

In this report, we show that hnRNPA2B1 is not a component of exosomes produced in HEp-2 or HEK293T cells. In herpes simplex virus 1 (HSV-1)-infected cells, hnRNPA2B1 was quantitatively translocated from the nucleus into the cytoplasm. In infected ΔhnRNPA2B1 cells, Golgi-dependent transport of virus from the apical surface to the extracellular medium was significantly reduced. In essence, this report supports the hypothesis that hnRNPA2B1 plays a key role in the egress of exosomes and HSV-1 from infected cells.
KEYWORDS: HSV-1, exosome, hnRNPA2B1, release
ABSTRACT
hnRNPA2B1, an abundant cellular protein, has been reported to recruit RNAs bearing a specific sequence (EXO motif) into exosomes. We characterized an exosome population averaging 100 ± 50 nm in diameter and containing a defined set of constitutive exosome markers. This population packages microRNAs (miRNAs) and can be directed to block targeted gene expression in a dose-dependent fashion. The objective of this study was to characterize the role of hnRNPA2B1 in the recruitment of miRNA. We report the following four key findings. (i) hnRNPA2B1 is not a component of exosomes produced in HEp-2 or HEK293T cells. Hence, hnRNPA2B1 carries its cargo, at most, to the site of exosome assembly, but it is not itself incorporated into exosomes. (ii) The accumulation of exosomes produced by cells in which the gene encoding hnRNPA2B1 has been knocked out (ΔhnRNPA2B1 cells) was reduced 3-fold. (iii) In uninfected HEp-2 cells, hnRNPA2B1 is localized in the nucleus. In cells infected with herpes simplex virus 1 (HSV-1), hnRNPA2B1 was quantitatively exported to the cytoplasm and at least a fraction of hnRNPA2B1 colocalized with a Golgi marker. (iv) Lastly, in ΔhnRNPA2B1 cells, there was a 2- to 3-fold reduction in virus yield but a significant (>10-fold) reduction in HSV-1 released through the apical surface into the extracellular environment. The absence of hnRNPA2B1 had no significant impact on the basolateral export of HSV-1 from infected to uninfected cells by direct cell-to-cell contact. The results suggest that hnRNPA2B1 plays a key role in the transport of enveloped virus from its site of assembly to the extracellular environment.
IMPORTANCE In this report, we show that hnRNPA2B1 is not a component of exosomes produced in HEp-2 or HEK293T cells. In herpes simplex virus 1 (HSV-1)-infected cells, hnRNPA2B1 was quantitatively translocated from the nucleus into the cytoplasm. In infected ΔhnRNPA2B1 cells, Golgi-dependent transport of virus from the apical surface to the extracellular medium was significantly reduced. In essence, this report supports the hypothesis that hnRNPA2B1 plays a key role in the egress of exosomes and HSV-1 from infected cells.
INTRODUCTION
Exosomes are relatively small extracellular vesicles carrying selected RNAs and proteins (1–4). They transport their cargo from the cells generating their cargo to recipient cells (5–10). Exosomes are currently recognized as a major avenue of cell-to-cell communication (11–15). A key component of exosome cargo is RNA that is selected for packaging on the basis of the presence of a short nucleotide sequence (EXO motif) by cognate heterogeneous nuclear RNA binding proteins (hnRNPs) (16, 17). It has been reported that the selection and packaging of exosome RNA cargo involves at least 30 hnRNPs (16, 18, 19). This report focuses on the function of hnRNPA2B1. hnRNPA2B1 is a member of a family containing two additional members, namely hnRNPA1 and hnRNPC (4, 16, 20, 21). All three have been reported to recruit RNAs containing a similar EXO motif into exosomes (16, 21). Exosomes have been reported to be heterogeneous in size. The studies carried out in this laboratory use exosome preparations ranging from 50 to 150 nm in diameter and containing CD9, annexin V, flotilin-1, Alix, and CD63 but not calnexin (22).
Interest in defining the function of hnRNPA2B1 stemmed from studies carried out in this laboratory and reported elsewhere (23). In brief, this laboratory designed a microRNA (miRNA) incorporating the EXO motif bound by hnRNPA2B1 and targeting the mRNA encoding ICP4, the major regulatory protein of herpes simplex virus 1 (HSV-1). The results were that the miRNA designated miR401 was packaged in the exosomes and that purified exosomes defined as described above and carrying miR401 effectively blocked HSV-1 replication in a dose-dependent fashion. The question posed in this study centered on the function of hnRNPA2B1. The key findings were as follows.
First, we report that exosomes derived from HEp-2 or HEK293T cells do not contain hnRNPA2B1 (ΔhnRNPA2B1 cells). Moreover, miR401 was packaged in exosomes produced by ΔhnRNPA2B1 cells. The key conclusions based on these results are that while hnRNPA2B1 may transport appropriately tagged RNAs to exosomes, it is not itself packaged into these bodies.
The second, unexpected finding concerns the role of hnRNPA2B1 in viral replication. Earlier studies have shown that HSV exits infected cells by two distinct pathways, i.e., by basolateral spread requiring contact between infected and uninfected cells and by Golgi-dependent transport from the site of envelopment to the extracellular medium via the apical surface (24, 25). The results presented in this report suggest that hnRNPA2B1 does not play a significant role in virus envelopment or basolateral spread but is required for efficient transport of HSV-1 from the site of envelopment to the apical surface for release into the extracellular environment.
RESULTS
Properties of the ΔhnRNPA2B1 cell line generated in this study.
