

Abstract
Autophagy, a multistep lysosomal degradation pathway that supports nutrient recycling and metabolic adaptation, has been implicated as a process that regulates cancer. Although autophagy induction may limit the development of tumors, evidence in mouse models demonstrates that autophagy inhibition can limit the growth of established tumors and improve response to cancer therapeutics. Certain cancer genotypes may be especially prone to autophagy inhibition. Different strategies for autophagy modulation may be needed depending on the cancer context. Here, we review new advances in the molecular control of autophagy, the role of selective autophagy in cancer, and the role of autophagy within the tumor microenvironment and tumor immunity. We also highlight clinical efforts to repurpose lysosomal inhibitors, such as hydroxychloroquine, as anticancer agents that block autophagy, as well as the development of more potent and specific autophagy inhibitors for cancer treatment, and review future directions for autophagy research.
INTRODUCTION
Macroautophagy (hereafter referred to autophagy) is the process by which cells form double-membraned autophagic vesicles (AV) that sequester organelles and proteins and target them for degradation in the lysosome. Although it was originally viewed as a “bulk degradation” process activated by cellular starvation, new findings demonstrate that autophagy can also be a highly selective quality-control mechanism that regulates levels of specific organelles and proteins. In cancer, autophagy may play a role in limiting the earliest stages of tumorigenesis; however, there is growing evidence that, in established cancers, autophagy can help cope with intracellular and environmental stresses, such as hypoxia, nutrient shortage, or cancer therapy, thereby favoring tumor progression. In this context, it is becoming increasingly clear that autophagy inhibition could improve therapeutic outcomes for patients with advanced cancer. Here we review advances in the molecular mechanisms underlying autophagy in different types and stages of cancer and efforts to translate these advances into specific autophagy inhibitors that could one day effectively treat or even cure advanced cancer.
AUTOPHAGY: LIST OF KEY AUTOPHAGY REGULATORS EXPANDS
In 2016, the Nobel Prize in Medicine was awarded to Yoshinori Ohsumi for his contributions in elucidating the genetic basis for autophagy in yeast (1). Many groups contributed to the effort in confirming that the core autophagy pathway is conserved from yeast to mammalian cells. As autophagy is a complex multistep process, understanding the details of autophagy is critical to developing effective tool compounds and eventually therapies to modulate autophagy potently and specifically. Our current understanding is that the autophagy pathway consists of at least seven steps, with conserved autophagy genes (ATG genes) regulating steps 1 to 5, whereas genes common to other endosomal/lysosomal pathways promote steps 6 and 7 (Fig. 1).
Figure 1.
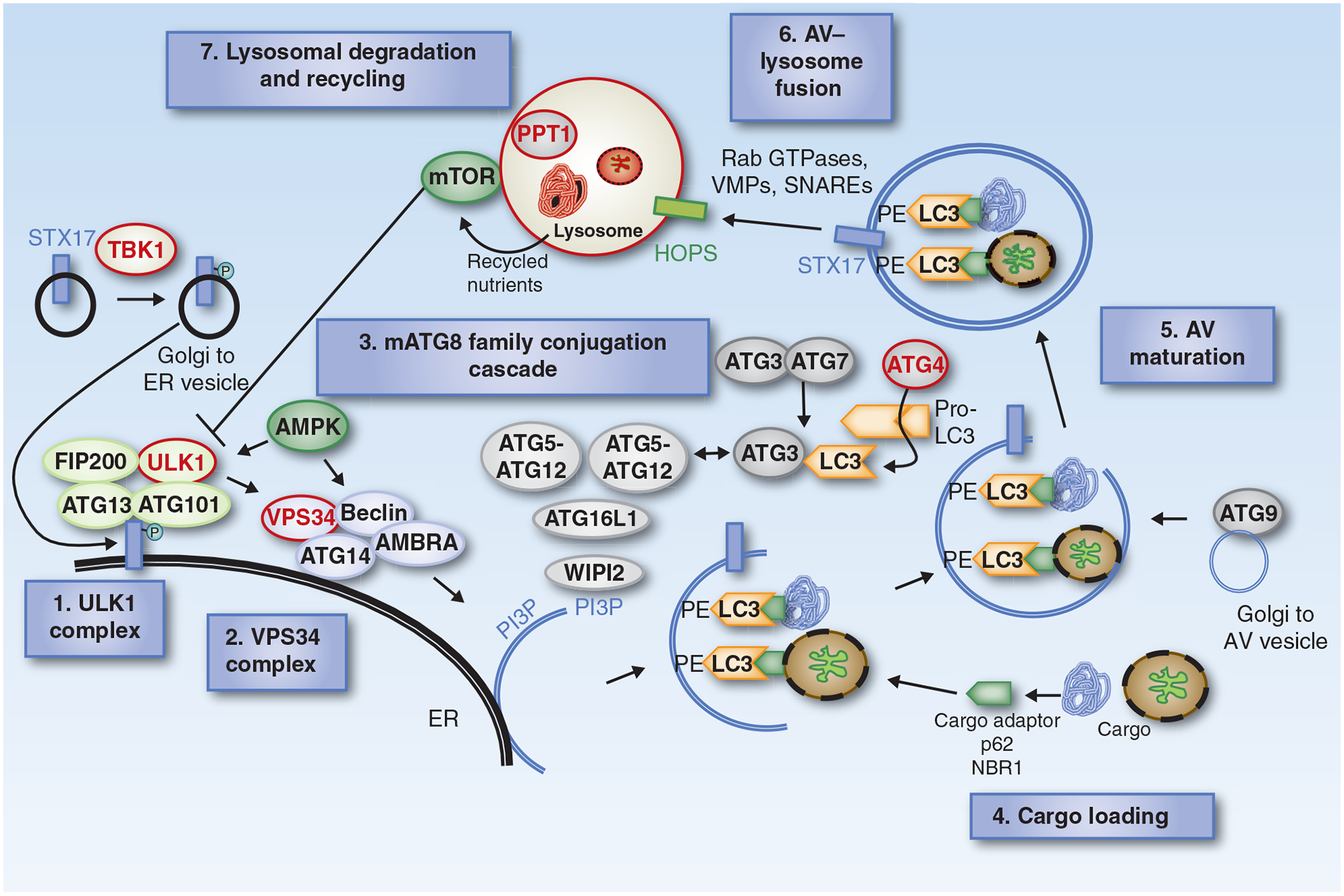
The autophagy pathway. There are 7 steps in the autophagy pathway. Steps 1 and 2 prepare intracellular membranes to form AVs by enriching the membrane for phosphatidylinositol 3 phosphate (PI3P). This lipid enrichment supports a complex ubiquitin-like conjugation system that results in the conjugation of LC3 family members to the lipid phosphatidylethanolamine (PE) on emerging AVs (Step 3). LC3 serves as a docking site for cargo adaptors that enable cargo loading into the AV (Step 4). AV maturation (Step 5) is followed by AV–lysosomal fusion (Step 6). Autophagic flux is completed with cargo degradation and recycling of nutrients (Step 7). Enzymes in the pathway that could serve as targets for drug therapy in cancer are highlighted in red.
Step 1: The Unc-51–Like Kinase Protein Kinase Complex Regulates Initiation of AV Formation
The Unc-51-like kinase (ULK) complex consists of an ULK family kinase, autophagy-related gene 13 (ATG13), and focal adhesion kinase interacting protein 200 kDa (FIP200). This complex is normally inactive, but becomes active when mTORC1 is inhibited or AMPK is activated. Thus, the ULK complex integrates nutrient and energy stress signals from the two master regulators of nutrient (mTOR) and energy stress (AMPK) in the cells. Notably, very recent work reveals that kinases other than ULK, namely tank binding kinase 1 (TBK1), may promote the assembly of ATG13–FIP200 protein complex via phosphorylation of Syntaxin17 (2).
Step 2: The VPS34 Lipid Kinase Complex Prepares the Membrane for Curvature
Once activated, the ULK1 kinase activity leads to activation of the Beclin1 (BECN1)–VPS34 complex, which includes BECN1, VPS34 (a class III PI3K), and other proteins such as VPS15, ATG14L, and autophagy and Beclin1 regulator 1 (AMBRA-1), depending on the subcellular localization of the complex (3). The VPS34 lipid kinase complex executes step 2 by forming phosphatidylinositol 3-phosphate (PI3P) on membranes most commonly from the endoplasmic reticulum (ER).
Step 3: LC3 Family Conjugation Cascade
A series of protein-to-lipid conjugation cascades attach a protein, an LC3 family member, to AV membrane lipid, which both identifies the vesicle as an AV and facilitates the receipt of cargo. Step 3 involves the binding of WIPI2B to PI3P (4), which is required to recruit and assemble two unique protein conjugation systems essential for AV formation. Once the WIPI2B scaffold is available, ATG7 and ATG10 conjugate ATG5 to ATG12, which forms a complex with ATG16L1. Both the ATG7–ATG3 and the ATG5–ATG12–ATG6L1 complex are required to conjugate LC3 (ATG8) family members (including GABARAPs) to the lipid phosphatidylethanolamine (PE), which is enriched in AV membranes (5, 6). Meanwhile, in order for LC3 to be conjugated to lipid by this cascade, the cysteine protease ATG4 is required to process pro-LC3 to its soluble form (LC3-I). Once LC3 is conjugated to lipid, it becomes inserted on the surface of the emerging AV (7). The lipidated form of LC3 (LC3-II) migrates faster than LC3-I on gel electrophoresis, allowing the ratio of lipidated to free LC3 to serve as an approximation of the number of AVs forming at any given time.
Step 4: Cargo Loading through Autophagy Cargo Adaptors
In addition to conjugation of LC3 on AV serving as a marker for AVs, LC3 acts as a docking site for a growing list of autophagy cargo receptors that bring autophagic cargo to the AVs (see below). Cargo receptors such as SQSTM1 (p62) and neighbor of BRCA1 (NBR1) bind to proteins and organelles marked for autophagic degradation through ubiquitin marks (8). Cargo receptors may provide selectivity to the autophagy process as specific cargo bind preferentially to a specific cargo receptor (9).
