

Respiratory tract infection by adenoviruses is among the most common diseases in children, attributing to approximately 20% of hospitalizations of children with acute respiratory infection (ARI). Adenovirus transmits by direct contact, but recurrent infection is common. Ever since its isolation, adenovirus has been known to have the ability to establish persistent or latent infection. We found 87.7% tonsillectomy specimens contained detectable amounts of adenoviral DNA. Isolated lymphocytes did not produce infectious adenoviruses without stimulation. By screening an epigenetic informer compound library, we identified several histone deacetylase inhibitors that promoted adenovirus reactivation that was evidenced by increased viral DNA replication and production of infectious viruses. The human tonsils are covered with bacterial pathogens that may utilize pathogen-associated pattern molecules or metabolites to cause epigenetic activation and proinflammatory gene transcription, which may lead to viral reactivation from latency. The study shows that recurrent adenovirus infection could arise from reactivation of residing virus from previous infections.
KEYWORDS: adenovirus, latency, reactivation, HDACi
ABSTRACT
Adenovirus (HAdV) infection is a common cause of illness among young children, immunocompromised patients, and transplant recipients. The majority of HAdV infections are self-limited, but recurring infection is frequently encountered in young children and may require hospitalization. In this study, we surveyed the presence of HAdV in tonsillectomy samples and investigated epigenetic conditions that contributed to HAdV reactivation. HAdV DNA was detected from 86.7% donors. The lymphocytes isolated from the samples failed to produce infectious HAdV after incubation, suggesting the viruses remained in a latent status. To determine whether epigenetic factors played a role in HAdV reactivation, isolated lymphocytes were treated with a small compound library. Viral DNA replication and infectious HAdV production were assayed by PCR and by a secondary infection assay. We identified several compounds, mainly pan- and selective histone deacetylase (HDAC) inhibitors, which showed activity to reactivate HAdV from latency. The viruses were isolated and were determined as species C HAdV. Using a model of HAdV lytic infection, we showed that the compounds promoted histone-3 acetylation and association with viral early gene promoters. In addition to demonstrate the palatine tonsils as a reservoir of latent HAdV, this study uncovers a critical role of histone acetylation in HAdV reactivation, linking HAdV latency to recurrent HAdV infection.
IMPORTANCE Respiratory tract infection by adenoviruses is among the most common diseases in children, attributing to approximately 20% of hospitalizations of children with acute respiratory infection (ARI). Adenovirus transmits by direct contact, but recurrent infection is common. Ever since its isolation, adenovirus has been known to have the ability to establish persistent or latent infection. We found 87.7% tonsillectomy specimens contained detectable amounts of adenoviral DNA. Isolated lymphocytes did not produce infectious adenoviruses without stimulation. By screening an epigenetic informer compound library, we identified several histone deacetylase inhibitors that promoted adenovirus reactivation that was evidenced by increased viral DNA replication and production of infectious viruses. The human tonsils are covered with bacterial pathogens that may utilize pathogen-associated pattern molecules or metabolites to cause epigenetic activation and proinflammatory gene transcription, which may lead to viral reactivation from latency. The study shows that recurrent adenovirus infection could arise from reactivation of residing virus from previous infections.
INTRODUCTION
The human adenoviruses (HAdVs) are a group of double-stranded DNA viruses that cause respiratory infection of children and adults (1–3). Typical symptoms include fever, pharyngitis, cough, sore throat, and tonsillitis. Although many infections are mild in immunocompetent individuals, the infection may lead to fatal pneumonia in young children (4–7). It was reported that more than 20% of hospitalized children with acute respiratory tract infections had HAdV (1, 8, 9), with species C as the unique pathogen in children with acute bronchiolitis or recurrent wheezing (8). HAdV infection transmits from person to person directly by coughing and sneezing or close contact, such as touching contaminated objects or surfaces. HAdV can multiply in the epithelial cells of the respiratory tract, pharynx, conjunctiva, or small intestine according to the mode of entry and then spreads to reginal lymph nodes. The virus has the property to establish persistent infection or latency in lymphoid organs, such as tonsils and Peyer’s patches (10–12). It is believed that exogenous infections occur in some instances, and the majority of invasive events appear to arise from viral reactivation from a previous infection (13). Latent infection can be reactivated in patients with underlying immunity, and the virus can switch to a lytic phase of replication (14). Adenovirus reactivation is increasingly being recognized as a significant problem and remains a clinical challenge in transplantation settings (15–19).
The human tissues, such as tonsils, serve as reservoirs for many medically important viruses, including Epstein-Barr virus (EBV) and HAdV. Several studies have reported that adenoviral DNA was detected in over 80% of human tonsil and adenoid lymphoid tissue samples (6, 20–23). Although HAdV was originally isolated from cultured tonsil fragments, adenovirus cannot be cultured from adenoids and tonsils immediately after excision (10). After various periods of cultivation of tonsil fragments or costimulation of isolated tonsil lymphocytes with anti-CD3 and CD28 antibodies, the specimens were able to produce infectious virus (10, 24), indicating T cell receptor (TCR) signaling may be involved in the regulation of HAdV reactivation.
Costimulation of T lymphocytes can promote a number of signaling cascades, leading to cell survival, proliferation, differentiation, and cytokine production (25). TCR signaling induces massive changes in histone modifications in human T cells that may mediate chromatin remodeling and T cell function (26–29). As obligate intracellular organisms, viruses rely, to a major extent, on the host cell for replication (30). The HAdV DNA is associated with viral proteins, such as core protein VII (pVII), but not histone proteins (31). Once in the nucleus, the incoming viral DNA undergoes remodeling by depositing host histone proteins to the pVII-DNA complex. The newly formed physiologically spaced nucleosomes were detectable within about 6 h of the infection (22, 32, 33). During the late phases of the infection, the association with histone-3 was dramatically reduced and the repeating of the nucleosome-like pattern was no longer evident (34). The adenoviral E1A alters global patterns of histone modification and regulates transcription and cellular transformation (35–37). Whether epigenetic regulation is involved in HAdV latency as well as its reactivation remains uninvestigated.
Accumulating evidence suggests that cellular and viral chromatin proteins are positive factors in the regulation of viral gene expression during the lytic cycle of infection (38). Chromatin dynamics may regulate adenoviral gene expression and infection since histone acetylation is commonly associated with actively transcribed genes (39, 40). Although human adenoviral DNA associates with nucleosomes containing histone variant H3.3 during the early phase of infection and chromatin immunoprecipitation (ChIP) assays also showed that HAdV2 promoters were associated with acetylated histone-3 during the lytic cycle of infection (33, 41), whether epigenetic factors play a role in HAdV reactivation remains unknown.
Given the significance of histone modifications in transcription and gene regulation, we asked if epigenetic factors played a role in HAdV reactivation. We screened an epigenetic compound library on tonsil samples and identified several histone deacetylase (HDAC) inhibitors that caused HAdV reactivation. Here, we report the characterization and identification of the compounds important for HAdV reactivation and infection.
RESULTS
Adenoviral DNA is common in tonsillectomy samples.
Fresh tonsil samples from patients undergoing routine tonsillectomies for chronic tonsillitis or hypertrophic tonsils were collected at Nanjing Children’s Hospital. A total of 65 samples from 45 donors (age, 1 to 11 years old) were analyzed by a PCR assay using panhexon-specific primers. The primers were designed to cover species C, the predominant serotype detected in human tonsils in previous studies, and species B, D, and E adenoviruses. Of the 65 tonsil samples, 57 contained detectable levels of the viral DNA, representing a positive rate of 87.6% of tonsil samples. Of those 57 positive samples, 19 were from those 25 patients who had 1 tonsil removed, while 38 were from 20 patients who had both tonsils removed. Of those 20 patients who had both tonsils removed, 18 had detectable HAdV DNA in both tonsils, while 2 patients had detectable viral DNA in 1 of their tonsils. Therefore, adenoviral DNA was commonly present in tonsillectomy samples.