The production of hnRNPA2B1-knockout cells used in these studies is detailed in Materials and Methods. The clone selected for further studies was designated ΔhnRNPA2B1. Three series of experiments were done to verify the absence of hnRNPA2B1 from ΔhnRNPA2B1 cells and the amounts and properties of the exosomes produced in parental and ΔhnRNPA2B1 cells.
Absence of hnRNPA2B1 from the knocked out cell line.
The absence of hnRNPA2B1 in the knocked out cell line was verified by immunoblotting. Specifically, HEp-2 or ΔhnRNPA2B1 cells grown on 12-well plates were harvested, solubilized, subjected to electrophoresis in a 10% denaturing gel, and treated with antibodies against hnRNPA2B1 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH). As shown in Fig. 1A, hnRNPA2B1 was present in ample amounts in HEp-2 cells but was absent from lysates of knocked out cells. GAPDH served as a loading control.
FIG 1.

(A) Validation of hnRNPA2B1-knockout cell line. HEp-2 cells or ΔhnRNPA2B1 cells in a 12-well plate were harvested, solubilized, subjected to electrophoresis in a 10% denaturing gel, and treated with antibodies against hnRNPA2B1 or GAPDH. (B) Particle size distribution and number of isolated exosomes extracted from ΔhnRNPA2B1, parental HEp-2, and HEK293T cells. Exosomes were isolated from HEK293T, HEp-2, or ΔhnRNPA2B1 cells as described in Materials and Methods. Particle size distribution and the number of isolated exosomes were analyzed by Izon’s qNano technology.
Production and properties of exosomes from HEK293T, HEp-2 parental, and ΔhnRNPA2B1 cell lines.
HEK293T, HEp-2 cells, or ΔhnRNPA2B1 cells were seeded in T150 flasks. To ensure the numbers of exosomes released into the extracellular medium were not statistically different from one batch to another, 5 × 106 cells were seeded into T150 flasks. The flasks were incubated for 24 ± 0.5 h, at which time the cultures were 90% confluent. The exosomes were then extracted from the extracellular medium. After 24 ± 0.5 h of incubation, the cells were thoroughly rinsed to remove accumulated exosomes and reincubated in serum-free medium. After 24 h, the spent medium was collected and the exosomes were isolated and analyzed by Izon’s qNano technology as described in Materials and Methods. The results (Fig. 1B) show that the size distributions of exosomes generated by the three cell lines were similar. However, while HEK293T and the parental HEp-2 cells produced 3.7 × 109 and 9.9 × 108 exosomes, respectively, the ΔhnRNPA2B1 cells produced 3.4 × 108 exosomes. On average, the parental and ΔhnRNPA2B1 cells produced 200 and 68 exosomes per cell per 24 h, respectively.
Impact of the knockout of hnRNPA2B1 on the distribution of selected exosome components in exosomes derived from parental and ΔhnRNPA2B1 cells.
In this series of experiments, 10-μg amounts of total cell lysates of HEK293T cells, parental HEp-2 cells, or ΔhnRNPA2B1 cells and of exosomes secreted from those cells were solubilized, subjected to electrophoresis in a denaturing gel, transferred to a polyvinylidene difluoride membranes, and treated with antibodies to CD9, annexin V, flotillin-1, Alix, calnexin, CD63, hnRNPA2B1, or GAPDH. The results, shown in Fig. 2, were as follows: (i) the relative amounts of proteins present in the lysates of the three cell lines or in exosomes derived from the cell lines show no significant differences and (ii) the relative amounts of proteins in exosomes and cell lines in which they are produced differ. CD9 is more abundant in exosomes than in cells in which they were produced. Calnexin was not detected in exosomes and virtually all tested proteins except that CD9 was more abundant in cells than in exosomes. The striking finding was the absence of hnRNPA2B1 from lysates of exosomes produced in HEp-2 or HEK293T cells.
FIG 2.
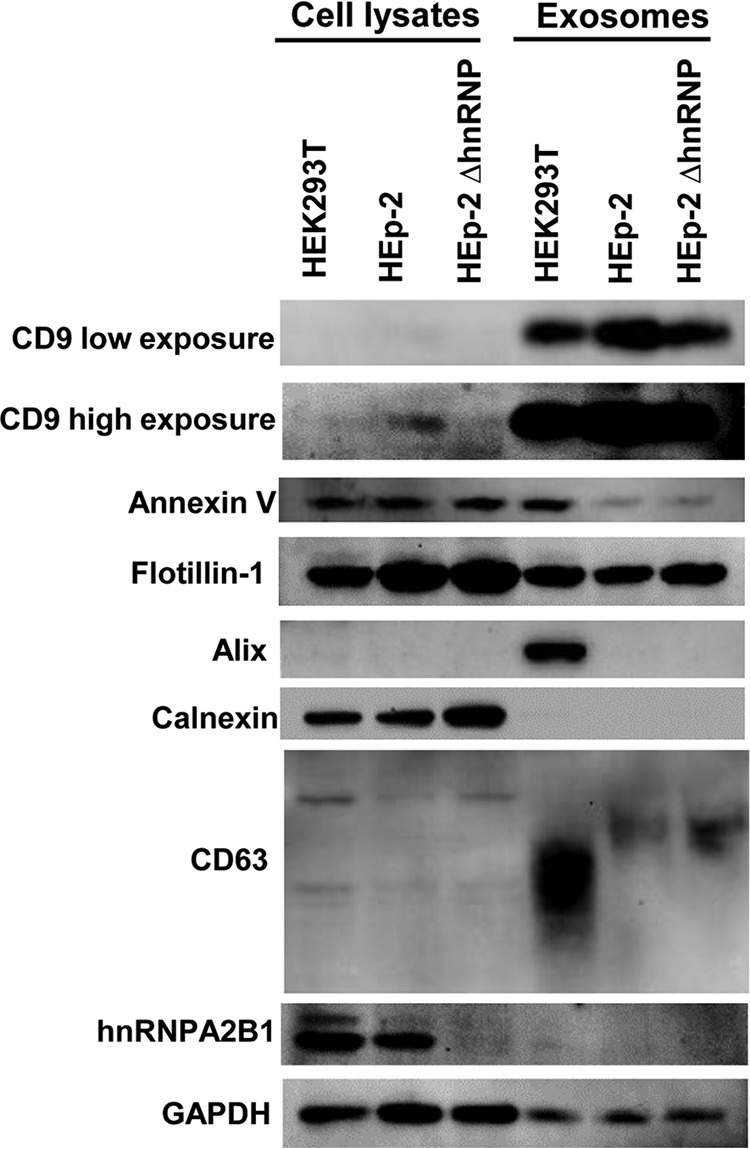
