

Human cytomegalovirus (HCMV), a large double-stranded DNA (dsDNA) virus, encodes more than 200 viral proteins. HCMV infection causes irreversible abnormalities of the central nervous system in newborns and severe syndromes in organ transplantation patients or AIDS patients. It has been demonstrated that HCMV has evolved multiple immune evasion strategies to establish latent infection. Previous studies pay more attention to the mechanism by which HCMV evades immune response in the early phase of infection. In this study, we identified UL94 as a negative regulator of the innate immune response, which functions in the late phase of HCMV infection.
KEYWORDS: HCMV, MITA/STING, UL94, antiviral innate immunity, immune evasion
ABSTRACT
Cyclic GMP-AMP synthase (cGAS) senses double-stranded DNA and synthesizes the second messenger cyclic GMP-AMP (cGAMP), which binds to mediator of IRF3 activation (MITA) and initiates MITA-mediated signaling, leading to induction of type I interferons (IFNs) and other antiviral effectors. Human cytomegalovirus (HCMV), a widespread and opportunistic pathogen, antagonizes the host antiviral immune response to establish latent infection. Here, we identified HCMV tegument protein UL94 as an inhibitor of the cGAS-MITA-mediated antiviral response. Ectopic expression of UL94 impaired cytosolic double-stranded DNA (dsDNA)- and DNA virus-triggered induction of type I IFNs and enhanced viral replication. Conversely, UL94 deficiency potentiated HCMV-induced transcription of type I IFNs and downstream antiviral effectors and impaired viral replication. UL94 interacted with MITA, disrupted the dimerization and translocation of MITA, and impaired the recruitment of TBK1 to the MITA signalsome. These results suggest that UL94 plays an important role in the immune evasion of HCMV.
IMPORTANCE Human cytomegalovirus (HCMV), a large double-stranded DNA (dsDNA) virus, encodes more than 200 viral proteins. HCMV infection causes irreversible abnormalities of the central nervous system in newborns and severe syndromes in organ transplantation patients or AIDS patients. It has been demonstrated that HCMV has evolved multiple immune evasion strategies to establish latent infection. Previous studies pay more attention to the mechanism by which HCMV evades immune response in the early phase of infection. In this study, we identified UL94 as a negative regulator of the innate immune response, which functions in the late phase of HCMV infection.
INTRODUCTION
The innate immune system is the first line of host defense against viral infection. Pathogen-derived components known as pathogen-associated molecular patterns (PAMPs) are recognized by host cellular pattern recognition receptors (PRRs), which initiate signaling cascades that lead to the induction of type I interferons (IFNs) and proinflammatory cytokines. Subsequently, these cytokines trigger a wide range of antiviral responses, including the suppression of viral replication, elimination of virus-infected cells, and facilitated activation of adaptive immune responses (1–3).
Viral nucleic acids, a type of potent PAMP, can be recognized by PRRs which sense cytoplasmic DNA and RNA (4, 5). Cyclic GMP-AMP synthase (cGAS), a member of nucleotidyltransferase family, has been demonstrated as a universal DNA sensor that detects cytosolic viral DNA in various cells and in mice (6, 7). Upon sensing cytosolic DNA, cGAS utilizes ATP and GTP as the substrates to synthesize the second messenger cyclic GMP-AMP (cGAMP), which then binds to the adaptor protein mediator of IRF3 activation (MITA; also called STING, ERIS, MPYS, and TMEM173) (8–11). MITA undergoes dimerization and then traffics from the endoplasmic reticulum (ER) via the Golgi apparatus to perinuclear microsomes. In this process, TBK1 and IRF3 are recruited to the MITA signalsome and activated. The activated IRF3 subsequently translocates into the nucleus and initiates the induction of type I IFNs (12, 13).
Human cytomegalovirus (HCMV), a typical double-stranded DNA (dsDNA) virus, is a widespread and opportunistic pathogen that belongs to the beta herpesvirus family and encodes more than 200 viral proteins (14). HCMV infection rarely causes severe diseases in immunocompetent individuals but leads to irreversible abnormalities of the central nervous system in newborns and severe syndromes in immunocompromised adults, such as organ transplantation patients or AIDS patients (15–17). During its evolution, HCMV obtained multiple immune evasion strategies to establish latent infection (18). Previous research has demonstrated that HCMV UL31 and UL83 impair the enzymatic activity of cGAS and inhibit the production of cGAMP (19, 20). It has also been reported that the tegument protein UL82 impedes the trafficking of MITA and assembly of MITA-TBK1-IRF3 complexes (21). In addition, the nucleus-localized UL44 suppresses IRF3- and NF-κB-mediated transcription of type I IFNs (22). However, these studies all focused on the early phase of infection, the mechanism by which HCMV evades immune response in the late phase of infection has not yet been elucidated.
HCMV UL94 is expressed with true late kinetics and acts as a structural component to facilitate the secondary envelopment of virions (23–25). In this study, we identified UL94 as an inhibitor of the MITA-mediated innate immune response. Overexpression of UL94 inhibited the viral DNA-triggered induction of type I IFNs and downstream antiviral genes, whereas UL94 deficiency had opposite effects. UL94 associated with MITA disrupted the dimerization of MITA and recruitment of TBK1 to the MITA signalsome. Therefore, UL94 impaired MITA-mediated antiviral signaling and contributed to HCMV replication. These findings expand our knowledge of HCMV immune evasion at the late phase of infection.
RESULTS
UL94 inhibits cGAS-MITA-mediated signaling.
It has been demonstrated that the cGAS-MITA axis plays a pivotal role in the antiviral response to HCMV infection (26). To identify HCMV proteins that have inhibitory functions on cGAS-MITA-mediated signaling, HEK293T cells were transfected with HCMV expression clones encoding individual proteins and expression plasmids of cGAS and MITA along with the IFN-β promoter, and reporter assays were performed to screen candidate proteins which inhibited cGAS-MITA-mediated activation of the IFN-β promoter. In this screen, we identified UL94 as a potential inhibitor. We found that overexpression of UL94 inhibited cGAS-MITA-mediated activation of the IFN-β promoter and NF-κB in a dose-dependent manner in HEK293T cells (Fig. 1A). In similar experiments, HCMV tegument protein UL82, which has been reported as an inhibitor of cGAS-MITA-mediated antiviral immune responses (21), also suppressed cGAS-MITA-triggered activation of IFN-β promoter, whereas the HCMV integral membrane protein UL50 had no marked effects on cGAS-MITA-mediated signaling (Fig. 1A). Quantitative PCR (qPCR) analysis indicated that overexpression of UL94 inhibited cGAS-MITA-triggered transcription of IFNB1, CXCL10, and RANTES in HEK293T cells (Fig. 1B). In contrast, UL94 showed little effects on IFN-γ-induced activation of IRF1 reporter or tumor necrosis factor alpha (TNF-α)-induced activation of NF-κB (Fig. 1C). Overexpression of UL94 also had no marked effects on IFN-β-triggered phosphorylation of STAT1 and STAT2 in HEK293T cells (Fig. 1D). These results suggest that UL94 specifically downregulates cGAS-MITA-mediated induction of type I IFNs and downstream antiviral genes.
