

Viral oncoproteins, including HPV16 E6 and E7, have been instrumental in elucidating the activities of cellular signaling networks, including those governed by the TP53 tumor suppressor. Our study demonstrates that the long noncoding RNA DINO is the long-sought missing link between HPV16 E7 and elevated TP53 levels. Importantly, the TP53-stabilizing DINO plays a critical role in the cell death response of HPV16 E7-expressing cells to metabolic stress or DNA damage.
KEYWORDS: human papillomavirus, E7, lncRNA, DINO, DINOL, TP53, metformin, metabolism, DNA damage, chemotherapy, KDM6A, long noncoding RNA
ABSTRACT
Human papillomavirus 16 (HPV16) E7 has long been known to stabilize the tumor suppressor TP53. However, the molecular mechanism of TP53 stabilization by HPV16 E7 has remained obscure, and this stabilization can occur independently of the E2F-regulated MDM2 inhibitor p14ARF. Here, we report that the damage-induced noncoding (DINO) lncRNA (DINOL) is the “missing link” between HPV16 E7 and increased TP53 levels. DINO levels are decreased in cells where TP53 is inactivated, either by HPV16 E6, by expression of a dominant negative TP53 minigene, or by TP53 depletion. DINO levels are increased in HPV16 E7-expressing cells. HPV16 E7 causes increased DINO expression independently of RB1 degradation and E2F1 activation. Similar to what is seen with the adjacent CDKN1A locus, DINO expression is regulated by the histone demethylase KDM6A. DINO stabilizes TP53 in HPV16 E7-expressing cells, and as it is a TP53 transcriptional target, DINO levels further increase. As with expression of other oncogenes, such as adenovirus E1A or MYC, HPV16 E7-expressing cells are sensitized to cell death under conditions of metabolic stress, which in the case of E7 has been linked to TP53 activation. Consistent with earlier studies, we show that HPV16 E7-expressing keratinocytes are highly sensitive to metabolic stress induced by starvation or the antidiabetic drug metformin. Sensitivity of HPV16 E7-expressing cells to metabolic stress is rescued by DINO depletion. Moreover, DINO depletion decreases sensitivity to the DNA damage-inducing chemotherapy agent doxorubicin. This work identifies DINO as a critical mediator of TP53 stabilization and activation in HPV16 E7-expressing cells.
IMPORTANCE Viral oncoproteins, including HPV16 E6 and E7, have been instrumental in elucidating the activities of cellular signaling networks, including those governed by the TP53 tumor suppressor. Our study demonstrates that the long noncoding RNA DINO is the long-sought missing link between HPV16 E7 and elevated TP53 levels. Importantly, the TP53-stabilizing DINO plays a critical role in the cell death response of HPV16 E7-expressing cells to metabolic stress or DNA damage.
INTRODUCTION
The E6 and E7 proteins of the cancer-associated, high-risk human papillomaviruses (HPVs) have oncogenic activities and are necessary for tumor induction and maintenance. These low-molecular-weight proteins lack intrinsic enzymatic activities and function by subverting host cellular regulatory networks (1, 2). Over the years, many potential cellular targets of the HPV E6 and E7 proteins have been identified. The main focus of these studies, however, has been the identification of host cellular protein targets and the resulting dysregulation of protein-coding mRNAs. Protein-coding genes, however, constitute only a minor fraction (<3%) of the human transcriptome (3), and advances in next-generation sequencing technologies have shown that noncoding RNAs, including microRNAs and long noncoding RNAs (lncRNAs), are also aberrantly expressed in HPV E6- and/or E7-expressing cells (4–7). It is now firmly established that noncoding RNAs are important regulators of a variety of cellular processes that drive specific biological phenotypes (8–11).
Many viral proteins, including the E6 and E7 proteins encoded by high-risk HPVs, have evolved to modulate the TP53 tumor suppressor pathway (12). TP53 is critically involved in sensing many forms of cellular stress, including those caused by viral infection, nutrient starvation, or DNA damage. Upon activation, TP53 is stabilized and elicits transcriptional programs that trigger cytostatic or cytotoxic responses, including cycle arrest, differentiation, senescence, or apoptosis (13). Hence, TP53 levels and activity are tightly regulated. A well-known regulator of TP53 is the MDM2 ubiquitin ligase. MDM2 is a TP53-responsive gene which in turns binds and targets TP53 for ubiquitin-dependent proteasomal degradation (14). MDM2 activity is regulated at multiple levels. One important inhibitor of MDM2 is the p14ARF protein, which is regulated by E2F transcription factors, the downstream effectors of the retinoblastoma (RB1) tumor suppressor.
The E6 and E7 proteins encoded by high-risk HPVs, including HPV16, each modulate TP53 levels and activity. HPV16 E6 targets TP53 for proteasome-mediated degradation through an MDM2-independent mechanism that involves the E6-associated ubiquitin ligase, UBE3A (E6AP) (15, 16). The ability of high-risk HPV E6 proteins to inhibit TP53 is thought to have evolved from the necessity to counteract TP53 stabilization and activation triggered by expression of the HPV E7 oncoprotein (17–22). Consequently, several other DNA tumor viruses, including adenoviruses and many polyomaviruses that encode proteins that inactivate the RB1 tumor suppressor, also encode proteins that dampen TP53 tumor suppressor activity. In the case of the adenovirus E1A protein, TP53 stabilization was shown to be mediated by the E2F-induced MDM2 inhibitor p14ARF (23). Surprisingly, however, HPV16 E7 has been shown to stabilize TP53 independently of p14ARF (21), and until now, the mediator of HPV16 E7-mediated TP53 stabilization has remained elusive.
In recent years, it has been appreciated that various host cellular noncoding RNAs also function as important regulators of the TP53 tumor suppressor pathway. In addition to microRNAs (24), a variety of cellular lncRNAs have been identified as components of the TP53 signaling network (25). lncRNAs are defined as transcripts longer than 200 bp with no or limited (<100 amino acids) protein coding capacity. lncRNAs are versatile molecules and can form complexes with RNA, DNA, and proteins. Some lncRNAs, including lincRNA-p21, PURPL, MEG3, and DINO, are transcriptional targets of TP53 and can also modulate TP53 levels and activity (26–29). DINO (damage-induced noncoding) lncRNA expression is induced by TP53 as a consequence of DNA damage. It has been reported that DINO binds TP53, thereby stabilizing and activating it. Hence, DINO functionally counteracts MDM2 (28). In the present article, we report that DINO is expressed at high levels in HPV16 E7-expressing cells and that this upregulation correlates with the ability of HPV16 E7 to cause TP53 stabilization. High-level DINO expression in HPV16 E7-expressing cells is driven by the histone demethylase KDM6A and TP53, and DINO, in turn, is necessary for maintaining elevated TP53 levels in HPV16 E7-expressing cells. Moreover, we show that the sensitivity of cells to undergo cell death in response to metabolic stress can be controlled by modulating DINO levels.
