

Gammaherpesviruses establish lifelong infection in over 95% of all adults and are associated with B cell lymphomas. The virus’s manipulation of the germinal center response and B cell differentiation to establish lifelong infection is thought to also precipitate malignant transformation, through a mechanism that remains poorly understood. The host transcription factor IRF-1, a well-established tumor suppressor, selectively attenuates MHV68-driven germinal center response, a phenotype that we originally hypothesized to occur in a B cell-intrinsic manner. In contrast, in testing, B cell-intrinsic IRF-1 expression promoted the MHV68-driven germinal center response and the establishment of chronic infection. Our report highlights the underappreciated multifaceted role of IRF-1 in MHV68 infection and pathogenesis.
KEYWORDS: IRF-1, chronic infection, gammaherpesvirus, germinal center B cell
ABSTRACT
Gammaherpesviruses are ubiquitous pathogens that are associated with cancers, including B cell lymphomas. These viruses are unique in that they infect naive B cells and subsequently drive a robust polyclonal germinal center response in order to amplify the latent reservoir and to establish lifelong infection in memory B cells. The gammaherpesvirus-driven germinal center response in combination with robust infection of germinal center B cells is thought to precipitate lymphomagenesis. Importantly, host and viral factors that selectively affect the gammaherpesvirus-driven germinal center response remain poorly understood. Global deficiency of antiviral tumor-suppressive interferon regulatory factor 1 (IRF-1) selectively promotes the murine gammaherpesvirus 68 (MHV68)-driven germinal center response and expansion of the viral latent reservoir. To determine the extent to which antiviral effects of IRF-1 are B cell intrinsic, we generated mice with conditional IRF-1 deficiency. Surprisingly, B cell-specific IRF-1 deficiency attenuated the establishment of chronic infection and the germinal center response, indicating that MHV68 may, in a B cell-intrinsic manner, usurp IRF-1 to promote the germinal center response and expansion of the latent reservoir. Further, we found that B cell-specific IRF-1 deficiency led to reduced levels of active tyrosine phosphatase SHP1, which plays a B cell-intrinsic proviral function during MHV68 infection. Finally, results of this study indicate that the antiviral functions of IRF-1 unveiled in MHV68-infected mice with global IRF-1 deficiency are mediated via IRF-1 expression by non-B cell populations.
IMPORTANCE Gammaherpesviruses establish lifelong infection in over 95% of all adults and are associated with B cell lymphomas. The virus’s manipulation of the germinal center response and B cell differentiation to establish lifelong infection is thought to also precipitate malignant transformation, through a mechanism that remains poorly understood. The host transcription factor IRF-1, a well-established tumor suppressor, selectively attenuates MHV68-driven germinal center response, a phenotype that we originally hypothesized to occur in a B cell-intrinsic manner. In contrast, in testing, B cell-intrinsic IRF-1 expression promoted the MHV68-driven germinal center response and the establishment of chronic infection. Our report highlights the underappreciated multifaceted role of IRF-1 in MHV68 infection and pathogenesis.
INTRODUCTION
Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) are two highly prevalent human gammaherpesviruses that are associated with several types of cancer, including B cell lymphomas (1). In vivo studies of EBV and KSHV are challenging given the species specificity of these viruses that has developed during coevolution with their host. To overcome this obstacle, the current study utilized murine gammaherpesvirus 68 (MHV68), a natural rodent pathogen that is genetically and biologically similar to EBV and KSHV (2–4) and offers a highly tractable experimental model to define virus-host interactions in vivo during chronic gammaherpesvirus infection.
Gammaherpesviruses commandeer B cell differentiation to establish a lifelong latent viral reservoir in memory B cells. To achieve this, gammaherpesviruses infect naive B cells and induce a robust germinal center response to expand the latent viral reservoir in germinal center B cells during early stages of chronic infection (5–7). In a T follicular helper cell-dependent manner, infected germinal center B cells differentiate into either memory B cells or plasma cells (8, 9). The long-term latent reservoir is maintained in the memory B cells, while differentiation of infected B cells into plasma cells triggers reactivation, the switch from latency to lytic replication (5, 10–12).
Intriguingly, the germinal center response induced by gammaherpesvirus infection is distinct from physiological B cell differentiation as it results in a robust, albeit transient increase in levels of class-switched polyclonal antibodies with reactivities against irrelevant nonvirus antigens (13, 14). These non-virus-specific self-reactive antibodies rapidly peak within the first 2 weeks of infection, whereas it takes at least a month for MHV68-specific class-switched antibodies to plateau (13, 14). Similarly, EBV acquisition generates a spike in non-virus-specific antibody responses, with high titers of antibodies against horse red blood cells used as a diagnostic test for recent EBV infection (15). Importantly, gammaherpesvirus-driven germinal center responses may lead to cellular transformation as germinal center B cells rapidly proliferate, with concomitant downregulation of tumor suppressors (16) and increased expression of mutagenic enzymes (17, 18). Not surprisingly, many gammaherpesvirus-driven B cell lymphomas are of germinal center or post-germinal center origin (19, 20). Despite the unique and likely pathogenic nature of the gammaherpesvirus-driven germinal center response, the host and viral mechanisms responsible for the increase in the levels of germinal center B cells and irrelevant B cell differentiation are poorly understood.
We showed that global expression of the interferon regulatory factor 1 (IRF-1) transcription factor selectively suppresses the MHV68-driven germinal center response and attenuates chronic infection (21). The mechanism by which IRF-1 selectively restricts the MHV68-driven germinal center response and establishment of chronic infection remains unclear. To determine the extent to which B cell-intrinsic expression of IRF-1 attenuates the MHV68-driven germinal center response, we generated mice with B cell-specific IRF-1 deficiency. IRF-1 expression by B cells was not required for B cell development and baseline immunoglobulin levels. However, in direct contrast to what was observed during global IRF-1 deficiency (21), B cell-specific IRF-1 deficiency resulted in reduced MHV68 latency and reactivation along with a significant reduction in the germinal center response; such a reduction was not observed following infection with an unrelated RNA virus. Thus, in an intriguing turn of events, B cell-intrinsic deficiency of IRF-1 produced effects opposite those seen with global IRF-1 deficiency, and those effects continued to be selective for MHV68 infection. Our findings suggest that while IRF-1 may be usurped by MHV68 in B cells for viral benefit, IRF-1 expression by other cellular populations, in a tug-of-war manner, attenuates MHV68-driven germinal center responses and chronic infection.
RESULTS
Generation of the mouse model of B cell-specific IRF-1 deficiency.
IRF-1 conditional ready mice [IRF1tm1a(ECOMM)Wtsi] were acquired from the Wellcome Trust Sanger Institute (Fig. 1A). The original targeted allele contained an FLP recombination target (FRT)-flanked LacZ/neomycin sequence (annotated as “lac+”; Fig. 1A) inserted in the intron between the first and second exons of IRF-1, followed by the loxP site with an additional loxP site downstream of exon 2. LacZ/neomycin sequence was first removed by crosses to a transgenic mouse strain with ubiquitous expression of Flp recombinase (22); germ line transmission was ensured by backcrossing to the C57BL/6J background (annotated as “BL6” in Fig. 1A). Finally, to generate the mouse model of B cell-specific IRF-1 deficiency, mice lacking the Flp recombinase transgene and positive for the germ line excision of the LacZ/neomycin cassette in the mutant IRF-1 allele were crossed to the mouse strain with Cre recombinase expression driven by the B cell-selective CD19 promoter (23). The presence of LoxP sites and excision of the LacZ/neomycin cassette were verified by PCR (Fig. 1B).
