is shown as the arrow.
Figure 2. The measured intensities for platelet and fibrin as a function of time for whole blood with PPACK, high CTI and high CTI with GPRP.
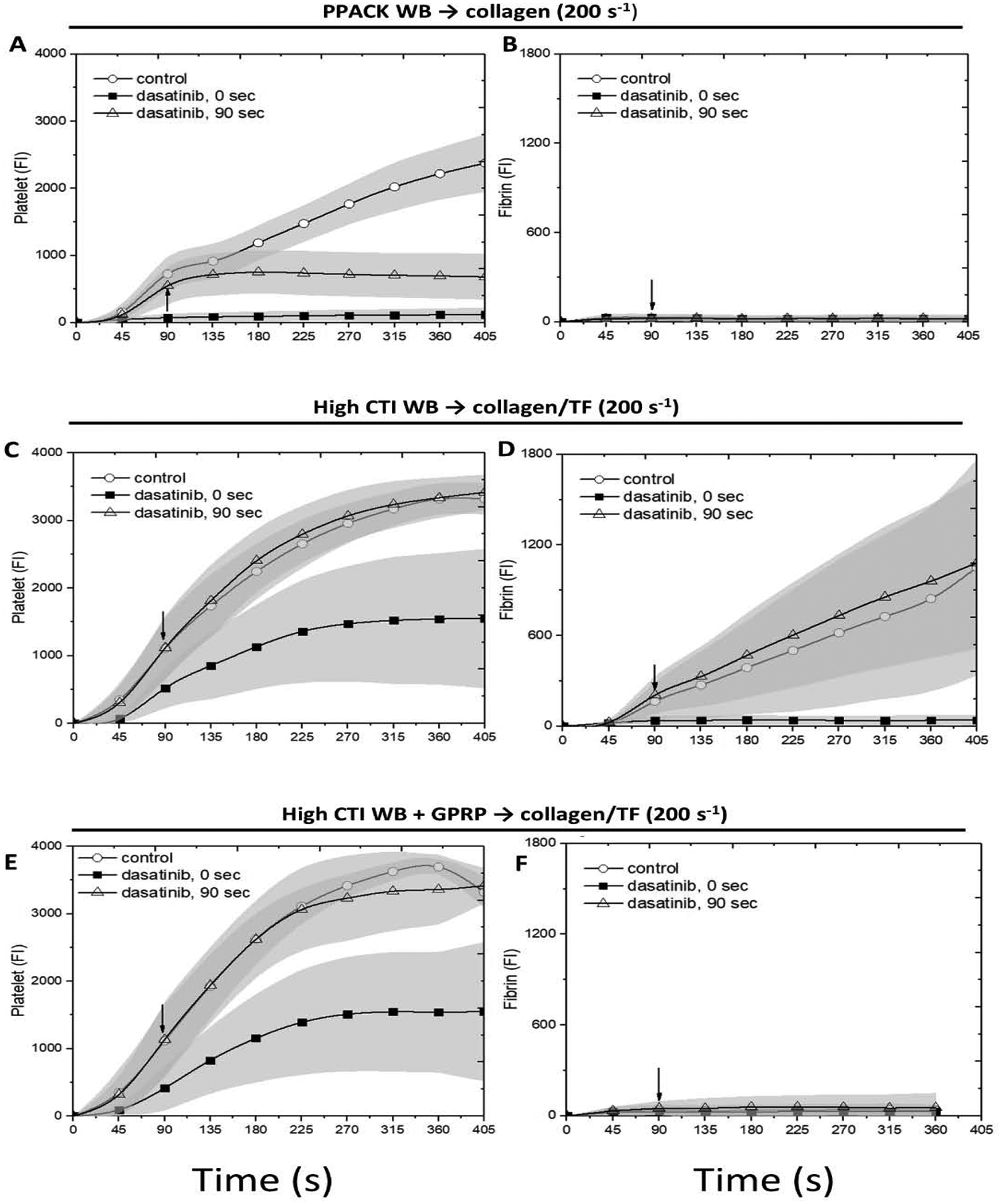
The experiment conditions are the same as Figure 1. Platelet (A, C, E) and fibrin (B, D, F) intensities are measured after imaging for all conditions, respectively. The arrow in each image shows the time where switching of blood takes place.
