

Abstract
Purpose of Review
Prostate cancer (PCa) demonstrates characteristic changes in metabolism and bioenergetics in the transition from benign to malignant tissue. It is feasible that some of these changes may be targetable for therapeutic purposes. This review will highlight some of the current metabolically targeted therapies being investigated for the treatment of prostate cancer.
Recent Findings
The transition from benign to malignant prostate cells is characterized by decreased intracellular zinc concentration and subsequent release of inhibition of the tricarboxylic acid cycle enzyme m-aconitase which leads to the decrease in citrate concentration within the cancer tissue. Instead of the largely glycolytic phenotype exhibited by most cancers, PCa relies on glutamine and lipids for survival and proliferation. Early studies are beginning to demonstrate that targeting some of the up-regulated pathways with inhibitors of key enzymes such as glutaminase, fatty acid synthase, HMG-CoA reductase, hexokinase, zinc transport, or complex I in the mitochondria may have significant metabolic effects and therapeutic potential.
Summary
The unique metabolic profile of PCa allows for many potential avenues of treatment. Future studies will continue to test if the metabolic characterization and treatment of PCa could be an important approach to provide personalized treatment for the disease.
MESH Keywords: Prostatic Neoplasms; Metabolomics; Metabolism; Therapies, Investigational; Metabolic Networks and Pathways; Precision Medicine
I. Introduction
Prostate cancer (PCa) is the most common cancer in men and second leading cause of cancer-related mortality in men worldwide [1]. Cancer cells exhibit altered metabolism, compared to their benign counterparts, partly because their continued growth and division requires a constant supply of energy and structural building blocks. To accommodate the altered energy requirements, cellular metabolism is altered through modifications of the activity and expression of key metabolic enzymes. These metabolic changes often occur downstream of various oncogenes or tumor suppressor genes such as c-Myc, Hif1α, PTEN, and RUNX2 [2–6]. This metabolic switch is not only an adaptive response of cancer cells, but can be a driver of tumorigenicity[3]. A single model cannot account for the sum of metabolic changes in cancer because of the vast heterogeneity that exists within tumors and the complexity of the microenvironments in which they grow. However, as the common metabolic changes involved in tumor progression from indolent to aggressive forms become better understood improved methods of risk-stratification, and ultimately personalized targeted therapy for specific metabolic dysfunctions, may be possible. Here, we will review current advances and future research directions regarding metabolism-focused therapeutic developments in PCa management.
II. Normal Prostate Metabolism
Prostate epithelial cells exhibit specific characteristics that distinguish them from other cells. It is notable that metabolic function in the prostate gland is particularly interesting as even benign prostate tissue demonstrates unique metabolism compared to most other tissues in the body. Perhaps the most significant of these differences is the ability to accumulate and secrete high levels of citrate and zinc into the prostatic fluid which mixes with sperm [7]. Seminal fluid citrate concentrations (40 – 150 mM) are considerably higher than blood plasma levels (0.1 – 0.3 mM) [7]. Citrate is believed to act as a buffering agent, a chelator of calcium and zinc, and a possible scavenger of free radicals [8,9]. In the normal prostate, citrate excretion is maintained by inhibition of m-aconitase, a tricarboxylic acid (TCA) cycle enzyme responsible for the conversion of citrate to isocitrate [10]. Thus the prostatic epithelial cells experience a buildup of mitochondrial citrate which is then transported to the cytoplasm and ultimately excreted into seminal fluid. Costello et al found that the aconitase inhibition is mediated by the unusually high levels of zinc in prostatic cells [10] [figure 1]. This is, in turn, regulated by an abundance of ZIP1 zinc transporters in prostatic epithelial cells [11]. Intriguingly, it has been well established that PCa loses the ability to concentrate zinc and citrate [11]. This results in the hallmark transformation of prostate epithelial cells from benign and citrate-secreting to malignant and citrate-oxidizing cells, which occurs through the TCA cycle.
Figure 1: Therapeutic Targets of Prostate Cancer Metabolism.