FIG 1.
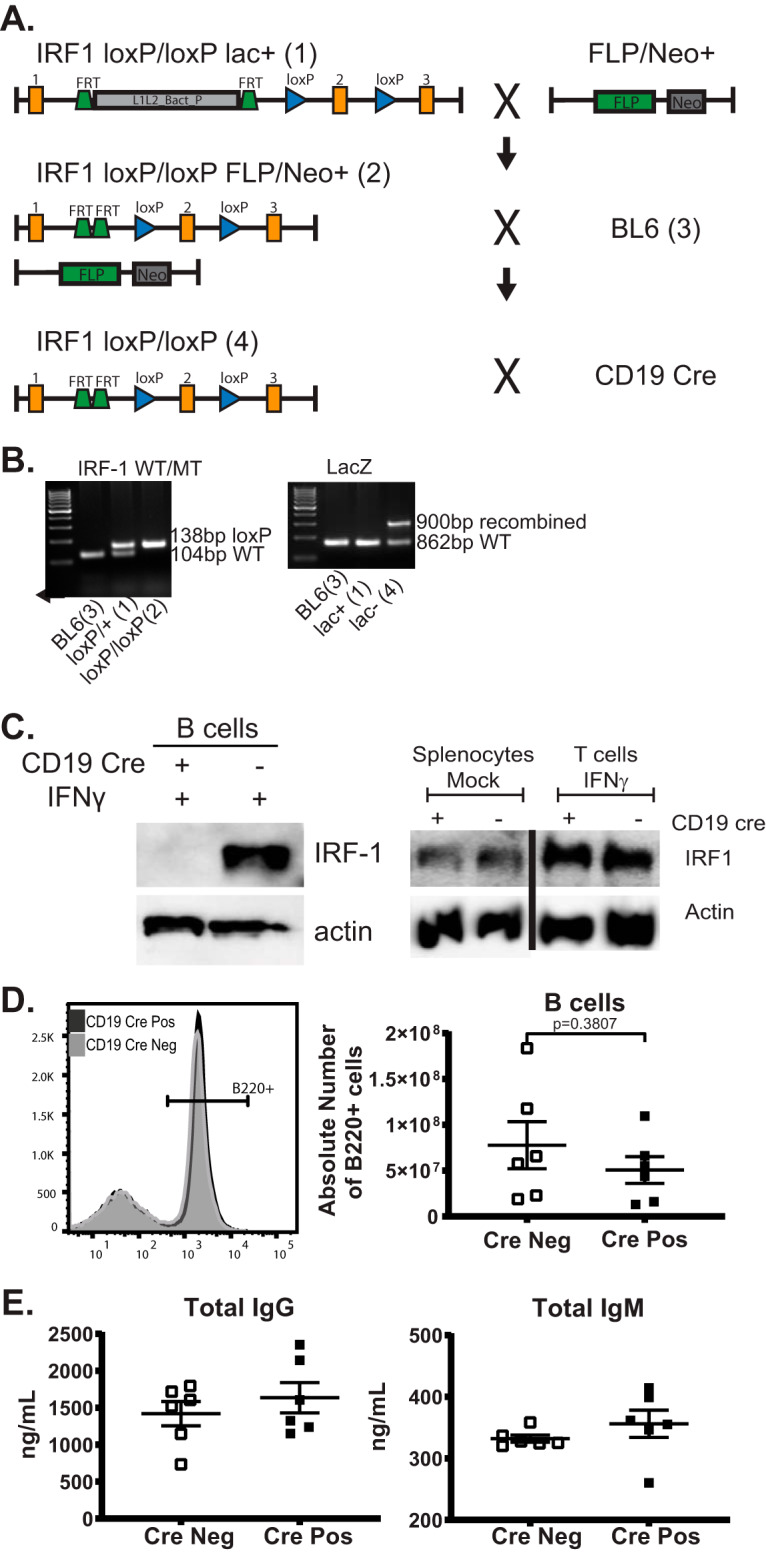
Generation of the mouse model of B cell-specific IRF-1 deficiency. (A) Graphical representation of genetic constructs (not to scale) and animal crosses used to generate IRF-1flox/flox mice. Numbers in parentheses correspond to the numbers identifying individual PCRs in panel B. (B) PCR products of genotyping reactions designed to detect the presence of the LoxP sites and the juxtaposed FRT sites within the conditional IRF-1 allele. (C) B cells, T cells, or splenocytes isolated from naive CD19 Cre-positive and Cre-negative littermates homozygous for the conditional IRF-1 allele were mock treated or stimulated with 10 U/ml IFN-γ for 5 h as indicated and subjected to Western analyses using the indicated antibodies. (D) Representative histogram of the B220-positive splenocytes isolated from CD19 Cre-positive (Pos) and Cre-negative (Neg) littermates, with data from multiple animals summarized in the adjacent graph. (E) Levels of total IgM (right panel) and IgG (left panel) in the sera of naive CD19 Cre-positive and Cre-negative littermates. Each symbol in panels D and E represents an individual animal. Error bars indicate means and standard errors of measurements. Data in panel E are also included in Fig. 4A and B.
Expression of IRF-1 was evaluated by measuring IRF-1 protein levels in sorted splenic B cells. To increase the sensitivity of residual IRF-1 detection, B cells were treated with interferon gamma (IFN-γ) to transcriptionally boost IRF-1 expression. IRF-1 protein levels were below the level of detection in IFN-γ-treated B cells isolated from CD19 Cre-positive IRF-1loxP/loxP mice compared to IRF-1loxP/loxP controls (Fig. 1C; referred to as CD19 Cre positive and Cre negative throughout this article). To determine if loss of IRF-1 in B cells impacted IRF-1 expression in other cell types, particularly T cells, IRF-1 protein levels were measured at baseline in total splenocytes and, following IFN-γ treatment, in sorted splenic T cells. As expected, there was no difference in the IRF-1 protein levels in total splenocytes and sorted T cells isolated from CD19 Cre-positive and Cre-negative mice (Fig. 1C). B cell-intrinsic IRF-1 deficiency had no effect on the frequency and total number of splenic B cells in naive CD19 Cre-positive and Cre-negative littermate controls (Fig. 1D), indicating that B cell-specific IRF-1 expression was not required for B cell development. Likewise, baseline serum levels of IgG and IgM were similar in CD19 Cre-positive and Cre-negative naive mice (Fig. 1E), indicating that B cell-intrinsic IRF-1 expression was not required for baseline immunoglobulin production.
B cell-intrinsic IRF-1 expression supports optimal establishment of MHV68 chronic infection.