Step 5: AV Maturation
Additional membrane is delivered to the forming AVs to close the vesicle; lipid membrane derived from mitochondria, plasma membrane, Golgi, or the endoplasmic reticulum is recruited to the forming AV by ATG9 (10–12). Once the isolation membrane is enclosed with trapped cargo, it is called the AV.
Step 6: AV-Lysosome Fusion: Docking and Fusion of Formed AVs with Cargo to the Lysosome
This step is regulated by Rab GTPases, membrane-tethering complexes (HOPS complex, VPS genes) and soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE; ref. 13).
Step 7: Lysosomal Degradation and Recycling of AV Cargo
Autophagic cargo are degraded by lysosomal enzymes, and recycled contents exit via nutrient transporters fueling growth of the cell (14).
Although these 7 steps of autophagy are well established, it is likely that additional regulators of autophagy will be discovered. Recently, novel autophagy regulators were identified using an siRNA screen in a pancreatic cancer cell line. Two of the promising candidates, MAGUK p55 subfamily member 7 (MPP7) and cytosolic malate dehydrogenase 1 (MDH1), were found to have roles in forming the autophagosome. MPP7 activates YAP1, inducing autophagy, and MDH1 regulates ULK1 levels (15).
BULK DEGRADATION AND SELECTIVE AUTOPHAGY CAN BOTH AFFECT THE CANCER PHENOTYPE
Autophagy has been considered a bulk degradation pathway and therefore represents a blunt tool the cell has to employ to escape stress. Bulk degradation of cytoplasmic contents provides cargo for the last two steps in autophagic flux, lysosomal degradation, and recycling. Importantly, nutrient transporters facilitate recycling of amino acids and sugars from autophago-lysosomes (16), and recent studies have begun to elucidate the specific metabolic contributions that autophagy provides to cancer cells, particularly in nutrient-stressed conditions (17).
Besides bulk degradation, which is in essence “taking out the trash,” recent evidence suggests that specific oncogenic proteins, or organelles, can be selectively degraded through the action of specific autophagy cargo receptors. Thus, selective autophagy may serve as a more fine-tuned, context-specific process that shapes the cancer phenotype. In a study focused on how the proteome is remodeled during autophagy, components of the innate immune response were found to be selectively degraded, whereas vesicle-trafficking proteins, which are important for autophagy, were not (18). A proteomics approach has been used to define the autophagy interaction network (3), and it is now recognized that there are multiple forms of selective autophagy, including mitophagy, ferritinophagy (19, 20), and ribophagy (21, 22). Mitophagy, the process by which mitochondria are selectively cleared by the autophagic machinery, is of special interest in cancer because of the mitochondria’s essential role as energy source, factory for generating building blocks for growth, and gatekeeper for apoptosis. Numerous molecular regulators of mitophagy have been identified (23), with the Parkin-PINK ubiquitin conjugation system being the most widely studied. Activation of this conjugation system promotes robust assembly of ubiquitin chains on various mitochondrial outer membrane proteins (24), tagging the organelle for recruitment to autophagy cargo receptors such as optineurin (OPTN). Endoplasmic reticulum autophagy (ER-phagy) is regulated by reticulophagy receptor 1 (RETREG1/FAM134B; ref. 25). In colorectal cancer, FAM134B functions as a tumor suppressor gene (26), whereas in isocitrate dehydrogenase–mutant gliomas, FAM134B has been identified as a synthetic lethal target (27). These findings reinforce the context-dependent role for selective autophagy programs in cancer. In addition to organelle-specific autophagy, protein-specific autophagy has been described. siRNA screens have identified lipid-binding proteins involved in positive (28) and negative (29) regulation of nonselective autophagy. A similar approach has been used to find that the PI3P-binding protein WDFY3/ALFY facilitates selective degradation of protein aggregates by autophagy (30, 31), including the oncogenic fusion protein PML–RARA, associated with acute myeloid leukemia (32).
THE COMPLEX ROLE OF AUTOPHAGY CARGO RECEPTORS IN DRIVING OR SUPPRESSING TUMOR GROWTH
The role of autophagy cargo adaptors in tumorigenesis has been a focus of recent research. SQSTM1/p62 has a rich domain architecture that serves to link several important signaling molecules involved in detoxification through autophagy and nuclear factor erythroid-derived 2-like 2 (NFE2L2/NRF2) activation, as well as inflammation through NFκB (33, 34). Expression of SQSTM1/p62 is significantly increased in human tumor tissue specimens including lung, prostate, pancreas, and liver cancers (35–37). Inhibition of SQSTM1/p62 in tumor cells significantly impairs tumor growth (36, 37). Furthermore, overexpression of p62 in hepatocytes is sufficient to drive carcinogenesis (37). Autophagy is a major regulator of SQSTM1/p62 levels, so it may be that one of the unwanted effects of inhibiting autophagy is sustained and increased levels of SQSTM1/p62 in tumor cells, which in some contexts may facilitate tumor progression and resistance to the autophagy inhibitor (36, 38).
However, although in aggressive tumors there is upregulation of p62 in the tumor cells, there is concurrently a significant downregulation of p62 in cancer-associated fibroblasts (CAF; refs. 39–41) that is essential for CAF-induced tumor progression. Moreover, when SQSTM1/p62 was selectively inactivated in adipocytes, there was an increase in the levels of osteopontin, resulting in enhanced fatty-acid oxidation in tumor cells, which became more invasive resulting in aggressive metastatic prostate cancer in vivo (42). A symbiotic cooperation was found in the metabolism of the tumor and the adipocytes whereby p62 deficiency triggered a general shutdown of energy-utilizing pathways in the adipose tissue through mTORC1 inhibition. This provided more nutrients for cancer cell fatty-acid oxidation to support the tumor’s aggressive phenotype. Therefore, p62 emerges as a dual molecule in cancer acting as a tumor promoter in the tumor cell but a tumor suppressor in the stromal and adipose tissue.
In addition to p62/SQSTM1, multiple autophagy cargo receptors promote selective autophagic degradation, including neighbor of BRCA1 (NBR1), OPTN, Tax1-binding protein 1 (TAX1BP1), and nuclear domain protein 52 (NDP52); moreover, early lines of evidence implicate these proteins in modulating tumorigenesis, both positively and negatively. For example, NBR1-mediated selective autophagy facilitates the clearance of midbodies during cell division and the disassembly of focal adhesions during cell adhesion and migration (43, 44). Although the resultant accumulation of midbodies and focal adhesions has been previously linked to enhanced tumor cell growth and proliferation, additional studies are needed to ascertain how NBR1-mediated selective autophagy influences tumor progression in vivo. In addition, the ubiquitylation of OPTN facilitates complex formation with p62/SQSTM1; the formation of this complex between two autophagy cargo receptors enhances autophagic flux and suppresses the growth of lung cancer xenografts, presumably by mitigating oxidative stress in tumor cells (45). Furthermore, the autophagic degradation of NDP52 and TAXBP1 activates noncanonical NFκB signaling in KRAS-mutant lung cancer cells. Although the precise mechanism downstream of these two mediators of selective autophagy remains unclear, the activation of prosurvival NFκB signaling presumably reinforces the reliance on basal autophagy commonly observed in RAS-transformed cells (46). Because the turnover of p62/SQSTM1 and other autophagy cargo receptors may dictate the efficacy of therapeutic autophagy inhibition in cancer, further delineating the contributions of these autophagy cargo receptors to cancer progression remains an important topic for future study.
AUTOPHAGY DRIVES TUMOR GROWTH OF ESTABLISHED TUMORS AND RESISTANCE TO THERAPY
To better understand the role autophagy plays during early tumorigenesis and once cancers are formed, mouse models of cancer in which autophagy genes were deleted in the tumor cells have been extensively studied. The first such model involved heterozygous deletion of Becn1, and in the absence of any other perturbation this led to an increased rate of spontaneous tumors arising compared with Becn1 wild-type mice (47). This genotype is the only autophagy-deficient genotype that produces spontaneous cancer and not simply benign polyps or adenomas. Complex interactions between BECN1, BRCA1 (48), and p53 (49) suggest that monoallelic loss of Becn1 may be associated with additional oncogenic events that do not occur when other autophagy genes are deficient. When other ATG genes have been deleted, tumor inhibition as opposed to promotion has been observed. The retention of autophagic flux in Becn1 heterozygous cells may explain some of the observed differences in comparison with the homozygous knockout of other autophagy genes. For instance, in a polyoma middle T–driven model of breast cancer with conditional knockout of the Fip200 autophagy gene in mammary tissue, a significant delay in tumor initiation and progression of established tumor was observed (50). Soon thereafter, a number of groups developed mouse models of RAS-driven cancers in which autophagy genes such as Atg5 or Atg7 were conditionally deleted in tumor cells (51–53). In many of these models, the incidence of premalignant lesions increases, but the growth of established malignant tumors is slowed or prevented, leading in most cases to increased survival of mice. One concern from these models is that the excessive outgrowth of premalignant lesions will lead to organ compromise; however, a critical control experiment was recently done where autophagy inhibition was induced genetically across the normal mouse pancreas, and no metaplasia or growth of benign lesions was observed (54). This study explains the findings from almost all previous autophagy knockout studies where autophagy inhibition promotes benign tumor growth preferentially or only in cells harboring additional oncogenic insults.