RESULTS
DINO levels correlate with TP53 levels in HPV oncoprotein-expressing cells.
Expression of high-risk HPV E6 and E7 proteins is known to modulate TP53 levels and activity. While HPV16 E7 expression causes TP53 stabilization, HPV16 E6 expression causes TP53 degradation. Hence, we determined whether the levels of DINO, a TP53-driven lncRNA, correlate with TP53 levels in HPV16 E6 and/or E7-expressing primary human foreskin keratinocytes (HFKs). HFKs with expression of a dominant negative TP53 (dnTP53) C-terminal fragment were used as a control (30). We assessed TP53 levels by immunoblotting and quantified DINO levels by quantitative reverse transcription-PCR (qRT-PCR). Compared to control HFKs, HFK populations where TP53 is inactivated either by the expression of the HPV16 E6 protein or by the dnTP53 C-terminal minigene had lower DINO levels (E6, 0.14 ± 0.01, P < 0.001; E6/E7, 0.22 ± 0.04, P < 0.001; dnTP53, 0.28 ± 0.01, P < 0.001) (Fig. 1). This is consistent with the original study that showed that DINO expression is controlled by TP53 (28). In contrast, HPV16 E7-expressing HFKs, which, as reported previously (17, 20, 21), express high levels of TP53 (Fig. 1), expressed significantly increased DINO levels (E7, 6.14 ± 0.14, P < 0.001) (Fig. 1B). Hence, DINO levels correlate with TP53 levels in HPV oncoprotein-expressing cells.
FIG 1.
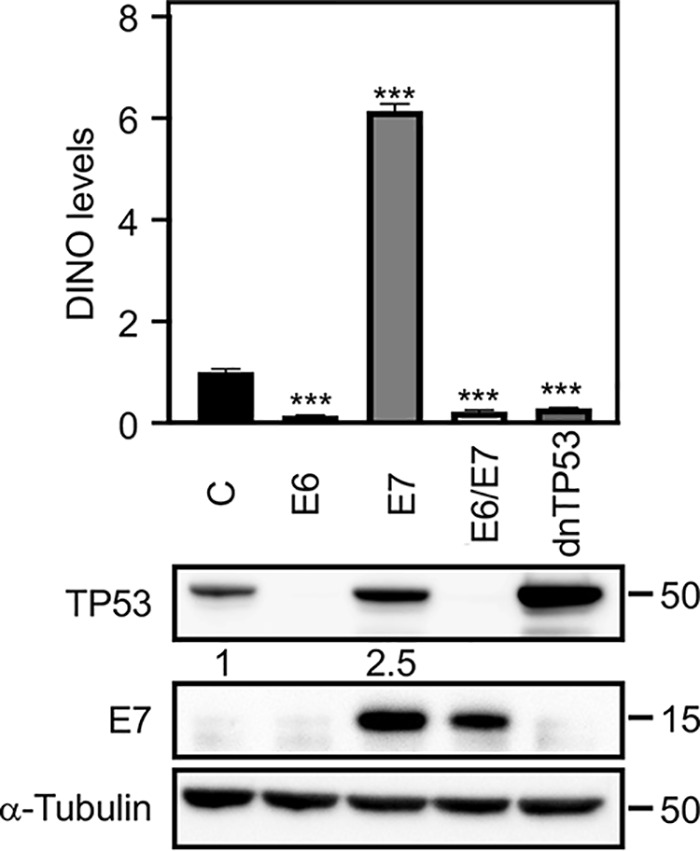
DINO levels correlate with TP53 levels in HPV oncoprotein-expressing cells. HFK populations with stable expression of HPV16 E6 and/or E7 or a dominant negative TP53 minigene (dnTP53) were validated by determining the levels of TP53 and E7 by Western blotting. DINO levels were assessed in these HFK populations by qRT-PCR assays. Levels shown are relative to those for control vector-transduced HFKs. The bar graph shows means and standard errors of the means (SEM; n = 3) calculated from a single representative experiment. ***, P < 0.001 (Student's t test). Similar results were obtained with three independently derived HFK populations.
TP53-driven DINO causes elevated TP53 levels in HPV16 E7-expressing cells.
Given the correlation between DINO and TP53 levels in E7-expressing cells, we next determined whether DINO expression in HPV16 E7-expressing cells was driven by TP53. We transiently depleted TP53 in control as well as in HPV16 E7-expressing human telomerase immortalized HFKs (iHFKs) with a pool of TP53 targeting small interfering RNAs (siRNAs). To assess the effect of TP53 depletion on expression of TP53 target genes, we analyzed expression of CDKN1A (p21CIP1) by Western blotting and qRT-PCR. As expected, CDKN1A protein and mRNA levels were decreased in control and HPV16 E7-expressing cells upon TP53 depletion (Fig. 2A to C). Similarly, TP53 depletion also caused a significant decrease of DINO in control as well as in HPV16 E7-expressing cells (control, 0.09 ± 0.01, P < 0.001; E7, 0.08 ± 0.02, P < 0.001) (Fig. 2B).
FIG 2.

DINO causes elevated TP53 levels in HPV16 E7-expressing cells. TP53 depletion in both control (C) and HPV16 E7-expressing (E7) iHFKs was validated by Western blotting and assessment of the canonical TP53 transcriptional target CKDN1A protein (A) and mRNA (B) levels. DINO levels were determined in TP53-depleted iHFKs by qRT-PCR assays, and results are relative to levels in control siRNA-transfected iHFKs (B). For acute depletion of DINO, HPV16 E7-expressing iHFKs harboring doxycycline-inducible DINO shRNA expression vectors were treated with 1 μg/ml doxycycline for 48 h. HPV16 E7 iHFKs with acute expression of nontargeting shRNA sequences were used as negative controls. Levels of TP53 protein were determined by Western blotting (C). Expression of the TP53-responsive CDKN1A mRNA and MDM2 mRNA, as well as DINO depletion, was assessed by qRT-PCR (D). The bar graphs present means and SEM (n = 3) calculated from a single representative experiment. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (Student's t test). Similar results were obtained in three independent experiments.
To determine whether DINO may be the “missing link” that mediates TP53 stabilization in E7-expressing cells, we acutely depleted DINO in HPV16 E7-expressing iHFKs using doxycycline-regulated short hairpin RNA (shRNA) expression. Depletion of DINO after doxycycline treatment for 48 h by two different shRNAs each caused an approximately 50% decrease in TP53 levels in E7-expressing cells (Fig. 2C). Similarly, mRNA levels of the TP53 transcriptional target gene, the CDKN1A gene, were also decreased (sh-DINO1, 0.61 ± 0.01, P < 0.01; sh-DINO2, 0.48 ± 0.02, P < 0.01) (Fig. 2D). A similar effect was observed when the expression of another TP53 transcriptional target gene, MDM2, was analyzed (sh-DINO1, 0.53 ± 0.01, P < 0.01; sh-DINO2: 0.44 ± 0.02, P < 0.03). Hence, DINO levels accumulate in HPV16 E7-expressing cells as a result of TP53 activation, and in addition, they amplify TP53 stabilization and presumably TP53 activation in HPV16 E7-expressing cells.
