

Within an individual living with HIV-1, factors that restrict the movement of HIV-1 between different compartments—such as between the blood and the male genital tract—could strongly influence which viruses reach sites in the body from which they can be transmitted. Using deep sequencing, we found strong evidence of restricted HIV-1 movements between the blood and genital tract in all 10 men that we studied. We additionally found that neither the degree to which particular genetic variants of HIV-1 proliferate (in blood or genital tract) nor an individual’s history of sexually transmitted infections detectably influenced the degree to which virus movements were restricted between the blood and genital tract. Last, we show evidence that viral replication gave rise to a large clonal amplification in semen in a donor with highly compartmentalized HIV-1 populations, raising the possibility that differential selection of HIV-1 variants in the genital tract may occur.
KEYWORDS: HIV-1, male genital tract, compartmentalization
ABSTRACT
Compartmentalization of HIV-1 between the systemic circulation and the male genital tract may have a substantial impact on which viruses are available for sexual transmission to new hosts. We studied compartmentalization and clonal amplification of HIV-1 populations between the blood and the genital tract from 10 antiretroviral-naive men using Illumina MiSeq with a PrimerID approach. We found evidence of some degree of compartmentalization in every study participant, unlike previous studies, which collectively showed that only ∼50% of analyzed individuals exhibited compartmentalization of HIV-1 lineages between the male genital tract (MGT) and blood. Using down-sampling simulations, we determined that this disparity can be explained by differences in sampling depth in that had we sequenced to a lower depth, we would also have found compartmentalization in only ∼50% of the study participants. For most study participants, phylogenetic trees were rooted in blood, suggesting that the male genital tract reservoir is seeded by incoming variants from the blood. Clonal amplification was observed in all study participants and was a characteristic of both blood and semen viral populations. We also show evidence for independent viral replication in the genital tract in the individual with the most severely compartmentalized HIV-1 populations. The degree of clonal amplification was not obviously associated with the extent of compartmentalization. We were also unable to detect any association between history of sexually transmitted infections and level of HIV-1 compartmentalization. Overall, our findings contribute to a better understanding of the dynamics that affect the composition of virus populations that are available for transmission.
IMPORTANCE Within an individual living with HIV-1, factors that restrict the movement of HIV-1 between different compartments—such as between the blood and the male genital tract—could strongly influence which viruses reach sites in the body from which they can be transmitted. Using deep sequencing, we found strong evidence of restricted HIV-1 movements between the blood and genital tract in all 10 men that we studied. We additionally found that neither the degree to which particular genetic variants of HIV-1 proliferate (in blood or genital tract) nor an individual’s history of sexually transmitted infections detectably influenced the degree to which virus movements were restricted between the blood and genital tract. Last, we show evidence that viral replication gave rise to a large clonal amplification in semen in a donor with highly compartmentalized HIV-1 populations, raising the possibility that differential selection of HIV-1 variants in the genital tract may occur.
INTRODUCTION
An estimated 80% of all the HIV-1 transmissions globally occur sexually and involve the transfer between individuals of viruses contained in genital secretions (1). It is therefore important to understand compartmentalization of HIV-1 (i.e., restriction of movement of viral variants between anatomical sites) between the genital compartment and the overall blood circulation. Compartmentalization of HIV-1 can be promoted by physical barriers to circulation between tissues and by differing selective pressures, including differences in host cell types and immune pressures.
Several studies on compartmentalization have relied upon heteroduplex tracking assays (HTAs) (2–5), analysis of viral sequences obtained by cloning from bulk PCRs (6–9), and, more recently, analyses of viral sequences obtained by single-genome amplification (SGA) (10–16). HTAs provide only qualitative information about the complexity of a virus population. Bulk PCR can generate artificial recombinants of similar sequences amplified in the same reaction and is subject to amplification biases (i.e., particular viral sequences are preferentially amplified over others). Both of these effects impede efforts to estimate the prevalences of different genetic variants of the virus (17). Although SGA was devised specifically to minimize artificial recombination (18) and PCR amplification biases (19), it is labor-intensive and hence results in limited sequencing depth and inability to detect low-frequency genetic variants within HIV-1 populations (20–22). The resulting underestimation of the sequence diversity of a target population (19, 23) can impact the power of phylogenetics-based approaches to detect and quantify evidence of viral population compartmentalization (23).
Next-generation sequencing (NGS) offers the opportunity to circumvent these shortcomings of HTA, bulk PCR, and SGA (24–31). By increasing the depth of sampling, NGS enables the sequencing of genetic variants that are present at low frequencies in the virus population, and NGS data should therefore better reflect the complexity of HIV-1 populations (32). Current NGS methods have been improved by the PrimerID (PID) approach, in which each cDNA is labeled with a unique barcode during the reverse transcription process (33–36). As a result, all DNA molecules in the PCR product carrying an identical barcode can be collapsed to a single consensus sequence representing a single original cDNA molecule. This simple step resolves most PCR artifacts, such as PCR-induced nucleotide substitutions, amplification biases (19), and artificial recombination (8, 37). Additionally, it is possible to distinguish sequences of sister PCR products from sequences of closely related but distinct RNA strands. In this study, we exploited the power of the PrimerID approach to more accurately quantify variant frequency and to better reconstruct HIV-1 populations as they occur in vivo within the blood and genital tract compartments. We used these approaches to test for evidence of restricted movements between these compartments in 10 individuals living with HIV-1.
RESULTS
Study subjects and relationship between blood and semen viral loads and STI.
Blood and semen samples were collected during a single visit from 43 seropositive men that had not started antiretroviral therapy (ART). The median log10 blood viral load was 4.10 copies (cp)/ml (interquartile range [IQR], 2.69 to 4.56), while the median semen log10 viral load was 2.72 cp/sample (IQR, 1.30 to 3.71). Viral loads in the two compartments were weakly associated (adjusted R2 = 0.28 [Fig. 1A]), with semen viral loads generally detectable in individuals with blood viral loads of >10,000 cp/ml (Fig. 1A) (38). We detected no difference in semen or blood viral loads between any pairs of these groups: participants who did not have a detected or recorded sexually transmitted infection (STI) (n = 25), those who had an STI that was treated more than a year before (STI > 1 year, n = 8), or those found to have a concomitant or recent sexually transmitted infection (STI < 1 yr, n = 11) (Fig. 1B).
FIG 1.

Relationship between donor blood and semen viral loads and sexually transmitted infection (STI) among the 43 study participants (38) (A). There was no detected difference in viral loads for participants with a concomitant sexually transmitted infection (STI < 1 year) or who had an STI treated more than a year prior (STI > 1 year) (B). ns, not significant.
Of the 21/44 study participants (48%) who had detectable viral loads in both semen and blood, we picked 10 individuals with semen viral loads of >4,000/sample for deep sequencing. The characteristics of these 10 study participants are summarized in Table 1. There was no difference in age for the 10 study subjects and those for whom deep sequencing was not performed (27.5 years [IQR, 25.5 to 33.25 years] versus 29 years [IQR, 25 to 37years]) (Mann-Whitney test, P = 0.9041). The CD4+ T cell counts for the 10 selected study subjects were significantly lower than those of study subjects whose viral populations were not deep sequenced (379 cells/μl [IQR, 221 to 488 cells/μl] versus 543 cells/μl [IQR. 425 to 669 cells/μl]) (t test, P = 0.015).
TABLE 1.
Characteristics of the 10 study participants used for evaluation of compartmentalization between blood and the male genital tract using deep sequencing
| Sample ID | Age (yrs) | CD4 count (cells/μl) | Blood viral load (log10 cp/ml) | Approximate semen viral load (log10 cp/sample)a | Recent STI history | Self-reported time since known to be HIV positive (yrs) |
|---|---|---|---|---|---|---|
| SVB0433 | 49 | 509 | 5.1 | 6.7 | None | 2 |
| SVB008 | 42 | 257 | 5.5 | 4.6 | None | 5 |
| SVB021 | 34 | 148 | 5.3 | 3.6 | Syphilis | 1 |
| SVB026 | 31 | 209 | 4.7 | 3.7 | Treated for gonorrhea >1 yr prior | 1 |
| SVB012 | 27 | 586 | 5.2 | 3.8 | Treated for gonorrhea and chlamydia >1 yr prior | 2 |
| SVB025 | 25 | 368 | 5.6 | 4.4 | None | 0.25 |
| SVB030 | 28 | 390 | 4.4 | 5.4 | None | 0.08 |
| SVB029 | 28 | 149 | 4.3 | 5.4 | None | 2 |
| SVB039 | 18 | 425 | 4.6 | 5.1 | None | 1 |
| SVB041 | 25 | 567 | 4.9 | 4.4 | None | 0.08 |
Viral load assumes 100% yield upon filtration and ultracentrifugation.