Following inoculation of a naive host, MHV68 replication occurs systemically for the first 10 to 12 days before lytic virus is cleared and the latent viral reservoir is expanded (24, 25). Peak viral latency in the spleen is observed around 16 days postinfection, with germinal center B cells hosting the majority of the latent viral reservoir at that time (5, 26). Global IRF-1 deficiency leads to a significant increase in the level of the peak latent MHV68 reservoir and, eventually, to increased viral reactivation in the spleens of long-term-infected mice (21).
To define the B cell-intrinsic role of IRF-1 during the establishment of chronic MHV68 infection, CD19 Cre-positive and Cre-negative mice were infected and the frequencies of MHV68 DNA-positive cells and ex vivo reactivation compared in the spleen at 16 days postinfection. In contrast to the results observed under conditions of global IRF-1 deficiency, there was a (∼3-fold) decrease in the frequency of MHV68 DNA-positive splenocytes (Fig. 2A), along with a (2.6-fold) decrease in the absolute number of MHV68 DNA-positive splenocytes (Fig. 2B) in the CD19 Cre-positive mice compared to the Cre-negative mice. Further, there was a significant reduction in the frequency of MHV68 ex vivo reactivation in the spleens of CD19 Cre-positive mice (Fig. 2C). Thus, unexpectedly, B cell-specific loss of IRF-1 resulted in attenuation of MHV68 latency and reactivation.
FIG 2.

B cell-intrinsic IRF-1 expression supports optimal establishment of MHV68 chronic infection. CD19 Cre-positive and Cre-negative littermates were infected with 1,000 PFU of MHV68. Spleens were collected at 16 days postinfection, processed into a single-cell suspension, and subjected to limiting dilution assays to determine the frequency (A) or absolute number (B) of MHV68 DNA-positive cells and the frequency of ex vivo viral reactivation (C). Each experimental group consisted of 3 to 4 animals; data were pooled from 2 to 3 independent experiments; standard errors of the means are displayed. In the data from the limiting dilution assays presented here and in Fig. 6, the dotted line is drawn at 62.5% and the x coordinate of the intersection of this line with the sigmoid graph represents the inverse of the frequency of positive events.
B cell-intrinsic IRF-1 expression supports MHV68-driven germinal center responses.
Gammaherpesviruses infect naive B cells, with subsequent entry of both infected and bystander B cells into the germinal center, where the viral latent reservoir is exponentially amplified via cellular proliferation. (5–7). Consistent with the increased MHV68 latent reservoir levels observed in mice with global IRF-1 deficiency, these mice also display a dramatic increase in levels of germinal center B cells (21). In contrast, there was a significant decrease in the frequency and number of germinal center B cells of CD19 Cre-positive mice following MHV68 infection (Fig. 3A to C). Consistent with the critical role of CD4 T follicular helper cells in MHV68-driven germinal center responses (9), the frequency and number of CD4 T follicular helper cells were also significantly decreased in the MHV68-infected CD19 Cre-positive mice (Fig. 3D to F). Differentiation of latently infected B cells into plasma cells triggers viral reactivation (10). Consistent with the decreased frequency of ex vivo MHV68 reactivation observed in CD19 Cre-positive mice (Fig. 2C), there was also a significant decrease in the frequency and number of class-switched plasma cells in mice with B cell-specific IRF-1 deficiency (Fig. 3G to I). In summary, B cell-intrinsic IRF-1 expression supported MHV68-driven germinal center responses and B cell differentiation.
FIG 3.

B cell-intrinsic IRF-1 expression supports gammaherpesvirus-driven germinal center response. CD19 Cre-positive and Cre-negative littermates were infected as described for Fig. 2. Spleens were collected at 16 days postinfection and processed into a single-cell suspension, and the germinal center response was measured, with germinal center B cells (A to C) defined as B220+ GL7+ CD95+ cells, T follicular helper cells (D to F) defined as CD3+ CD4+ CXCR5+ PD-1+ cells, and plasma cells (G to I) defined as B220+ IgD− GL7− IRF4+ cells. Data are pooled from 2 to 3 independent experiments, with each symbol representing an individual mouse. Means and standard errors of the means are shown. *, P < 0.05; **, P < 0.001.
B cell-intrinsic IRF-1 deficiency attenuates generation of MHV68- and dsDNA-specific antibodies.
Gammaherpesviruses are unique as they drive irrelevant B cell differentiation, which leads to an early increase in the levels of non-virus-specific, self-directed antibodies (13, 27). Given this unique manipulation of B cell differentiation and the attenuated levels of germinal center B cells and plasma cells in MHV68-infected CD19 Cre-positive mice (Fig. 3), humoral responses were assessed. While some heterogeneity was observed, there were no statistically significant differences in IgG and IgM serum levels following infection of CD19 Cre-positive and Cre-negative littermates (Fig. 4A and B; data for uninfected mice are the same as those in Fig. 1E). In contrast, there was a significant reduction in the levels of MHV68-specific IgG (Fig. 4C) and IgM (Fig. 4D) in the sera of infected CD19 Cre-positive mice. Next, levels of antibodies against double-stranded DNA (dsDNA) were assessed as a readout of the self-directed antibody response. Surprisingly, and in spite of having similar total IgG levels, MHV68-infected CD19 Cre-positive mice had significantly reduced anti-dsDNA IgG titers compared to Cre-negative littermates (Fig. 4E). Thus, B cell-intrinsic IRF-1 expression supported the generation of both MHV68- and dsDNA-specific class switched antibodies.
FIG 4.

B cell-intrinsic IRF-1 deficiency attenuates generation of MHV68- and dsDNA-specific antibodies. CD19 Cre-positive and Cre-negative littermates were infected as described for Fig. 2. Serum was collected from these and mock-infected mice at 16 days postinfection to determine total IgG (A) and IgM (B), MHV68-specific IgG (C) and IgM (D), and dsDNA (E) antibody titers. Data are pooled from 2 to 3 independent experiments, with each symbol representing an individual mouse. Means and standard errors of the means are shown. *, P < 0.05; **, P < 0.001.
B cell-intrinsic IRF-1 deficiency selectively attenuates MHV68-driven germinal center responses.
Restriction of the germinal center responses observed in mice with global IRF-1 deficiency was selective for MHV68 infection, as germinal center responses following immunization or infection with lymphocytic choriomeningitis virus (LCMV) were similar in control and IRF-1−/− mice (21). To determine the extent to which B cell-intrinsic IRF-1 expression is selective for MHV68-driven germinal center response, CD19 Cre-positive and Cre-negative mice were infected with LCMV clone 13 and the peak germinal center response was examined at 14 days postinfection. In contrast to the results of observed during MHV68 infection (Fig. 3A to F), there was no difference in the frequency and number of germinal center B cells (Fig. 5A and B) in the LCMV-infected CD19 Cre-positive and Cre-negative groups. Further, despite the similar frequencies, the absolute number of CD4 T follicular helper cells was increased in LCMV-infected CD19 Cre-positive mice (Fig. 5C and D). Finally, there were no differences in the abundances of LCMV-specific IgG (Fig. 5E) and IgM (Fig. 5F) antibodies. Thus, B cell-intrinsic IRF-1 expression selectively promoted MHV68-driven but not LCMV-driven germinal center and humoral responses.
FIG 5.