A similar perfusion-switch experiment was repeated with CTI-treated blood (± dasatinib) perfused over collagen/TF, a condition that robustly generates thrombin and fibrin by engagement of the extrinsic pathway. With thrombin generation promoted in the assay, dasatinib present at t = 0 no longer strongly limited platelet deposition to a monolyer, but instead caused a marked delay and attenuation in platelet accumulation (Figs. 1B, 2C) as well as a striking inhibition of fibrin deposition (Figs. 1B, 2C–D). The inhibition of fibrin formation may indicate that dasatinib reduced the procoagulant activity of collagen-bound platelets (to be discussed later). In complete contrast, addition of dasatinib by perfusion-switch at 90 sec had no effect on platelet deposition or fibrin generation. Clearly, the generation of thrombin and/or fibrin modulated the potency of dasatinib (with thrombin being the likely modulator because essentially no fibrin was made with dasatinib present initially at t = 0).
To investigate whether such loss-of-potency behavior was induced by thrombin or fibrin, the experiment was repeated with CTI-treated blood with added GPRP to allow thrombin generation without fibrin polymerization (Figs. 1C, 2E–F). As seen in the prior experiment with thrombin generation, dasatinib had no effect when added at 90 sec by perfusion switch. As expected, GPRP blocked fibrin polymerization in the clot.
With thrombin generation (± GPRP), dasatinib present at t=0 reduced, but did not limit platelet deposition to a monolayer on collagen/TF. However, once primary deposition had progressed for 90 sec with full SFK signaling (no dasatinib), the subsequent platelet deposition and fibrin generation was completely unaffected by SFK inhibition at 90 sec, implicating the dominance of PAR-1/4 signaling in the growing clot after 90 sec relative to SFK-dependent signaling. In the absence of dasatinib, blocking fibrin polymerization with GPRP had no effect on platelet deposition under flow (Fig. 2C vs. 2E). Also, see Fig. S2.
The Syk inhibitor GS-9973 inhibited platelet deposition and fibrin generation on collagen
GPVI signaling results in activation of SFKs that drive Syk phosphorylation. We explored the role of direct Syk inhibition using GS-9973 (Kd = 7.6 nM, no other kinase < 100 nM [31]) at clot initiation or added at later stages of clot growth using perfusion-switch. For PPACK-treated whole blood perfused over collagen (no thrombin/fibrin), GS-9973 present initially caused a reduction in platelet deposition (Figs. 3A, 4A), although clearly not with the potency of dasatinib. However, in contrast to dasatinib, a perfusion-switch at 90 sec to GS-9973-treated blood resulted in a modest reduction in platelet deposition, indicating a role for Syk at later stages of platelet deposition in the absence of thrombin and/or fibrin-driven signaling (Fig. 3A, 4B).