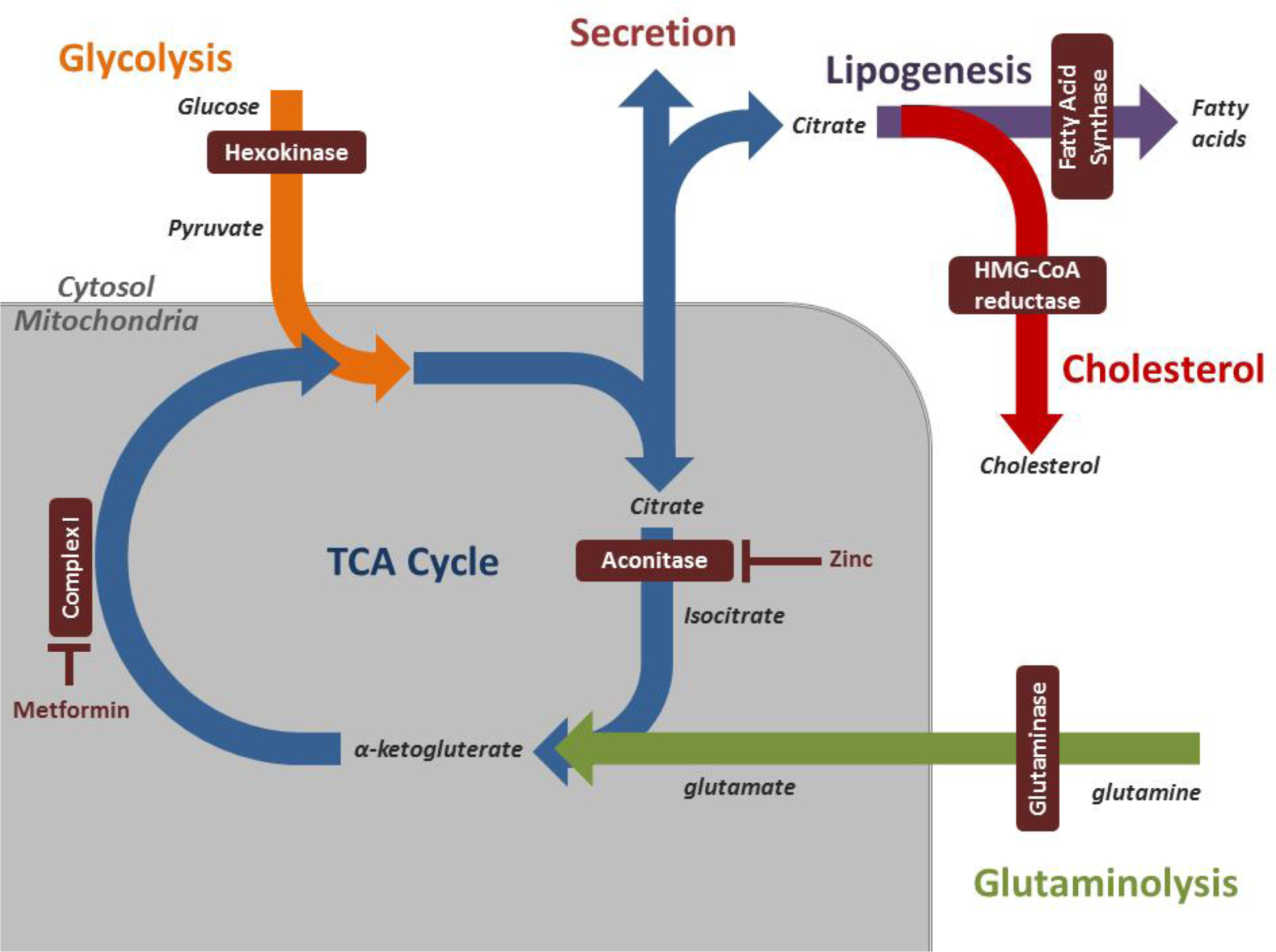
Prostate cancer is characterized by changes in glycolysis, the tricarboxylic acid (TCA) cycle, lipogenesis, cholesterol metabolism, and glutaminolysis which are inter-related as demonstrated here. Benign prostate tissue demonstrates increased zinc accumulation, which leads to inhibition of m-aconitase and citrate secretion. This zinc buildup is lost with tumorigenesis leading to loss of citrate secretion and numerous other metabolic changes within the cell. Targetable enzymes in prostate cancer metabolism are highlighted (maroon boxes).