Selection of sequences for analysis of compartmentalization.
We obtained a total of 20,987 PID consensus sequences (median, 1,130; IQR, 547 to 1,355/compartment/donor) after data processing and cleanup (Table 2). There were more PID consensus sequences from blood than from semen (median blood, 1,451 [IQR, 1,126 to 1,726]; median semen, 670 [IQR, 283 to 1,102]) (Wilcoxon matched-pairs signed-rank test P = 0.0186). For compartmentalization analysis, we selected the maximum possible equal number of PID consensus sequences (median, 514 [IQR, 283 to 861]) by randomly selecting sequences from the compartment with the larger number of PID consensus sequences to match the number obtained from the other compartment (Table 2).
TABLE 2.
Summary of numbers of sequences obtained (PID consensus), the numbers used for analysis of compartmentalization per compartment (equal number from blood or semen), and the numbers of unique sequences
| Sample IDa | No. of: |
||
|---|---|---|---|
| PID consensus sequences | Consensus sequences used for compartmentalization analysis | Unique sequences | |
| SVB008_BP | 1,643 | 448 | 848 |
| SVB008_SP | 448 | 448 | 160 |
| SVB012_BP | 1,139 | 81 | 286 |
| SVB012_SP | 81 | 81 | 26 |
| SVB021_BP | 1,645 | 165 | 667 |
| SVB021_SP | 165 | 165 | 45 |
| SVB025_BP | 1,753 | 246 | 164 |
| SVB025_SP | 246 | 246 | 52 |
| SVB026_BP | 2,209 | 394 | 919 |
| SVB026_SP | 394 | 394 | 96 |
| SVB029_BP | 580 | 580 | 256 |
| SVB029_SP | 892 | 580 | 255 |
| SVB030_BP | 1,259 | 1,201 | 378 |
| SVB030_SP | 1,201 | 1,201 | 247 |
| SVB039_BP | 2,234 | 1,193 | 351 |
| SVB039_SP | 1,193 | 1,193 | 191 |
| SVB041_BP | 1,121 | 912 | 267 |
| SVB041_SP | 912 | 912 | 140 |
| SVB043_BP | 707 | 707 | 185 |
| SVB043_SP | 1,165 | 707 | 113 |
| Total | 20,987 | 11,854 | 5,646 |
| Median (IQR) | 1,130 (547–1,355) | 514 (283–861) | 219 (133–302) |
| Median blood (IQR) | 1,451 (1,126–1,726) | 319 (259–599) | |
| Median semen (IQR) | 670 (283–1,102) | 127 (63–183) | |
BP, blood plasma; SP, semen plasma.
Statistical analysis reveals compartmentalization in all the study subjects.
We found evidence of compartmentalization in all 10 study participants (Fig. 2) using three statistical tests: Wright’s measure of population subdivision FST (39) (Fig. 2A), nearest neighbor statistic (Snn) (40) (Fig. 2B), and the Slatkin-Maddison test (SMT) (41) (Fig. 2C). A donor was identified as having compartmentalized blood and semen HIV-1 populations if all three tests yielded significant P values (<0.05). The P values were calculated either by using population-structure randomization (FST and Snn) or by permutations wherein the compartment designations of sequences were randomized (SMT) (42). As a negative control, we performed an SMT analysis with intermixed populations: 100 permutations per donor of the sequences from blood compartment randomly assigned to two equal-size groups, for a total of 1,000 tests. None of the tests resulted in a significant P value; the lowest P value encountered was 0.42, and 38/1,000 tests resulted in a P value of <0.9 (data not shown), all suggesting that false positives in intermixed populations are very rare.
FIG 2.

Various extents of compartmentalization were observed among all 10 participants that were studied using three standard techniques, two distance-based (Wright’s FST [A] and nearest neighbor statistic [B] tests) and one tree-based (Slatkin-Maddison test [C]). The x axis represents the 10 study subjects ranked by FST, P values were all significant (P < 0.001). SVB043 had the most compartmentalized viral populations between the blood and the genital compartments. Inferred migration events between blood and semen from Slatkin-Maddison test are indicated in panel C. Viral loads and CD4+ T cell counts did not detectably correlate with the extent of compartmentalization (D).
The degrees of compartmentalization varied among the donors (Fig. 2) with all three tests, with donor SVB043 showing the most highly compartmentalized blood and semen viral populations (Fig. 2). In addition, there was no association between the degree of compartmentalization and either blood viral load or CD4+ T cell count (Fig. 2D). We considered that semen sequences may have come from heterogenous sources within the genital tract, and that subsampling of the semen sequences might favor sequences from one or more particular anatomical source. To test this possibility, we resubsampled semen sequences 10 times from donors SVB029 and SVB043, the only two from whom we subsampled semen sequences (Table 2). We found that using different subsampled sequences did not change the FST, Snn or SMT coefficients. If the semen sequences were from heterogeneous sources and our subsampling favored a subset, this did not alter the overall compartmentalization result. Out of necessity, we selected samples with higher viral loads in semen in order to ensure successful sequencing. This also resulted in selecting samples with higher blood viral loads (Fig. 3).
FIG 3.
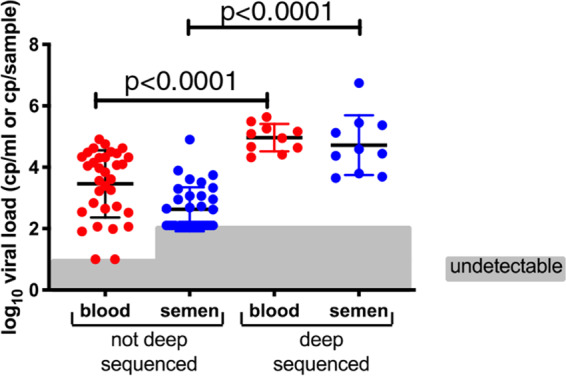
Selection of study participants for deep sequencing was based on higher viral load in semen, to help ensure successful deep sequencing. The subjects also had significantly higher viral loads in blood as well. Viral load in semen is reported per sample, while that in blood is reported as copies per milliliter of blood.
Some sequence clusters reveal compartmentalization, even in donors with minimal compartmentalization.
Visual inspection of phylogenetic trees has previously been used to evaluate the compartmentalization of HIV-1 populations between the blood and other anatomical sites (6, 7, 13, 16, 43). This relies on the guiding hypothesis that sequences from blood and the other anatomical compartments (in our case, semen), if distinct enough, would visibly cluster together within phylogenetic trees according to their anatomical compartment. However, it is difficult to identify compartmentalization in this way when only some of the variants in the tissue in question display evidence of compartmentalization (44, 45). Further, when large numbers of sequences generated by NGS are displayed, the geometry of the branches may be less clear by eye, especially with many clusters of nearly identical sequences.
We therefore determined the proportion of blood and semen sequences within clusters represented by 10 or more sequences. For donor SVB043, who displayed the most obvious phylogenetic evidence of compartmentalization (i.e., with minimal intermingling of sequences from blood and semen within the tree), we observed two main clusters: one containing only sequences from the blood and the other only sequences from semen (Fig. 4). However, for donors displaying less obvious evidence of compartmentalization, the largest clusters frequently contained closer to a 1:1 ratio of blood and semen sequences (Fig. 4). Interestingly, even for these donors, some phylogenetic clusters contained exclusively blood or semen sequences, suggesting that some variants are not homogeneously distributed between the two anatomical sites. These “nondistributed” variants could be due to local replication of the variants within one of the anatomical sites coupled from which they tend not to move or have not yet moved.
FIG 4.

Some variants were found primarily in one compartment, even in study participants with minimal compartmentalization. Shown are bubble plots for the study subjects who showed clonally amplified clusters at the terminal nodes of phylogenetic trees. The samples are arranged by the extent of blood versus genital tract viral compartmentalization, with SVB043 and SVB041 being the most and least compartmentalized donors, respectively. Each bubble represent a terminal node in a blood-semen phylogenetic tree carrying identical sequences, and the size represent the number of sequences present in that node. The y axis shows the relative composition of either blood or semen sequences in these node clusters, with red and blue representing proportions of blood and semen sequences, respectively. The x axis shows the sample IDs of the study participants whose HIV-1 populations were analyzed. Only clusters of 10 or more sequences were analyzed. Participants SVB021 and SVB026 did not have clusters containing 10 sequences or more, and their results are not shown.