Figure 3. The montage image of microfluidic assay at 0.75, 3.75 and 6.75 minute for platelet and fibrin under 10×.

Whole blood with (A) PPACK, (B) high CTI and (C) high CTI with GPRP were perfused over (A) collagen or (B, C) collagen incubated with TF under venous shear rate (200 s−1). The left two channels are control conditions, the next two channels are control conditions with GS-9973 at t = 0 sec, and the right four channels are control conditions switched to blood with GS-9973 at t = 90 sec. Direction of blood flow is shown as the arrow.
Figure 4. The measured intensities for platelet and fibrin as a function of time for whole blood with PPACK, high CTI and high CTI with GPRP.
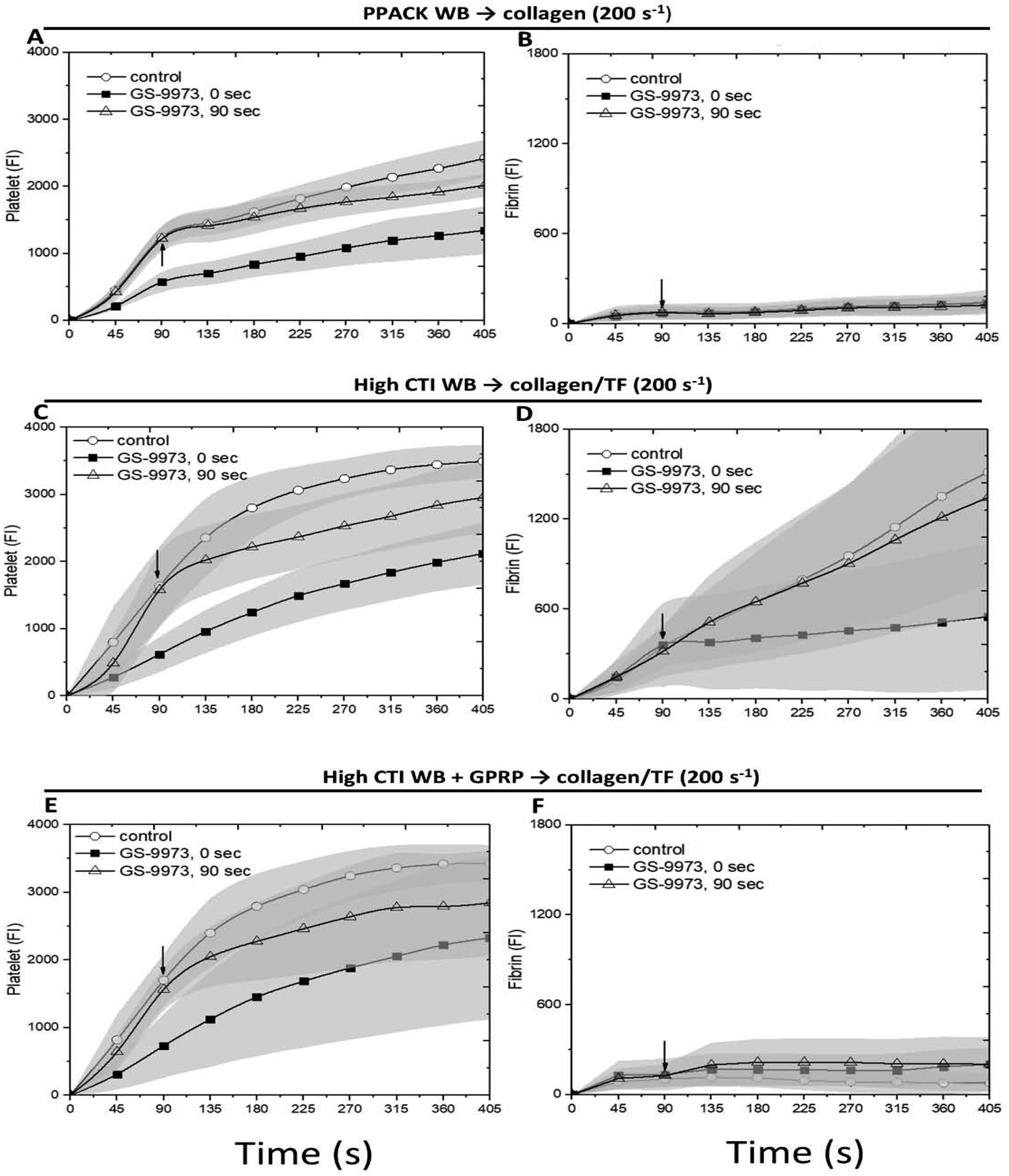
The experiment conditions are the same as Figure 3. Platelet (A, C, E) and fibrin (B, D, F) intensities are measured after imaging for all conditions, respectively. The arrow in each image shows the time where switching of blood takes place.
Repeating this experiment with CTI-treated blood perfused over collagen/TF, GS-9973 reduced platelet deposition and fibrin deposition when the drug was present initially (Figs. 3B, 4C–D). In contrast to dasatinib, switch to GS-9973-treated blood at 90 sec also reduced platelet secondary accumulation without effect on fibrin deposition.
To investigate the role of thrombin generation without fibrin polymerization, CTI-treated blood with added GPRP was perfused over collagen/TF (Figs. 3C, 4E–F). GS-9973 reduced platelet deposition when present initiation or when added at 90 sec by perfusion-switch. We conclude that the Syk inhibitor GS-9973 reduced platelet deposition by inhibiting Syk signaling in both collagen-adherent platelets and subsequently arriving platelets, regardless of the presence or absence of either thrombin or fibrin mediated signaling. Again, GPRP to block fibrin generation had no inhibitory effect on platelet deposition (as shown in Fig. 4C vs. 4E).
Dasatinib and GS-9973 reduce phosphatidylserine (PS) exposure by collagen adherent platelets
Since thrombin and collagen in combination potently induces PS exposure in platelets, CTI-whole blood was perfused over collagen/TF. In some experiments, secondary accumulation was blocked with GR144053 to inhibit fibrinogen binding to αIIbβ3. Both dasatinib and GR144053 reduced deposition to a single sparse monolayer of platelets (Fig. 5). GR144053 had only moderate effects on PS exposure, while SFK inhibition with dasatinib caused a substantial reduction in PS exposure, even on a per-platelet basis (Fig. 5F). This could be the source of poor fibrin production in the presence of dasatinib as seen in Fig. 2D. While GS-9973 was not as potent of blocker of platelet deposition compared to dasatinib, GS-9973 did cause a marked reduction in PS exposure (Fig. 6).
Figure 5. The intensities of platelet, fibrin and Annexin V for all conditions under venous shear rate (200 s−1).