RAS activation promotes tumor cell reliance on autophagy (55–58), which can serve to maintain the pool of functioning mitochondria to support metabolism during nutrient deprivation and tumorigenesis (52). In a Kras-driven lung cancer model, autophagy is required to maintain mitochondrial function and tumor cell growth and survival, and autophagy inhibition converts carcinomas to benign oncocytoma-like tumors (52, 59). Autophagy inhibition causes defective fatty acid oxidation (FAO) in tumor cells when p53 is deleted (59). These findings suggest that autophagy inhibition or dual inhibition of autophagy and FAO might be a potential cancer therapy depending on the context of oncogene activation or tumor suppressor inactivation. Autophagy is required to recycle metabolites to maintain redox state and energy homeostasis, and to prevent fatal nucleotide pool depletion for Kras-driven lung cancer cells to survive starvation (17). Similar findings were made when Atg5 was deleted in a Kras-driven Trp53+/− pancreatic cancer model. In this context, autophagy inhibition impaired the progression of premalignant lesions to invasive cancer, in part due to the maintenance of mitochondrial metabolism (60). Importantly, when Atg5 was deleted in the pancreas of animals in the setting of wild-type Kras, there was no tumorigenesis observed. This finding adds to a growing body of literature that demonstrates that the Becn1 heterozygous mouse is unique in comparison to the loss of other ATG genes.
Although these studies collectively demonstrate the role of autophagy in supporting the growth of established cancers in mouse models, a very large body of literature has emerged that supports the induction of autophagy during cancer therapy as a key resistance mechanism in multiple cancer types. As described below, many of the stress-sensing pathways that induce autophagy are engaged by cancer therapies used in the clinic. Although complete and prolonged autophagy can produce cell death in vitro, in vivo the major role of autophagy induction is to enable survival of the cancer cell during therapy. Cytotoxic chemotherapy, targeted therapy, and radiotherapy can all activate cytoprotective autophagy. The role of autophagy in therapy resistance is reviewed extensively in ref. 61. Here, we focus on pathways that can be activated by multiple therapies to induce cytoprotective autophagy and engender resistance.
PATHWAYS AND STRESSORS THAT ACTIVATE AUTOPHAGY IN CANCER
Cancer cells grow amidst constant stress conditions within the tumor microenvironment. Without any treatment, dysregulated vasculature and surveillance from the immune system impose nutrient and energy stress on cancer cells and stroma. Once cancer therapy is applied, stress signaling in cancer cells and malignant stroma is amplified. There are a number of common mechanistic pathways that are activated by metabolic or therapeutic stress that in turn activate autophagy. Here we provide an overview of a few key examples. Numerous other regulators of autophagy have been described as well.
Direct Regulators
AMPK/Energy Stress
Nutrient limitation or inhibitors of cancer cell metabolism can induce energy stress in cancer cells impinging on the energy sensor 5′-AMP activated protein kinase 1 (AMPK1) and nutrient sensor mTORC1. A drop in ATP:AMP ratio results in activation of AMPK1, which in turn phosphorylates and inhibits the mTORC1 regulators Raptor and tuberous sclerosis complex 2 (TSC2), while directly phosphorylating and activating the ULK1 complex (62, 63). This coordinately ensures activation of autophagy.
mTORC/Nutrient Stress and Growth Factor Kinase Inhibitors
Recruitment of mTOR to the surface of the lysosome promotes the activation of mTOR through phosphorylation by lysosome-bound RHEB. The vacuolar ATPase that acidifies the lysosome serves as a scaffold for the Ragulator protein complex which docks RAG GTPases. When amino acids are present, RAG GTPases directly interact with the Raptor component of mTORC1, resulting in the lysosomal recruitment of mTORC1 (64, 65). Once on the lysosomal surface, mTORC1 is fully activated by RHEB (66). RHEB, the master activator of mTORC1, is negatively regulated by the tuberous sclerosis complex 1 (TSC1/2). Growth factor (GF) signaling through the PI3K pathway regulates TSC2, which in turn either activates or inhibits RHEB (67). Therefore, with the lysosomal residence of the RAG GTPases and RHEB, considered the most direct regulators of mTORC1, the lysosomal surface represents a critical signaling conduit where global cellular health information is integrated and translated into the activation status of mTORC1. Unlike AMPK-induced phosphorylation of ULK1, which activates autophagy, mTORC1-induced phosphorylation of ULK1 inhibits its downstream activation of the VPS34 complex (68, 69). Inhibition of mTORC1 signaling through nutrient withdrawal, allosteric inhibitors (e.g., rapamycin derivatives), or direct mTORC1 kinase inhibitors, PI3K inhibitors, or AKT inhibitors, will activate autophagy through ULK1.
Transcriptional Regulators of Autophagy Activated by Cancer Therapies
TFE Family and PI3K/mTOR Inhibitors
In addition to post-translational regulation of autophagy, mTORC1 also regulates autophagy at the transcriptional level by controlling the subcellular localization of the transcription factor TFEB. The TFEB/TFE3/MITF family of transcription factors are phosphorylated by activated mTORC1 and sequestered in the cytoplasm. When mTORC1 is inactivated by the PI3K pathway or mTOR inhibitors, TFE family members translocate to the nucleus and activate the transcription of the CLEAR network of genes that regulate lysosome and autophagy genes (70).
p53 Activation and DNA-Damaging Agents
As the guardian of the genome, p53 activates autophagy through transcription of multiple p53 targets (71). DNA damage response checkpoint proteins have been shown to be critical for DNA damage–induced autophagy both as direct regulators of p53 and through downstream effects on AMPK1 and mTORC1 signaling.
BRD4 and Epigenetic Modulators
A recent study suggested that lysosomal function is under the control of the chromatin reader protein BRD4 (72). This has particular significance for tumor therapy due to the development of bromodomain inhibitors for cancer treatment. Inhibition of BRD4 promoted prosurvival autophagy that may be significant for the use of these new therapeutics in the clinic.
The ER Stress Response/Targeted Therapies
Previous work has established that ER stress can activate autophagy (73). For instance, MYC expression activates an ER stress response consisting of protein kinase R (PKR)–like endoplasmic reticulum kinase (PERK)–mediated regulation of LC3 and increased autophagic flux (74). PERK-dependent phosphorylation of eIF2α results in selective translation of ATF4, a transcription factor that regulates the expression of the LC3 gene family, ATG5, and ATG7 (75, 76). In inositol-requiring enzyme 1α–dependent signaling, c-Jun N-terminal kinase (JNK) is a known regulator of autophagy both at the transcriptional level and through the phosphorylation of B-cell lymphoma 2 (BCL2) and displacement of BECN1–BCL2 binding (77). The transcription factor X-box binding protein 1 (XBP1) is known to regulate BECN1 levels, and ATF6α promotes increased expression of death-associated protein kinase 1 (DAPK1), which directly regulates autophagy at multiple steps (78). Recently, it has been appreciated that the ER stress response can function as an intermediary between mitogenic signaling and autophagy. In BRAF-mutant melanoma, combined BRAF and MEK inhibition activates the canonical ER stress response that is associated with activation of autophagy (79). More recently, the molecular mechanism of this resistance pathway connecting MAPK pathway inhibition to autophagy was further elucidated in BRAF-mutant melanoma. MAPK inhibitors activate ER translocation of ERK through the Sec61 translocase. ERK rephosphorylation (reactivation) has been described as a common resistance mechanism to MAPK inhibition, although the mechanism is poorly understood. ER translocation was found to be necessary for ERK reactivation. In turn, one of the key roles of ERK reactivation was to phosphorylate and stabilize ATF4, which transcribes a number of autophagy genes. Thus, the canonical and noncanonical ER stress response programs link autophagy and ER reactivation as a unified pathway of resistance to MAPK inhibitors (80).
AUTOPHAGY PLAYS A CRITICAL ROLE IN BOTH TUMOR AND HOST CELLS TO SUPPORT TUMOR GROWTH
Although the initial studies of the effects of deletion of autophagy genes focused on cell-intrinsic autophagy, more recent studies using more complex mouse models have started to study the effects of genetic autophagy inhibition in both tumor and host cells. In a model of mutant Kras-driven pancreas cancer, it was demonstrated that autophagy in the stromal compartment of pancreatic cancers supports tumor growth. Pancreatic stellate cells secrete the nonessential amino acid alanine in part through the autophagy/lysosome system (81). Pancreatic tumor cells were able to use the alanine to fuel mitochondrial metabolism, allowing them to thrive in an austere microenvironment.
Mouse models have been developed to test the effects of whole-body genetic inhibition of autophagy through Atg7 deletion after mice reached adulthood. After several months of whole-body Atg7 deletion, mice developed a number of metabolic disorders including starvation intolerance, gradual loss of white adipose tissue, liver glycogen, and muscle mass (82). As mentioned above, deletion of Atg7 within tumor cells impairs their growth in multiple models (14, 83). However, in comparison, acute, systemic deletion of Atg7 in all of the cells in the mouse induced greater regression of RAS-driven tumors than autophagy deletion only in the tumor cells, suggesting a role for host autophagy in promoting tumor growth (54, 82). Importantly, tumor regression occurred far faster than the systemic metabolic and neurologic deterioration that eventually takes the animal’s life, supporting a therapeutic window for autophagy inhibition. The majority of mice succumbed to neurodegenerative disease, which suggests that a substantial fraction of the toxicity of autophagy inhibition may be mitigated by creating inhibitors that cannot cross the blood–brain barrier. In a follow-up study, host-specific Atg7 deletion impaired the growth of some but not all allografted tumors. Deletion of autophagy genes in the host cells was associated with a reduction in circulating arginine. It was found that tumor cell lines sensitive to host autophagy deletion were in fact arginine auxotrophs that did not express the enzyme argininosuccinate synthase. When Atg7 or Atg5 was deleted in the host cells, the arginine-degrading enzyme arginase I (ARG1) was released by the liver into the circulation, and this results in degradation of serum arginine, limiting the growth of arginine auxotrophic tumors. Thus, inhibition of host autophagy could result in an ARG1-dependent limitation of tumor growth (84).
The importance of autophagy to maintain tumor growth in both the tumor and host tissues has also been demonstrated in other model organisms. In a RAS-driven tumor model in Drosophila melanogaster, autophagy was found to be activated in tumor cells and systemically, even in distant tissues. Chemical or genetic autophagy inhibition delivered systemically blunted tumor growth and invasion. Stunted tumors from autophagy-deficient flies were able to resume exponential growth when transplanted into autophagy-proficient flies. The studies suggest that tumor cells require proficient autophagy in normal cells to grow properly, further supporting the paradigm of targeting autophagy systemically to limit cancer growth (85).