III. Metabolic Characteristics of Prostate Cancer
Prostate tumorigenesis is a highly complex process. In addition to the aforementioned changes in zinc and citrate metabolism, prostatic carcinoma exhibits aberrations in nearly all major aspects of metabolism. There is a dynamic reliance on glucose, increased glutamine demand, extensive alterations in lipid metabolism and a strong reliance on the pentose phosphate pathway (PPP) [5]. These alterations in metabolomics allows for novel targeted approaches for diagnosis, prognosis, monitoring, and anti-cancer therapy.
Prostate cancer metabolism remains a nascent field. Several challenges remain that impede characterization of changes that occur with prostate tumorigenesis. These include the heterogeneous nature of the disease and a shifting metabolic picture depending on the grade of the disease. The vast majority of solid cancers undergo the Warburg effect, a phenomenon of increased glucose uptake and glycolysis resulting in pyruvate and lactic acid build up despite aerobic conditions [12]. This change in energy metabolism seems almost counterintuitive considering the heightened energy requirements of cancer and the lower ATP-producing efficiency of glycolysis relative to oxidative phosphorylation. One of the leading theories as to why this occurs is that increased glycolysis allows for increased production of intermediates which are required for anaplerosis [13]. Another reason is the more rapid flux of glucose through glycolytic pathways relative to oxidative phosphorylation [14]. However, PCa does not seem to undergo Warburg metabolic changes [5]. While glucose and glutamine metabolism are usually noted as some of the chief energy sources for cellular respiration, PCa is not heavily reliant on glucose. Rather, what seems to occur is an increased reliance on oxidative phosphorylation. This is mediated by the decreased activity of ZIP1 transporters in PCa, with the resultant decrease in zinc concentrations leading to increased activity of m-aconitase and the TCA cycle. Although not fully understood, the metabolic pathways of greatest importance with prostate cancer seem to be more fatty acid metabolism and glutamine metabolism [5]. We will next discuss various metabolically targeted therapies under consideration for treatment of prostate cancer.
IV. Metabolic Therapeutic Targets
There are a variety of modalities for the treatment of PCa. Although optimal treatment is at times difficult to determine, options range from surgical excision and radiation in localized disease to hormone therapy and chemotherapy in advanced disease. The mainstay of advanced PCa is androgen deprivation therapy, usually involving luteinizing hormone-releasing hormone (LHRH) agonists and oral anti-androgens. The majority of cancers respond initially, followed by progression to Castrate Resistant PCa (CRPC), with overexpression of androgen receptors and up regulation of androgen biosynthesis enzymes being among some of the important pathways leading to the CRPC phenotype [15,16]. Newer anti-androgens and androgen biosynthesis inhibitors, such as enzalutamide and abiraterone acetate, have been developed to enhance efficacy in advanced cancers and can improve overall survival. Although chemotherapy can also improve overall survival, the median survival for men with CRPC remains in the 2- to3-year range despite these treatments [16, 17]. Thus, there is a great need for new therapeutic modalities. Because of the significant metabolic changes in PCa, metabolic-targeted therapy and research focused on the metabolic alterations of prostate cancer is warranted. Below we will discuss the role of zinc metabolism, AMP kinase activators, lipogenesis inhibitors, steroid inhibitors, and glutamine pathway inhibitors as promising therapeutic modalities.
Zinc
Zinc is an essential element of great interest in cancer research due to its anti-oxidant and anti-inflammatory properties and its ability to improve cell-mediated immune functions and inhibit NF-kB [18]. As previously described, normal prostate epithelial cells have the highest concentrations of zinc and changes in zinc metabolism are crucial to PCa pathogenesis. There is much experimental support for the anti-neoplastic effects of zinc. Specifically, many studies show an increase in PCa cell apoptosis with zinc administration [19,20]. Ku et al specifically have reported a dose-dependent apoptotic effect in both androgen-dependent and -independent cell lines [21]. Furthermore, their study was one of the first to show specific changes in apoptotic and anti-apoptotic molecules, which inhibited cell proliferation. Zinc administration was associated with increased expression of BAX mRNA and diminished expression of Bcl-2 and survivin mRNAs [21]. Animal trials have allowed development of various methods of zinc administration, with 5-chloro-7-iodo-8-quinolinol (Clioquinol) being one of the newer forms. Clioquinol is a zinc ionophore that allows intracellular zinc accumulation despite the absence of ZIP zinc transporters in malignant prostate cells [22]. Costello et al demonstrated over 70% suppression of tumor growth in mice treated with Clioquinol, with minimal to no side effects [22]. Epidemiological data on the effect of zinc on PCa has been largely inconsistent [23–25]. There have been over 10 such studies on dietary and supplemental zinc intake, mostly assessing PCa prevention [26,27]. In a more recent study, however, Epstein et al demonstrated that Swedish men with high zinc intake exhibited lower disease-specific mortality, especially in men with localized tumors [27].