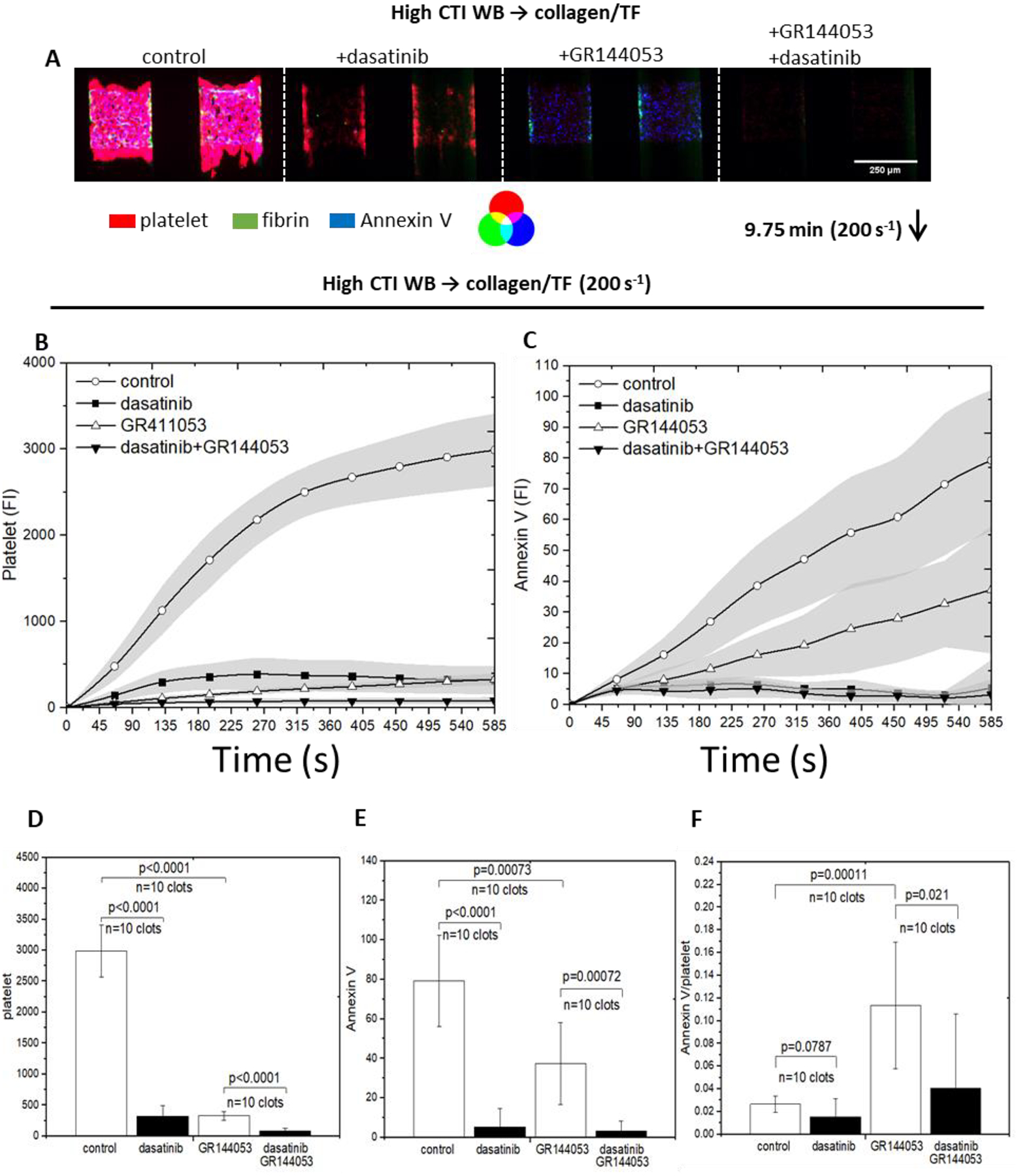
High CTI blood was perfused over collagen incubated with TF. Dasatinib was added to inhibit Src and GR144053 was added to form monolayers. The montage images of clots/monolayers were taken for platelet, fibrin and Annexin V at 9.75 minutes under 10× (A), direction of blood flow was shown as the arrow. Fluorescent intensities of platelet (B) and Annexin V (C) were plotted over 9.75 minutes for all conditions. Platelet intensities were inhibited for both presence of dasatinib and GR144053, but Annexin V intensity was not inhibited by the presence of GR144053. Bar charts of fluorescent intensities of platelet (D) and Annexin V (E) at 9.75 minutes for all conditions illustrate the end point comparison; Annexin V signal on per-platelet basis was achieved by obtaining the ratio of Annexin V/platelet signal (C); the highest ratio is obtained by monolayer without dasatinib, implies that primary platelet deposition is crucial for clot development.
Figure 6. The intensities of platelet, fibrin and Annexin V for all conditions under venous shear rate (200s−1).
