

The membrane deformation activity of the herpesvirus nuclear egress complex (NEC) allows capsids to transit through both nuclear membranes into the cytoplasm. NEC activity must be precisely controlled during viral infection, and yet our knowledge of how NEC activity is controlled is incomplete. To determine how pUL16 and pUL21, two viral proteins required for nuclear egress of herpes simplex virus 2, function in nuclear egress, we examined how the lack of each protein impacted NEC distribution. These analyses revealed a function of pUL16 in nuclear egress distinct from that of pUL21, uncovered a novel role for pUL21 in regulating NEC activity, and shed new light on how a viral kinase, pUs3, regulates nuclear egress. Nuclear egress of capsids is required for all herpesviruses. A complete understanding of all aspects of nuclear egress, including how viral NEC activity is controlled, may yield strategies to disrupt this process and aid the development of herpes-specific antiviral therapies.
KEYWORDS: herpes simplex virus, nuclear egress, nuclear envelope, pUL16, pUL21, pUs3
ABSTRACT
Viral proteins pUL16 and pUL21 are required for efficient nuclear egress of herpes simplex virus 2 capsids. To better understand the role of these proteins in nuclear egress, we established whether nuclear egress complex (NEC) distribution and/or function was altered in the absence of either pUL16 or pUL21. NEC distribution in cells infected with pUL16-deficient viruses was indistinguishable from that observed in cells infected with wild-type viruses. In contrast, NEC distribution was aberrant in cells infected with pUL21-deficient virus and, instead, showed some similarity to the aberrant NEC distribution pattern observed in cells infected with pUs3-deficient virus. These results indicated that pUL16 plays a role in nuclear egress that is distinct from that of pUL21 and pUs3. Higher-resolution examination of nuclear envelope ultrastructure in cells infected with pUL21-deficient viruses by transmission electron microscopy showed different types of nuclear envelope perturbations, including some that were not observed in cells infected with pUs3 deficient virus. The formation of the nuclear envelope perturbations observed in pUL21-deficient virus infections was dependent on a functional NEC, revealing a novel role for pUL21 in regulating NEC activity. The results of comparisons of nuclear envelope ultrastructure in cells infected with viruses lacking pUs3, pUL16, or both pUs3 and pUL16 were consistent with a role for pUL16 in advance of primary capsid envelopment and shed new light on how pUs3 functions in nuclear egress.
IMPORTANCE The membrane deformation activity of the herpesvirus nuclear egress complex (NEC) allows capsids to transit through both nuclear membranes into the cytoplasm. NEC activity must be precisely controlled during viral infection, and yet our knowledge of how NEC activity is controlled is incomplete. To determine how pUL16 and pUL21, two viral proteins required for nuclear egress of herpes simplex virus 2, function in nuclear egress, we examined how the lack of each protein impacted NEC distribution. These analyses revealed a function of pUL16 in nuclear egress distinct from that of pUL21, uncovered a novel role for pUL21 in regulating NEC activity, and shed new light on how a viral kinase, pUs3, regulates nuclear egress. Nuclear egress of capsids is required for all herpesviruses. A complete understanding of all aspects of nuclear egress, including how viral NEC activity is controlled, may yield strategies to disrupt this process and aid the development of herpes-specific antiviral therapies.
INTRODUCTION
Herpes virions are complex structures composed of a large DNA genome encapsulated in an icosahedral capsid surrounded by a proteinaceous layer of tegument, which in turn is surrounded by a lipid envelope studded with numerous membrane glycoproteins. Befitting this structural complexity, the morphogenesis of these particles is equally complex and is not fully understood. The virion assembly pathway begins in the infected cell nucleus, where newly replicated viral genomes are packaged into preformed procapsids. DNA-containing capsids are then preferentially selected for further maturation and acquire a primary envelope by budding at the inner nuclear membrane (INM), resulting in the formation of a primary enveloped virion (PEV) residing in the perinuclear space. These PEVs then fuse with the outer nuclear membrane (ONM), releasing a capsid into the cytoplasm that is subsequently enveloped at a post-Golgi compartment, and the mature virion is secreted from the cell (1).
The present study concerns the nuclear egress of capsids from the nucleoplasm to the cytoplasm. Key viral factors required for this process are the components of the nuclear egress complex (NEC). Components of the NEC, the orthologs of the herpes simplex virus (HSV) proteins pUL31 and pUL34, localize predominantly to the INM in virally infected cells (2, 3). Studies from several laboratories have described the molecular structure of NEC complexes derived from a number of herpesviruses, and these structures have provided important insight into the molecular mechanisms by which the NEC promotes capsid envelopment at the INM (4–9). Deletion of UL31 or UL34 from HSV-1 results in a 4-log reduction in virus replication in most cell types and the accumulation of capsids in the nuclei of infected cells (10, 11). pUL34 is a type II membrane protein with a C-terminal transmembrane domain. The N terminus of pUL34 is localized on the cytoplasmic or nucleoplasmic face of the endoplasmic reticulum and the INM, respectively. pUL31, a soluble nucleoplasmic phosphoprotein, can interact with the N terminus of pUL34 and is recruited to the INM in the presence of pUL34. Expression of pUL31 and pUL34 in the absence of other viral proteins leads to vesiculation of the INM at the nuclear periphery (12), and the interaction of purified pUL31 and pUL34 with synthetic membranes in vitro results in the induction of membrane vesiculation (13). Artificial targeting of purified pUL31 to membranes in the absence of pUL34 also leads to membrane vesiculation in vitro, suggesting that a key function of pUL34 is to recruit pUL31 to the membrane surface (14). In infected cells, however, pUL31 and pUL34 are distributed evenly throughout the INM, and vesiculation of the INM is not observed in the absence of capsids (2, 12). These findings suggest that the INM vesiculation activity of the NEC is negatively regulated by a protein(s) found in infected cells.
One protein that is capable of regulating NEC activity is the viral serine/threonine kinase pUs3. In cells infected with pUs3 mutants of pseudorabies virus (PRV), HSV-1, equine herpesvirus 1 (EHV-1), and Marek’s disease virus (MDV) PEVs accumulate in herniations of the INM (3, 15–17). HSV-1 pUs3 phosphorylates pUL31 at a number of serine residues in its N terminus, and it has been proposed that these modifications are required for efficient de-envelopment of PEVs at the ONM because mutation of these pUL31 serine residues to alanine recapitulates the pUs3 mutant phenotype (18). Moreover, mutation of the serine residues in the N terminus of HSV-1 pUL31 to glutamic acid, to mimic the hyperphosphorylated pUL31 state, resulted in a failure of capsids to be efficiently enveloped at the INM (18). Taken together, these findings suggest that a hypophosphorylated form of pUL31 is required for NEC-mediated envelopment of capsids and that pUs3 may act to restrict NEC-mediated INM vesiculation by phosphorylating pUL31.