Quantification of exosomes purified from HEK293T, parental HEp-2, and ΔhnRNPA2B1 cells. HEK293T, HEp-2, or ΔhnRNPA2B1 cultures each containing 5 × 106 cells were rinsed with PBS and incubated in serum-free medium for 24 h. The exosomes were isolated from culture medium as described in Materials and Methods. Exosomes purified from equal amounts of cells and 10 μg of cell pellets were analyzed for the presence of exosomal marker proteins (CD9, annexin V, flotillin-1, Alix, calnexin, CD63, hnRNPA2B1, and GAPDH) by immunoblotting as detailed in Materials and Methods. Calnexin was used as a negative exosome marker protein.
The two key conclusions of these studies are (i) that exosomes purified as detailed in Materials and Methods and derived from parental HEp-2 cells or HEK293T cells do not contain detectable levels of hnRNPA2B1 and (ii) that with respect to the markers tested, exosomes derived from ΔhnRNPA2B1 cells cannot be differentiated from those derived from parental cells.
In HSV-1-infected cells, hnRNPA2B1 is displaced from a predominantly nuclear localization to the cytoplasm and colocalizes, at least in part, with Golgi vesicles.
In these experiments, HEp-2 cells were mock infected or exposed to 10 PFU of HSV-1 per cell for 1 h. At the intervals shown in Fig. 3, the cultures were fixed and treated with antibodies to TGN46 (green), hnRNPA2B1 (red), or 4′,6-diamidino-2-phenylindole (DAPI; blue). The results of the immunofluorescence studies illustrated in Fig. 3 may be summarized as follows. (i) In uninfected cells, hnRNPA2B1 was localized primarily in the nucleus (Fig. 3A and B). Trace amounts of hnRNPA2B1 could be detected in association with the Golgi marker (Fig. 3B). (ii) Between 6 and 18 h after infection (Fig. 3C, D, and E), nearly all detectable hnRNPA2B1 was translocated to the cytoplasm. At least some of the hnRNPA2B1 appeared to colocalize with Golgi structures, as pointed out by arrows in Fig. 3D and E.
FIG 3.

Intracellular localization of Golgi marker protein TGN46 and hnRNPA2B1. HEp-2 cells were mock infected or exposed to 10 PFU of HSV-1 per cell for 1 h. The inoculum was replaced with fresh culture medium. The cells were fixed at the times shown and were costained with an antibody to TGN46 (green, a trans-Golgi marker protein) as well as those to hnRNPA2B1 (red) and DAPI (blue, for nuclei). The images were captured and processed using a confocal laser-scanning microscope (magnification, ×63 and ×200).
The immunofluorescence studies suggest that hnRNPA2B1 is primarily localized in the nucleus and that in uninfected cells it shuttles to the cytoplasm to a site at or near Golgi structures. In infected cells, there is a mass exodus of hnRNPA2B1 from the nucleus. The translocation of hnRNPA2B1 is barely perceptible by 6 h after infection and is virtually complete between 12 and 18 h.
Impact of the knockout of hnRNPA2B1 on virus replication in parental and ΔhnRNPA2B1 cells.
In this series of experiments, replicate cultures of HEp-2 or ΔhnRNPA2B1 cells were exposed to 10 PFU of HSV-1(F) per cell. The cells were then rinsed and overlaid with fresh medium. At times indicated in Fig. 4, the cultures were harvested, the infected cells were pelleted by centrifugation, and the virus titers in medium and cells were determined in Vero cells. The results illustrated in Fig. 4 were as follows. (i) The intracellular yields of virus in HEp-2 cells were virus multiplicity independent. The intracellular yields of virus accumulating in ΔhnRNPA2B1 cells were 3-fold lower in cells infected with 10 PFU/cell. The results suggest there is not a significant reduction in cell-to-cell transmission of virus at higher multiplicities. (ii) Significant differences emerged on analyses of virus released from cells. Thus, the amounts of extracellular virus recovered from infected HEp-2 cells were 10- to 100-fold lower than those of the intracellular virus. There was little or no accumulation of extracellular virus in infected ΔhnRNPA2B1 cells. In these cells, the amounts of extracellular virus were 100- to more than a 1,000-fold lower than the amounts contained in the infected cells.
FIG 4.
