

Middle East respiratory syndrome coronavirus (MERS-CoV) is a major emerging infectious disease with zoonotic potential and has reservoirs in dromedary camels and bats. Since its first outbreak in 2012, the virus has repeatedly transmitted from camels to humans, with 2,468 confirmed cases causing 851 deaths. To date, there are no efficacious drugs and vaccines against MERS-CoV, increasing its potential to cause a public health emergency. In order to develop novel drugs and vaccines, it is important to understand the molecular mechanisms that enable the virus to infect host cells. Our data have found that calcium is an important regulator of viral fusion by interacting with negatively charged residues in the MERS-CoV FP region. This information can guide therapeutic solutions to block this calcium interaction and also repurpose already approved drugs for this use for a fast response to MERS-CoV outbreaks.
KEYWORDS: MERS, spike protein, coronavirus, fusion, fusion peptide, membrane fusion, viral fusion
ABSTRACT
Fusion with, and subsequent entry into, the host cell is one of the critical steps in the life cycle of enveloped viruses. For Middle East respiratory syndrome coronavirus (MERS-CoV), the spike (S) protein is the main determinant of viral entry. Proteolytic cleavage of the S protein exposes its fusion peptide (FP), which initiates the process of membrane fusion. Previous studies on the related severe acute respiratory syndrome coronavirus (SARS-CoV) FP have shown that calcium ions (Ca2+) play an important role in fusogenic activity via a Ca2+ binding pocket with conserved glutamic acid (E) and aspartic acid (D) residues. SARS-CoV and MERS-CoV FPs share a high sequence homology, and here, we investigated whether Ca2+ is required for MERS-CoV fusion by screening a mutant array in which E and D residues in the MERS-CoV FP were substituted with neutrally charged alanines (A). Upon verifying mutant cell surface expression and proteolytic cleavage, we tested their ability to mediate pseudoparticle (PP) infection of host cells in modulating Ca2+ environments. Our results demonstrate that intracellular Ca2+ enhances MERS-CoV wild-type (WT) PP infection by approximately 2-fold and that E891 is a crucial residue for Ca2+ interaction. Subsequent electron spin resonance (ESR) experiments revealed that this enhancement could be attributed to Ca2+ increasing MERS-CoV FP fusion-relevant membrane ordering. Intriguingly, isothermal calorimetry showed an approximate 1:1 MERS-CoV FP to Ca2+ ratio, as opposed to an 1:2 SARS-CoV FP to Ca2+ ratio, suggesting significant differences in FP Ca2+ interactions of MERS-CoV and SARS-CoV FP despite their high sequence similarity.
IMPORTANCE Middle East respiratory syndrome coronavirus (MERS-CoV) is a major emerging infectious disease with zoonotic potential and has reservoirs in dromedary camels and bats. Since its first outbreak in 2012, the virus has repeatedly transmitted from camels to humans, with 2,468 confirmed cases causing 851 deaths. To date, there are no efficacious drugs and vaccines against MERS-CoV, increasing its potential to cause a public health emergency. In order to develop novel drugs and vaccines, it is important to understand the molecular mechanisms that enable the virus to infect host cells. Our data have found that calcium is an important regulator of viral fusion by interacting with negatively charged residues in the MERS-CoV FP region. This information can guide therapeutic solutions to block this calcium interaction and also repurpose already approved drugs for this use for a fast response to MERS-CoV outbreaks.
INTRODUCTION
Coronaviruses (CoVs) comprise a family of enveloped viruses causing respiratory and/or enteric tract infections across a variety of hosts. Recently, the emergence of Middle East respiratory syndrome coronavirus (MERS-CoV), first isolated in 2012 from a 60-year-old man in the Kingdom of Saudi Arabia, has highlighted the pathogenic potential of CoVs (1). To date, MERS-CoV has caused 2,468 laboratory-confirmed human infections worldwide, resulting in 851 deaths (case fatality, 34.48%) (2). However, human MERS-CoV originated in bats from where it transitioned into intermediate hosts, such as dromedary camels (3–5). It is currently believed that disease outbreaks in humans result from interactions with dromedary camels. Although transmission between humans is rare, this route was responsible for a widespread outbreak of MERS-CoV in 2015 in South Korea (6). Moreover, it was demonstrated that MERS-like bat CoV could potentially infect human tissues in the absence of an intermediate host when the respective tissue expresses a protease that is able to prime the virus for fusion (7). More recently, the World Health Organization (WHO) listed MERS-CoV as one of ten diseases that requires urgent accelerated research and development because of its potential to cause a public health emergency with few or no efficacious drugs and/or vaccines to counter them (8). To address these needs, it is important to have a more nuanced understanding of the MERS-CoV infection process to develop therapeutics to target crucial steps in MERS-CoV infection.
MERS-CoV is an enveloped virus that utilizes a class I fusion protein for viral entry into the host cell (9). The MERS-CoV spike (S) protein contains a receptor binding subunit (S1) and a membrane fusion subunit (S2), which facilitate binding to the host cell surface receptor dipeptidyl peptidase 4 (DPP4) and fusion with the host cell membrane, respectively (10, 11). To mediate fusion with the host cell membrane, the S protein needs to be cleaved by either host cell surface proteases or endosomal proteases (such as transmembrane protease serine 2 [TMPRSS2], furin, or cathepsins) (12–16). Cleavage can occur at two different sites, namely, the S1 and S2 boundary (S1/S2) and adjacent to a fusion peptide (FP) within the S2 domain (S2’) (17, 18). It is suggested that both cleavage events act in concert to promote membrane fusion and viral infectivity. The first cleavage event takes place at the S1/S2 site, which then is believed to open up the S2’ site for further proteolytic processing, resulting in the exposure of the FP (19). As a consequence, the FP inserts into the host membrane, mediating membrane fusion (19).
MERS-CoV viral fusion can occur at two different locations in the host cell, namely, at the plasma membrane or at the endosomal membrane. Host cell surface proteases, such as TMPRSS2, can cleave MERS-CoV S protein for entry directly at the plasma membrane (13). In the absence of cell surface proteases, MERS-CoV is endocytosed, and endosomal proteases, such as cathepsins, can cleave the S protein for endosomal entry, with an additional role for furin likely. (19, 20). However, in contrast to other viruses, such as influenza A virus, MERS-CoV fusion does not seem to depend on a pH change in the endosomes, as receptor binding in conjunction with proteolytic activation are sufficient to trigger fusion (21). A more thorough description of the fusion pathways is summarized in several excellent reviews (20–23).
A recent study on the FP of severe acute respiratory syndrome coronavirus (SARS-CoV) suggested that the FP region begins immediately downstream of the S2’ cleavage site and that it can be separated into two distinct domains, namely, FP1 and FP2 (24). We showed that both FP1 and FP2 increase membrane ordering and proposed that FP1 and FP2 together form an extended FP which acts as a bipartite fusion platform. Intriguingly, both FP subdomains required calcium ions (Ca2+) to exert their function in membrane entry and fusion. It was proposed that negatively charged aspartic acid (D) and glutamic acid (E) residues coordinate with the positively charged Ca2+ within the fusion platform to promote greater membrane ordering. Furthermore, we showed that depletion of extracellular as well as intracellular Ca2+ pools resulted in significantly reduced infectivity of SARS-CoV pseudoparticles, suggesting that both the plasma membrane and endosomal cell entry pathways are regulated by Ca2+. Other studies have also revealed an important role for Ca2+ in the fusion dynamics of enveloped viruses. Rubella virus (RuV), for example, requires Ca2+ for virus entry and infection (25). Ca2+ is coordinated in between the two fusion loops of the RuV E1 fusion protein by asparagine (N) and aspartic acid, and both residues were shown to be essential for virus viability. More recently, Ca2+ was reported to enhance the entry of Ebola virus (EBOV) by directly interacting with the FP of the Ebola virus fusion protein (26). We were able to identify two negatively charged residues (D522 and E540) in the FP that interact with Ca2+, and this interaction is critical for viral fusion with the host cell.
Here, we addressed the question whether Ca2+ is required for viral entry and infectivity of MERS-CoV by creating a set of S protein mutants in which negatively charged residues in the FP were mutated to alanines (A). We used the different mutants in pseudoparticle infection assays to demonstrate that fusion of MERS-CoV is promoted but not dependent on Ca2+. Biophysical analysis demonstrated that Ca2+ interactions with the MERS-CoV increases membrane ordering in a Ca2+-dependent manner. Our data suggest that a single glutamic acid residue in a region complementary to the SARS-CoV FP1 domain is involved in Ca2+ coordination.