The DNA sequences of the samples were analyzed. All the samples belonged to species C adenoviruses, including HAdV2 (34/57, 59.6%), HAdV5 (20/57, 35.1%), and HAdV1 (3/57, 5.3%). No HAdV6 was detected. The distribution of the serotypes was consistent with results from other studies (1, 23, 42). In addition, no patients had more than one serotype of HAdV detected.
Table 1 summarizes HAdV distribution information and information of the patients, including counts of white blood cells, lymphocytes, neutrophils, platelets, hemoglobin, and C-reactive protein (CRP). The parameters were within normal ranges and were not significantly different from those of healthy donors.
TABLE 1.
The demographic characteristics of the participants and HAdV DNA statusa
| Characteristic | Ad-positive samples (n = 39, 86.7%) | Ad-negative samples (n = 6, 13.3%) |
|---|---|---|
| Gender (n [%]) | ||
| Male | 25 (64.1) | 2 (33.3) |
| Female | 14 (35.9) | 4 (66.7) |
| Age range (n [%]) | ||
| 0 to <3 | 3 (7.7) | 1 (16.7) |
| 3 to <6 | 26 (66.7) | 3 (50) |
| 6 to <9 | 6 (15.3) | 2 (33.3) |
| 9 to <12 | 4 (10.3) | 0 |
| Mean ± interquartile range | 5.06 ± 1.76 | 5.58 ± 2.19 |
| Blood test (median ± interquartile range) | ||
| WBC (109 cells/liter) | 9.25 ± 2.16 | 8.36 ± 1.90 |
| Leukopenia (<4,000/μl) | 56.92 ± 11.94 | 43.82 ± 9.42 |
| Lymphocyte (%) | 34.70 ± 11.55 | 44.15 ± 9.45 |
| Neutrophils (%) | 56.92 ± 11.72 | 43.82 ± 8.60 |
| Platelets (109 cells/liter) | 270.24 ± 77.69 | 258.16 ± 43.64 |
| Hemoglobin (g/liter) | 127.00 ± 9.19 | 124.00 ± 13.94 |
| CRP, >8 mg/liter (n [%]) | 6 (15.4) | 1 (16.7) |
Data shown are from 45 donors. HAdV-positive rates were determined as 86.7% (39/45) in patients or 87.7% (57/65) of tonsil specimens. Ad, adenovirus; WBC, white blood cells; CRP, C-reactive protein.
An informer compound library screening identifies HDAC inhibitors that promote HAdV reactivation from tonsil lymphocytes.
The HAdV was first isolated by the cultivation of tonsil fragments (10, 43) or identified following TCR activation (23). We reasoned that epigenetic regulation might have played a role in HAdV reactivation since TCR signaling has been reported to induce massive changes in histone modifications that mediate chromatin remodeling and gene activation in human T lymphocytes (26, 44). We, therefore, screened an epigenetic compound library on freshly isolated tonsil lymphocytes. In this regard, freshly isolated, HAdV-positive tonsil lymphocytes were vehicle-treated or treated with 50 ng/ml phorbol myristate acetate (PMA), a compound that previously was reported to reactivate HAdV, or with a test compound at the half-maximal nontoxic concentrations. After treatment for 48 h, the cells and culture medium were collected. The mixtures were subjected to freeze and thaw for 3 cycles in liquid nitrogen and a 37°C water bath to release the virus and viral DNA.
To determine whether the compounds caused latent HAdV reactivation, we first performed a PCR assay to check for DNA amplification. For this purpose, a portion of the supernatants was first treated with proteinase K to remove viral DNA-associated proteins. The samples were tested for HAdV DNA accumulation by PCR assay. We found that PMA treatment weakly promoted HAdV DNA accumulation. Instead, we identified several compounds that markedly induced HAdV DNA accumulation (Fig. 1B). The observation was independently validated on HAdV-positive lymphocytes from 3 different patients. Compounds such as abexinostat (PCI24781), belinostat (PXD101), dacinostat (LAQ824), trichostatin A (TSA), pan- or selective HDAC inhibitors, and decitabine, a methyltransferase inhibitor, consistently promoted HAdV DNA accumulation (Fig. 1B). We also identified vorinostat (SAHA) with moderate activity in promoting HAdV DNA production (Fig. 1B). A recent study showed that at 10 μM concentration both TSA and SAHA suppressed human adenovirus-mediated gene expression and replication (45). We did not follow this course since the concentration used in the study was significantly toxic to the system we used.
FIG 1.
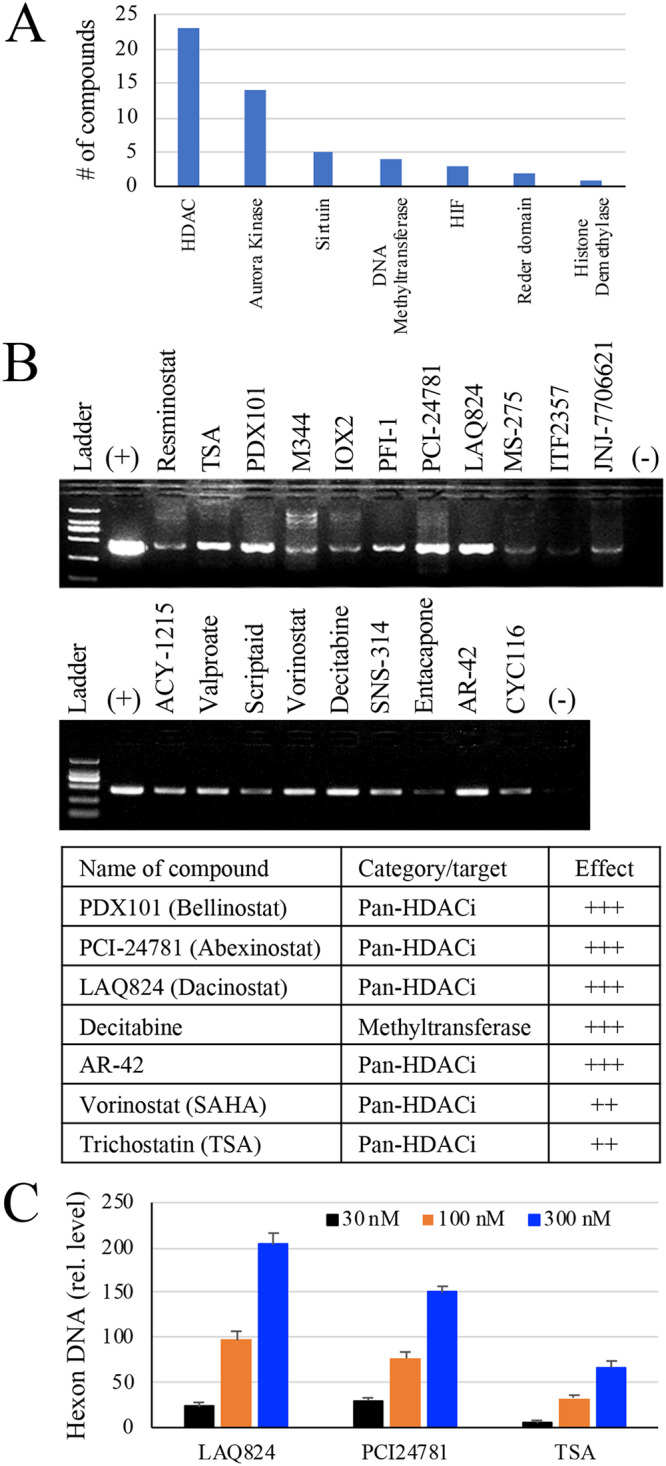
PCR results from epigenetic compound library screening. (A) The primary categories of the epigenetic informer compounds tested. (B) Compound screening on isolated tonsil lymphocytes. Tonsil lymphocytes were first examined for the presence of HAdV DNA using a pair of primers that anneal to the hexon gene of species B, C, D, and E HAdV. HAdV-positive lymphocytes in 96-well plates were vehicle treated (0.1% DMSO in culture medium) or treated with a compound for approximately 48 h. The cells and culture medium were collected and used for the detection of HAdV by PCR. The induction of HAdV DNA accumulation in the treated samples was demonstrated by PCR analysis and verified using 3 different tonsil samples. The PCR result from a representative experiment is presented, and compounds that showed more potent effects were listed. +, positive control for the PCR. (C) Dose effect of HDACi on HAdV reactivation. HAdV2-positive tonsil lymphocytes were treated with various doses of PCI24781, LAQ824, or TSA. The cells and culture medium were collected 48 h later and used for detection of HAdV2 hexon DNA by qPCR. The data were normalized against GAPDH in the samples and plotted as relative abundance against vehicle-treated controls.