FIG 1.

Identification of HCMV UL94 as an inhibitor of cGAS-MITA-mediated signaling. (A) UL94 inhibits cGAS-MITA-mediated activation of the IFN-β promoter and NF-κB in a dose-dependent manner. HEK293T cells (1 × 105) were transfected with the luciferase reporter of IFN-β promoter (0.05 μg) or NF-κB (0.005 μg) plus expression plasmids for cGAS (0.01 μg), MITA (0.02 μg), or an empty vector (0.03 μg; Vec) as well as the indicated amounts of UL94, UL82, and UL50 plasmids for 24 h before luciferase assays. The levels of transfected proteins were examined by immunoblots. (B) UL94 inhibits cGAS-MITA-induced transcription of antiviral genes in a dose-dependent manner. HEK293T cells (4 × 105) were transfected with cGAS (0.05 μg) and MITA (0.1 μg) or an empty vector plus the indicated amounts of UL94 plasmid for 24 h before qPCR analysis. The levels of transfected proteins were examined by immunoblots. (C) Effects of UL94 on IFN-γ- or TNF-α-induced signaling. HEK293T cells (1 × 105) were transfected with the IRF1 (0.05 μg) or NF-κB (0.005 μg) luciferase reporter and the indicated amounts of UL94 plasmid for 24 h. The cells were then left untreated or treated with IFN-γ (100 ng/ml) or TNF-α (10 ng/ml) for 12 h before luciferase assays. The levels of transfected proteins were examined by immunoblots. (D) Effects of UL94 on IFN-β-induced phosphorylation of downstream components. HEK293T cells (4 × 105) were transfected with UL94 plasmid (0.5 μg) for 24 h. The cells were then treated with IFN-β (100 ng/ml) for the indicated times before immunoblot analysis were performed with the indicated antibodies. Graphs show means ± standard deviations (SDs), n = 3. *, P < 0.05; **, P < 0.01; ns, not significant (unpaired t test).
UL94 antagonizes the viral DNA-triggered antiviral immune response.
It was previously reported that human primary foreskin fibroblasts (HFFs) are able to activate cGAS-MITA signaling and express type I IFNs in response to HCMV infection (27). We established HFF cells stably expressing UL94 (HFF-UL94) by lentiviral transduction. Following HCMV infection, transcription of IFNB1, IL6, and CXCL10 genes in HFF-UL94 cells was significantly impaired in comparison with that in the control cells (Fig. 2A). In addition, transcription of the antiviral genes triggered by other DNA viruses such as herpes simplex virus 1 (HSV-1) and vaccinia virus (VACV) as well as by various transfected dsDNAs that represent the genome fragments of HSV-1 and VACV, was also markedly impaired in HFF-UL94 cells (Fig. 2A and B). In contrast, UL94 showed no effects on the RNA virus Sendai virus (SeV)-triggered induction of antiviral genes (Fig. 2C). Additionally, secretion of IFN-β was also remarkably impaired in HFF-UL94 cells following HCMV and HSV-1 but not SeV infection (Fig. 2D). Since the phosphorylations of TBK1 and IRF3 are key events in the activation of cGAS-MITA-mediated signaling, we further investigated the effects of UL94 on these processes. As shown in Fig. 2E and F, UL94 significantly inhibited HCMV- and transfected HSV120-triggered but not SeV-triggered phosphorylations of TBK1 and IRF3. These results suggest that UL94 acts as an inhibitor for DNA virus-triggered induction of type I IFNs and downstream antiviral genes.
FIG 2.

UL94 negatively regulates viral DNA-triggered signaling. (A) UL94 inhibits HCMV, HSV-1, or VACV-triggered transcription of antiviral genes in HFFs. HFFs stably expressing UL94 (4 × 105) were left uninfected or infected with HCMV (multiplicity of infection [MOI] = 1), HSV-1 (MOI = 1), or VACV (MOI = 1) for 6 h before qPCR analysis. (B) Effects of UL94 on transcription of antiviral genes induced by transfected dsDNA in HFFs. HFFs stably expressing UL94 (4 × 105) were transfected with the synthesized dsDNA representing a 60-bp or 120-bp fragment of HSV-1 genome (HSV60 or HSV120, respectively) or a 70-bp fragment of VACV genome (VACV70) (1 μg/ml) for 6 h before qPCR analysis. (C) Effects of UL94 on SeV-induced transcription of antiviral genes in HFFs. HFFs stably expressing UL94 (4 × 105) were left uninfected or infected with SeV (MOI = 1) for the indicated times before qPCR analysis. (D) Effects of UL94 on secreted IFN-β in HFFs. HFFs stably expressing UL94 (4 × 105) were left uninfected or infected with HCMV (MOI = 1), HSV-1 (MOI = 1), or SeV (MOI = 1) for 12 h before measurement of IFN-β by ELISA. (E) Effects of UL94 on the phosphorylation of TBK1 and IRF3 induced by HCMV and transfected HSV120 in HFFs. HFFs stably expressing UL94 (4 × 105) were left uninfected or infected with HCMV (MOI = 1), or transfected with HSV120 (1 μg/ml) for the indicated times before immunoblot analyses were performed with the indicated antibodies. (F) Effects of UL94 on SeV-induced phosphorylation of TBK1 and IRF3 in HFFs. HFFs stably expressing UL94 (4 × 105) were left uninfected or infected with SeV (MOI = 1) for the indicated times before immunoblot analyses were performed with the indicated antibodies. Graphs show means ± SDs, n = 3. *, P < 0.05; **, P < 0.01 (unpaired t test).
UL94 deficiency enhances the innate antiviral response against HCMV.
It was previously reported that UL94 is a true late protein (25). To better detect the protein of UL94, we generated a recombinant HCMV strain (HCMV-UL94-HA) by BAC, in which a hemagglutinin (HA) tag was inserted at the C terminus of UL94 without interruption of the reading frame. Following HCMV infection, the mRNA of UL94 was detected at 24 h postinfection and rapidly enriched at 48 h postinfection (Fig. 3A). Consistently, the protein of UL94 was not detectable before 24 h postinfection but readily detected at 48 and 72 h postinfection (Fig. 3B). To determine whether UL94 is present in the virion, we performed an immunoblot analysis with purified viruses. As expected, UL94, UL82, and UL44, which all have been reported as HCMV structural proteins (24, 28, 29), were detected in virions (Fig. 3C). To confirm the function of UL94 in HCMV immune evasion, we constructed two UL94 short hairpin RNA (shRNA) plasmids that markedly inhibited the expression of UL94 but not UL82 or UL44 (Fig. 3D). qPCR analysis showed that the UL94 shRNAs remarkably knocked down the transcription of HCMV UL94 (Fig. 3E). Knockdown of UL94 obviously increased HCMV-induced transcription of IFNB1, CXCL10, and ISG56 genes (Fig. 3E) and enhanced phosphorylation of TBK1 and IRF3 (Fig. 3F) in HFFs. In contrast, knockdown of UL94 showed no effects on HSV-1-induced transcription of antiviral genes (Fig. 3G). These results suggest that UL94 negatively regulates the innate immune response to HCMV.
FIG 3.

