

Abstract
Defects in biliary transport proteins, MDR3 in humans and Mdr2 in mice, can lead to a spectrum of cholestatic liver disorders. Although B cell disorders and the aberrant antibody production are the leading extrahepatic manifestations of cholestatic liver diseases, the mechanism underlying this phenomenon is incompletely understood. Using mice with deficiency of Mdr2, that progressively develop cholestatic liver disease, we investigated the contributions of B cell activating factor (BAFF) to aberrant IgG, autoantibody production, and hepatic fibrosis. In Mdr2−/− mice, hepatic B lymphocytes constitutively produced IgG during fibrosis progression, which correlated with elevated serum levels of BAFF, anti-nuclear antibodies (ANA) and immune complexes. The elevated BAFF and ANA titers were also detected in human patients with primary sclerosing cholangitis (PSC) and hepatobiliary cholangiopathies. Consistent with the higher BAFF levels, liver-specific selection of focused BCR heavy chain (IgH) repertoire was found on hepatic B cells in Mdr2−/− mice. Interestingly, the administration of anti-BAFF mAb in Mdr2−/− mice altered the BCR repertoire on hepatic B lymphocytes, and resulted in reduced ANA and immune complex titers. However, anti-BAFF treatment did not attenuate hepatic fibrosis as measured by collagen deposition, hepatic expressions of collagen-1a, alpha-smooth muscle actin, and mononuclear cell infiltration (CD11b+Ly6chi, monocytes and CD11b+ Gr1+, neutrophils). Importantly, depletion of B cells by anti-CD20 mAb reduced both hepatic fibrosis and serum levels of ANA and immune complexes. Our findings implicate B cells as the potential therapeutic targets for hepatic fibrosis, and targeting BAFF specifically for attenuating the autoantibody production associated with cholestatic liver disease.
Introduction
Aberrant antibody production and autoimmune disorders ranging from mixed cryoglobulinemia (MC) to B cell lymphoma are among the most common extrahepatic manifestations of advanced liver disease (1–4). Specifically, cholestatic liver diseases are the complex liver disorders that involve immunological dysregulations, including B cell disorders and autoantibody production, as the potential etiological factors (5, 6). Disruption of ABCB4 locus or Mdr2 locus in mice which encodes biliary transport protein, i.e. multidrug resistance gene 2, results in inability to transport phospholipids and emulsification of toxic bile salts, that consequently develop intrahepatic sclerosing cholangitis, hepatic fibrosis, and hepatocellular carcinoma (6, 7). Likewise, humans that lack a functional MDR3 protein, a homolog of mouse Mdr2, are unable to emulsify toxic bile salts, thereon developing a progressive familial intrahepatic cholangitis disease (6–8). In particular, primary sclerosing cholangitis (PSC) is an autoimmune liver disorder, characterized by the elevated serum aminotransaminases, immunoglobulin G (IgG) and autoantibodies, that can progress into end-stage cirrhosis in children and adults (9–13). Diverse autoantibodies against biliary epithelial antigens, perinuclear anti-neutrophil cytoplasmic antibodies (pANCA), and ubiquitous self-antigens have been strongly associated with the PSC with increasing incidences of autoimmune disorders in PSC patients (13–15).
B-cell activating factor (BAFF; also known as BLyS, TANK, TALL-1, and TNFSF-13b) is a survival and maturation factor for B cells (16–21). It is primarily expressed by macrophages, monocytes, neutrophils, and dendritic cells, and binds three TNF ligand superfamily receptors; BAFF-specific receptor BAFF-R (TNFSR13C), transmembrane activator and CAML-interactor (TACI; TNFSR13B), and B-cell maturation antigen (BCMA; TNFSF17 or CD269) (16, 18, 20, 22). The interferon gamma (IFN-γ) and interleukin 10 (IL-10) have been shown to stimulate the BAFF expression by macrophage and dendritic cells (23). The increased serum levels of BAFF have been linked to B cell disorders in humans during chronic hepatitis C virus (HCV) infection (1–4), primary biliary cholangitis (PBC) (24, 25), Sjogren’s syndrome (26), rheumatoid arthritis (27), autoimmune hepatitis (AIH) (28), and in murine models with autoimmune diseases including systemic lupus erythromatosus (SLE) and experimental autoimmune encephalopathy (EAE) (29, 30). In addition, the overexpression of BAFF has been shown to augment B cell expression of toll like receptors (TLRs), rescue B cells from apoptosis or death, promote immune responses to nucleic acids (i.e. dsDNA, RNA etc.), and cause autoimmune disorders and autoreactivity (31, 32). Moreover, the binding of BAFF to TACI receptor has been demonstrated to promote T cell-independent differentiation of autoreactive B cells (33–35). Taken together, these studies implicate BAFF as the modulator of B cell-IgG aberrations and autoantibody production (1–4, 24, 25, 28). However, the contribution of BAFF to progression of hepatic fibrosis, B cell disorder and autoantibody production remain unclear during advancement of cholestatic liver disease.
In this study, we found that intrahepatic B cells constitutively produced IgG during progression of hepatic fibrosis in Mdr2−/− mice, and it correlated with increased serum levels of BAFF, ANA and production of immune complexes. BAFF and ANA levels were also elevated in human patients with PSC or hepatobiliary cholangiopathies. Although antibody-mediated blocking of BAFF did not attenuate hepatic fibrosis in Mdr2−/− mice, it did however alter the BCR repertoire on hepatic B lymphocytes and reduced serum titers for ANA and immune complexes. On the other hand, the depletion of B cells by anti-CD20 monoclonal antibodies (mAb) diminished both hepatic fibrosis and autoimmune antibody titers in Mdr2−/− mice. Our result suggests B-cell-targeted therapy as a potential therapeutic option for hepatic fibrosis and autoimmunity, and BAFF specifically for attenuating autoantibody production associated with cholestatic liver disease.
Materials and Methods
Mice
FVB/NJ (WT) and Mdr2−/+ (FVB.129P2-Abcb4tm1Bor/J) heterozygotes mice were purchased from the Jackson Laboratory and housed in specific pathogen-free environment per Emory University IACUC guidelines. Mice were backcrossed to generate Mdr2−/−, Mdr2−/+ and WT littermate controls, and confirmed by PCR. Where applicable, anti-mouse BAFF mAb (SANDY-2, Adipogen Corporation, San Diego, CA) were administered as 250μg in sterile PBS I.P. every 7 days starting at 8 weeks; continued until 25 weeks of age. In some experiments, anti-mouse CD20 (18B12, Biogen Idec, Cambridge, MA) mAb or isotype control (2B8, IgG2a, Biogen) were administered as 250 μg in sterile PBS I.P. every 7 days starting at 8 weeks until 25 weeks of age.
Human Subjects
A total of 13 patients undergoing orthotopic liver transplantation (n = 13) for either primary sclerosing cholangitis or hepatobiliary cholangiopathy (PSC; n = 5) or hepatitis C virus (HCV; n = 5) related end-stage liver disease, or non-HCV, non-alcoholic liver disease, non-nonalcoholic fatty liver liver-related liver resections (nonfibrotic control; n = 3) at The Emory Transplant Center were enrolled in the study in accordance with the Emory University Institutional Review Board (IRB) approval (IRB# 00006248). Patient characteristics with clinical information and relevant biological variables are summarized in Supplementary Table 1. Written informed consent was obtained from each patient and IRB #00006248 conforms to the guidelines of the 1975 Declaration of Helsinki (revised 2013).
Histology and Immunohistochemistry
Upon animal sacrifice, liver biopsies were immediately fixed in 10% formalin overnight and paraffin embedded. Sirius Red and Hematoxylin & Eosin (H&E) staining were performed by the Pathology Core of Yerkes National Primate Research Center at Emory University. Quantification of liver fibrosis was performed by a senior pathologist and provided score at Yerkes based on the criteria outlined by Ishak et al (36).
Isolation of Liver Cells and Flow Cytometry
Upon animal sacrifice, the livers were perfused with 1X PBS and harvested. Tissues were dissociated with scalpel in serum-free DMEM-F12 (Lonza) medium containing 2mg/mL type IV collagenase (Worthington, NY). Tissues were processed with enzymatic digestion at 370C for 45 mins as described previously (6, 37, 38). Following incubation, tissue digests were strained through sterile cheesecloth to remove particular matter and liver lymphocytes were isolated by Percoll gradient. B cells were purified by using CD19-positive selection kit (Miltenyi). For staining, following antibodies were used: anti-CD19 (1D3), CD3 (17A2), CD4 (RM4-5), CD8 (53-6.7), CD11b (M1/70), CD11c (N418), Ly6C (HK1.4), GR1 (RB6-8C5), I/A-I/E (M5/114.15.2), CD45.1 (A20) (Tonbo Biosciences, USA). Cells were analyzed using BD LSRII and FlowJo9.6.4 (TreeStar, Oregon, USA), and dead cells were excluded with Ghost red dye- 780 (Tonbo). For the analysis of myeloid populations and non-parenchymal liver cells, we gated out auto-fluorescent parenchymal cells such as hepatocytes in liver as described previously (39).
Quantification of hepatic stellate cells (HSCs)
After density gradient purification, liver cells were stained with antibodies to CD45 (104, BD Biosciences, San Jose, CA), and intracellularly stained with antibodies to alpha-smooth muscle actin (α-SMA; 1A4, R&D Systems). HSCs were identified as UV autofluorescence-positive (UVAF+) and CD45-ve population, and α-SMA expression was determined on UVAF+ CD45-ve HSCs population.
Quantitative Polymerase Chain Reaction
Quantitative PCR was performed as described previously by our laboratory (6). Briefly, liver tissues were homogenized in 1mL Trizol (Zymo Research) and processed to isolate mRNA, and reverse-transcription PCR to generate cDNA using high-capacity cDNA reverse transcription kit (Applied Biosystem). PCR reactions used were as follows; initial 950C for 10 min followed by 40 cycles of 950C for 15 sec, 600C for 60 sec and 720C for 30 sec and run using 7900HT Fast Real-Time PCR system (Applied Biosystem). Gene expressions were normalized to housekeeping genes GAPDH and calculated using the 2-ΔΔCt method. Primer sequences used: GAPDH, F-GTGCCAGCCTCGTCCC and R-ACTGTGCCGTTGAATTTGCC; Collagen1a, F-GGAGAGTACTGGATCGACCCTAAC and R-ACACAGGTCTGACCTGTCTCCAT; α-smooth muscle actin (α-SMA), F-CGGGAGAAAATGACCCAGATT and R-AGGGACAGCACAGCCTGAATAG.