The effect of the compounds on HAdV reactivation was then confirmed with a secondary infection assay. To this end, the supernatant (30 μl) from vehicle-treated or PMA-, PCI24781-, or LAQ824-treated tonsil lymphocytes was used to infect A549 cells in 96-well plates. The cells and culture medium were collected after 60 h to 72 h of incubation, and the recovered HAdV was used to infect A549 cells for another round of amplification. The titers of the viruses in PCI24781- and LAQ824-treated samples were determined to be approximately 1 × 102 and 7 × 102 PFU/ml, respectively. For comparison, no infectious virus was detected in the vehicle-treated samples, while those treated with PMA did not produce a detectable amount of HAdV. The results together demonstrated that HDAC inhibitors are a novel class of small compounds that reactivate HAdV from latency.
The dose effect of the HDACi on HAdV reactivation was also tested on freshly isolated tonsil lymphocytes by measuring HAdV DNA accumulation using qPCR. PCI24781, LAQ824, or TSA showed potent effects in promoting HAdV reactivation. At submicromolar concentrations, the compounds dose-dependently promoted HAdV DNA accumulation (Fig. 1C). The effect of LAQ824 was more potent than the other compounds tested in both DNA amplification and in virus recovery assays. In subsequent studies, we therefore used LAQ824, a pan-HDAC inhibitor, to treat tonsil lymphocytes and isolated 15 cultivated viruses. Sequence analysis of those isolates confirmed that the viruses were HAdV2 (10/15, 67%) and HAdV5 (5/15, 33%).
Reactivation of HAdV by HDACi in a xenograft mouse model.
Next, we tested whether the compounds were able to reactivate HAdV in vivo using a xenograft model system. HAdV-positive tonsil fragments were implanted in nude mice. The animals were then treated with a vehicle, with LAQ824, or with TSA, a well-studied HDACi, on the 3rd, 4th, and 5th day after tonsil implantation. On the 7th day, tonsil implants were removed and used to extract DNA. HAdV DNA production was determined by measuring viral DNA production with qPCR. As shown in Fig. 2, HDACi treatment significantly enhanced viral DNA replication in the xenograft model, indicating epigenetic factors had a role in HAdV reactivation from latent infection.
FIG 2.
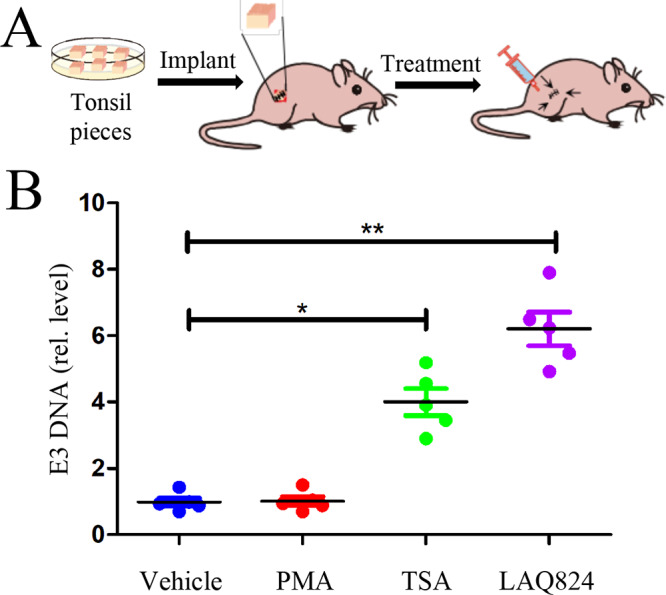
Reactivation of HAdV from tonsil fragments in a xenograft model. Tonsil fragments of approximately 2 by 2 by 2 mm in size were surgically implanted in the rear flanks of athymic nude mice (3 mice or 6 pieces per group). The mice were then treated by subcutaneous injection near the sites of the implants (as the arrows indicated) with a vehicle (10% DMSO in normal saline) or with a test compound (in 30 μl volume per site) on days 3, 4, and 5. TSA (10 mg/kg), LAQ824 (10 mg/kg), and PMA (30 μg/kg) as a reference, were tested. The tonsil implants were removed on day 7, and viral DNA was quantitatively measured by qPCR. The experiments were performed 2 times independently. The data are presented as mean ± SD (n = 6) of one experiment; *, denotes P < 0.05 and **, denotes P < 0.01.
Role of histone acetylation in HAdV lytic infection.
Since the majority of the compounds identified in this study were HDAC inhibitors, we therefore studied HAdV infection on histone-3 modifications. Due to the relatively low abundance of viral DNA in the primary tonsil lymphocytes and possibly low activity of cell proliferation/differentiation, we were unable to demonstrate histone modifications in freshly isolated tonsil lymphocytes by immunoblotting studies. We, therefore, used a lytic infection model to address whether histone acetylation promoted HAdV infection. We found that HAdV2 infection promoted H3K9 and H3K18 acetylation at the early stages of an infection (Fig. 3A). At later stages, H3K9 and H3K18 acetylation were reduced. For comparison, the level of histone-3 phosphorylation (Ser10) remained relatively unchanged during the course of the infection.
FIG 3.

Effect of HAdV infection on histone H3 modifications. (A) HAdV2 infection induces histone-3 acetylation. Monolayers of A549 cells were infected with HAdV2 at an MOI of 1 for times as indicated. The levels of acetylated and phosphorylated histome-3 (Ac-H3K9, Ac-H3K18, and p-H3 Ser 10) were determined by immunoblotting studies. Histone-3 expression and hexon production were also examined as loading and infection controls, respectively. TSA, Ad2 infected for 24 h in the presence of 150 nM TSA. The experiment was performed 2 times independently. (B and C) Histone-3 acetylation during HAdV2 infection. Monolayers of A549 cells were uninfected or infected with HAdV2 at 5 to 15 MOIs for 3 h or 6 h. TSA, Ad2 at 10 MOI for 6 h in the presence of 150 nM TSA. Histone-3 acetylation and phosphorylation was detected by immunoblotting assay using anti-Ac-H3K9, Ac-H3K18, or anti-p-Histone-3 (Ser10) antibody. (B) The expression of histone-3 and production of hexon was included as controls. (C) The relative intensities of Ac-H3K9 and anti-Ac-H3K18 were quantitatively measured and plotted.