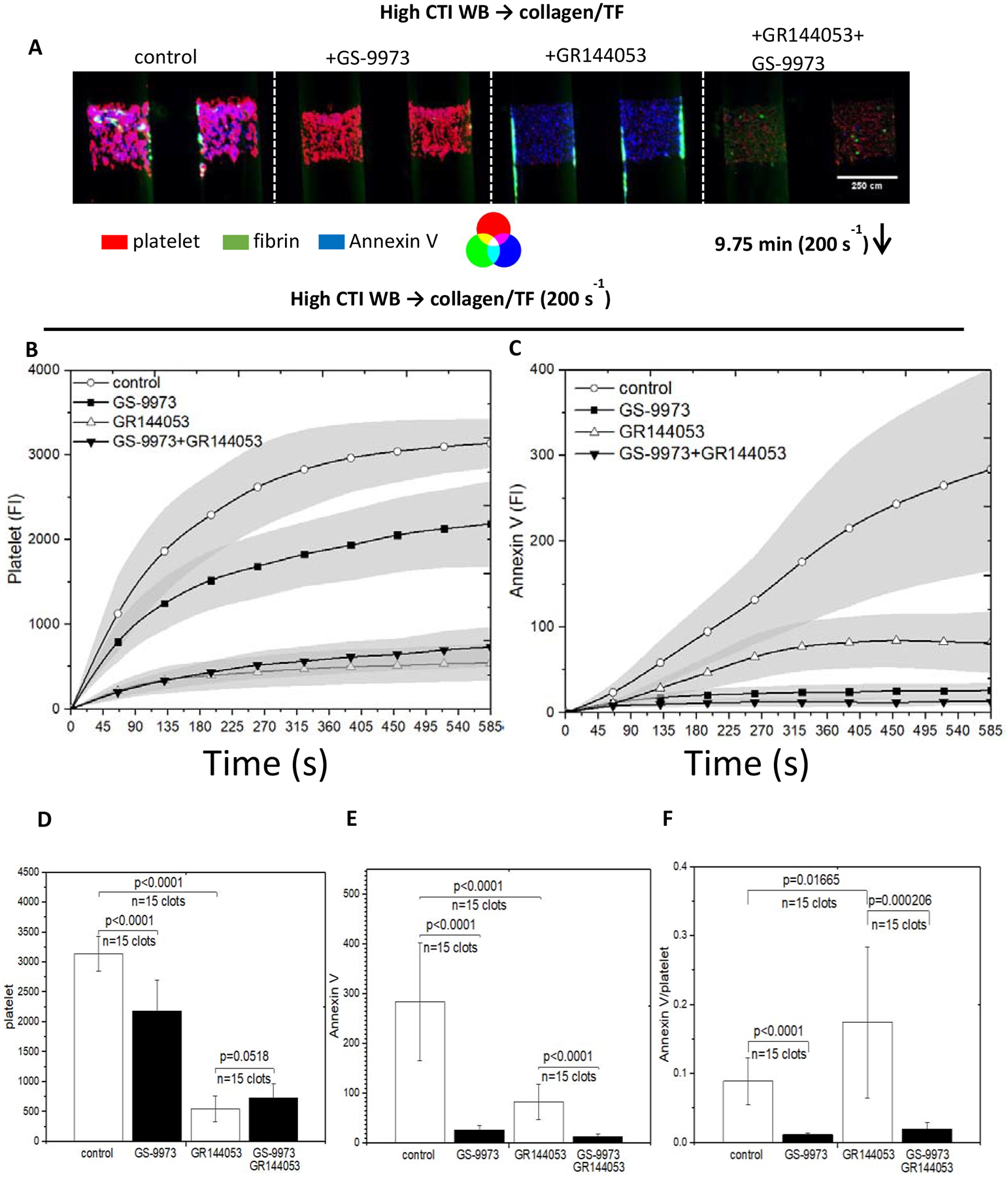
High CTI blood was perfused over collagen incubated with TF. GS-9973 was added to inhibit Syk and GR144053 was added to form monolayers. The montage images of clots/monolayers were taken for platelet, fibrin and Annexin V at 9.75 minutes under 10× (A), direction of blood flow was shown as the arrow. and fluorescent intensities of platelet (B) and Annexin V (C) were plotted over 9.75 minutes for all conditions. Platelet intensities were inhibited for both presence of GS-9973 and GR144053, but Annexin V intensity was not inhibited by the presence of GR144053. Bar charts of fluorescent intensities of platelet (D) and Annexin V (E) at 9.75 minutes for all conditions illustrate the end point comparison; Annexin V signal on per-platelet basis was achieved by obtaining the ratio of Annexin V/platelet signal (C); the highest ratio is obtained by monolayer without GS-9973, implies that primary platelet deposition is crucial for clot development.