RESULTS
S protein mutants with amino acid substitutions in the FP are expressed and cleaved as WT proteins.
In order to test if Ca2+ is required and which residues within the MERS-CoV FP are necessary for its coordination, we generated a set of mutated S proteins in which we substituted negatively charged aspartic acid and glutamic acid residues in the FP with alanine (A) (Fig. 1A). We defined the FP region based on a sequence comparison with the SARS-CoV FP that was described previously (24) (Fig. 1A). The S protein mutant plasmids were generated via site-directed mutagenesis and verified by sequencing (data not shown). We next tested whether the mutated S proteins were expressed by transfecting the plasmid DNA into HEK293T cells. Since the MERS-CoV S protein can be cleaved by constitutively expressed proteases during the maturation process, we added a protease inhibitor, dec-RVKR-CMK, immediately after transfection to block this cleavage (Fig. 2A). Dec-RVKR-CMK was shown to inhibit a wide variety of proteases, such as furin, cathepsins, trypsin, and TMPRSS2, to block S protein cleavage (27). As we wanted to confirm that the mutant S proteins are properly expressed and trafficked to the cell membrane surface, like the wild-type (WT) S protein, we isolated proteins 18 h posttransfection via cell-surface biotinylation. Western blot analysis of the cell surface proteins showed that the S protein mutants E891A, D892A, E891A/D892A, D896A, D902A, D910A, D911A, D910A/D911A, and D922A were expressed to similar levels as the MERS-CoV WT S protein (Fig. 2B). With all mutant S proteins expressed, we next wanted to investigate whether the mutants exhibit the same cleavage pattern as WT S since proteolytic cleavage is an important and crucial trigger for viral fusion. We accomplished this by treating transfected cells with trypsin prior to protein isolation. Western blot analysis revealed that the cleavage pattern of the abovementioned mutated S proteins was similar to that of the WT S protein, suggesting that the introduced mutations in the FP did not impair protease accessibility and cleavage (Fig. 2B).
FIG 1.

Sequence and model of MERS-CoV S protein fusion loop. (A) Sequences of SARS-CoV S Urbani and MERS-CoV S EMC/2012 fusion peptides (FPs). FP1 and FP2 designate the two different domains in the FP. Sequences illustrate the mutations that were introduced in the MERS-CoV S protein via site-directed mutagenesis. In red are the negatively charged residues D and E; in green are the A substitutions. (B) Modeling of the MERS-CoV S protein monomer with an emphasis on the FP. Negatively charges D and E are depicted as atomic bonds in red. The S2’ site is orange, and the FP1 and FP2 domains are labeled blue and pink, respectively.
FIG 2.
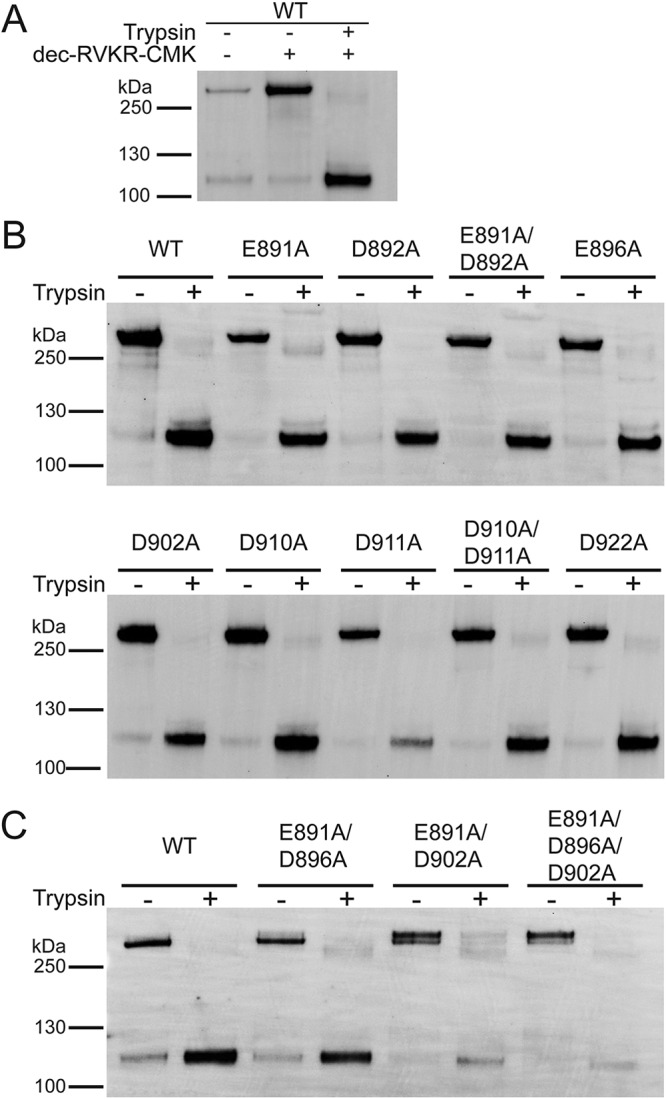
Protein expression and trypsin-mediated cleavage of MERS-CoV S protein WT and mutants. (A) Plasmid DNA encoding MERS-CoV S protein WT EMC/2012 was transfected in HEK293T cells. The protease inhibitor dec-RVKR-CMK at a concentration of 75 μM was added at the time of transfection, as indicated. After 18 h, transfected cells were treated with 0.8 nM TPCK-treated trypsin, as indicated. Proteins were subsequently isolated via cell-surface biotinylation. The cell surface proteins were analyzed using SDS-PAGE and detected using a Western blot with MERS-CoV S antibodies. (B and C) MERS-CoV S mutant proteins with indicated A substitutions were expressed in HEK293T cells. Protease inhibitor dec-RVKR-CMK was added at the time of transfection, and after 18 h, cells were treated with TPCK-treated trypsin, as indicated. Cell surface proteins were isolated and analyzed as described above. Full-length S proteins are visible at approximately 250 kDa. S1/S2-cleaved S proteins are visible at approximately 115 kDa.
Residue E891 in the FP1 region seems critical for the fusogenic activity of the MERS-CoV S protein.
After we established that the mutated versions of the MERS-CoV S protein described in the previous paragraph are expressed and exhibit a similar cleavage pattern as the WT S protein, we initially screened whether the different mutant proteins are compromised in their ability to induce cell-cell fusion. Therefore, we coexpressed the S protein together with MERS-CoV receptor DPP4 in Vero cells for 18 h and visualized syncytium formation using an immunofluorescence assay (IFA). Trypsin treatment of transfected cells was not required, as the cells natively express proteases that cleave WT and mutated S proteins for cell-cell fusion. As a control, we added the protease inhibitor dec-RVKR-CMK to cells expressing the WT S protein to prevent S protein cleavage and block MERS-CoV entry (27). We analyzed the cells by fluorescence microscopy and quantified cell-cell fusion by counting the number of nuclei per syncytium. Cells expressing WT S protein exhibited strong syncytium formation with multiple nuclei, and the addition of dec-RVKR-CMK as a control efficiently prevented cell-cell fusion (Fig. 3A and B). Strikingly, in the absence of the inhibitor, the E891A and E891A/D892A mutants were impaired in their ability to trigger cell-cell fusion to the same extent as cells treated with dec-RVKR-CMK (Fig. 3A and B). However, since the cells expressing D892A formed syncytia similar to those of cells expressing the WT S protein, this finding suggests that E891 is a critical residue for the fusogenic activity of the MERS-CoV S protein and was responsible for the inability of E891A/D892A mutant to mediate cell-cell fusion. All other tested mutants showed attenuated cell-cell fusion at various degrees; however, none of the mutants, with the exception of the E891A/D892A double mutant, was found to be as defective as the E891A S protein in mediating cell-cell fusion (Fig. 3A and B).
FIG 3.

Immunofluorescence assay of MERS-CoV S protein WT and mutants. (A) Vero cells were transfected with plasmid DNA encoding the respective MERS-CoV S protein variants and the DPP4 binding receptor and grown for 18 h. As Vero cells express endogenous proteases, which cleave MERS-CoV S proteins for fusion, no further protease treatment was needed to induce syncytium formation. WT + furin inhibitor (FI) indicates the condition in which protease inhibitor dec-RVKR-CMK at a concentration of 75 μM was added at the time of transfection to block fusion. Syncytia were visualized using immunofluorescence microscopy by staining the MERS-CoV S protein with a polyclonal anti-S antibody (in green) and the nuclei with 4′,6-diamidino-2-phenylindole (DAPI; in blue). Images were taken at a magnification of ×25. (B) Quantification of syncytia. Nuclei of 9 syncytia were counted, and the average number of nuclei per syncytium was calculated. Error bars represent standard deviations (n = 9). Statistical analysis was performed using an unpaired Student’s t test comparing the WT against each of the respective mutant *, P > 0.5; **, P > 0.05; ***, P > 0.005.