Finally, using a model of autophagy inhibition that could be induced in space and time, acute autophagy inhibition in a fully formed Kras-driven pancreatic tumor promoted marked tumor regression (54). This model used a dominant-negative Atg4b mutant under the control of a tetracycline-inducible promoter, allowing autophagy to be turned on and off in both tumor cells and host cells, again akin to drug therapy. Response was seen even under intermittent autophagy inhibition, suggesting that antiautophagy therapies may not need to be given continuously in the clinic. This study also affirmed the importance of host autophagy in tumor growth in a series of experiments in which autophagy was inhibited in various combinations of the host and the tumor cells. Both host and tumor cell autophagy were shown to contribute to the ability of tumors to form.
AUTOPHAGY AND TUMOR IMMUNITY
With the widespread success of anti–PD-1 antibody and other immune checkpoint inhibitors, it is clear that harnessing the immune system to combat cancer is not only possible but can yield durable tumor control. This raises the question of how autophagy inhibition would interact with cancer treatments in the age of immunotherapy. There is evidence that inhibition of autophagy would impair hematopoiesis and/or systemic immunity. For example, there is a steady decrease in autophagy levels in aging T lymphocytes (86). Autophagy maintains hematopoietic stem cells (87, 88), and the survival of memory T cells is also reliant on autophagy (89). In the myeloid compartment, autophagy provides free fatty acids to differentiating neutrophils (90) and promotes self-renewal in B1 cells (91). Autophagy has been shown to be important for priming of tumor-specific CD8+ T cells (92). In addition, autophagy has been shown to be essential in effector and memory T-cell activation (93). Finally, autophagy has been shown to dictate the immunogenicity of cell death in certain tumors treated with chemotherapy (94), although these phenotypes appear highly context-dependent.
More recent work, however, suggests that autophagy inhibition does not impair T-cell function in preclinical models of melanoma and breast cancer, including chemotherapy-treated cells (95). In addition, a number of groups have demonstrated that autophagy may play a tumor-protective role in tumor immunity. Autophagy promotes degradation of the cytolytic granules produced by CD8+ T cells and natural killer (NK) cells (96, 97). In melanoma, the hypoxic tumor microenvironment upregulates autophagy in tumor cells, limiting cell death induced by immune effectors. Treatment with hydroxychloroquine (HCQ) enhanced T-cell killing in the context of hypoxia (97). A recent report demonstrated that in melanoma, lysosomes were responsible for limiting the anticancer efficacy of CD8+ T cells (98). Certain subsets of suppressor immune cells may be more or less susceptible to autophagy inhibition. A recent report indicated that immunosuppressive Tregs are critically dependent on autophagy (99). Work from DeVorkin and colleagues showed that deletion of Atg5 or Atg7 in T cells produced striking rejection of tumor implants in syngeneic mouse tumor models (100). Work by Mgrditchian and colleagues has shown that genetic inhibition of BECN1 augments infiltration of T cells into the immune microenvironment (101). Furthermore, recent studies have demonstrated that treatment with lysosomal inhibitors such as chloroquine derivatives can augment antitumor immunity by eliciting an M2 to M1 macrophage polarization switch, which in turn enables tumor cell killing by cytotoxic T cells (102, 103). In addition, the previously mentioned dominant-negative Atg4b mouse model was used to demonstrate that the antitumor effects of inhibiting autophagy in pancreas tumors were, in part, mediated by the immune system. In particular, there was an influx of antitumor macrophages that was critical for these effects (54).
Although a greater understanding of autophagy’s role in tumor immunity is emerging, it is becoming clear that investigators need to be aware of an important distinction between canonical autophagy and autophagy-related programs. Phagocytosis and macroautophagy are actually related processes that share common machinery in specific contexts. Phagocytic cells can utilize LC3-associated phagocytosis (LAP), in which the process of phagocytosis activates certain components of the autophagy machinery to associate with the phagosome, promoting its fusion to lysosomes (phagosome maturation; ref. 104). Like autophagy, LAP requires BECN1, VPS34-generated PI3P, ATG5, and ATG7, but unlike autophagy, in LAP LC3 associates with the single phagosome membrane, rather than the double membrane of autophagosomes. Furthermore, unlike autophagy, LAP does not require the first autophagy protein complex (ULK1, ATG13, and FIP200) and involves a BECN1–VPS34 complex that includes the protein Rubicon. Recently, LAP in the tumor microenvironment has been shown to be an essential mechanism of immunosuppression (105). This raises an interesting possibility: Some of the inflammatory diseases that have become associated with polymorphisms in essential autophagy genes may actually be a consequence of defective LAP. Going forward, investigators working in models involving an intact immune microenvironment need to consider dissecting out the roles of LAP versus autophagy by systematically inhibiting multiple steps in the autophagy pathway, specifically including ULK1 complex inhibition in their studies to differentiate autophagy from LAP. Putting together the information about autophagy’s role in tumor–host interactions and tumor immunity, there is an emerging appreciation for the effects autophagy inhibition may have on many cell types besides the cancer cell, but further work is clearly needed (Fig. 2).
Figure 2.
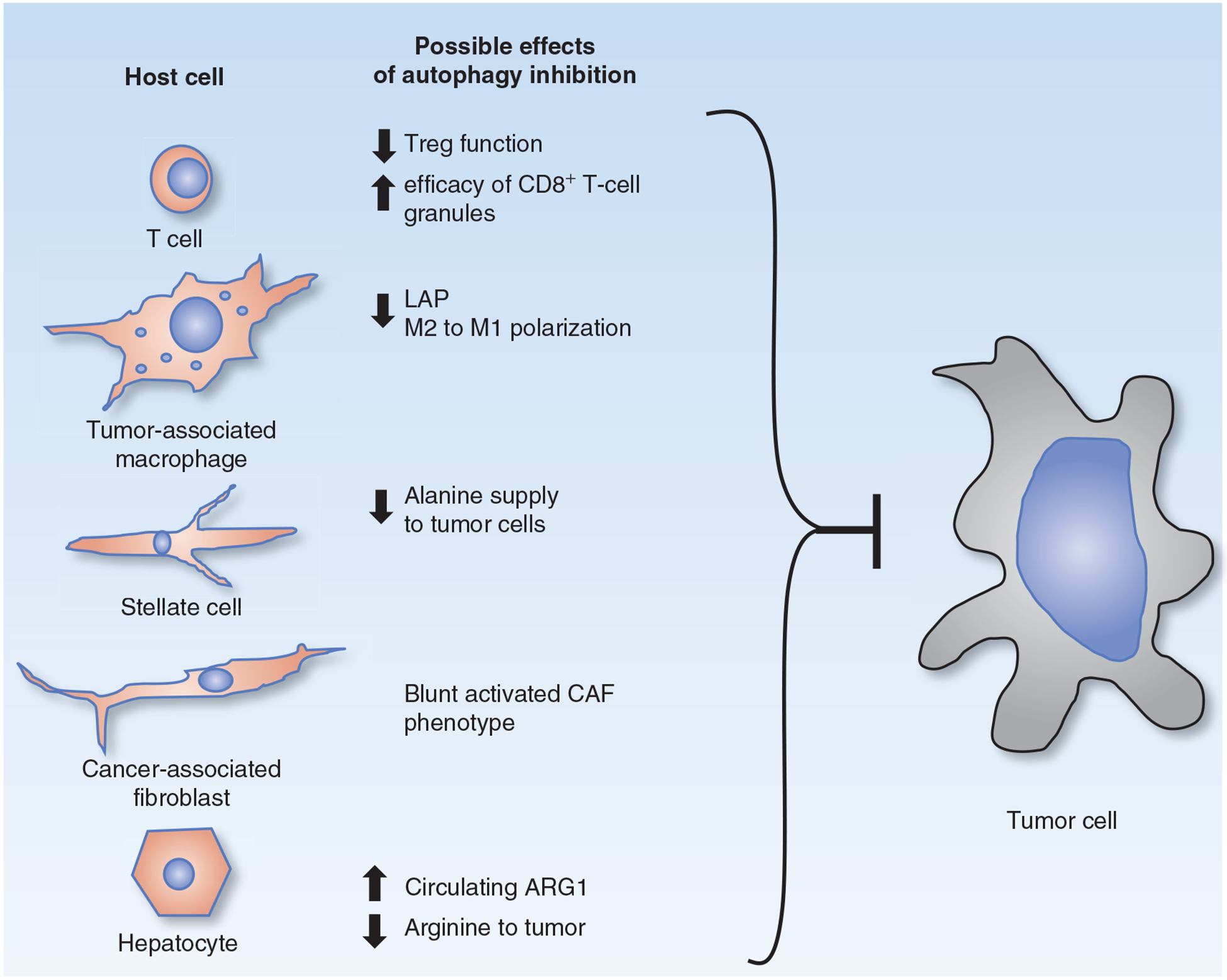
The role of autophagy in host–tumor cell interactions. Nontumor cells within the tumor microenvironment and outside of the tumor undergo profound changes when autophagy is inhibited in a systemic manner. Evidence from a number of laboratories suggests that autophagy in host cells enables tumor growth in specific ways. Autophagy inhibition in host cells impairs the growth of tumor cells independently of impairment of tumor growth achieved by autophagy inhibition in the tumor cells themselves.
DETERMINANTS OF AUTOPHAGY DEPENDENCY IN CANCER
The mechanisms by which autophagy inhibition leads to cell death have only recently started to be elucidated. When autophagy is inhibited, FOXO3A levels increase expression of the PUMA/BBC3 gene that sensitizes tumor cells to apoptosis induced by an anticancer agent (106). Surprisingly, although other transcription factors also control the PUMA gene and FOXO3 regulates many other genes, mutation of a single FOXO-binding site in the PUMA gene was sufficient to abolish the ability of either HCQ or genetic inhibition of ATG5 or ATG7 to sensitize to anticancer drugs. Although this may be a common mechanism of cell death when autophagy inhibitors are combined with anticancer drugs, not all cancer cells die in response to therapeutic regimens involving autophagy inhibition.