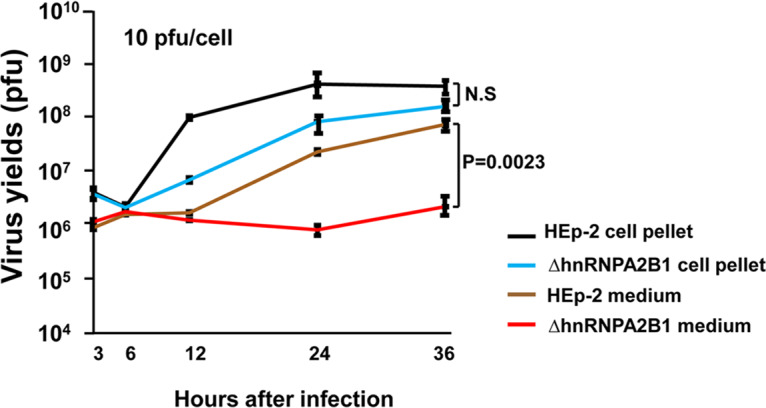
Accumulation of intracellular and extracellular virus in HEp-2 and ΔhnRNPA2B1 cell cultures exposed to 10 PFU of virus per cell. The virus progeny in the cell pellet and medium were harvested at indicated time points, and the titers were determined in Vero cells.
The key conclusion of these studies is that virus release via apical surface to the extracellular medium is impaired in infected ΔhNRNPA2B1 cells.
Effect of hnRNPA2B1 deletion on basolateral spread of virus from cell to cell.
In principle, HSV-1 spreads from cell to cell by two mechanisms, i.e., by direct basolateral transmission of virus from infected to noninfected cells and by infection of uninfected cells by virus released into the extracellular medium. The results shown in Fig. 5 indicate that the release of virus into the extracellular medium is impaired. The results do not explain the relatively low differences in the total yields of virus from cells infected at different ratios of virus particles per cell. One hypothesis that could explain the data is that the basolateral spread of virus from cell to cell is not impaired in ΔhnRNPA2B1-infected cells. The simplest procedure to test this hypothesis is to compare the sizes of the plaques formed by HSV-1 in HEp-2 and ΔhnRNPA2B1 cells. In principle, plaques are formed by virus transmitted basolaterally from an infected to uninfected cells in direct contact. If basolateral spread was impaired in the absence of hnRNPA2B1, the size of the plaques would be predicted to be much smaller. As illustrated in Fig. 5, the plaques formed by HSV-1 in HEp-2 cells could not be differentiated with respect to size from those made in ΔhnRNPA2B1 cells.
FIG 5.
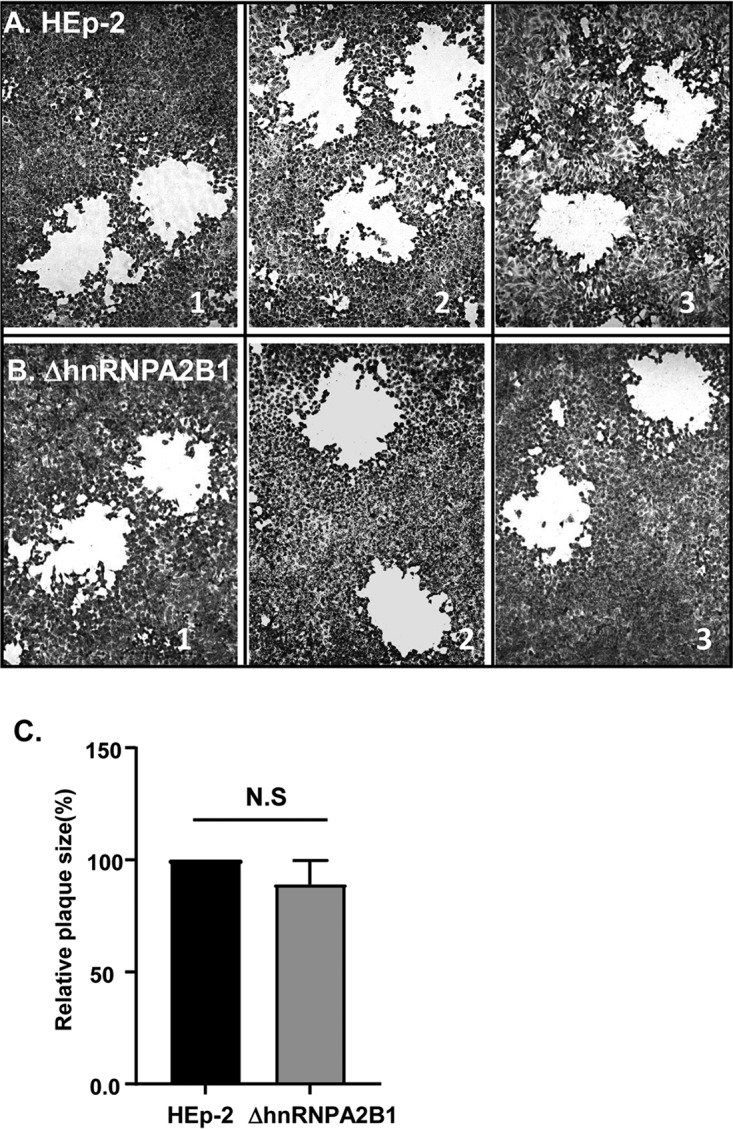
Photographs (A and B) and average plaque size (C) of plaques produced by HSV-1 in HEp-2 and ΔhnRNPA2B1 cell cultures. HEp-2 cells and ΔhnRNPA2B1 cells grown in T25 flasks were exposed to 0.01 PFU of HSV-1(F) and then overlaid with DMEM supplemented with 1% FBS plus 0.05% (wt/vol) immunoglobulin. At 72 h postinfection, the cells were fixed with 4% (wt/vol) paraformaldehyde for 30 min, stained with Giemsa for 30 min, and photographed at ×20 magnification with the aid of an inverted microscope. The size of the plaques formed by HSV-1 in HEp-2 and ΔhnRNPA2B1 cell cultures was calculated as a percentage of the plaque size in HEp-2 cells.