UL94 deficiency potentiates HCMV-induced antiviral response. (A and B) Expression of UL94 following HCMV infection. HFF cells (2 × 107) were infected with HCMV-UL94-HA (MOI = 3) for the indicated times before qPCR (A) or immunoblot (B) analyses were performed. (C) UL94 is present in the virion. Purified HCMV-UL94-HA viruses were analyzed by immunoblot assay with the indicated antibodies. (D) Effects of UL94-shRNAs on expression of UL94. HFFs stably expressing UL94 shRNA (2 × 107) were infected with HCMV-UL94-HA (MOI = 3) for 48 h before immunoblot analyses were performed with the indicated antibodies. (E) Knockdown of UL94 enhances HCMV-induced transcription of antiviral genes in HFFs. HFFs stably expressing UL94 shRNA (4 × 105) were left uninfected or infected with HCMV (MOI = 1) for the indicated times before qPCR analysis. (F) Knockdown of UL94 potentiates HCMV-induced phosphorylation of TBK1 and IRF3 in HFFs. HFFs stably expressing UL94 shRNA (4 × 105) were left uninfected or infected with HCMV (MOI = 1) for the indicated times before immunoblot analyses were performed with the indicated antibodies. (G) Effects of UL94 knockdown on HSV-1-induced transcription of antiviral genes in HFFs. HFFs stably expressing UL94 shRNA (4 × 105) were left uninfected or infected with HSV-1 (MOI = 1) for 10 h before qPCR analysis. (H) Confirmation of UL94 deficiency of HCMV-ΔUL94. HFFs (2 × 107) were infected with HCMV-UL94-HA (MOI = 3) or HCMV-ΔUL94 (MOI = 3) for 48 h before immunoblot analyses were performed with the indicated antibodies. (I) UL94 deficiency enhances HCMV-induced transcription of antiviral genes in HFFs. HFFs (4 × 105) were infected with HCMV-UL94-HA (MOI = 1) or HCMV-ΔUL94 (MOI = 1) for the indicated times before qPCR analysis. (J) UL94 deficiency enhances HCMV-induced phosphorylation of TBK1 and IRF3 in HFFs. HFFs (4 × 105) were infected with HCMV-UL94-HA (MOI = 1) or HCMV-ΔUL94 (MOI = 1) for the indicated times before immunoblot analyses were performed with the indicated antibodies. (K) UL94 inhibits UV-inactivated HCMV-triggered transcription of antiviral genes in HFFs. HFFs stably expressing UL94 (4 × 105) were left uninfected or infected with untreated or UV-inactivated HCMV (MOI = 1) for the indicated times before qPCR analysis. Graphs show means ± SDs, n = 3. *, P < 0.05; **, P < 0.01 (unpaired t test).
To further confirm the role of UL94, we generated a UL94-deficient HCMV strain (HCMV-ΔUL94) by introducing a C-to-A point mutation at 24 bp of the UL94 coding sequence, which resulted in a premature stop codon at this position. The recombinant virus was confirmed by sequencing. Additionally, immunoblot analysis confirmed the knockout of UL94 (Fig. 3H). We next examined the effects of UL94 deficiency on HCMV-triggered induction of antiviral genes. qPCR analysis indicated that mRNA levels of IFNB1, IL6, ISG56, and RANTES genes induced by UL94-deficient HCMV were significantly higher at 48 and 72 h postinfection than that induced by HCMV-UL94-HA in HFFs (Fig. 3I). The expression of antiviral genes induced by HCMV was weakened at 72 h postinfection, suggesting that induction of cytokines is regulated by elaborate mechanisms to prevent excessive immune responses. Consistently, UL94-deficient HCMV also induced higher levels of TBK1 and IRF3 phosphorylation at the late phase of infection (Fig. 3J). Furthermore, we treated HCMV with UV. UV-treated HCMV, which is capable of infection but not transcription or translation, induced higher transcription of antiviral genes such as RANTES and CXCL10 than untreated viruses. Notably, UL94 also inhibited transcription of antiviral genes triggered by UV-treated HCMV, indicating that UL94 directly affects HCMV-induced transcription of antiviral genes (Fig. 3K). Taken together, these results suggest that UL94 plays an important role in the inhibition of the HCMV-triggered innate immune response.
UL94 mediates HCMV immune evasion.
Since UL94 inhibits HCMV-triggered induction of type I IFNs, we next investigated the role of UL94 in HCMV immune evasion. PCR analysis showed that at the early phase of HCMV infection, the levels of viral genome indicated by UL44, UL83, UL122, and UL123 genes showed little difference in control and UL94-expressing cells (Fig. 4A), suggesting that UL94 has no marked effects on HCMV infection at the early phase. UL94 enhanced expression of HCMV proteins at late phase of infection, whereas UL94 deficiency showed the opposite effect (Fig. 4B and C). Consistently, 50% tissue culture infective dose (TCID50) assays indicated that UL94 markedly increased HCMV replication at 5 days postinfection (Fig. 4D). Next, to further investigate whether cGAS-MITA axis is involved in UL94-mediated immune evasion, we generated MITA-deficient HFFs by the CRISPR-Cas9 system and infected control and MITA-deficient cells with HCMV-UL94-HA or HCMV-ΔUL94 viruses. TCID50 assays showed that the progeny viruses of HCMV-ΔUL94 were not detected in either control or MITA-deficient HFFs (Fig. 4E), confirming from a previous report that UL94 is crucial for virion envelopment and thus is essential for viral replication (25). We further examined the role of UL94 in viral genome replication by analyzing levels of UL82 and UL44 genes at different time points after infection. As shown in Fig. 4F, in control cells, UL94 deficiency significantly impaired viral genome replication at 72 and 96 h postinfection. In MITA-deficient cells, UL94 deficiency had no marked effects on HCMV genome replication at all times examined except 96 h. The difference at 96 h postinfection may have partially been caused by the generation of a large number of progeny viruses by HCMV-UL94-HA (Fig. 4F). Notably, as shown in Fig. 4G, UL94 markedly increased HCMV replication in control but not MITA-deficient HFF cells, suggesting that UL94 enhances HCMV replication by interrupting MITA-mediated signaling. Taken together, these results suggest an important role of UL94 in viral immune evasion.
FIG 4.