We then tried to determine an optimal multiplicity of infection (MOI) for H3K9 and H3K18 acetylation since we would like to perform ChIP assays to demonstrate acetylated histone association with viral early gene promoters. Since the ChIP assay tends to require relatively high infection multiplicity to determine the early event of an infection, we infected A549 cells with HAdV2 at 5 to 15 MOIs for either 3 h or 6 h. At 10 and 15 MOIs, HAdV2 infection caused strong acetylation of H3K9 and H3K18 at 3 h and 6 h postinfection (p.i.) (Fig. 3B and C).
Association of acetylated histone-3 with viral early genes or gene promoters.
Since both cellular and viral chromatin proteins are positive factors in the regulation of HAdV gene expression (38), we therefore performed ChIP assays to investigate whether acetylated histone was associated with viral gene promoters. A549 cells were uninfected or infected with HAdV2 at an MOI of 15 for 3 h and 6 h. The samples were then processed and immunoprecipitated by incubation with anti-Ac-H3K9 or anti-Ac-H3K18. The presence of E1A, E2, E3, E4, or IVa2, a later gene as a control, in the anti-H3K9 or H3K18 immunocomplexes was detected by qPCR. As shown in Fig. 4, the acetylated histones were associated with viral early gene promoters. No presence of gene IVa2 DNA was detected in the immunoprecipitation complexes (data not shown).
FIG 4.

Association of acetylated histone-3 and viral early genes or gene promoters. Monolayers of A549 cells were uninfected or infected with HAdV2 (15 MOI) for 6 h. The samples were used to detect acetylated histone-3 to viral gene promoters by ChIP assay. Adenoviral E1, E2, E3, and E4 early genes as well as a late gene IVa2 in the anti-Ac-H3K9 and anti-Ac-H3K18 immunocomplexes were determined by qPCR measurement. Data for IVa2 were not included since they were not detected. The experiment was performed 2 times independently. The data are presented from a representative experiment.
LAQ824 on histone acetylation and acetylated histone association with viral genes or gene promoters.
Next, we addressed whether the LAQ824 effect on HAdV infection correlated with histone modifications. A549 cells were treated with LAQ824 prior to HAdV2 infection. LAQ824 treatment alone promoted Ac-H3K9 and Ac-H3K18 (Fig. 5A, lane 3). In HAdV2-infected samples, the levels of acetylated H3K9 and H3K18 were significantly higher than those in mock-treated samples (Fig. 5A, lane 2), which were determined by immunoblotting assay and confirmed by immunofluorescence staining studies (Fig. 5B).
FIG 5.

Effect of HDACi LAQ824 on histone-3 acetylation and association with viral early gene promoters. (A and B) LAQ824 treatment enhances Ac-H3K9 and Ac-H3K18. A549 cells were mock treated or treated with LAQ824 at indicated concentrations prior to HAdV2 infection (MOI, 15). The levels of hexon, Ac-H3K9, Ac-H3K18, and p-H3 were detected at 6 h p.i. by immunoblotting (A). In parallel experiments, the samples were fixed at 6 h p.i. and were used to detect histone modification by immunofluorescence (B). The experiments were performed 2 times independently; representative data are presented. (C) LAQ824 promotes Ac-H3K9 and Ac-H3K18 association with viral genes or gene promoters. A549 cells were uninfected or infected with HAdV2 (15 MOI) for 6 h in the absence or presence of 100 or 300 nM LAQ824. The samples were processed for ChIP assay by immunoprecipitating for Ac-H3K9 and Ac-H3K18. Adenoviral E1, E2, E3, and E4 early genes and late gene IVa2 in the immunocomplexes were determined by qPCR measurement. Data for IVa2 were not included since they were not detected. The experiment was performed 2 times independently. The data are presented from a representative experiment.
We then studied LAQ824 on acetylated histone-3 association with viral early gene promoters. Treatment with LAQ824 resulted in enhanced detection of viral DNA of the E1A, E2, E3, and E4 regions in the anti-H3K9 immunocomplexes by approximately 34- to 190-fold (Fig. 5C), while those in the anti-Ac-H3K18 immunocomplexes were increased by 3- to 24-fold compared with those in the dimethyl sulfoxide (DMSO)-treated controls. As a control of the assay, IVa2 was not detected in anti-Ac-H3K9 nor the anti-Ac-H3K18 immunocomplexes. These results, therefore, indicated that treatment with LAQ824 boosted histone modifications and acetylated histone association with viral early gene promoters. Since histone acetylation tends to be linked to gene activation, this result suggests that HDACi like LAQ824 promoted HAdV reactivation through epigenetic regulation.
DISCUSSION
The human adenoviruses are members of the Adenoviridae family and Mastadenovirus genus that consists of seven species/subgroups (A to G) and over 50 serotypes known to cause human infection. Adenovirus infections of the upper respiratory tract are common and often subclinical but may cause common cold symptoms, pharyngitis, tonsillitis, otitis media, and pharyngoconjunctival fever. The infections are usually mild and self-limited but can be contagious in close-contact settings and be more serious in children and in people with weakened immune systems. Among HAdVs, species C adenoviruses (serotypes HAdV1, 2, 5, and 6) are common pathogens causing childhood respiratory tract illness. Other serotypes, including HAdV3, HAdV4, HAdV7, and HAdV14, have also been reported (7). After entry into the human body, the viruses actively replicate in the epithelial cells to produce lytic effects. In most cases, the symptoms disappear within 3 to 5 days, but recurrent infection occurs at times, indicating the existence of persistent or latent infection (14). We reasoned that the human tonsils could be a reservoir for HAdV that might undergo reactivation. We surveyed 65 tonsil samples from 45 donors and found HAdV DNA was prevalent in the samples. The majority of the samples seemed to have HAdV in latent status since cultured lymphocytes did not produce infectious viruses without stimulation. Both in vitro and in vivo stimulation of isolated lymphocytes or tissue fragments with HDAC inhibitors resulted in HAdV DNA replication, indicating epigenetic factors regulate viral reactivation and possibly the maintenance of latency.
Despite the frequent detection of significant quantities of adenoviral DNA in tonsil and adenoid tissues, infectious virus was rarely present, as measured by coculture with permissive cells (10, 12, 14, 21). The molecular details by which viral latency and reactivation are regulated have been less investigated. The adenoviral DNA is wrapped into a repeating nucleosome-like array containing histone during the early phase of an infection (33, 46). Garnett and colleagues reported that, following in vitro activation with PMA/ionomycin along with interleukin-2 (IL-2) and anti-CD3/CD28 antibodies, infectious virus was produced from purified tonsil T lymphocytes (24), suggesting TCR activation leads to HAdV reactivation. TCR signaling induces massive changes in histone modifications that modulates global chromatin remodeling and gene activation in human T cells. We questioned whether epigenetic factors played a role in HAdV latency and reactivation and screened an epigenetic compound library. We identified several compounds that promoted HAdV reactivation from human tonsil lymphocytes at submicromolar concentrations. The compounds were classified as HDAC inhibitors, or those that target histone modification and transcription activation. We showed that the compounds promoted HAdV DNA replication both in vitro and in vivo. In addition, we demonstrated that acetylated histones were associated with viral early gene promoters, underlining the importance of histone modifications in HAdV reactivation and lytic infections. This argues that HAdVs from reactivation could be a contributing factor for recurrent infection.