Mutation of E891 diminishes infectivity of MERS-CoV pseudoparticles.
The cell-cell fusion results strongly suggest that E891 is an important residue for the fusogenic activity of the MERS-CoV S protein. With the exception of D892, the data obtained for the other mutants indicate that they may play a minor role to trigger fusion. However, the mechanisms of cell-cell fusion and virus-cell fusion may significantly differ. Factors such as membrane curvature and/or density of viral S proteins may impact virus-cell fusion differently than cell-cell fusion (19). Hence, the cell-cell fusion results provide an indication about the role of the individual D and E residues in the FP region, but they need to be verified by viral infection of host cells. As MERS-CoV is a risk group 3 agent that needs to be handled in a biosafety level 3 (BSL-3) setting, we employed a viral pseudotyping technique to generate pseudoparticles (PPs) that serve as surrogates of native virions and are suitable for infection in a BSL-2 setting. These pseudoparticles consist of a murine leukemia virus (MLV) core and are decorated with their respective S glycoprotein so that they recapitulate the entry steps (binding and fusion) of the native virus (28). Furthermore, the PPs carry a genome-encoding luciferase, and upon successful infection of cells, the luciferase reporter is integrated in the host cell genome and drives luciferase production within the cell, which can be used to quantify the degree of infectivity. We generated PPs carrying the mutant S proteins and infected Huh-7 cells, a permissive cell line for MERS-CoV infection with high DPP4 expression (15). After 72 h, the cells were lysed and briefly incubated with the luciferin substrate, which can be oxidized with cellular luciferase to produce luminescence. The measured relative luminescence values provide a quantitative read-out for the infectivity of each mutant S protein-mediated infection.
To ensure that the infectivity of PPs carrying different S protein variants can be compared to each other, we performed a Western blot analysis to detect the amount of S protein that was incorporated into the PPs (Fig. 4). Because the cells were not treated with the protease inhibitor dec-RVKR-CMK, the S proteins were cleaved by endogenously expressed proteases. PPs with WT S proteins and the mutant S proteins E891A, D892A, E891A/D892D, D896A, D902A, D910A, D911A, D910A/D911A, and D922A exhibited comparable levels of incorporated full-length protein (∼250 kDa) and protein cleaved at the S1/S2 site (∼130 kDa). Cleavage at the S2’ site seemed slightly reduced for the D896A, D902A, D910A, D911A, D910A/D911A, and D922A S protein mutants (∼115 kDa). As the Western blot data show that the PP incorporate full-length and cleaved S proteins, for all S variants, we continued to analyze the infectivity of PPs carrying these mutated S proteins.
FIG 4.

Western blot analysis of S proteins incorporated into PPs. A total of 1 ml of DMEM containing PPs per each tested S protein was ultracentrifuged, washed in PBS, and resuspended in SDS Laemmli buffer. Incorporated S proteins were analyzed using SDS-PAGE and detected using a Western blot with MERS-CoV S protein antibodies.
In agreement with the IFA observations, E891A and E891A/D892A PPs exhibited a 5-fold reduction in the infectivity compared with the WT S protein PPs, while D892A PPs were not significantly different from WT S protein PPs (Fig. 5A). In addition, we also observed that infectivity of PPs carrying D911A was significantly lower than WT S protein PPs. However, infectivity was restored in the D911A/D912A double mutant (Fig. 5A), suggesting that the D911 residue is not as critical for fusion as the E891 residue. If D911 was crucial for fusion, we would have expected the double mutant to display impaired infectivity as well. None of the other tested mutants exhibited significant differences compared with infections with WT S protein PPs (Fig. 5A). D922A seemed to exhibit enhanced infection compared with the WT, but we did find it to be not statistically significant (Fig. 5B).
FIG 5.

Pseudoparticle assays of MERS-CoV S protein WT and mutants. Huh-7 cells were infected with MLV-based pseudoparticles (PPs) carrying MERS-CoV S protein WT or one of the respective S mutants. After 72 h, infected cells were lysed and assessed for luciferase activity. (A) PP infectivity of Huh 7 cells. (B) Infectivity of PP carrying the D922A S protein. Δenv and VSV-G served as representative controls for all PP assays. (C) Impact of intracellular Ca2+ on MERS-CoV fusion. Cells were pretreated with growth medium containing either 50 μM calcium chelator BAPTA-AM or dimethyl sulfoxide (DMSO) for 1 h. Cells were then infected with their respective PPs in the presence of BAPTA-AM or DMSO for 2 h and grown for 72 h before assessment for luciferase activity. (D) Impact of extracellular Ca2+ on MERS-CoV fusion. Cells were pretreated with growth medium either with or without 1.8 mM Ca2+ for 1 h. The infection protocol is as described above except PPs were treated with 1.5 mM EGTA for calcium chelation. Infectivity was normalized such that WT PP infectivity is 1. Error bars represent standard deviations (n = 3). Statistical analysis was performed using an unpaired Student’s t test, as indicated. *, P > 0.5; **, P > 0.05; ***, P > 0.005.
Depletion of intracellular and extracellular Ca2+ ions abates viral infectivity.
SARS-CoV was previously shown to depend on both extracellular and intracellular Ca2+ pools for successful infection, and it is believed that this dependency is due to Ca2+ interactions with the SARS-CoV FP (24). Because of the high sequence similarity between the SARS-CoV and MERS-CoV FP (Fig. 1A), we were interested in determining if extracellular and/or intracellular Ca2+ also plays a role in MERS-CoV fusion. Our analysis of the negatively charged residues in the MERS-CoV S FP region thus far suggest that E891 is a crucial residue for viral fusion, perhaps for coordinating with Ca2+. We are further interested in observing if the fusion behavior of E891 mutants would differ from that of WT in various calcium concentration environments. Thus, we focused only on WT S and S proteins carrying the amino acid substitutions E891A, D892A, and E891A/D892A. Although the D911A expressed decreased infectivity, we did not proceed for reasons previously mentioned.
First, we depleted intracellular Ca2+ by incubating the cells with 50 μM of 1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’,-tetraacetic acid tetrakis (BAPTA-AM) prior to infection with PPs. BAPTA-AM is a Ca2+ chelator that is only active intracellularly and does not significantly impact cell viability at this concentration (24, 29). Chelating intracellular Ca2+ resulted in a 2-fold drop in infectivity of MERS-CoV WT PPs (Fig. 5C). Infectivity was also reduced in the E891A and the D892A mutants. However, we did not detect a significant reduction of infectivity in the E891A/D892A double mutant when depleting intracellular Ca2+. We next tested the impact of extracellular Ca2+ on MERS-CoV fusion with the host cell membrane by incubating the cells with 150 μM of ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA) prior to infection. Infectivity of MERS-CoV WT PPs was reduced by 25% when extracellular Ca2+ was depleted (Fig. 5D). There was no difference in infectivity with any of the PPs carrying the mutated S proteins.
E891A/D902A mutation impairs S protein incorporation into PPs.
The results described above suggest that Ca2+ promotes entry of MERS-CoV into the host cell, with a greater dependency on intracellular Ca2+ concentration than extracellular Ca2+ concentration. Since we hypothesized that E891 is a critical residue in Ca2+ coordination, we would have expected its knockout variant, E891A, to have infectivity levels that are independent of Ca2+ concentrations. However, the intracellular Ca2+ studies show that single-mutant E891A is impacted by Ca2+. A possible explanation for this apparent contradiction could be due to FP flexibility. Perhaps, if E891 is not available for Ca2+ binding, the adjacent D892 can compensate for E891 and maintain Ca2+ coordination in the FP. In support of this explanation is that the infectivity of the E891A/D892A double mutant is not detectably Ca2+ dependent, as both possible residues that can mediate Ca2+ interactions have been knocked out. Thus, we believe that E891 does mediate Ca2+ binding in the MERS-CoV FP1 region. However, previous studies with RuV and EBOV showed that two distinct opposing residues in the FP region are required to coordinate Ca2+ (25, 26), and so we proceeded to determine the other residues with which Ca2+ can coordinate.