Role of UL94 in HCMV immune evasion. (A) Effects of UL94 on HCMV initial infection. HFFs stably expressing UL94 (4 × 105) were left uninfected or infected with HCMV (MOI = 3) for the indicated times. Genomic DNA was extracted at the indicated times postinfection for analysis of HCMV DNA with PCR assays. (B) UL94 enhances viral gene expression during HCMV infection in HFFs. HFFs stably expressing UL94 (4 × 105) were infected with HCMV (MOI = 3) for the indicated times before immunoblot analyses were performed with the indicated antibodies. (C) Effects of UL94 deficiency on HCMV gene expression. HFFs (2 × 107) were infected with HCMV-UL94-HA or HCMV-ΔUL94 (MOI = 3) for the indicated times before immunoblot analyses were performed with the indicated antibodies. (D) UL94 enhances HCMV replication in HFFs. HFFs stably expressing UL94 (1 × 106) were infected with HCMV (MOI = 1), and the supernatants were harvested at the indicated times postinfection for measurements of viral titers with standard TCID50 assays. (E) Effects of UL94-deficiency on HCMV replication. HFFs transduced with a control gRNA or MITA gRNA were infected with HCMV-UL94-HA or HCMV-ΔUL94 (MOI = 1). The supernatants were harvested at the indicated times postinfection for measurements of viral titers with standard TCID50 assays in HFFs stably expressing UL94. (F) Effects of UL94 deficiency on viral genome replication during HCMV infection in HFFs. HFFs stably expressing MITA gRNA (4 × 105) were infected with HCMV-UL94-HA or HCMV-ΔUL94 (MOI = 1), and genomic DNA was extracted at the indicated times postinfection for quantification of HCMV DNA with qPCR assays. (G) UL94 facilitates HCMV replication via the MITA-mediated innate antiviral response. HFFs transduced with UL94 and a control gRNA or MITA gRNA were infected with HCMV (MOI = 1), and the supernatants were harvested at the indicated times postinfection for measurements of viral titers with standard TCID50 assays. The protein levels of MITA and UL94 were examined with immunoblot assays. Graphs show means ± SDs, n = 3. *, P < 0.05; **, P < 0.01 (unpaired t test).
UL94 interacts with MITA.
Ectopic expression of UL94 impaired the second messenger cGAMP-induced transcription of IFNB, CXCL10, RANTES, and ISG56 genes as well as phosphorylation of TBK1 and IRF3 (Fig. 5A and B). Co-immunoprecipitation experiments indicated that UL94 specifically associated with MITA but not other components of the signaling pathway such as cGAS, TBK1, IRF3, IKKα, IKKβ, or p65 (Fig. 5C). To further confirm the association of UL94 with MITA under physiological conditions, we established HFF cells that stably express Flag-tagged MITA by lentivirus-mediated transduction. Semiendogenous co-immunoprecipitation experiments indicated that UL94 was associated with MITA following HCMV-UL94-HA infection (Fig. 5D). In addition, in vitro pulldown assays showed that UL94 directly bound to MITA (151 to 379), a truncation of MITA in which the N-terminal transmembrane domains have been deleted, in a dose-dependent manner (Fig. 5E). Domain-mapping experiments indicated that the N terminus of UL94 (amino acids [aa] 1 to 172) was essential for its interaction with MITA (Fig. 5F). Notably, qPCR analysis indicated that UL94 and UL94 (aa 1 to 172) but not the MITA binding-defective mutant UL94 (aa 173 to 345) inhibited HCMV-induced transcription of IFNB1 and CXCL10 genes in HFFs (Fig. 5G). Taken together, these findings suggest that UL94 inhibits the antiviral innate immune response by targeting MITA.
FIG 5.

UL94 targets MITA. (A) UL94 inhibits cGAMP-induced transcription of antiviral genes in HFFs. HFFs stably expressing UL94 (4 × 105) were transfected with cGAMP (0.2 μg/ml) for 3 h before qPCR analysis. (B) UL94 inhibits cGAMP-induced phosphorylation of TBK1 and IRF3 in HFFs. HFFs stably expressing UL94 (4 × 105) were transfected with cGAMP (0.2 μg/ml) for 3 h before immunoblot analyses were performed with the indicated antibodies. (C) Association of UL94 with MITA. HEK293T cells (2 × 106) were transfected with the indicated plasmids for 24 h. Co-immunoprecipitation and immunoblot analyses were performed with the indicated antibodies. (D) Endogenous association of UL94 with MITA following HCMV infection. HFFs stably expressing Flag-tagged MITA (2 × 107) were infected with HCMV-UL94-HA (MOI = 3) for 72 h. Co-immunoprecipitation and immunoblot analyses were performed with the indicated antibodies. (E) UL94 directly binds to MITA. Purified GST-MITA (151 to 379) was bound to glutathione agarose beads and incubated with purified His-UL94 for 3 h. Immunoblot analyses were performed with the indicated antibodies. (F) Domain mapping of the UL94-MITA association. HEK293T cells (2 × 106) were transfected with the indicated UL94 truncation mutants for 24 h before co-immunoprecipitation and immunoblot analyses were performed with the indicated antibodies. (G) Effects of UL94 truncations on HCMV-induced transcription of antiviral genes in HFFs. HFFs stably expressing UL94 or the indicated UL94 truncations (4 × 105) were left uninfected or infected with HCMV (MOI = 1) for 12 h before qPCR analysis. The protein levels of UL94 and its truncations were examined with immunoblot assays. Graphs show means ± SDs, n = 3. *, P < 0.05; **, P < 0.01 (unpaired t test).
UL94 impairs dimerization and trafficking of MITA.
It has been demonstrated that MITA dimerization and its trafficking from the ER via the Golgi apparatus to perinuclear microsomes are crucial for its activation (9, 30). We then determined the effects of UL94 on these events. Co-immunoprecipitation experiments indicated that UL94 inhibited self-association of MITA (Fig. 6A). Consistently, UL94 also impaired HCMV- and transfected HSV120-induced dimerization of MITA in HFFs (Fig. 6B). In addition, confocal microscopy showed that compared with that in the control cells, HSV120-induced accumulation of MITA in perinuclear microsomes was disrupted in cells ectopically expressing UL94 (Fig. 6C). Previously, it was reported that TRAPβ, iRhom2, and SNX8 form a translocon complex which is involved in the trafficking of MITA (8, 31, 32). Surprisingly, UL94 showed no effects on the association of MITA with TRAPβ, iRhom2, or SNX8 in co-immunoprecipitation experiments (Fig. 6D). It has been reported that the G158 of MITA is essential for its dimerization (33). Interestingly, accumulation of MITA in perinuclear microsomes induced by dsDNA was dramatically hindered in MITA-deficient cells reconstituted with the MITA G158L mutant (Fig. 6E), indicating that dimerization of MITA is a prerequisite for its trafficking. Taken together, these findings suggest that UL94 inhibits dimerization of MITA and consequently disrupts the translocation of MITA to the perinuclear microsomes.
FIG 6.

UL94 disrupts dimerization and translocation of MITA. (A) UL94 inhibits self-interaction of MITA. HEK293T cells (2 × 106) were transfected with the indicated plasmids for 24 h. Co-immunoprecipitation and immunoblot analyses were performed with the indicated antibodies. (B) UL94 disrupts dimerization of MITA. HFFs stably expressing UL94 (2 × 107) were left uninfected, infected with HCMV (MOI = 1), or transfected with HSV120 (1 μg/ml) for the indicated times. Immunoblot analyses were performed with the indicated antibodies. (C) UL94 impairs translocation of MITA to perinuclear microsome. Mita−/− MLFs that had been reconstituted with MITA-Flag and stably transduced with UL94-HA were transfected with HSV120 (1 μg/ml) for 3 h before confocal microscopy. The aggregation ratio of MITA is shown in the histogram on the right. (D) UL94 has no effect on association of MITA with the components of translocon complex. HEK293T cells (2 × 106) were transfected with the indicated plasmids for 24 h. Co-immunoprecipitation and immunoblot analyses were performed with the indicated antibodies. (E) Dimerization of MITA is required for its translocation. Mita−/− MLFs that had been reconstituted with MITA-WT or MITA-G158L mutant were transfected with HSV120 (1 μg/ml) for 3 h before confocal microscopy.