ELISpot
ELISpot for total IgG antibody secreting cells was performed as described previously (38). In brief, Milipore multiscreen plates (Milipore, MA) were coated with 2 μg/mL Goat Anti-mouse Ig (Southern Biotech). The following day, plates were washed and blocked with complete medium. CD19 magnetic bead (Miltenyi) enriched B cells (50,000 cells) were added to the plate and incubated for 18 hours at 370C and 5% CO2. Standard procedures using biotinylated anti-mouse IgG (Sigma) was followed with streptavidin-ALP (mABTech), and assay was developed using NBT/BCIP substrate (Thermo-Scientific), and analyzed using CTL Immunospot 5.0 software (Cellular Tech Ltd.). Data are represented as IgG antibody secreting cells (ASC) spots per 106 cells (IgG ASC/106 cells).
IgG ELISA
For determination of total serum IgG, Nunc Maxi-sorp plates (Thermo-Scientific) were coated with 1 μg/mL anti-mouse Ig (Southern Biotech) overnight at 40C. Goat anti-mouse IgG-HRP (Southern Biotech) was as the secondary antibody, and BD OPT-EIA kit was used for detection (BD Biosciences). Known concentrations of mouse IgG was used for the generation of standard curves. For determination of IgG production by intrahepatic B cells, CD19-enriched intrahepatic B cells (200,000 cells) were plated with complete IMDM medium with or without stimulation for 5 days. Supernatants were collected following incubation and used for IgG ELISA as described for total serum IgG.
ANA analysis
Upon animal sacrifice, serum samples collected from animals were serially diluted (1:50, 1:100, 1:100, 1:200, and 1:400), and ANA titers were detected using an immunofluorescence staining of human epithelioid cell line (HEp-2) as described previously (40). The Hep-2 cells (Antibodies Inc.) were stained with different dilutions of serum according to manufacturer’s instructions. The secondary detection antibody FITC-conjugated F(ab’)2 goat anti-mouse IgG (Jackson Immunoresearch) were used, and cells were mounted with ProLong Gold anti-fade reagent (Invitrogen). Cells were photographed using a Zeiss Immunofluorescence microscope (Zeiss). Similarly, detection of serum ANA titers from human patients were performed using Hep-2 substrate staining using secondary human IgG detection antibody.
Detection of immune complexes
Upon animal sacrifice, serum samples collected from animals were serially diluted (1:30, 1:60, 1:120, 1:240, 1:240, 1:480, and 1:960 etc.), and serum titers for immune complexes were detected using a standard C1q ELISA. Similarly, detection of serum C1q titers from human patients were performed using C1q ELISA using human IgG detection antibody.
BCR IgH sequencing
B lymphocytes were sorted from the spleens and livers of 8-weeks and 25-weeks old WT and Mdr2−/− mice, and anti-BAFF or PBS treated Mdr2−/− animals using a FACSAria II and kept frozen. Cells were shipped to Adaptive Biotechnologies (Seattle, WA) for gDNA extraction and survey sequenced for BCR IgH. For analysis, out of frame and stop codon sequences were filtered out within the CDR3 region as described previously (41). In addition, clonotypes with sequence counts less than average count per cell were also removed. BCR clonotypes were determined based on their frequency as follows; dominant clonotypes (red, frequency >0.1%), sub dominant (blue, frequency 0.05-0.1%), low dominant (green, frequency 0.01-0.05%) and lowest dominant (white, frequency <0.01). Somatic hypermutation (V gene mutation) frequency was calculated using percentage representing the number of substitution per 100 base pairs as described previously (41). Comparison of BCR repertoire from anti-BAFF and PBS treated Mdr2−/− animals were performed using Simpson’s clonality estimation (a method of quantifying the shape of a repertoire, value ranging between 0 and 1) (42), Daley Smith Estimator (a nonparametric empirical estimator of repertoire richness) (43) and Morisita overlap analysis (a measurement of no similarity or similarity between two data sets, value ranging between 0 and 1) (44).
Statistical analysis
All statistical data was obtained using a two-tailed Mann Whitney U test, and one-way ANOVA analysis of variance using Graph Pad Prism 6 software (GraphPad, La Jolla, CA). Two-tailed Student t test was used to determine statistical significance where stated. *P<.05, **P<.001, and ***P<.0001.
Results
Aberrant IgG production during hepatic fibrogenesis in Mdr2−/− mice
In this study, our aim was to identify the contributions of aberrant antibody production to progression of cholestatic liver disease in Mdr2−/− mice. As characterized previously (6–8), we found that the hepatic fibrosis typically peaks around 25 weeks of age in Mdr2−/− animals, with a characteristic collagen deposition around hepatic bile duct and moderate to severe bridging fibrosis (average fibrosis score 3) (Fig. 1A). Fibrosis level was also corroborated with an elevated serum alanine transaminase (ALT) and alkaline phosphatase (ALP) levels, and mononuclear cells infiltrations (CD11b+Gr1+, neutrophils; and CD11b+Ly6chi, monocytes) into the liver (Supplemental Fig. 1A–B). Analysis of hepatic stellate cells (HSCs), based on FACS sorting of vitamin A autofluorescence and alpha-smooth muscle actin (α-SMA) expression (37), and immunofluorescence analysis for liver α-SMA also revealed an increased activation state of HSCs in Mdr2−/− animals (Supplemental Fig. 1C–D). Interestingly, an increased number and frequency of CD19+MHCII+CD5- B lymphocytes were found in fibrotic livers of 25-weeks old Mdr2−/− mice compared to age-matched WT mice (Fig. 1B, Supplemental Fig. 1B). These phenomena were not observed in the livers of 8-weeks old Mdr2−/− mice when fibrosis had not yet been evident to the extent seen at 25 week. Moreover, these elevated frequencies of B cells were not evident in from splenic (Fig. 1C) or peripheral blood compartments (data not shown). Furthermore, CD19-enriched total B lymphocytes from the fibrotic livers, but not spleens of 25-weeks old Mdr2−/− mice produced elevated IgG antibody levels in the absence of any stimulation as determined by ELISA (Fig. 1D–E). This was also verified with total IgG antibody secreting cells (ASC/106 cells) by ELISpot (Fig. 1F). Taken together, hepatic B lymphocytes constitutively produce IgG when hepatic fibrosis in Mdr2−/− mice occurs at 25 weeks of age.
FIGURE 1.
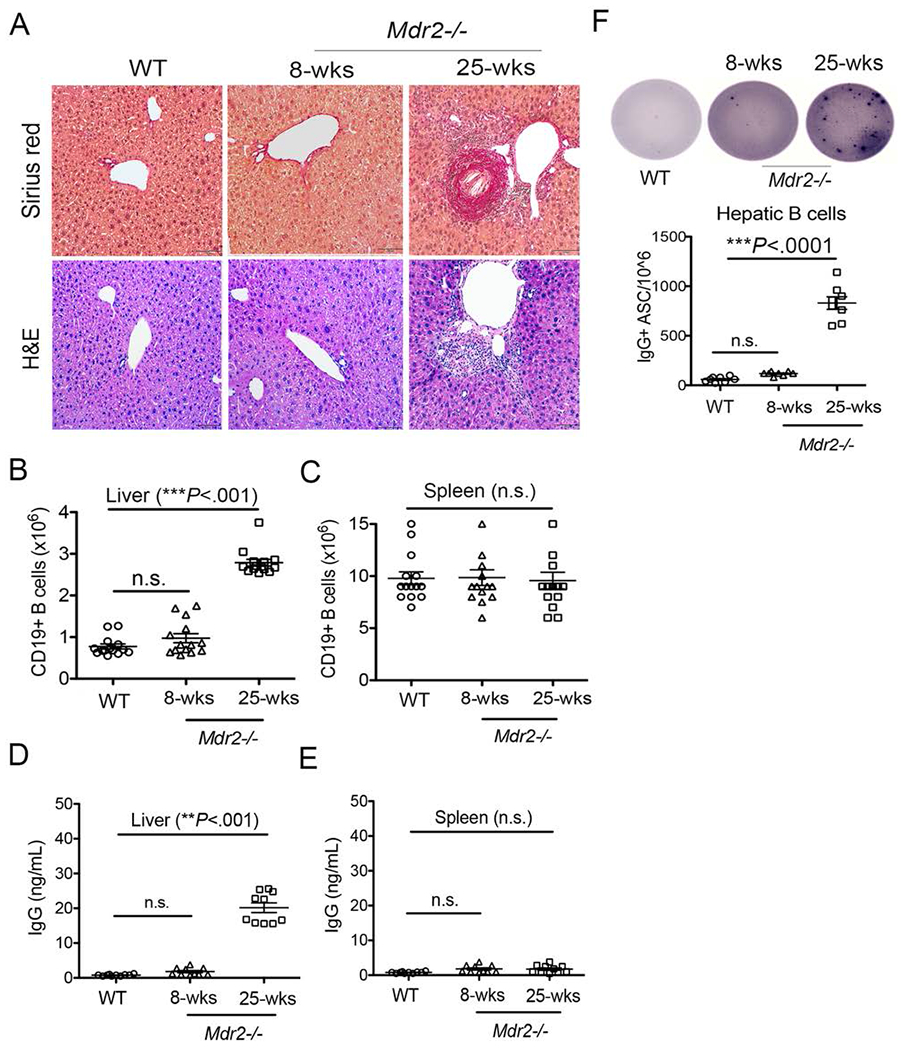
Aberrant IgG production during hepatic fibrosis. (A) Upon animal sacrifice, liver specimens from 8-weeks old and 25-weeks-old WT and Mdr2−/− mice were collected, fixed overnight in 10% formalin, and processed for histological analysis. Representative liver sections stained with Sirius red and H&E staining (Mag. 100×). are shown. (B-C) Total number of intrahepatic B lymphocytes (CD19+MHCII+) in the livers (B) and spleens (C) of WT and Mdr2−/− mice are shown. (D-E) B lymphocytes were purified and enriched using CD19 microbeads from the livers (D) and spleens (E) of WT and Mdr2−/− mice, and cultured (2 ×105 cells) in a complete IMDM medium without any stimulation for 5 days. Culture supernatants were collected and processed for total IgG detection using a standard ELISA technique. (F) Purified B lymphocytes (50,000 cells) were added to the culture plate coated overnight with anti-mouse IgG antibody and total IgG+ antibody secreting cells (IgG+ ASC/106) were detected using ELISpot technique. Data are representative of more than 3 independent experiments with 3-5 mice per group. Statistical significance was determined by a two-tailed Mann-Whitney Test, *P<.05, **P<.001, ***P<.0001. Error bars reflect the SEM.