We decided to test if E891 can coordinate with D896 or D902 as these are the next closest negative residues to E891. The prefusion MERS-CoV FP structure does show that these two residues might be too far from E891 (Fig. 1B). However, it is possible that during the fusion process, the FP undergoes conformational changes that will enable E891 to coordinate with these residues under the presence of Ca2+. Therefore, we created MERS-CoV S proteins carrying E891A/D896A, E891A/D902A, and E891A/D896A/D902A substitutions. We confirmed that all three mutated proteins are expressed and trafficked to the plasma membrane in HEK293T cells as proteins were extracted by cell-surface biotinylation (Fig. 2C). E891A/D902A and E891A/D896A/D902A, however, showed a reduced proper cleavability in response to trypsin treatment. The IFA results show that cells expressing E891A/D896A, E891A/D902A, and E891A/D896A/D902A are impaired in their ability to form syncytia to a similar extent as the cells expressing WT S proteins treated with dec-RVKR-CMK or the E891A single mutant (Fig. 3A and B). However, when we assessed protein incorporation into PPs, we found that E891A/D896A PPs carried less amounts of protein compared with WT and that there was no protein incorporation in E891A/D902A and the E891A/D896A/D902A PPs (Fig. 4). When cells were infected with PPs equipped with S protein mutations, we found that infectivity of E891A/D896A was similar to the E891A single mutant (Fig. 6A). Depleting intracellular Ca2+ levels using BAPTA-AM did not result in statistically significant changes in the infectivity of the tested PPs carrying S with the E891A/D896A substitution (Fig. 6B). When chelating extracellular Ca2+ using EGTA, infectivity was also not altered in the mutant (Fig. 6C). In summary, our further mutant analysis to identify the second binding partner of Ca2+ is inconclusive. The data suggest that E896A is likely not the second binding partner because the E891A/D896A infectivity is similar to that of the E891A single mutant.
FIG 6.

Pseudoparticle assays of MERS-CoV S protein WT and E891A/D896A. Huh-7 cells were infected with MLV-based pseudoparticles (PPs) carrying MERS-CoV S WT or one of the respective mutants. Infectivity was normalized to the WT sample. Error bars represent standard deviations (n = 3). Statistical analysis was performed using an unpaired Student’s t test comparing the WT against the respective mutant (for B and C, the untreated WT was compared to each sample). *, P > 0.5; **, P > 0.05; ***, P > 0.005. (A) Infectivity of PPs without pretreatment of cells. (B) Impact of intracellular Ca2+ on MERS-CoV fusion. Cells and PPs were treated as described for Fig. 5C. (C) Impact of extracellular Ca2+ on MERS-CoV fusion. Cells and PPs were treated as described for Fig. 5D.
Ca2+ increases MERS-CoV FP membrane insertion and lipid ordering.
We used electron spin resonance (ESR) spectroscopy to examine the effect of MERS-CoV FP on membranes and to test whether membrane fusion of the peptide requires Ca2+. In this technique, spin labels are incorporated into lipids at various positions, acting as depth probes. Multilamellar vesicles (MLVs) containing dipalmitoylphosphatidyl-tempo-choline (DPPTC) are spin labeled in the head region, whereas those with phosphocholine are labeled in either the upper tail region (5PC) or lower tail region (14PC). The ordering parameter S0 is an indication of the amount of membrane ordering at a given depth and serves as a readout for FP membrane penetration depth (30–32). Changes in membrane ordering may reduce the energy barrier between two membranes and, thus, promote fusion (31, 33). Previously studied FPs from different viruses induced membrane ordering illustrated as an S-shaped curve of S0 as a function of increasing FP-to-lipid (P/L) ratio, while nonfunctional FPs and random peptides do not have a membrane-ordering effect and produce a scrambled signal (24, 30–32, 34). Therefore, examining the membrane-ordering effect of a peptide allows us to determine whether it is a FP (24, 34).
As shown in Fig. 7A and B, MERS-CoV FP induced the greatest ordering effect of the head (DPPTC) and upper tail (5PC) regions with 1 mM Ca2+. When we tested the deep hydrophobic region (14PC), no membrane ordering was observed (data not shown). When no Ca2+ is supplemented and trace Ca2+ is chelated with 1 mM EGTA at the same time, no such ordering effect is measured. This finding indicates that the function of the MERS-CoV FP is Ca2+ dependent. Compared with the SARS-CoV FP, however, the MERS-CoV FP has a weaker effect. For the DPPTC case, the maximal S0 for MERS-CoV FP is smaller (approximately 0.472) than that for SARS-CoV FP (approximately 0.479) at the highest P/L ratio in our experiment (5%). We fitted the S-shape curve using a four-parameter logistic equation Y = S0,min + (S0,max − S0,min)/(1 + (X − X0)p), which is a built-in function of Origin (OriginLab), where the X0 is the P/L ratio (X) that induces half of maximal membrane order change (S0,max − S0,min), and p is the Hill’s slope. The Hill’s slope is an indicator for cooperativity in a ligand-receptor binding reaction. The X0 for SARS-CoV FP is 0.25% and for MERS-CoV FP is 0.61%, indicating the MERS-CoV FP is less effective than the SARS-CoV FP. The Hill’s slope for SARS-CoV FP is 2.39 and for MERS-CoV FP is 1.44, indicating SARS-CoV FP induces membrane ordering in a more cooperative fashion than MERS-CoV FP at 1 mM Ca2+. The scrambled peptide shows no effect on membrane order. Although the scrambled control peptide is based on the SARS-CoV FP sequence, it is used as a control for MERS-CoV FP, too, because of the high sequence similarity of MERS-CoV and SARS-CoV FPs (Fig. 1A).
FIG 7.

ESR and ITC analysis of the MERS-CoV FP. (A and B) Plots of order parameters of DPPTC (A), and 5PC (B) versus peptide:lipid ratio (P/L ratio) of MERS FP or SARS FP in POPC/POPS/Chol of 3/1/1 MLVs in buffer with 150 mM NaCl at 25°C. Black, MERS FP, 1 mM Ca2+ and at pH 5; red, MERS FP calcium-less buffer with 1 mM EGTA and at pH 5; blue, SARS FP, 1 mM Ca2+ at pH 5; and purple, scrambled peptide, 1 mM Ca2+ and at pH 5. (C) Plot of the difference of order parameters of DPPTC with and without 1% peptide binding (ΔS0) versus Ca2+ concentration in POPC/POPS/Chol of 3/1/1 MLVs in buffer with 150 mM NaCl at 25°C. Black, MERS FP; blue, SARS FP; and green, scrambled peptide. The experiments were typically repeated two to three times. The typical uncertainties found for S0 ranges from 1 × 10−3 to 5 × 10−3, while the uncertainties from repeated experiments were 5 × 10−3 to 8 × 10−3 or less than ±0.01. We show the standard deviation bars in A and B. (D) ITC analysis of Ca2+ binding to MERS-CoV FP. The peptides were titrated with CaCl2. The integrated data represent the enthalpy change per mole of injectant, ΔH, in units of kJ/mol as a function of the molar ratio. Data points and fitted data are overlaid. The fitting is based on the one-site model.
To further test the effect of Ca2+ on S0, we varied the Ca2+ concentration while fixing the P/L ratio at 1%. The effects of any ordering caused by Ca2+ interacting with the liposomes rather than the fusion peptide was removed by subtracting S0 of a liposome and Ca2+-only (no peptide) mixture from the measurement of S0 when all three components (liposomes, peptides, and Ca2+) were present, yielding ΔS0. This method proved to be a solid approach to single out the effect of FP under the influence of Ca2+ in a complex membrane-peptide-Ca2+ system (24, 26). As shown in Fig. 7C, ΔS0 increased with increasing Ca2+, indicating that there is more FP-induced membrane ordering at higher Ca2+ concentrations in the lipid head group regions. When fitted with the same logistic equation, we calculated that the concentration of Ca2+ to enable half of the maximal FP-induced ordering effect (X0,Ca) for SARS-CoV is 0.32 mM. X0,Ca for MERS-CoV has a very large standard error and, thus, is not reliable, due to the fact that the ΔS0-[Ca2+] curve has not been saturated within our experimental range (0 to 3 mM). But, X0,Ca for MERS-CoV is obviously larger than that for SARS-CoV. The Hill’s coefficients for SARS-CoV FP and MERS-CoV FP are 1.56 and 0.70, respectively, indicating that Ca2+ ions bind to the SARS-CoV FP more cooperatively than to MERS-CoV FP. For the SARS-CoV FP case, a hypothesis for this cooperativity is that the SARS-CoV FP has two Ca2+ binding sites and binding a Ca2+ ion in one site will stabilize the secondary structure and help to bind another Ca2+ ion in the other site. The lack of cooperativity in the case of the MERS-CoV FP suggests that it may have only one binding site.