Down-sampling simulations: modeling lower depth of sequencing from NGS data.
Previous studies that have tested for compartmentalization of HIV-1 between blood and the male genital tract (MGT) have collectively reported evidence of compartmentalization in an average of approximately 50% of study participants (10, 11, 13, 15, 16, 46). While it is possible that the donors we analyzed were more prone to displaying compartmentalization of HIV-1 populations between blood and semen than those analyzed in other studies, it is also plausible that the discrepancy may simply be a consequence of our study involving the analysis of a far greater number of sequences than those examined in these previous studies. To test the latter possibility, we randomly sampled 35 sequences from each compartment to simulate the SGA data sets used in these previous compartmentalization studies (with a relatively high number of sequences analyzed compared to those in these studies) and then retested for compartmentalization using FST (Table 3). The random subsampling was repeated 1,000 times, with FST scores being calculated for each of the 1,000 simulated SGA data sets obtained from each of the 10 donors. The proportion of the 1,000 simulated SGA data sets in which compartmentalization was detected for a given donor should be approximately the probability that compartmentalization would have been observed in that donor using an SGA-based approach.
TABLE 3.
Proportion of sequence subsamples that were compartmentalized by FST analysis, of 1,000 iterations
| Sample ID | Compartmentalized iterations (%)a |
|---|---|
| SVB043 | 99.1 |
| SVB008 | 99.5 |
| SVB021 | 50.0 |
| SVB026 | 72.6 |
| SVB012 | 15.8 |
| SVB025 | 6.9 |
| SVB030 | 27.1 |
| SVB029 | 17.6 |
| SVB039 | 8.1 |
| SVB041 | 0.1 |
A thousand random subsamples, each with 35 sequences per compartment, were generated from blood or semen of every study participant. Each subsample was analyzed for compartmentalization using FST, and the proportion of iterations that scored as compartmentalized is shown. Proportions over 50% are underlined.
We analyzed subsamples of 35 sequences per compartment in order to approximate an analysis without deep sequencing and considered it likely that an analysis with limited depth of sequencing would have detected compartmentalization if more than 50% of the subsamples were detectably compartmentalized. We found that this was true for only 4 out of the 10 study participants’ viral populations (Table 3). We concluded therefore that differences in sequencing depth could alone explain the fact that we found evidence of compartmentalization in 100% of our study participants while other studies found such evidence in only ∼50% of analyzed individuals.
Donors displaying high degrees of compartmentalization have viral populations in their blood that are more diverse than those in their semen.
Overall, we did not find differences in median pairwise genetic distances between viral sequences derived from blood and those derived from semen (P = 0.1250 [Fig. 5]). However, the HIV-1 populations of three of the donors displaying the highest degree of compartmentalization (SVB043, SVB008, and SVB021) displayed more genetic diversity in blood than in semen (Fig. 5).
FIG 5.

Overall there was no significant differences in median pairwise genetic distances between the variants from blood and those from the genital compartment (P = 0.125). However, three of the most compartmentalized sample pairs had substantially higher diversity in blood than in the genital compartment (SVB043, SVB008, and SVB021).
Most semen viral variants were derived from the blood compartment.
Maximum likelihood trees (Fig. 6) and maximum clade credibility (MCC; Bayesian) trees (Fig. 7) generally appeared to be rooted in the blood. This suggests that ancestral sequences from the original focus of infection likely established themselves in blood after transmission (assuming that the donor’s HIV-1 was acquired via sexual transmission) and then trafficked to the genital tract later, overwhelming any remaining foci of infection if they were there. This is further supported by evidence of back-and-forth movements of viruses between semen and blood (Fig. 2C) that were revealed by counting numbers of inferred minimum migration events obtained from the SMTs that we performed (41). The inferred migration events from these tests have previously been shown to correlate with Bayesian Markov jump counts (12). The minimum number of inferred migration events gathered from the SMTs showed that although there was bidirectional movement of viruses, there were more inferred migration events from blood into the genital compartment than vice versa (Fig. 2C) (P = 0.0098), presumably due to much larger absolute HIV-1 population sizes in blood than in semen.
FIG 6.

Bubble trees derived from RaxML maximum likelihood trees, showing clonal amplifications. The trees are arranged by the extent of compartmentalization as determined using FST. Clonal amplification was found in all the study participants and was a characteristic of both semen and blood compartments. The trees are rooted by HIV-1 subtype C consensus sequence of 2004. Sequences of ≥99% identity from the same compartment were collapsed into the bubbles.
FIG 7.

Maximum clade credibility (MCC) trees derived from a Bayesian analysis of the HIV-1 sequence sets from the donors. Red represents sequences from blood, while blue represents sequences from semen. The Bayesian analysis for SVB026 never converged, and no MCC tree could be generated.
Evidence of clonal amplification was found in all the study participants.
Clonal amplification was determined by collapsing the PID consensus sequences into haplotypes containing sequences that all shared >99% identity. Although this revealed anatomical-site-specific clonal amplification in all of the donors (Fig. 6), it also indicated that the extent of clonal amplification varied substantially between donors. For example, more than half of the semen sequences from SVB043, whose HIV-1 populations displayed a high degree of compartmentalization, appeared to have been derived from a single clone (Fig. 6A). Importantly, clonal amplification was found in both blood and semen populations and was observed both in donors displaying highly compartmentalized HIV-1 populations and donors displaying minimally compartmentalized HIV-1 populations.
The large clonal expansion in SVB043 arose at least partly from error-prone replication.
We considered the possibility that the very large apparent clonal amplification in the semen of SVB043 may have arisen for reasons independent of the HIV-1 sequences that this subject possessed, particularly due to proliferation of one or a few T cell clones harboring the corresponding HIV-1 genome or closely related genomes (47). If so, it would be unlikely that the sequences in the clonal amplification were selected in the male genital tract, based upon viral characteristics. To explore whether this clonal amplification arose because of expansion of HIV-1 genomes while integrated in proliferating T cells, we generated 10 full-length env clones by single-genome amplification (SGA)-based techniques (23, 48, 49). Of these 10 sequences (Fig. 8), 8 mapped within the large cluster using the region that was deep sequenced (all except clones SVB043_SGA_9 and SVB043_SGA_10). These eight full-length env sequences displayed notable sequence diversity (Fig. 8), suggesting that the sequences within this cluster likely arose at least partly due to error prone (i.e., viral) replication.
FIG 8.

Highlighter plot of 10 full-length HIV-1 envelope semen-derived sequences of donor SVB043. All except clones SVB043_SGA_9 and SVB043_SGA_10 were found in the large clonal amplification observed with the deep sequencing. The seminal sequences from donor SVB043 exhibit mutation most consistent with error-prone viral replication rather than expansion of virally infected antigen-specific T cells. The red rectangle represents the region of the envelope that was deep sequenced.
DISCUSSION
In this study, next-generation sequencing (NGS) was used for the first time to evaluate HIV-1 population compartmentalization between semen and blood during chronic HIV-1 infection. The use of PrimerID and high fidelity Taq polymerase allowed us to remove several sequencing artifacts which would have otherwise confounded subsequent analyses, thereby allowing us to study clonal amplification in more detail and with higher confidence than has been previously achievable. Accordingly, we detected convincing evidence of both HIV-1 population compartmentalization between blood and semen and clonal amplification in both body compartments in all of the subjects that were studied. Further, in the study participant with the most severely compartmentalized HIV-1 populations, there was a large apparent clonal amplification. Because the replication appeared error-prone when we analyzed full-length env clones that map within the cluster, we conclude that the cluster of closely related sequences that dominated the semen HIV-1 population did not expand simply because of proliferation of an infected T cell in the genital tract, which would not have been error-prone. Thus, there were opportunities for HIV-1 sequences to be selected in the genital tract due to viral characteristics. It also appears that analyzing only ∼400 bp of sequence can result in an underestimate of the mutation rate for the full-length gene.
There is no universally accepted standard for analyzing compartmentalization (42). We used three different statistical approaches (two distance based and one phylogenetic tree based), which all yielded the same conclusions: that HIV-1 sequences sampled from all 0 donors exhibited clear evidence of compartmentalization. This is in contrast with previous studies that showed compartmentalization in only ∼50% of examined individuals. Using SGA-based approaches or database sequences, several studies (10–16, 43, 46) showed an aggregate proportion of 55% of men living with HIV-1 having viral populations that displayed evidence of compartmentalization between blood and semen. Strikingly, we obtained a similar proportion of study participants displaying evidence of compartmentalized HIV-1 populations (4/10) when we used down-sampling simulations, i.e., we simulated SGA data sets based on subsampling of our NGS data (Table 3). Thus, the fundamental difference between our result (all donors displaying compartmentalization) and an aggregate of prior results (∼55% of donors displaying compartmentalization) can be explained by differences in sequencing depth. There is therefore no evidence that our donor population was different from previously analyzed populations.