Elevated autoantibodies and immune complexes in Mdr2−/− mice
The deposition of autoimmune antibodies and immune complexes in circulation and systemic organs are the prominent features of autoimmune diseases (40, 45). To determine whether aberrant IgG production by intrahepatic B cells leads to autoantibody production, we measured serum IgG, ANA and immune complexes in Mdr2−/− mice. Consistent with constitutive IgG production by intrahepatic B cells, the total serum IgG titers were higher in 25-weeks-old Mdr2−/− mice compared to age-matched WT mice (Fig. 2A). Once more, this observation was not evident in 8 week-old Mdr2−/− mice. To measure immune complexes in Mdr2−/− mice, we employed an approach to analyze serum C1q, a component of classical complement pathway that specifically binds to antigen-antibody complexes. We analyzed serum C1q titers in 8-week and 25-week-old Mdr2−/− mice and age-matched WT mice by using standard ELISA technique. Compared to WT mice (serum C1q titer <30), C1q titers were detected in the range of 120-480 (average 300) in 25-weeks-old Mdr2−/− mice (Fig. 2B). Importantly, 25-week-old Mdr2−/− mice showed higher titers of serum ANA (average 200) compared to WT mice (C1q titer <50) (Fig. 2C–D) with characteristic peripheral staining of nuclear speckled patterns; suggestive of dsDNA, ssDNA, DNP, and nuclear antigens (46). Altogether, these data indicate that the constitutive IgG production by activated hepatic B lymphocytes lead to extrahepatic autoimmune antibodies and immune complexes formation in Mdr2−/− mice.
FIGURE 2.
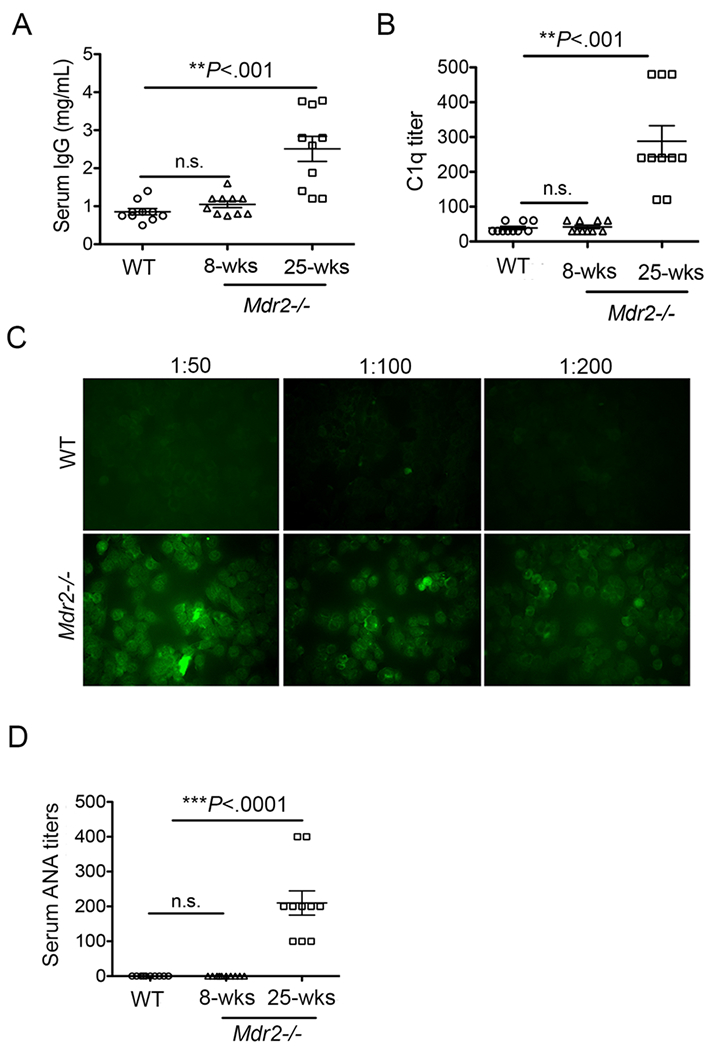
Elevated autoantibodies and immune complexes in Mdr2−/− mice. Serum specimens were collected from 8-weeks old and 25-weeks old Mdr2−/− and WT animals, and processed for detection of (A) total IgG by using a standard IgG ELISA technique, (B) total C1q titers by using a modified ELISA technique, and (C) Representative ANA images by using immunofluorescence staining of Hep-2 substrate are shown. (D) ANA titers are shown in 8-weeks old and 25-weeks old Mdr2−/− mice and WT controls. Data are representative of at least 3 independent experiments (n=3-5 per group).
Statistical significance was determined by a two-tailed Mann-Whitney Test, *P<.05, **P<.005, ***P<.0001. Error bars represent the SEM.
Elevated BAFF levels in Mdr2−/− mice during hepatic fibrosis progression
Studies have demonstrated that the excess production of soluble BAFF subverts B cell self-tolerance and rescues auto-reactive B cells (33–35). To understand how BAFF alters the balance between B cell tolerance and B cell dysregulation in regards to antibody production during cholestatic liver disease, we analyzed serum titers of BAFF in Mdr2−/− mice. Importantly, the serum BAFF levels were significantly higher in 25-week-old but not in 8-week-old Mdr2−/− and WT mice (Fig. 3A). This increased BAFF level is correlated with higher frequency of inflammatory myeloid populations such as neutrophils (CD11b+Gr1+) and/or monocytes (CD11b+ Ly6chi) in the fibrotic livers of Mdr2−/− mice (Supplemental Fig. 1B). Interestingly, the serum BAFF levels in human patients with PSC or related hepatobiliary cholangiopathies/cryptogenic liver pathology that closely resembles cholangitis seen in Mdr2−/− mice, were higher when compared to healthy controls without any known liver cirrhotic pathology (Fig. 3B). As controls, we also included human patients with chronic HCV infection, which also demonstrated an increased serum levels of BAFF (Fig. 3B). In addition, a higher ANA titer (average 400) were found in both PSC and HCV patients in comparison to healthy controls (Fig. 3C). Altogether, these findings suggest that serum BAFF levels increase during progression of hepatic fibrosis in Mdr2−/− mice and human patients with advanced sclerosing cholangitis.
FIGURE 3.
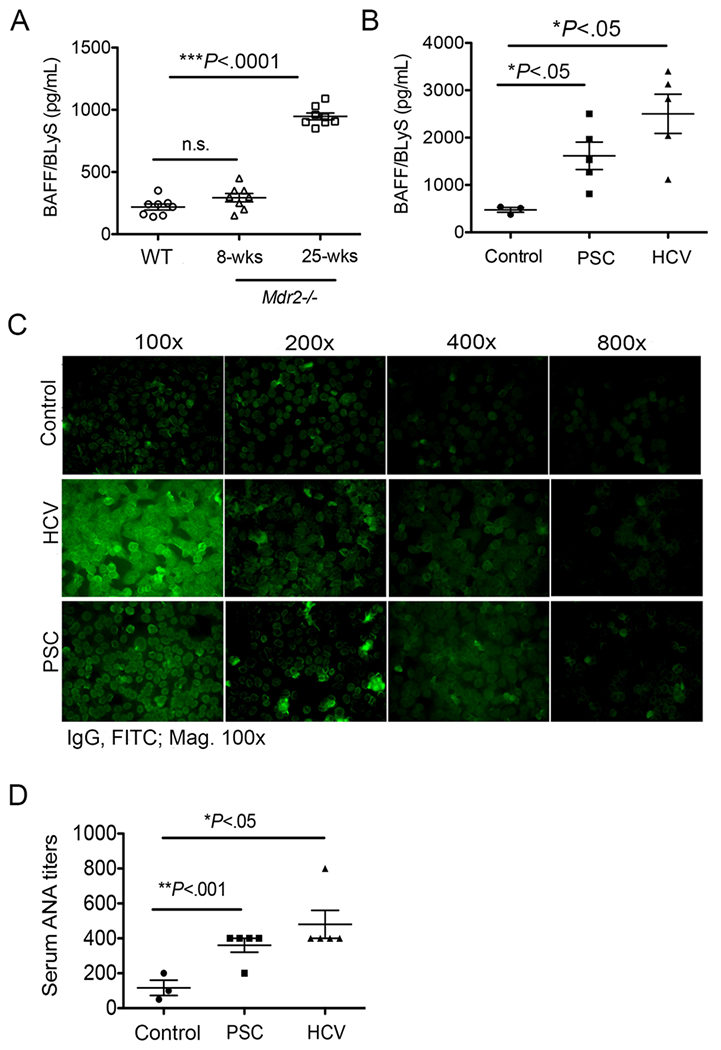
Elevated BAFF in Mdr2−/− mice and PSC human patients. (A) Serum specimens were collected from 8-weeks old and 25-weeks old Mdr2−/− and WT animals, and processed for detection of BAFF levels by using a standard mouse BAFF ELISA technique. Data are representative of at least 2 independent experiments (n = 3-5). (B) Plasma specimens from human subjects with end-stage liver disease (ESLD) as a result of PSC (n=5) or HCV (n=5) or non-HCV, non-alcoholic liver disease, non-nonalcoholic fatty liver liver-related liver resectioning (nonfibrotic control; n = 3) were processed for detection of BAFF levels by using a standard human BAFF ELISA method. (C) Representative ANA images in human patients by using human specific-IgG immunofluorescence staining of Hep-2 substrate are shown. (D) ANA titers are shown in control, PSC and HCV human patients. Statistical significance was determined by one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison test. *P<.05, **P<.001, ***P<.0001. Error bars reflect SEM.