The binding stoichiometry of Ca2+ and MERS CoV FP is close to one.
To directly investigate the interaction between Ca2+ and MERS-CoV FP, we used isothermal titration calorimetry (ITC). In this experiment, 1 ml of 0.05 mM synthetic MERS-CoV FP was added into the reaction cell at neutral buffer conditions (pH 7), and a total volume of 97 μl of 1 mM CaCl2 (pH 7) was titrated into the cell in a stepwise manner consisting of 5 μl per injection, except that the first injection was 2 μl. The heat flows generated by the reaction were recorded, and the reaction heat for each injection (ΔH) is calculated by integrating the heat flow over the time. After the subtraction of dilution heat, which is measured by titrating 1 ml CaCl2 into the pH 7 buffer, the reaction heat between the Ca2+ and the FP for each injection versus the molar ratio of Ca2+ to FP was plotted. As shown in Fig. 7D, the reaction heats are positive, indicating the reaction is endothermic. The reaction heats decrease during the titration, indicating the FPs have been saturated by the Ca2+ ions toward the end of the titration. The reason for the endothermic reaction may be due to the Na+ ions interacting with the FPs being replaced by the Ca2+ ions. An endothermic reaction was also observed when we injected CaCl2 into the SARS CoV FP in a similar experimental setup (24). Fitting the data with the one-site model, which assumes all binding site(s) have the same binding affinity, we calculated the binding constant Kb = 1.14 ± 0.23 × 104 M−1, the reaction enthalpy ΔH = 3.12 ± 0.45 kCal/mol, and the binding stoichiometry (Ca2+ to the FP) n = 1.12 ± 0.08. The stoichiometry of the Ca2+ to the MERS-CoV FP is significantly different from that for the FP1_2 of SARS-CoV FP (n = 1.7) and similar to FP1 or FP2 of the SARS-CoV FP (n = 0.92 and 1.02, respectively) (24). The results strongly support the hypothesis that the MERS-CoV FP binds only one Ca2+ ion, while the SARS-CoV FP binds two, one on FP1 and one on FP2. The weaker Ca2+ binding energies of the MERS-CoV FP is also consistent with the ESR results, which shows that the MERS-CoV FP induces a maximal membrane-ordering effect in a higher Ca2+ concentration than the SARS-CoV FP.
DISCUSSION
Viral entry into the host cell is one of the most critical steps in the life cycle of a virus. Enveloped viruses, such as MERS-CoV and SARS-CoV, utilize class I fusion proteins that facilitate binding to and fusion with the host cell membrane. A central factor that allows the viral and host membrane to merge is the FP within these proteins that is exposed upon proteolytic cleavage. Recently, reports demonstrated a crucial role for Ca2+ in viral fusion, as it coordinates with negatively charged residues, such as D and E within the FP (24–26).
Here, we show that Ca2+ promotes viral entry of MERS-CoV into the host cell. Of the investigated residues, E891 seems to be the major residue for Ca2+ binding because the E891A substitution dramatically decreased viral infectivity (Fig. 5A).
Our attempts to identify the binding partner by generating double mutants of E891 with neighboring residues D896 and D902 were not successful, as we observed poor S protein incorporation into the PPs. It is intriguing that the Western blots and cell-cell fusion results show that the E891A/D902A S mutants are properly expressed and trafficked to the cell membrane but are not integrated into the virion surrogates. One possible explanation is that these mutations impact the integrity of the FP domain in a manner that hinders virion incorporation.
The role of Ca2+ for viral fusion has been explored for other enveloped viruses, such as RuV (25), SARS-CoV (24), EBOV (26, 35), human immunodeficiency virus type 1 (HIV-1) (36), and influenza A virus (24). There seem to be significant differences regarding the role of Ca2+ in mediating viral host cell membrane fusion. Studies showed that Ca2+ does not bind to the FPs of influenza A virus or HIV-1 during the fusion event. Our prior observations of influenza fusion protein, which does not show increased membrane interaction in the presence of Ca2+ (24), indicate that Ca2+ does not act as a blanket promoter of viral FP insertion into target membranes. (24). In the case of HIV, this leads to structural changes of the FP, which forms an α-helix in the absence of Ca2+, but changes conformation to an antiparallel β-sheet structure in the presence of Ca2+ (36). However, it is important to note that while Ca2+ enhances fusion for the abovementioned viruses, it is attenuated in its absence but is not abrogated. In contrast, Ca2+ directly interacts with the FPs of RuV, SARS-CoV, and EBOV, and depletion of Ca2+ results in the incapability of the virus to fuse with the host cell (24–26). For both RuV and EBOV, key residues within the FP were identified that coordinate with the Ca2+ and, in concert with low pH, resulted in conformational changes of the FP and subsequent fusion. Here, we demonstrate that MERS-CoV FP has a similar interaction with Ca2+ as SARS-CoV FP but with distinct differences.
Our data show that Ca2+ significantly promotes MERS-CoV fusion and that fusion in the absence of Ca2+ is attenuated but not completely abrogated. This could be due to the fact that the MERS-CoV FP binds only one Ca2+ ion (which is discussed in greater detail below). In contrast, host cell entry of SARS-CoV which binds two Ca2+ in its FP region is entirely abrogated when depleting intracellular Ca2+ levels (24). Given the high sequence similarity between the FP regions of the two viruses, this finding implicates that the stoichiometry between FP and Ca2+ has a significant impact on the fusion dynamics of the virus. However, because MERS-CoV shows a similar fusion behavior as influenza A virus or HIV-1 in the absence of Ca2+, it could be argued that fusion of MERS-CoV is regulated by Ca2+ binding to the membrane. We believe that we can rule out this possibility because our data obtained with the E891A mutant, the ESR, and ITC provide good evidence that Ca2+ interacts directly with the MERS-CoV FP.
By using ESR, we have demonstrated that the MERS-CoV FP also exerts a membrane-ordering effect similar to that observed for SARS-CoV FP as well as for other viral FPs, which further strengthens our hypothesis that the membrane-ordering effect is a required step for viral entry (24). The membrane-ordering effect, in turn, serves as a useful biophysical indicator for examining the conditions required for the FP activity. Using this method, we have previously demonstrated that SARS-CoV and EBOV FPs are Ca2+ dependent and now show the MERS-CoV FP is Ca2+ dependent too. However, although the S-shaped curve of the MERS-CoV FP is similar to the SARS-CoV FP, MERS-CoV FP has a smaller membrane-ordering effect than SARS-CoV FP, and at 1.0 mM Ca2+, the membrane-ordering effect is saturated by a higher P/L ratio of MERS-CoV FP than SARS-CoV FP (Fig. 7A and B). By keeping the P/L ratio constant at 1%, the MERS-CoV FP is also saturated at a higher concentration of Ca2+ (an increase to half of its maximum at around 1.5 mM, compared with around 0.5 mM for SARS-CoV FP) (Fig. 7C). Those results suggest that the MERS-CoV FP has a weaker interaction with Ca2+ and, thus, requires a higher Ca2+ concentration to reach its maximal activity. This hypothesis is further supported by the ITC experiment, which shows that the reaction energies between the Ca2+ and the MERS-CoV FP are weaker than those of the SARS-CoV FP. The ITC experiments also demonstrate that the stoichiometry of Ca2+ to the MERS-CoV FP is close to one (1.15). This result is different than that of the SARS-CoV case, in which SARS-CoV binds two Ca2+ in its FP region, one in each FP1 and FP2 (24). We interpret this to indicate that one Ca2+ interacts with the MERS-CoV FP.
Thus, although the sequences of SARS and MERS-CoV FP are similar, they may interact with Ca2+ differently. We believe that this interaction occurs in the MERS-CoV FP1 region, as our data identified a residue in the FP1 region that interacts with Ca2+, suggesting that this domain is important for Ca2+ binding. None of the tested mutants in the FP2 region provided clear evidence that Ca2+ binding occurs in this domain, which is different from SARS-CoV FP. It is striking that despite the high sequence similarity between SARS-CoV and MERS-CoV FP, there are significant differences in their FP Ca2+ interactions. The ESR results indicate that the ordering effect is sequence specific. It is possible that the small difference in the sequence of the FP2 region of MERS-CoV and SARS-CoV is responsible for this difference in the Ca2+ interaction.