Multiple studies are underway to assess the efficacy of combination therapy with zinc and various anti-neoplastic drugs for the treatment of prostate cancer. One such drug is Sorafenib, a multi-kinase inhibitor that is FDA approved for the treatment of renal cell and hepatocellular cancers [28,29] and has displayed anti-neoplastic properties in PCa [30–32]. Chen et al reported increased sensitivity of PCa cells to Sorafenib-induced apoptosis following zinc administration [33]. Thus, it may be possible to maintain the therapeutic efficacy of Sorafenib with reduced dosage. This is of specific interest as Sorafeib therapy is associated with a variety of significant side effects. Another drug similarly potentiated by zinc is paclitaxel. Uzzo et al have shown zinc to sensitize PCa cells to paclitaxel-mediated cell death [34].
AMPK Activators
Adenosine monophosphate-activated protein kinase is a central cellular energy regulator that responds to increased AMP/ATP ratio by enhancing energy producing pathways and down-regulating energy consuming pathways such as lipogenesis and mTORC1 pathways [35]. With its extensive involvement in cellular metabolism, AMPK has enormous potential for anti-cancer therapy. In fact, various AMPK activators have been shown to reduce cancer risk [36,37]. They are divided into two groups, 1) indirect activators, including biguanides such as metformin and 2) direct activators, including 5-aminoimidazole-4carboxamide-1-β-ribofuranoside (AICAR) and other more recently developed small molecule AMPK activators [5].
Metformin is a guanidine derivative widely used as first-line therapy for type II diabetes mellitus that decreases hepatic gluconeogenesis and enhances peripheral insulin sensitivity. Bowker et al showed that diabetics treated with metformin exhibited lower incidence of cancer and cancer-related mortality compared to patients treated with insulin and sulfonylureas [38]. Another study showed metformin to be associated with a 44% risk reduction in PCa cases [39]. Biguanide’s anti-cancer mechanism involves inhibition of Complex 1 of the electron transport chain. This results in reduced ATP synthesis and increased AMP/ATP ratio and subsequent activation of AMPK leading to inhibition of mTORC1 activity and decreased translational efficiency in PCa cells [40]. Although its anti-neoplastic mechanism is yet to be fully illuminated, the existing data may stimulate doctor-patient discussion about starting metformin in hesitant diabetic patients. There are approximately 25 clinical trials currently ongoing to utilize metformin as an adjuvant treatment in a range of PCa patients, either as a stand-alone medication or in combination with other treatments such as surgery, radiation, enzalutamide and docetaxel [clinicaltrials.gov].
The nucleoside AICAR was the first agent reported to activate AMPK in intact cells and in vivo [5]. AICAR is taken up by adenosine transporters and converted to ZMP, a 5’-AMP analog. AICAR has been shown to diminish proliferation of PCa in xenograft models; however, a lack of specificity and poor oral bioavailability has led to a search for more specific and potent direct AMPK activators [5,41,42]. Small molecule AMPK activators are newer, subunit-specific drugs that are under active investigation. Examples include A-769662, PT1 and OSU-53 [5]. These newer agents show potent anti-cancer activity in tumor bearing-mice with OSU-53 resulting in 47–49% growth suppression [43]. Unfortunately, the limitations of poor oral availability and non-specificity remain, and therefore efforts to develop more targeted direct AMPK activators continue.
Inhibitors of lipogenesis
In normal tissues, lipogenesis is inhibited by the presence of dietary fatty acids, with the exception of the liver, adipose tissue and lactating breast tissue [5,44]. In cancer, however, there is an over expression of the major lipogenic enzymes, namely ATP citrate lyase (ACLY), acetyl-Coa carboxylase (ACC), and fatty acid synthase (FASN) [45]. Additionally, expression of many of these enzymes is stimulated by the c-Myc oncogene, which also increases expression of TCA cycle genes responsible for citrate synthesis, a precursor for fatty acids and cholesterol [46]. FASN overexpression is an early event in PCa tumorigenesis and is associated with cancer progression and bone metastasis [47–49]. Additionally, in experiments with prostate epithelial cells, ectopic expression of FASN was associated with development of PIN and progression to invasive tumors [50].