Liver-specific selection of a focused BCR repertoire during cholestatic liver disease progression
The overexpression of BAFF has been demonstrated to regulate BCR repertoire selection (47). Moreover, our recent findings from chronically infected patients with HCV revealed that the BCR repertoire of antigen-specific B lymphocytes is focused (41). To better understand how elevation of BAFF influences BCR repertoire (clonotype) selection during cholestatic liver disease progression in Mdr2−/− mice, we sequenced the BCR heavy chain (IgH) of sorted intrahepatic B lymphocytes (CD19+MHCII+CD5-) from 25-weeks old Mdr2−/− mice (Fig. 4A). Corresponding splenic B lymphocytes from both Mdr2−/− and WT mice and non-fibrotic hepatic B lymphocytes from WT mice were also sorted for analysis. The sorted B lymphocytes were sequenced for the BCR IgH locus. Interestingly, the selection and amplification of B cell clonotypes implicated a focused BCR repertoire in fibrotic livers of Mdr2−/− mice. Dominant clonotypes (red, as determined by their frequency) were uniquely present in intrahepatic B cells from fibrotic livers (n=3) of Mdr2−/− mice, compared to its corresponding splenic compartment (n=3) and non-fibrotic liver-derived B cells (n=3) from WT mice (Fig. 4B). The BCR repertoire of splenic B cells showed no selection of dominant clonotypes and comprised of lowest abundance clonotypes (>84%), while non-fibrotic hepatic B cells showed sub-dominant clonotypes (>64%), and few dominant clonotypes (>15%). However, hepatic B cells from fibrotic Mdr2−/− mice showed mainly dominant clonotypes (>60%), and a few sub-dominant clonotypes (>11%). Interestingly, dominant clonotypes in Mdr2−/− mice were selected late during progression of liver fibrosis in the 25-week old Mdr2−/− mice as these dominant clonotypes were absent in the 8-week old Mdr2−/− mice when fibrotic lesion (score 0) was far less evident as compared to the extent seen in the 25-week old mice (mean score 3) (Fig. 4B). In addition, the somatic hypermutation i.e. V gene mutation rate in hepatic B cells from 25-week old Mdr2−/− mice was also increased compared to 8-week old mice Mdr2−/− and WT controls (Fig. 4C). Taken together, these findings suggest a liver-specific selection of BCR repertoire occur during hepatic fibrosis.
FIGURE 4.
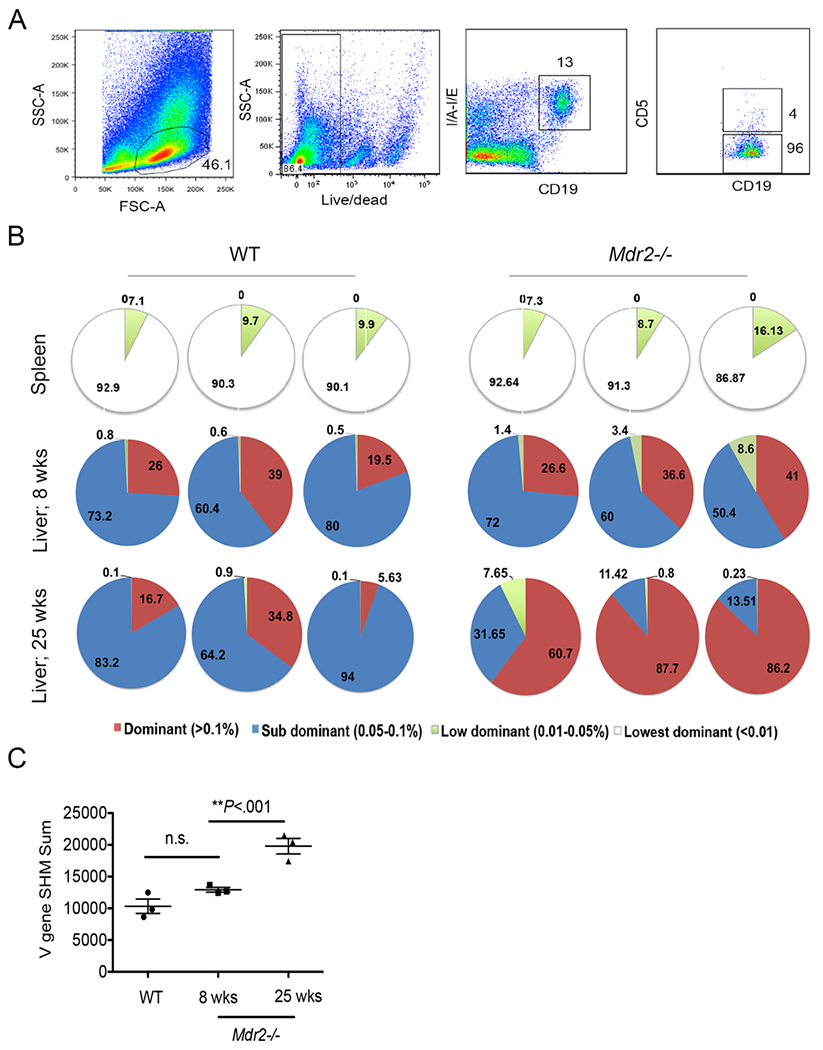
Liver-specific selection of focused BCR repertoire during hepatic fibrosis. (A) WT (n=3), 8-weeks old Mdr2−/− (n=3), and 25-weeks-old) Mdr2−/− mice (n=3) were harvested to purify liver and splenic B lymphocytes, and stained and sorted based on their expressions of CD19, MHCII and CD5 as shown (CD19+MHCII+CD5-). (B) The sorted B lymphocytes were survey sequenced for the BCR IgH. BCR clonotypes were determined based on their frequency as follows; dominant clonotypes (red, frequency >0.1%), Sub dominant (blue, frequency 0.05-0.1%), Low dominant (green, frequency 0.01-0.05%) and lowest dominant (white, frequency <0.01). (C) The somatic hypermutation rate in sorted hepatic B cells from WT and Mdr2−/− mice is shown. Statistical analysis was performed by one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison test for C. *P<.05, **P<.001. Error bars reflect SEM.
Blockade of BAFF reshapes the hepatic B-cell receptor (BCR) repertoire during progression of hepatic fibrosis
To understand the role of BAFF in BCR repertoire selection, we administered anti-BAFF mAb (SANDY-2) intraperitoneally (i.p.) once a week to Mdr2−/− mice starting from 8 weeks to 25 weeks of age (Supplemental Fig. 2A). Antibody treatment reduced the soluble BAFF levels and the total frequencies of B cells in the peripheral blood of Mdr2−/− mice (Supplemental Fig. 2B–C). Compared to peripheral blood however, B cell frequencies were not reduced to the same extent in the fibrotic livers and the spleens of Mdr2−/− mice (Supplemental Fig. 2C). We sorted intrahepatic B lymphocytes (CD19+MHCII+CD5-) from anti-BAFF treated Mdr2−/− mice and PBS controls, and sequenced the BCR IgH as described above. Importantly, anti-BAFF treatment significantly altered the BCR clonotypes on hepatic B cells (Fig. 5). Although total productive templates (clones) were less in number with anti-BAFF treatment, Simpson’s clonality measurement (a method of quantifying the shape of a repertoire) and Daley Smith Estimator (a nonparametric estimation of repertoire richness) indicated a higher clonality and more focused repertoire in the anti-BAFF treated Mdr2−/− animals as opposed to control animals (Fig. 5A–C). Interestingly, comparison of top ten clones indicated a higher frequency of dominant clonotypes in anti-BAFF treated Mdr2−/− animals compared to controls (Fig. 5D). Moreover, Morisita analysis (a measurement of similarity and/or dissimilarity between two data sets) showing heat map values all close to zero (blue squares) and therefore indicate no overlap of clonotypes among anti-BAFF treated and control animals (Fig. 5E). Taken together, these findings suggest that blockade of BAFF reshaped the BCR repertoire during hepatic fibrosis progression in cholestatic liver disease.
FIGURE 5.
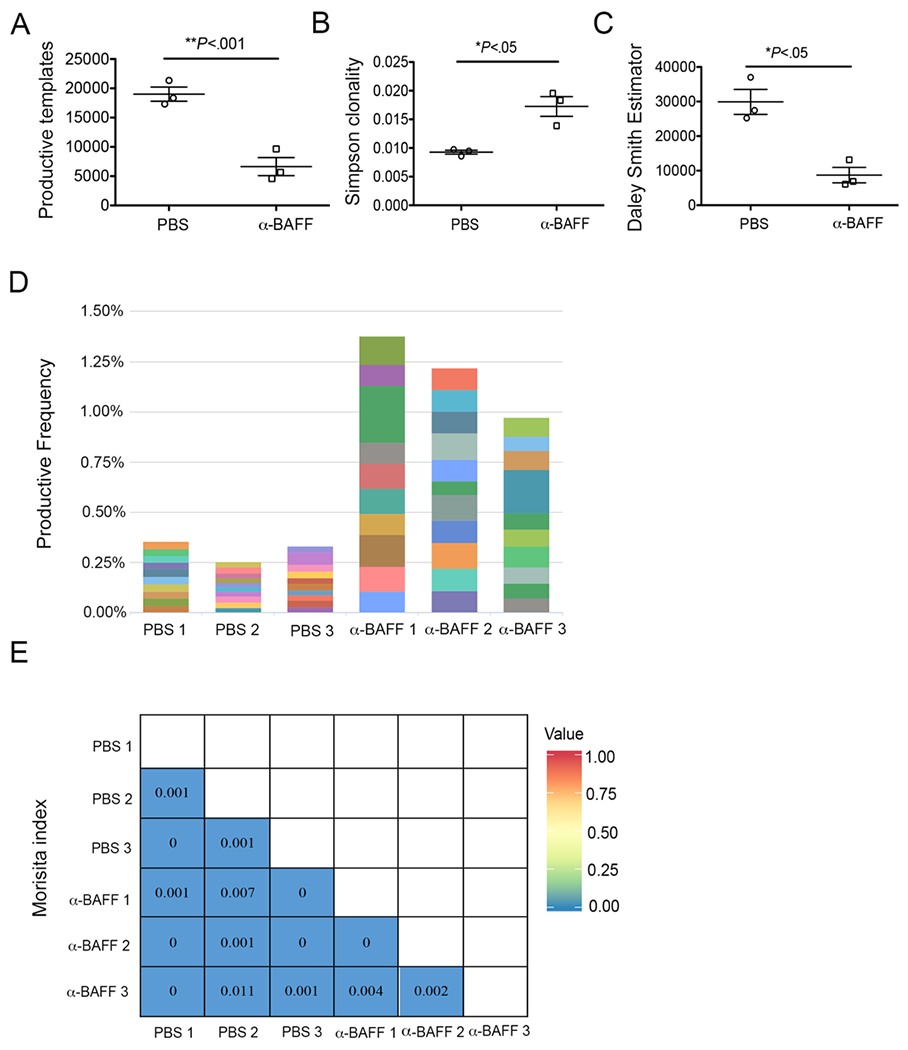
Blockade of BAFF alters BCR repertoire during hepatic fibrosis. Anti-BAFF mAb or sterile PBS were administered into Mdr2−/− mice once a week starting at 8 weeks until 25 weeks of age. Total B lymphocytes (CD19+MHCII+CD5-) were enriched by FACS sorting and analyzed for BCR IgH repertoire. Comparison of BCR repertoire from anti-BAFF (n=3) and PBS (n=3) treated Mdr2−/− animals were performed using (A) productive templates analysis, (B) Simpson’s clonality estimation, and (C) Daley Smith Estimator. (D) The stacked bar graph showing the frequency of top ten clonotypes found in anti-BAFF and PBS treated Mdr2−/− animals. (E) Morisita analysis showing heatmap of overlap among top ten clonotypes found in anti-BAFF and PBS treated Mdr2−/− animals. Statistical analysis was performed by unpaired Student t test for panel A. *P<.05, **P<.001. Error bars reflect SEM.