HPV16 E7 stabilizes TP53 independently of p14ARF.
It has been reported that HPV16 E7 can stabilize TP53 in p19Arf-deficient mouse embryo fibroblasts (21). To determine whether p14ARF contributed to E7-mediated TP53 stabilization in human keratinocytes, we transiently depleted p14ARF in HPV16 E7-expressing and control vector-transduced HFKs. As expected, p14ARF levels were increased in HPV16 E7-expressing HFKs at the protein (2.1-fold) (Fig. 3A) and mRNA (1.42 ± 0.10-fold) (Fig. 3B) levels. Depletion of p14ARF caused an approximately 50% decrease in p14ARF protein levels (Fig. 3A) and mRNA levels (sip14, 0.51 ± 0.09, P > 0.01) (Fig. 3B) in HPV16 E7-expressing HFKs. Depletion of p14ARF in HPV16 E7-expressing HFKs, however, did not cause a significant decrease in TP53 protein levels (Fig. 3A). Moreover, there was no significant decrease in the mRNA levels of the TP53 transcriptional target gene, CKDN1A (siRNA control, 1.51 ± 0.02; sip14, 1.41 ± 0.10, nonsignificant) (Fig. 3C). Hence, in agreement with an earlier report (21), our experiments showed that HPV16 E7 can stabilize TP53 in human keratinocytes independent of p14ARF.
FIG 3.
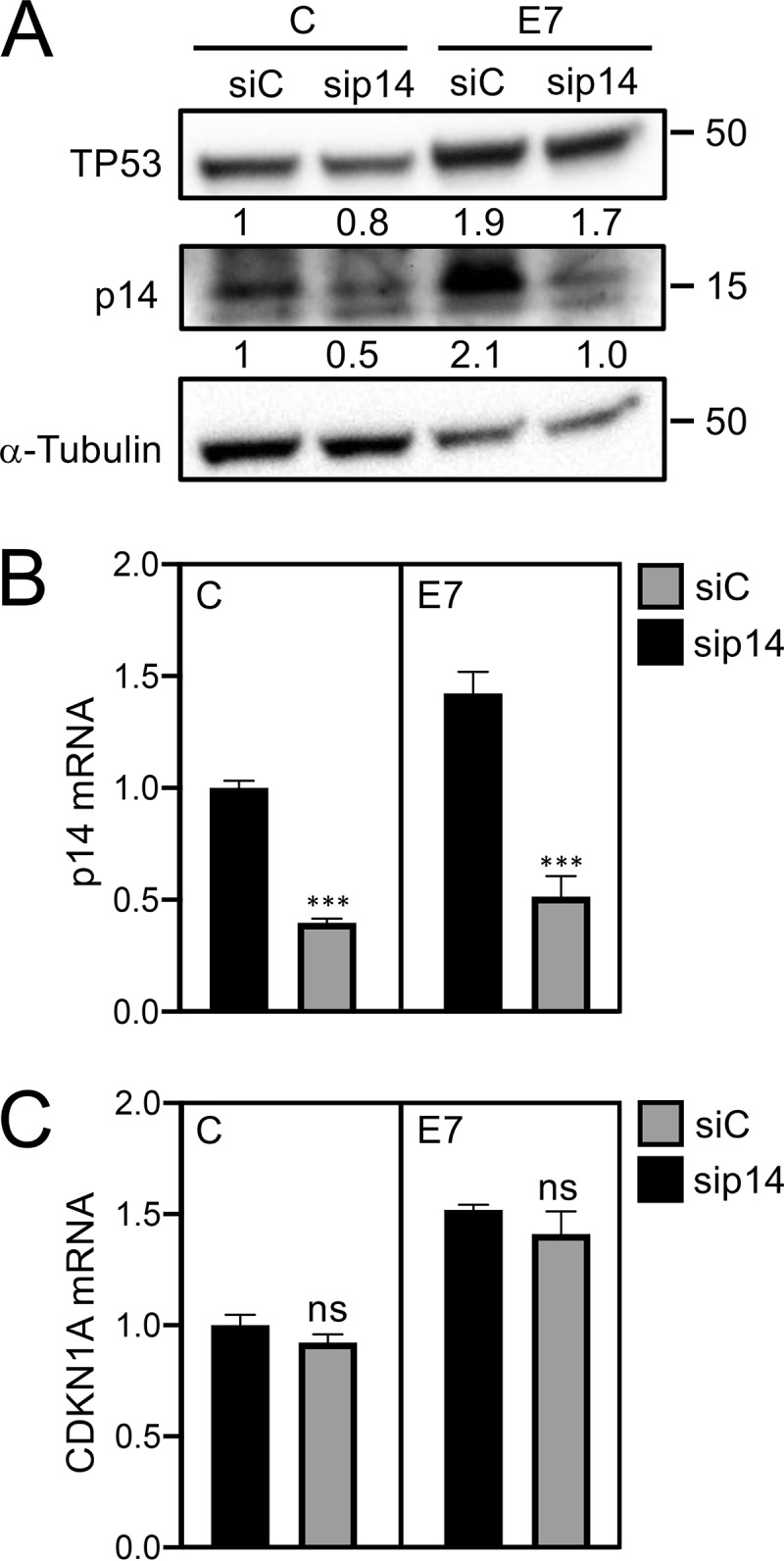
HPV16 E7 stabilizes TP53 independently of p14ARF. Validation of p14ARF depletion in control (C) and HPV16 E7-expressing (E7) HFKs by Western blotting (A) and qRT-PCR (B). The mRNA levels of the TP53-responsive gene CDKN1A were also accessed in p14-depleted HFKs (C). The bar graph presents means and SEM (n = 3) calculated from a single representative experiment. ***, P < 0.001; ns, nonsignificant (Student's t test). Similar results were obtained in two independently derived HFK populations.
TP53 and KDM6A, but not RB1 and E2F1, are the upstream regulators of DINO.