Inhibition of FASN results in suppressed cell adhesion, reduced migration, and attenuation of growth and apoptosis [51]. Several inhibitors of FASN have been described and studied to date, including Orlistat, Cerulenin, C75, and C93. Orlistat, initially marketed as an anti-obesity drug, has been found to inhibit FASN by forming a covalent bond with the enzyme [52]. Flavin et al showed Orlistat inhibits proliferation and induces apoptosis in PC3 cells [53]. Unfortunately, pharmacological limitations for this drug are multiple and include poor oral availability, poor metabolic stability, low solubility, low cell permeability, and lack of selectivity [53]. Development of C75, Cerulenin, and C93 for cancer treatment has been limited by side effects such as dramatic anorexia and weight loss in animal trials [5,54].
Recently, newer and more potent FASN inhibitors were identified through medicinal chemistry and high-throughput screening: Astra Zeneca developed a series of bisamide derivatives, Merck a series of 3-aryl-4-hydroxyquinolin-2(1H)-one derivatives, and GlaxoSmithKline produced GSK837149, a FASN inhibitor with nanomolar potency [54]. In addition to FASN, other lipogenic enzymes represent promising targets for prostate cancer therapy. ACLY inhibition by RNA interference (RNAi) or with chemical inhibitors SB-204990 and simvastatin inhibited proliferation and survival of tumor cells displaying aerobic glycolysis, which reduced tumor growth in vivo [55].
Cholesterol metabolism inhibitors
The tumorigenesis, maintenance, and progression of PCa are largely associated with abnormalities in cholesterol metabolism. There is an epidemiological correlation between hypercholesterolemia and aggressive prostate cancer [56]. Additionally, animal models have linked hypercholesterolemia to increased PCa growth, AKT activation, and progression to metastasis [57,58]. Multiple studies have shown statins to reduce the risk of prostate cancer [56]. Statins inhibit HMG-CoA reductase (HMGCR) which catalyzes the conversion of HMG-CoA to mevalonate, the rate limiting step of steroidogenesis. Treatment of androgen-dependent and - independent PCa cell lines with simvastatin significantly reduced cell migration, invasion and proliferation [59]. Furthermore, treatment of PC3 xenografts with the same drug yielded a reduction in tumor growth [59]. Long term studies have shown reduced risk of advanced PCa with statin therapy; however, data on overall risk for PCa is inconclusive [56]. Multiple studies are underway to elaborate this relationship.
Glutamine inhibitors
Glutamine is an abundant nutrient, essential for cell survival. It functions as a major source of energy, a source of nitrogen for amino acids, and in the synthesis of the antioxidant glutathione. In cancer, glutamine is increasingly converted to glutamate and then alpha keto-glutarate which maintains the TCA cycle to satisfy the elevated energy requirements of cancer cells [5]. Glutaminase (GLS) catalyzes the conversion of glutamine to glutamate, exists in multiple forms (GLS-1, GLS-2), and is under the control of the c-Myc oncogene [46]. GLS-1 is found in the kidney and also present in prostate cancer cells [60]. Pan et al showed that GLS-1 promotes cell proliferation and survival; furthermore, t elevated GLS correlates with higher grade and more advanced tumors [61]. Reduced expression of GLS-1 by siRNA or bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES), a direct inhibitor, resulted in reduced glucose uptake, diminished ATP levels, and a resultant inhibition of cell proliferation and progression [61]. These findings indicate GLS-1 may be a viable therapeutic target in PCa treatment.
Inhibitors of other metabolic enzymes
Several metabolic enzymes such as hexokinase (HK), lactate dehydrogenase A(LDHA) and pyruvate dehydrogenase kinase 1(PDK1) are direct targets of oncogenic transcription factors, such as c-Myc and hypoxia-inducible factor-1a (HIF1a) [46]. These enzymes represent opportunities for targeted therapy. Most notably, HK has an important role in both glycolysis and apoptosis, and its inhibitors, such as 2-deoxyglucose (2-DG), 3-bromopyruvate (3-BrPA), and Lonidamine (LND), are in pre-clinical and early phase clinical trials [62].