Blockade of BAFF reduces ANA and immune complexes titers during hepatic fibrosis
Because anti-BAFF antibody treatment altered the BCR repertoire, we next asked whether it could have any significant consequences on the autoimmune antibody production and immune complex formation. To this end, we analyzed serum levels of ANA and immune complexes in Mdr2−/− mice following anti-BAFF antibody treatment. Interestingly, antibody-mediated blockade of BAFF significantly reduced the serum ANA titers in Mdr2−/− mice (Fig. 6A–B). Likewise, the serum titers of total IgG and the immune complexes (i.e. C1q) were also reduced following antibody treatment in Mdr2−/− mice (Fig. 6C–D). Taken together, anti-BAFF antibody treatment reduced the rampant IgG production, autoimmune antibodies and immune complexes (i.e. ANA and C1q titers) during hepatic fibrosis progression in Mdr2−/− mice.
FIGURE 6.
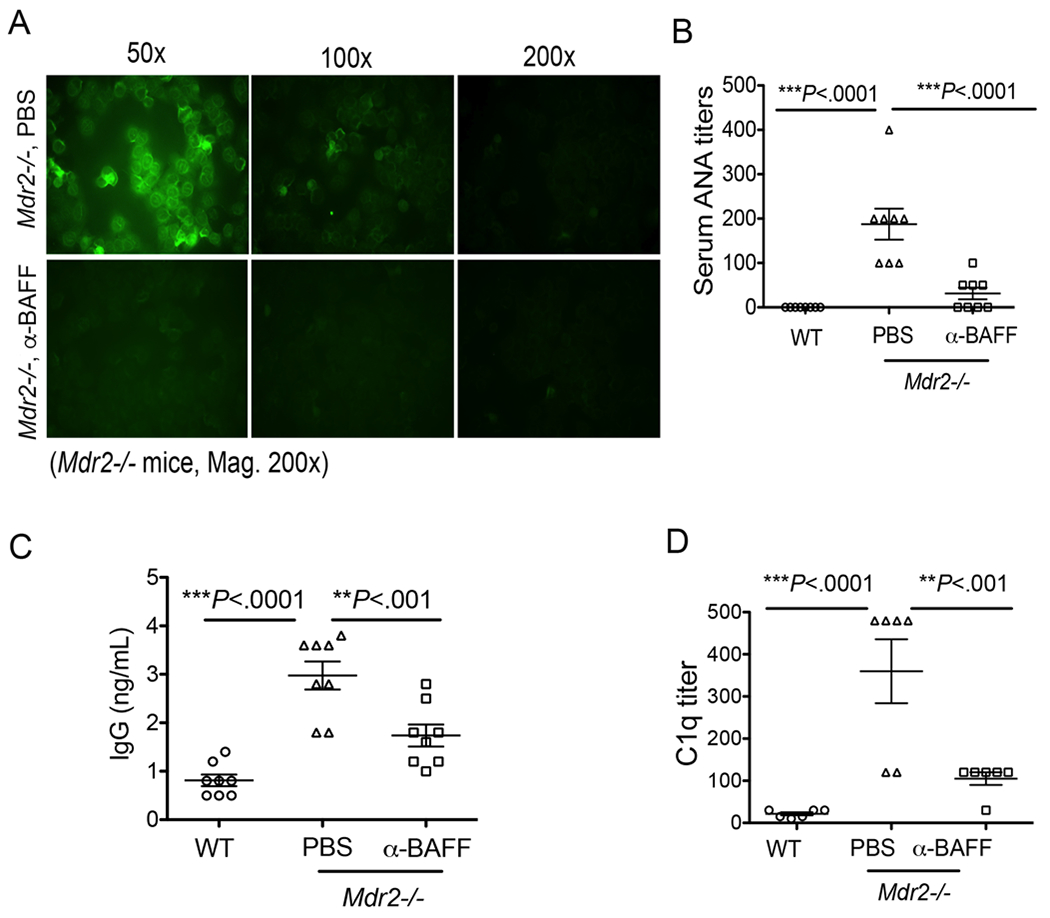
Blockade of BAFF reduces autoantibodies and immune complexes. Mdr2−/− mice were administered anti-mouse BAFF (SANDY-2) mAb or sterile PBS as described above. (A) Serum samples collected from animals were serially diluted (1:50, 1:100, and 1:200), and ANA titers were detected using an immunofluorescence staining of human epithelioid cell line (HEp-2) as shown. (B) Serum ANA titers in PBS- and anti-BAFF-treated Mdr2−/− and WT animals are shown. (C) Serum samples collected from animals were processed for determination of total IgG by a standard ELISA. (D) Serum samples were serially diluted (1:30, 1:60, 1:120, 1:240, 1:480, and 1:960 etc.), and serum titers for immune complexes were detected using C1q ELISA. Statistical significance was determined by a two-tailed Mann-Whitney Test. **P<.001, ***P<.0001. Error bars reflect SEM.
Blockade of BAFF does not attenuate hepatic fibrosis in Mdr2−/− mice
Next, to understand whether BAFF is required for the pathogenesis of hepatic fibrosis in our model, we administered anti-BAFF mAb (SANDY-2) i.p. once a week to Mdr2−/− mice starting from 8 weeks to 25 weeks of age as described above. Interestingly, antibody-mediated blockade of BAFF did not alter the collagen deposition in the liver as shown by Sirius red staining (Fig. 7A), and the quantification of hepatic fibrosis by blind scoring (average score 3) (Fig. 7B). Likewise, the relative gene expressions of hepatic collagen1a and α-SMA were not altered as measured by quantitative PCR (Fig. 7C). In addition, the total frequencies of inflammatory cells such as CD11b+Ly6c++ (monocytes) and CD11b+Gr1+ (neutrophils) were not significantly reduced in the livers of Mdr2−/− mice (Fig. 7D–E). Altogether, anti-BAFF antibody treatment did not alter the progression of hepatic fibrosis during cholestatic liver disease progression in Mdr2−/− mice.
FIGURE 7.
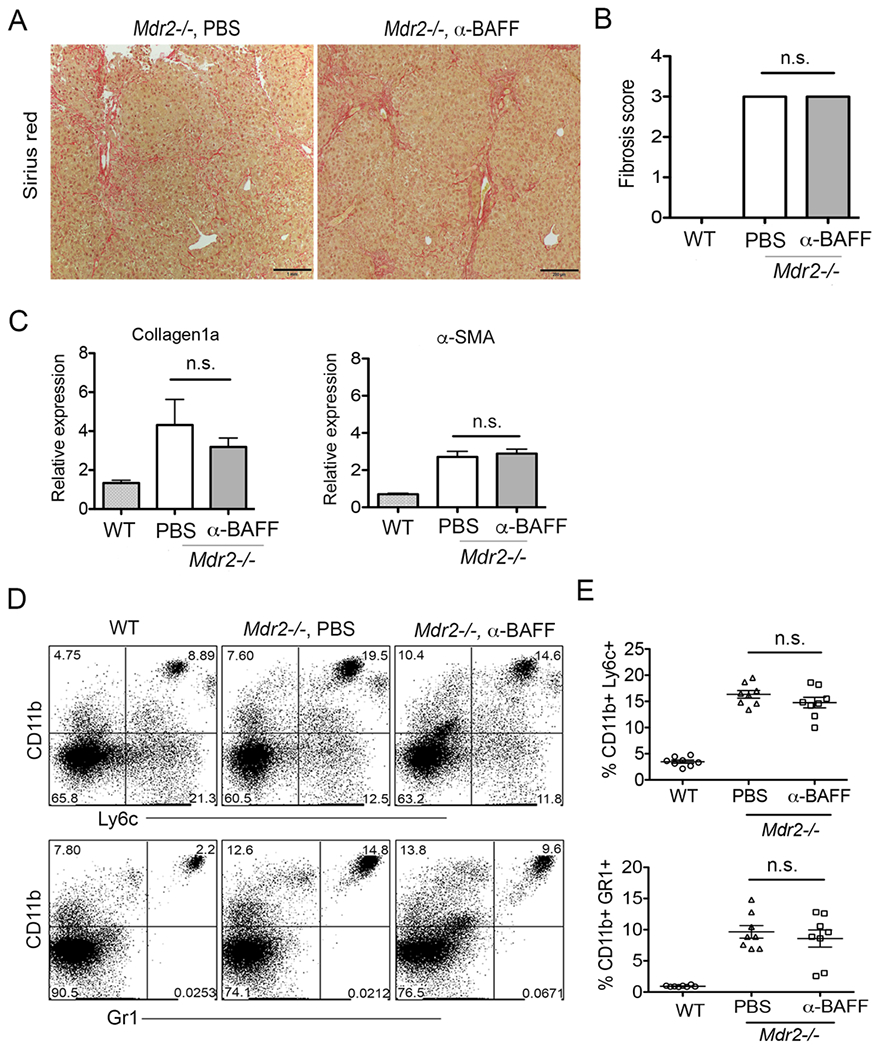
Blockade of BAFF do not attenuate hepatic fibrosis. (A) Mdr2−/− mice were administered anti-BAFF mAb as 250μg in sterile PBS i.p. every 7 days starting at 8 weeks continued until 25 weeks of age. Histologic analysis of representative liver specimens for Sirius red staining (mag. 100×) are shown. (B) Blind fibrosis scoring of Mdr2−/− animals following antibody treatment is shown. (C) Relative gene expression of liver collagen1a and α-SMA by PCR analysis following antibody treatment is shown. (D) Representative FACS plots of intrahepatic myeloid populations monocytes (CD11b+Ly6c++), and neutrophils (CD11b+Gr1+) are shown. (E) Frequencies of myeloid populations monocytes and neutrophils are shown. Statistical significance was determined by a two-tailed Mann-Whitney Test. *P<.05. Error bars represent SEM.