We show here that while SARS-CoV and MERS-CoV both functionally interact with Ca2+, there appear to be significant positional and stoichiometric differences between the two spike proteins. Prior mutagenesis experiments were carried out on the conserved E and D residues in the SARS-CoV FP1 and FP2 regions before the recognition of the bipartite nature of the FP and prior to any consideration of their potential for Ca2+ interactions (37, 38). For SARS-CoV, the residue equivalent to MERS-CoV E891 (E801) could not be incorporated into PPs when mutated. Instead, the FP1 SARS-CoV residue D812 showed a major reduction in fusion activity when mutated. The equivalent MERS-CoV residue (D902) showed a much more modest effect when mutated. Notably, SARS-CoV D830 (in what is now defined as FP2) also showed a major reduction in fusion activity when mutated, in contrast to the equivalent residue in MERS-CoV (D922). D922A actually showed increased infectivity when mutated, and the reasons for this are currently unclear. Overall, these prior studies are in broad agreement with the interaction of Ca2+ with only FP1 for MERS-CoV, compared with both FP1 and FP2 for SARS-CoV.
In a biological scenario, a possible explanation for the differences might be that Ca2+ primes the FP for different steps in the host cell entry process. For SARS-CoV, extracellular Ca2+ binding through the FP2 domain may prepare the fusion loop for subsequent intracellular Ca2+ in the FP1. For MERS-CoV, the extracellular Ca2+ binding step may not be required, as none of the tested mutants in the FP2 region showed a striking reduction of infectivity, whereas intracellular Ca2+ depletion significantly reduced infectivity. Future studies will be designed to explore how swapping different residues or entire regions between MERS-CoV and SARS-CoV FP would impact membrane fusion, entry pathways, and calcium interactions. It would be interesting to see if MERS-CoV will adopt SARS-CoV fusion behavior and vice versa. Different cell types, however, could also affect the fusion behavior of both viruses. Previous studies explored the fusion dynamics of SARS-CoV and MERS-CoV PP using Vero E6 cells (15, 24), while we and others used Huh-7 cells to study MERS-CoV PP infections which exhibit a higher expression of furin and DPP4 and seem to promote entry mainly via the endocytic route (15, 16). Addressing the impact of cell type on the fusion dynamics of MERS-CoV and SARS-CoV will complement our studies on the FP domains of the two viruses.
MATERIALS AND METHODS
Cells, plasmids, and reagents.
HEK293T and Vero E6 cells were obtained from the American Type Culture Collection. Huh-7 cells were obtained from the Japan Health Science Research Resources Bank. All cells were maintained in Dulbecco’s modified Eagle medium (DMEM) (Cellgro) supplemented with 25 mM HEPES (Cellgro) and 10% HyClone FetalClone II (GE) (cDMEM) at 37°C and 5% CO2. The MERS-CoV S EMC/2012 gene was codon optimized for mammalian expression and cloned into pDNA3.1+ (Invitrogen) as described previously (15). The pCMV-MLV gag-pol murine leukemia virus (MLV) packaging construct, the pTG-Luc transfer vector-encoding luciferase reporter, and pCAGGS/VSV-G plasmid were described before (15, 39).
Recombinant l-1-tosylamide-2-phenylethyl chloromethyl ketone (TPCK)-treated trypsin was purchased from Sigma. The protease inhibitor dec-RVKR-CMK was obtained from Tocris. Calcium chelators BAPTA-AM and EGTA were purchased from Tocris and VWR, respectively.
Site-directed mutagenesis.
All mutated MERS-CoV S protein constructs were generated by site-directed mutagenesis using the QuikChange Lightning site-directed mutagenesis kit (Agilent). pDNA3.1+/MERS-CoV S EMC/2012 served as the template for all the mutagenesis performed. PCRs and Escherichia coli XL 10 Gold transformations were carried out according to the manufacturer’s recommendations. Primers (IDT) were designed using the primer design tool from Agilent. The following primers were used: E891A (ATCGAACAACAGATCCGCGATCGCTGATCGTGC/GCACGATCAGCGATCGCGGATCTGTTGTTCGAT), D892A (CCTTATCGAACAACAGAGCCTCGATCGCTGATCGT/ACGATCAGCGATCGAGGCTCTGTTGTTCGATAAGG), E891A/D892A (CCTTATCGAACAACAGAGCCGCGATCGCTGATCGTGCG/CGCACGATCAGCGATCGCGGCTCTGTTGTTCGATAAGG), D896A (CGGCAATGGTCACCTTAGCGAACAACAGATCCTCG/CGAGGATCTGTTGTTCGCTAAGGTGACCATTGCCG), D902A (GCATATAGCCCGGAGCGGCAATGGTCACC/GGTGACCATTGCCGCTCCGGGCTATATGC), D910A (CCTGCTGCATGCAGTCAGCGTAACCTTGCATATAG/CTATATGCAAGGTTACGCTGACTGCATGCAGCAGG), D911A (CCCTGCTGCATGCAGGCATCGTAACCTTGCA/TGCAAGGTTACGATGCCTGCATGCAGCAGGG), D910A/D911A (GCCCCTGCTGCATGCAGGCAGCGTAACCTTGCATATAG/CTATATGCAAGGTTACGCTGCCTGCATGCAGCAGGGGC), D922A (GGCGCAGATAAGGGCTCTGGCGCTGGC/GCCAGCGCCAGAGCCCTTATCTGCGCC), E891A/D896A (GGCAATGGTCACCTTAGCGAACAACAGATCCGC/GCGGATCTGTTGTTCGCTAAGGTGACCATTGCC), E891A/D902A (GCATATAGCCCGGAGCGGCAATGGTCACC/GGTGACCATTGCCGCTCCGGGCTATATGC), and E891A/D896A/D902A (GCATATAGCCCGGAGCGGCAATGGTCACC/GCGGATCTGTTGTTCGCTAAGGTGACCATTGCC/GGCAATGGTCACCTTAGCGAACAACAGATCCGC/GGTGACCATTGCCGCTCCGGGCTATATGC). All constructs were verified by Sanger sequencing.
Western blot analysis of MERS-CoV S protein WT and mutants.
HEK293T cells were grown in 6-well plates and transfected with pCDNA3.1+ encoding the respective S protein variant using polyethylenimine (PEI) (Fisher). For each well, 2 μg DNA and 6 μl PEI were incubated with 50 μl Opti-MEM (Gibco) separately for 5 minutes before being combined and incubated for 20 minutes. After 20 minutes, 2 ml cDMEM was added to the transfection mixture. Cells were washed with phosphate-buffered saline (PBS) (Cellgro) once, and the DNA/PEI/DMEM mix was added. For cells treated with protease inhibitor, 75 μM dec-RVKR-CMK was added to cells at the time of transfection. Cells were then incubated for 18 h. For cells treated with TPCK-trypsin, they were washed with PBS once, and 1 ml PBS supplemented with 0.8 nM TPCK-trypsin was added to the cells. Cells were then incubated for 10 min at 37°C and 5% CO2. Cell-surface biotinylation, Western blotting, and antibody analysis were performed as previously described (40). S protein was detected using the MERS-CoV S rabbit polyclonal antibody (catalog no. 40069-RP01; Sino Biological) as the primary antibody and Alexa Fluor 488-labeled anti-rabbit secondary antibody (Invitrogen).
Immunofluorescence assay of MERS-CoV S protein WT and mutants.
Vero cells were grown in microscopy chamber slides (Millipore) and transfected with the respective pCDNA3.1+/MERS-CoV S construct using Lipofectamine 2000 (Invitrogen). Transfections were performed according to the manufacturer’s protocol. At 18 h posttransfection, the immunofluorescence assay was carried out as previously described, with the exception that membranes were not permeabilized with Triton X-100 (15). The MERS-CoV S protein was detected using the same antibodies as described for the Western blot analysis. For quantification, images of at least five randomly selected fields were acquired. The nuclei of nine syncytia for each condition were counted manually to calculate the average nuclei/syncytia. The average and standard deviation were calculated using Microsoft Excel and GraphPad Prism 7. Data were visualized using GraphPad Prism 7.
Pseudoparticle assays.