Many viruses have both lytic and latent status of infection. Like their cellular host counterparts, many invading viral pathogens must contend with, modulate, and utilize the host cell chromatin machinery to promote efficient lytic infection or control persistent-latent states (32). The adenoviral E1A protein interacts with histone acetyl transferases p300/CBP and tumor-suppressor retinoblastoma protein (Rb) to repress select host genes and promote productive virus infection (35, 37). Thus, viruses may utilize encoded proteins or different immediate early (IE) gene products to drive their replication in response to cellular signaling, including mitogen-activated protein kinase (MAPK) in conjunction with the NF-κB pathway activation (47–49). The mechanisms by which the establishment, maintenance, and reactivation from latency are regulated are complex and diverse among virus families, species, and strains (50, 51). It is reasonable to speculate that a possible unifying theme for the molecular trigger from within the cells may initiate virus reactivation (30). Among the exogenic factors that may cause virus reactivation, stress almost always seems to be a major factor leading to a reactivation process. Stress, along with various other growth factors/inflammatory cytokines, has the propensity to trigger cellular signaling and epigenetic regulation. Histone acetylation causes destabilization of chromatin, leading to a loose, open structure of the promoter, so that it becomes easily accessible to basic transcription factors (52). There exists a complicated set of interactions between these cellular events, leading to chromatin remodeling and action of transcription, ultimately resulting in cell proliferation/differentiation or in viral reactivation.
Recurrent HAdV infection is common among young children. The human tonsils are exposed to diverse viral and bacterial pathogens (53). For example, bacterial pathogens such as Streptococcus spp., Neisseria sp., Staphylococcus spp., and Haemophilus influenzae are among the tonsillar microbiota and have been shown to be associated with recurrent tonsillitis (54, 55). The host utilizes pattern-recognition receptors to sense pattern molecules from Gram-positive and Gram-negative bacteria to trigger proinflammatory responses. In addition, the host-adapted bacterial strains may release glycolysis metabolites, such as succinate, in the airway to promote inflammation through activation of inflammasome signaling (56, 57). Proinflammatory response leads to epigenetic modification and gene transcription that may cause virus reactivation (58, 59). Indeed, we found stimulation of tonsil lymphocytes with lipopolysaccharide-induced HAdV DNA amplification. Another molecular relationship is the so-called cross talk between microbiota and their metabolites, including the production of short-chain fatty acids, which are known to exhibit activity as HDAC inhibitors (60). It is possible that bacterial infection instigates recurrent tonsillitis through the production of metabolites or bacterial pattern molecules.
MATERIALS AND METHODS
Cells and viruses.
Human lung alveolar epithelial A549 cells (ATCC, CCL-185) and HAdV2 were purchased from ATCC (Manassas, VA). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; high glucose; catalog number 11965-092) supplemented with 10% fetal bovine serum (FBS; number 10099-141), 10 mM HEPES, sodium pyruvate, nonessential amino acids, and penicillin-streptomycin (Life Technologies) in a humidified incubator with 5% CO2 at 37°C. Lymphocytes were cultured in RPMI 1640 (number 670089) with 10% FBS, penicillin-streptomycin, glutamax (number 35030-061), and 10 mM HEPES. The adenoviruses were propagated and titrated in A549 cells.
Tissue samples.
The studies were approved by the Ethics Committees of Nanjing Medical University and Nanjing University Medical School, and informed consent was obtained from legal guardians involved in this study in accordance with the Declaration of Helsinki. Fresh tonsil samples from patients undergoing routine tonsillectomies for chronic tonsillitis or hypertrophic tonsils were collected at Nanjing Children’s Hospital. Laboratory data on admission and during hospitalization were collected (Table 1). None of the patients had symptoms that would indicate acute adenovirus infection. The tissue samples were processed within 4 h after surgeries. After being thoroughly rinsed with ice-cold DMEM medium, the samples were used immediately or stored away after being snap-frozen for further studies.
Reagents and antibodies.
A compound library of epigenetics was purchased from Selleck Chemicals (catalog number L1900; Shanghai, China). The library contains 52 cell-permeable inhibitors targeting the histone deacetylases (HDACs), histone acetyltransferases (HATs), DNA methyltransferases (DNMTs), and inhibitors of the epigenetic reader domain or aurora kinase (Fig. 1A). Rabbit polyclonal antibodies to acetyl-histone H3K9 (A7255) and anti-acetyl-H3K18 (A7257) were purchased from ABclonal (Wuhan, China), to histone H3 (ab176842) from Abcam, and to phospho-histone H3 (Ser10, number 3377) from Cell Signaling; a rabbit control IgG (A7016; Beyotime, Shanghai, Chinga) was obtained commercially, and rabbit anti-HAdV2 hexon (anti-hexon) was prepared by ABmart (Shanghai, China). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Bio-Rad Laboratories (Shanghai, China). Alexa Fluor 568-conjugated anti-rabbit IgG and phalloidin 488 (Green) were purchased from Life Technologies. 4′,6-Diamidino-2-phenylindole (DAPI; C1002) and Protein G agarose beads (P2009) were purchased from Beyotime. Phorbol-12-myristate-13-acetate (PMA) and PCR grade proteinase K (Roche; number 3115879001) were purchased from Sigma-Aldrich (Shanghai, China).
Lymphocyte preparation and compound library screening studies.
Tissue samples were rinsed in RPMI 1640 medium supplemented with penicillin-streptomycin and were minced into fine pieces using razor blades (61). The mix was passed through a Falcon cell strainer, and the cells were then washed twice with RPMI 1640 before being used for reactivation or for virus recovery studies.
Prior to compound screening studies, the cytotoxicity of the test compounds was determined on Vero cells using a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) method (62). To get an estimated nontoxic concentration of a test compound, we used the reported 50% inhibitory concentration (IC50) data of individual compounds as the highest concentration and diluted down the compound by series dilution. An optical density (OD) reading within 10 percentage points of DMSO treated-controls was derived as the maximal nontoxic concentration. To reactivate adenoviruses, freshly isolated tonsil lymphocytes were plated in 96-well plates at 1 × 105 cells/well using complete RPMI 1640 medium. The cells were vehicle-treated (DMSO to a final concentration of 0.1%) or treated with a test compound and were cultured in a 37°C cell culture incubator for 48 h. At the end, the cells and the culture medium were collected. After 3 freeze-thaw cycles (frozen in liquid nitrogen and thawed in a 37°C water bath), the samples were used for PCR studies or for virus recovery studies.
To recover HAdV, a portion of the supernatants from compound-stimulated lymphocytes was first used to infect the permissive A549 cells in 96-well plates for 2 to 3 days. The samples were used to infect A549 cells for a second round. During this time, the presence of HAdV DNA in the culture medium was monitored by PCR, while the cells were monitored for cytopathic effect (CPE). Those with obvious CPE were harvested for virus characterization and for virus titer determination to preliminarily determine a relative potency of representative compounds. The isolated viruses were subjected to sequence analysis.
DNA extraction.
Freshly removed tonsil tissues (approximately 20 mg) or isolated tonsil lymphocytes (1 × 107) were lysed in 100 μl buffer containing 150 mM NaCl, 0.5% NP-40, 0.1% SDS, and 20 mM Tris-HCl (pH 7.6). The lysate was then treated with proteinase K (0.2 mg/ml) at 55°C overnight, followed by heat inactivation at 95°C for 10 min. DNA was purified by phenol-chloroform extraction, followed by ethanol precipitation. Total DNA was resuspended in Tris-EDTA (TE) buffer and the concentrations were determined using a Nanodrop 2000 instrument.
PCR and real-time PCR.
For the first-round screening, we used the following pair of primers that align to the hexon gene of serotypes B, C, D, and E of HAdV: HexF1, 5′-AACTTCCAGCCCATGAGC and HexR1, 5′-GAAGGGCGTGCGCAGGTA (477 bp; nucleotide position 21274 to 21720 of HAdV2; GenBank accession number J01917.1). Typically, the PCR was carried out in 25-μl volume using Taq DNA polymerase (P211-01; Vazyme, Nanjing, China) and 1 μg total DNA for amplification. PCR amplification started with denaturation for 3 min at 94°C; followed by 30 cycles of 30 s at 94°C, 30 s at 58°C, and 30 s at 72°C; followed by a final extension at 72°C for 10 min. The products were analyzed by electrophoresis and automated DNA sequencing analysis.