V. Conclusion
The unique metabolic characteristics of prostate cancer provide opportunities for advanced, more targeted therapy. Perhaps the most distinct characteristic of prostate epithelial cells is their ability to concentrate zinc and its involvement in tumorigenesis. Although data on the therapeutic efficacy of zinc supplementation is inconsistent, new work shows its potential benefit in terms of disease-specific mortality. A variety of lipid metabolism aberrations are present in PCa, specifically the upregulation of FASN and steroidogenesis, which suggests promising avenues for treatment. The most exciting and clearly beneficial AMPK activator is metformin. Although further clarification of its effects on PCa is necessary, there is a clear reduction in PCa incidence in metformin-treated diabetics. Finally glutaminase-1 inhibitors show strong potential as therapeutic metabolic targets as they inhibit prostate cancer progression. Continued research into how metabolic dysfunction is associated with prostate cancer tumorigenesis and progression may lay the foundation for future refined research on potential metabolic targets of interest.
Key Points.
Unlike most solid tumors, prostate cancer delays the onset of the Warburg effect, resulting in a greater reliance on glutamine and lipids for energy.
Prostate epithelial cells are unique in terms of zinc metabolism and the relative suppression of the TCA cycle.
There is much experimental data supporting the antineoplastic effects of zinc on prostate cancer and, more specifically, the safe use of zinc ionophore as treatment.
Metformin reduces all cancer-related mortality and reduces prostate cancer incidence.
Statins reduce risk of aggressive prostate cancer and trials are underway to further clarify their effect on the full range of prostate cancer disease states.
Acknowledgements:
Part of this work was supported by a Merit Review Award, Dept of Veterans Affairs (A.H., A.P.).
Footnotes
Conflict of Interests: There are no conflicts of interest.
REFERENCES
- 1.Skolarus TA, Wolf AMD, Erb NL et al. American Cancer Society prostate cancer survivorship care guidelines. CA. Cancer J. Clin 2014; 64:225–249. [DOI] [PubMed] [Google Scholar]
- 2.Koh CM, Bieberich CJ, Dang CV et al. MYC and Prostate Cancer. Genes Cancer 2010; 1:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dang CV. Links between metabolism and cancer. Genes Dev. 2012; 26:877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akech J, Wixted JJ, Bedard K, et al. Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene 2010; 29:811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 2013; 1831:1518–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choe M, Brusgard JL, Chumsri S et al. The RUNX2 Transcription Factor Negatively Regulates SIRT6 Expression to Alter Glucose Metabolism in Breast Cancer Cells. J. Cell. Biochem 2015; 116:2210–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costello LC, Liu Y, Franklin RB, Kennedy MC. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J. Biol. Chem 1997; 272:28875–81. [DOI] [PubMed] [Google Scholar]
- 8.Zinc Arver S. and zinc ligands in human seminal plasma. III. The principal low molecular weight zinc ligand in prostatic secretion and seminal plasma. Acta Physiol. Scand 1982; 116:67–73. [DOI] [PubMed] [Google Scholar]
- 9.Ford WC, Harrison A. The role of citrate in determining the activity of calcium ions in human semen. Int J Androl 1984; 7:198–202. [DOI] [PubMed] [Google Scholar]
- 10.Costello LC, Franklin RB. Novel role of zinc in the regulation of prostate citrate metabolism and its implications in prostate cancer. Prostate 1998; 35:285–296. [DOI] [PubMed] [Google Scholar]
- 11.Costello LC, Franklin RB, Zou Jet al. Human prostate cancer ZIP1/zinc/citrate genetic/metabolic relationship in the TRAMP prostate cancer animal model. Cancer Biol. Ther 2011; 12:1078–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *12.Pacini N, Borziani F. Cancer stem cell theory and the warburg effect, two sides of the same Coin? Int. J. Mol. Sci 2014; 15:8893–8930. [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive review of cancer stem cell theory and warburg effect, describing them as one homogenous pattern. The Warburg effect is shown to be an aberrant expression of the metabolic layout which is characterestic of the undifferentiated cell.