Previous studies from our laboratory demonstrated that HSV-2 strain 186 lacking either pUL21 or pUL16 showed deficiencies in the nuclear egress of capsids (19–21). Since pUL16 and pUL21 can form a complex (22), we hypothesized that this complex was required for efficient nuclear egress of capsids (19). To define the roles of pUL16 and pUL21 in nuclear egress and to determine whether these proteins function individually or as a complex in nuclear egress, we analyzed the distribution and function of the NEC in cells infected with multiple HSV strains deficient in either pUL16 or pUL21. Our analyses revealed that pUL16 mutants were phenotypically distinct from pUL21 mutants in terms of NEC distribution in infected cells and indicated that pUL16 functions in advance of pUL21 and pUs3 in nuclear egress. Our analyses also revealed a novel role for pUL21 in regulating the activity of the NEC. Based on the results of further phenotypic analyses of viruses lacking both pUL16 and pUs3, we also propose a new interpretation of pUs3 function in nuclear egress.
RESULTS
The NEC is irregularly distributed in cells infected with HSV strains lacking pUL21 or pUs3.
One possible explanation for the capsid nuclear egress deficiencies seen with HSV-2 pUL21 and pUL16 mutants is that these proteins, individually or as a complex, are required for the appropriate distribution and/or function of the viral nuclear egress complex (NEC), comprised of pUL31 and pUL34. To enable investigation of HSV-2 NEC distribution and function, antisera reactive against HSV-2 pUL31 and pUL34 were produced in rats and chickens, respectively. The antisera produced were specific in both Western blot analyses (Fig. 1A) and in indirect immunofluorescence microscopy experiments (Fig. 1B) and were reactive against pUL31 and pUL34 orthologs from both HSV-2 and HSV-1 (see below).
FIG 1.

Characterization of antisera generated against HSV-2 pUL31 and HSV-2 pUL34. (A) Western blot analysis. Equal volumes of whole-cell protein extracts prepared at 24 h posttransfection from 293T cells transfected with plasmids encoding the proteins indicated at the top of each panel, or from untransfected 293T cells, were electrophoresed through 8% polyacrylamide gels and transferred to PVDF membranes. Membranes were probed with antisera indicated at the bottom of each panel. Molecular weight markers in kDa are indicated on the left side of the panels. (B) Indirect immunofluorescence microscopy. HeLa cells were transfected with plasmids encoding the proteins indicated at the left side of each panel and then fixed at 18 h posttransfection and incubated with either pUL31 antisera (top panels) or pUL34 antisera (bottom panels), followed by incubation with the appropriate Alexa Fluor 568-conjugated secondary antibody. Nuclei were stained with Hoechst 33342. Scale bars, 10 μm.
Using these antisera we investigated the distribution of the NEC in cells infected with pUL21-, pUL16-, and pUs3-deficient viruses derived from HSV-2 strain 186 (designated here as Δ21, Δ16, and ΔUs3). Whereas NEC distribution was uniform and smooth at the nuclear rim of cells infected with WT and Δ16 strains, the NEC distribution was irregular in cells infected with Δ21 and ΔUs3 (Fig. 2). In the case of Δ21-infected cells, pUL31 and pUL34 still colocalized at the nuclear rim; however, the distribution pattern was uneven, with puncta of various sizes distributed irregularly on both the nucleoplasmic and the cytoplasmic sides of the nuclear envelope. In the case of ΔUs3-infected cells, colocalization of pUL31 and pUL34 was also observed, but the NEC distribution appeared distinct from that observed in Δ21-infected cells, with a more uniform, regularly spaced arrangement of puncta along the nuclear rim. A similar pattern of NEC distribution has been observed in cells infected with Us3-deficient strains of several alphaherpesviruses (3, 15–17). The differences in NEC distribution observed between Δ16, Δ21, and ΔUs3 HSV-2 strains suggests that pUL16, pUL21, and pUs3 play roles that are distinct from one another in the nuclear egress of HSV-2.
FIG 2.

Distribution of the NEC in cells infected with WT and mutant HSV-2 strains. Vero cells were infected with the indicated virus strains, all of which are in an HSV-2 strain 186 background, or mock infected. At 8 hpi, cells were fixed and stained with pUL31 and pUL34 antisera and Alexa Fluor 488-conjugated and Alexa Fluor 568-conjugated secondary antibodies, respectively. Nuclei were stained with Hoechst 33342. Scale bar, 10 μm.
To investigate whether the irregular distribution of the NEC observed in cells infected with Δ21 was a phenotype common to other strains of HSV-2 and HSV-1, we examined the distribution of the NEC in cells infected with two additional strains of HSV-2 (HG52 and SD90e) and two strains of HSV-1 (F and KOS) alongside their corresponding pUL21 mutants (23). Cells infected with pUL21-deficient viruses, regardless of background strain, showed similar aberrant NEC distribution in comparison to cells infected with parental viruses (Fig. 3). These findings indicate that irregular NEC distribution is a conserved property of pUL21 mutants across HSV species and strains.
FIG 3.

Distribution of the NEC is aberrant in cells infected with virus lacking pUL21 regardless of background strain. Vero cells were infected with the parental strains indicated in the center of the figure or their corresponding mutants lacking pUL21. At 8 hpi, cells were fixed and stained with pUL31 antisera and Alexa Fluor 488-conjugated secondary antibodies. Nuclei were stained with Hoechst 33342. Z-series projection images, generated with FV10-ASW version 04.01 software, are shown. Scale bars, 10 μm.
To ensure that this irregular NEC distribution was due to the loss of pUL21 and not due to unintended mutations introduced during strain construction, HaCaT cells stably expressing HSV-2 pUL21, HaCaT21, were isolated and infected with wild-type (WT) and pUL21 mutant HSV strains derived from HSV-2 (Δ21) and HSV-1 (FFS62/34). Whereas parental HaCaT cells infected with pUL21 mutants displayed irregular NEC distribution similar to that observed in infected Vero cells (compare Fig. 4A with Fig. 3), HaCaT21 cells infected with pUL21 mutants displayed smooth NEC distribution (Fig. 4A). The proportion of infected cells displaying smooth versus perturbed NEC distribution was quantified in two independent experiments (Fig. 4B). Whereas expression of HSV-2 pUL21 in HaCaT cells had no effect on the distribution of the NEC in WT virus-infected cells, it restored the normal, smooth, distribution of the NEC in pUL21 mutant-infected cells from 37.5 to 84.5% in Δ21-infected cells and from 7.5 to 41% in FFS62/34-infected cells. The superior complementation observed with the HSV-2 versus the HSV-1 UL21 mutant may be due to the expression of a HSV-2 pUL21 ortholog in HaCaT21 cells rather than a HSV-1-derived pUL21. Taken together, these data confirm that the perturbation in NEC distribution observed in cells infected with pUL21 mutant strains was due to the loss of pUL21.