UL94 impairs recruitment of TBK1 to the MITA complex.
MITA functions as a scaffold protein to recruit TBK1 and IRF3 to the MITA signalsome in which IRF3 is phosphorylated by TBK1 and activated, leading to subsequent induction of type I IFNs (13, 34). We next examined whether UL94 affects recruitment of TBK1 and IRF3 to MITA. Co-immunoprecipitation experiments indicated that UL94 impaired the association of MITA with TBK1 but showed no effects on the interaction between MITA and IRF3 (Fig. 7A and B). Consistently, in vitro glutathione transferase (GST) pulldown assays indicated that UL94 disrupted binding of MITA (151 to 379) to TBK1 but not IRF3 (Fig. 7C and D). Taken together, these results suggest that UL94 selectively impairs recruitment of TBK1 to the MITA signalsome, leading to the inhibition of downstream signal transduction.
FIG 7.

UL94 hinders the recruitment of TBK1 to MITA. (A and B) Effect of UL94 on the association between MITA and TBK1 or IRF3. HEK293T cells (2 × 106) were transfected with the indicated plasmids for 24 h. Co-immunoprecipitation and immunoblot analyses were performed with the indicated antibodies. (C and D) Effects of UL94 on binding of MITA to TBK1 or IRF3 in vitro. Purified GST-MITA (151 to 379) was bound to glutathione agarose beads and incubated with lysates of HEK293T cells transiently expressing Flag-TBK1 (C) or Flag-IRF3 (D). Immunoblot analyses were performed with the indicated antibodies.
DISCUSSION
Herpesviruses are a large family of enveloped double-stranded DNA viruses, which usually establish latent infection in hosts. The viral DNA can be recognized by cGAS, which subsequently leads to induction of type I IFNs and antiviral effectors, resulting in suppression of viral replication and elimination of virus-infected cells (6, 7, 35). However, herpesviruses have evolved multiple strategies to antagonize the innate immune response, which is important for the establishment of life-long latent infection. For example, it has been reported that the HSV-1 tegument protein UL37 and the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF52 inhibit the enzymatic activity of cGAS and thus disrupt synthesis of cGAMP (36, 37). In addition, HSV-1 ICP27 and KSHV vIRF1 have been shown to target the MITA signalsome, thus inhibiting phosphorylation and activation of the transcription factor IRF3 (38, 39). In the case of HCMV, multiple viral proteins such as UL31, UL83, UL82, and UL42 have been demonstrated to antagonize cGAS-MITA-mediated signaling at an early stage of infection (19–21, 40). However, the mechanisms by which HCMV evades immune response in the late phase of infection need more investigation.
The HCMV protein UL94 has been demonstrated as a true late protein (24). In our study, UL94 was readily detected in the late phase (48 and 72 h postinfection) of infection in both qPCR and immunoblot experiments (Fig. 3A and B). Previous studies have reported that UL94 localizes in the cytoplasm and nuclei in transfected cells and was detected predominantly in a juxtanuclear structure during HCMV infection (25). Functionally, UL94 has been demonstrated to direct UL99 to the assembly complex and facilitate secondary envelopment of virions, which is essential for viral replication (25, 41).
In a screen for HCMV proteins that inhibit the antiviral innate immune response, we identified UL94 as a candidate. Overexpression of UL94 inhibited cGAS-MITA-, HCMV-, and cytosolic dsDNA-induced transcription of type I IFNs and downstream antiviral genes (Fig. 1B and 2A and B). Conversely, UL94 deficiency promoted HCMV-triggered induction of antiviral genes (Fig. 3I). In addition, we found that UL94 enhanced HCMV replication, whereas UL94 deficiency impaired viral replication (Fig. 4D to F). Several evidences suggest that UL94 targets MITA for immune evasion. First, ectopic expression of UL94 impaired cGAMP-induced downstream signaling (Fig. 5A and B). Second, both co-immunoprecipitation experiments and in vitro pulldown assays showed that UL94 interacted with MITA (Fig. 5C to E). Third, the UL94 truncation mutant which lost its ability to interact with MITA also failed to inhibit HCMV-induced transcription of type I IFNs and downstream genes (Fig. 5F and G). Fourth, UL94 failed to enhance viral replication in MITA-deficient cells (Fig. 4G). These data suggest that UL94 impairs the innate immune response to DNA viruses by targeting MITA.
Further studies suggest that UL94 acts in two distinct mechanisms to inhibit the function of MITA (Fig. 8). First, UL94 suppresses the dimerization of MITA in response to HCMV infection (Fig. 6B), disrupting the translocation of MITA to perinuclear microsomes (Fig. 6C). In addition, as demonstrated in both co-immunoprecipitation experiments and in vitro GST pulldown assays (Fig. 7A and C), UL94 directly impairs binding of TBK1 to MITA, which is essential for the activation of IRF3 and induction of type I IFNs.
FIG 8.
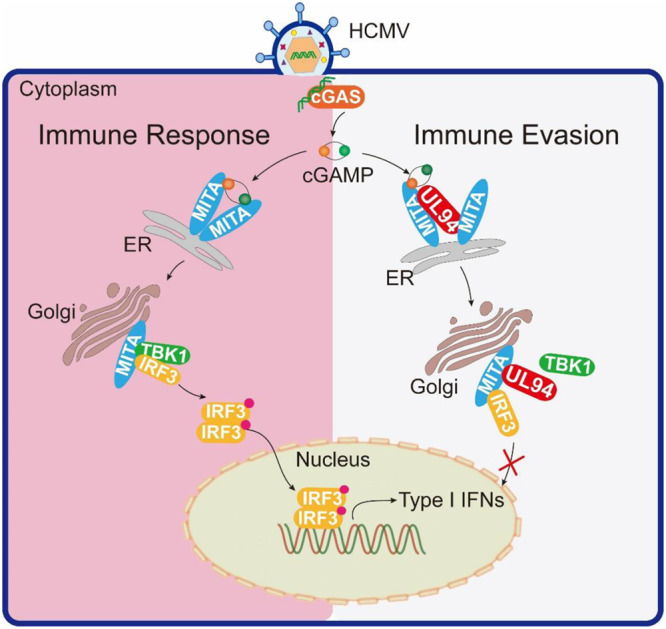
A working model on UL94-mediated HCMV immune evasion. Cytosolic DNA sensor cGAS recognizes HCMV DNA and synthesizes the second messenger cGAMP. cGAMP binds to MITA, facilitating the dimerization and oligomerization of MITA, and initiates translocation of MITA from the ER to perinuclear microsomes. During this process, MITA recruits TBK1 and IRF3, leading to phosphorylation and activation of IRF3 by TBK1. HCMV protein UL94 disrupts MITA dimerization and recruitment of TBK1 and therefore inhibits the MITA-mediated innate immune response against HCMV infection.