DISCUSSION
We used microfluidic assay of human blood to evaluate the potency of kinase inhibitors present at the start of clotting or added acutely at 90 sec once clotting was engaged. This novel experimental design allowed the interrogation of signaling pathways utilized by the initial platelets engaging collagen (± thrombin) relative to the signaling utilized by later arriving platelets (no collagen, ± thrombin). The intent was not to mimic therapy, but rather to use inhibitors to explore signaling pathways at various times and locations during a clotting event with and without thrombin or fibrin. Prior in vitro work has deployed 10 μM concentration [28]. Interestingly, the anti-platelet action of high concentration dasatinib was mitigated when thrombin is generated.
Strong signaling from the Src/SFK is expected in collagen-adherent platelets via GPVI activation pathway, but other receptors can also drive SFK activation such as α2β1, αIIbβ3, P2Y12, and even PAR-4 during clotting [18]. In the absence of thrombin, dasatinib strongly limited the initial deposition of platelets to a sparse monolayer (Fig. 2A). Dasatinib also reduced Src-phosphorylation in collagen-adherent platelet monolayers (Fig. S1) and blocked platelet deposition when added after 90 sec of clotting in the absence of thrombin. However, dasatinib had a striking lack of potency when added after 90 sec of clotting in the presence of thrombin generation. We conclude that thrombin-driven signaling via Gαq drives platelet activation in a manner to substantially bypass SFK inhibition by dasatinib (See Fig. S3 for schematic model). For human blood clotting from 0 to 500 sec, there was little evidence of any role for fibrin-mediated activation of GPVI signaling and SFK signaling: under no conditions did the absence of fibrin using GPRP cause a reduction in platelet deposition. Syk inhibition with GS-9973 in the absence of thrombin was not as potent as dasatinib. GS-9973 may have more Syk selectivity than other small molecule inhibitors [32–34]. Interestingly, GS-9973 showed a stronger inhibition effect at later stages of clotting than dasatinib, even in the presence of thrombin. Again, none of the observations with GS-9973 indicated a substantial role for fibrin-mediated signaling through platelet GPVI. We also conducted the same experiments with R-406 with only 2 of 5 blood samples responding to R-406 (Fig. S4). The cause of donor variation is unknown, but suggests the possible utility of platelet testing to gage patient-specific risk to kinase inhibitor. Based on our experiment results, the potency of a kinase inhibitor can depend on both the timing of its addition as well as the nature of the triggering surface to generate thrombin. Fibrin has substantial anti-thrombin I activity. Blocking fibrin with GPRP may increase local PAR1/4 signaling, thrombin-bound GPIb-dependent SFK signaling, or thrombin-mediated feedback pathways (eg. FXI activation). Our observations with thrombin generation modulating the potency of kinase inhibitors on clotting are relevant to both off-target bleeding effects during cancer treatment as well as antithrombotic therapy. These observations may bring more insights on the mechanisms of excessive bleeding during cancer treatment, so undesired phenomena could be better expected and regulated. However, even with fluorescence staining, the true spatiotemporal dynamics of Src phosphorylation and dephosphoryaltion remain challenging to quantify within the small clots formed under flow using microfluidics. At present, microfluidics does not have the molecular resolution expected with western blotting or LCMS phosphoproteomics.
While dasatinib and other kinase inhibitors are known to reduce GPVI-dependent clot growth under flow [11], their effects on late arriving platelets has not been thoroughly studied, and also challenging to conduct in in vivo models. For example, inhibitor tests under flow have been previously described [35] where blood pretreated with the BCR-abl inhibitor. The “drug perfusion-switch” allowed interrogation of clot progression after the initial platelets already engaged in surface-driven signaling. This novel assay allows an interrogation of a particular pathway in late arriving platelets that are not interacting with collagen.
Further experiment can be done regarding concurrent medication since other BCR-ABL inhibitors could be used at the same time with dasatinib, and relative work has been done on ponatinib inhibited whole blood under flow [35]. Interaction between dasatinib and other BCR-ABL inhibitors may cause a different form of bleeding episode. Some concerns that the specificity of CD61 as platelet marker may not be as good as CD41, since CD61 could bind with cells such as leukocytes. However, the cell count of platelets is 40 times higher than that of leukocytes, and leukocytes do not bind with collagen. Therefore, all the CD61 signal shown on collagen strip could be confidently attributed to platelets. Besides, previous works done on specificity of platelet markers [36] mentioned that in practice CD41 and CD61 can be used interchangeably.
Supplementary Material
Highlights.