FIG 4.

HaCaT21 cells complement the aberrant NEC distribution phenotype of viruses lacking pUL21. (A) HaCaT or HaCaT21 cells were infected with the indicated virus strains. FFS62/34 is an HSV-1 strain F virus lacking pUL21 described previously (23). At 18 hpi, the cells were fixed and stained with pUL31 antisera and pUL34 antisera and Alexa Fluor 488-conjugated and Alexa Fluor 568-conjugated secondary antibodies, respectively. Nuclei were stained with Hoechst 33342. Scale bars, 10 μm. (B) To quantify NEC distribution (perturbed versus smooth), 100 infected cells were scored for the appearance of pUL34 around the nuclear rim. Two independent experiments were scored.
Cells infected with HSV strains lacking pUL21 display a variety of nuclear envelope perturbations.
Since the NEC is anchored within the INM by its pUL34 component, the NEC labeled irregular puncta in UL21 mutant-infected cells that appeared to protrude into and out of the nucleus likely represented perturbations of the nuclear envelope. To examine this at higher resolution, transmission electron microscopy (TEM) analyses were carried out on Vero cells infected with three strains of HSV-2 (186, HG52, and SD90e) and two strains of HSV-1 (F and KOS) alongside their corresponding pUL21 mutants. The appearances of the nuclear envelope in cells infected with WT strains were similar to each other, characterized by a smooth appearance occasionally punctuated by small, localized perturbations associated with egressing capsids (Fig. 5, arrowheads). By contrast, cells infected with UL21 mutant strains displayed a variety of nuclear envelope perturbations (Fig. 6). Extravagations of the nuclear envelope into the cytoplasm were frequently observed and often were comprised of multiple layers of the nuclear envelope arranged in a concentric fashion (Fig. 6, white arrowheads), occasionally surrounding cytoplasmic contents that included virus capsids (Fig. 6, white arrows). Invaginations of the nuclear envelope into the nucleoplasm, resembling type II nucleoplasmic reticulum were also observed (24) (Fig. 6, black arrowheads) as were invaginations of the INM (Fig. 6, black arrows), resembling type I nucleoplasmic reticulum that occasionally contained PEVs (24) (Fig. 6, white asterisks). These findings are quantified in Fig. 7, and higher-magnification images of these features are shown in Fig. S1 in the supplemental material. These analyses demonstrated that perturbations of the nuclear envelope were more readily observed in cells infected with pUL21-deficient viruses, regardless of background strain, and explained the perturbations in NEC distribution observed in Fig. 2 to 4.
FIG 5.

Ultrastructural analysis of cells infected with WT HSV strains. Vero cells were infected with the virus strains indicated on the left side of the figure at an MOI of 3. At 16 hpi, the cells were fixed and processed for TEM as described in Materials and Methods. Representative images are shown. Arrowheads indicate small, localized nuclear membrane perturbations associated with egressing capsids that were occasionally observed in WT infected cells. Nu, nucleus.
FIG 6.

Ultrastructural analysis of cells infected with pUL21-deficient HSV strains. Vero cells were infected with the virus strains indicated on the left side of the figure at an MOI of 3. At 16 hpi, the cells were fixed and processed for TEM as described in Materials and Methods. A montage of images depicting the variety of nuclear perturbations that were readily observed in cells infected with pUL21 deficient viruses is shown. White arrowheads indicate extravagations of the nuclear envelope into the cytoplasm, white arrows indicate extravagations surrounding cytoplasmic capsids, black arrowheads indicate invaginations of both membranes of the nuclear envelope into the nucleoplasm, black arrows indicate invaginations of the INM, and white asterisks indicate invaginations of the INM containing PEVs. Nu, nucleus.
FIG 7.
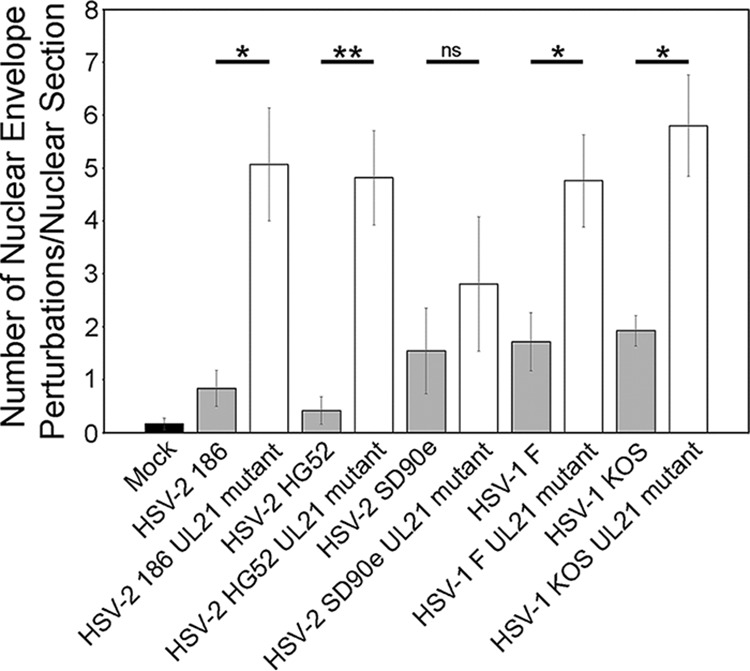
Quantitation of ultrastructural analysis of cells infected with WT or pUL21-deficient HSV. The total number of nuclear envelope perturbations observed in nuclear sections was scored in mock-infected Vero cells and in Vero cells infected with the virus strains indicated on the x axis. More nuclear envelope perturbations were observed in cells infected with pUL21 deficient HSV in comparison to cells infected with the corresponding WT HSV, regardless of background strain. The numbers of nuclear sections scored were as follows: Mock (n = 12); WT 186 (n = 12); pUL21-deficient 186 (n = 13); WT HG52 (n = 12); pUL21-deficient HG52 (n = 11); WT SD90e (n = 11); pUL21-deficient SD90e (n = 16); WT F (n = 14); pUL21-deficient F (n = 17); WT KOS (n = 13); and pUL21-deficient KOS (n = 15). Error bars indicate the standard errors of the mean. *, P < 0.05; **, P < 0.01; ns, not significant.
The nuclear envelope perturbations associated with Δ21 infection require pUL31.