In our recent studies, we have identified HCMV UL82 and UL42 as MITA inhibitors. However, these proteins and UL94 function by distinct mechanisms. First, both UL82 and UL42 are early proteins that were detected at 6 to 12 h postinfection. Nevertheless, UL94 has been demonstrated to be a true late protein which could only be readily detected at 48 h postinfection. Given the differences in the timing of expression, it is reasonable to speculate that these proteins function at different stages following HCMV infection, which collaborate to prolong the suppression of the host innate immune response. Second, although UL82, UL42, and UL94 all impair the trafficking of MITA, the detailed mechanisms are different. UL82 impedes trafficking of MITA by disrupting the assembly of the MITA translocon complex, while UL42 impairs translocation of MITA by facilitating the degradation of TRAPβ, which is a key component of the translocon complex (21, 40). Notably, UL94 exhibited no effects on the formation or stability of the MITA translocon complex. Instead, UL94 disrupts the dimerization of MITA, which is a prerequisite for its translocation.
Taken together, in this study, we identified previously uncharacterized roles of UL94 in the immune evasion of HCMV during the late phase of infection. Combined with results from a previous study, it is likely that UL94 is involved in both secondary envelopment of virions and immune evasion. These findings expand our knowledge on the mechanisms used by HCMV to ensure persistent immune suppression and enhance our understanding of the virus-host interplay.
MATERIALS AND METHODS
Reagents, antibodies, cells and viruses.
The following reagents and antibodies were purchased from the indicated manufacturers: dual-specific luciferase assay kit (Promega); recombinant human IFN-γ, IFN-β, and TNF-α (R&D Systems); Polybrene (Millipore); puromycin and RNase inhibitor (Thermo); SYBR (Bio-Rad); 2′3′-cGAMP and lipofectamine 2000 (Invitrogen); digitonin (Sigma); enzyme-linked immunosorbent assay (ELISA) kits for human IFN-β (4A Biotech); mouse antibodies against Flag and β-actin (Sigma), His and HA (OriGene), MyC (9B11), IKKβ (8943S), and MITA (13647S) (Cell Signaling Technology); and rabbit antibodies against TBK1 (ab109735; Abcam), IRF3 (11312-1-AP; Proteintech), phosphor-TBK1 (S172) (5483S), phosphor-IRF3 (S396) (4947S), phosphor-IKKα (Ser176)/β (Ser177) (2078S), phosphor-STAT1 (Y701) (P1675), phosphor-STAT2 (Y690) (88410S), STAT1 (14994S), and STAT2 (72604S) (Cell Signaling Technology). Antisera against UL94, UL82, and UL44 were generated by immunizing rabbits or mice with purified recombinant UL94, UL82, and UL44 proteins.
HEK293T cells (transformed human embryonic kidney 293 cell line) were purchased from ATCC. HFFs (human primary foreskin fibroblasts) were provided by Min-Hua Luo (Wuhan Institute of Virology, CAS). Reconstituted Mita−/− mouse lymphatic fibroblast (MLF) cells were generated as previously described (31). HCMV (AD169) was provided by Min-Hua Luo (Wuhan Institute of Virology, CAS). SeV (Cantell strain) was obtained from Charles River Laboratories. HSV-1 (KOS strain) and VACV (Tian-Tan strain) were purchased from China Center for Type Culture Collection, Wuhan, China.
Constructs.
Mammalian expression plasmids for Flag-, HA-, His-, and Myc-tagged UL94 and its truncation mutants were constructed by standard molecular cloning procedures. Expression plasmids for HA-tagged cGAS, MITA, TBK1, IRF3, IKKα, IKKβ, and p65 and the luciferase reporter plasmids of IFN-β, IRF1, and NF-κB were previously described (11, 42, 43).
DNA oligonucleotides.
The following oligonucleotides were used to stimulate cells: HSV120, 5′-AGACGGTATATTTTTGCGTTATCACTGTCCCGGATTGGACACGGTCTTGTGGGATAGGCATGCCCAGAAGGCATATTGGGTTAACCCCTTTTTATTTGTGGCGGGTTTTTTGGAGGACTT-3′; VACV70, 5′-CCATCAGAAAGAGGTTTAATATTTTTGTGAGACCATCGAAGAGAGAAAGAGATAAAACTTTTTTACGACT-3′; HSV60, 5′-TAAGACACGATGCGATAAAATCTGTTTGTAAAATTTATTAAGGGTACAAATTGCCCTAGC-3′.
shRNA.
Double-stranded oligonucleotides corresponding to the targeting sequences were cloned into the pSuper-Retro RNAi plasmid (Oligoengine). The following sequences were targeted: UL94-shRNA 1, 5′-GCGACGGCGAATGGATCATCT-3′; UL94-shRNA 2, 5′-GGGAGACGGCCTGGATTATCA-3′; and control-shRNA, 5′-GGAAGATGTATGGAGACATGG-3′.
Transfection and reporter assays.
HEK293T cells were transfected by the standard calcium phosphate precipitation method. HFFs and MLFs were transfected with Lipofectamine 2000 according to the manufacturer’s instructions. Empty control plasmid was added to ensure that each transfection received the same amount of total DNA. To normalize for transfection efficiency, pRL-TK (Renilla luciferase) reporter plasmid (0.01 μg) was included in each transfection. Luciferase assays were performed using a dual-specific luciferase assay kit. Firefly luciferase activities were normalized on the basis of Renilla luciferase activities.
Transduction of HFF cells.
For generation of HFF cells stably expressing UL94-shRNAs, HEK293T cells were transfected with two packaging plasmids, pGag-Pol (10 μg) and pVSV-G (3 μg) along with a control or UL94-shRNA retroviral plasmid (10 μg). After 12 h, cells were incubated with new medium without antibiotics for another 24 h. The recombinant virus-containing medium was filtered and then used to infect HFF cells in the presence of Polybrene (6 μg/ml). The infected HFF cells were selected with puromycin (1 μg/ml) for 2 weeks. Similar methods were utilized to establish HFFs stably expressing UL94 and HFFs transduced with MITA genomic RNA (gRNA).
Real-time PCR.
Total RNA was isolated with TRIzol reagent (TaKaRa) and reverse transcribed to cDNA for real-time PCR analysis. The mRNA abundances of targeted genes were normalized to human GAPDH. Gene-specific primer sequences were as follows: GAPDH, GAGTCAACGGATTTGGTCGT (forward), GACAAGCTTCCCGTTCTCAG (reverse); IFNB1, TTGTTGAGAACCTCCTGGCT (forward), TGACTATGGTCCAGGCACAG (reverse); IL6, TTCTCCACAAGCGCCTTCGGTC (forward), TCTGTGTGGGGCGGCTACATCT (reverse); CXCL10, GGTGAGAAGAGATGTCTGAATCC (forward), GTCCATCCTTGGAAGCACTGCA (reverse); RANTES, GGCAGCCCTCGCTGTCATCC (forward), GCAGCAGGGTGTGGTGTCCG (reverse); ISG56, TCATCAGGTCAAGGATAGTC (forward), CCACACTGTATTTGGTGTCTAGG (reverse); UL44, TCGTGCTCCGTAACTATA (forward), GCTGAAGAACTTCTACCA (reverse); UL82, ACGACACCGTAGACCTGACC (forward), AAAGAGGTGCAGTCCGCTAA (reverse); UL83, GCAGAACCAGTGGAAAGAGC (forward), GTCCTCTTCCACGTCAGAGC (reverse); UL94, CTTCGGAGGTCCTTTGGGTC (forward), ACGTCGGGCGTAATACCATC (reverse); UL122, ACCATGCAGGTGAACAACAA (forward), GCAATTCTTTGAGGCTCCAC (reverse); UL123, CAAGGAGCACATGCTGAAAA (forward), CATCCACATCTCCCGCTTAT (reverse).