It is noteworthy that one previous study using next-generation sequencing (Roche 454) to evaluate evidence of HIV-1 population compartmentalization between blood and semen found compartmentalization in only two of six donors at initial sampling (50). However, this study involved donors in the acute phase of infection, in which the viral variants are expected to be more homogeneous.
Klein et al. (51) found higher sequence diversity in the vaginal tracts of recently infected women living with HIV-1, suggesting that sequences from the initial HIV-1 inoculum persisted in the vaginal tract but that only a small proportion of the virions from the genital tract resulted in the disseminated HIV-1 infection. We observed no evidence for a similar effect in this study: sequence diversity was not higher in semen in any individual studied (Fig. 5), and the semen sequences were generally embedded within clusters of blood sequences in phylogenetic trees (Fig. 6 and 7). Additionally, viral loads in the semen correlated with the viral loads in the much larger blood/circulation compartment (Fig. 1). In this case, we do not know the route of infection. The clinic at which we recruited attracted many men who have sex with men (MSM), and they have a high rate of intravenous drug use: 80% within the past 3 months in one survey (52). We therefore cannot rule out inoculation of HIV-1 by the rectal or intravenous route, and in such cases, seeding of the male genital tract would presumably be via the general circulation. If there are cases in which the initial inoculation was via the male genital tract, any sequences from the original inoculum would presumably have been overwhelmed by blood-derived imports before we collected our samples.
Clonal amplification is an indicator of local and disproportional replication of one viral variant within a virus population. Clonal amplification has been reported for various anatomical sites in the body, including cerebrospinal fluid (CSF) (53), breast milk (54–56), and the genital tract (10). Deep sequencing and utilization of a PrimerID approach helped us to study clonal amplification in greater detail than has previously been reported for semen. We found evidence of clonal amplification in both the blood and semen of all 10 individuals whose HIV-1 populations were sequenced (Fig. 6). Interestingly, these clonally amplified variants exhibited relatively limited movement between compartments (Fig. 4). This was even true in donors displaying the lowest degrees of HIV-1 population compartmentalization between the blood and semen (Fig. 2).
There are several non-mutually exclusive explanations for the observed compartmentalization of clonally amplified variants. First, variants may have been amplified in a secluded physical compartment with limited access to blood flow. Second, certain variants in blood or the genital tract may be selected for more efficiently in one compartment, leading to a paucity of variants that are least adapted to the respective compartment. Third, it is possible that the most recent common ancestors of the clonally amplified variants may have existed so recently that few of its descendants had sufficient time to move between the sampled anatomical sites.
Clonally amplified viral variants may arise as a consequence of increased replicative fitness of particular variants with a given anatomical compartment. If the anatomical compartment is the male genital tract, then both the abundance and increased fitness of such variants might increase their probability of being transmitted to a new host. In support of this idea, one study has shown that clonally amplified drug-resistant HIV-1 variants in breast milk tend to be those which are transmitted from lactating mothers to their infants (54). We considered the possibility that clonal amplifications may have arisen primarily due to proliferation of T cell clones that have integrated HIV-1 genomes (47). Even if so, the cells producing virus and the resulting viral sequences did not generally move from blood to semen, at least in the time frame from their production to the collection of the sample. Had movement between the compartments been free in some donors, we ought not to have detected the compartmentalization of these clones. However, it is clear that even very large apparent clonal expansions in the male genital tract, such as that in SVB043 (Fig. 6A), can arise from error-prone viral replication (Fig. 8), suggesting that sequences therein could have been selected based upon viral characteristics (57, 58).
We were unable to detect an association between history of sexually transmitted infections (STIs) and CD4+ T cell counts, blood or semen viral load, or viral diversity or extent of HIV-1 population compartmentalization. Other studies that have reported increased shedding of virus in the genital tract when a donor is concomitantly infected with an STI (59–61) and that treatment of STI reduces the viral shedding in the genital tract (61–63). Our small sample size may not have been sufficient to identify any such association.
In summary, this is the first study to probe HIV-1 compartmentalization between the systemic circulation and the male genital tract in chronically infected individuals utilizing deep sequencing. In contrast to previous studies, this investigation showed that compartmentalization was present in all individuals studied. In addition, deep sequencing revealed clonal amplification in blood in a higher proportion of donors than has been previously reported. Both apparent discrepancies are likely due to the increased sensitivity to detect these phenomena that is enabled by deep sequencing. The extent of compartmentalization was not obviously related to CD4+ T cell counts, viral loads, or recent history of sexually transmitted infections. This information furthers our understanding of how HIV-1 populations in the male genital tract are shaped prior to sexual transmission.
MATERIALS AND METHODS
Ethics statement.
This study was approved by the Human Research Ethics Committee of the Faculty of Health Sciences of the University of Cape Town (HREC 727/2014). All the study participants provided written informed consent before interview and sample collection. All study participants were offered antiretroviral therapy during the recruitment process in line with the test-and-treat guidelines instituted nationally in South Africa during the study period.
Study participants and sample collection.
HIV-1-positive, antiretroviral therapy-naive study participants were approached for recruitment at ANOVA Health’s Ivan Tom Health4Men clinic in Woodstock or Green Point, or at their satellite clinic in Khayelitsha, all in Cape Town, South Africa. Those who expressed interest were given an appointment to meet with study staff for consent and sample collection in the clinic while it was otherwise closed. Study recruitment and sample collection occurred from June 2015 to January 2017. In total, blood and semen samples were collected from 43 ART-naive individuals. Each study participant’s age, nationality, and racial group information was collected. In addition, history of sexually transmitted infection was also obtained from the participant’s medical records and also through oral interviews with a clinician. The reported date of the study participant’s last negative HIV-1 test was also recorded. The study participant characteristics are described in Table 1.
DNA extractions and cDNA synthesis.
Viral RNA was extracted from blood plasma and semen plasma samples using the QIAamp viral RNA minikit (Qiagen). The extracted RNA was reverse transcribed into cDNA using the SuperScript III First Strand system for reverse transcription-PCR (RT-PCR) (Invitrogen). The cDNA gene-specific long primers were used at a final concentration of 0.25 μM. The cDNA long primer was comprised of a gene-specific region of interest (C3-V5 or V3) at the 3′ prime end, a 15-random-nucleotide stretch (PrimerID), and a site for binding the universal primer at the 5′ end. The cDNA primers sequences that were used in each case are in Table 4. The mixture contained 0.5 mM (each) deoxynucleotide triphosphates (dNTPs). First, the extracted RNA was heated to 65°C for 5 min to denature the RNA secondary structures. This was followed by addition of 35 μl of a mixture made of 1× of 5× buffer, 5 mM dithiothreitol (DTT), 2 U/μl of RNase inhibitor, and 10 U/μl of Superscript III reverse transcriptase. The reaction mixture was then incubated at 45°C and then at 50°C for an hour each. To deactivate the enzyme, the mixture was incubated at 70°C for 15 min. The synthesized cDNA was removed from the DNA-RNA hybrid by addition of 1 μl of 5-U/μl Rnase H and incubation at 37°C for 20 min. The cDNA was then purified from unused primers and nucleotides using Agencourt RNAClean XP beads (Beckman Coulter). The beads were washed 4 times with 70% ethanol, and DNA was eluted in 50 μl of RNase-free water.
TABLE 4.
Primers used for amplification for the miSeq library and for SGA-based PCR amplification of full-length env
| Primer name | Sequence (5′–3′) | Purpose | Region |
|---|---|---|---|
| Env_C5_cDNA | GAGATGTGTATAAGAGACAGNNNNNNNNNNNNNNNGTCCYTCATATYTCCTCCTYCAGG | cDNA primer | C3-V-5 |
| Env_V3_F | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGNNNNNNNGGACCAGGGAGAGCATTTGTTAC | Forward primer | C3-V5 |
| Env_C3_cDNA | GAGATGTGTATAAAGAGACAGNNNNNNNNNNNNNNNTGTGTTGTAAYTTCTAGRTC | cDNA primer | V3 |
| Env_C2_F | TGGACGGCAGCGTCAGATGTGTATAAGAGACAGNNNNNNNGCTGGTTATGCGATTGTAAACTG | Forward primer | V3 |
| Univ_i7_Rev | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG | Reverse primer | Universal reverse |
| OFM19 | GCACTCAAGGCAAGCTTTATTGAGGCTTA | Reverse primer | full env |
| VIF-1 | GGGTTTATTACAGGGACAGCAGAG | Forward primer | full env |
| ENV-N | CTGCCAATCAGGGAAGTAGCCTTGTGT | Reverse primer | Full env |
| ENV-A | GGCTTAGGCATCTCCTATGGCAGGAAGAA | Forward primer | Full env |
Amplification of the region of interest.