The key conclusion of these studies is that hnRNPA2B1 plays a key role in the exocytosis of HSV-1 via Golgi to the apical cell surface. There is no evidence in support of the hypothesis that the absence of hnRNPA2B1 impairs the basolateral spread of virus by direct contact between infected and uninfected cells.
Accumulation of viral protein in infected cells reflects the reduced spread of virus from cell to cell in hnRNPA2B1 cells infected at low multiplicities.
One prediction of the low rate of spread of virus released into the extracellular medium is a decrease of virus accumulation in cultures infected at relatively low ratios of virus/cell. We report two series of experiments. In the first experiment, HEp-2 and ΔhnRNPA2B1 cells were each infected with 1 PFU per cell. The cells were harvested, solubilized, subjected to electrophoresis in a denaturing gel, and probed with antibody to viral proteins made at different stages of the replicative cycle and to hnRNPA2B1. The results (Fig. 6A) show, as expected, a decreased accumulation of viral protein in ΔhnRNPA2B1 cells and the absence of hnRNPA2B1 in these cells.
FIG 6.

(A) Accumulation of viral proteins in HEp-2 cells and ΔhnRNPA2/B1 cells. HEp-2 or ΔhnRNPA2/B1 cells were mock infected or infected with one PFU/cell of HSV-1(F) for 24 h. The cell lysates were harvested, electrophoretically separated in a 10% denaturing gel, and treated with indicated antibodies. (B) The efficiency of hnRNPA2B1-specific siRNA. HEp-2 cells were transfected with nontarget (NT) siRNA or siRNA targeting hnRNPA2B1 (Si-hnRNP-1 and Si-hnRNP-2). After 72 h of incubation, the cells were harvested, electrophoretically separated in a 10% denaturing gel, and treated with indicated antibodies. (C) The accumulation of viral proteins in HEp-2 cells transfected with the hnRNPA2B1-specific siRNA. HEp-2 cells were transfected with nontarget (NT) siRNA or Si-hnRNP-2 siRNA. After 72 h of incubation, the cells were mock infected or infected with 0.1 PFU/cell for 24 or 36 h. At indicated times, the cell lysates were harvested, electrophoretically separated in a 10% denaturing gel, and treated with indicated antibodies.
The objective of the second series of experiments was to verify the causative link between the deletion of hnRNPA2B1 and reduced accumulation of viral proteins in cells infected at a low multiplicity of infection. We first confirmed the efficiency of hnRNPA2B1-specific small interfering RNA (siRNA). HEp-2 cells were transfected with hnRNPA2B1 siRNAs (Si-hnRNP-1 and Si-hnRNP-2) or a nontargeting control siRNA (Si-NT). The sequences of siRNAs designated are listed in Materials and Methods. After transfection for 72 h, the knockdown efficiency was evaluated by blotting for the hnRNPA2B1 protein. The results show the knockdown of hnRNPA2B1 protein expression by hnRNPA2B1-specific siRNAs, especially Si-hnRNP-2 (Fig. 6B). Thus, we chose Si-hnRNP-2 for the next study. In the next experiment, HEp-2 cells were transfected with nontarget (NT) siRNA or siRNA targeting the mRNAs encoding hnRNPA2B1 (Si-hnRNP-2). After 72 h, the cultures were mock infected or infected with 0.1 PFU of virus per cell and incubated for an additional 24 or 36 h. The cells were then harvested, solubilized, electrophoretically separated in a denaturing gel, and treated with indicated antibodies. The results (Fig. 6C) show a diminished accumulation of viral proteins in HEp-2 cells transfected with the Si-hnRNP-2 siRNA. The results support the conclusion that the diminished accumulation of viral proteins in ΔhnRNPA2B1 cells is related to the absence of hnRNPA2B1 rather than to a nontarget effect of hnRNPA2B1 knockout.
DISCUSSION
Numerous publications have reported that hnRNPA2B1 plays an important role in the packaging of RNAs bearing a specific nucleotide signature (the EXO motif) into exosomes (4, 16, 17). The results presented in this report cast a new light on the function of this protein. We report two sets of data.
The first set concerns the role of hnRNPA2B1 in the recruitment of RNA into exosomes. We have used preparations of exosomes produced in HEp-2 or HEK293T cells and defined by size and protein content. These exosomes package microRNAs bearing the appropriate EXO motifs present in targeted mRNAs. We report the following: in uninfected cells, hnRNPA2B1 accumulated in relatively large amounts in the nucleus and only trace amounts could be detected in the cytoplasm. In infected cells, virtually all hnRNPA2B1 was translocated into the cytoplasm within a span of 12 h, starting approximately 4 to 6 h after infection. In some but not all cells, patches containing the protein colocalized with Golgi markers. The key finding reported here is that hnRNPA2B1 carried from the nucleus to cytoplasm was not packaged in exosomes formed in either HEp-2 or HEK293T cells. This finding is consistent with a report that hnRNPA2B1 present in neurons transports RNA to the axons, but it is not incorporated into the vesicles generated by the neurons and which carry RNA to recipient cells (17).
The key question raised by these findings is the mechanism by which the hnRNPA2B1 RNA cargo is translocated to the exosomes. One hypothesis that remains to be tested is that the cargo contains proteins in addition to the RNA and that the cargo is deposited at the sites of exosome assembly.