B cell depletion by anti-CD20 mAb attenuates hepatic fibrosis and autoimmune antibodies in Mdr2−/− mice
Next, we sought to determine whether directly targeting B cell depletion with anti-CD20 mAb could have a significant alteration on hepatic fibrosis mechanisms in Mdr2−/− mice. To this end, we administered anti-mouse CD20 mAb (clone 18B12, IgG2a, Biogen) i.p. into Mdr2−/− mice every 7 days starting at 8 weeks and continued until 25 weeks of age (Fig. 8A). Anti-CD20 treatment significantly depleted the B cells (CD19+MHCII+) in the peripheral blood, spleen and the liver compartment of Mdr2−/− mice when compared to isotype-treated animals (Supplemental Fig. 3A–B). Interestingly, anti-CD20 treated animals gained significant body weight during the treatment from 16 to 25 weeks (Supplemental Fig. 3C). We postulate that B cell depletion significantly improved the liver pathology and disease outcome at a time when fibrosis begins to develop robustly. Indeed, anti-CD20 treated animals exhibited a reduced fibrosis lesion (score 1 to 2) compared to isotype controls (mean score 3) (Fig. 8B). Likewise, collagen deposition and mononuclear cells infiltration were also significantly reduced in the livers of anti-CD20-treated Mdr2−/− mice (Fig. 8C–D). In support of this observation, we also detected decreased levels of α-SMA expression on HSC thus correlating with reduced HSC activation (Supplemental Fig. 3D). Importantly, antibody treatment also reduced total IgG in serum (Fig. 8E), and diminished both serum ANA titers (Fig. 8F) and immune complexes (Fig. 8G) in Mdr2−/− mice. Combined together, the depletion of B lymphocytes using anti-CD20 mAb attenuated both the hepatic fibrosis and rampant IgG production during cholestatic liver disease progression in Mdr2−/− mice.
FIGURE 8.
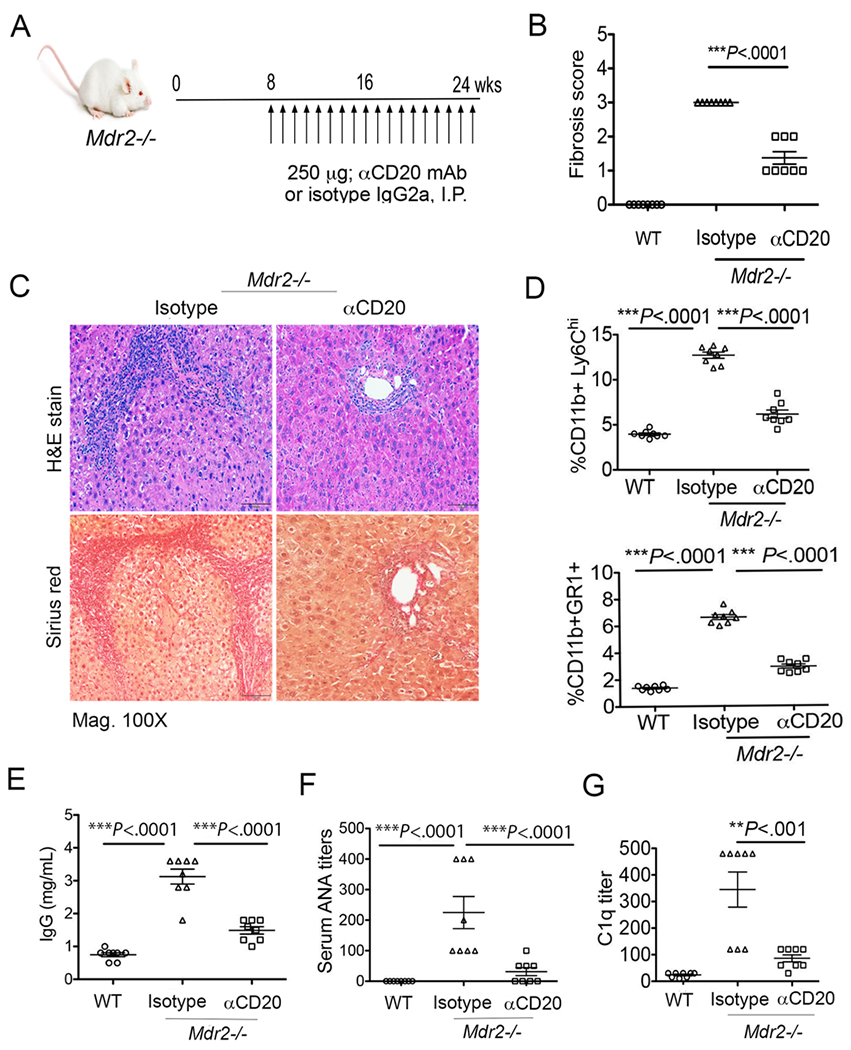
Anti-CD20-mediated depletion of B cells attenuates hepatic fibrosis and autoantibodies in Mdr2−/− mice. (A) Mdr2−/− mice were administered either anti-mouse CD20 (clone 18B12, IgG2a, Biogen) mAb or isotype control (2B8, IgG2a) as 250μg in sterile PBS i.p. every 7 days starting at 8 weeks continued until 25 weeks of age as demonstrated. (B) Blind quantification of liver fibrosis following antibody treatment is shown. (C) Histologic analysis of representative liver specimens for H&E and Sirius red staining (mag. 100×) are shown. (D) Frequencies of intrahepatic myeloid populations monocytes (CD11b+Ly6c++) and neutrophils (CD11b+Gr1+) are shown. (E-G) Serum specimens collected from animals were analyzed for IgG (E), ANA titers (F) and C1q titers (G) as described in Materials and Methods section. Statistical significance was determined by one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison test. **P<.001, ***P<.0001. Error bars reflect SEM.
Discussion
There is an established association between advanced liver disease and proliferative B cell disorders, ranging from mixed cryoglobulinemia to lymphoma (1–3). Studies involving HCV patients have observed a clonal expansion of B cells present in the peripheral blood of HCV+MC+ patients, and the selective activation of memory B cell subsets (48, 49). It is suggested that chronic B cell stimulation plays a significant role in their dysregulation, as the resolution of both MC and non-Hodgkin’s lymphoma has been observed following HCV eradication (50). Dysregulation of B cell IgG-production is not exclusive to HCV-mediated liver disease; recent evidence in non-alcoholic steatohepatitis (NASH) and alcohol liver disease (ALD) patients suggests that aberrant Ig-production occurs as a result of diseased liver (51–53). In agreement with this phenomenon, we demonstrate here that a rampant IgG production by activated intrahepatic B cells occur during progression of hepatic fibrosis that contributes to elevated autoimmune antibodies and immune complex formation during cholestatic liver disease progression.
The elevated BAFF levels have been linked to B cell disorders in humans during chronic HCV infection (1–4), and PBC pathogenesis (24, 25). Previous studies have implicated an association between the elevated BAFF levels with MC in chronic HCV individuals, but not in HCV without MC or chronic HBV patients (4). Although our study did not stratify between HCV patients with or without MC, we did notice that chronic HCV patients had a trend toward higher BAFF level expression than PSC patients. However, this finding did not reach statistical significance. We did however find a liver-specific selection of focused BCR repertoire during hepatic fibrosis progression in Mdr2−/− mice. The somatic hypermutation i.e. V gene mutation rate on hepatic B cells were also increased during disease progression thus implicating the importance of these mutations in selection of dominant repertoire with particular specificity and/or affinity (54, 55). We found that BAFF plays a significant role in this process as the blockade of BAFF molecule with mAb markedly altered the dominant BCR clonotypes in the liver of these animals. Specifically, a higher frequency of dominant clonotypes following the blockade of BAFF was observed. Morisita analysis, which depicts similarity and/or dissimilarity between clonotypes, suggested no overlap of clonotypes between anti-BAFF treated Mdr2−/− animals and controls. Although our findings suggested that there were no direct role for BAFF in modulating hepatic fibrosis, the antibody-mediated blockade of BAFF did however reduce the serum titers of ANA and immune complexes in Mdr2−/− mice. A possible explanation for these findings is that anti-BAFF blockade altered the autoimmune BCR repertoire of hepatic B cells during disease progression. In addition, the reduction of B cell frequencies in the peripheral, splenic and hepatic microenvironments during anti-BAFF treatment, along with an impaired B cell humoral response (i.e. IgG production) may also contribute to this phenomenon (31). Interestingly, the activation of TLR7/9, but not TLR4, has been demonstrated to strongly upregulate the BAFF receptor, TACI, which plays a critical role in autoantibody production (34).