Previous mutational studies with HPV16 E7 revealed that TP53 stabilization by HPV16 E7 required sequences similar to those that are necessary for RB1 destabilization (20). Consistent with these earlier studies, the RB1 binding and degradation defective HPV16 E7 ΔDLYC mutant failed to stabilize TP53 (Fig. 4A). Similarly, unlike wild-type HPV16 E7, expression of HPV16 E7 ΔDLYC also failed to upregulate DINO in iHFKs (E7, 5.88 ± 0.09, P < 0.01; E7 ΔDLYC, 1.09 ± 0.10, nonsignificant) (Fig. 4B). To determine whether DINO levels in HPV16 E7-expressing cells were increased as a consequence of RB1 tumor suppressor degradation and E2F activation, we transiently depleted RB1 and E2F1 in control and HPV16 E7-expressing iHFKs by transfecting the corresponding siRNA pools. Transfections of a TP53-targeting siRNA pool and a nontargeting siRNA pool were used as positive and negative controls, respectively. Depletion of the corresponding proteins was assessed by Western blotting (Fig. 4C) and qRT-PCR assays (Fig. 4D). Whereas, as expected, TP53 depletion caused a significant decrease of DINO levels, RB1 and E2F1 depletion did not cause significant changes of DINO levels in control cells (siRNA control, 1.00 ± 0.12; siRB1, 1.15 ± 0.03, nonsignificant; siE2F1, 0.96 ± 0.14, nonsignificant) or HPV16 E7-expressing cells (siRNA control, 6.64 ± 0.52; siRB1, 5.58 ± 0.56, nonsignificant; siE2F1, 6.15 ± 0.04, nonsignificant) (Fig. 4E). The DINO locus is in close proximity to the CDKN1A locus (28), which is subject to epigenetic derepression by the histone H3 lysine 27 (H3K27) demethylase, KDM6A (31, 32). Hence, we next tested whether DINO expression may also be regulated by KDM6A. Consistent with previous reports (32, 33), we found that KDM6A expression was increased in E7-expressing cells (1.7-fold) (Fig. 4C). Moreover, DINO expression was significantly decreased upon KDM6A depletion (control, 0.61 ± 0.09, P < 0.01; E7, 2.83 ± 0.17, P < 0.001) (Fig. 4E). Hence, HPV16 E7 causes increased DINO expression, at least in part, through a mechanism that involves KDM6A-mediated derepression.
FIG 4.

KDM6A and TP53 but not RB1 and E2F1 are the upstream regulators of DINO. iHFKs expressing either control, wild-type HPV16 E7, or the RB1 binding- and degradation-defective HPV16 E7 ΔDLYC mutant were assessed for RB1 degradation, TP53 stabilization, and E2F1 expression by Western blotting (A). DINO levels were assessed by qRT-PCR (B). Levels shown are relative to those for control vector-transduced iHFKs. TP53, RB1, E2F1, and KDM6A depletion in control (C) and HPV16 E7-expressing (E7) iHFKs was validated by Western blotting (C), as well as by qRT-PCR (D). DINO levels in each of these lines were determined by qRT-PCR (D). Bar graphs present means and SEM (n = 3) calculated from a single representative experiment. ***, P < 0.001; **, P < 0.01; ns, nonsignificant (Student's t test). Similar results were obtained in three independent experiments.
Depletion of DINO protects HPV16 E7-expressing cells from cell death induced by metabolic stress and DNA damage.
Our research group previously reported that HPV16 E7-expressing fibroblasts are sensitized to undergo cell death under conditions of serum starvation. This response was shown to be TP53 dependent, since it was abrogated by coexpression of the HPV16 E6 protein or a dominant negative TP53 minigene (18, 20). Given our results that DINO is critical to TP53 stabilization and activation in HPV16 E7-expressing cells, we next wanted to determine whether cell death responses of HPV16 E7-expressing cells may be controlled by modulating DINO levels. Since keratinocytes are grown in serum-free medium, we first needed to define conditions that might mimic growth factor deprivation in keratinocytes. We evaluated metformin, a drug that induces metabolic stress by activating AMPK, inhibiting mTOR and mitochondrial respiratory complex I, and depleting glycolytic and tricarboxylic acid (TCA) cycle intermediates (34–37). Our results show that HPV16 E7-expressing HFKs are more vulnerable than control HFKs when subjected to metformin treatment (control, 24.78 ± 1.12; E7, 68.48 ± 0.38, P < 0.001) (Fig. 5A). Heightened metformin vulnerability of HPV16 E7-expressing cells was abrogated upon HPV16 E6 coexpression (−0.46 ± 0.20, P < 0.001), consistent with the model that the vulnerability to metformin may be controlled by TP53. To determine whether DINO may modulate metformin-induced cell death, we depleted DINO in HPV16 E7-expressing primary HFKs and treated them with metformin. As expected, DINO depletion caused reduced levels of TP53 and decreased expression of the TP53 transcriptional target CDKN1A (Fig. 5B and C). DINO depletion did not affect the viability of HPV16 E7-expressing HFKs when they were grown under standard tissue culture conditions. In contrast, however, DINO-depleted HPV16 E7-expressing HFKs were significantly more resistant (shRNA control, 50.76 ± 0.78; sh-DINO1, −1.71 ± 0.47, P < 0.001; sh-DINO2, −10.21 ± 0.68, P < 0.001) to metformin treatment than control shRNA-transduced HPV16 E7-expressing HFKs (Fig. 5D). Hence, metformin can be utilized to investigate the metabolic vulnerability of HPV16 E7-expressing cells, and DINO regulates the cellular response to metformin-induced metabolic stress. As an alternate method of inducing metabolic stress, we starved control and DINO-depleted HPV16 E7-expressing HFKs by not changing the cell culture medium for 7 days. Consistent with the metformin experiments, DINO-depleted cells were more resistant (shRNA control, 45.99 ± 0.39; sh-DINO1: 34.86 ± 1.22, P < 0.001; sh-DINO2, 30.68 ± 1.08, P < 0.001) to cell death induced by starvation than control-shRNA transduced HPV16 E7-expressing HFKs (Fig. 5E). Last, we also showed that DINO depletion reduced cell death of HPV16 E7-expressing cells in response to treatment with the DNA damage-inducing chemotherapy agent doxorubicin (shRNA control: 56.71 ± 4.61; sh-DINO1: 33.07 ± 2.84, P < 0.01; sh-DINO2: 28.97 ± 2.74, P < 0.001), a previously described potent inducer of DINO (28) (Fig. 5F).
FIG 5.

Depletion of DINO protects HPV16 E7-expressing cells from cell death induced by metabolic stress and DNA damage. Cell viability was assessed by resazurin assays of control, HPV16 E7-expressing, and HPV16 E6/E7-expressing HFKs that were either untreated (mock) or treated with 20 mM metformin for 96 h (A). TP53 and CDKN1A expression in HPV16 E7-expressing HFKs was determined by Western blotting (B). Expression of CDKN1A mRNA and validation of DINO depletion in these HFK populations were determined by qRT-PCR (C). Viabilities of HPV16 E7-expressing keratinocytes with expression of either of two DINO-specific shRNAs or a scrambled control shRNA were assessed in response to treatment with metformin for 3 days (D), to being fed regularly or being starved for 7 days (E), and to treatment with 0.125 μg/ml of the DNA-damaging chemotherapy agent doxorubicin for 3 days (F). Bar graphs present means and SEM (n = 3) calculated from a single representative experiment. ***, P < 0.001; **, P < 0.01; *, P < 0.05; #, P < 0.001 (Student's t test). Similar results were obtained in three independent experiments.