Dasatinib inhibits platelet deposition on collagen, but is modulated by thrombin
GS-9973 inhibits platelet deposition and fibrin generation on collagen
Blocking fibrin polymerization had no effect on platelet deposition under flow
Both inhibitors reduce phosphatidylserine exposure by collagen adherent platelets
ACKNOWLEDGEMENTS
Y.Z. and S.L.D. design the research, analyzed the data, and wrote the manuscript. Y.Z. performed the experiments. This work was supported by R01-HL-103419 and U01-HL-131053 (S.L.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in Imatinib-Resistant Philadelphia Chromosome–Positive Leukemias. N Engl J Med. 2006;354:2531–41. [DOI] [PubMed] [Google Scholar]
- 2.Keating GM. Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia. Drugs. 2017;77:85–96. [DOI] [PubMed] [Google Scholar]
- 3.Sasaki K, Jabbour EJ, Ravandi F, Short NJ, Thomas DA, Garcia-Manero G, et al. Hyper-CVAD plus ponatinib versus hyper-CVAD plus dasatinib as frontline therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: A propensity score analysis. Cancer. 2016;122:3650–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li R, Grosser T, Diamond SL. Microfluidic whole blood testing of platelet response to pharmacological agents. Platelets. 2017;28:457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andorsky DJ, Kolibaba KS, Assouline S, Forero-Torres A, Jones V, Klein LM, et al. An open-label phase 2 trial of entospletinib in indolent non-Hodgkin lymphoma and mantle cell lymphoma. Br J Haematol. 2019;184:215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flinn IW, Bartlett NL, Blum KA, Ardeshna KM, Lacasce AS, Flowers CR, et al. A phase II trial to evaluate the efficacy of fostamatinib in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). Eur J Cancer. 2016;54:11–7. [DOI] [PubMed] [Google Scholar]
- 7.Fostamatinib Bajpai M., a Syk inhibitor prodrug for the treatment of inflammatory diseases. IDrugs. 2009;12:174–85. [PubMed] [Google Scholar]
- 8.Burke JM, Shustov A, Essell J, Patel-Donnelly D, Yang J, Chen R, et al. An Open-label, Phase II Trial of Entospletinib (GS-9973), a Selective Spleen Tyrosine Kinase Inhibitor, in Diffuse Large B-cell Lymphoma. Clin Lymphoma, Myeloma Leuk. 2018;18:e327–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duran GE, Sikic BI. The Syk inhibitor R406 is a modulator of P-glycoprotein (ABCB1)-mediated multidrug resistance. PLoS One. 2019;14:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kostos L, Burbury K, Srivastava G, Prince HM. Gastrointestinal bleeding in a chronic myeloid leukaemia patient precipitated by dasatinib-induced platelet dysfunction: Case report. Platelets. 2015;26:809–11. [DOI] [PubMed] [Google Scholar]
- 11.Gratacap M-P, Martin V, Valera M-C, Sophie A, Garcia C, Sie P, et al. The new tyrosine-kinase inhibitor and anticancer drug dasatinib reversibly affects platelet activation in vitro and in vivo. Blood. 2009;114:1884–92. [DOI] [PubMed] [Google Scholar]
- 12.Totani L, Amore C, Di Santo A, Dell’Elba G, Piccoli A, Martelli N, et al. Roflumilast inhibits leukocyte-platelet interactions and prevents the prothrombotic functions of polymorphonuclear leukocytes and monocytes. J Thromb Haemost. 2016;14:191–204. [DOI] [PubMed] [Google Scholar]
- 13.Van Eeuwijk JMM, Stegner D, Lamb DJ, Kraft P, Beck S, Thielmann I, et al. The novel oral Syk inhibitor, Bl1002494, protects mice from arterial thrombosis and thromboinflammatory brain infarction. Arterioscler Thromb Vasc Biol. 2016;36:1247–53. [DOI] [PubMed] [Google Scholar]
- 14.Clarke AS, Rousseau E, Wang K, Kim JY, Murray BP, Bannister R, et al. Effects of GS-9876, a novel spleen tyrosine kinase inhibitor, on platelet function and systemic hemostasis. Thromb Res. 2018;170:109–18. [DOI] [PubMed] [Google Scholar]
- 15.Andre P, Morooka T, Sim D, Abe K, Lowell C, Nanda N, et al. Critical role for Syk in responses to vascular injury. Blood. 2011;118:5000–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stalker TJ, Wu J, Morgans A, Traxler EA, Wang L, Chatterjee MS, et al. Endothelial cell specific adhesion molecule (ESAM) localizes to platelet-platelet contacts and regulates thrombus formation in vivo. J Thromb Haemost. 