To investigate whether the nuclear envelope perturbation phenotype observed in the absence of pUL21 required pUL31, we measured the effect of pUL31 knockdown on nuclear envelope perturbations. The production of pUL31 was specifically knocked down by Dicer-substrate siRNA (DsiRNA) 19 directed against the UL31 transcript (Fig. 8A and B). Knockdown of pUL31 had no effect on the distribution of pUL34 at the nuclear rim in WT-infected cells since HSV-2 pUL34, like other pUL34 homologues, can localize to the nuclear rim in a smooth distribution in the absence of other viral proteins (see, for example, the lower portion of Fig. 1). In contrast, knockdown of pUL31 had an effect on pUL34 distribution in Δ21-infected cells. This effect is most clearly demonstrated in Fig. 8C. The cell that escaped pUL31 knockdown by DsiRNA 19 (arrowhead) displayed perturbed pUL34 distribution, whereas the cell knocked down for pUL31 (arrow) displayed smooth pUL34 distribution. The proportion of infected cells displaying smooth versus perturbed pUL34 distribution was quantified in two independent experiments (Fig. 8D). In WT virus-infected cells, smooth distribution of pUL34 predominated (>90%) in the presence of either DsiRNA 19 or nonsilencing control DsiRNA. In Δ21-infected cells, smooth distribution of pUL34 was observed at a level comparable to that in WT-infected cells in the presence of DsiRNA 19 (92%) but not in the presence of nonsilencing control DsiRNA (45.5%). These data are consistent with a requirement for a functional NEC to produce the nuclear envelope perturbations observed in Δ21-infected cells.
FIG 8.

The nuclear membrane perturbation phenotype of viruses lacking pUL21 requires pUL31. Vero cells were transfected with DsiRNAs specific for UL31 (DsiRNA 19) or nonsilencing control DsiRNAs (NC) and infected with wild-type HSV-2 strain 186 (WT) or the corresponding mutant lacking pUL21 (Δ21) at an MOI of 0.5 at 24 h posttransfection. (A) At 18 hpi, whole-cell extracts were prepared, electrophoresed through 10% polyacrylamide gels, and transferred to PVDF membranes. Membranes were probed with antisera indicated on the right side of each panel. Molecular weight markers in kDa are indicated on the left side of each panel. (B) At 18 hpi, cells were fixed and stained with pUL31 antisera and pUL34 antisera and Alexa Fluor 488-conjugated and Alexa Fluor 568-conjugated secondary antibodies, respectively. Nuclei were stained with Hoechst 33342. Scale bars, 50 μm. (C) Enlarged images of cells transfected with DsiRNA 19 and infected with Δ21 are shown. The arrow indicates a cell in which the production of pUL31 was knocked down, and the arrowhead indicates a cell that produced pUL31. Scale bar, 10 μm. (D) Quantitation of the effect of UL31 knockdown on the distribution of pUL34 around the nuclear rim. A total of 100 pUL34-positive cells were scored for the appearance of pUL34 around the nuclear rim (perturbed versus smooth). Two independent experiments were scored.
HSV-2 pUL16 functions in advance of pUs3 in nuclear egress.
As stated above, the smooth NEC distribution pattern observed in Δ16-infected cells (Fig. 2) supports the notion that pUL16 functions in advance of pUs3 and pUL21 in capsid nuclear egress. A smooth NEC distribution pattern was also observed in cells infected with other strains of HSV-2 and HSV-1 lacking pUL16 (20) (Fig. 9), confirming that this NEC distribution phenotype is conserved among HSV species and strains. To further test the idea that pUL16 functions in advance of pUs3, we analyzed the phenotype of two independently isolated HSV-2 strain 186 viruses carrying deficiencies in both proteins (Δ16/ΔUs3). We predicted that if pUL16 was functioning in advance of pUs3 in capsid nuclear egress, the NEC distribution in cells infected with the Δ16/ΔUs3 mutants would resemble that of cells infected with Δ16. Western blot analysis of cells infected with Δ16/ΔUs3 viruses alongside cells infected with WT, Δ16, or ΔUs3 viruses demonstrated the absence of both pUL16 and pUs3 in the double mutants (Fig. 10). The distribution of the NEC in cells infected with Δ16/ΔUs3 was mostly smooth (Fig. 2), similar to what was observed in WT- and Δ16-infected cells (Fig. 2 and 9). However, closer examination of the nuclear envelope by TEM revealed herniations of the INM that were rarely seen in Δ16-infected cells (Fig. 11). These herniations resembled the INM structures formed in ΔUs3-infected cells (Fig. 11A and B) except that in Δ16/ΔUs3-infected cells they lacked PEVs (Fig. 11C and D). Collectively, these findings are consistent with the idea that pUL16 functions in advance of capsid primary envelopment. Moreover, the data suggest that it is the lack of pUs3 that is the cause of INM herniation rather than the accumulation of PEVs in the perinuclear space.
FIG 9.

Distribution of the NEC is smooth in cells infected with virus lacking pUL16 regardless of background strain. Vero cells were infected with the parental strains indicated on the left side of the figure and their corresponding mutants lacking pUL16. At 8 hpi, the cells were fixed and stained with pUL31 antisera and Alexa Fluor 488-conjugated secondary antibodies. Nuclei were stained with Hoechst 33342 reagent. Scale bar, 10 μm.
FIG 10.

Characterization of Δ16/ΔUs3 mutant virus strains. Vero cells were infected with the indicated viruses, all of which are in an HSV-2 strain 186 background, at an MOI of 0.1. At 18 hpi, whole-cell extracts were prepared and electrophoresed through 10% polyacrylamide gels and transferred to PVDF membranes. Membranes were probed with antisera indicated on the right side of each panel. Molecular weight markers in kDa are indicated on the left side of each panel.
FIG 11.

Ultrastructural analysis of cells infected with pUs3- and pUL16-deficient HSV-2 strains. Vero cells were infected with the viruses indicated on the x axis, all of which are in an HSV-2 strain 186 background, at an MOI of 3. At 16 hpi, the cells were fixed and processed for TEM as described in Materials and Methods. (A) Representative image of INM herniations containing PEVs, indicated by asterisks, observed in cells infected with virus deficient in pUs3. An enlarged example of an INM herniation containing PEVs is shown in the inset image. Nu, nucleus. (B) Quantitation of INM herniations containing PEVs in infected cells. INM herniations containing PEVs were more frequently observed in cells infected with pUs3-deficient virus than in cells infected with WT virus or pUL16-deficient viruses. The numbers of nuclear sections scored were as follows: WT 186 (n = 12); ΔUs3 (n = 28); Δ16 (n = 14); Δ16/ΔUs3 isolate 1 (n = 38); and Δ16/ΔUs3 isolate 2 (n = 10). Error bars indicate the standard errors of the mean. *, P < 0.05. (C) Representative image of INM herniations lacking PEVs, indicated by black arrows, observed in cells infected with virus deficient in both pUs3 and pUL16. An enlarged example of an INM herniation lacking PEVs is shown in the inset image. Nu, nucleus. (D) Quantitation of INM herniations. INM herniations containing PEVs and those lacking PEVs were scored together. INM herniations were more frequently observed in cells infected with viruses deficient in pUs3 than in cells infected with WT virus or pUL16-deficient virus. The numbers of nuclear sections scored were as follows: WT 186 (n = 12); ΔUs3 (n = 28); Δ16 (n = 14); Δ16/ΔUs3 isolate 1 (n = 38); and Δ16/ΔUs3 isolate 2 (n = 10). Error bars indicate the standard errors of the mean. *, P < 0.05; ns, not significant.