DISCUSSION
The TP53 tumor suppressor is a central hub that integrates various cellular stress signals and triggers appropriate cytostatic and cytotoxic responses. In normal cells, TP53 has a very short half-life and is present at low levels, because the TP53-regulated MDM2 ubiquitin ligase targets TP53 for proteasomal degradation. Oncogenic insults can trigger TP53 activation. In response to RB1 tumor suppressor inactivation, the E2F-regulated p14ARF protein inhibits MDM2 and causes TP53 activation. However, it became clear early on that there must be additional regulators of TP53, since TP53 was stabilized and activated in p19Arf-null mouse embryo fibroblasts in response to DNA damage (38). Similarly, even though HPV16 E7 causes E2F activation, it can stabilize and activate TP53 in p19Arf-null mouse embryo fibroblasts, and the mechanism of HPV16 E7-mediated TP53 stabilization and activation has remained elusive (21).
DINO is a TP53-responsive gene, and DINO levels increase in response to DNA damage. DINO was reported to bind and stabilize TP53, thereby amplifying the TP53 transcriptional response to DNA damage. Hence, DINO functionally counteracts MDM2 (28, 39). Here, we show that DINO expression correlates with TP53 expression in HPV16 oncoprotein-expressing cells and is highly expressed in HPV16 E7-expressing keratinocytes. DINO depletion causes a decrease in TP53 levels in HPV16 E7-expressing cells, and thus, DINO is the missing link between HPV16 E7 expression and TP53 stabilization. The RB1 binding- and degradation-defective HPV16 E7 ΔDLYC mutant, which was previously shown to be defective for TP53 stabilization (20), does not cause increased DINO expression. Hence, we were quite surprised that we did not detect any changes in DINO expression when RB1 was depleted by RNA interference. This suggests that RB1 degradation and the ensuing activation of E2F are not the primary triggers of DINO expression. In contrast, however, depletion of the H3K27 demethylase KDM6A, which is expressed at higher levels in E7-expressing cells, causes a significant decrease in DINO. Hence, our results suggest a model whereby E7-mediated upregulation of KDM6A may provide an initial trigger for DINO induction (Fig. 6). However, our current results cannot distinguish whether, as is seen with the proximal CDKN1A locus (28, 32), DINO expression is directly derepressed through KDM6A-mediated H3K27 demethylation or whether this involves an indirect mechanism. It is noted, however, that DINO and CDKN1A are distinct genetic loci (28). According to our model, the initial KDM6A-mediated increase in DINO expression causes TP53 stabilization and activation, which in turn causes a further increase in DINO and TP53. Given that E7 causes increased p14ARF expression presumably as a consequence of RB1 degradation and E2F activation, this may impair MDM2 activity and cause a further increase in TP53 and DINO. However, p14ARF depletion did not cause significant changes in TP53 levels in our experiments. Importantly, our model, where the initial trigger of DINO expression is at least in part through KDM6A, accommodates the finding that HPV16 E7 expression can cause increased TP53 levels in p19Arf-deficient mouse embryo fibroblasts (21) (Fig. 6).
FIG 6.
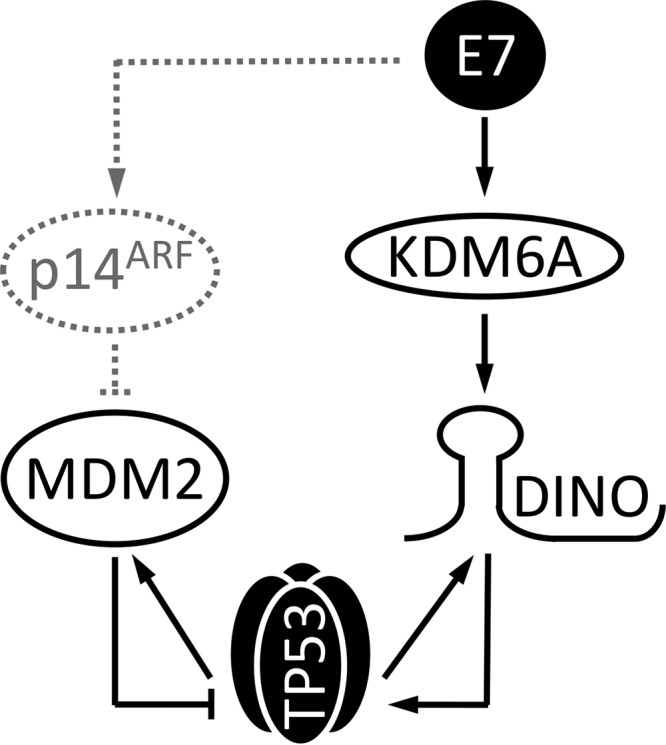
Model of TP53 stabilization and activation by the HPV16 E7 oncoprotein. HPV16 E7 expression triggers DINO expression at least in part through a KDM6A-mediated pathway. This causes TP53 activation, which results in further TP53-mediated DINO transcription. Despite the induction of the MDM2 ubiquitin ligase inhibitor p14ARF as a consequence of HPV16-mediated RB1 degradation, p14ARF has no or minimal effect on E7-mediated TP53 stabilization in keratinocytes. See the text for details.
Having established DINO as the key mediator of elevated TP53 levels in E7-expressing cells, we next wanted to determine whether DINO may modulate not only TP53 levels but also a known TP53-dependent biological phenotype of HPV16 E7-expressing cells. We previously discovered that, as with adenovirus E1A or MYC, HPV16 E7-expressing primary cells undergo cell death when deprived of growth factors (17, 20, 40–42). This form of cell death is triggered by conflicting growth signals; the proliferative signal generated by oncogene expression clashes with the antiproliferative signal as a consequence of serum deprivation. This likely represents a cell-intrinsic, innate tumor-suppressive response that has been dubbed the “trophic sentinel response” (43). In the case of HPV16 E7, this response is TP53 dependent and is overridden by coexpression of the HPV16 E6 oncoprotein (18, 20).
Serum deprivation is not a practicable approach to induce metabolic stress in keratinocytes, the normal host cells of HPVs, since they are grown in serum-free medium. Hence, we evaluated other means of generating metabolic stress and used metformin, one of the antidiabetic biguanide compounds that are being evaluated for repurposing as cancer-therapeutic and/or chemoprevention drugs. Metformin treatment of control and HPV16 E7-expressing keratinocytes revealed that, compared to parental cells, HPV16 E7-expressing cells are more sensitive to metformin treatment and that DINO depletion abrogates this sensitivity. Moreover, DINO-depleted HPV16 E7-expressing cells also showed decreased sensitivity to cell death in response to starvation and treatment with the DNA damage-inducing chemotherapy agent doxorubicin.