- 13.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat. Rev. Drug Discov 2011; 10:671–684. [DOI] [PubMed] [Google Scholar]
- 14.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mostaghel EA, Page ST, Lin DW et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: Therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007; 67:5033–5041. [DOI] [PubMed] [Google Scholar]
- 16.Dillard PR, Lin MF, Khan SA. Androgen-independent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Mol. Cell. Endocrinol 2008; 295:115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sartor AO. Progression of metastatic castrate-resistant prostate cancer: impact of therapeutic intervention in the post-docetaxel space. J. Hematol. Oncol 2011; 4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *18.Prasad AS. Impact of the discovery of human zinc deficiency on health. J. Trace Elem. Med. Biol 2014; 28:357–363. [DOI] [PubMed] [Google Scholar]; Review of the vast functions of zinc in humans and developments in zinc use for therapy.
- 19.Cortesi M, Fridman E, Volkov A et al. : Clinical assessment of the cancer diagnostic value of prostatic zinc: A comprehensive needle-biopsy study. Prostate 2008; 68:994–1006. [DOI] [PubMed] [Google Scholar]
- 20.Costello LC, Franklin RB, Feng P et al. Zinc and prostate cancer: A critical scientific, medical, and public interest issue (United States). Cancer Causes Control 2005; 16:901–915. [DOI] [PubMed] [Google Scholar]
- 21.Ku JH, Seo SY, Kwak C, Kim HH. The role of survivin and Bcl-2 in zinc-induced apoptosis in prostate cancer cells. Urol. Oncol. Semin. Orig. Investig 2012; 30:562–568. [DOI] [PubMed] [Google Scholar]
- *22.Costello LC, Franklin RB, Zou J, Naslund MJ. Evidence that Human Prostate Cancer is a ZIP1-Deficient Malignancy that could be Effectively Treated with a Zinc Ionophore (Clioquinol) Approach. Chemotherapy 2015; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]; Review of prior studies on zinc and prostate cancer prevention and treatment. Addresses feasibility and safety of clinical trials with zinc therapy.
- 23.Gallus S, Foschi R, Negri E et al. Dietary Zinc and Prostate Cancer Risk: A Case-Control Study from Italy. Eur. Urol 2007; 52:1052–1057. [DOI] [PubMed] [Google Scholar]
- 24.Key TJ, Silcocks PB, Davey GK et al. A case-control study of diet and prostate cancer. Br. J. Cancer 1997; 76:678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez A, Peters U, Lampe JW, White E. Zinc intake from supplements and diet and prostate cancer. Nutr. Cancer 2009, 61:206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kristal AR, Arnold KB, Neuhouser ML et al. Diet, supplement use, and prostate cancer risk: Results from the prostate cancer prevention trial. Am. J. Epidemiol 2010; 172:566–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epstein MM, Kasperzyk JL, Andren O et al. Dietary zinc and prostate cancer survival in a Swedish cohort. Am. J. Clin. Nutr 2011; 93:586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kane RC, Farrell AT, Saber H et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res 2006; 12:7271–8. [DOI] [PubMed] [Google Scholar]
- 29.Lang L FDA approves sorafenib for patients with inoperable liver cancer. [Internet]. Gastroenterology 2008; 134:379. [DOI] [PubMed] [Google Scholar]
- 30.Ullén A, Farnebo M, Thyrell L et al. Sorafenib induces apoptosis and autophagy in prostate cancer cells in vitro. Int. J. Oncol 2010; 37:15–20. [DOI] [PubMed] [Google Scholar]
- 31.Oh SJ, Erb HHH, Hobisch A et al. Sorafenib decreases proliferation and induces apoptosis of prostate cancer cells by inhibition of the androgen receptor and Akt signaling pathways. Endocr. Relat. Cancer 2012; 19:305–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang R, Chen X-Q, Huang Y et al. The multikinase inhibitor sorafenib induces caspase-dependent apoptosis in PC-3 prostate cancer cells. Asian J. Androl 2010; 12:527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X, Che X, Wang J et al. Zinc sensitizes prostate cancer cells to sorafenib and regulates the expression of Livin. Acta Biochim. Biophys. Sin. (Shanghai) 2013; 45:353–8. [DOI] [PubMed] [Google Scholar]
- 34.Uzzo RG, Leavis P, Hatch W et al. Zinc inhibits nuclear factor-kappa B activation and sensitizes prostate cancer cells to cytotoxic agents. Clin. Cancer Res 2002; 8:3579–83. [PubMed] [Google Scholar]
- 35.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. (Oxf) 2009; 196:65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim. Biophys. Acta 2010; 1804:581–91. [DOI] [PubMed] [Google Scholar]