DISCUSSION
Since pUL21 and pUL16 both function in the nuclear egress of HSV-2, this study was initiated to determine the effect of deleting these proteins on the distribution of NEC components. Since pUL21 and pUL16 are capable of forming a complex (22), we anticipated that the NEC distribution phenotype would be similar between the Δ21 and Δ16 strains. This was not the case (Fig. 2). Whereas Δ21 demonstrated irregular NEC distribution, the NEC distribution in Δ16-infected cells was indistinguishable from that observed in WT-infected cells. These findings suggested that pUL21 and pUL16 were functioning in mechanistically distinct ways to promote nuclear egress and that it was unlikely that a pUL21/pUL16 complex was the functional unit enabling efficient nuclear egress.
How might pUL16 function in HSV-2 nuclear egress? A failure to produce DNA-containing capsids in pUL16 mutant-infected cells cannot explain this defect (Fig. 11C) (19, 20). It is equally unlikely that the lack of pUL16 impacts the transport of DNA-containing capsids to the INM since capsid movement within the nucleus is thought to occur primarily by diffusion (25, 26). It may be that the absence of pUL16 influences the composition of nuclear capsids such that they are unable to efficiently engage the NEC.
Analysis of cells infected with UL21 mutants derived from multiple HSV-2 and HSV-1 strains indicated that the impact of pUL21 loss on NEC distribution was conserved between HSV strains and species. Ultrastructural examination of UL21 mutant strains revealed a number of nuclear envelope perturbations, including extravagations of the nuclear envelope into the cytoplasm, as well as invaginations of the nuclear envelope and the INM into the nucleoplasm (Fig. 6). When INM invaginations were observed, PEVs were often seen accumulating in these structures that were reminiscent of INM herniations seen in cells infected with Us3 mutants derived from PRV (17), EHV-1 (15), MDV (16), HSV-1 (3), and HSV-2 (Fig. 11A). Knockdown of pUL31 in cells infected with Δ21 prevented these nuclear envelope perturbations, indicating that pUL31 activity was required to drive the formation of these structures and further suggesting that NEC activity is dysregulated in the absence of pUL21.
Coexpression of pUL31 and pUL34 in the absence of other viral proteins leads to vesiculation of the INM at the nuclear periphery leading to the formation of perinuclear vesicles (12), and the interaction of purified pUL31 and pUL34 with synthetic membranes also results in membrane vesiculation (8, 13). Evidence is mounting that the membrane vesiculation activity of the NEC is regulated in the context of viral infection (27). In cells infected with WT strains of HSV-1 (Fig. 3) (2), PRV (28), and HSV-2 (Fig. 3) (29), pUL31 and pUL34 localize in a smooth and even distribution at the nuclear membrane and INM vesiculation is rarely apparent, suggesting that factors found in virally infected cells regulate NEC activity such that it is only active when capsids are presented for envelopment. One such regulator of the NEC is the viral serine/threonine kinase pUs3. Although pUs3 functions in multiple facets of the virus-host interaction (30), the best characterized is its role in nuclear egress. PEVs were observed to accumulate in INM herniations in cells infected with a PRV Us3 mutant virus (17). The interpretation of this observation was that PEV accumulations result as a consequence of the failure of PEVs to de-envelop at the ONM. This interpretation has been widely utilized to explain the phenotypes of Us3 mutant strains derived from other herpesviruses, including HSV-1, EHV-1, and MDV (3, 15, 16). The data presented here confound this interpretation. Our analysis of two independently constructed Δ16/ΔUs3 mutants revealed the presence of INM herniations that lack PEVs (Fig. 11C). Collectively, these findings suggest that formation of INM herniations is not driven by the accumulation of PEVs in the perinuclear space but rather by the absence of pUs3.
Baines and coworkers demonstrated that pUs3 phosphorylates multiple serine residues in the N terminus of pUL31 and that serine-to-alanine substitution of these residues recapitulates the Us3-null phenotype (i.e., the accumulation of PEVs in the perinuclear space) (18). We hypothesize that the absence of pUs3-mediated phosphorylation of pUL31 “activates” the NEC to form INM herniations. We suggest that, by breaching the lamina and marginalized chromatin, INM herniations serve as attractive sites for primary envelopment of capsids. However, this might position PEVs too far away from the ONM, where de-envelopment must take place, and thereby leads to their accumulation in the perinuclear space. In other words, if the separation between the site of primary envelopment and the site of de-envelopment is too large, this can impair nuclear egress. This idea is supported by observations from the Mettenleiter laboratory showing that when the perinuclear space was enlarged through expression of a dominant negative SUN protein, PRV PEVs accumulated within this compartment (31). Since the formation of INM invaginations is a feature associated with UL21 mutants (Fig. 6), separation of the sites of primary envelopment and de-envelopment may also explain why UL21 mutants can display nuclear egress impairment even in the presence of functional pUs3.
Our observation that knockdown of pUL31 in Δ21-infected cells prevented irregular NEC distribution implies that NEC activity is required for this irregular distribution and further, similar to pUs3, pUL21 can regulate NEC activity. In addition to other nuclear envelope perturbations, we observed the accumulation of PEVs within extensive INM invaginations, particularly in HSV-1 strains harboring UL21 deletions (Fig. 6; Fig. S1). As mentioned above, a likely explanation for the Us3 mutant PEV accumulation phenotype is the hypophosphorylation of pUL31 observed in the absence of pUs3 (18). It may be that pUL21 enhances pUs3 activity or, alternatively, regulates the phosphorylation status of pUL31 by other means such as by preventing its dephosphorylation. Our earlier observations that pUL21 localizes to the nuclear rim during virus infection would position pUL21 in the appropriate location to perform such activities (21). Determining the mechanism by which pUL21 localizes to the nuclear rim and examination of pUL31 phosphorylation status in UL21 mutant-infected cells should provide clearer insight into how pUL21 regulates NEC activity.