2009;7:1886–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muthard RW, Welsh JD, Brass LF, Diamond SL. Fibrin, γ′-Fibrinogen, and Transclot Pressure Gradient Control Hemostatic Clot Growth during Human Blood Flow over a Collagen/Tissue Factor Wound. Arterioscler Thromb Vasc Biol. 2015;35:645–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Senis YA, Mazharian A, Mori J. Src family kinases: At the forefront of platelet activation. Blood.2014;124:2013–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmaier AA, Zou Z, Kazlauskas A, Emert-Sedlak L, Fong KP, Neeves KB, et al. Molecular priming of Lyn by GPVI enables an immune receptor to adopt a hemostatic role. Proc Natl Acad Sci. 2009;106:21167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jooss NJ, Simone I De, Provenzale I, Fern DI, Brouns SLN, Farndale RW, et al. Role of Platelet Glycoprotein VI and Tyrosine Kinase Syk in Thrombus Formation on Collagen-Like Surfaces. Int J Mol Sci. 2019;20:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belair DG, Le NN, Murphy WL. Regulating VEGF signaling in platelet concentrates: Via specific VEGF sequestering. Biomater Sci. 2016;4:819–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruggeri ZM, Zarpellon A, Roberts JR, Mc Clintock RA, Jing H, Mendolicchio GL. Unravelling the mechanism and significance of thrombin binding to platelet glycoprotein Ib. Thromb Haemost. 2010;104:894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alshehri OM, Hughes CE, Montague S, Watson SK, Frampton J, Bender M, et al. Fibrin activates GPVI in human and mouse platelets. Blood. 2015;126:1601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onselaer M, Hardy AT, Wilson C, Sanchez X, Babar AK, Miller JLC, et al. Fibrin and D-dimer bind to monomeric GPVI. Blood Adv. 2017;1:1495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollitt AY, Poulter NS, Gitz E, Navarro-Nuñez L, Wang YJ, Hughes CE, et al. Syk and src family kinases regulate c-type lectin receptor 2 (clec-2)-mediated clustering of podoplanin and platelet adhesion to lymphatic endothelial cells. J Biol Chem. 2014;289:35695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu S, Travers RJ, Morrissey JH, Diamond SL. FXIa and platelet polyphosphate as therapeutic targets during human blood clotting on collagen/tissue factor surfaces under flow. Blood. 2015;126:1494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu S, Chen J, Diamond SL. Establishing the Transient Mass Balance of ThrombosisHighlights. Arterioscler Thromb Vasc Biol. 2018;38:1528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mazharian A, Ghevaert C, Zhang L, Massberg S, Watson SP. Dasatinib enhances megakaryocyte differentiation but inhibits platelet formation. Blood. 2011;117:5198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maloney SF, Brass LF, Diamond SL. P2Y12 or P2Y1 inhibitors reduce platelet deposition in a microfluidic model of thrombosis while apyrase lacks efficacy under flow conditions. Integr Biol. 2010;2:183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colace TV, Muthard RW, Diamond SL. Thrombus growth and embolism on tissue factor-bearing collagen surfaces under flow: Role of thrombin with and without fibrin. Arterioscler Thromb Vasc Biol. 2012;32:1466–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharman J, Hawkins M, Kolibaba K, Boxer M, Klein L, Wu M, et al. An open-label phase 2 trial of entospletinib (GS-9973), a selective spleen tyrosine kinase inhibitor, in chronic lymphocytic leukemia. Blood. 2015;125:2336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spalton JC, Mori J, Pollitt AY, Hughes CE, Eble JA, Watson SP. The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets. J Thromb Haemost. 2009;7:1192–9. [DOI] [PubMed] [Google Scholar]
- 33.Currie KS, Kropf JE, Lee T, Blomgren P, Xu J, Zhao Z, et al. Discovery of GS-9973, a selective and orally efficacious inhibitor of spleen tyrosine kinase. J Med Chem. 2014;57:3856–73. [DOI] [PubMed] [Google Scholar]
- 34.Fasbender F, Claus M, Wingert S, Sandusky M, Watzl C. Differential requirements for Src-family kinases in SYK or ZAP70-mediated SLP-76 phosphorylation in lymphocytes. Front Immunol. 2017;8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loren CP, Aslan JE, Rigg RA, Nowak MS, Healy LD, Gruber A, et al. The BCR-ABL inhibitor ponatinib inhibits platelet immunoreceptor tyrosine-based activation motif (ITAM) signaling, platelet activation and aggregate formation under shear. Thromb Res. 2015;135:155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blair TA III F AL. Platelet surface marker analysis by mass cytometry. Platelets. 2019;0:1–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.