Studies from many labs have concluded that some, but not all, cancer cells are dependent on autophagy. However, other studies challenge this idea, because even supposedly autophagy-dependent cancer cells can grow as well as parental cells after complete inactivation of the autophagy regulator ATG7 (107). In this study, using a large panel of cell lines grown in two-dimensional (2-D) culture under ideal conditions, no particular tumor type or genotype was found to be entirely dependent on specific autophagy genes. In contrast, a subset of cancer cell lines was found to be sensitive to lysosomal inhibition with chloroquine derivatives. The conclusion was that autophagy genes are dispensable for growth. There are a number of possible explanations for the discrepancies between this report and other reports in the literature (15, 51, 52, 55–58), including adaptations during selection of knockout clones, increased reliance on other lysosomal scavenging pathways in adapted clones, use of short-term 2-D assays, reliance on nutrient-rich conditions, and exclusive use of immune-incompetent hosts. Indeed, mouse model experiments discussed above support the notion that the role of autophagy becomes more critical within the context of a physiologic tumor microenvironment.
In addition to autophagy itself, evidence also supports specific determinants of susceptibility to lysosomal inhibitors. For example, a recent study showed that bladder cancer cell lines passaged and selected to develop a higher tendency to metastasize in vivo were more susceptible to lysosomal inhibitors in vitro than parental cell lines (108). To determine whether a gene signature predicts sensitivity to chloroquine derivatives, differentially expressed genes from HCQ-sensitive (S) compared with HCQ-resistant (R) cancer cell lines were identified and a predictive classification tree for sensitivity to HCQ was developed (109). Specific patterns of aldehyde dehydrogenase 1A1 (ALDH1A1) and helicase-like transcription factor (HLTF) expression were strong predictors of sensitivity or resistance to HCQ. On the basis of these results, cancer cells expressing high levels of ALDH1A1 and low levels of HLTF would be especially vulnerable to lysosomal inhibitors.
TUMOR GENOTYPES ESPECIALLY SENSITIVE TO COMBINATION STRATEGIES INVOLVING AUTOPHAGY INHIBITORS
Besides tumor types, stage, and gene-expression signatures, somatic mutations that are critical for rewiring cellular metabolism may predispose sensitivity to autophagy inhibition.
RAS-Mutant Cancers
Recently, a series of articles from separate groups, building on the initial identification that autophagy was important for the growth of Ras-mutant tumors, have found that either MEK or ERK inhibition in multiple models of RAS-mutant cancers further activates autophagy as a key resistance mechanism. Combining HCQ (or genetic autophagy inhibition) with MEK (110), ERK (111), or genetic MAPK inhibition (112) produced potent synergistic cytotoxicity in multiple mutant RAS-driven models, including xenografts derived from the KPC mouse model of pancreas cancer, patient-derived xenografts of pancreatic cancer, an NRAS-mutant melanoma xenograft model, and a KRAS-mutant lung cancer model. Treatment of a highly refractory patient with metastatic pancreatic cancer with trametinib plus HCQ resulted in a partial but nonetheless striking disease response. These exciting data have led to the launch of a clinical trial of trametinib and HCQ in pancreatic cancer (NCT03825289).
BRAF-Mutant Cancers
Multiple groups have found that autophagy may play an especially important role in supporting growth and resistance to targeted therapy in BRAF-mutant melanoma (113). BRAF and/or combined BRAF and MEK inhibition activates autophagy as a resistance mechanism in BRAF-mutant melanoma and other BRAF-mutant cancers (79, 114, 115). More recently, the relative importance of autophagy as a major resistance mechanism to BRAF-targeted therapies was demonstrated when it was found that ERK reactivation, the other major mechanism of resistance to BRAF and MEK inhibition, drives autophagy through transcriptional regulation (80). In melanoma, a clinical trial has been launched combining dabrafenib, trametinib, and HCQ (BAMM trial; NCT02257424).
BRAFV600E pediatric brain tumors are another tumor type that appears to be susceptible to autophagy inhibition with chloroquine (CQ) derivatives. Following treatment with BRAF inhibitor, BRAFV600E-mutant gliomas exhibit an increase in autophagy and an addiction to this survival pathway (114, 116). Autophagy inhibition was found to overcome mechanistically distinct forms of resistance to BRAF inhibitor (115), not only in preclinical models but in a pediatric patient with a brain tumor. On the basis of these findings and the preliminary safety of the regimen established in adults, the first pediatric trial of dabrafenib, trametinib, and HCQ is opening in 2019 in collaboration with the Pediatric Brain Tumor Consortium.
TP53-Mutant Cancers
TP53 is the most frequently mutated gene in cancer, so the effects of autophagy inhibition in TP53-mutant tumors is salient. In a model of pancreas cancer in which all pancreatic cells harbored the Kras-mutant; Trp53−/− genotype, genetic ablation of Atg7 or Atg5 within pancreatic cells or treatment with the lysosomal inhibitor HCQ accelerated the formation of pancreatic cancer in mice (117). An important caveat to this finding is the manner in which this mouse model was genetically engineered to express the oncogenic drivers and delete p53 in every cell of the pancreas during embryogenesis, resulting in the development of advanced carcinoma at an early age. As these pancreata develop without ever having functional p53, it is likely that they do not possess a completely functional autophagy program even from birth (p53 is critical for the expression of multiple autophagy genes; ref. 118). In addition, the very rapid growth of pancreatic cancer observed in this mouse model does not match the slower stepwise progression from pancreatic intraepithelial neoplasia to advanced pancreatic cancer found in humans. This stepwise progression from benign to malignant was observed more faithfully in a pancreas-specific Kras-mutant; Trp53+/− conditional knockout model. When ATG5 was deleted in pancreas cells in this model, tumor progression was significantly impaired (60). Impressive tumor growth reduction was consistently observed in KRAS-mutant; TP53-mutant pancreatic patient-derived xenograft lines with pharmacologic autophagy inhibition. Taken together, these studies and others indicate that TP53 mutation status does not discriminate tumor sensitivity to autophagy inhibition.
LKB1-Mutant Cancers
A recent report demonstrates that KRAS-mutant; LKB1 mutant (KL) non–small cell lung cancers are uniformly resistant to immunotherapy, indicating an unmet need to develop new therapeutic approaches for this genotype (119). Loss of tumor suppressor liver kinase B1 (LKB1) promotes cancer cell proliferation, but also leads to decreased metabolic plasticity in dealing with energy crises. A recent report of a mouse model for Kras-mutant; Lkb1-deficient lung cancer (KL model) with conditional deletion of Atg7 found that autophagy ablation was synthetically lethal during both tumor initiation and tumor growth. In the KL model, autophagy deficiency causes defective intracellular recycling, which limits amino acids to support mitochondrial energy production in starved cancer cells and causes autophagy-deficient cells to be more dependent on FAO for energy production. Importantly, the extent of tumor growth inhibition by autophagy inhibition was more pronounced in KL tumors than in Kras-mutant; Trp53-mutant (KP) tumors. These findings strongly suggest that autophagy inhibition could be an effective therapeutic strategy specifically for treating KL lung cancer (120).
REPURPOSING HCQ IN CLINICAL ONCOLOGY TRIALS
Currently, the only clinically available inhibitors of autophagy are the chloroquine derivatives. Because of their long history of use in humans for the treatment of malaria and rheumatologic disorders, these agents, most notably HCQ, have been repurposed in numerous clinical trials for the treatment of diverse cancers. Certain findings from these ongoing trials, some of which are in phase II, warrant special mention (Table 1).
Table 1.
Published phase II clinical trials involving HCQ
| Trial | Disease | Comments | Reference |
|---|---|---|---|
| Phase II HCQ | Previously treated stage IV pancreatic cancer | 0% response rate | Wolpin Oncologist 2014 (121) |
| Phase I/II temozolomide/radiation + HCQ | Frontline glioma | Addition of HCQ provided no survival benefit | Rosenfeld Autophagy 2014 (129) |
| Phase I/II neoadjuvant gemcitabine + HCQ | Borderline resectable pancreatic cancer | Well tolerated; 61% CA19–9 decrease; 70% R0 resection; modulation of autophagy correlated with disease-free survival | Boone Ann Surg Oncol 2015 (135) |
| Phase II vorinostat + HCQ | Heavily pretreated stage IV colon cancer | 5/19 patients stable disease, Decrease in Tregs in serial biopsies compared to baseline | Patel Oncotarget 2016 (132) |
| Phase II trial of everolimus and HCQ | Previously treated stage IV renal cell carcinoma | Well tolerated; modest increase in PFS compared to historical control | Haas Clin Cancer Res 2019 (133) |
| Randomized phase II trial of gemcitabine abraxane with or without HCQ | Stage IV frontline pancreas cancer | Well tolerated; significant increase in response rate in HCQ treated patients, no difference in OS | Karasic JAMA Oncol 2019 (137) |
A single-arm phase II study determined the safety and activity of single-agent HCQ in third-line treatment-refractory stage IV pancreatic cancer patients (121). No activity for single-agent HCQ was observed, with the major caveat being that the majority of patients in this study were extremely poor performance status patients with terminal cancer. All subsequent trials have used HCQ in combination with other agents. The first phase I combination trials combined HCQ with temozolomide in melanoma and glioma, HCQ with temsirolimus in melanoma, HCQ with vorinostat in refractory solid tumors, and HCQ with bortezomib in relapsed refractory myeloma (121–131). In these studies, conducted in a highly treatment-refractory phase I population, there was a <10% grade 3–4 nonhematologic adverse event rate, demonstrating the safety of the approach. Response rates were not high in individual trials; however, multiple patients experienced partial responses or prolonged stable disease on these HCQ combinations, showing an encouraging degree of activity in some patients. Pharmacokinetic–pharmacodynamic studies in these trials demonstrated high-dose HCQ was able to produce a modest but reproducible degree of autophagy inhibition in peripheral blood mononuclear cells (121–130). In the temsirolimus and HCQ study, evidence of autophagy inhibition in serial tumor biopsies was observed by electron microscopy (127).