Our observation (Fig. 2 and 11A) that the NEC was irregularly distributed and that PEVs accumulated in INM herniations in ΔUs3-infected cells, similar to what has been observed for other alphaherpesviruses, lies in contrast with work from the Kawaguchi laboratory, which suggested that nuclear egress is unperturbed in HSV-2 Us3 mutants (29). Since the strain of HSV-2 and the cell type utilized in the two laboratories were identical, the nature of the mutations may be responsible for this discrepancy. The ΔUs3 strain used here contains tandem nonsense codons after the second Us3 codon and infections with ΔUs3 produce no pUs3 (Fig. 10; Fig. S2). By contrast, the strains utilized by Morimoto et al. express catalytically inactive forms of pUs3, raising the possibility that pUs3 is important for HSV-2 nuclear egress but that its kinase activity is not (29). Interestingly, replacement of HSV-1 Us3 with HSV-2 Us3 resulted in a chimeric strain that displayed nuclear egress deficiencies (32). These findings indicated that HSV-2 Us3 could not complement the loss of HSV-1 Us3 and further supported the idea that HSV-2 pUs3 does not function in nuclear egress. However, based on our findings that the absence of pUs3 in HSV-2 perturbs nuclear egress (Fig. 2 and Fig. 11A and B), it may be that HSV-2 pUs3 expressed in the HSV-1 background strain is unable to engage other, as-yet-unidentified, viral or cellular proteins required for pUs3 nuclear egress activity.
In conclusion, we have provided evidence that pUL16, pUL21 and pUs3 play distinct roles in the nuclear egress of HSV-2 capsids. Whereas pUL16 appears to function in advance of capsid primary envelopment, both pUs3 and pUL21 appear to function by regulating NEC activity through distinct mechanisms.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney cells (Vero), 293T cells, HeLa cells, human keratinocytes (HaCaT), and murine L fibroblasts were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum in a 5% CO2 environment. L cells and HaCaT cells that stably produce pUL21 of HSV-2 strain 186 (referred to here as L21 and HaCaT21) and L cells and HaCaT cells that stably produce pUL16 of HSV-2 strain 186 (referred to here as L16 and HaCaT16) were isolated by retroviral transduction using an amphotropic Phoenix-Moloney murine leukemia virus system, as described previously (19, 21). HSV-2 strains SD90e, HG52, and 186 were acquired from D. M. Knipe (Harvard University), D. J. McGeoch (MRC Virology Unit, University of Glasgow), and Y. Kawaguchi (University of Tokyo), respectively; HSV-1 strains KOS and F were acquired from L. W. Enquist (Princeton University). Viruses deficient in pUL21 or pUL16 were constructed by two-step Red-mediated mutagenesis of a bacterial artificial chromosome (BAC) or by CRISPR/Cas9 mutagenesis as described previously (19–21, 23). HSV-2 186 virus deficient in pUs3 (ΔUs3) was constructed by two-step Red-mediated mutagenesis (33), using pYEbac373 (21) in Escherichia coli GS1783. Primers 5′-CCCGTCGCTCGGGGTGCTCGTTGGTTGGCACGCGCGACGCGGCGAATGGCCTGATGAAAGAGGATGACGACGATAAGTAGGG-3′ and 5′-CTTGTCGGGTCTACGGTAGACCCCACAGAACTTTCATCAGGCCATTCGCCGCGTCGCGCGCAACCAATTAACCAATTCTGATTAG-3′ were used to amplify a PCR product from pEP-Kan-S2, kindly provided by N. Osterrieder (Freie Universität Berlin), to introduce two tandem stop codons (boldface) after the first two codons of Us3 (underlined).
Restriction fragment length polymorphism analysis was used to confirm the integrity of each recombinant BAC clone in comparison to the wild-type (WT) BAC by digestion with EcoRI. In addition, a PCR fragment that spanned the region of interest was amplified and sequenced to confirm the presence of the tandem stop codons in Us3. Virus was reconstituted from ΔUs3 BAC DNA as described previously (21). To repair the Us3 deletion mutant, the primers 5′-CCCGTCGCTCGGGGTGCTCGTTGGTTGGCACGCGCGACGCGGCGAATGGCCTGTCGTAAGAGGATGACGACGATAAGTAGGG-3′ and 5′-CTTGTCGGGTCTACGGTAGACCCCACAGAACTTACGACAGGCCATTCGCCGCGTCGCGCGCAACCAATTAACCAATTCTGATTAG-3′ were used to amplify a PCR product from pEP-Kan-S2, which was used to restore the complete Us3 gene in the ΔUs3 BAC as described above. Western blot analysis confirmed that pUs3 was not produced in cells infected with ΔUs3 (Fig. S2) and was restored in the repaired strain ΔUs3R. HSV-2 186 viruses deficient in both pUs3 and pUL16 (Δ16/ΔUs3) were constructed by CRISPR/Cas9 mutagenesis with HSV-2 UL16 specific guide RNAs on viral genomic DNA isolated from ΔUs3 as described previously (20). Two separate Δ16/ΔUs3 viruses, isolated from independent cotransfections, were utilized in this study. In both Δ16/ΔUs3 viruses, the UL16 gene contains an in-frame deletion of codons 10 through 360. WT viruses and pUs3-deficient virus were propagated in either Vero or HaCaT cells. All pUL21-deficient viruses were propagated in either L21 or HaCaT21 cells. All pUL16-deficient viruses were propagated in L16 cells or HaCaT16 cells. The times postinfection, reported as hours postinfection (hpi), refer to the time elapsed following medium replacement after a 1-h inoculation period.
Production of antisera against HSV-2 pUL31 and HSV-2 pUL34.
Recombinant full-length HSV-2 GST-UL31 and HSV-2 GST-UL34 fusion proteins were produced in E. coli strain Rosetta(DE3). Bacteria were lysed, and inclusion bodies were purified using a B-Per protein purification kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s instructions. Proteins in inclusion bodies were separated on preparative SDS-PAGE gels, the bands corresponding to glutathione S-transferase fusions were excised and sent to Cedarlane (Burlington, Ontario, Canada) to immunize rats for pUL31 antiserum production or to Virusys (Taneytown, MD) to immunize chickens for pUL34 antiserum production. Rat polyclonal antiserum against pUL31 was used for indirect immunofluorescence microscopy at a dilution of 1:500 and for Western blotting at a dilution of 1:200; chicken polyclonal antiserum against pUL34 was used for indirect immunofluorescence microscopy at a dilution of 1:500 and for Western blotting at a dilution of 1:500.
Other immunological reagents.