The production of and infection with pseudoparticles (PPs) were performed as previously described, with minor modifications (28). In brief, PPs of each individual construct were produced by transfecting HEK293T cells with 600 μg of the respective plasmid DNA, 600 μg pTG-Luc, and 800 μg pCMV-MLVgag-pol using PEI as described above. At 48 h posttransfection, the supernatants were harvested, centrifuged at 290 × g for 7 minutes to remove cell debris, filtered through a 0.45-μm syringe filter, and stored at –80°C in small aliquots. For infection assays, Huh-7 cells were seeded in 24-well plates. Cells were washed with 500 μl PBS, and 200 μl PPs were added and incubated for 2 h at 37°C and 5% CO2 with rocking. After 2 h, the cells were supplemented with 300 μl cDMEM and incubated for 72 h at 37°C and 5% CO2. Cells were then lysed, and luciferase activity was measured using a luciferase assay kit (Promega). Readings were performed with a Glomax 20/20 system (Promega). For each experiment, three technical replicates were prepared. Each experiment was repeated at least three times. Data were analyzed using GraphPad Prism 7.
To chelate intracellular calcium, cells were washed with 500 μl PBS and, subsequently, pretreated with 200 μl of 50 μM BAPTA-AM (dissolved in DMSO) in DMEM+ (2% HyClone, 10 mM HEPES, and 1.8 mM Ca2+) for 1 h at 37°C and 5% CO2. After 1 h, DMEM+ was removed and cells were infected with 200 μl PP containing 50 μM BAPTA-AM and incubated for 2 h at 37°C and 5% CO2 with rocking. After 2 h, cells were supplemented with 300 μl cDMEM and incubated for 72 h. The analyses of infected cells are as described above. Negative controls were conducted by pretreating cells in DMEM+ and infecting them with PP with an equivalent volume of DMSO.
To chelate extracellular calcium, cells were preincubated with 200 μl of DMEM- (2% HyClone and 10 mM HEPES) for 1 h at 37°C and 5% CO2. After 1 h, DMEM− was aspirated and cells were infected with 200 μl PP containing 50 μM EGTA for 2 h at 37°C and 5% CO2 with rocking. The analysis of infected cells is as described above. Negative controls were controls conducted by preincubating the cells in DMEM+ and infecting them with PP without treatment. For Western blot analysis of the incorporated S protein, 1 ml of harvested PP was spun down at 43,000 × g for 2 h at 4°C. The supernatant was aspirated, and the protein pellet was resuspended in 2× Laemmli buffer. All protein was loaded on an SDS-PAGE gel, and Western blot analysis was carried out as described above.
Modeling of MERS-CoV S protein monomer.
The structural model of the MERS-CoV S protein was based on the MERS-CoV prefusion structure (PDB 6Q05). Protein alignment of the MERS-CoV S protein (GenBank accession no. AFS88936.1) and MERS-CoV structure was performed with Geneious (v.2020.1.1). The structural model of the MERS-CoV S protein was generated using the Modeller comparative modeling tool (v.9.23) in Chimera software (v. 1.14; University of California) (41–43). Images were created using Adobe Illustrator CC v.24.03.
Lipids and peptides.
The lipids POPC and POPS; the chain spin labels 5PC, 10PC, and 14PC; and a head group spin label, dipalmitoylphospatidyl-tempo-choline (DPPTC), were purchased from Avanti Polar Lipids (Alabaster, AL) or synthesized by our laboratory according to previous protocols. Cholesterol was purchased from Sigma (St. Louis, MO). All peptides were synthesized by SynBioSci Co. (Livermore, CA). The sequence of SARS FP and its corresponding shuffle peptide are the same as those in reference 24.
Vesicle preparation.
The composition of membranes used in this study is consistent with our previous study (24). The desired amount of POPC, POPS, cholesterol, and 0.5% (mol:mol) spin-labeled lipids in chloroform were mixed well and dried by N2 flow. The mixture was evacuated in a vacuum drier overnight to remove any trace of chloroform. To prepare MLVs, the lipids were resuspended and fully hydrated using 1 ml of pH 7 or pH 5 buffer (5 mM HEPES, 10 mM 4-morpholineethanesulfonic acid [MES], 150 mM NaCl, and 0.1 mM EDTA [pH 7 or pH 5]) at room temperature (RT) for 2 h. To prepare small, unilamellar vesicles (SUVs), the lipids were resuspended in pH 7 or pH 5 buffer and sonicated in an ice bath for 20 minutes or when the suspension became clear. The SUV solution was then further clarified by centrifugation at 13,000 rpm for 10 min.
Isothermal titration calorimetry.
ITC experiments were performed in an N-ITC III calorimeter (TA Instrument, New Castle, DE). A total 97 μl of 1 mM CaCl2 in pH 7 fusion buffer was injected into 1 ml of 0.05 mM FPs in pH 7 fusion buffer at 37°C in a stepwise manner, consisting of 5 μl per injection except that the first injection was 2 μl. The injection time was 15 sec for each injection, and the interval time was 10 min. The data were analyzed with Origin (OriginLab Corp., Northampton, MA). The one-site model was used in the fitting to calculate the thermodynamic parameters. The protein concentration was determined by dry weight and UV spectroscopy.
ESR spectroscopy and nonlinear least-squares fit of ESR spectra.
To prepare the samples for lipid ESR study, the desired amounts of FPs (1 mg/ml) were added into the lipid MLV dispersion. After 20 min of incubation, the dispersion was spun at 13,000 rpm for 10 min. The concentrations of peptides were measured using UV to ensure complete binding of the peptides. The pellet was transferred to a quartz capillary tube for ESR measurement. ESR spectra were collected on an Elexsys ESR spectrometer (Bruker Instruments, Billerica, MA) at X-band (9.5 GHz) at 25°C using an N2 temperature controller (Bruker Instruments).
The ESR spectra from the labeled lipids were analyzed using the Non-Linear Least Squares (NLLS)-fitting program based on the stochastic Liouville equation using the microscopic order macroscopic disorder (MOMD) model as described in previous studies (31). The fitting strategy is described below. We employed the Budil et al. NLLS fitting program (44) to obtain convergence to optimum parameters. The g-tensor and A-tensor parameters used in the simulations were determined from rigid limit spectra (24). The ordering tensor parameters S0 and S2 were extracted from the spectra, which are defined as follows: S0 = (D2,00) = (1/2[3cos2θ − 1]) and S2 = (D2,02 + D2,0-2) = (√[3/2]sin2θcos2φ), where D2,00, D2,02, and D2,0-2 are the Wigner rotation matrix elements and θ and φ are the polar and azimuthal angles for the orientation of the rotating axes of the nitroxide bonded to the lipid relative to the director of the bilayer, i.e., the preferential orientation of lipid molecules; the angular brackets imply ensemble averaging. S0 and its uncertainty were then calculated in a well-known fashion from its definition and the dimensionless ordering potentials C20 and C22, and its uncertainty was then found in the fitting. The typical uncertainties we find for S0 range from 1× 10−3 to 5 × 10−3, while the uncertainties from repeated experiments are 5× 10−3 to 8 × 10−3 or less than ±0.01. S0 indicates how strongly the chain segment to which the nitroxide is attached is aligned along the normal to the lipid bilayer, which is strongly correlated with hydration/dehydration of the lipid bilayers (31, 45).
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health research grants R01AI35270, R01GM123779, and P41GM103521. T.T. acknowledges support from the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1650441.
We thank David Eliezer (Weill Cornell Medicine) and Javier A. Jaimes (College for Veterinary Medicine, Cornell University) for their critical input. We thank Brian Crane (Cornell University, Department of Chemistry and Chemical Biology) for sharing equipment.