We also designed the following set of primers for serotype analysis of species C HAdV: HexF2, 5′-GACAGCTATGATCCAGATGT and HexR2, 5′-GCTCATGGGCTGGAAGTT. These primers anneal to the hexon gene of species C HAdV (nucleotide position 20047 to 21291 of HAdV2; GenBank accession number J01917.1), and generated an amplicon of 1,245 bp (Table 2). The products were sequenced using automated DNA analyzer (Sangong Biotech, Nanjing, China). The sequences were subjected to a BLAST search against the GenBank database and aligned using the ClustalW2 program.
TABLE 2.
List of primers used for HAdV characterization and ChIP assaysa
| Primer name | Forward sequence | Reverse sequence | Amplicon size (bp) | Amplicon nucleotide position |
|---|---|---|---|---|
| HexF1/R1 | 5′-AACTTCCAGCCCATGAGC | 5′-GAAGGGCGTGCGCAGGTA | 477 | 21274–21720 |
| HexF2/R2 | 5′-GACAGCTATGATCCAGATGT | 5′-GCTCATGGGCTGGAAGTT | 1,245 | 20047–21291 |
| E1 | 5′-CCACCTACCCTTCACGAACT | 5′-CGCCAACATTACAGACTCGG | 102 | 676–777 |
| E2 | 5′-AAGCCCAAGAAATCCACAGC | 5′-CCTATTTCTAAGCTCGCGGG | 96 | 26840–26935 |
| E3 | 5′-ACCACTGCTACCGGACTAAC | 5′-AAACCACCACATGTCCAAGC | 90 | 29510–29599 |
| E4 | 5′-CGGTAAACACATCAGGTTGGT | 5′-GCTGTAATGTTGTCTCTACGCC | 100 | 35304–35403 |
| IVa2 | 5′-GGGAGATCAGCTGGGAAGAA | 5′-GTTGCAGCCGGTAATAGGTG | 92 | 4857–4948 |
The primers were designed based on the sequence of HAdV2 (GenBank accession number J01917.1). Their relative positions on the sequence with accession number J01917.1 were given. The E1 and E4 primer pairs anneal to the E1a and E4 gene introns, respectively; E2 and IVa2 to the corresponding gene promoter regions; and E3 to the E3 gene-coding region. HexF1/R1 refers to the hexon gene and the primers anneal to species B, C, D, and E HAdV, while HexF2/R2 anneal to species C HAdV. IVa2, DNA encapsidation gene.
Real-time PCR were carried out using the Applied Biosystems 7300 real-time PCR system with SYBR green PCR master mix (Q711-02-AA; Vazyme, Nanjing, China). Data analysis was carried out with the with 2−ΔΔCT value method to obtain values of relative abundances. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for normalization.
Xenograft model and in vivo activation studies.
We used a xenograft model to investigate HAdV reactivation in vivo (63). All experimental procedures were carried out strictly in accordance with the guide for the care and use of laboratory animals and the related ethical regulations instilled at Nanjing University Medical School. Female BALB/c athymic mice (catalog number D000521) of 4 to 6 weeks of age were purchased from Model Animal Research Center of Nanjing University and were housed in microisolator cages under specific-pathogen-free conditions on a 12:12-h light:dark cycle with free access to food and water.
The tonsils were thoroughly washed with serum-free RPMI 1640 containing antibiotics. The tissue samples were cut into small cubes of approximately 2 mm in each dimension. Mice were anesthetized with ketamine. Under aseptic conditions, tissue fragments were surgically implanted in the rear flanks (one on each side). The surgery procedure resulted in minimal postoperative morbidity and mortality. Some of the tissue samples were snap-frozen and stored at −70°C as preimplantation controls.
Forty-eight hours after the surgery, mice (3 per group) were randomly grouped into vehicle (10% DMSO in normal saline), TSA (10 mg/kg of body weight), LAQ824 (10 mg/kg), or PMA (30 μg/kg) groups. On day 3, the mice were treated by subcutaneous (s.c.) injection near the sites of the implants for 3 consecutive days. The compounds were dissolved in DMSO and were freshly diluted with normal saline prior to each injection (30 μl; DMSO at approximately 10%). The tissues were removed on day 7 and were tested for DNA replication by qPCR.
Chromatin immunoprecipitation (ChIP) assay.
The ChIP assay was carried out to determine histone and viral DNA interactions following a previous report (41). Briefly, A549 cells in 10-cm dishes were uninfected or infected with HAdV2 (MOI, 15) for 6 h. The cells were then rinsed with ice-cold phosphate-buffered saline (PBS) three times and subsequently fixed with 1% paraformaldehyde for 10 min at room temperature. After removal of the fixation solution, the cells were then incubated with 10 ml of 125 mM glycine in PBS for 5 min to quench excess paraformaldehyde, followed by washing with ice-cold PBS. The cells (1 × 107) were collected into 1 ml cell lysis buffer containing 1% SDS, 1 mM EDTA, and protease inhibitors with a cell scraper. The mixture was placed on ice for 15 min followed by sufficient sonication at an amplitude of 30% on a 10-s on-and-off cycle for 20 min to shear DNA. For immunoprecipitation, the chromatin preparation was diluted with 10× ChIP dilution buffer and precleared with 70-μl protein G agarose beads coated with sheared herring sperm DNA (SS-DNA) and bovine serum albumin (BSA). The chromatin preparation was incubated at 4°C overnight with an antibody and protein G beads. The immune complexes were washed once with low salt wash buffer, a high salt wash buffer, then lithium chloride, and twice with Tris-EDTA buffer. The complexes were eluted with 1% SDS and 100 mM sodium bicarbonate (250 μl). After cross-link reversal by incubating at 65°C for overnight, the eluants were treated with proteinase K (0.2 mg/ml). The DNA was purified by phenol-chloroform extraction and ethanol precipitation. The recovered DNA was detected by real-time PCR and PCR using primers targeting the genes or gene promoters of the HAdV early genes and the IVa2 late gene (Table 2).
Western blot.
Cell lysates were subjected to SDS-PAGE. The proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore). After blocking for 1 h with 3% milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T), the membrane was incubated with a primary antibody diluted in TBS-T containing 1% milk. The membranes were then incubated with a corresponding secondary antibody conjugated with HRP. The blots were detected using an ECL reagent kit (Beyotime) and the ChemiScope 6000 Touch imaging system (Clinx, China).
Statistical analysis.
Statistical analysis was performed by the one-way analysis of variance (ANOVA) method using the SPSS 17.0 software package. Data were analyzed by Student’s t test. A P value of equal to or less than 0.05 was considered statistically significant for all statistical tests.
ACKNOWLEDGMENTS
The work was supported by grants from the NSFC (81871636 and 81071859), by Key Project of Research and Development of Ningxia Hui Autonomous Region (2017-BN04), and a grant from Central Universities Fundamental Research Funds (14380456) to E.L.
We thank members from Jiangsu Key Laboratory of Molecular Medicine for technical assistance.