The PrimerID-labeled cDNA obtained was divided into 5-μl aliquots and used for the first-round PCR amplification using Platinum HIFI Taq polymerase (Life Technologies). First-round PCR for the C3-V5 or V3 region was done using forward primers env_V3_subC_F and env_C2_subC_F (Table 4). A universal reverse primer (Univ_i7_REV [Table 4]) which binds to the primer binding site previously introduced by the cDNA long primer during the cDNA synthesis was used. The PCR mixture contained final concentrations of 10× buffer, 2 mM MgSO4, 0.5 mM (each) dNTPs and 0.2 μM (each) forward and reverse primers. The reaction mixture was incubated at 94°C for 2 min, followed by 25 cycles of 94°C for 30 s, 58°C for 15 s, and 68°C for 2 min. A final extension step was done at 68°C for 10 min.
The first-round PCR products were then purified using SPRISelect beads (Beckman Coulter), with a bead-to-PCR product volume ratio of 0.6. The beads were then washed 3 times using 80% ethanol, and DNA was eluted in 50 μl of nuclease-free water.
Indexing PCR.
Indexing PCR was performed using 5 μl of the first-round product. The indexing primers contained a 6-nucleotide-long index region complementary to the Illumina sequencing adapters fixed on the sequencing chip. To allow multiplexing of the samples in the same Miseq sequencing run, different combinations of various indexing primers were used per sample. Our sequenced region was designed to exploit paired-end sequencing without information being lost at the intertwining region between first (R1) and second (R2) reads. The PrimerID region was designed to always be sequenced at the second end (R2). The PCR mixture contained 1× of 10× buffer, 2 mM MgSO4, 0.5 mM (each) dNTPs, and a 0.2 μM conecntration of each primer. We used Platinum HIFI Taq polymerase (Invitrogen) for the PCRs. The cycling conditions involved an initial temperature of 94°C for 2 min followed by 25 cycles at 94°C for 30 s of denaturation, 58°C for 15 s of annealing, and 68°C for 2 min of extension. Final extension was done at 68°C for 10 min, followed by holding at 4°C. The indexed PCR products were then bead-purified using SPRISelect beads and washed three times using 80% ethanol.
Single genome amplification.
Single genome sequences were obtained by utilizing the endpoint dilution technique and sequencing (23, 49). PCR amplification was done using Platinum HIFI Taq polymerase to keep the PCR mismatches as low as was practical.
Illumina Miseq sequencing.
The size of the C3-V5 env amplicon was approximately 423 bp, covering HXB2 positions 7179 to 7602 (64) on the HIV-1 genome, while that of V3 region was approximately 416 bp, covering positions 6909 to 7325 of HXB2. We used 2 × 300-bp paired-end multiplex Illumina Miseq to sequence the constructed libraries.
Template consensus pipeline.
An in-house pipeline (available at https://github.com/HIVDiversity/NGS_processing_pipeline) was used to process the fastq format files into PID consensus sequences. PID consensus sequences were made by collapsing all sequences with the same PrimerID motif into a consensus sequence, using a “majority vote” rule at each nucleotide position. The pipeline utilized tools for assessing the quality of the read data from both R1 and R2 reads supplied. The reads that did not match the required read lengths and sequences that contained more than 2% ambiguous nucleotides or obvious primer dimers were discarded. The numbers of consensus PID sequences obtained after employing the pipeline are summarized in Table 2.
Bayesian phylogenetic analysis.
From blood and semen PrimerID consensus sequences, unique sequences were constructed using an in-house hyplotyper tool, this time allowing 100% similarity. The blood and semen unique sequences were then combined into one fasta file. Alignments were done using MAFFT (65) and inspected and manually edited in Aliview (66). Bayesian Evolutionary Analysis by Sampling Trees (BEAST 1) software (67) was then used to perform Bayesian inference to resolve the phylogenies. Briefly, five replicates each with 100 million generations were run from the same XML file generated in BEAUTI. Each blood-semen pair alignment was run under the GTR model of nucleotide substitution. The rest of the parameters were left as defaults, i.e., a strict clock and assuming a fixed population size for the tree priors. The trees were sampled at every 10,000th tree and convergence was inspected in Tracer (68) with an expected sample size (ESS) of above 200. If any replicate had achieved this convergence, we only took the trees sampled in this replicate to construct the maximum clade credibility (MCC) tree using the TreeAnnotator. If none of the replicates had achieved convergence after running 100 million generations, we combined the logs of the 5 replicates using LogCombiner and resampled the trees logs at a lower frequency (i.e., 50,000th tree log). We then checked in Tracer whether convergence was achieved. If so, the same sampling of the trees was performed at the same frequency to construct the MCC trees. Trees were visualized in FigTree (69).
Compartmentalization analysis.
First, we selected the same number of sequences from blood and semen to minimize intercompartment sampling bias. For the compartment of each donor with the smaller number of PID sequences, all were used, and for the other compartment, the same number was chosen at random. Compartmentalization analyses was then done using three statistical tools: two distance-based methods, Wright’s measure of population subdivision (FST) (42, 70) and nearest neighbor statistic (Snn) (40), and one tree-based method, the Slatkin-Maddison test (41). These tests were all implemented in HYPHY (70). The FST computes mean genetic distances between and within compartments using Tamura-Nei-93 algorithm and computes a ratio, F-statistic (FST). The closer to 1 the FST is, the more compartmentalized the populations are. Nearest neighbor is another distance-based method that computes a ratio between the observed mean genetic distances and compares them to expected ones in a hypothetical randomly distributed neighbor. On the other hand, SMT is a tree-based technique that counts the number of minimum migration events from one compartment to the other and compares this with the number of migration events expected in a fully mixed population. This random distribution set was made from the 1,000 permutations. Additionally, in order to query any possible effects from subsampling the sequences from the compartment with more sequences, we resampled the larger compartment 10 times for each donor and repeated the FST analysis.
In order to detect any bias inherent in the tree structure, we subjected each donor’s tree sequences to a randomized tip swap (71, 72), i.e., random assignment of sequences to compartment A or B in the same proportions as were present for the blood and semen compartments. None of the trees exhibited detectable compartmentalization after being subjected to this randomized tip swap, indicating that it is unlikely that positive scores were due to bias in the tree geometry (data not shown).
Clonal amplification analysis.
To evaluate clonal amplification, the PrimerID consensus sequences were first collapsed into haplotypes with the frequency of the sequences used tagged to the name of the sequence. This was made using Vsearch (73), clustering the sequences at 0.99 identity. Alignment was done using MAFFT (65). Phylogenetic trees were then constructed using RaxML (74). Bubble trees were plotted with the python library ete3 (75), with the size of the bubble corresponding to the number of sequences found in the haplotype relative to the total PID consensus sequences per compartment. Sequences were collapsed into single bubbles if they were from the same compartment and were ≥99% identical.
To study the distribution of variants in the blood and genital compartments, phylogenetic trees were first constructed from the PID consensus sequences using RaxML. An R-based package (Analyses of Phylogenetics and Evolution [APE]) (76) was used to extract the terminal node leaves of the phylogenetic trees. The numbers of blood and semen sequences from nodes with 10 or more sequences were then counted and tabulated in bubble plots.
Diversity analysis.
To analyze the HIV-1 diversity between the blood and the male genital compartments, blood and semen unique sequences were first separately constructed from PrimerID consensus sequences with 100% similarity. The median pairwise genetic distances between variants from blood and semen were determined using Molecular Evolutionary Genetics Analysis (MEGA) version 7 (77). The differences in diversity between blood and semen were compared using the Wilcoxon paired signed-rank test.
Movement of viruses between blood and semen.
We estimated the minimum number of viruses moving between the blood and semen compartments after normalizing for equal numbers of sequences using the Slatkin-Maddison test, which allows counting of the number of inferred migration events from blood to semen and vice versa. The difference between the migration events from blood to semen and semen to blood were analyzed using the nonparametric Wilcoxon signed-rank test. The inferred migration events from SMT have previously been shown to correlate with Bayesian Markov jump counts (12).
SGA of full-length env clones.