ELISA.
HFFs were infected with viruses for 12 h. The culture media were collected for measurement of IFN-β by ELISA.
Co-immunoprecipitation and immunoblot analysis.
HEK293T cells or HFFs were lysed with 1 ml NP-40 lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride). For each immunoprecipitation, a 0.45-ml aliquot of lysate was incubated with the control IgG or indicated antibody (0.5 μg), and protein G Sepharose beads (25 μl) at 4 °C for 3 h. The Sepharose beads were collected and washed four times with 0.8 ml NP-40 lysis buffer containing 500 mM NaCl. Immunoblot analyses were performed as previously described (42).
GST pulldown assay.
GST-MITA (151-379) was bound to glutathione agarose beads and incubated with purified His-UL94 or lysates of HEK293T cells transiently expressing Flag-TBK1 or Flag-IRF3 along with an empty or HA-UL94 plasmid for 3 h. The beads were collected and washed three times with lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride). The beads were then mixed with SDS loading buffer and boiled for 10 min. The input/elutes were resolved by SDS-PAGE and analyzed by Coomassie staining and/or immunoblot analysis.
Confocal microscopy.
MLFs were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 15 min on ice. The cells were then blocked with 1% bovine serum albumin (BSA) in PBS and stained with the designated primary and secondary antibodies for 1 h. The nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) for 2 min. The stained cells were observed and imaged with a Nikon confocal microscope under a 60× oil lens objective (44).
MITA dimerization assay.
Analysis of MITA dimerization was performed as described previously (45). HFFs were lysed in 0.8 ml lysis buffer [10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES)-KOH buffer (pH 7.0), 50 mM NaCl, 5 mM MgCl2, 5 mM EGTA, 10% glycerol, and a mixture of protease and phosphatase inhibitors] containing 1% Nonidet P-40 and centrifuged at 20,000 × g for 15 min. The cell lysates were mixed with 5 × SDS buffer (200 mM Tris-HCl buffer [pH 6.8], 10% SDS, 25% glycerol, and 0.05% bromphenol blue) without 2-mercaptoethanol (2-ME), and samples were analyzed by SDS-PAGE.
Generation of recombinant virus.
Bacterial artificial chromosome (BAC) pAD-GFP carrying the green fluorescent protein (GFP)-tagged genome of the HCMV AD169 strain was constructed by inserting the BAC sequence containing GFP expression cassette following the US28 open reading frame without the deletion of any viral sequence and used to reconstitute recombinant HCMV viruses (46). pAD-GFP-UL94HA and pAD-GFP-UL94stop BACs were generated by using a two-step markerless recombination system in the GS1783 Escherichia coli strain as previously described (47). The marker cassette linear fragment was amplified with the oligonucleotides primers UL94HA (forward, GATAGTGTGTTCCTGTCCGGTGCTTAAGAACCTAGTGCACTACCCATACGACGTACCAGATTACGCTTAACGGGGTCTGACAGTTCAAGGATGACGACGATAAGTAGGG; reverse, TTTCTTGTTTCTTCTCCCCGTGAACTGTCAGACCCCGTTAAGCGTAATCTGGTACGTCGTATGGGTAGTGCACTAGGTTCTTAAGCACAACCAATTAACCAATTCTGATTAG) and UL94stop (forward, TCGCGCGTCACTCTTCATGGCTTGGCGCAGCGGGCTTTGAGAGACCGATTCCAGAACTTTAGGATGACGACGATAAGTAGGG; reverse, CCTCTTGCAAGAACTGCTTCAAAGTTCTGGAATCGGTCTCTCAAAGCCCGCTGCGCCAAGCAACCAATTAACCAATTCTGATTAG). The PCR products of the marker cassette were electroporated into recombination-competent GS1783 cells. The expression of the inducible I-SceI resulted in cleavage of the complete marker cassette and production of the desired sequence modification. Recombinant viruses were generated by transfecting pAD-GFP-UL94HA BAC DNA into HFFs or pAD-GFP-UL94stop BAC DNA into HFFs stably expressing UL94. The cell-free culture supernatant was collected when the entire monolayer of HFF cells was lysed.
HCMV genome sequencing and read mapping.
Genomic DNAs of HCMV-UL94-HA and HCMV-ΔUL94 were extracted and sequenced with an Illumina MiSeq platform. The mapping of sequencing reads was analyzed by Burrows-Wheeler Aligner (BWA) against the reference sequence (FJ527563.1) (48). The results of alignments were generated in the Sequence Alignment/Map (SAM) format, and SAMtools was utilized for sorting, merging, and indexing of the alignments (49). Finally, Integrative Genomics Viewer (IGV) supported the visualization of these results (50).
ACKNOWLEDGMENTS
We thank Ke Lan and Zhi-Kang Qian for help with the construction of HCMV recombinant virus. We also thank Ding Gao and Lei Zhang of the Core Facility and Technical Support Facility of Wuhan Institute of Virology for help with the confocal microscopy and HCMV genome sequencing, respectively.
This study was supported by the National Science Fund for Distinguished Young Scholars (31425010), the National Natural Science Foundation of China (31621061, 31800732), and Key Research Programs of Frontier Sciences funded by the Chinese Academy of Sciences.