None of the combinations tested in the earlier series of phase I studies were pursued in phase II studies except a phase II study of vorinostat and HCQ in metastatic colon cancer, which did show a reasonable progression-free survival (PFS) and safety profile for highly treatment-refractory colorectal cancer (132). More recently, a phase I/II trial of everolimus 10 mg daily and HCQ in patients with advanced clear cell renal cell carcinoma found no dose-limiting toxicity in the phase I trial. Disease control was achieved in 67% of evaluable patients. PFS ≥ 6 months was achieved in 45% of patients who achieved disease control. This study showed that combined HCQ and everolimus was tolerable and showed some degree of activity, but not enough to warrant further study of this regimen when compared with contemporary combination therapies available for renal cell carcinoma (133).
Recently, a series of HCQ clinical trials in pancreatic cancer have been reported that suggest that HCQ may have activity in this disease when used in combination with chemotherapy (51, 134). A phase I/II trial of neoadjuvant gemcitabine and HCQ in patients with borderline resectable pancreatic adenocarcinomas found that a full dose of 1,200 mg/day of HCQ in combination with gemcitabine was well tolerated in the neoadjuvant setting (135). Resection rates in this single-arm study looked very encouraging when compared with historical controls from the same institution. A randomized phase II trial with neoadjuvant gemcitabine, abraxane, with or without HCQ in patients with resectable pancreatic cancer has been completed (NCT01978184). Preliminary results presented in abstract form indicate that the primary endpoint, pathologic response in the resection specimen, was significantly increased in the HCQ arm (136). In addition, patients receiving HCQ had a greater decrease in CA-19–9, fewer lymph nodes involved at the time of surgery, and an increased infiltration of T cells.
In an open-label, randomized phase II study conducted in patients with previously untreated metastatic or advanced pancreatic ductal adenocarcinoma, patients were randomized in a 1:1 ratio to gemcitabine and nab-paclitaxel with or without HCQ 600 mg twice daily. The addition of HCQ to chemotherapy did not significantly increase PFS or overall survival, the primary endpoint. However, similar to the neoadjuvant pancreas trial described above, HCQ did significantly increase the response rate of gemcitabine and nab-paclitaxel. This was accompanied by a mild increase in the rates of typical chemotherapy side effects, but not enough to be dose-limiting. The addition of HCQ to block autophagy did not improve the primary endpoint of OS at 12 months, so these data do not support the routine use of gemcitabine and nab-paclitaxel plus HCQ in metastatic pancreatic cancer in the absence of a biomarker. However, improvement in the overall response rate with HCQ may indicate a role for HCQ in the locally advanced setting, where tumor response may permit resection (137).
Table 1 summarizes findings from multiple published HCQ clinical trials to date. Although there is evidence of autophagy inhibition in many of these combinations, the potency and pharmacology of HCQ may limit responses. There is a growing awareness that cationic amphiphilic drugs such as HCQ have variable cell penetration in the acidic tumor microenvironment due to their inherent basicity (138). Efforts to improve drug delivery of HCQ using liposomal or nanoparticle encapsulation technology to address this issue as well as stromal interference are under way (139). Meanwhile, dimeric chloroquines and dimeric quinacrines (see below) may address this issue because they have superior cell penetration and lysosomal localization compared with HCQ in the acidic tumor microenvironment (140).
NOVEL AUTOPHAGY INHIBITORS FOR CANCER
Although these ongoing trials with CQ derivatives have undoubtedly broached the promise of autophagy inhibition as a therapeutic strategy, they have also illuminated the crucial need to develop new compounds targeting autophagy, including both tool compounds, and, ultimately, clinical drug candidates. In this regard, certain targets are showing initial promise.
ULK1 Inhibitors
The ULK1 inhibitor SBI-0206965 (141) was one of the first small-molecule inhibitors targeting ULK1. Although this inhibitor is a potent ULK1 inhibitor, it has off-target liabilities including FAK. A recent structure-based study revealed that many compounds that would inhibit ULK1 would likely inhibit ULK2 and Aurora kinase as well (142). A more recently developed ULK1 inhibitor, ULK101, with increased specificity has been reported (143). The increasing number of tool compounds will allow for more specific interrogation of autophagy compared with LAP and other autophagy-related processes.
VPS34 Inhibitors
Numerous potent and specific VPS34 inhibitors have been reported, including SAR405 (85, 144, 145) and compound 13 (146). Recently, SB02024, developed by Sprint Biosciences, was reported as a highly potent VPS34 inhibitor with a favorable pharmacokinetic profile and an excellent selectivity toward the kinome that makes it suitable for further profiling toward a clinical candidate (147).
ATG4B Inhibitors
ATG4B inhibitors that have been reported, such as S130 (148) and FMK-9a (149), with biochemical and in vitro activity. In addition, the ATG4B inhibitor NSC185058 (150) has been shown to have both in vitro and in vivo antitumor activity. Further work is needed to develop more potent and specific ATG4B inhibitors with pharmacologic properties for clinical trials.
Palmitoyl-Protein Thioesterase 1 Inhibitors
As discussed above, although several HCQ clinical trials are showing promising results, effective repurposing of existing CQ derivatives such as HCQ for cancer has been limited by a missing molecular target. A series of more potent dimeric compounds based on chloroquines and quinacrines have been generated including Lys05 (151), DQ661 (152), and DC661 (140). The dimeric chloroquine Lys05 has been shown to have significant antitumor activity in vivo in multiple tumor models. The activity of the first-generation dimer Lys05 has resulted in widespread adoption of this compound as a tool compound to study autophagy in many cancer types (103, 107, 138, 153). Recently, it was shown that all of these compounds bind to and inhibit the lysosomal enzyme palmitoyl-protein thioesterase 1 (PPT1), which regulates palmitoylation-mediated intracellular trafficking of a substantial number of proteins, including receptors and secreted proteins. Many of the autophagy, mTOR, metabolic enzymes, solute carriers, and IFN signaling proteins are regulated by palmitoylation (154). Therefore, HCQ and more potent derivatives can be considered targeted therapies and not simply weak bases. Development of potent and specific PPT1 inhibitors for cancer is currently being pursued.
PIKFYVE Inhibitors
The phosphoinositide kinase, FYVE-type zinc finger–containing kinase (PIKFYVE) is a lipid kinase that regulates vesicle trafficking related to and beyond autophagy. The PIKFYVE inhibitor apilimod and newer more specific PIKFYVE inhibitors have been shown to disrupt lysosomal homeostasis, thereby blocking autophagy (155, 156). However, due to the multiple locations of PIKFYVE activity in the cell, there are conflicting reports that PKFYVE inhibition induces autophagy. Apilimod was first developed as an anti-inflammatory due to effects on toll-like receptor, and a phase I clinical trial of apilimod has been launched in B-cell malignancies (NCT02594384). Further work is needed to understand how best to position PIKFYVE inhibitors for cancer therapy.
Pros and Cons of Targeting Upstream Autophagy Genes versus the Lysosome
One of the unanswered conceptual questions of how to target autophagy is at what level of the pathway would inhibition be most optimal. For example, inhibition of the later steps, such as the lysosome, may have the advantage of inhibiting other metabolic scavenging pathways such as macropinocytosis, which has also been shown to be critical for tumor metabolism and growth (157). Furthermore, targeting the distal stages of the autophagy pathway results in an accumulation of undigested autophagosomes, which itself could potentially be toxic to tumor cells. In contrast, inhibition of earlier phases of the process, such as those involved in autophagosome biogenesis, would allow for the buildup of toxic protein aggregates and damaged mitochondria that would no longer be encompassed by the autophagosome and allow the tumor cells to be continuously exposed to these toxic insults. Another important concept is whether one needs to target the general macroautophagy pathway or whether inhibiting certain selective autophagy pathways will be sufficient. Targeting specific cargo adaptors required for the various forms of selective autophagy may allow for therapeutic efficacy with decreased potential for toxicity. At this point, the optimal inhibition strategy has not been systematically studied. The development of more selective and potent inhibitors of the autophagy/lysosome pathway at different levels will allow for the essential preclinical validation studies to occur.
SUMMARY AND FUTURE DIRECTIONS
The deepening understanding of regulation of autophagy and autophagy-related pathways such as LAP that co-opt subsets of the autophagy machinery requires redoubled efforts in preclinical studies to interrogate multiple steps in the autophagy pathway genetically. The role of cargo receptors and selective autophagy remains a major focus in the field, which could lead to new therapeutic approaches in the future. New data from mouse models contributes to the growing literature that autophagy inhibition does not cause progression to fully invasive cancer in normal tissues lacking driver oncogene mutations. These previous concerns restricted the focus of autophagy inhibition studies to targeting advanced cancers, but in light of the new findings, there is a critical need to understand the role of autophagy inhibition in earlier-stage disease, either to prevent metastases or to prevent the initial development of cancer. The role of autophagy in tumor–host interactions and tumor immunity mandates the use of immunocompetent animal models as the mainstay of autophagy research. The role of autophagy in sustaining stem-like cancer cells and metastases has seen some progress, but much remains to be learned. The mechanism of action of HCQ and more potent and specific PPT1 inhibitors will allow the dissection of the autophagy-dependent and autophagy-independent cell death mechanisms elicited by these agents. Currently, there are dozens of HCQ clinical trials in cancer active on clinicalTrials.gov. Although some HCQ clinical trials show encouraging activity, there may be a limit to the activity of HCQ combinations in unselected populations. Because of compelling preclinical evidence from multiple groups, there is particular interest in seeing the published results of clinical trials combining MAPK inhibitors with HCQ in RAS- or BRAF-mutant cancers, as autophagy induction by MAPK inhibitors in these cancers seems to be a major resistance mechanism and the combinations could be a synthetic lethal approach to genetically defined tumors. Further research into identifying other potential synthetic lethal approaches, marrying our growing understanding of cancer genetics with autophagy inhibition, may be the most fruitful approach. Meanwhile, it is clear that more potent and specific autophagy inhibitors are needed both as tool compounds and as clinical drug candidates.