The second key finding concerns the function of hnRNPA2B1 in the course of HSV infection. The studies presented in this report show that in the absence of hnRNPA2B1, the total yield of HSV-1 decreases 2- to 3-fold. However, most of the infectious virus is retained in the cytoplasm and is not released into the extracellular medium. This finding suggests that hnRNPA2B1 plays a key role in egress, i.e., in the transport of vesicles containing the enveloped particle rather than in the process of envelopment itself.
The results presented in this report suggest that the primary function of hnRNPA2B1 is to deliver exosome cargo to the nucleus, presumably at the sites of exosome assembly. An unexpected finding is that components of the cargo play a critical role in virus release from infected cells and a rather minor role in the release of exosomes. One hypothesis that remains to be resolved is whether the cargo essential for release of exosomes is delivered by other hnRNPs present in abundant numbers in cells.
MATERIALS AND METHODS
Cell lines and virus.
HEp-2, HEK293T, and Vero cells were obtained from the American Type Culture Collection. HEp-2 cells and the knockout cell lines were routinely cultured in Dulbecco’s modified Eagle medium (DMEM) (Life Technologies) supplemented with 5% (vol/vol) fetal bovine serum (FBS). HEK293T cells were maintained in DMEM supplemented with 10% (vol/vol) FBS. Vero cells were maintained in 5% (vol/vol) newborn bovine serum. HSV-1(F), the prototype HSV-1 strain used in this laboratory (26), was propagated and the titer determined on Vero cells.
Antibodies.
Antibodies used in this study were anti-CD9 (catalog [cat.] number 13174s; Cell Signaling Technology), anti-CD63 (cat. number AF1471; Beyotime), anti-annexin V (cat. number 8555; Cell Signaling Technology), anti-flotillin-1 (cat. number 18634; Cell Signaling Technology), anti-Alix (cat. number 2171S; Cell Signaling Technology), anti-calnexin (cat. number AC018; Beyotime), anti-hnRNPA2B1 (cat. number BS6196; Bioworld), anti-GAPDH (cat. number KM9002; Sungene Biotech), and anti-TGN46 (cat. number AHP500G; AbD Serotec). Antibodies against ICP0, ICP27 (27), ICP8 (Rumbaugh Goodwin Institute for Cancer Research, Inc.), VP16, and US11 (23) have been described in detail elsewhere.
Exosome isolation and quantification.
Exosomes were purified by differential centrifugation processes (28). Cells were seeded in T150 flasks at the same time and produced cultures of identical density. Then, the cells were rinsed with phosphate-buffered saline (PBS) three times to exclude potential contamination of exosomes in serum and were cultured in fetal bovine serum-free medium for another 24 h. The supernatant medium was collected and spun down at 300 × g for 10 min at 4°C to remove nonadherent cells. Then, the supernatant medium was centrifuged at 12,000 × g for 30 min at 4°C. The supernatants were transferred into a clean polycarbonate bottle for ultracentrifugation at 120,000 × g for 70 min at 4°C. The pelleted exosomes were then resuspended in 100 μl of PBS or were lysed in radioimmunoprecipitation assay (RIPA) buffer and then quantified by a bicinchoninic acid (BCA) assay using the Enhanced BCA protein assay kit (Beyotime Biotechnology, China) according to the manufacturer’s instructions. Exosome protein content was determined by calibration against a standard curve, which was prepared by plotting the absorbance at 562 nm versus the bovine serum albumin (BSA) standard concentration.
Exosome size analysis.
Exosome size distribution analysis was done using the qNano system (Izon, Christchurch, New Zealand). Izon’s qNano technology (http://izon.com) was employed to detect exosomes passing through a nanopore by way of single-molecule electrophoresis (29). In practice, it enables accurate particle-by-particle characterization of vesicles from 50 to 300 nm in size of exosomes, without averaging the particle sizes. Purified exosomes were eluted with PBS, vigorously shaken, and measured by using an NP150 (A48844) nanopore aperture. The samples were measured at a 45.9 mm stretch, with a voltage of 0.60 V at a pressure of 8 as the standard, according to the manufacturer’s instructions. Data processing and analysis were carried out on Izon Control Suite software v3.3 (Izon Science).
Immunoblotting assays.
Immunoblotting analysis was performed as previously described (30). For exosome marker protein detection, 10 micrograms of proteins from cell lysates and exosomal proteins purified from equal amount of cells were loaded in each lane. Cells and purified exosomes were harvested and lysed with RIPA lysis buffer (Beyotime) supplemented with 1 mM protease inhibitor phenylmethylsulfonyl fluoride (PMSF; Beyotime) and phosphatase inhibitor (Beyotime). Cell and exosome lysates were heat denatured, separated by SDS-PAGE, and transferred to polyvinylidene difluoride membranes (Millipore). The proteins were detected by incubation with an appropriate primary antibody, followed by incubation with a horseradish peroxidase-conjugated secondary antibody (Invitrogen) and the ECL reagent (Pierce). Images were captured using a ChemiDoc touch imaging system (Bio-Rad) and processed using ImageLab software.
Virus titer determination.
HEp-2 cells or ΔhnRNPA2B1 cells were seeded in 6-well plate at a density of 1 × 106 cells per well for 24 h and then were exposed to 10 PFU of HSV-1(F) per cell for 1 h. The inoculum then was replaced with fresh medium. The virus progeny in the cell pellet and medium were harvested at indicated time points, and then titers were determined on Vero cells after three freeze-thaw cycles.
Plaque assay.