The single-genome amplification (SGA) technique was used to amplify single genomes from cDNA generated from blood and semen. First-stage PCRs (35 cycles) were performed in a 20-μl reaction mixture containing buffer, 2 mM MgSO4, 0.2 mM (each) dNTPs, a 0.2 μM concentration of each primer, and 0.025 U/μl of Platinum HIFI Taq. The first-round primers were forward primer VIF-1 (5′- GGGTTTATTACAGGGACAGCAGAG-3′) and reverse primer OFM19 (5′- GCACTCAAGGCAAGCTTTATTGAGGCTTA-3′), representing HXB2 positions 4903 to 4923 and 9604 to 9632, respectively. Two microliters of the first-round PCR product was amplified in a second-stage PCR (45 cycles) using the following primers: forward primer ENV-A (5′- GGCTTAGGCATCTCCTATGGCAGGAAGAA-3′) and reverse primer ENV-N (5′- CTGCCAATCAGGGAAGTAGCCTTGTGT-3′), representing HXB2 positions 5954 to 5982 and 9145 to 9171, respectively. This generated an amplicon of approximately 3 kb. This was done by serially diluting the cDNA templates and distributing the dilutions in a 96-well plate, followed by the first and second stages of the nested PCR. We used the dilution of cDNA that resulted in less than 30% positive wells from the total reactions after gel electrophoresis was targeted. Assuming a Poisson distribution of cDNA templates, when 30% of wells are positive, the likelihood that the PCR product in any one positive well was amplified from only one amplified template is approximately 84% (23, 48, 49). Use of a single template was confirmed to the extent possible by sequencing of the amplicons and checking for mixed bases from the chromatograms. Clones were obtained from several independent PCRs for each sample to minimize the sampling bias. The PCR amplifications were performed using Platinum HIFI Taq (Invitrogen) to keep the likelihood of PCR mismatch errors as low as was practical.
Data availability.
Sequences of full-length env clones may be found in GenBank under accession numbers MT260117 to MT260126. MiSeq sequence data are available under BioProject accession number PRJNA613850.
ACKNOWLEDGMENTS
Author contributions are as follows: conception of the study, J.R.D., S.M.K., P.S., K.K.A., and K.R.; participant recruitment, participant interviews, and sample handling, K.R.; sample processing and performance of the experiments, S.M.K.; design of the data analysis approaches, J.R.D., C.W., C.A., D.M., D.P.M., and S.M.K.; analysis and interpretation of the data, S.M.K., P.S., C.A., D.M., M.-R.A., D.P.M., K.K.A., and J.R.D.; writng of the first draft of the manuscript, J.R.D., S.M.K., and P.S.; editing of the manuscript, all authors.
We thank the donors for this project. We thank Sandra Tshisa, Louise Suka, and Johan Human for help with recruitment and study participant interviews. We thank Leo Heyndrickx for technical support and advice. Computations were performed using facilities provided by the University of Cape Town’s ICTS High Performance Computing Team (hpc.uct.ac.za).
We thank the South Africa National Research Foundation, the Poliomyelitis Research Foundation, and the International Centre for Genetic Engineering and Biotechnology for financial support. K.K.A. is supported by the Fund for Scientific Research Flanders (FWO).
REFERENCES
- 1.Royce RA, Sena A, Cates W Jr, Cohen MS. 1997. Sexual transmission of HIV. N Engl J Med 336:1072–1078. doi: 10.1056/NEJM199704103361507. [DOI] [PubMed] [Google Scholar]
- 2.Long E, Martin H, Kreiss J, Rainwater S, Lavreys L, Jackson D, Rakwar J, Mandaliya K, Overbaugh J. 2000. Gender differences in HIV-1 diversity at time of infection. Nat Med 6:71–75. doi: 10.1038/71563. [DOI] [PubMed] [Google Scholar]
- 3.Learn GH, Muthui D, Brodie SJ, Zhu T, Diem K, Mullins JI, Corey L. 2002. Virus population homogenization following acute human immunodeficiency virus type 1 infection. J Virol 76:11953–11959. doi: 10.1128/jvi.76.23.11953-11959.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ritola K, Pilcher CD, Fiscus SA, Hoffman NG, Nelson JA, Kitrinos KM, Hicks CB, Eron JJ Jr, Swanstrom R. 2004. Multiple V1/V2 env variants are frequently present during primary infection with human immunodeficiency virus type 1. J Virol 78:11208–11218. doi: 10.1128/JVI.78.20.11208-11218.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poss M, Martin HL, Kreiss JK, Granville L, Chohan B, Nyange P, Mandaliya K, Overbaugh J. 1995. Diversity in virus populations from genital secretions and peripheral blood from women recently infected with human immunodeficiency virus type 1. J Virol 69:8118–8122. doi: 10.1128/JVI.69.12.8118-8122.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta P, Leroux C, Patterson BK, Kingsley L, Rinaldo C, Ding M, Chen Y, Kulka K, Buchanan W, McKeon B, Montelaro R. 2000. Human immunodeficiency virus type 1 shedding pattern in semen correlates with the compartmentalization of viral quasi species between blood and semen. J Infect Dis 182:79–87. doi: 10.1086/315644. [DOI] [PubMed] [Google Scholar]
- 7.Ghosn J, Viard JP, Katlama C, de Almeida M, Tubiana R, Letourneur F, Aaron L, Goujard C, Salmon D, Leruez-Ville M, Rouzioux C, Chaix ML. 2004. Evidence of genotypic resistance diversity of archived and circulating viral strains in blood and semen of pre-treated HIV-infected men. AIDS 18:447–457. doi: 10.1097/00002030-200402200-00011. [DOI] [PubMed] [Google Scholar]
- 8.Meyerhans A, Vartanian JP, Wain-Hobson S. 1990. DNA recombination during PCR. Nucleic Acids Res 18:1687–1691. doi: 10.1093/nar/18.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gianella S, Mehta SR, Strain MC, Young JA, Vargas MV, Little SJ, Richman DD, Kosakovsky Pond SL, Smith DM. 2012. Impact of seminal cytomegalovirus replication on HIV-1 dynamics between blood and semen. J Med Virol 84:1703–1709. doi: 10.1002/jmv.23398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson JA, Ping LH, Dibben O, Jabara CB, Arney L, Kincer L, Tang Y, Hobbs M, Hoffman I, Kazembe P, Jones CD, Borrow P, Fiscus S, Cohen MS, Swanstrom R, Center for HIV/AIDS Vaccine Immunology. 2010. HIV-1 populations in semen arise through multiple mechanisms. PLoS Pathog 6:e1001053. doi: 10.1371/journal.ppat.1001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diem K, Nickle DC, Motoshige A, Fox A, Ross S, Mullins JI, Corey L, Coombs RW, Krieger JN. 2008. Male genital tract compartmentalization of human immunodeficiency virus type 1 (HIV). AIDS Res Hum Retroviruses 24:561–571. doi: 10.1089/aid.2007.0115. [DOI] [PubMed] [Google Scholar]
- 12.Chaillon A, Gianella S, Wertheim JO, Richman DD, Mehta SR, Smith DM. 2014. HIV migration between blood and cerebrospinal fluid or semen over time. J Infect Dis 209:1642–1652. doi: 10.1093/infdis/jit678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curran R, Ball JK. 2002. Concordance between semen-derived HIV-1 proviral DNA and viral RNA hypervariable region 3 (V3) envelope sequences in cases where semen populations are distinct from those present in blood. J Med Virol 67:9–19. doi: 10.1002/jmv.2186. [DOI] [PubMed] [Google Scholar]
- 14.Butler DM, Delport W, Kosakovsky Pond SL, Lakdawala MK, Cheng PM, Little SJ, Richman DD, Smith DM. 2010. The origins of sexually transmitted HIV among men who have sex with men. Sci Transl Med 2:18re11. doi: 10.1126/scitranslmed.3000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boeras DI, Hraber PT, Hurlston M, Evans-Strickfaden T, Bhattacharya T, Giorgi EE, Mulenga J, Karita E, Korber BT, Allen S, Hart CE, Derdeyn CA, Hunter E. 2011. Role of donor genital tract HIV-1 diversity in the transmission bottleneck. Proc Natl Acad Sci U S A 108:E1156–E1163. doi: 10.1073/pnas.1103764108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown RJ, Peters PJ, Caron C, Gonzalez-Perez MP, Stones L, Ankghuambom C, Pondei K, McClure CP, Alemnji G, Taylor S, Sharp PM, Clapham PR, Ball JK. 2011. Intercompartmental recombination of HIV-1 contributes to env intrahost diversity and modulates viral tropism and sensitivity to entry inhibitors. J Virol 85:6024–6037. doi: 10.1128/JVI.00131-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmer S, Kearney M, Maldarelli F, Halvas EK, Bixby CJ, Bazmi H, Rock D, Falloon J, Davey RT Jr, Dewar RL, Metcalf JA, Hammer S, Mellors JW, Coffin JM. 2005. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J Clin Microbiol 43:406–413. doi: 10.1128/JCM.43.1.406-413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson JR, Marcelino LA, Polz MF. 2002. Heteroduplexes in mixed-template amplifications: formation, consequence and elimination by ‘reconditioning PCR’. Nucleic Acids Res 30:2083–2088. doi: 10.1093/nar/30.9.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu SL, Rodrigo AG, Shankarappa R, Learn GH, Hsu L, Davidov O, Zhao LP, Mullins JI. 1996. HIV quasispecies and resampling. Science 273:415–416. doi: 10.1126/science.273.5274.415. [DOI] [PubMed] [Google Scholar]