A number of alternative drugs such as obeticholic acid (a farnesoid X receptor, FXR agonist), bezafibrate (a peroxisome proliferator-activated receptor, PPAR agonist), and ursodeoxycholic acid (UDCA), are in advanced clinical trials for PBC (56), yet the efficacy of these drugs is unexplored or limited in PSC patients. Importantly, however, our results reveal that B-cell targeted therapy as the potential therapeutic option in sclerosing cholangitis and fibrosis sequelae. Anti-CD20 mAb completely eliminated B cells populations, including activated hepatic B cells, and resulted in reduced infiltration of inflammatory myeloid populations into the livers of Mdr2−/− mice, and hence reduced liver pathology (Fig. 8). These results are consistent with our previously published data (37), and the findings from Novobrantseva (57) and Faggioli et al. studies (58), which demonstrated that ablation of B cells, but not CD4+/CD8+ T cells, promote senescence-mediated fibrosis resolution. Faggioli’s study indicated that the lack of B lymphocytes changed the intrahepatic metabolic and oxidative environment and resulted in macrophage polarization into anti-inflammatory M2 phenotype in Mdr2−/− mice (58). Our study further suggested that ablation of B cells also diminished autoantibodies and immune complex titers (Fig. 8E). Clinical studies involving anti-CD20 (rituximab)-mediated B cell depletion have also suggested improvements in biliary enzymes, serum IgM, and anti-mitochondrial antibodies (AMA) production in PBC patients (59) (60). Likewise, in murine models, a beneficial role of anti-CD20 was found in early autoimmune cholangitis development in transforming growth factor-β receptor II dominant negative mice (dnTGF-βRII) mice, however, an exacerbation of inflammatory bowel disease was noted in these animals (61). In dnTGF-βRII mice expressing humanized CD20 and human Fcγ, a reduction in portal inflammation, and mononuclear cells and CD8+ T cell infiltration was found after 16 weeks of treatment with anti-hCD20 antibody (62). Although studies involving anti-CD20 therapy have resulted in clear outcomes in PBC patients and murine models, further efficacy studies are warranted in PSC patients.
Overall, our findings presented here provide further insight into BAFF-mediated IgG aberration during cholestatic liver disease progression in a relevant mouse model. Furthermore, translational studies linking human patients with PSC or hepatobiliary cholangiopathies strengthen these observations. Our results highlight BAFF as the modulator of BCR repertoire during cholestatic disease progression and implicate BAFF as the potential target specifically for autoimmune antibodies associated with the disease outcome. Caveats in our study do include a lack of phenotypic analysis of the autoreactive and/or anergic B cells and determination of the auto-specificity. Future studies will focus on these issues and also the characterization of the dominant clonotypes detected in the control and anti-BAFF treated Mdr2−/− animals in order to determine the specificity of these clonotypes.
Supplementary Material
KEY POINTS.
Liver-specific selection of focused BCR repertoire in cholestatic liver disease.
Blockade of BAFF reshapes BCR repertoire and reduces autoantibody production.
Depletion of B cells reduces both the hepatic fibrosis and autoantibody production.
Acknowledgements
We are grateful to the patient donors who were undergoing liver transplantation at Emory University Hospital Transplant Center who kindly shared their invaluable samples with us. We are thankful to Emory Transplant Center team: surgeons and surgical staff, nursing staff, and coordinators for their assistance and help during this study. We also thank Eduardo Salinas, Dan Chopyk, Patrick Speck, Dennis Jang, Sarah Chewing and Fengzhi Jin for their technical help and support. We are also grateful to the Flow Cytometry, Immunology and Pathology Cores of Emory Vaccine Center, and veterinary and animal care staff of Yerkes National Primate Research Center, Emory University for their assistance.
This project was supported by NIH grants R01AI136533, R01AI124680, R01AI096882 and R01AI126890 to A.G., and ORIP/OD P51OD011132 (formerly NCRR P51RR000165) to the Yerkes National Primate Research Center. M.T. is supported by National Institute of Diabetes, and Digestive and Kidney Diseases (NIDDK) Mentored Career Development Award (K01DK109025). This work was facilitated by the Immunology and Flow Cytometry Core of the Center for AIDS Research at Emory University (P30AI050409). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Abbreviations used in this article:
- α-SMA
α-smooth muscle actin
- ANA
anti-nuclear antibodies
- BAFF/BLyS
B-cell activating factor/B lymphocyte stimulator
- BCR
B cell receptor
- ESLD
End-stage liver disease
- HCV
hepatitis C virus
- HSC
hepatic stellate cell
- IgG
immunoglobulin G
- IgH
Immunoglobulin heavy chain
- i.p.
intraperitoneally
- mAb
monoclonal antibody
- MC
mixed cryoglobulinemia
- Mdr2
multidrug resistance gene 2
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- SEM
standard error of mean
Footnotes
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Dammacco F, Sansonno D, Piccoli C, Tucci FA, and Racanelli V. 2001. The cryoglobulins: an overview. Eur J Clin Invest 31: 628–638. [DOI] [PubMed] [Google Scholar]
- 2.Pozzato G, Mazzaro C, Crovatto M, Modolo ML, Ceselli S, Mazzi G, Sulfaro S, Franzin F, Tulissi P, Moretti M, and et al. 1994. Low-grade malignant lymphoma, hepatitis C virus infection, and mixed cryoglobulinemia. Blood 84: 3047–3053. [PubMed] [Google Scholar]
- 3.Rasul I, Shepherd FA, Kamel-Reid S, Krajden M, Pantalony D, and Heathcote EJ. 1999. Detection of occult low-grade b-cell non-Hodgkin’s lymphoma in patients with chronic hepatitis C infection and mixed cryoglobulinemia. Hepatology 29: 543–547. [DOI] [PubMed] [Google Scholar]
- 4.Lake-Bakaar G, Jacobson I, and Talal A. 2012. B cell activating factor (BAFF) in the natural history of chronic hepatitis C virus liver disease and mixed cryoglobulinaemia. Clinical and experimental immunology 170: 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirschfield GM, Heathcote EJ, and Gershwin ME. 2010. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology 139: 1481–1496. [DOI] [PubMed] [Google Scholar]
- 6.Tedesco D, Thapa M, Chin CY, Ge Y, Gong M, Li J, Gumber S, Speck P, Elrod EJ, Burd EM, Kitchens WH, Magliocca JF, Adams AB, Weiss DS, Mohamadzadeh M, and Grakoui A. 2018. Alterations in Intestinal Microbiota Lead to Production of Interleukin 17 by Intrahepatic gammadelta T-Cell Receptor-Positive Cells and Pathogenesis of Cholestatic Liver Disease. Gastroenterology 154: 2178–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, Mol CA, Ottenhoff R, van der Lugt NM, van Roon MA, and et al. 1993. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 75: 451–462. [DOI] [PubMed] [Google Scholar]
- 8.Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, Krause R, Lammert F, Langner C, Zatloukal K, Marschall HU, Denk H, and Trauner M. 2004. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 127: 261–274. [DOI] [PubMed] [Google Scholar]
- 9.Lazaridis KN, and LaRusso NF. 2016. Primary Sclerosing Cholangitis. The New England journal of medicine 375: 1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Folseraas T, and Boberg KM. 2016. Cancer Risk and Surveillance in Primary Sclerosing Cholangitis. Clinics in liver disease 20: 79–98. [DOI] [PubMed] [Google Scholar]
- 11.Charatcharoenwitthaya P, and Lindor KD. 2006. Primary sclerosing cholangitis: diagnosis and management. Current gastroenterology reports 8: 75–82. [DOI] [PubMed] [Google Scholar]
- 12.Tabibian JH, Enders F, Imam MH, Kolar G, Lindor KD, and Talwalkar JA. 2014. Association between serum IgE level and adverse clinical endpoints in primary sclerosing cholangitis. Annals of hepatology 13: 384–389. [PMC free article] [PubMed] [Google Scholar]
- 13.Abdo AA, Bain VG, Kichian K, and Lee SS. 2002. Evolution of autoimmune hepatitis to primary sclerosing cholangitis: A sequential syndrome. Hepatology 36: 1393–1399. [DOI] [PubMed] [Google Scholar]
- 14.Chung BK, Henriksen EKK, Jorgensen KK, Karlsen TH, Hirschfield GM, and Liaskou E. 2018. Gut and Liver B Cells of Common Clonal Origin in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease. Hepatology communications 2: 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hov JR, Boberg KM, and Karlsen TH. 2008. Autoantibodies in primary sclerosing cholangitis. World journal of gastroenterology 14: 3781–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, Ambrose C, Lawton P, Bixler S, Acha-Orbea H, Valmori D, Romero P, Werner-Favre C, Zubler RH, Browning JL, and Tschopp J. 1999. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. The Journal of experimental medicine 189: 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stadanlick JE, and Cancro MP. 2008. BAFF and the plasticity of peripheral B cell tolerance. Current opinion in immunology 20: 158–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider P, and Tschopp J. 2003. BAFF and the regulation of B cell survival. Immunology letters 88: 57–62. [DOI] [PubMed] [Google Scholar]
- 19.Mackay F, and Browning JL. 2002. BAFF: a fundamental survival factor for B cells. Nature reviews. Immunology 2: 465–475. [DOI] [PubMed] [Google Scholar]
- 20.Mackay F, Schneider P, Rennert P, and Browning J. 2003. BAFF AND APRIL: a tutorial on B cell survival. Annual review of immunology 21: 231–264. [DOI] [PubMed] [Google Scholar]
- 21.Do RK, and Chen-Kiang S. 2002. Mechanism of BLyS action in B cell immunity. Cytokine & growth factor reviews 13: 19–25. [DOI] [PubMed] [Google Scholar]
- 22.Cancro MP 2009. Signalling crosstalk in B cells: managing worth and need. Nature reviews. Immunology 9: 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogden CA, Pound JD, Batth BK, Owens S, Johannessen I, Wood K, and Gregory CD. 2005. Enhanced apoptotic cell clearance capacity and B cell survival factor production by IL-10-activated macrophages: implications for Burkitt’s lymphoma. Journal of immunology 174: 3015–3023. [DOI] [PubMed] [Google Scholar]