B cell-intrinsic IRF-1 deficiency selectively attenuates MHV68-driven germinal center response. (A to D) CD19 Cre-positive and Cre-negative littermates were intravenously infected with 2 × 106 PFU of LCMV clone 13 (CL13). Spleens were collected at 14 days postinfection and processed into a single-cell suspension, and the germinal center response was measured with germinal center B cells (A and B) defined as B220+ GL7+ CD95+ cells and T follicular helper cells (C and D) defined as CD3+ CD4+ CXCR5+ PD-1+ cells. (E and F) Abundance of LCMV-specific IgG (E) and IgM (F) antibodies in the sera. Each symbol represents an individual spleen. P values are as indicated. **, P < 0.001.
B cell-intrinsic IRF-1 function is dispensable for long-term MHV68 infection.
Long-term gammaherpesvirus infection is established by 42 days postinfection concurrent with a return of the germinal center and self-directed antibody responses to nearly baseline levels. Long-term MHV68 infection remains poorly controlled under conditions of global IRF-1 deficiency, with sustained increase in the latent viral reservoir, viral reactivation, and further exacerbation of the germinal center response (21). To determine the B cell-intrinsic role of IRF-1 during long-term infection, parameters of MHV68 latency were compared between CD19 Cre-positive and Cre-negative littermates at 42 days postinfection. Interestingly, there was no longer a difference in the frequencies (Fig. 6A) or absolute numbers (Fig. 6B) of MHV68 DNA-positive splenocytes in CD19 Cre-positive and Cre-negative mice and the levels of viral reactivation in the spleen were equivalently undetectable (Fig. 6C). Intriguingly, while there were no differences with respect to the latent viral reservoir and viral reactivation between CD19 Cre-positive and Cre-negative mice at 42 days postinfection, there was a slight attenuation of the germinal center response seen as a decrease in the frequency and number of germinal center B cells (Fig. 6D to F) as well as in the frequency of CD4 T follicular helper cells (Fig. 6G to I) in CD19 Cre-positive mice. Therefore, while IRF-1 B cell-intrinsic expression aided the establishment of peak viral latent reservoir levels (Fig. 2 and 3), it was dispensable during long-term infection, despite sustained IRF-1-dependent differences in the germinal center responses.
FIG 6.

B cell-intrinsic IRF-1 function is dispensable for long-term gammaherpesvirus infection. (A to C) CD19 Cre-positive and Cre-negative littermates were infected as described for Fig. 2. Spleens were collected at 42 days postinfection, processed into a single-cell suspension, and subjected to limiting dilution assays to determine the frequency (A) or absolute number (B) of MHV68 DNA-positive cells and the frequency of ex vivo viral reactivation (C). (D to I) The germinal center response was measured, with germinal center B cells (D to F) defined as B220+ GL7+ CD95+ cells and T follicular helper cells (G to I) defined as CD3+ CD4+ CXCR5+ PD-1+ cells. Frequencies of cells, absolute numbers of cells, and reactivation frequencies were determined for panels D and G, E and H, and F and I as described for panels A, B, and C, respectively. Each experimental group consisted of 3 to 4 animals; data were pooled from 2 to 3 independent experiments; standard errors of the means are displayed.
B cell-intrinsic IRF-1 deficiency leads to reduced levels of active SHP1.
We recently showed that B cell-intrinsic expression of the tyrosine phosphatase SHP1 supports the MHV68-driven germinal center response and the establishment of peak viral latency (28). Interestingly, IRF-1 promotes SHP1 expression in glia (29). To determine the extent to which B cell-specific IRF-1 deficiency affects total protein levels of SHP1, B cells were isolated from spleens of naive CD19 Cre-positive and Cre-negative mice and cultured in the presence of IFN-γ to induce IRF-1 expression. Surprisingly, Cre-positive, IRF-1-deficient B cells had the same levels of SHP1 protein as wild-type B cells, regardless of IFN-γ treatment (Fig. 7A), indicating that in contrast to glia, IRF-1 does not promote SHP1 expression in splenic B cells.
FIG 7.
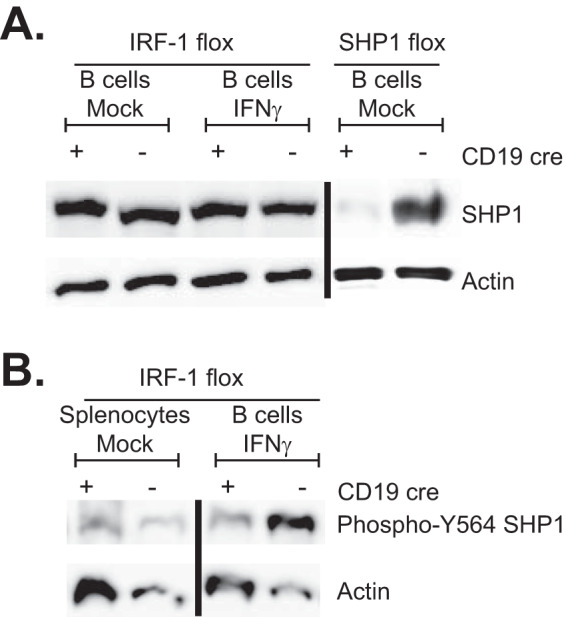
B cell-intrinsic IRF-1 deficiency leads to reduced levels of active SHP1. (A) Spleens were collected from CD19 Cre-positive and Cre-negative IRF-1 conditional littermates, and B cells were purified via negative selection and mock treated or stimulated with 10 U/ml IFN-γ for 5 h prior to Western analysis of total SHP1. Splenic B cells isolated from CD19 Cre-positive and Cre-negative SHP1flox/flox mice were used as an antibody specificity control. (B) Total splenocytes and IFN-γ-stimulated purified B cells from CD19 Cre-positive and Cre-negative mice were subjected to Western analyses using the indicated antibodies.
At baseline, SHP1 exists in an autoinhibited state. Activation of several kinases leads to phosphorylation of SHP1 on either of two C-terminal tyrosines, including Y564, thus increasing its phosphatase activity by relieving autoinhibition and allowing interaction with downstream adaptor proteins (30, 31). Having observed similar levels of total SHP1 protein in CD19 Cre-positive and Cre-negative B cells, levels of active SHP1 were measured in total splenocytes and purified splenic B cells stimulated with IFN-γ. Interestingly, the CD19 Cre-positive B cells had lower levels of Y564-phosphorylated SHP1 following activation with IFN-γ (Fig. 7B). Thus, B cell-intrinsic IRF-1 expression, while dispensable for overall SHP1 protein levels, promoted SHP1 activation.
DISCUSSION
Role of IRF-1 in the germinal center response.