Similar to microRNAs, lncRNA activity can be modulated by nucleic acid-based inhibitors and mimics. Our study suggests that such DINO-based compounds may be efficacious in modulating the clinical response to drugs that cause metabolic stress or DNA damage in HPV-associated cancers and other tumor types where TP53 is not mutated.
MATERIALS AND METHODS
Cell culture.
Primary human foreskin keratinocytes (HFKs) were prepared from a pool of 3 to 5 neonatal foreskins obtained anonymously from the obstetrics and gynecology department at Tufts Medical Center. Human telomerase (hTERT)-immortalized HFKs (iHFKs) were a generous gift from Aloysius Klingelhutz (44). HFKs and iHFKs were grown and maintained in keratinocyte serum-free medium (KSFM) supplemented with human recombinant epidermal growth factor and bovine pituitary extract (Invitrogen). HPV16 E6 and/or E7 and dominant negative TP53 (dnTP53) C-terminal minigene-expressing HFKs and/or iHFKs were created by retroviral infection with recombinant retroviruses provided kindly by Denise Galloway, Paul Lambert (HPV oncogenes [45, 46]), and Moshe Oren (dnTP53 [30]). Cells were selected in 300 μg/ml G418 (Gibco) or 1 μg/ml puromycin (Sigma), and G418- or puromycin-resistant pooled cell populations were used for analyses. Donor- and passage-matched HFKs (<8 passages) were used. Doxycycline, doxorubicin, and Polybrene were purchased from Sigma. Metformin was purchased from Cayman Chemicals. Doxycycline and metformin were dissolved in phosphate-buffered saline (PBS) and freshly prepared prior to use.
Western blotting and antibodies.
Lysates were prepared by incubating the cells in radioimmunoprecipitation assay (RIPA) lysis buffer supplemented with Pierce protease inhibitor (Thermo Scientific) and Pierce phosphatase inhibitor (Thermo Scientific) at 4°C for 30 min. Protein extracts were cleared by centrifugation at 15,000 × g for 15 min. Equal amounts (50 μg) of proteins were loaded and fractionated on 4 to 12% NuPAGE bis-Tris gels (Invitrogen). Protein was transferred to polyvinylidene difluoride (PVDF) membranes (Millipore) and blocked with TNET buffer (200 mM Tris-HCl, 1 M NaCl, 50 mM EDTA, 0.1% Tween 20, pH 7.5) containing 5% nonfat dry milk at room temperature for 1 h. Blots were incubated with the following primary antibodies at 4°C overnight: TP53 (OP43, 1:1,000; Calbiochem), RB1 (Ab-5, 1:100; Millipore), E2F1 (sc-251, 1:500; Santa Cruz), KDM6A (ab36938, 1:300; Abcam), CDKN1A (ab109520, 1:1,000; Abcam), p14 (sc-8613, 1:200; Santa Cruz), α-tubulin (ab18251, 1:1,000; Abcam), and actin (Ab-1501, 1:1,000; Millipore). Membranes were rinsed in TNET and incubated with the corresponding secondary antibodies, horseradish peroxidase (HRP)-conjugated anti-mouse antibody (NA931, 1:10,000; GE Healthcare Life Sciences), HRP-conjugated anti-rabbit antibody (NA934V, 1:10,000; GE Healthcare Life Sciences), or HRP-conjugated anti-goat antibody (sc-2020, 1:6,666; Santa Cruz) for 1 h at room temperature. Antigen-antibody complexes were visualized by enhanced chemiluminescence (Life Technologies), and signals were digitally acquired on a G:Box Chemi-XX6 imager with Genesys software (Syngene). Protein bands were quantified using GeneTools software (Syngene).
Lentiviral expression plasmids.
Lentiviral vectors encoding two separate DINO-targeting shRNA hairpin sequences were kind gifts from Howard Chang (28). Doxycycline-inducible DINO knockdown vectors were created by inserting these DINO-targeting hairpin sequences into the Tet-pLKO-puro vector backbone (Addgene plasmid no. 21915) (47). Oligonucleotides used for cloning were sh-DINO1 oligo (5′-3′, (CCGGCACAGAAGAATTGGACATTGAACTCGAGTTCAATGTCCAATTCTTCTGTGTTTTT) and sh-DINO2 oligo (5′-3′, CCGGCTGGTTTATGGAGATGACATAACTCGAGTTATGTCATCTCCATAAACCAGTTTTT). As a negative control, the nonmammalian shRNA sequence (SHC002; Sigma) (5′-3′, CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT) was cloned into Tet-pLKO-puro vectors. All inserted sequences were verified by DNA sequencing.
RNA interference and lentiviral transduction.
Control and HPV16 E7 iHFKs (3 × 106 each) were seeded on 10-cm cell culture dishes and allowed to adhere overnight. The next day, cells were transfected with 30 nM target-specific siRNAs (ON-TARGETplus SMARTpools; Thermo Scientific Dharmacon) or a negative-control siRNA (nontargeting pool; Thermo Scientific Dharmacon) using the Lipofectamine RNAiMax reagent (Invitrogen) per the manufacturer’s instructions. Dharmacon references for gene-specific siRNAs used in this study are as follows: TP53, L-003329-00; RB1, L-003296-02; E2F1, L-003259-00; KDM6A, L-014140-01; and nontargeting control, D-001810-10. The custom-designed p14ARF siRNAs 5′-CGCGGAAGGUCCCUCAGAC-3′, 5′-GAACAUGGUGCGCAGGUUC-3′, and 5′-GAACCUGCGCACCAUGUUC-3′ were also purchased from Thermo Scientific Dharmacon. Recombinant lentiviruses expressing either nontargeting control or DINO shRNA sequences were made by transfecting HEK293T cells with the corresponding lentiviral vector, psPAX2 packaging (Addgene no. 12260), and pMD2.G (Addgene no. 12259) envelope plasmid DNA at a ratio of 4:3:1, respectively. Culture medium was collected at 48 h posttransfection and used for infection in conjunction with 0.4 μg/ml Polybrene. At 4 h postinfection, the inoculum was removed and replaced with fresh medium. Stable cell populations were generated after selection in 1 μg/ml puromycin.
RNA isolation and quantitative PCR.