REFERENCES
- 1.Janeway CA Jr, Medzhitov R. 2002. Innate immune recognition. Annu Rev Immunol 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Barbalat R, Ewald SE, Mouchess ML, Barton GM. 2011. Nucleic acid recognition by the innate immune system. Annu Rev Immunol 29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi O, Akira S. 2009. Innate immunity to virus infection. Immunological Rev 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paludan SR, Bowie AG. 2013. Immune sensing of DNA. Immunity 38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X-D, Wu J, Gao D, Wang H, Sun L, Chen ZJ. 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. 2009. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A 106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC. 2008. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol 28:5014–5026. doi: 10.1128/MCB.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, Chen ZJ. 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka Y, Chen ZJ. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Science Signaling 5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stern-Ginossar N, Weisburd B, Michalski A, Le VTK, Hein MY, Huang S-X, Ma M, Shen B, Qian S-B, Hengel H, Mann M, Ingolia NT, Weissman JS. 2012. Decoding human cytomegalovirus. Science 338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheeran MC, Lokensgard JR, Schleiss MR. 2009. Neuropathogenesis of congenital cytomegalovirus infection: disease mechanisms and prospects for intervention. Clin Microbiol Rev 22:99–126. doi: 10.1128/CMR.00023-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fishman JA. 2007. Infection in solid-organ transplant recipients. N Engl J Med 357:2601–2614. doi: 10.1056/NEJMra064928. [DOI] [PubMed] [Google Scholar]
- 17.Drew WL. 1992. Cytomegalovirus infection in patients with AIDS. Clinical Infectious Diseases 14:608–615. doi: 10.1093/clinids/14.2.608-a. [DOI] [PubMed] [Google Scholar]
- 18.Powers C, DeFilippis V, Malouli D, Früh K. 2008. Cytomegalovirus immune evasion. Curr Top Microbiol Immunol 325:333–359. doi: 10.1007/978-3-540-77349-8_19. [DOI] [PubMed] [Google Scholar]
- 19.Huang ZF, Zou HM, Liao BW, Zhang HY, Yang Y, Fu YZ, Wang SY, Luo MH, Wang YY. 2018. Human cytomegalovirus protein UL31 inhibits DNA sensing of cGAS to mediate immune evasion. Cell Host Microbe 24:69.e4–80.e4. doi: 10.1016/j.chom.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Biolatti M, Dell'Oste V, Pautasso S, Gugliesi F, von Einem J, Krapp C, Jakobsen MR, Borgogna C, Gariglio M, De Andrea M, Landolfo S. 2017. Human cytomegalovirus tegument protein pp65 (pUL83) dampens type I interferon production by inactivating the DNA sensor cGAS without affecting STING. J Virol 92:e01774. doi: 10.1128/JVI.01774-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu YZ, Su S, Gao YQ, Wang PP, Huang ZF, Hu MM, Luo WW, Li S, Luo MH, Wang YY, Shu HB. 2017. Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe 21:231–243. doi: 10.1016/j.chom.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Fu YZ, Su S, Zou HM, Guo Y, Wang SY, Li S, Luo MH, Wang YY. 2019. Human cytomegalovirus DNA polymerase subunit UL44 antagonizes antiviral immune responses by suppressing IRF3- and NF-kappaB-mediated transcription. J Virol 93:e00181-19. doi: 10.1128/JVI.00181-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wing BA, Huang ES. 1995. Analysis and mapping of a family of 3′-coterminal transcripts containing coding sequences for human cytomegalovirus open reading frames UL93 through UL99. J Virol 69:1521–1531. doi: 10.1128/JVI.69.3.1521-1531.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wing BA, Lee GC, Huang ES. 1996. The human cytomegalovirus UL94 open reading frame encodes a conserved herpesvirus capsid/tegument-associated virion protein that is expressed with true late kinetics. J Virol 70:3339–3345. doi: 10.1128/JVI.70.6.3339-3345.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips SL, Bresnahan WA. 2012. The human cytomegalovirus (HCMV) tegument protein UL94 is essential for secondary envelopment of HCMV virions. J Virol 86:2523–2532. doi: 10.1128/JVI.06548-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paijo J, Doring M, Spanier J, Grabski E, Nooruzzaman M, Schmidt T, Witte G, Messerle M, Hornung V, Kaever V, Kalinke U. 2016. cGAS senses human cytomegalovirus and induces type I interferon responses in human monocyte-derived cells. PLoS Pathog 12:e1005546. doi: 10.1371/journal.ppat.1005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gray EE, Winship D, Snyder JM, Child SJ, Geballe AP, Stetson DB. 2016. The AIM2-like receptors are dispensable for the interferon response to intracellular DNA. Immunity 45:255–266. doi: 10.1016/j.immuni.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nowak B, Gmeiner A, Sarnow P, Levine AJ, Fleckenstein B. 1984. Physical mapping of human cytomegalovirus genes: identification of DNA sequences coding for a virion phosphoprotein of 71 kDa and a viral 65-kDa polypeptide. Virology 134:91–102. doi: 10.1016/0042-6822(84)90275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S. 2009. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A 106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo WW, Li S, Li C, Lian H, Yang Q, Zhong B, Shu HB. 2016. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat Immunol 17:1057–1066. doi: 10.1038/ni.3510. [DOI] [PubMed] [Google Scholar]
- 32.Wei J, Lian H, Guo W, Chen YD, Zhang XN, Zang R, Zhong L, Yang Q, Hu MM, Luo WW, Shu HB, Li S. 2018. SNX8 modulates innate immune response to DNA virus by mediating trafficking and activation of MITA. PLoS Pathog 14:e1007336. doi: 10.1371/journal.ppat.1007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ouyang S, Song X, Wang Y, Ru H, Shaw N, Jiang Y, Niu F, Zhu Y, Qiu W, Parvatiyar K, Li Y, Zhang R, Cheng G, Liu ZJ. 2012. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 36:1073–1086. doi: 10.1016/j.immuni.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowie A. 2012. The STING in the tail for cytosolic DNA-dependent activation of IRF3. Science Signaling 5:pe9. doi: 10.1126/scisignal.2002919. [DOI] [PubMed] [Google Scholar]
- 35.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. 2013. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, Ma S, Liu X, Miley W, Konrad A, Neipel F, Sturzl M, Whitby D, Li H, Zhu F. 2015. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18:333–344. doi: 10.1016/j.chom.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Zhao J, Xu S, Li J, He S, Zeng Y, Xie L, Xie N, Liu T, Lee K, Seo GJ, Chen L, Stabell AC, Xia Z, Sawyer SL, Jung J, Huang C, Feng P. 2018. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe 24:234.e5–248.e5. doi: 10.1016/j.chom.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, Mettenleiter T, Chen ZJ, Knipe DM, Sandri-Goldin RM, Enquist LW, Hartmann R, Mogensen TH, Rice SA, Nyman TA, Matikainen S, Paludan SR. 2016. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J 35:1385–1399. doi: 10.15252/embj.201593458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, Barber GN, Glaunsinger BA, Dittmer DP, Damania B. 2015. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci U S A 112:E4306–E4315. doi: 10.1073/pnas.1503831112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu YZ, Guo Y, Zou HM, Su S, Wang SY, Yang Q, Luo MH, Wang YY. 2019. Human cytomegalovirus protein UL42 antagonizes cGAS/MITA-mediated innate antiviral response. PLoS Pathog 15:e1007691. doi: 10.1371/journal.ppat.1007691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips SL, Cygnar D, Thomas A, Bresnahan WA. 2012. Interaction between the human cytomegalovirus tegument proteins UL94 and UL99 is essential for virus replication. J Virol 86:9995–10005. doi: 10.1128/JVI.01078-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 43.Zhou Q, Lin H, Wang S, Wang S, Ran Y, Liu Y, Ye W, Xiong X, Zhong B, Shu HB, Wang YY. 2014. The ER-associated protein ZDHHC1 is a positive regulator of DNA virus-triggered, MITA/STING-dependent innate immune signaling. Cell Host Microbe 16:450–461. doi: 10.1016/j.chom.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Yang Y, Wang SY, Huang ZF, Zou HM, Yan BR, Luo WW, Wang YY. 2016. The RNA-binding protein Mex3B is a coreceptor of Toll-like receptor 3 in innate antiviral response. Cell Res 26:288–303. doi: 10.1038/cr.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Motani K, Ito S, Nagata S. 2015. DNA-mediated cyclic GMP-AMP synthase-dependent and -independent regulation of innate immune responses. J Immunol 194:4914–4923. doi: 10.4049/jimmunol.1402705. [DOI] [PubMed] [Google Scholar]
- 46.Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol 76:2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 48.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]