- 37.Evans JMM, Donnelly LA et al. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005; 330:1304–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin: Response to Farooki and Schneider. Diabetes Care 2006; 29:1990–1. [DOI] [PubMed] [Google Scholar]
- 39.Wright JL, Stanford JL. Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer Causes Control 2009; 20:1617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben Sahra I, Laurent K, Loubat A et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008; 27:3576–3586. [DOI] [PubMed] [Google Scholar]
- 41.Xiang X, Saha AK, Wen R et al. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem. Biophys. Res. Commun 2004; 321:161–7. [DOI] [PubMed] [Google Scholar]
- 42.Xiao B, Heath R, Saiu P et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007; 449:496–500. [DOI] [PubMed] [Google Scholar]
- 43.Lee K-H, Hsu E-C, Guh J-H et al. Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer by a novel adenosine monophosphate-activated protein kinase (AMPK) activator. J. Biol. Chem 2011; 286:39247–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jayakumar A, Tai MH, Huang WY et al. Human fatty acid synthase: properties and molecular cloning. Proc. Natl. Acad. Sci. U. S. A 1995; 92:8695–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953; 13:27–9. [PubMed] [Google Scholar]
- *46.Stine ZE, Walton ZE, Altman BJ et al. MYC, Metabolism, and Cancer. Cancer Discov. 2015; 5:1024–39. [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive review of metabolic profile of cancer with emphasis on oncogene mutations and downstream effects.
- 47.Rossi S, Graner E, Febbo P et al. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol. Cancer Res 2003; 1:707–15. [PubMed] [Google Scholar]
- 48.Swinnen JV, Roskams T, Joniau S et al. Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer. Int. J. Cancer 2002; 98:19–22. [DOI] [PubMed] [Google Scholar]
- 49.Swinnen JV, Vanderhoydonc F, Elgamal et al. Selective activation of the fatty acid synthesis pathway in human prostate cancer. Int. J. Cancer 2000; 88:176–9. [DOI] [PubMed] [Google Scholar]
- 50.Loda M, Migita T, Ruiz S et al. : Fatty acid synthase: A metabolic enzyme and candidate oncogene in prostate cancer. J. Natl. Cancer Inst 2009; 101:519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu X, Daniels G, Lee P, Monaco ME. Lipid metabolism in prostate cancer. [Internet]. Am. J. Clin. Exp. Urol 2014; 2:111–20. [PMC free article] [PubMed] [Google Scholar]
- 52.Pemble CW, Johnson LC, Kridel SJ, Lowther WT. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. [Internet]. Nat. Struct. Mol. Biol 2007, 14;704–9. [DOI] [PubMed] [Google Scholar]
- 53.Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010; 6:551–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flavin R, Zadra G, Loda M. Metabolic alterations and targeted therapies in prostate cancer. J. Pathol 2011; 223:283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hatzivassiliou G, Zhao F, Bauer DE et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005; 8:311–321. [DOI] [PubMed] [Google Scholar]
- 56.Pelton K, Freeman MR, Solomon KR. Cholesterol and prostate cancer. Curr. Opin. Pharmacol 2012; 12:751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhuang L, Kim J, Adam RM et al. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J. Clin. Invest 2005; 115:959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Llaverias G, Danilo C, Wang Y et al. A Western-type diet accelerates tumor progression in an autochthonous mouse model of prostate cancer. Am. J. Pathol 2010; 177:3180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kochuparambil ST, Al-Husein B, Goc A et al. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J. Pharmacol. Exp. Ther 2011; 336:496–505. [DOI] [PubMed] [Google Scholar]
- 60.Curthoys NP, Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu. Rev. Nutr 1995; 15:133–59. [DOI] [PubMed] [Google Scholar]
- *61.Pan T, Gao L, Wu G et al. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem. Biophys. Res. Commun 2015; 456:452–8. [DOI] [PubMed] [Google Scholar]; Review of glutamine utilization in prostate cancer focusing interactions between glutaminase, TCA cycle, and glucose uptake.
- 62.Sadeghi RN, Karami-Tehrani F, Salami S. Targeting prostate cancer cell metabolism: impact of hexokinase and CPT-1 enzymes. Tumor Biol. 2015; 36:2893–2905. [DOI] [PubMed] [Google Scholar]