

The rapid spread of mosquito-borne viral diseases in humans puts a huge economic burden on developing countries. For many of these infections, including those caused by chikungunya virus (CHIKV), there are no specific treatment possibilities to alleviate disease symptoms. Understanding the virus-host interactions that are involved in the viral replication cycle is imperative for the rational design of therapeutic strategies. In this study, we discovered an antiviral compound, elucidated its mechanism of action, and propose serotonergic drugs as potential host-directed antivirals for CHIKV.
KEYWORDS: 5-NT, chikungunya virus, MM, serotonin receptors, host-directed antivirals, virus-host interactions
ABSTRACT
Chikungunya virus (CHIKV) is an important reemerging human pathogen transmitted by mosquitoes. The virus causes an acute febrile illness, chikungunya fever, which is characterized by headache, rash, and debilitating (poly)arthralgia that can reside for months to years after infection. Currently, effective antiviral therapies and vaccines are lacking. Due to the high morbidity and economic burden in the countries affected by CHIKV, there is a strong need for new strategies to inhibit CHIKV replication. The serotonergic drug 5-nonyloxytryptamine (5-NT) was previously identified as a potential host-directed inhibitor for CHIKV infection. In this study, we determined the mechanism of action by which the serotonin receptor agonist 5-NT controls CHIKV infection. Using time-of-addition and entry bypass assays, we found that 5-NT predominantly inhibits CHIKV in the early phases of the replication cycle, at a step prior to RNA translation and genome replication. Intriguingly, however, no effect was seen during virus-cell binding, internalization, membrane fusion and genomic RNA (gRNA) release into the cell cytosol. In addition, we show that the serotonin receptor antagonist methiothepin mesylate (MM) also has antiviral properties toward CHIKV and specifically interferes with the cell entry process and/or membrane fusion. Taken together, pharmacological targeting of 5-HT receptors may represent a potent way to limit viral spread and disease severity.
IMPORTANCE The rapid spread of mosquito-borne viral diseases in humans puts a huge economic burden on developing countries. For many of these infections, including those caused by chikungunya virus (CHIKV), there are no specific treatment possibilities to alleviate disease symptoms. Understanding the virus-host interactions that are involved in the viral replication cycle is imperative for the rational design of therapeutic strategies. In this study, we discovered an antiviral compound, elucidated its mechanism of action, and propose serotonergic drugs as potential host-directed antivirals for CHIKV.
INTRODUCTION
Chikungunya fever is an important reemerging mosquito-borne human disease caused by chikungunya virus (CHIKV). Over the past decade, the virus has continued to spread throughout the Americas and Asia, thereby infecting millions of people (1). Chikungunya fever is characterized by fever, headache, rash, and myalgia. A potential long-lasting and debilitating feature of CHIKV infection is the onset of (poly)arthralgia and/or polyarthritis, which can last for months to years after infection (2, 3). Roughly 85% of all infected individuals develop chikungunya fever, of which approximately 30 to 40% develop long lasting (poly)arthralgia/arthritis (1, 4). Consequently, CHIKV has a high morbidity and economic burden in the countries affected, especially since there are no vaccines or antiviral therapies available.
Antiviral therapies against CHIKV should be designed with the aim to lower viral burden and/or to prevent the onset of chronic disease. There are two classes of antivirals, (i) direct-acting drugs, which target the virus itself, and (ii) host-directed drugs, which target cellular factors important for the replication cycle of the virus (5–7). An advantage of direct-acting antivirals is that these are more specific; however, viral resistance is often obtained quickly (8). Host-directed antivirals, on the other hand, are less specific and may cause more side effects, yet the development of viral resistance is greatly reduced (9). To identify novel host-directed antivirals, it is imperative to understand the dynamic and temporal interactions of the virus with the host during infection.