Manipulation of B cells is critical for gammaherpesvirus infection, as the virus infects naive B cells and then drives a robust germinal center response in order to establish lifelong infection in memory B cells. Our previous study identified transcription factor IRF-1 as the first host factor to selectively restrict MHV68-driven germinal center response throughout chronic infection (21). Given the significance of B cell differentiation for gammaherpesvirus infection and the observed viral and host phenotypes in mice with global IRF-1 deficiency, we hypothesized that IRF-1 must function in a B cell-intrinsic manner to suppress MHV68-driven germinal center response and viral latency. To test this hypothesis, we generated mice with IRF-1 deficiency restricted to B cells (CD19 Cre-positive mice) (Fig. 1), to our knowledge, the first reported mouse model with conditional IRF-1 alleles. To our surprise and in direct contrast to what was found under conditions of global IRF-1 deficiency (21), there was a significant reduction in the MHV68-driven germinal center response following infection of the CD19 Cre-positive mice (Fig. 3 and 6). Interestingly, no attenuation of germinal center responses was observed in CD19 Cre-positive mice following LCMV infection, indicating a selective role of B cell-intrinsic IRF-1 expression in promoting the MHV68-driven germinal center response. Thus, in an intriguing turn of events, B cell-specific IRF-1 expression appears to be proviral, whereas expression of IRF-1 by another, yet-to-be-defined cellular population is likely to be responsible for the IRF-1-dependent attenuation of MHV68-driven germinal center response.
The findings of our study highlight the fact that IRF-1 functions are modified in a cell type-dependent manner, an expected phenotype as IRF family members can associate with cell type-specific transcription factors to affect gene expression (32). IRF-1 is constitutively expressed and active in a majority of cell types, with baseline levels of expression further enhanced transcriptionally, most notably by interferons, and posttranslationally, by regulation of protein stability. Thus, IRF-1-dependent gene targets are likely to be further modified by the infection status of the tissue, changes in the cytokine milieu, and cellular differentiation, all of which are likely to affect IRF-1-interacting cellular partners and downstream gene expression.
We were intrigued by the possibility that PTPN6 (encoding SHP1) could be one of what are likely many relevant IRF-1 gene targets, as we have recently shown that B cell-specific expression of SHP1 supports the establishment of latent MHV68 infection and MHV68-driven germinal center response (28) and the reported positive regulation of SHP1 expression by IRF-1 in glial cells (29). However, SHP1 protein levels were similar in CD19 Cre-positive and Cre-negative sorted B cells (Fig. 7A). Despite similar total SHP1 levels, levels of active SHP1, as measured by Y564 phosphorylation, were increased in IFN-γ-stimulated sorted B cells from CD19 Cre-negative mice compared to those from CD19 Cre-positive mice (Fig. 7B). Many tyrosine kinases can phosphorylate Y564 of SHP1, including Lck and Lyn (31, 33), two tyrosine kinases expressed to high levels in T cells and B cells, respectively. Lck expression was not affected in IRF-1−/− thymocytes (34). However, it is not clear whether IRF-1 regulates expression of Lyn or other tyrosine kinases that can activate SHP1, potentially contributing to cell type-dependent phenotypes of IRF-1 in vivo.
We have observed that positive effects of B cell-intrinsic IRF-1 expression on germinal center responses were evidenced following MHV68 infection but not LCMV infection. This further suggests that IRF-1-dependent changes in B cell-intrinsic host gene expression, while dispensable for the physiological germinal center response, are likely critical for the unique nature of the gammaherpesvirus-driven germinal center response. In addition to their benefitting from “conventional” IRF-1 effects on B cell-intrinsic host gene expression, it is tempting to speculate that gammaherpesviruses may usurp and/or redirect IRF-1 to affect expression of host and/or viral genes. IRF-1 attenuates lytic MHV68 replication in primary macrophages (35); however, MHV68 infection is latent in most B cells (with the exception of plasma cells). Interestingly, KSHV and closely related rhadinoviruses of nonhuman primates encode several viral IRFs that share limited homology with the DNA binding domain defining the cellular members of the IRF family (reviewed in references 36 and 37). Some of these viral IRFs are expressed during KSHV latency and interact with IRF-1, although the role of KSHV viral IRFs during chronic infection remains undefined. In contrast, lack of viral IRF clusters in rhesus macaque rhadinovirus led to attenuated infection and decreased B cell hyperplasia in infected animals, along with robust induction of proinflammatory and T cell responses (38). EBV and MHV68 do not encode proteins with detectable homology to cellular IRFs, and viral regulators of IRF-1 have not been described for these two viruses. However, consistent with our current study demonstrating a proviral role of B cell-intrinsic IRF-1 during the establishment of latent MHV68 infection, IRF-1 and IRF-2 constitutively activate promoters of EBV EBNA1 (39), a critical viral latent gene, suggesting a proviral role of IRF-1 during latent EBV infection of B cells.
B cell-intrinsic function of IRF-1 during gammaherpesvirus infection and pathogenesis.
Although B cell-intrinsic IRF-1 expression promoted the MHV68-driven germinal center response both at the peak of viral latency and during long-term infection, the proviral effects of B cell-intrinsic IRF-1 expression were limited to the establishment of viral latency. While the germinal center response is critical for the optimal amplification of latent viral load during peak viral latency, long-term infection is associated with a decrease in and stabilization of the latent viral load, maintenance of which becomes more dependent on the infection of developing B cell populations (40). As B cell-specific IRF-1 deficiency had no effect on the peripheral B cell numbers and, therefore, on B cell development, it is perhaps not very surprising that the proviral effects of IRF-1 were limited to the stage of latent infection that is highly dependent on the germinal center response.
Uncovering the exact mechanism by which IRF-1 B cell-intrinsic function impacts MHV68 latency and gene expression is going to be challenging, as there is no in vitro experimental system to study MHV68 infection of germinal center B cells. B cell differentiation through the germinal center cannot be recapitulated in vitro as it depends on the still incompletely understood interactions between B cells, CD4 T cells, and antigen-presenting cells that are regulated temporally and that intimately depend on the architectural features of lymphoid tissue in vivo, features that cannot yet be reproduced in culture. Not surprisingly, given these limitations, infection of cultured splenic B cells by MHV68 is notoriously inefficient and/or abortive, in contrast to in vivo infection, where MHV68 genome is present in up to 20% of all germinal center B cells at the peak of viral latency (26). In spite of this rather high latent viral presence in the germinal center B cells of infected animals, conventional transcriptome sequencing (RNA-seq) approaches are biased toward detection of much more abundant cellular transcripts; further, these approaches have to be significantly modified to detect expression of multiple noncoding viral RNAs or complex protein-coding viral transcripts. Thus, future studies defining regulation of latent MHV8 infection and comprehensive viral gene expression in germinal center B cells await technological advances.
Gammaherpesviruses are associated with B cell lymphomas, and IRF-1 is an established tumor suppressor, at least in the context of breast cancer, endometrial carcinoma, and esophageal cancer (41–43). We previously showed that IRF-1 protein levels are selectively decreased in EBV-positive but not EBV-negative posttransplant lymphoproliferative disorder (PTLD) (21), suggesting a dichotomous role of B cell-intrinsic IRF-1 expression which supports the efficient establishment of chronic gammaherpesvirus infection but likely suppresses viral lymphomagenesis during long-term latency. It is tempting to speculate that certain B cell-intrinsic IRF-1-regulated cellular (and perhaps viral) genes support the establishment of MHV68 latency either directly or indirectly, the latter by mechanisms uniquely required for efficient MHV68-driven germinal center responses. In this scenario, loss of B cell-intrinsic IRF-1 expression is detrimental for the establishment of chronic infection, as our study demonstrated. However, other well-established IRF-1 functions may be particularly important to antagonize virus-driven lymphomagenesis. For example, IRF-1 cooperates with p53 to enforce cell cycle checkpoints in response to DNA damage to allow sufficient time for DNA repair or to facilitate apoptosis if the damage exceeds the repair capacity (44, 45). Thus, decreased expression of IRF-1 in B cells of an immunocompromised host may select for more-efficient viral transformation of germinal center B cells that already undergo exaggerated mutagenesis as a part of the physiological differentiation process. Dissecting out IRF-1-regulated genes that are differentially involved in viral lymphomagenesis versus the establishment of chronic infection in an immunocompetent host is an important focus of future studies.