Significance:
Autophagy plays a complex role in cancer, but autophagy inhibition may be an effective therapeutic strategy in advanced cancer. A deeper understanding of autophagy within the tumor microenvironment has enabled the development of novel inhibitors and clinical trial strategies. Challenges and opportunities remain to identify patients most likely to benefit from this approach.
Acknowledgments
We would like to thank Rebecca Leshan and the Banbury Center at the Cold Spring Harbor Laboratory for facilitating discussion. We would like to thank Drs. Nathan Bahary, Daniel Flynn, Douglas Green, Jun-Lin Guan, Yanxiang Guo, Wade Harper, Bassam Janji, Jessica Martinsson, Jorge Moscat, Peter O’Dwyer, Rushika Perera, Kevin Ryan, Christopher Sevin, Reuben Shaw, Tor Erik Rusten, Anne Simonsen, Andrew Thorburn, Sharon Tooze, Eileen White, and Roberto Zoncu for vigorous discussions that helped shape this review. This work was supported by NCI grants P01CA114046; P30 CA016520; SPORE P50 CA174523; 1R01CA198015, R01CA157490, R01CA188048, P01CA117969, R35CA232124; ACS Research Scholar Grant RSG-13-298-01-TBG; NIH grant R01GM095567; and the Lustgarten Foundation (to A.C. Kimmelman); R01CA201849, R01CA126792, R01CA213775, and the Samuel Waxman Cancer Research Foundation (to J. Debnath).
Footnotes
Disclosure of Potential Conflicts of Interest
R.K. Amaravadi reports receiving a commercial research grant from Incyte, reports receiving other commercial research support from Novartis, has ownership interest (including stock, patents, etc.) in Pinpoint Therapeutics, and is a consultant/advisory board member for Sprint Biosciences, Immunaccel, and Array Biosciences. A.C. Kimmelman has ownership interest (including stock, patents, etc.) in Vescor Therapeutics and Raphael Pharma and is a consultant/advisory board member for Vescor Therapeutics and Raphael Pharma. J. Debnath is a consultant/advisory board member for Vescor Therapeutics.
REFERENCES
- 1.Levine B, Klionsky DJ. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: breakthroughs in baker’s yeast fuel advances in biomedical research. Proc Natl Acad Sci U S A 2017;114:201–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar S, Gu Y, Abudu YP, Bruun JA, Jain A, Farzam F, et al. Phosphorylation of syntaxin 17 by TBK1 controls autophagy initiation. Dev Cell 2019;49:130–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010;466:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12–5–16L1. Mol Cell 2014;55:238–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 2013;9:424–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010;12:747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature 2000;408:488–92. [DOI] [PubMed] [Google Scholar]
- 8.Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009;8:1986–90. [DOI] [PubMed] [Google Scholar]
- 9.Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol 2018;20:233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 2006;119:3888–900. [DOI] [PubMed] [Google Scholar]
- 11.Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 2012;23:1860–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shibutani ST, Yoshimori T. A current perspective of autophagosome biogenesis. Cell Res 2014;24:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura S, Yoshimori T. New insights into autophagosomelysosome fusion. J Cell Sci 2017;130:1209–16. [DOI] [PubMed] [Google Scholar]
- 14.Kimmelman AC, White E. Autophagy and Tumor Metabolism. Cell Metab 2017;25:1037–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tooze SA, New M, Van Acker T, Sakamaki JI, Jiang M, Saunders RE, et al. MDH1 and MPP7 regulate autophagy in pancreatic ductal adenocarcinoma. Cancer Res 2019;79:1884–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017;358:807–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev 2016;30:1704–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathew R, Khor S, Hackett SR, Rabinowitz JD, Perlman DH, White E. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol Cell 2014;55: 916–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014;509:105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wyant GA, Abu-Remaileh M, Frenkel EM, Laqtom NN, Dharamdasani V, Lewis CA, et al. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 2018;360:751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.An H, Harper JW. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol 2018;20:135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drake LE, Springer MZ, Poole LP, Kim CJ, Macleod KF. Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol 2017;47:110–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harper JW, Ordureau A, Heo J-M. Building and decoding ubiquitin chains for mitophagy. Nat Rev Cell Mol Biol 2018;19:93–108. [DOI] [PubMed] [Google Scholar]
- 25.Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015;522:354–8. [DOI] [PubMed] [Google Scholar]
- 26.Islam F, Gopalan V, Pillai S, Lu CT, Kasem K, Lam AK. Promoter hypermethylation inactivate tumor suppressor FAM134B and is associated with poor prognosis in colorectal cancer. Genes Chromosomes Cancer 2018;57:240–51. [DOI] [PubMed] [Google Scholar]
- 27.Viswanath P, Radoul M, Izquierdo-Garcia JL, Ong WQ, Luchman HA, Cairncross JG, et al. 2-Hydroxyglutarate-mediated autophagy of the endoplasmic reticulum leads to an unusual downregulation of phospholipid biosynthesis in mutant IDH1 gliomas. Cancer Res 2018;78:2290–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knaevelsrud H, Soreng K, Raiborg C, Haberg K, Rasmuson F, Brech A, et al. Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation. J Cell Biol 2013;202:331–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holland P, Knaevelsrud H, Soreng K, Mathai BJ, Lystad AH, Pankiv S, et al. HS1BP3 negatively regulates autophagy by modulation of phosphatidic acid levels. Nat Commun 2016;7:13889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 2010;38:265–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lystad AH, Ichimura Y, Takagi K, Yang Y, Pankiv S, Kanegae Y, et al. Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B-positive structures. EMBO Rep 2014;15:557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlafli AM, Isakson P, Garattini E, Simonsen A, Tschan MP. The autophagy scaffold protein ALFY is critical for the granulocytic differentiation of AML cells. Sci Rep 2017;7:12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009;137:1001–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moscat J, Karin M, Diaz-Meco MT. p62 in cancer: signaling adaptor beyond autophagy. Cell 2016;167:606–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008;13:343–54. [DOI] [PubMed] [Google Scholar]
- 36.Todoric J, Antonucci L, Di Caro G, Li N, Wu X, Lytle NK, et al. Stress-activated NRF2-MDM2 cascade controls neoplastic progression in pancreas. Cancer Cell 2017;32:824–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, et al. p62, Upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016;29:935–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li N, Wu X, Holzer RG, Lee JH, Todoric J, Park EJ, et al. Loss of acinar cell IKKalpha triggers spontaneous pancreatitis in mice. J Clin Invest 2013;123:2231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 2014;26:121–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linares JF, Cordes T, Duran A, Reina-Campos M, Valencia T, Ahn CS, et al. ATF4-induced metabolic reprograming is a synthetic vulnerability of the p62-deficient tumor stroma. Cell Metab 2017;26:817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duran A, Hernandez ED, Reina-Campos M, Castilla EA, Subramaniam S, Raghunandan S, et al. p62/SQSTM1 by binding to vitamin D receptor inhibits hepatic stellate cell activity, fibrosis, and liver cancer. Cancer Cell 2016;30:595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang J, Duran A, Reina-Campos M, Valencia T, Castilla EA, Muller TD, et al. Adipocyte p62/SQSTM1 suppresses tumorigenesis through opposite regulations of metabolism in adipose tissue and tumor. Cancer Cell 2018;33:770–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuo TC, Chen CT, Baron D, Onder TT, Loewer S, Almeida S, et al. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol 2011;13:1214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kenific CM, Stehbens SJ, Goldsmith J, Leidal AM, Faure N, Ye J, et al. NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol 2016;212:577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Z, Chen P, Gao H, Gu Y, Yang J, Peng H, et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell 2014;26:106–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newman AC, Scholefield CL, Kemp AJ, Newman M, McIver EG, Kamal A, et al. TBK1 kinase addiction in lung cancer cells is mediated via autophagy of Tax1bp1/Ndp52 and non-canonical NF-kappaB signalling. PLoS One 2012;7:e50672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003;112:1809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laddha SV, Ganesan S, Chan CS, White E. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol Cancer Res 2014;12:485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beer LA, Wang H, Tang HY, Cao Z, Chang-Wong T, Tanyi JL, et al. Identification of multiple novel protein biomarkers shed by human serous ovarian tumors into the blood of immunocompromised mice and verified in patient sera. PLoS One 2013;8:e60129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011;25:1510–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011;25:717–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011;25:460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang A, Kimmelman AC. Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy 2014;10:1683–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov 2018;8:276–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perez E, Das G, Bergmann A, Baehrecke EH. Autophagy regulates tissue overgrowth in a context-dependent manner. Oncogene 2015;34:3369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, et al. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem 2011;286:12924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 2011;22:165–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kinsey C, Balakrishnan V, O’Dell MR, Huang JL, Newman L, Whitney-Miller CL, et al. Plac8 links oncogenic mutations to regulation of autophagy and is critical to pancreatic cancer progression. Cell Rep 2014;7:1143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 2013;27:1447–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov 2014;4:905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene 2016;35:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011;331:456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13:132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010;141:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012;150:1196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carroll B, Maetzel D, Maddocks OD, Otten G, Ratcliff M, Smith GR, et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. Elife 2016;5:pii:e11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006;126: 955–68. [DOI] [PubMed] [Google Scholar]
- 68.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009;20:1981–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011;7:643–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Puertollano R, Ferguson SM, Brugarolas J, Ballabio A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J 2018;37:pii:e98804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 2013;27:1016–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sakamaki JI, Wilkinson S, Hahn M, Tasdemir N, O’Prey J, Clark W, et al. Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol Cell 2017;66:517–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem 2006;281:30299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 2012;122:4621–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, et al. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010;29:4424–35. [DOI] [PubMed] [Google Scholar]