To initiate infection, CHIKV interacts with cellular receptors expressed at the plasma membrane. Among others, the cell adhesion molecule Mxra8 and N-sulfated heparan sulfate have been proposed as putative receptors for CHIKV, thereby facilitating virus internalization via clathrin-mediated endocytosis (6, 10). The acidic lumen of the early endosome subsequently triggers conformational changes in the E2 and E1 viral spike glycoproteins, leading to E1-mediated membrane fusion (11, 12). Thereafter, the viral nucleocapsid is dissociated by an as yet ill-understood process, and the positive-sense RNA is translated to form the nonstructural proteins of the virus. These nonstructural proteins interact with multiple cellular factors to facilitate (i) RNA replication, (ii) translation of structural proteins from the viral subgenomic mRNA, and (iii) production of new genomic RNA. The structural protein E1 and a precursor, E2, are translocated to the endoplasmic reticulum (ER), where heterodimerization of E1/E2 occurs. Maturation of the E1/E2 viral spike complex occurs via transit through the cellular secretory pathway. Progeny genomic RNA interacts with newly produced viral capsid proteins to form a nucleocapsid, which is transported to the plasma membrane, where virion assembly and budding occur (13, 14).
Serotonin (5-hydroxytryptamine; 5-HT) receptors are expressed at the plasma membrane and are known to facilitate or alter the infectivity of different classes of viruses (15–17). Most of the 5-HT receptors are G-protein coupled receptors and regulate important physiological functions and signaling pathways, including the cycling AMP (cAMP), calcium, and phosphatidylinositol pathways (18). There are multiple subtypes of 5-HT receptors, and these are divided into 7 families (19). The 5-HT2 receptor family has been described to facilitate cell entry of JC polyomavirus (20), and a 5-HT1 receptor is suggested to be involved in HIV-1 replication (21). Furthermore, the infectivity of multiple RNA viruses was found to be controlled by 5-HT receptor agonists and antagonists (9, 22–24). Indeed, reovirus and CHIKV infectivity was found to be reduced in the presence of the 5-HT receptor agonist 5-nonyloxytryptamine (5-NT), which is described as a specific 5-HT1B/5-HT1D receptor agonist, although it has also low-level affinity to other 5-HT receptor families (24, 25). 5-NT was shown to interfere with reovirus intracellular transport and disassembly kinetics during cell entry (24). Contrary to the agonist, the 5-HT receptor antagonist methiothepin mesylate (MM) increased reovirus infectivity. However, it is yet unclear whether the mechanism of action of 5-HT receptor stimulation with this 5-HT receptor agonist and antagonist is the same for CHIKV.
In this study, we confirmed the antiviral properties of 5-NT and unraveled the mode of action in CHIKV infection. Also, and in contrast to observations for reovirus, we found an antiviral effect of the serotonin receptor antagonist MM toward CHIKV. We show that the serotonergic drugs 5-NT and MM target distinct steps during CHIKV cell entry, and we conclude that targeting 5-HT receptors may be a novel strategy to alleviate CHIKV disease.
RESULTS
5-NT strongly inhibits CHIKV infection and virus particle production in U-2 OS cells.
The effect of 5-NT on CHIKV infectivity was analyzed in human bone osteosarcoma (OS) epithelial U-2 OS cells, as epithelial cells are natural targets during human CHIKV infection (26, 27). Also, these cells were used by Mainou and coworkers, who previously identified 5-NT as an inhibitor of CHIKV and reovirus infection (24). First, the mRNA expression levels of 10 distinct 5-HT receptor subtypes were determined in U-2 OS cells. We confirmed expression of 8 distinct 5-HT receptor subtypes, which included the 5-HT1B and 5-HT1D receptors to which 5-NT binds with high affinity (Fig. 1A). Next, we assessed the cellular cytotoxicity of 5-NT in U-2 OS cells by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay, which revealed no significant cytotoxicity up to a concentration of 5 μM 5-NT (Fig. 1B). The highest dose in our infectivity experiments was therefore set at 5 μM 5-NT. 5-NT was dissolved in dimethyl sulfoxide (DMSO), and the final DMSO concentration was below 1% in all experiments. Then, we analyzed CHIKV strain La Reunion (CHIKV-LR) infectivity and infectious viral particle production in U-2 OS cells in the presence of increasing concentrations of 5-NT. Cells were pretreated with increasing doses of 5-NT or NH4Cl, a lysosomotropic agent known to neutralize the endosomal pH and thereby inhibit the membrane fusion activity of CHIKV, for 1 h and infected with CHIKV-LR 5′ green fluorescent protein (5′GFP) at a multiplicity of infection (MOI) of 5. At 20 h postinfection (h p.i.), cells and supernatants were collected and analyzed for GFP expression by flow cytometry and for the production of infectious virus particles by plaque assay. A clear dose-dependent reduction in the number of CHIKV-infected cells was observed (Fig. 1C). Importantly, the vehicle control DMSO had no significant effect on the number of infected cells (Fig. 1C). CHIKV infection was reduced from 50.8% ± 3.0% to 9.2% ± 2.9% (corresponding to an 82% ± 6.4% reduction) in the presence of 5 μM 5-NT (Fig. 1C). The 50% effective concentration (EC50), i.e., the concentration at which 50% reduction is achieved, was found to be 2.8 μM 5-NT (95% confidence interval, 2.2 to 3.6 μM). In line with these results, we also observed a reduction in infectious virus particle production. At a concentration of 5 μM 5-NT, infectious virus particle production was reduced by >1 log10 (94% ± 4.4%) compared to that in the vehicle control (Fig. 1D). These results correspond to the findings of Mainou and colleagues and confirm that 5-NT interferes with productive CHIKV infection (24).