REFERENCES
- 1.Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus A, Fouchier R. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 2.WHO. 2019. Middle East respiratory syndrome coronavirus (MERS-CoV). WHO, Geneva, Switzerland. [Google Scholar]
- 3.Wang Q, Qi J, Yuan Y, Xuan Y, Han P, Wan Y, Ji W, Li Y, Wu Y, Wang J, Iwamoto A, Woo PCY, Yuen KY, Yan J, Lu G, Gao GF. 2014. Bat origins of MERS-CoV supported by bat Coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe 16:328–337. doi: 10.1016/j.chom.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azhar EI, El-Kafrawy SA, Farraj SA, Hassan AM, Al-Saeed MS, Hashem AM, Madani TA. 2014. Evidence for camel-to-human transmission of MERS coronavirus. N Engl J Med 370:2499–2505. doi: 10.1056/NEJMoa1401505. [DOI] [PubMed] [Google Scholar]
- 5.Mohd HA, Al-Tawfiq JA, Memish ZA. 2016. Middle East respiratory syndrome coronavirus (MERS-CoV) origin and animal reservoir. Virol J 13:87. doi: 10.1186/s12985-016-0544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh MD, Park WB, Park SW, Choe PG, Bang JH, Song KH, Kim ES, Kim HB, Kim NJ. 2018. Middle east respiratory syndrome: what we learned from the 2015 outbreak in the republic of Korea. Korean J Intern Med 33:233–246. doi: 10.3904/kjim.2018.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menachery VD, Dinnon KH, Yount BL, McAnarney ET, Gralinski LE, Hale A, Graham RL, Scobey T, Anthony SJ, Wang L, Graham B, Randell SH, Lipkin WI, Baric RS. 2019. Trypsin treatment unlocks barrier for zoonotic bat coronaviruses infection. J Virol 94:e01774-19. doi: 10.1128/JVI.01774-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Si Mehand M, Millett P, Al-Shorbaji F, Roth C, Kieny MP, Murgue B. 2018. World Health Organization methodology to prioritize emerging infectious diseases in need of research and development. Emerg Infect Dis 24:e1–e9. doi: 10.3201/eid2409.171427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosch BJ, van der Zee R, de Haan CAM, Rottier P. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77:8801–8811. doi: 10.1128/jvi.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raj VS, Mou H, Smits SL, Dekkers DHW, Müller MA, Dijkman R, Muth D, Demmers JAA, Zaki A, Fouchier RAM, Thiel V, Drosten C, Rottier PJM, Osterhaus A, Bosch BJ, Haagmans BL. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barlan A, Zhao J, Sarkar MK, Li K, McCray PB, Perlman S, Gallagher T. 2014. Receptor variation and susceptibility to Middle East respiratory syndrome coronavirus infection. J Virol 88:4953–4961. doi: 10.1128/JVI.00161-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shirato K, Kawase M, Matsuyama S. 2013. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol 87:12552–12561. doi: 10.1128/JVI.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Krämer-Kühl A, Welsch K, Winkler M, Meyer B, Drosten C, Dittmer U, von Hahn T, Simmons G, Hofmann H, Pöhlmann S. 2013. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol 87:5502–5511. doi: 10.1128/JVI.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian Z, Dominguez SR, Holmes KV. 2013. Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS-CoV) in virus entry and syncytia formation. PLoS One 8:e76469. doi: 10.1371/journal.pone.0076469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millet JK, Whittaker GR. 2014. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci U S A 111:15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park JE, Li K, Barlan A, Fehr AR, Perlman S, McCray PB, Gallagher T. 2016. Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc Natl Acad Sci U S A 113:12262–12267. doi: 10.1073/pnas.1608147113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belouzard S, Chu VC, Whittaker GR. 2009. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A 106:5871–5876. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heald-Sargent T, Gallagher T. 2012. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 4:557–580. doi: 10.3390/v4040557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belouzard S, Millet JK, Licitra BN, Whittaker GR. 2012. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4:1011–1033. doi: 10.3390/v4061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Millet JK, Whittaker GR. 2014. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res 16:120–134. doi: 10.1016/j.virusres.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White JM, Whittaker GR. 2016. Fusion of enveloped viruses in endosomes. Traffic 17:593–614. doi: 10.1111/tra.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kielian M. 2014. Mechanisms of virus membrane fusion proteins. Annu Rev Virol 1:171–189. doi: 10.1146/annurev-virology-031413-085521. [DOI] [PubMed] [Google Scholar]
- 23.Harrison SC. 2015. Viral membrane fusion. Virology 479–480:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai AL, Millet JK, Daniel S, Freed JH, Whittaker GR. 2017. The SARS-CoV fusion peptide forms an extended bipartite fusion platform that perturbs membrane order in a calcium-dependent manner. J Mol Biol 429:3875–3892. doi: 10.1016/j.jmb.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubé M, Rey FA, Kielian M. 2014. Rubella virus: first calcium-requiring viral fusion protein. PLoS Pathog 10:e1004530. doi: 10.1371/journal.ppat.1004530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nathan L, Lai A, Millet J, Straus M, Whittaker GR, Freed JH, Daniel S. 2019. Calcium ions directly interact with the Ebola virus fusion peptide to promote structure-function changes that enhance infection. ACS Infect Dis 6:250–260. doi: 10.1021/acsinfecdis.9b00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuyama S, Shirato K, Kawase M, Terada Y, Kawachi K, Fukushi S, Kamitani W. 2018. Middle East respiratory syndrome coronavirus spike protein is not activated directly by cellular furin during viral entry into target cells. J Virol 92:e00683-18. doi: 10.1128/JVI.00683-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Millet JK, Tang T, Nathan L, Jaimes JA, Hsu HL, Daniel S, Whittaker GR. 2019. Production of pseudotyped particles to study highly pathogenic coronaviruses in a biosafety level 2 setting. J Vis Exp doi: 10.3791/59010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tymianski M, Spigelman I, Zhang L, Carlen PL, Tator CH, Charlton MP, Wallace MC. 1994. Mechanism of action and persistence of neuroprotection by cell-permeant Ca2+ chelators. J Cereb Blood Flow Metab 14:911–923. doi: 10.1038/jcbfm.1994.122. [DOI] [PubMed] [Google Scholar]
- 30.Ge M, Freed JH. 2009. Fusion peptide from influenza hemagglutinin increases membrane surface order: an electron-spin resonance study. Biophys J 96:4925–4934. doi: 10.1016/j.bpj.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai AL, Freed JH. 2015. Interaction between the influenza HA fusion peptide and transmembrane domain affects membrane structure. Biophys J 109:2523–2536. doi: 10.1016/j.bpj.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai AL, Freed JH. 2014. HIV gp41 fusion peptide increases membrane ordering in a cholesterol-dependent fashion. Biophys J 106:172–181. doi: 10.1016/j.bpj.2013.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ge M, Freed JH. 2011. Two conserved residues are important for inducing highly ordered membrane domains by the transmembrane domain of influenza hemagglutinin. Biophys J 100:90–97. doi: 10.1016/j.bpj.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinello JF, Lai AL, Millet JK, Cassidy-Hanley D, Freed JH, Clark TG. 2017. Structure-function studies link class II viral fusogens with the ancestral gamete fusion protein HAP2. Curr Biol 27:651–660. doi: 10.1016/j.cub.2017.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das DK, Bulow U, Diehl WE, Durham ND, Senjobe F, Chandran K, Luban J, Munro JB. 2020. Conformational changes in the Ebola virus membrane fusion machine induced by pH, Ca2+, and receptor binding. PLoS Biol 18:e3000626. doi: 10.1371/journal.pbio.3000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nieva JL, Nir S, Muga A, Félix I, Goñiji M, Wilschut J. 1994. Interaction of the HIV-1 fusion peptide with phospholipid vesicles: different structural requirements for fusion and leakage. Biochemistry 33:3201–3209. doi: 10.1021/bi00177a009. [DOI] [PubMed] [Google Scholar]
- 37.Madu IG, Belouzard S, Whittaker GR. 2009. SARS-coronavirus spike S2 domain flanked by cysteine residues C822 and C833 is important for activation of membrane fusion. Virology 393:265–271. doi: 10.1016/j.virol.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madu IG, Roth SL, Belouzard S, Whittaker GR. 2009. Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein S2 domain with characteristics of a viral fusion peptide. J Virol 83:7411–7421. doi: 10.1128/JVI.00079-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartosch B, Dubuisson J, Cosset F-L. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1–E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Straus MR, Kinder JT, Segall M, Dutch RE, Whittaker GR. 2020. SPINT2 inhibits proteases involved in activation of both influenza viruses and metapneumoviruses. Virology 543:43–53. doi: 10.1016/j.virol.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 42.Jaimes JA, Whittaker GR. 2018. Feline coronavirus: insights into viral pathogenesis based on the spike protein structure and function. Virology 517:108–121. doi: 10.1016/j.virol.2017.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Šali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 44.Budil DE, Sanghyuk L, Saxena S, Freed JH. 1996. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J Magn Reson 120:155–189. doi: 10.1006/jmra.1996.0113. [DOI] [Google Scholar]
- 45.Ge M, Freed JH. 2003. Hydration, structure, and molecular interactions in the headgroup region of dioleoylphosphatidylcholine bilayers: an electron spin resonance study. Biophys J 85:4023–4040. doi: 10.1016/S0006-3495(03)74816-4. [DOI] [PMC free article] [PubMed] [Google Scholar]