MATERIALS AND METHODS
Animals.
All experimental manipulations of mice were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin (MCW) (AUA971). All mice were housed and bred in a specific-pathogen-free facility. The IRF-1loxP/loxP mice were generated as described for Fig. 1. Briefly, mice with loxP sites flanking exon 2 of the IRF1 gene with an additional FRT site-flanked β-lactamase/neomycin resistance reporter cassette inserted in the first intron (L1L2_Bact_P) were acquired from the Wellcome Trust Sanger Institute [Irf1tm1a(EUCOMM)Wtsi]. These mice were subsequently bred to a FLP recombinase transgenic mouse obtained from Jackson Laboratories [B6.129S4-Gt(Rosa)26Sortm1(FLP1)Dym/RainJ] (22). The resultant offspring were backcrossed to C57BL/6J mice (Jackson Laboratories) to achieve germ line recombination of the FRT sites and loss of the FLP transgene. To generate mice with B cell-specific IRF-1 deficiency, mice were further bred to CD19 Cre-positive [B6.129P2-Cd19tm1(cre)Cgn/J] mice (23) from The Jackson Laboratories (Bar Harbor, ME). Excision of the FRT-flanked cassette was monitored by PCR using the following primer sequences: AGTGGTGGTGGTAAGAGCCT (forward) and GATGTCCCAGCCGTGCTTAG (reverse). The presence of the conditional IRF-1 allele was detected using the following primer pair: TGTTCTAGCAAGTTCTCAGAGG (forward) and TGGTACCCTGACTCACAACTG (reverse). CD19 Cre recombinase allele was detected using ACGTACTGACGGTGGAGAA (forward) and CAAAAATCCCTTCCAGGGCG (reverse).
Infections.
Mice between 6 and 10 weeks of age were intranasally infected with 1,000 PFU of MHV68 (WUMS) diluted in sterile serum-free Dulbecco’s modified Eagle’s medium (DMEM) (15 μl/mouse), under light anesthesia. The spleen was harvested from euthanized mock-infected and MHV68-infected animals at indicated time points postinfection. MHV68 virus stock was grown and titers were determined using NIH 3T12 cells. For LCMV infections, mice between 6 and 10 weeks of age were infected intravenously with 2 × 106 PFU of LCMV clone 13. LCMV was prepared by a single passage on BHK21 cells, with viral titers being determined via plaque assay on Vero cells. Mice were euthanized by CO2 inhalation from a compressed gas source in a nonovercrowded chamber. Mice were bled prior to euthanasia via the submandibular route, and serum was isolated using BD Microtainer blood collection tubes (Becton, Dickinson and Company, Franklin Lakes, NJ).
Limiting dilution assays.
The frequency of cells harboring viral DNA and the frequency of ex vivo reactivation of MHV68 were determined as previously described (46). Briefly, to determine the frequency of virally infected cells, splenocytes from 3 to 4 mice per experimental group were pooled and 3-fold serial dilutions were subjected to nested PCR (12 replicates/dilution) using primers against the viral genome. The frequency of ex vivo reactivation of MHV68 was determined by pooling infected splenocytes from 3 to 4 mice per group and plating 8 serial 2-fold dilutions onto a monolayer of mouse embryonic fibroblasts (MEFs) at 24 replicates per dilution. MHV68 was allowed to reactivate and further replicate within MEFs for 21 days, followed by assessment of cytopathic effect in every well. The limit of detection for this assay is a single plaque-forming unit.
Flow cytometry.
Single-cell suspensions of splenocytes from both mock-infected and infected mice were prepared in fluorescence-activated cell sorter (FACS) buffer (phosphate-buffered saline [PBS], 2% fetal calf serum [FCS], 0.05% sodium azide) at 1 × 107 nucleated cells/ml. A total of 1 × 106 cells were treated with Fc block (24G2) prior to extracellular staining with an optimal antibody concentration for 30 min on ice. Data acquisition was performed on a LSR II flow cytometer (BD Biosciences, San Jose, CA), and data were analyzed using FlowJo software (Tree Star, Ashland, OR). The following antibodies used in this study were purchased from BioLegend (San Diego, CA): CD3 (17A2), CD4 (RM4-5), CD8a (53-7.3), CD44 (IM7), CD95 (Jo2), PD-1 (29f.1A12), B220 (RA3-6B2), and GL7 (GL-7). CXCR5 staining was done by triple amplification using CXCR5-rat anti-mouse, biotin-goat anti-rat, and Streptavidin-allophycocyanin (Streptavidin-APC) antibodies.
Enzyme-linked immunosorbent assay (ELISA).
Total, MHV68-specific, LCMV-specific, and dsDNA immunoglobulin levels were determined as previously described (14, 21, 47–50). Briefly, Nunc MaxiSorp plates (Fisher Scientific, Pittsburgh, PA) were coated with either anti-mouse IgG antibodies (heavy plus light chain) or anti-mouse IgM antibodies (total IgG and IgM levels) (Jackson ImmunoResearch, West Grove, PA), UV-irradiated MHV68 virus stock–PBS (Stratalinker UV Crosslinker 1800; Agilent Technologies, Santa Clara, CA) (740,000 μJ/cm2 × 2), LCMV clone 13-infected BHK cell lysates, or dsDNA from Escherichia coli (12.5 μg/ml; Sigma-Aldrich, St. Louis, MO) overnight at 4°C. Plates were washed with PBS–Tween (0.05%) and blocked for 1 h with PBS–Tween (0.05%)–bovine serum albumin (BSA) (3%), incubated with 5-fold serial dilutions of serum in PBS–Tween (0.05%)–BSA (1.5%) for 2 h, and then washed with PBS–Tween (0.05%). Bound antibody was detected with horseradish peroxidase (HRP)-conjugated goat anti-mouse total IgG (heavy plus light chain) or IgM (Jackson ImmunoResearch, West Grove, PA) using 3,3′,5,5′-tetramethylbenzidine substrate (Life Technologies, Gaithersburg, MD). HRP enzymatic activity was stopped by the addition of 1 N HCl (Sigma-Aldrich, St. Louis, MO), and the absorbance was read at 450 nm on a model 1420 Victor3V multilabel plate reader (PerkinElmer, Waltham, MA).
Western blotting.