- 24.Liang Y, Yang ZX, Zhu Y, Wang Y, Zeng XM, Deng AM, and Zhong RQ. 2007. [Study on association of BAFF receptors gene expression and primary biliary cirrhosis]. Zhonghua yi xue za zhi 87: 128–130. [PubMed] [Google Scholar]
- 25.Migita K, Ilyassova B, Kovzel EF, Nersesov A, Abiru S, Maeda Y, Komori A, Ito M, Yano K, Yatsuhashi H, Shimoda S, Ishibashi H, and Nakamura M. 2010. Serum BAFF and APRIL levels in patients with PBC. Clinical immunology 134: 217–225. [DOI] [PubMed] [Google Scholar]
- 26.Nilsson AM, Tufvesson E, Hesselstrand R, Olsson P, Wollmer P, and Mandl T. 2018. Increased B-cell activating factor, interleukin-6, and interleukin-8 in induced sputum from primary Sjogren’s syndrome patients. Scandinavian journal of rheumatology: 1–8. [DOI] [PubMed] [Google Scholar]
- 27.Wei F, Chang Y, and Wei W. 2015. The role of BAFF in the progression of rheumatoid arthritis. Cytokine 76: 537–544. [DOI] [PubMed] [Google Scholar]
- 28.Migita K, Abiru S, Maeda Y, Nakamura M, Komori A, Ito M, Fujiwara S, Yano K, Yatsuhashi H, Eguchi K, and Ishibashi H. 2007. Elevated serum BAFF levels in patients with autoimmune hepatitis. Human immunology 68: 586–591. [DOI] [PubMed] [Google Scholar]
- 29.Moisini I, and Davidson A. 2009. BAFF: a local and systemic target in autoimmune diseases. Clinical and experimental immunology 158: 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Li J, Zhang YM, Zhang XM, and Tao J. 2015. Effect of TACI signaling on humoral immunity and autoimmune diseases. Journal of immunology research 2015: 247426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Treml LS, Carlesso G, Hoek KL, Stadanlick JE, Kambayashi T, Bram RJ, Cancro MP, and Khan WN. 2007. TLR stimulation modifies BLyS receptor expression in follicular and marginal zone B cells. Journal of immunology 178: 7531–7539. [DOI] [PubMed] [Google Scholar]
- 32.Sindhava VJ, Oropallo MA, Moody K, Naradikian M, Higdon LE, Zhou L, Myles A, Green N, Nundel K, Stohl W, Schmidt AM, Cao W, Dorta-Estremera S, Kambayashi T, Marshak-Rothstein A, and Cancro MP. 2017. A TLR9-dependent checkpoint governs B cell responses to DNA-containing antigens. The Journal of clinical investigation 127: 1651–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, and Mackay F. 2007. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. The Journal of experimental medicine 204: 1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobs HM, Thouvenel CD, Leach S, Arkatkar T, Metzler G, Scharping NE, Kolhatkar NS, Rawlings DJ, and Jackson SW. 2016. Cutting Edge: BAFF Promotes Autoantibody Production via TACI-Dependent Activation of Transitional B Cells. Journal of immunology 196: 3525–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fairfax KA, Tsantikos E, Figgett WA, Vincent FB, Quah PS, LePage M, Hibbs ML, and Mackay F. 2015. BAFF-driven autoimmunity requires CD19 expression. Journal of autoimmunity 62: 1–10. [DOI] [PubMed] [Google Scholar]
- 36.Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, Denk H, Desmet V, Korb G, MacSween RN, and et al. 1995. Histological grading and staging of chronic hepatitis. J Hepatol 22: 696–699. [DOI] [PubMed] [Google Scholar]
- 37.Thapa M, Chinnadurai R, Velazquez VM, Tedesco D, Elrod E, Han JH, Sharma P, Ibegbu C, Gewirtz A, Anania F, Pulendran B, Suthar MS, and Grakoui A. 2015. Liver fibrosis occurs through dysregulation of MyD88-dependent innate B-cell activity. Hepatology 61: 2067–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tedesco D, Thapa M, Gumber S, Elrod EJ, Rahman K, Ibegbu CC, Magliocca JF, Adams AB, Anania F, and Grakoui A. 2017. CD4(+) Foxp3(+) T cells promote aberrant immunoglobulin G production and maintain CD8(+) T-cell suppression during chronic liver disease. Hepatology 65: 661–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu YR, O’Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER, Que L, and Gunn MD. 2016. A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PloS one 11: e0150606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, Wortis HH, Kearney JF, Ucci AA, and Imanishi-Kari T. 2006. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity 25: 429–440. [DOI] [PubMed] [Google Scholar]
- 41.Boisvert M, Zhang W, Elrod EJ, Bernard NF, Villeneuve JP, Bruneau J, Marcotrigiano J, Shoukry NH, and Grakoui A. 2016. Novel E2 Glycoprotein Tetramer Detects Hepatitis C Virus-Specific Memory B Cells. Journal of immunology 197: 4848–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simpson EH 1949. Measurement of diversity. Nature 163:688. [Google Scholar]
- 43.Daley T, and Smith AD. 2013. Predicting the molecular complexity of sequencing libraries. Nature methods 10: 325–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morisita M 1959. Measuring of the dispersion and analysis of distribution patterns. Memories of the Faculty of Science, Kyushu University. Series E: Biology, 2, 215–235. [Google Scholar]
- 45.Hampe CS 2012. B Cell in Autoimmune Diseases. Scientifica 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Damoiseaux J, von Muhlen CA, Garcia-De La Torre I, Carballo OG, de Melo Cruvinel W, Francescantonio PL, Fritzler MJ, Herold M, Mimori T, Satoh M, Andrade LE, Chan EK, and Conrad K. 2016. International consensus on ANA patterns (ICAP): the bumpy road towards a consensus on reporting ANA results. Auto- immunity highlights 7: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ota M, Duong BH, Torkamani A, Doyle CM, Gavin AL, Ota T, and Nemazee D. 2010. Regulation of the B cell receptor repertoire and self-reactivity by BAFF. Journal of immunology 185: 4128–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Charles ED, Green RM, Marukian S, Talal AH, Lake-Bakaar GV, Jacobson IM, Rice CM, and Dustin LB. 2008. Clonal expansion of immunoglobulin M+CD27+ B cells in HCV-associated mixed cryoglobulinemia. Blood 111: 1344–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santer DM, Ma MM, Hockman D, Landi A, Tyrrell DL, and Houghton M. 2013. Enhanced activation of memory, but not naive, B cells in chronic hepatitis C virus-infected patients with cryoglobulinemia and advanced liver fibrosis. PloS one 8: e68308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arcaini L, Merli M, Volpetti S, Rattotti S, Gotti M, and Zaja F. 2012. Indolent B-cell lymphomas associated with HCV infection: clinical and virological features and role of antiviral therapy. Clinical & developmental immunology 2012: 638185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McPherson S, Hardy T, Henderson E, Burt AD, Day CP, and Anstee QM. 2015. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol 62: 1148–1155. [DOI] [PubMed] [Google Scholar]
- 52.Kobata T, Jacquot S, Kozlowski S, Agematsu K, Schlossman SF, and Morimoto C. 1995. CD27-CD70 interactions regulate B-cell activation by T cells. Proceedings of the National Academy of Sciences of the United States of America 92: 11249–11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doi H, Iyer TK, Carpenter E, Li H, Chang KM, Vonderheide RH, and Kaplan DE. 2012. Dysfunctional B-cell activation in cirrhosis resulting from hepatitis C infection associated with disappearance of CD27-positive B-cell population. Hepatology 55: 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Storb U 1996. The molecular basis of somatic hypermutation of immunoglobulin genes. Current opinion in immunology 8: 206–214. [DOI] [PubMed] [Google Scholar]
- 55.Kleinstein SH, Louzoun Y, and Shlomchik MJ. 2003. Estimating hypermutation rates from clonal tree data. Journal of immunology 171: 4639–4649. [DOI] [PubMed] [Google Scholar]
- 56.Harms MH, van Buuren HR, and van der Meer AJ. 2018. Improving prognosis in primary biliary cholangitis - Therapeutic options and strategy. Best Pract Res Clin Gastroenterol 34-35: 85–94. [DOI] [PubMed] [Google Scholar]
- 57.Novobrantseva TI, Majeau GR, Amatucci A, Kogan S, Brenner I, Casola S, Shlomchik MJ, Koteliansky V, Hochman PS, and Ibraghimov A. 2005. Attenuated liver fibrosis in the absence of B cells. The Journal of clinical investigation 115: 3072–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Faggioli F, Palagano E, Di Tommaso L, Donadon M, Marrella V, Recordati C, Mantero S, Villa A, Vezzoni P, and Cassani B. 2018. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology 67: 1970–1985. [DOI] [PubMed] [Google Scholar]
- 59.Myers RP, Swain MG, Lee SS, Shaheen AA, and Burak KW. 2013. B-cell depletion with rituximab in patients with primary biliary cirrhosis refractory to ursodeoxycholic acid. Am J Gastroenterol 108: 933–941. [DOI] [PubMed] [Google Scholar]
- 60.Tsuda M, Moritoki Y, Lian ZX, Zhang W, Yoshida K, Wakabayashi K, Yang GX, Nakatani T, Vierling J, Lindor K, Gershwin ME, and Bowlus CL. 2012. Biochemical and immunologic effects of rituximab in patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Hepatology 55: 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moritoki Y, Lian ZX, Lindor K, Tuscano J, Tsuneyama K, Zhang W, Ueno Y, Dunn R, Kehry M, Coppel RL, Mackay IR, and Gershwin ME. 2009. B-cell depletion with anti-CD20 ameliorates autoimmune cholangitis but exacerbates colitis in transforming growth factor-beta receptor II dominant negative mice. Hepatology 50: 1893–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moritoki Y, Tsuneyama K, Nakamura Y, Kikuchi K, Shiota A, Ohsugi Y, Lian ZX, Zhang W, Yang GX, Ueki S, Takeda M, Omokawa A, Saga T, Saga A, Watanabe D, Miura M, Ueno Y, Leung PSC, Tanaka A, Gershwin ME, and Hirokawa M. 2018. Anti-drug Antibodies Against a Novel Humanized Anti-CD20 Antibody Impair Its Therapeutic Effect on Primary Biliary Cholangitis in Human CD20- and FcgammaR-Expressing Mice. Frontiers in immunology 9: 2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.