Total RNA was isolated using Quick-RNA Miniprep (Zymo Research), and 1 μg of total RNA was reverse transcribed using the QuantiTect reverse transcription kit (Qiagen) per the manufacturer’s instructions. Quantitative PCR (qPCR) was performed in triplicate using SYBR green PCR Master Mix (Applied Biosystems) reagents in a StepOne Plus (Applied Biosystems) thermocycler system. For all qPCRs in this study, thermocycler settings of 20 s at 95°C followed by 40 cycles of 3 s at 95°C and 30 s at 60°C were used. The following qPCR primer sequences were used in this study: DINO, 5′-GGAGGCAAAAGTCCTGTGTT-3′ (forward) and 5′-GGGCTCAGAGAAGTCTGGTG-3′ (reverse); CDKN1A, 5′-CATGTGGACCTGTCACTGTCTTGTA-3′ (forward) and 5′-GAAGATCAGCCGGCGTTTG-3′ (reverse); MDM2, 5′-CAGTGGCGATTGGAGGGTAG-3′ (forward) and 5′-GACTACTACCAAGTTCCTGTAG-3′ (reverse); p14, 5′-CCCTCGTGCTGATGCTACTG-3′ (forward) and 5′-ACCTGGTCTTCTAGGAAGCGG-3′ (reverse); E2F1, 5′-GACCTCTTCGACTGTGACTTT-3′ (forward) and 5′-AAGGTGAGCATCTCTGGAAAC-3′ (reverse); KDM6A, 5′-TTTGTCAATTAGGTCASCTTCACCTC-3′ (forward) and 5′-AAAAAGGCAGCATTCTTCCAGTAGTC-3′ (reverse); RPLP0,5′-ATCAACGGGTACAAACGAGTC-3′ (forward) and 5′-CAGATGGATCAGCCAAGAAGG-3′ (reverse). The expression data shown were quantified using the ΔΔCT method by normalizing all the qPCR targets against a housekeeping gene, RPLP0.
Cell viability.
Cell viability was assessed using a resazurin assay, as previously described (48). On the day of cell viability reading, cells were incubated with fresh medium containing 10 μg/ml resazurin (Sigma). After a 1-h incubation, fluorescence readings were recorded on a Synergy H1 microplate reader (BioTek) using 560-nm excitation and 590-nm emission filters.
ACKNOWLEDGMENTS
We thank Howard Chang (Stanford University) for generously sharing DINO reagents, Al Klingelhutz (University of Iowa) for providing telomerase-immortalized human foreskin keratinocytes, Amy Yee, Philip Hinds, Peter Juo, Claire Moore, and members of the Munger lab for stimulating discussions and valuable suggestions throughout the course of this work, and the two anonymous reviewers for their constructive comments and suggestions on the manuscript.
This work was supported by PHS grants AI147176, CA066980, and CA228543 (K.M.) and a Dean’s Fellowship and a Tufts Collaborative Cancer Biology Award from the Tufts Graduate School of Biomedical Sciences (S.S.).
REFERENCES
- 1.Rozenblatt-Rosen O, Deo RC, Padi M, Adelmant G, Calderwood MA, Rolland T, Grace M, Dricot A, Askenazi M, Tavares M, Pevzner SJ, Abderazzaq F, Byrdsong D, Carvunis A-R, Chen AA, Cheng J, Correll M, Duarte M, Fan C, Feltkamp MC, Ficarro SB, Franchi R, Garg BK, Gulbahce N, Hao T, Holthaus AM, James R, Korkhin A, Litovchick L, Mar JC, Pak TR, Rabello S, Rubio R, Shen Y, Singh S, Spangle JM, Tasan M, Wanamaker S, Webber JT, Roecklein-Canfield J, Johannsen E, Barabási A-L, Beroukhim R, Kieff E, Cusick ME, Hill DE, Münger K, Marto JA, Quackenbush J, Roth FP, DeCaprio JA, Vidal M. 2012. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487:491–495. doi: 10.1038/nature11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu SC, Canarte V, Beeravolu H, Grace M, Sharma S, Munger K. 2020. Finding how human papillomaviruses alter the biochemistry and identity of infected epithelial cells, p 53–65. In Jenkins D, Bosch FX (ed), Human papillomavirus. Academic Press, New York, NY. [Google Scholar]
- 3.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See L-H, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigó R, Gingeras TR. 2012. Landscape of transcription in human cells. Nature 489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gunasekharan V, Laimins LA. 2013. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J Virol 87:6037–6043. doi: 10.1128/JVI.00153-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harden ME, Prasad N, Griffiths A, Munger K. 2017. Modulation of microRNA-mRNA target pairs by human papillomavirus 16 oncoproteins. mBio 8:e02170-16. doi: 10.1128/mBio.02170-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He H, Liu X, Liu Y, Zhang M, Lai Y, Hao Y, Wang Q, Shi D, Wang N, Luo XG, Ma W, Zhang TC. 2019. Human papillomavirus E6/E7 and long noncoding RNA TMPOP2 mutually upregulated gene expression in cervical cancer cells. J Virol 93:e01808-18. doi: 10.1128/JVI.01808-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma S, Munger K. 2018. Expression of the cervical carcinoma expressed PCNA regulatory (CCEPR) long noncoding RNA is driven by the human papillomavirus E6 protein and modulates cell proliferation independent of PCNA. Virology 518:8–13. doi: 10.1016/j.virol.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hutvagner G, Simard MJ, Mello CC, Zamore PD. 2004. Sequence-specific inhibition of small RNA function. PLoS Biol 2:E98. doi: 10.1371/journal.pbio.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien J, Hayder H, Zayed Y, Peng C. 2018. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne) 9:402. doi: 10.3389/fendo.2018.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rinn JL, Chang HY. 2012. Genome regulation by long noncoding RNAs. Annu Rev Biochem 81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilusz JE, Sunwoo H, Spector DL. 2009. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev 23:1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aloni-Grinstein R, Charni-Natan M, Solomon H, Rotter V. 2018. p53 and the viral connection: back into the future. Cancers (Basel) 10:178. doi: 10.3390/cancers10060178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lane DP. 1992. p53, guardian of the genome. Nature 358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 14.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. 1992. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69:1237–1245. doi: 10.1016/0092-8674(92)90644-R. [DOI] [PubMed] [Google Scholar]
- 15.Hengstermann A, Linares LK, Ciechanover A, Whitaker NJ, Scheffner M. 2001. Complete switch from Mdm2 to human papillomavirus E6-mediated degradation of p53 in cervical cancer cells. Proc Natl Acad Sci U S A 98:1218–1223. doi: 10.1073/pnas.98.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 17.Demers GW, Halbert CL, Galloway DA. 1994. Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene. Virology 198:169–174. doi: 10.1006/viro.1994.1019. [DOI] [PubMed] [Google Scholar]
- 18.Eichten A, Rud DS, Grace M, Piboonniyom SO, Zacny V, Munger K. 2004. Molecular pathways executing the “trophic sentinel” response in HPV-16 E7-expressing normal human diploid fibroblasts upon growth factor deprivation. Virology 319:81–93. doi: 10.1016/j.virol.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 19.Eichten A, Westfall M, Pietenpol JA, Munger K. 2002. Stabilization and functional impairment of the tumor suppressor p53 by the human papillomavirus type 16 E7 oncoprotein. Virology 295:74–85. doi: 10.1006/viro.2002.1375. [DOI] [PubMed] [Google Scholar]