- 20.Schnell G, Joseph S, Spudich S, Price RW, Swanstrom R. 2011. HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog 7:e1002286. doi: 10.1371/journal.ppat.1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sturdevant CB, Dow A, Jabara CB, Joseph SB, Schnell G, Takamune N, Mallewa M, Heyderman RS, Van Rie A, Swanstrom R. 2012. Central nervous system compartmentalization of HIV-1 subtype C variants early and late in infection in young children. PLoS Pathog 8:e1003094. doi: 10.1371/journal.ppat.1003094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ping LH, Joseph SB, Anderson JA, Abrahams MR, Salazar-Gonzalez JF, Kincer LP, Treurnicht FK, Arney L, Ojeda S, Zhang M, Keys J, Potter EL, Chu H, Moore P, Salazar MG, Iyer S, Jabara C, Kirchherr J, Mapanje C, Ngandu N, Seoighe C, Hoffman I, Gao F, Tang Y, Labranche C, Lee B, Saville A, Vermeulen M, Fiscus S, Morris L, Karim SA, Haynes BF, Shaw GM, Korber BT, Hahn BH, Cohen MS, Montefiori D, Williamson C, Swanstrom R, CAPRISA Acute Infection Study and the Center for HIV-AIDS Vaccine Immunology Consortium. 2013. Comparison of viral Env proteins from acute and chronic infections with subtype C human immunodeficiency virus type 1 identifies differences in glycosylation and CCR5 utilization and suggests a new strategy for immunogen design. J Virol 87:7218–7233. doi: 10.1128/JVI.03577-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S, Manigart O, Mulenga J, Anderson JA, Swanstrom R, Haynes BF, Athreya GS, Korber BT, Sharp PM, Shaw GM, Hahn BH. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol 82:3952–3970. doi: 10.1128/JVI.02660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Archer J, Weber J, Henry K, Winner D, Gibson R, Lee L, Paxinos E, Arts EJ, Robertson DL, Mimms L, Quinones-Mateu ME. 2012. Use of four next-generation sequencing platforms to determine HIV-1 coreceptor tropism. PLoS One 7:e49602. doi: 10.1371/journal.pone.0049602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Archer J, Braverman MS, Taillon BE, Desany B, James I, Harrigan PR, Lewis M, Robertson DL. 2009. Detection of low-frequency pretherapy chemokine (CXC motif) receptor 4 (CXCR4)-using HIV-1 with ultra-deep pyrosequencing. AIDS 23:1209–1218. doi: 10.1097/QAD.0b013e32832b4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swenson LC, Mo T, Dong WW, Zhong X, Woods CK, Jensen MA, Thielen A, Chapman D, Lewis M, James I, Heera J, Valdez H, Harrigan PR. 2011. Deep sequencing to infer HIV-1 co-receptor usage: application to three clinical trials of maraviroc in treatment-experienced patients. J Infect Dis 203:237–245. doi: 10.1093/infdis/jiq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsibris AM, Korber B, Arnaout R, Russ C, Lo CC, Leitner T, Gaschen B, Theiler J, Paredes R, Su Z, Hughes MD, Gulick RM, Greaves W, Coakley E, Flexner C, Nusbaum C, Kuritzkes DR. 2009. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One 4:e5683. doi: 10.1371/journal.pone.0005683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dybowski JN, Heider D, Hoffmann D. 2010. Structure of HIV-1 quasi-species as early indicator for switches of co-receptor tropism. AIDS Res Ther 7:41. doi: 10.1186/1742-6405-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bunnik EM, Swenson LC, Edo-Matas D, Huang W, Dong W, Frantzell A, Petropoulos CJ, Coakley E, Schuitemaker H, Harrigan PR, van ‘T Wout AB. 2011. Detection of inferred CCR5- and CXCR4-using HIV-1 variants and evolutionary intermediates using ultra-deep pyrosequencing. PLoS Pathog 7:e1002106. doi: 10.1371/journal.ppat.1002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raymond S, Saliou A, Nicot F, Delobel P, Dubois M, Cazabat M, Sandres-Saune K, Marchou B, Massip P, Izopet J. 2011. Frequency of CXCR4-using viruses in primary HIV-1 infections using ultra-deep pyrosequencing. AIDS 25:1668–1670. doi: 10.1097/QAD.0b013e3283498305. [DOI] [PubMed] [Google Scholar]
- 31.van Zyl G, Bale MJ, Kearney MF. 2018. HIV evolution and diversity in ART-treated patients. Retrovirology 15:14. doi: 10.1186/s12977-018-0395-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seifert D, Di Giallonardo F, Töpfer A, Singer J, Schmutz S, Günthard HF, Beerenwinkel N, Metzner KJ. 2016. A comprehensive analysis of primer IDs to study heterogeneous HIV-1 populations. J Mol Biol 428:238–250. doi: 10.1016/j.jmb.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 33.Zhou S, Jones C, Mieczkowski P, Swanstrom R. 2015. Primer ID validates template sampling depth and greatly reduces the error rate of next-generation sequencing of HIV-1 genomic RNA populations. J Virol 89:8540–8555. doi: 10.1128/JVI.00522-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keys JR, Zhou S, Anderson JA, Eron JJ Jr, Rackoff LA, Jabara C, Swanstrom R. 2015. Primer ID informs next-generation sequencing platforms and reveals preexisting drug resistance mutations in the HIV-1 reverse transcriptase coding domain. AIDS Res Hum Retroviruses 31:658–668. doi: 10.1089/AID.2014.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. 2011. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci U S A 108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boltz VF, Rausch J, Shao W, Hattori J, Luke B, Maldarelli F, Mellors JW, Kearney MF, Coffin JM. 2016. Ultrasensitive single-genome sequencing: accurate, targeted, next generation sequencing of HIV-1 RNA. Retrovirology 13:87. doi: 10.1186/s12977-016-0321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Görzer I, Guelly C, Trajanoski S, Puchhammer-Stöckl E. 2010. The impact of PCR-generated recombination on diversity estimation of mixed viral populations by deep sequencing. J Virol Methods 169:248–252. doi: 10.1016/j.jviromet.2010.07.040. [DOI] [PubMed] [Google Scholar]
- 38.Kariuki SM, Selhorst P, Norman J, Cohen K, Rebe K, Williamson C, Dorfman JR. 2020. Detectable viral loads in semen in individuals with very low blood viral loads. Virol J 17:29–33. doi: 10.1186/s12985-020-01300-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hudson RR, Slatkin M, Maddison WP. 1992. Estimation of levels of gene flow from DNA sequence data. Genetics 132:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudson RR, Boos DD, Kaplan NL. 1992. A statistical test for detecting geographic subdivision. Mol Biol Evol 9:138–151. doi: 10.1093/oxfordjournals.molbev.a040703. [DOI] [PubMed] [Google Scholar]
- 41.Slatkin M, Maddison WP. 1989. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 123:603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zárate S, Pond SL, Shapshak P, Frost SD. 2007. Comparative study of methods for detecting sequence compartmentalization in human immunodeficiency virus type 1. J Virol 81:6643–6651. doi: 10.1128/JVI.02268-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paranjpe S, Craigo J, Patterson B, Ding M, Barroso P, Harrison L, Montelaro R, Gupta P. 2002. Subcompartmentalization of HIV-1 quasispecies between seminal cells and seminal plasma indicates their origin in distinct genital tissues. AIDS Res Hum Retroviruses 18:1271–1280. doi: 10.1089/088922202320886316. [DOI] [PubMed] [Google Scholar]
- 44.Blackard JT. 2012. HIV compartmentalization: a review on a clinically important phenomenon. Curr HIV Res 10:133–142. doi: 10.2174/157016212799937245. [DOI] [PubMed] [Google Scholar]