HEp-2 cells or ΔhnRNPA2B1 cells seeded in T25 flasks were exposed to 0.01 PFU of HSV-1(F) per cell for 2 h and were maintained in DMEM supplemented with 1% FBS plus 0.05% (wt/vol) human pooled immunoglobulin for 72 h. Immunoglobulin is routinely incorporated into the medium to neutralize virus released from the apical surface. It has no effect on the transmission of virus from cell to cell by basolateral contact between infected and uninfected cells. The cells were fixed with 4% (wt/vol) of paraformaldehyde for 30 min, rinsed three times with PBS, and stained with Giemsa. The images were captured by an inverted Leica microscope. Plaque size was measured using ImageJ software, and values were calculated by normalization to the plaque size in HEp-2, which was set at 100%.
Immunofluorescence.
HEp-2 cells (5 × 104) were seeded into culture slides for 16 h. And then, the cells were mock infected or exposed to 10 PFU of HSV-1 per cell for 1 h. The inoculum was replaced with fresh culture medium. Cells were rinsed with PBS and fixed with 4% paraformaldehyde for 30 min at room temperature at the indicated times, followed by permeabilization with 0.1% Triton X-100. The cells were costained with an antibody to TGN46 (cat. AHP500G; AbD Serotec) as well as those to hnRNPA2B1 (cat. BS6196; Bioworld) overnight at 4°C. The cells were then incubated with Alexa Fluor 488-conjugated anti-sheep (cat. A11015; Invitrogen) and Alexa Fluor 568-conjugated anti-rabbit (cat. A11036; Invitrogen) secondary antibodies at room temperature for 1 h. They were then washed with PBS and embedded in DAPI-containing mounting medium (cat. 18961S; Cell Signaling Technology). The images were captured and processed using a confocal laser-scanning microscope (magnification, ×63 and ×200).
Generation of hnRNPA2B1-knockout cell line using CRISPR.
The hnRNPA2B1-knockout cell lines were generated by using a single-guide RNA (sgRNA) targeting exon 4 or exon 5 of the hnRNPA2B1 gene, which produced ideal lines with abolished hnRNPA2B1 protein expression. The sgRNA targeting hnRNPA2B1 was cloned into the plasmid pSpCas9(BB)-2A-GFP (PX458), which expresses EGFP and human codon-optimized Cas9. pSpCas9(BB)-2A-GFP (PX458) was purchased from Addgene (cat. 48138). Parental HEp-2 cells were cotransfected with the sgRNA vector plasmid and a donor vector with a selection cassette expressing GFP. GFP-positive cells were selected using fluorescence-activated cell sorting and were serially diluted to form single cell-derived colonies that expanded. The knockouts were further verified by genomic DNA sequencing and immunoblotting.
The sgRNA sequences are as follows: CACCGGCAAGACCTCATTCAATTGAT (HNRNPA2B1 sgRNA1-F), AAACATCAATTGAATGAGGTCTTGC (HNRNPA2B1 sgRNA1-R), CACCGGTTTTGATGACCATGATCCTG (HNRNPA2B1 sgRNA2-F), and AAACCAGGATCATGGTCATCAAAAC (HNRNPA2B1 sgRNA2-R).
Knockdown of hnRNPA2B1 in HEp-2 Cells by siRNA.
The siRNA transfections were performed using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. The sequences for the siRNA were as follows: 5′-GGCGGAAUUAAAGAAGAUATT-3′ (hnRNPA2B1 siRNA [Si-hnRNP-1]), 5′-GGACCAGGAAGUAACUUUATT-3′ (hnRNPA2B1 siRNA [Si-hnRNP-2]), and 5′-UUCUCCGAACGUGUCACGUTT-3′ (negative control [siNT]).
ACKNOWLEDGMENTS
This study was supported by grants from the Shenzhen Overseas High-Caliber Peacock Foundation (KQTD2015071414385495), Shenzhen Science and Innovation Commission project grant JCYJ20170411094933148 to the Shenzhen International Institute for Biomedical Research, and Guangdong Nature Science Foundation grant 2016A030308007 to Guangzhou Medical University.
X.Z., X.C., B.R., and G.G.Z. designed the research; X.Z., L.W., and W.Z. performed the research; and X.Z., B.R., and G.G.Z. analyzed data and wrote the paper.
We declare no conflict of interest.