- 20.Jones DL, Thompson DA, Munger K. 1997. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology 239:97–107. doi: 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- 21.Seavey SE, Holubar M, Saucedo LJ, Perry ME. 1999. The E7 oncoprotein of human papillomavirus type 16 stabilizes p53 through a mechanism independent of p19(ARF). J Virol 73:7590–7598. doi: 10.1128/JVI.73.9.7590-7598.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou X, Munger K. 2009. Expression of the human papillomavirus type 16 E7 oncoprotein induces an autophagy-related process and sensitizes normal human keratinocytes to cell death in response to growth factor deprivation. Virology 385:192–197. doi: 10.1016/j.virol.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Stanchina E, McCurrach ME, Zindy F, Shieh SY, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. 1998. E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes Dev 12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng Z, Zhang C, Wu R, Hu W. 2011. Tumor suppressor p53 meets microRNAs. J Mol Cell Biol 3:44–50. doi: 10.1093/jmcb/mjq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaudhary R, Lal A. 2017. Long noncoding RNAs in the p53 network. Wiley Interdiscip Rev RNA 8:e01808-18. doi: 10.1002/wrna.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, Attardi LD, Regev A, Lander ES, Jacks T, Rinn JL. 2010. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li XL, Subramanian M, Jones MF, Chaudhary R, Singh DK, Zong X, Gryder B, Sindri S, Mo M, Schetter A, Wen X, Parvathaneni S, Kazandjian D, Jenkins LM, Tang W, Elloumi F, Martindale JL, Huarte M, Zhu Y, Robles AI, Frier SM, Rigo F, Cam M, Ambs S, Sharma S, Harris CC, Dasso M, Prasanth KV, Lal A. 2017. Long noncoding RNA PURPL suppresses basal p53 levels and promotes tumorigenicity in colorectal cancer. Cell Rep 20:2408–2423. doi: 10.1016/j.celrep.2017.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmitt AM, Garcia JT, Hung T, Flynn RA, Shen Y, Qu K, Payumo AY, Peres-da-Silva A, Broz DK, Baum R, Guo S, Chen JK, Attardi LD, Chang HY. 2016. An inducible long noncoding RNA amplifies DNA damage signaling. Nat Genet 48:1370–1376. doi: 10.1038/ng.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R, Ansell PJ, Zhao J, Weng C, Klibanski A. 2007. Activation of p53 by MEG3 non-coding RNA. J Biol Chem 282:24731–24742. doi: 10.1074/jbc.M702029200. [DOI] [PubMed] [Google Scholar]
- 30.Gottlieb E, Haffner R, von Ruden T, Wagner EF, Oren M. 1994. Down-regulation of wild-type p53 activity interferes with apoptosis of IL-3-dependent hematopoietic cells following IL-3 withdrawal. EMBO J 13:1368–1374. doi: 10.1002/j.1460-2075.1994.tb06390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim D-H, Tang Z, Shimada M, Fierz B, Houck-Loomis B, Bar-Dagen M, Lee S, Lee S-K, Muir TW, Roeder RG, Lee JW. 2013. Histone H3K27 trimethylation inhibits H3 binding and function of SET1-like H3K4 methyltransferase complexes. Mol Cell Biol 33:4936–4946. doi: 10.1128/MCB.00601-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soto DR, Barton C, Munger K, McLaughlin-Drubin ME. 2017. KDM6A addiction of cervical carcinoma cell lines is triggered by E7 and mediated by p21CIP1 suppression of replication stress. PLoS Pathog 13:e1006661. doi: 10.1371/journal.ppat.1006661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLaughlin-Drubin ME, Crum CP, Münger K. 2011. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci U S A 108:2130–2135. doi: 10.1073/pnas.1009933108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinke SA, Martens GA, Cai Y, Finsi J, Heimberg H, Pipeleers D, Casteele M. 2009. Methyl succinate antagonises biguanide-induced AMPK-activation and death of pancreatic beta-cells through restoration of mitochondrial electron transfer. Br J Pharmacol 150:1031–1043. doi: 10.1038/sj.bjp.0707189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, Marette A, Kozma SC, Thomas G. 2010. Metformin, independent of AMPK, inhibits mTORC1 in a Rag GTPase-dependent manner. Cell Metab 11:390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaw RJ. 2009. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, Viollet B, Guigas B. 2011. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 54:3101–3110. doi: 10.1007/s00125-011-2311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamijo T, van de Kamp E, Chong MJ, Zindy F, Diehl JA, Sherr CJ, McKinnon PJ. 1999. Loss of the ARF tumor suppressor reverses premature replicative arrest but not radiation hypersensitivity arising from disabled atm function. Cancer Res 59:2464–2469. [PubMed] [Google Scholar]
- 39.Khanal T, Leung YK, Jiang W, Timchenko N, Ho SM, Kim K. 2019. NR2E3 is a key component in p53 activation by regulating a long noncoding RNA DINO in acute liver injuries. FASEB J 33:8335–8348. doi: 10.1096/fj.201801881RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hermeking H, Eick D. 1994. Mediation of c-Myc-induced apoptosis by p53. Science 265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- 41.Lowe SW, Ruley HE. 1993. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev 7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- 42.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 43.Evan GI, Vousden KH. 2001. Proliferation, cell cycle and apoptosis in cancer. Nature 411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 44.Farwell DG, Shera KA, Koop JI, Bonnet GA, Matthews CP, Reuther GW, Coltrera MD, McDougall JK, Klingelhutz AJ. 2000. Genetic and epigenetic changes in human epithelial cells immortalized by telomerase. Am J Pathol 156:1537–1547. doi: 10.1016/S0002-9440(10)65025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Demers GW, Espling E, Harry JB, Etscheid BG, Galloway DA. 1996. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J Virol 70:6862–6869. doi: 10.1128/JVI.70.10.6862-6869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halbert CL, Demers GW, Galloway DA. 1991. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol 65:473–478. doi: 10.1128/JVI.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao Y-M, Lengauer C, Sellers WR, Benson JD. 2009. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8:498–504. doi: 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- 48.Riss TL, Moravec RA, Niles AL, Duellman S, Benink HA, Worzella TJ, Minor L. 2004. Cell viability assays In Sittampalam GS, Grossman A, Brimacombe K, Arkin M, Auld D, Austin CP, Baell J, Bejcek B, Caaveiro JMM, Chung TDY, Coussens NP, Dahlin JL, Devanaryan V, Foley TL, Glicksman M, Hall MD, Haas JV, Hoare SRJ, Inglese J, Iversen PW, Kahl SD, Kales SC, Kirshner S, Lal-Nag M, Li Z, McGee J, McManus O, Riss T, Saradjian P, Trask OJ, Jr., Weidner JR, Wildey MJ, Xia M, Xu X (ed), Assay guidance manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda, MD. [Google Scholar]