- 45.Houzet L, Perez-Losada M, Matusali G, Deleage C, Dereuddre-Bosquet N, Satie AP, Aubry F, Becker E, Jegou B, Le Grand R, Keele BF, Crandall KA, Dejucq-Rainsford N. 2018. Seminal simian immunodeficiency virus in chronically infected cynomolgus macaques is dominated by virus originating from multiple genital organs. J Virol 92:e00133-18. doi: 10.1128/JVI.00133-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams-Wietzikoski CA, Campbell MS, Payant R, Lam A, Zhao H, Huang H, Wald A, Stevens W, Gray G, Farquhar C, Rees H, Celum C, Mullins JI, Lingappa JR, Frenkel LM. 2019. Comparisons of human immunodeficiency virus type 1 envelope variants in blood and genital fluids near the time of male-to-female transmission. J Virol 93:e01769-18. doi: 10.1128/JVI.01769-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hosmane NN, Kwon KJ, Bruner KM, Capoferri AA, Beg S, Rosenbloom DI, Keele BF, Ho YC, Siliciano JD, Siliciano RF. 2017. Proliferation of latently infected CD4(+) T cells carrying replication-competent HIV-1: potential role in latent reservoir dynamics. J Exp Med 214:959–972. doi: 10.1084/jem.20170193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abrahams MR, Center for HIV-AIDS Vaccine Immunology Consortium, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping LH, Athreya GS, Treurnicht FK, Keele BF, Wood N, Salazar-Gonzalez JF, Bhattacharya T, Chu H, Hoffman I, Galvin S, Mapanje C, Kazembe P, Thebus R, Fiscus S, Hide W, Cohen MS, Karim SA, Haynes BF, Shaw GM, Hahn BH, Korber BT, Swanstrom R, Williamson C. 2009. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-Poisson distribution of transmitted variants. J Virol 83:3556–3567. doi: 10.1128/JVI.02132-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A 105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaillon A, Smith DM, Vanpouille C, Lisco A, Jordan P, Caballero G, Vargas M, Gianella S, Mehta SR. 2017. HIV trafficking between blood and semen during early untreated HIV infection. J Acquir Immune Defic Syndr 74:95–102. doi: 10.1097/QAI.0000000000001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klein K, Nickel G, Nankya I, Kyeyune F, Demers K, Ndashimye E, Kwok C, Chen PL, Rwambuya S, Poon A, Munjoma M, Chipato T, Byamugisha J, Mugyenyi P, Salata RA, Morrison CS, Arts EJ. 2018. Higher sequence diversity in the vaginal tract than in blood at early HIV-1 infection. PLoS Pathog 14:e1006754. doi: 10.1371/journal.ppat.1006754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Semugoma NP, Rebe K, Sonderup MW, Kamkeumah M, De Swardt G, Struthers H, Eksen H, McIntyre J. 2017. Hepatitis C: a South African literature review and results from a burden of disease study among a cohort of drug-using men who have sex with men in Cape Town, South Africa. S Afr Med J 107:1116–1120. doi: 10.7196/SAMJ.2017.v107i12.12623. [DOI] [PubMed] [Google Scholar]
- 53.Schnell G, Price RW, Swanstrom R, Spudich S. 2010. Compartmentalization and clonal amplification of HIV-1 variants in the cerebrospinal fluid during primary infection. J Virol 84:2395–2407. doi: 10.1128/JVI.01863-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Permar SR, Salazar MG, Gao F, Cai F, Learn GH, Kalilani L, Hahn BH, Shaw GM, Salazar-Gonzalez JF. 2013. Clonal amplification and maternal-infant transmission of nevirapine-resistant HIV-1 variants in breast milk following single-dose nevirapine prophylaxis. Retrovirology 10:88. doi: 10.1186/1742-4690-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salazar-Gonzalez JF, Salazar MG, Learn GH, Fouda GG, Kang HH, Mahlokozera T, Wilks AB, Lovingood RV, Stacey A, Kalilani L, Meshnick SR, Borrow P, Montefiori DC, Denny TN, Letvin NL, Shaw GM, Hahn BH, Permar SR, Center for HIV/AIDS Vaccine Immunology A0167854. 2011. Origin and evolution of HIV-1 in breast milk determined by single-genome amplification and sequencing. J Virol 85:2751–2763. doi: 10.1128/JVI.02316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Becquart P, Chomont N, Roques P, Ayouba A, Kazatchkine MD, Belec L, Hocini H. 2002. Compartmentalization of HIV-1 between breast milk and blood of HIV-infected mothers. Virology 300:109–117. doi: 10.1006/viro.2002.1537. [DOI] [PubMed] [Google Scholar]
- 57.Joseph SB, Swanstrom R, Kashuba AD, Cohen MS. 2015. Bottlenecks in HIV-1 transmission: insights from the study of founder viruses. Nat Rev Microbiol 13:414–425. doi: 10.1038/nrmicro3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kariuki SM, Selhorst P, Arien KK, Dorfman JR. 2017. The HIV-1 transmission bottleneck. Retrovirology 14:22. doi: 10.1186/s12977-017-0343-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson LF, Lewis DA. 2008. The effect of genital tract infections on HIV-1 shedding in the genital tract: a systematic review and meta-analysis. Sex Transm Dis 35:946–959. doi: 10.1097/OLQ.0b013e3181812d15. [DOI] [PubMed] [Google Scholar]
- 60.Dyer JR, Eron JJ, Hoffman IF, Kazembe P, Vernazza PL, Nkata E, Costello Daly C, Fiscus SA, Cohen MS. 1998. Association of CD4 cell depletion and elevated blood and seminal plasma human immunodeficiency virus type 1 (HIV-1) RNA concentrations with genital ulcer disease in HIV-1-infected men in Malawi. J Infect Dis 177:224–227. doi: 10.1086/517359. [DOI] [PubMed] [Google Scholar]
- 61.Eron JJ Jr, Gilliam B, Fiscus S, Dyer J, Cohen MS. 1996. HIV-1 shedding and chlamydial urethritis. JAMA 275:36. doi: 10.1001/jama.1996.03530250040022. [DOI] [PubMed] [Google Scholar]
- 62.Rotchford K, Strum AW, Wilkinson D. 2000. Effect of coinfection with STDs and of STD treatment on HIV shedding in genital-tract secretions: systematic review and data synthesis. Sex Transm Dis 27:243–248. doi: 10.1097/00007435-200005000-00001. [DOI] [PubMed] [Google Scholar]
- 63.Cohen MS, Hoffman IF, Royce RA, Kazembe P, Dyer JR, Daly CC, Zimba D, Vernazza PL, Maida M, Fiscus SA, Eron JJ Jr, AIDSCAP Malawi Research Group. 1997. Reduction of concentration of HIV-1 in semen after treatment of urethritis: implications for prevention of sexual transmission of HIV-1. Lancet 349:1868–1873. doi: 10.1016/S0140-6736(97)02190-9. [DOI] [PubMed] [Google Scholar]
- 64.Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K. 1985. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 313:277–284. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- 65.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Larsson A. 2014. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30:3276–3278. doi: 10.1093/bioinformatics/btu531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. 2018. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rambaut A. 2010. FigTree v1.3.1. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, United Kingdom. [Google Scholar]
- 70.Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 71.Edwards CJ, Suchard MA, Lemey P, Welch JJ, Barnes I, Fulton TL, Barnett R, O’Connell TC, Coxon P, Monaghan N, Valdiosera CE, Lorenzen ED, Willerslev E, Baryshnikov GF, Rambaut A, Thomas MG, Bradley DG, Shapiro B. 2011. Ancient hybridization and an Irish origin for the modern polar bear matriline. Curr Biol 21:1251–1258. doi: 10.1016/j.cub.2011.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tongo M, Essomba RG, Nindo F, Abrahams F, Nanfack AJ, Fokam J, Takou D, Torimiro JN, Mpoudi-Ngole E, Burgers WA, Martin DP, Dorfman JR. 2015. Phylogenetics of HIV-1 subtype G env: greater complexity and older origins than previously reported. Infect Genet Evol 35:9–18. doi: 10.1016/j.meegid.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 73.Rognes T, Flouri T, Nichols B, Quince C, Mahe F. 2016. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huerta-Cepas J, Serra F, Bork P. 2016. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol 33:1635–1638. doi: 10.1093/molbev/msw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Paradis E, Claude J, Strimmer K. 2004. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 77.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequences of full-length env clones may be found in GenBank under accession numbers MT260117 to MT260126. MiSeq sequence data are available under BioProject accession number PRJNA613850.