Alexa Fluor 488-conjugated donkey anti-rat IgG, Alexa Fluor 568-conjugated goat anti-rat IgG, and Alexa Fluor 568-conjugated goat anti-chicken IgG (Molecular Probes, Eugene, OR) were all used for indirect immunofluorescence microscopy at a dilution of 1:500. Rabbit polyclonal antiserum against pUL16 (34), kindly provided by J. W. Wills (The Pennsylvania State University College of Medicine), was used for Western blotting at a dilution of 1:3,000. Rat polyclonal antiserum against pUL21 (21) was used for Western blotting at a dilution of 1:600. Rat polyclonal antiserum against pUs3 (35) was used for Western blotting at a dilution of 1:1,000. Mouse monoclonal antibody against ICP27 (Virusys) was used for Western blotting at a dilution of 1:500. Mouse monoclonal antibody against EGFP (Clontech, Mountain View, CA) was used for Western blotting at a dilution of 1:1,000. Mouse monoclonal antibody against ICP8 (Virusys) was used for Western blotting at a dilution of 1:4,000, and mouse monoclonal antibody against β-actin (Sigma, St. Louis, MO) was used for Western blotting at a dilution of 1:2,000. Horseradish peroxidase-conjugated goat anti-mouse IgG, horseradish peroxidase-conjugated goat anti-chicken IgY, horseradish peroxidase-conjugated rabbit anti-rat IgG, and horseradish peroxidase-conjugated goat anti-rabbit IgG (Sigma) were used for Western blotting at dilutions of 1:10,000, 1:30,000, 1:80,000, and 1:5,000, respectively.
Indirect immunofluorescence microscopy.
Cells for microscopic analyses were grown on 35 mm glass bottom dishes (MatTek, Ashland, MA), fixed with 4% paraformaldehyde and stained as described previously (35). Images were captured with an Olympus FV1000 laser scanning confocal microscope using a 60× (1.42 NA) oil immersion objective and Fluoview 1.7.3.0 software. Composites of representative images were prepared using Adobe Photoshop software.
Transmission electron microscopy.
Vero cells growing in 100-mm dishes were infected with virus at a multiplicity of infection (MOI) of 3 and processed for TEM at 16 hpi. Infected cells were rinsed with phosphate-buffered saline (PBS) three times before fixing in 1.5 ml of 2.5% electron microscopy-grade glutaraldehyde (Ted Pella, Redding, CA) in 0.1 M sodium cacodylate buffer (pH 7.4) for 60 min. Cells were collected by scraping into fixative and centrifugation at 300 × g for 5 min. Cell pellets were carefully enrobed in an equal volume of molten 5% low-melting-temperature agarose (Lonza, Rockland, ME) and allowed to cool. Specimens in agarose were incubated in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 1.5 h and postfixed in 1% osmium tetroxide for 1 h. The fixed cells in agarose were rinsed with distilled water three times and stained in 0.5% uranyl acetate overnight before dehydration in ascending grades of ethanol (30 to 100%). Samples were transitioned from ethanol to infiltration with propylene oxide and embedded in Embed-812 hard resin (Electron Microscopy Sciences, Hatfield, PA). Blocks were sectioned at 50 to 60 ηm and stained with uranyl acetate and Reynolds’ lead citrate. Images were collected using a Hitachi H-7000 transmission electron microscope.
DsiRNA knockdown.
Dicer-substrate siRNAs (DsiRNAs) directed against HSV-2 strain 186 UL31 were custom designed using the Integrated DNA Technologies design tool (Integrated DNA Technologies, Coralville, IA). DsiRNAs were transfected into Vero cells growing on 35-mm glass-bottom dishes or on standard 35-mm dishes using PepMute transfection reagent (SignaGen Laboratories, Rockville, MD) according to the manufacturer’s protocols. At 24 h after transfection, the transfected cells were infected with virus at an MOI of 0.5. At 18 hpi, infected cells were fixed in preparation for indirect immunofluorescent staining, or whole-cell lysates were prepared for Western blot analysis.
Plasmids and transfections.
The construction of a plasmid encoding EGFP-UL31 was described previously (36). To construct EGFP-UL34, PCR utilizing the forward primer 5′-AGTTCGAATTCTATGGCGGGGATGGGGAAGCCCTACG-3′ and the reverse primer 5′-GATCGTCGACTCATATAGGCGCGCGCCAACCGCC-3′ were used to amplify the UL34 gene using purified HSV-2 DNA as the template. The product was digested with EcoRI and SalI and ligated into similarly digested pEGFP-C1 (Clontech). All plasmids constructed utilizing PCR were sequenced to ensure that no unintended mutations were introduced. For preparing whole-cell extracts of transfected cells for Western blot analysis, plasmids were transfected into 293T cells using the calcium phosphate coprecipitation method (37). To examine the localization of EGFP fusion proteins by microscopy, plasmids were transfected into HeLa cells growing on 35-mm glass-bottom dishes using X-treme GENE HP DNA transfection reagent (Roche, Laval, Quebec, Canada) according to the manufacturer’s protocols.
Preparation and analysis of whole-cell protein extracts.
To prepare whole-cell protein extracts of transfected or infected cells for Western blot analyses, cells were washed with cold PBS and then scraped into cold PBS containing protease inhibitors (Roche) and 5 mM sodium fluoride (New England Biolabs, Ipswich, MA) and 1 mM sodium orthovanadate (New England Biolabs) to inhibit phosphatases. Harvested cells were transferred to a 1.5-ml microcentrifuge tube containing 3× SDS-PAGE loading buffer. The lysate was repeatedly passed through a 28-1/2 gauge needle to reduce viscosity and then heated at 100°C for 5 min. For Western blot analysis, 10 to 20 μl of whole-cell extract was electrophoresed through SDS-PAGE gels. Separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA) and probed with appropriate dilutions of primary antibody followed by appropriate dilutions of horseradish peroxidase-conjugated secondary antibody. The membranes were treated with Pierce ECL Western blotting substrate (Thermo Scientific) and exposed to film.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Canadian Institutes of Health Research operating grant 93804, Natural Sciences and Engineering Council of Canada Discovery Grant 418719, and Canada Foundation for Innovation award 16389 to B.W.B. J.G. was supported in part by an award from the China Scholarship Council. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank Xiaohu Yan (Queen’s University) for assistance with preparing samples for TEM analyses and Kristin Piche for technical assistance with plasmid construction.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15:170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 2.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/jvi.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76:8939–8952. doi: 10.1128/jvi.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bigalke JM, Heldwein EE. 2015. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J 34:2921–2936. doi: 10.15252/embj.201592359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leigh KE, Sharma M, Mansueto MS, Boeszoermenyi A, Filman DJ, Hogle JM, Wagner G, Coen DM, Arthanari H. 2015. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc Natl Acad Sci U S A 112:9010–9015. doi: 10.1073/pnas.1511140112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lye MF, Sharma M, El Omari K, Filman DJ, Schuermann JP, Hogle JM, Coen DM. 2015. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J 34:2937–2952. doi: 10.15252/embj.201592651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walzer SA, Egerer-Sieber C, Sticht H, Sevvana M, Hohl K, Milbradt J, Muller YA, Marschall M. 2015. Crystal structure of the human cytomegalovirus pUL50-pUL53 core nuclear egress complex provides insight into a unique assembly scaffold for virus-host protein interactions. J Biol Chem 290:27452–27458. doi: 10.1074/jbc.C115.686527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bäuerlein FJB, Cheleski J, Werner S, Guttmann P, Rehbein S, Henzler K, Demmerle J, Adler B, Koszinowski U, Schermelleh L, Schneider G, Enquist LW, Plitzko JM, Mettenleiter TC, Grünewald K. 2015. Structural basis of vesicle formation at the inner nuclear membrane. Cell 163:1692–1701. doi: 10.1016/j.cell.2015.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeev-Ben-Mordehai T, Weberruß M, Lorenz M, Cheleski J, Hellberg T, Whittle C, El Omari K, Vasishtan D, Dent KC, Harlos K, Franzke K, Hagen C, Klupp BG, Antonin W, Mettenleiter TC, Grünewald K. 2015. Crystal structure of the herpesvirus nuclear egress complex provides insights into inner nuclear membrane remodeling. Cell Rep 13:2645–2652. doi: 10.1016/j.celrep.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang YE, Van Sant C, Krug PW, Sears AE, Roizman B. 1997. The null mutant of the U(L)31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J Virol 71:8307–8315. doi: 10.1128/JVI.71.11.8307-8315.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. 2000. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J Virol 74:117–129. doi: 10.1128/jvi.74.1.117-129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci U S A 104:7241–7246. doi: 10.1073/pnas.0701757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. doi: 10.1038/ncomms5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia-Saez AJ, Mettenleiter TC, Antonin W. 2015. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J Biol Chem 290:6962–6974. doi: 10.1074/jbc.M114.627521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Proft A, Spiesschaert B, Izume S, Taferner S, Lehmann MJ, Azab W. 2016. The role of the equine herpesvirus type 1 (EHV-1) US3-encoded protein kinase in actin reorganization and nuclear egress. Viruses 8:275. doi: 10.3390/v8100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schumacher D, Tischer BK, Trapp S, Osterrieder N. 2005. The protein encoded by the US3 orthologue of Marek’s disease virus is required for efficient de-envelopment of perinuclear virions and involved in actin stress fiber breakdown. J Virol 79:3987–3997. doi: 10.1128/JVI.79.7.3987-3997.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagenaar F, Pol JM, Peeters B, Gielkens AL, de Wind N, Kimman TG. 1995. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J Gen Virol 76:1851–1859. doi: 10.1099/0022-1317-76-7-1851. [DOI] [PubMed] [Google Scholar]
- 18.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol 83:5181–5191. doi: 10.1128/JVI.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao J, Hay TJM, Banfield BW. 2017. The product of the herpes simplex virus 2 UL16 gene is critical for the egress of capsids from the nuclei of infected cells. J Virol 91:e00350-17. doi: 10.1128/JVI.00350-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Yan X, Banfield BW. 2018. Comparative analysis of UL16 mutants derived from multiple strains of HSV-2 and HSV-1 reveals species-specific requirements for the UL16 protein. J Virol 92:e00629-18. doi: 10.1128/JVI.00629-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Sage V, Jung M, Alter JD, Wills EG, Johnston SM, Kawaguchi Y, Baines JD, Banfield BW. 2013. The herpes simplex virus 2 UL21 protein is essential for virus propagation. J Virol 87:5904–5915. doi: 10.1128/JVI.03489-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harper AL, Meckes DG Jr, Marsh JA, Ward MD, Yeh PC, Baird NL, Wilson CB, Semmes OJ, Wills JW. 2010. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J Virol 84:2963–2971. doi: 10.1128/JVI.02015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finnen RL, Banfield BW. 2018. CRISPR/Cas9 mutagenesis of UL21 in multiple strains of herpes simplex virus reveals differential requirements for pUL21 in viral replication. Viruses 10:258. doi: 10.3390/v10050258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malhas A, Goulbourne C, Vaux DJ. 2011. The nucleoplasmic reticulum: form and function. Trends Cell Biol 21:362–373. doi: 10.1016/j.tcb.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Bosse JB, Enquist LW. 2016. The diffusive way out: herpesviruses remodel the host nucleus, enabling capsids to access the inner nuclear membrane. Nucleus 7:13–19. doi: 10.1080/19491034.2016.1149665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosse JB, Hogue IB, Feric M, Thiberge SY, Sodeik B, Brangwynne CP, Enquist LW. 2015. Remodeling nuclear architecture allows efficient transport of herpesvirus capsids by diffusion. Proc Natl Acad Sci U S A 112:E5725–E5733. doi: 10.1073/pnas.1513876112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banfield BW. 2019. Beyond the NEC: modulation of herpes simplex virus nuclear egress by viral and cellular components. Curr Clin Micro Rpt 6:1–9. doi: 10.1007/s40588-019-0112-7. [DOI] [Google Scholar]
- 28.Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J Virol 76:364–378. doi: 10.1128/JVI.76.1.364-378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J Virol 83:11624–11634. doi: 10.1128/JVI.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deruelle MJ, Favoreel HW. 2011. Keep it in the subfamily: the conserved alphaherpesvirus US3 protein kinase. J Gen Virol 92:18–30. doi: 10.1099/vir.0.025593-0. [DOI] [PubMed] [Google Scholar]
- 31.Klupp BG, Hellberg T, Granzow H, Franzke K, Dominguez Gonzalez B, Goodchild RE, Mettenleiter TC. 2017. Integrity of the linker of nucleoskeleton and cytoskeleton is required for efficient herpesvirus nuclear egress. J Virol 91:e00330-17. doi: 10.1128/JVI.00330-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shindo K, Kato A, Koyanagi N, Sagara H, Arii J, Kawaguchi Y. 2016. Characterization of a herpes simplex virus 1 (HSV-1) chimera in which the Us3 protein kinase gene is replaced with the HSV-2 Us3 gene. J Virol 90:457–473. doi: 10.1128/JVI.02376-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 34.Han J, Chadha P, Starkey JL, Wills JW. 2012. Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc Natl Acad Sci U S A 109:19798–19803. doi: 10.1073/pnas.1212900109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finnen RL, Roy BB, Zhang H, Banfield BW. 2010. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 397:23–33. doi: 10.1016/j.virol.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sherry MR, Hay TJM, Gulak MA, Nassiri A, Finnen RL, Banfield BW. 2017. The herpesvirus nuclear egress complex component, UL31, can be recruited to sites of DNA damage through poly-ADP ribose binding. Sci Rep 7:1882. doi: 10.1038/s41598-017-02109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham FL, van der Eb AJ. 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.