Splenic B and T cells were negatively selected (Stem Cell EasySep mouse B cell isolation kit [Stem Cell Technologies Inc., Cambridge, MA] and MojoSort mouse CD3 T cell isolation kit [BioLegend, San Diego, CA]) according to the instructions of the manufacturers. Splenic B cells isolated from CD19 Cre-positive and Cre-negative SHP1flox/flox mice (28) were used to assess SHP1 antibody specificity. B and T cells were lysed in Laemmli buffer and subjected to Western analyses as previously described (51). The following antibodies were used: anti-IRF-1 (1:1,000), anti-phospho-SHP1 (Y564) (1:1,000), and anti-β actin (1:40,000) (Cell Signaling Technologies, Danvers, MA); anti-SHP1 (1:5,000) (Santa Cruz Biotechnology, Santa Cruz, CA); and a secondary goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:15,000) (Jackson ImmunoResearch, West Grove, PA).
Statistical analyses.
Statistical analyses were performed using Student's t test (Prism; GraphPad Software, Inc.).
ACKNOWLEDGMENTS
This study was supported by CA183593 and CA203923 (V.L.T.), by an American Cancer Society Postdoctoral Award (134165-PF-19-176-01-MPC, C.N.J.), and by F31 CA243364 (K.E.J.) and 20PRE35200108 (P.A.S.).
C.N.J. and V.L.T. led the conceptual design of the study and wrote the manuscript. C.N.J., K.E.J., A.A.U., P.A.S., and C.N. designed, performed, and analyzed experiments. W.C. supervised the design of the LCMV experiments.
We declare that we have no competing interests.
REFERENCES
- 1.Cesarman E. 2014. Gammaherpesviruses and lymphoproliferative disorders. Annu Rev Pathol 9:349–372. doi: 10.1146/annurev-pathol-012513-104656. [DOI] [PubMed] [Google Scholar]
- 2.Efstathiou S, Ho YM, Hall S, Styles CJ, Scott SD, Gompels UA. 1990. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J Gen Virol 71:1365–1372. doi: 10.1099/0022-1317-71-6-1365. [DOI] [PubMed] [Google Scholar]
- 3.Virgin HW IV, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol 71:5894–5904. doi: 10.1128/JVI.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tarakanova VL, Suarez FS, Tibbetts SA, Jacoby M, Weck KE, Hess JH, Speck SH, Virgin HW IV. 2005. Murine gammaherpesvirus 68 infection is associated with lymphoproliferative disease and lymphoma in BALB beta2 microglobulin-deficient mice. J Virol 79:14668–14679. doi: 10.1128/JVI.79.23.14668-14679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flano E, Kim IJ, Woodland DL, Blackman MA. 2002. Gamma-herpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J Exp Med 196:1363–1372. doi: 10.1084/jem.20020890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willer DO, Speck SH. 2005. Establishment and maintenance of long-term murine gammaherpesvirus 68 latency in B cells in the absence of CD40. J Virol 79:2891–2899. doi: 10.1128/JVI.79.5.2891-2899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Babcock GJ, Decker LL, Freeman RB, Thorley-Lawson DA. 1999. Epstein-Barr virus-infected resting memory B cells, not proliferating lymphoblasts, accumulate in the peripheral blood of immunosuppressed patients. J Exp Med 190:567–576. doi: 10.1084/jem.190.4.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins CM, Speck SH. 2015. Interleukin 21 signaling in B cells is required for efficient establishment of murine gammaherpesvirus latency. PLoS Pathog 11:e1004831. doi: 10.1371/journal.ppat.1004831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins CM, Speck SH. 2014. Expansion of murine gammaherpesvirus latently infected B cells requires T follicular help. PLoS Pathog 10:e1004106. doi: 10.1371/journal.ppat.1004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang X, Collins CM, Mendel JB, Iwakoshi NN, Speck SH. 2009. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog 5:e1000677. doi: 10.1371/journal.ppat.1000677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson SJ, Tsao EH, Webb BL, Ye H, Dalton-Griffin L, Tsantoulas C, Gale CV, Du MQ, Whitehouse A, Kellam P. 2007. X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J Virol 81:13578–13586. doi: 10.1128/JVI.01663-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol 79:1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sangster MY, Topham DJ, D'Costa S, Cardin RD, Marion TN, Myers LK, Doherty PC. 2000. Analysis of the virus-specific and nonspecific B cell response to a persistent B-lymphotropic gammaherpesvirus. J Immunol 164:1820–1828. doi: 10.4049/jimmunol.164.4.1820. [DOI] [PubMed] [Google Scholar]
- 14.Gauld SB, De Santis JL, Kulinski JM, McGraw JA, Leonardo SM, Ruder EA, Maier W, Tarakanova VL. 2013. Modulation of B-cell tolerance by murine gammaherpesvirus 68 infection: requirement for Orf73 viral gene expression and follicular helper T cells. Immunology 139:197–204. doi: 10.1111/imm.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleisher GR, Collins M, Fager S. 1983. Limitations of available tests for diagnosis of infectious mononucleosis. J Clin Microbiol 17:619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phan RT, Dalla-Favera R. 2004. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 432:635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 17.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem 274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 18.Martin A, Bardwell PD, Woo CJ, Fan M, Shulman MJ, Scharff MD. 2002. Activation-induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature 415:802–806. doi: 10.1038/nature714. [DOI] [PubMed] [Google Scholar]
- 19.Küppers R. 2003. B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol 3:801–812. doi: 10.1038/nri1201. [DOI] [PubMed] [Google Scholar]
- 20.Küppers R, Klein U, Hansmann ML, Rajewsky K. 1999. Cellular origin of human B-cell lymphomas. N Engl J Med 341:1520–1529. doi: 10.1056/NEJM199911113412007. [DOI] [PubMed] [Google Scholar]
- 21.Mboko WP, Olteanu H, Ray A, Xin G, Darrah EJ, Kumar SN, Kulinski JM, Cui W, Dittel BN, Gauld SB, Tarakanova VL. 2015. Tumor suppressor interferon-regulatory factor 1 counteracts the germinal center reaction driven by a cancer-associated gammaherpesvirus. J Virol 90:2818–2829. doi: 10.1128/JVI.02774-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farley FW, Soriano P, Steffen LS, Dymecki SM. 2000. Widespread recombinase expression using FLPeR (flipper) mice. Genesis 28:106–110. doi: 10.1002/1526-968X(200011/12)28:3/4<106::AID-GENE30>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 23.Rickert RC, Roes J, Rajewsky K. 1997. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res 25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weck KE, Kim SS, Virgin HW IV, Speck SH. 1999. B cells regulate murine gammaherpesvirus 68 latency. J Virol 73:4651–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weck KE, Kim SS, Virgin HW IV, Speck SH. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J Virol 73:3273–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins CM, Speck SH. 2012. Tracking murine gammaherpesvirus 68 infection of germinal center B cells in vivo. PLoS One 7:e33230. doi: 10.1371/journal.pone.0033230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Getahun A, Smith MJ, Kogut I, van Dyk LF, Cambier JC. 2012. Retention of anergy and inhibition of antibody responses during acute γ herpesvirus 68 infection. J Immunol 189:2965–2974. doi: 10.4049/jimmunol.1201407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson KE, Lange PT, Jondle CN, Volberding PJ, Lorenz UM, Cui W, Dittel BN, Tarakanova VL. 12 December 2019, posting date B cell-intrinsic SHP1 expression promotes gammaherpesvirus-driven germinal center response and the establishment of chronic infection. J Virol doi: 10.1128/JVI.01232-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christophi GP, Hudson CA, Gruber R, Christophi CP, Massa PT. 2008. Promoter-specific induction of the phosphatase SHP-1 by viral infection and cytokines in CNS glia. J Neurochem 105:2511–2523. doi: 10.1111/j.1471-4159.2008.05337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Shen K, Lu W, Cole PA. 2003. The role of C-terminal tyrosine phosphorylation in the regulation of SHP-1 explored via expressed protein ligation. J Biol Chem 278:4668–4674. doi: 10.1074/jbc.M210028200. [DOI] [PubMed] [Google Scholar]
- 31.Yoshida K, Kharbanda S, Kufe D. 1999. Functional interaction between SHPTP1 and the Lyn tyrosine kinase in the apoptotic response to DNA damage. J Biol Chem 274:34663–34668. doi: 10.1074/jbc.274.49.34663. [DOI] [PubMed] [Google Scholar]
- 32.Langlais D, Barreiro LB, Gros P. 2016. The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J Exp Med 213:585–603. doi: 10.1084/jem.20151764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lorenz U, Ravichandran KS, Pei D, Walsh CT, Burakoff SJ, Neel BG. 1994. Lck-dependent tyrosyl phosphorylation of the phosphotyrosine phosphatase SH-PTP1 in murine T cells. Mol Cell Biol 14:1824–1834. doi: 10.1128/mcb.14.3.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Penninger JM, Sirard C, Mittrucker HW, Chidgey A, Kozieradzki I, Nghiem M, Hakem A, Kimura T, Timms E, Boyd R, Taniguchi T, Matsuyama T, Mak TW. 1997. The interferon regulatory transcription factor IRF-1 controls positive and negative selection of CD8+ thymocytes. Immunity 7:243–254. doi: 10.1016/s1074-7613(00)80527-0. [DOI] [PubMed] [Google Scholar]
- 35.Mboko WP, Mounce BC, Emmer J, Darrah E, Patel SB, Tarakanova VL. 9 April 2014, posting date Interferon regulatory factor-1 restricts gammaherpesvirus replication in primary immune cells. J Virol doi: 10.1128/JVI.00638-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baresova P, Pitha PM, Lubyova B. 2013. Distinct roles of Kaposi’s sarcoma-associated herpesvirus-encoded viral interferon regulatory factors in inflammatory response and cancer. J Virol 87:9398–9410. doi: 10.1128/JVI.03315-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koch S, Schulz TF. 2017. Rhadinoviral interferon regulatory factor homologues. Biol Chem 398:857–870. doi: 10.1515/hsz-2017-0111. [DOI] [PubMed] [Google Scholar]
- 38.Robinson BA, O'Connor MA, Li H, Engelmann F, Poland B, Grant R, DeFilippis V, Estep RD, Axthelm MK, Messaoudi I, Wong SW. 2012. Viral interferon regulatory factors are critical for delay of the host immune response against rhesus macaque rhadinovirus infection. J Virol 86:2769–2779. doi: 10.1128/JVI.05657-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaefer BC, Paulson E, Strominger JL, Speck SH. 1997. Constitutive activation of Epstein-Barr virus (EBV) nuclear antigen 1 gene transcription by IRF1 and IRF2 during restricted EBV latency. Mol Cell Biol 17:873–886. doi: 10.1128/mcb.17.2.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coleman CB, McGraw JE, Feldman ER, Roth AN, Keyes LR, Grau KR, Cochran SL, Waldschmidt TJ, Liang C, Forrest JC, Tibbetts SA. 2014. A gammaherpesvirus Bcl-2 ortholog blocks B cell receptor-mediated apoptosis and promotes the survival of developing B cells in vivo. PLoS Pathog 10:e1003916. doi: 10.1371/journal.ppat.1003916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Connett JM, Badri L, Giordano TJ, Connett WC, Doherty GM. 2005. Interferon regulatory factor 1 (IRF-1) and IRF-2 expression in breast cancer tissue microarrays. J Interferon Cytokine Res 25:587–594. doi: 10.1089/jir.2005.25.587. [DOI] [PubMed] [Google Scholar]
- 42.Kuroboshi H, Okubo T, Kitaya K, Nakayama T, Daikoku N, Fushiki S, Honjo H. 2003. Interferon regulatory factor-1 expression in human uterine endometrial carcinoma. Gynecol Oncol 91:354–358. doi: 10.1016/s0090-8258(03)00515-8. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Liu DP, Chen PP, Koeffler HP, Tong XJ, Xie D. 2007. Involvement of IFN regulatory factor (IRF)-1 and IRF-2 in the formation and progression of human esophageal cancers. Cancer Res 67:2535–2543. doi: 10.1158/0008-5472.CAN-06-3530. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka N, Ishihara M, Lamphier MS, Nozawa H, Matsuyama T, Mak TW, Aizawa S, Tokino T, Oren M, Taniguchi T. 1996. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature 382:816–818. doi: 10.1038/382816a0. [DOI] [PubMed] [Google Scholar]
- 45.Tamura T, Ishihara M, Lamphier MS, Tanaka N, Oishi I, Aizawa S, Matsuyama T, Mak TW, Taki S, Taniguchi T. 1995. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature 376:596–599. doi: 10.1038/376596a0. [DOI] [PubMed] [Google Scholar]
- 46.Tarakanova VL, Stanitsa E, Leonardo SM, Bigley TM, Gauld SB. 2010. Conserved gammaherpesvirus kinase and histone variant H2AX facilitate gammaherpesvirus latency in vivo. Virology 405:50–61. doi: 10.1016/j.virol.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 47.Xin G, Zander R, Schauder DM, Chen Y, Weinstein JS, Drobyski WR, Tarakanova V, Craft J, Cui W. 2018. Single-cell RNA sequencing unveils an IL-10-producing helper subset that sustains humoral immunity during persistent infection. Nat Commun 9:5037. doi: 10.1038/s41467-018-07492-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Darrah EJ, Kulinski JM, Mboko WP, Xin G, Malherbe LP, Gauld SB, Cui W, Tarakanova VL. 12 September 2017, posting date B cell-specific expression of ataxia-telangiectasia mutated protein kinase promotes chronic gammaherpesvirus infection. J Virol doi: 10.1128/JVI.01103-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kulinski JM, Darrah EJ, Broniowska KA, Mboko WP, Mounce BC, Malherbe LP, Corbett JA, Gauld SB, Tarakanova VL. 2015. ATM facilitates mouse gammaherpesvirus reactivation from myeloid cells during chronic infection. Virology 483:264–274. doi: 10.1016/j.virol.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Darrah EJ, Jondle CN, Johnson KE, Xin G, Lange PT, Cui W, Olteanu H, Tarakanova VL. 3 April 2019, posting date Conserved gammaherpesvirus protein kinase selectively promotes irrelevant B cell responses. J Virol doi: 10.1128/JVI.01760-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mounce BC, Tsan FC, Droit L, Kohler S, Reitsma JM, Cirillo LA, Tarakanova VL. 2011. Gammaherpesvirus gene expression and DNA synthesis are facilitated by viral protein kinase and histone variant H2AX. Virology 420:73–81. doi: 10.1016/j.virol.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]