- 76.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 2010;120:127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008;30:678–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ojha R, Amaravadi RK.Targeting the unfolded protein response in cancer. Pharmacol Res 2017;120:258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest 2014;124:1406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ojha R, Leli NM, Onorati A, Piao S, Verginadis II, Tameire F, et al. ER translocation of the MAPK pathway drives therapy resistance in BRAF mutant melanoma. Cancer Discov 2018;9:396–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016;536:479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 2014;4:914–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev 2016;30:1913–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, et al. Autophagy maintains tumour growth through circulating arginine. Nature 2018;563:569–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Rahman MM, et al. Microenvironmental autophagy promotes tumour growth. Nature 2017;541:417–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Phadwal K, Alegre-Abarrategui J, Watson AS, Pike L, Anbalagan S, Hammond EM, et al. A novel method for autophagy detection in primary cells: impaired levels of macroautophagy in immunosenes-cent T cells. Autophagy 2012;8:677–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med 2011;208:455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Watson AS, Riffelmacher T, Stranks A, Williams O, De Boer J, Cain K, et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov 2015;1:pii:15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, et al. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife 2014;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, et al. Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity 2017;47: 466–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Clarke AJ, Riffelmacher T, Braas D, Cornall RJ, Simon AK. B1a B cells require autophagy for metabolic homeostasis and self-renewal. J Exp Med 2018;215:399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Uhl M, Kepp O, Jusforgues-Saklani H, Vicencio JM, Kroemer G, Albert ML. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ 2009;16:991–1005. [DOI] [PubMed] [Google Scholar]
- 93.Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol 2014;15:1152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015;28:690–714. [DOI] [PubMed] [Google Scholar]
- 95.Starobinets H, Ye J, Broz M, Barry K, Goldsmith J, Marsh T, et al. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J Clin Invest 2016;126:4417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A 2013;110:17450–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res 2011;71:5976–86. [DOI] [PubMed] [Google Scholar]
- 98.Khazen R, Muller S, Gaudenzio N, Espinosa E, Puissegur MP, Valitutti S. Melanoma cell lysosome secretory burst neutralizes the CTL-mediated cytotoxicity at the lytic synapse. Nat Commun 2016;7:10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol 2016;17:277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, et al. Autophagy regulation of metabolism is required for CD8(+) T cell anti-tumor immunity. Cell Rep 2019;27:502–13. [DOI] [PubMed] [Google Scholar]
- 101.Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A 2017;114:E9271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J, et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun 2018;9:873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Frazier J, Bertout JA, Kerwin WS, Moreno-Gonzalez A, Casalini JR, Grenley MO, et al. Multi-drug analyses in patients distinguish efficacious cancer agents based on both tumor cell killing and immunomodulation. Cancer Res 2017;77:2869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol 2015;17:893–906. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 105.Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell 2018;175:429–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fitzwalter BE, Thorburn A. FOXO3 links autophagy to apoptosis. Autophagy 2018;14:1467–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A 2016;113:182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Morgan MJ, Fitzwalter BE, Owens CR, Powers RK, Sottnik JL, Gamez G, et al. Metastatic cells are preferentially vulnerable to lysosomal inhibition. Proc Natl Acad Sci U S A 2018;115:E8479–E88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Piao S, Ojha R, Rebecca VW, Samanta A, Ma XH, McAfee Q, et al. ALDH1A1 and HLTF modulate the activity of lysosomal autophagy inhibitors in cancer cells. Autophagy 2017;13:2056–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kinsey CG, Camolotto SA, Boespflug AM, Gullien KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 2019;25:620–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 2019;25:628–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci U S A. 2019. Feb 1 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xie X, Koh JY, Price S, White E, Mehnert JM. Atg7 Overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov 2015;5:410–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Levy JMM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, et al. Autophagy inhibition improves chemosensitivity in BRAFV600E brain tumors. Cancer Discov 2014;4:773–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mulcahy Levy JM, Zahedi S, Griesinger AM, Morin A, Davies KD, Aisner DL, et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 2017;6:pii:e19671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mulcahy Levy JM, Foreman NK, Thorburn A. Using BRAF(V600E) as a marker of autophagy dependence in pediatric brain tumors. Autophagy 2014;10:2077–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013;504:296–300. [DOI] [PubMed] [Google Scholar]
- 118.Kenzelmann Broz D, Attardi LD. TRP53 activates a global autophagy program to promote tumor suppression. Autophagy 2013;9:1440–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 2018;8:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bhatt V, Khayati K, Hu ZS, Lee A, Kamran W, Su X, et al. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev 2019;33:150–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014;19:637–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shi TT, Yu XX, Yan LJ, Xiao HT. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol 2017;79:287–94. [DOI] [PubMed] [Google Scholar]
- 123.Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 2014;10:1415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Donohue E, Tovey A, Vogl AW, Arns S, Sternberg E, Young RN, et al. Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J Biol Chem 2011;286:7290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007;14:500–10. [DOI] [PubMed] [Google Scholar]
- 126.Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014;10:1380–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014;10:1391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014;10:1369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014;10:1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014;10:1403–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Scott EC, Maziarz RT, Spurgeon SE, Medvedova E, Gajewski J, Reasor-Heard S, et al. Double autophagy stimulation using chemotherapy and mTOR inhibition combined with hydroxychloroquine for autophagy modulation in patients with relapsed or refractory multiple myeloma. Haematologica 2017;102:e261–e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Patel S, Hurez V, Nawrocki ST, Goros M, Michalek J, Sarantopoulos J, et al. Vorinostat and hydroxychloroquine improve immunity and inhibit autophagy in metastatic colorectal cancer. Oncotarget 2016;7:59087–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Haas NB, Appleman LJ, Stein M, Redlinger M, Wilks M, Xu X, et al. Autophagy inhibition to augment mTOR inhibition: a phase I/II trial of everolimus and hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin Cancer Res 2019;25:2080–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015;524:361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, Wu WC, et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol 2015;22:4402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Miller-Ocuin JL, Bahary NS, Singhi AD, Normolle DP, Lembersky B, Stoller R, et al. Inhibition of autophagy improves pathologic and bio-marker response to preoperative gemcitabine/nab-paclitaxel in potentially resectable pancreatic cancer: A phase II randomized controlled trial. 2017. Society of Surgical Oncology Annual Cancer Symposium, Seattle, WA, March 15–18, 2017. Presented March 17, 2017; Abstract 3. [Google Scholar]
- 137.Karasic TB, O’Hara MH, Loaiza-Bonilla A, Reiss KA, Teitelbaum UR, Borazanci E, et al. Effect of gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: a phase 2 randomized clinical trial. JAMA Oncol 2019;5: 993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pellegrini P, Strambi A, Zipoli C, Hagg-Olofsson M, Buoncervello M, Linder S, et al. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy 2014;10:562–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang Y, Shi K, Zhang L, Hu G, Wan J, Tang J, et al. Significantly enhanced tumor cellular and lysosomal hydroxychloroquine delivery by smart liposomes for optimal autophagy inhibition and improved antitumor efficiency with liposomal doxorubicin. Autophagy 2016;12:949–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rebecca VW, Nicastri MC, Fennelly C, Chude CI, Barber-Rotenberg JS, Ronghe A, et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov 2019;9:220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell 2015;59:285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chaikuad A, Koschade SE, Stolz A, Zivkovic K, Pohl C, Shaid S, et al. Conservation of structure, function and inhibitor binding in UNC-51-like kinase 1 and 2 (ULK1/2). Biochem J 2019;476:875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Martin KR, Celano SL, Solitro AR, Gunaydin H, Scott M, O’Hagan RC, et al. A potent and selective ULK1 inhibitor suppresses autophagy and sensitizes cancer cells to nutrient stress. iScience 2018;8:74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pasquier B SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy 2015;11:725–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol 2014;10:1013–9. [DOI] [PubMed] [Google Scholar]
- 146.Pasquier B, El-Ahmad Y, Filoche-Romme B, Dureuil C, Fassy F, Abecassis PY, et al. Discovery of (2S)-8-[(3R)-3-methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)-3,4-dihydro-2H-pyrimido [1,2-a]pyrimidin-6-one: a novel potent and selective inhibitor of Vps34 for the treatment of solid tumors. J Med Chem 2015;58: 376–400. [DOI] [PubMed] [Google Scholar]
- 147.Dyczynski M, Yu Y, Otrocka M, Parpal S, Braga T, Henley AB, et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to sunitinib. Cancer Lett 2018;435:32–43. [DOI] [PubMed] [Google Scholar]
- 148.Fu Y, Hong L, Xu J, Zhong G, Gu Q, Gu Q, et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2018;15:295–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chu J, Fu Y, Xu J, Zheng X, Gu Q, Luo X, et al. ATG4B inhibitor FMK-9a induces autophagy independent on its enzyme inhibition. Arch Biochem Biophys 2018;644:29–36. [DOI] [PubMed] [Google Scholar]
- 150.Akin D, Wang SK, Habibzadegah-Tari P, Law B, Ostrov D, Li M, et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014;10:2021–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A 2012;109:8253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rebecca VW, Nicastri MC, McLaughlin N, Fennelly C, McAfee Q, Ronghe A, et al. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov 2017;7:1266–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.DeVorkin L, Hattersley M, Kim P, Ries J, Spowart J, Anglesio MS, et al. Autophagy inhibition enhances sunitinib efficacy in clear cell ovarian carcinoma. Mol Cancer Res 2016;15:250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sanders SS, Martin DD, Butland SL, Lavallee-Adam M, Calzolari D, Kay C, et al. Curation of the mammalian palmitoylome indicates a pivotal role for palmitoylation in diseases and disorders of the nervous system and cancers. PLoS Comput Biol 2015;11:e1004405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gayle S, Landrette S, Beeharry N, Conrad C, Hernandez M, Beckett P, et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood 2017;129:1768–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sharma G, Guardia CM, Roy A, Vassilev A, Saric A, Griner LN, et al. A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis. Autophagy 2019;1–25 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013;497:633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]