REFERENCES
- 1.Lynch JP III, Kajon AE. 2016. Adenovirus: epidemiology, global spread of novel serotypes, and advances in treatment and prevention. Semin Respir Crit Care Med 37:586–602. doi: 10.1055/s-0036-1584923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hasegawa K, Goto T, Hirayama A, Laham FR, Mansbach JM, Piedra PA, Camargo CA Jr. 2019. respiratory virus epidemiology among US infants with severe bronchiolitis: analysis of 2 multicenter, multiyear cohort studies. Pediatr Infect Dis J 38:e180–e183. doi: 10.1097/INF.0000000000002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain S, Williams DJ, Arnold SR, Ampofo K, Bramley AM, Reed C, Stockmann C, Anderson EJ, Grijalva CG, Self WH, Zhu Y, Patel A, Hymas W, Chappell JD, Kaufman RA, Kan JH, Dansie D, Lenny N, Hillyard DR, Haynes LM, Levine M, Lindstrom S, Winchell JM, Katz JM, Erdman D, Schneider E, Hicks LA, Wunderink RG, Edwards KM, Pavia AT, McCullers JA, Finelli L, CDC EPIC Study Team. 2015. Community-acquired pneumonia requiring hospitalization among U.S. children. N Engl J Med 372:835–845. doi: 10.1056/NEJMoa1405870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ladenheim HS, Mistchenko AS, Drut R. 1995. Expression of early and late adenoviral proteins in fatal adenovirus bronchopneumonia. Pediatr Pathol Lab Med 15:291–298. doi: 10.3109/15513819509026964. [DOI] [PubMed] [Google Scholar]
- 5.Rosman FC, Mistchenko AS, Ladenheim HS, do Nascimento JP, Outani HN, Madi K, Lenzi HL. 1996. Acute and chronic human adenovirus pneumonia: cellular and extracellular matrix components. Pediatr Pathol Lab Med 16:521–541. doi: 10.1080/15513819609168688. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi S, Hogg JC. 2007. Adenovirus infections and lung disease. Curr Opin Pharmacol 7:237–243. doi: 10.1016/j.coph.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kajon AE, Lamson DM, St. George K. 2019. Emergence and re-emergence of respiratory adenoviruses in the United States. Curr Opin Virol 34:63–69. doi: 10.1016/j.coviro.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Tortora RP, Guimaraes M, de Souza LM, Santos IA, Varella RB, de Fatima Pombo March M, da Cunha A, Sant’ Anna CC. 2015. Adenovirus species C detection in children under four years of age with acute bronchiolitis or recurrent wheezing. J Clin Virol 73:77–80. doi: 10.1016/j.jcv.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Zhou W, Zhao Y, Wang Y, Xie Z, Lou Y, Tan W. 2015. Molecular typing and epidemiology profiles of human adenovirus infection among paediatric patients with severe acute respiratory infection in China. PLoS One 10:e0123234. doi: 10.1371/journal.pone.0123234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Veen J, Lambriex M. 1973. Relationship of adenovirus to lymphocytes in naturally infected human tonsils and adenoids. Infect Immun 7:604–609. doi: 10.1128/IAI.7.4.604-609.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox JP, Hall CE, Cooney MK. 1977. The Seattle Virus Watch. VII. Observations of adenovirus infections. Am J Epidemiol 105:362–386. doi: 10.1093/oxfordjournals.aje.a112394. [DOI] [PubMed] [Google Scholar]
- 12.Evans AS, Evans B, Gogat G. 1958. Latent adenovirus infections of the human respiratory tract. Am J Hyg 67:256–266. doi: 10.1093/oxfordjournals.aje.a119932. [DOI] [PubMed] [Google Scholar]
- 13.Lion T. 2019. Adenovirus persistence, reactivation, and clinical management. FEBS Lett 593:3571–3582. doi: 10.1002/1873-3468.13576. [DOI] [PubMed] [Google Scholar]
- 14.Radke JR, Cook JL. 2018. Human adenovirus infections: update and consideration of mechanisms of viral persistence. Curr Opin Infect Dis 31:251–256. doi: 10.1097/QCO.0000000000000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walls T, Shankar AG, Shingadia D. 2003. Adenovirus: an increasingly important pathogen in paediatric bone marrow transplant patients. Lancet Infect Dis 3:79–86. doi: 10.1016/S1473-3099(03)00515-2. [DOI] [PubMed] [Google Scholar]
- 16.Sandherr M, Einsele H, Hebart H, Kahl C, Kern W, Kiehl M, Massenkeil G, Penack O, Schiel X, Schuettrumpf S, Ullmann AJ, Cornely OA. 2006. Antiviral prophylaxis in patients with haematological malignancies and solid tumours: guidelines of the Infectious Diseases Working Party (AGIHO) of the German Society for Hematology and Oncology (DGHO). Ann Oncol 17:1051–1059. doi: 10.1093/annonc/mdj132. [DOI] [PubMed] [Google Scholar]
- 17.Ullmann AJ, Schmidt-Hieber M, Bertz H, Heinz WJ, Kiehl M, Kruger W, Mousset S, Neuburger S, Neumann S, Penack O, Silling G, Vehreschild JJ, Einsele H, Maschmeyer G; Infectious Diseases Working Party of the German Society for Hematology and Medical Oncology (AGIHO/DGHO) and the DAG-KBT (German Working Group for Blood and Marrow Transplantation). 2016. Infectious diseases in allogeneic haematopoietic stem cell transplantation: prevention and prophylaxis strategy guidelines 2016. Ann Hematol 95:1435–1455. doi: 10.1007/s00277-016-2711-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kosulin K, Geiger E, Vecsei A, Huber WD, Rauch M, Brenner E, Wrba F, Hammer K, Innerhofer A, Potschger U, Lawitschka A, Matthes-Leodolter S, Fritsch G, Lion T. 2016. Persistence and reactivation of human adenoviruses in the gastrointestinal tract. Clin Microbiol Infect 22:381.e1–381.e8. doi: 10.1016/j.cmi.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 19.Hum RM, Deambrosis D, Lum SH, Davies E, Bonney D, Guiver M, Turner A, Wynn RF, Hiwarkar P. 2018. Molecular monitoring of adenovirus reactivation in faeces after haematopoietic stem-cell transplantation to predict systemic infection: a retrospective cohort study. Lancet Haematol 5:e422–e429. doi: 10.1016/S2352-3026(18)30130-3. [DOI] [PubMed] [Google Scholar]
- 20.Neumann R, Genersch E, Eggers HJ. 1987. Detection of adenovirus nucleic acid sequences in human tonsils in the absence of infectious virus. Virus Res 7:93–97. doi: 10.1016/0168-1702(87)90060-8. [DOI] [PubMed] [Google Scholar]
- 21.Matsuse T, Hayashi S, Kuwano K, Keunecke H, Jefferies WA, Hogg JC. 1992. Latent adenoviral infection in the pathogenesis of chronic airways obstruction. Am Rev Respir Dis 146:177–184. doi: 10.1164/ajrccm/146.1.177. [DOI] [PubMed] [Google Scholar]
- 22.Ross PJ, Kennedy MA, Christou C, Risco Quiroz M, Poulin KL, Parks RJ. 2011. Assembly of helper-dependent adenovirus DNA into chromatin promotes efficient gene expression. J Virol 85:3950–3958. doi: 10.1128/JVI.01787-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garnett CT, Talekar G, Mahr JA, Huang W, Zhang Y, Ornelles DA, Gooding LR. 2009. Latent species C adenoviruses in human tonsil tissues. J Virol 83:2417–2428. doi: 10.1128/JVI.02392-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garnett CT, Erdman D, Xu W, Gooding LR. 2002. Prevalence and quantitation of species C adenovirus DNA in human mucosal lymphocytes. J Virol 76:10608–10616. doi: 10.1128/JVI.76.21.10608-10616.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akimova T, Beier UH, Liu Y, Wang L, Hancock WW. 2012. Histone/protein deacetylases and T-cell immune responses. Blood 119:2443–2451. doi: 10.1182/blood-2011-10-292003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roh TY, Cuddapah S, Cui K, Zhao K. 2006. The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci U S A 103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, Hellmann MD, Wolchok JD, Leslie CS, Schietinger A. 2017. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pereira RM, Hogan PG, Rao A, Martinez GJ. 2017. Transcriptional and epigenetic regulation of T cell hyporesponsiveness. J Leukoc Biol 102:601–615. doi: 10.1189/jlb.2RI0317-097R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Courtney AH, Lo WL, Weiss A. 2018. TCR signaling: mechanisms of initiation and propagation. Trends Biochem Sci 43:108–123. doi: 10.1016/j.tibs.