REFERENCES
- 1.Thery C, Zitvogel L, Amigorena S. 2002. Exosomes: composition, biogenesis and function. Nat Rev Immunol 2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 2.Colombo M, Raposo G, Thery C. 2014. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- 3.van Niel G, D'Angelo G, Raposo G. 2018. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19:213–228. doi: 10.1038/nrm.2017.125. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Li S, Li L, Li M, Guo C, Yao J, Mi S. 2015. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinformatics 13:17–24. doi: 10.1016/j.gpb.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahadi A, Brennan S, Kennedy PJ, Hutvagner G, Tran N. 2016. Long non-coding RNAs harboring miRNA seed regions are enriched in prostate cancer exosomes. Sci Rep 6:24922. doi: 10.1038/srep24922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beltrami C, Besnier M, Shantikumar S, Shearn AI, Rajakaruna C, Laftah A, Sessa F, Spinetti G, Petretto E, Angelini GD, Emanueli C. 2017. Human pericardial fluid contains exosomes enriched with cardiovascular-expressed microRNAs and promotes therapeutic angiogenesis. Mol Ther 25:679–693. doi: 10.1016/j.ymthe.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casadei L, Calore F, Creighton CJ, Guescini M, Batte K, Iwenofu OH, Zewdu A, Braggio DA, Bill KL, Fadda P, Lovat F, Lopez G, Gasparini P, Chen JL, Kladney RD, Leone G, Lev D, Croce CM, Pollock RE. 2017. Exosome-derived miR-25-3p and miR-92a-3p stimulate liposarcoma progression. Cancer Res 77:3846–3856. doi: 10.1158/0008-5472.CAN-16-2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu Y, Zhang L, Zhang F, Tang T, Zhou Q, Feng C, Jin Y, Wu Z. 2017. Exosome-mediated miR-146a transfer suppresses type I interferon response and facilitates EV71 infection. PLoS Pathog 13:e1006611. doi: 10.1371/journal.ppat.1006611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi J. 2016. Considering exosomal miR-21 as a biomarker for cancer. J Clin Med 5:42. doi: 10.3390/jcm5040042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C, Zhang C, Liu L, A X, Chen B, Li Y, Du J. 2017. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol Ther 25:192–204. doi: 10.1016/j.ymthe.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan Q, Yang L, Zhang X, Peng X, Wei S, Su D, Zhai Z, Hua X, Li H. 2018. The emerging role of exosome-derived non-coding RNAs in cancer biology. Cancer Lett 414:107–115. doi: 10.1016/j.canlet.2017.10.040. [DOI] [PubMed] [Google Scholar]
- 12.Kalamvoki M, Du T, Roizman B. 2014. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc Natl Acad Sci U S A 111:E4991–E4996. doi: 10.1073/pnas.1419338111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y, Chen W, Liu F, Sun W, Li XF, Wang X, Wang Y, Xu ZY, Gao L, Yang Q, Xu B, Li YM, Fang ZY, Xu ZP, Bao Y, Wu DS, Miao X, Sun HY, Sun YH, Wang HY, Wang LH. 2016. Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell 29:653–668. doi: 10.1016/j.ccell.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Raposo G, Stoorvogel W. 2013. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simons M, Raposo G. 2009. Exosomes—vesicular carriers for intercellular communication. Curr Opin Cell Biol 21:575–581. doi: 10.1016/j.ceb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 16.Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, Martinez-Herrera DJ, Pascual-Montano A, Mittelbrunn M, Sánchez-Madrid F. 2013. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun 4:2980. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoek KS, Kidd GJ, Carson JH, Smith R. 1998. hnRNP A2 selectively binds the cytoplasmic transport sequence of myelin basic protein mRNA. Biochemistry 37:7021–7029. doi: 10.1021/bi9800247. [DOI] [PubMed] [Google Scholar]
- 18.Hobor F, Dallmann A, Ball NJ, Cicchini C, Battistelli C, Ogrodowicz RW, Christodoulou E, Martin SR, Castello A, Tripodi M, Taylor IA, Ramos A. 2018. A cryptic RNA-binding domain mediates Syncrip recognition and exosomal partitioning of miRNA targets. Nat Commun 9:831. doi: 10.1038/s41467-018-03182-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santangelo L, Giurato G, Cicchini C, Montaldo C, Mancone C, Tarallo R, Battistelli C, Alonzi T, Weisz A, Tripodi M. 2016. The RNA-binding protein SYNCRIP is a component of the hepatocyte exosomal machinery controlling MicroRNA sorting. Cell Rep 17:799–808. doi: 10.1016/j.celrep.2016.09.031. [DOI] [PubMed] [Google Scholar]
- 20.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP. 2013. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koloteva-Levine N, Amichay M, Elroy-Stein O. 2002. Interaction of hnRNP-C1/C2 proteins with RNA: analysis using the yeast three-hybrid system. FEBS Lett 523:73–78. doi: 10.1016/s0014-5793(02)02938-1. [DOI] [PubMed] [Google Scholar]
- 22.Konoshenko MY, Lekchnov EA, Vlassov AV, Laktionov PP. 2018. Isolation of extracellular vesicles: general methodologies and latest trends. Biomed Res Int 2018:8545347. doi: 10.1155/2018/8545347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Chen X, Zhou X, Roizman B, Zhou GG. 2018. miRNAs targeting ICP4 and delivered to susceptible cells in exosomes block HSV-1 replication in a dose-dependent manner. Mol Ther 26:1032–1039. doi: 10.1016/j.ymthe.2018.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farnsworth A, Johnson DC. 2006. Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J Virol 80:3167–3179. doi: 10.1128/JVI.80.7.3167-3179.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM (ed), Fields virology, 6th ed. Wolters Kluwer/Lippincott-Williams and Wilkins, New York, NY. [Google Scholar]
- 26.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 27.Ackermann M, Braun DK, Pereira L, Roizman B. 1984. Characterization of herpes simplex virus 1 alpha proteins 0, 4, and 27 with monoclonal antibodies. J Virol 52:108–118. doi: 10.1128/JVI.52.1.108-118.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, Alvarez-Erviti L, Sargent IL, Wood MJ. 2012. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc 7:2112–2126. doi: 10.1038/nprot.2012.131. [DOI] [PubMed] [Google Scholar]
- 29.Momen-Heravi F, Balaj L, Alian S, Tigges J, Toxavidis V, Ericsson M, Distel RJ, Ivanov AR, Skog J, Kuo WP. 2012. Alternative methods for characterization of extracellular vesicles. Front Physiol 3:354. doi: 10.3389/fphys.2012.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou G, Te D, Roizman B. 2010. The CoREST/REST repressor is both necessary and inimical for expression of herpes simplex virus genes. mBio 2:e00313-10. doi: 10.1128/mBio.00313-10. [DOI] [PMC free article] [PubMed] [Google Scholar]