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Traylen CM, Patel HR, Fondaw W, Mahatme S, Williams JF, Walker LR, Dyson OF, Arce S, Akula SM. 2011. Virus reactivation: a panoramic view in human infections. Future Virol 6:451–463. doi: 10.2217/fvl.11.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoeben RC, Uil TG. 2013. Adenovirus DNA replication. Cold Spring Harb Perspect Biol 5:a013003. doi: 10.1101/cshperspect.a013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knipe DM, Lieberman PM, Jung JU, McBride AA, Morris KV, Ott M, Margolis D, Nieto A, Nevels M, Parks RJ, Kristie TM. 2013. Snapshots: chromatin control of viral infection. Virology 435:141–156. doi: 10.1016/j.virol.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giberson AN, Saha B, Campbell K, Christou C, Poulin KL, Parks RJ. 2018. Human adenoviral DNA association with nucleosomes containing histone variant H3.3 during the early phase of infection is not dependent on viral transcription or replication. Biochem Cell Biol 96:797–807. doi: 10.1139/bcb-2018-0117. [DOI] [PubMed] [Google Scholar]
- 34.Giberson AN, Davidson AR, Parks RJ. 2012. Chromatin structure of adenovirus DNA throughout infection. Nucleic Acids Res 40:2369–2376. doi: 10.1093/nar/gkr1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horwitz GA, Zhang K, McBrian MA, Grunstein M, Kurdistani SK, Berk AJ. 2008. Adenovirus small e1a alters global patterns of histone modification. Science 321:1084–1085. doi: 10.1126/science.1155544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrari R, Pellegrini M, Horwitz GA, Xie W, Berk AJ, Kurdistani SK. 2008. Epigenetic reprogramming by adenovirus e1a. Science 321:1086–1088. doi: 10.1126/science.1155546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrari R, Gou D, Jawdekar G, Johnson SA, Nava M, Su T, Yousef AF, Zemke NR, Pellegrini M, Kurdistani SK, Berk AJ. 2014. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 16:663–676. doi: 10.1016/j.chom.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komatsu T, Haruki H, Nagata K. 2011. Cellular and viral chromatin proteins are positive factors in the regulation of adenovirus gene expression. Nucleic Acids Res 39:889–901. doi: 10.1093/nar/gkq783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 40.Bode AM, Dong Z. 2005. Inducible covalent posttranslational modification of histone H3. Sci STKE 2005:re4. doi: 10.1126/stke.2812005re4. [DOI] [PubMed] [Google Scholar]
- 41.Hsu E, Pennella MA, Zemke NR, Eng C, Berk AJ. 2018. Adenovirus E1A activation domain regulates H3 acetylation affecting varied steps in transcription at different viral promoters. J Virol 92:e00805-18. doi: 10.1128/JVI.00805-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Binder AM, Biggs HM, Haynes AK, Chommanard C, Lu X, Erdman DD, Watson JT, Gerber SI. 2017. Human adenovirus surveillance—United States, 2003–2016. MMWR Morb Mortal Wkly Rep 66:1039–1042. doi: 10.15585/mmwr.mm6639a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rowe WP, Huebner RJ, Gilmore LK, Parrott RH, Ward TG. 1953. Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Proc Soc Exp Biol Med 84:570–573. doi: 10.3181/00379727-84-20714. [DOI] [PubMed] [Google Scholar]
- 44.Roh TY, Cuddapah S, Zhao K. 2005. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev 19:542–552. doi: 10.1101/gad.1272505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saha B, Parks RJ. 2019. Histone deacetylase inhibitor suberoylanilide hydroxamic acid suppresses human adenovirus gene expression and replication. J Virol 93:e00088-19. doi: 10.1128/JVI.00088-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Komatsu T, Quentin-Froignant C, Carlon-Andres I, Lagadec F, Rayne F, Ragues J, Kehlenbach RH, Zhang W, Ehrhardt A, Bystricky K, Morin R, Lagarde JM, Gallardo F, Wodrich H. 2018. In vivo labelling of adenovirus DNA identifies chromatin anchoring and biphasic genome replication. J Virol 92:e00795-18. doi: 10.1128/JVI.00795-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herrmann CH, Gold MO, Rice AP. 1996. Viral transactivators specifically target distinct cellular protein kinases that phosphorylate the RNA polymerase II C-terminal domain. Nucleic Acids Res 24:501–508. doi: 10.1093/nar/24.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gold MO, Tassan JP, Nigg EA, Rice AP, Herrmann CH. 1996. Viral transactivators E1A and VP16 interact with a large complex that is associated with CTD kinase activity and contains CDK8. Nucleic Acids Res 24:3771–3777. doi: 10.1093/nar/24.19.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fonseca GJ, Thillainadesan G, Yousef AF, Ablack JN, Mossman KL, Torchia J, Mymryk JS. 2012. Adenovirus evasion of interferon-mediated innate immunity by direct antagonism of a cellular histone posttranslational modification. Cell Host Microbe 11:597–606. doi: 10.1016/j.chom.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 50.Lieberman PM. 2016. Epigenetics and genetics of viral latency. Cell Host Microbe 19:619–628. doi: 10.1016/j.chom.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cary DC, Fujinaga K, Peterlin BM. 2016. Molecular mechanisms of HIV latency. J Clin Invest 126:448–454. doi: 10.1172/JCI80565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Q, Yao H, Vo N, Goodman RH. 2000. Acetylation of adenovirus E1A regulates binding of the transcriptional corepressor CtBP. Proc Natl Acad Sci U S A 97:14323–14328. doi: 10.1073/pnas.011283598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perry M, Whyte A. 1998. Immunology of the tonsils. Immunol Today 19:414–421. doi: 10.1016/S0167-5699(98)01307-3. [DOI] [PubMed] [Google Scholar]
- 54.Jensen A, Fagö-Olsen H, Sørensen CH, Kilian M. 2013. Molecular mapping to species level of the tonsillar crypt microbiota associated with health and recurrent tonsillitis. PLoS One 8:e56418. doi: 10.1371/journal.pone.0056418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding T, Schloss PD. 2014. Dynamics and associations of microbial community types across the human body. Nature 509:357–360. doi: 10.1038/nature13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen TS, Prince AS. 2013. Activation of inflammasome signaling mediates pathology of acute P aeruginosa pneumonia. J Clin Invest 123:1630–1637. doi: 10.1172/JCI66142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O'Neill LA. 2016. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167:457–470.e13. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smale ST. 2010. Selective transcription in response to an inflammatory stimulus. Cell 140:833–844. doi: 10.1016/j.cell.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Riquelme SA, Wong Fok Lung T, Prince A. 2020. Pulmonary pathogens adapt to immune signaling metabolites in the airway. Front Immunol 11:385. doi: 10.3389/fimmu.2020.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Licciardi PV, Wong SS, Tang ML, Karagiannis TC. 2010. Epigenome targeting by probiotic metabolites. Gut Pathog 2:24. doi: 10.1186/1757-4749-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Assadian F, Sandstrom K, Laurell G, Svensson C, Akusjarvi G, Punga T. 2015. Efficient isolation protocol for B and T lymphocytes from human palatine tonsils. J Vis Exp doi: 10.3791/53374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 63.Inoue T, Terada N, Kobayashi T, Ogawa O. 2017. Patient-derived xenografts as in vivo models for research in urological malignancies. Nat Rev Urol 14:267–283. doi: 10.1038/nrurol.2017.19. [DOI] [PubMed] [Google Scholar]