FIG 1.

Serotonin receptor agonist 5-NT strongly inhibits CHIKV infection. (A) Threshold cycle (ΔCT) values between 5-HT receptors and GAPDH mRNA expression in U-2 osteosarcoma (OS) cells. RNA derived from U-2 OS cell lysates was reverse transcribed into cDNA and subjected to quantitative PCR (qPCR) with specific primers for 10 subtypes of the serotonin receptor family and GAPDH (1:10 dilution) as the reference gene. Three independent experiments were performed, each in duplicate. Each dot represents the average of an independent experiment. (B) MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay to determine the cytotoxicity of 5-NT in U-2 OS cells. Cells were treated for 21 h in the absence or presence of increasing concentrations of the inhibitor to mimic conditions during the infectivity assay. Dotted line indicates 75% cell survival. Three independent experiments were performed, each in sextuplicate. (C, D) U-2 OS cells were pretreated for 1 h with the vehicle control dimethyl sulfoxide (DMSO), 75 mM NH4Cl, or increasing concentrations of 5-NT and were subsequently challenged with CHIKV-LR 5′ green fluorescent protein (5′GFP) at a multiplicity of infection (MOI) of 5 for 20 h. Three independent experiments were performed with one replicate per experiment. (C) Cells were collected for analysis with flow cytometry for GFP-positive cells, then (D) supernatants were harvested and virus particle production was analyzed by plaque assay on Vero-WHO cells. (A to D) Each dot represents an independent experiment. Bars and error bars represent means and standard deviations (SDs) of the experiments, respectively. Statistics were done by use of Student’s t test (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ***, P ≤ 0.0001). NT, nontreated; ns, nonsignificant.
5-NT inhibits CHIKV infection in the first stages of the replication cycle.
To unravel the mode of action of 5-NT in controlling CHIKV infection, we first investigated the potential viricidal activity of 5-NT on CHIKV. To this end, CHIKV-LR 5′GFP virions were incubated with 5 μM 5-NT for 1.5 h, after which viral infectivity was measured by a plaque assay. In the plaque assay, the final end concentration of 5-NT was below 0.5 μM as the highest dilution used was 1:10. Incubation with 5-NT did not reduce viral infectivity, demonstrating that 5-NT does not have a direct negative effect on the infectivity of CHIKV particles (Fig. 2A). Thereafter, a time-of-addition experiment was performed to delineate where 5-NT acts in the replicative cycle. In these experiments, it is important to analyze the results within one round of replication, and we therefore first performed a growth curve analysis on U-2 OS cells. Figure 2B shows that initial GFP fluorescence was detected at 6 h p.i. (Fig. 2B, light gray bars), and robust infectious virus particle production was seen at 8 h p.i. (Fig. 2B, dark gray bars). To increase the sensitivity of the readout, we decided to analyze the effect at 10 h p.i., which still represents one round of replication. In the time-of-addition assay, cells were treated with 5-NT or DMSO for 1 h prior to infection (pre), during the adsorption of the virus (during), 1.5 h after adsorption (post), or with a combination of treatments (Fig. 2C). The results were normalized to the DMSO vehicle control. The strongest inhibition of infection was observed when 5-NT was present prior to and during virus adsorption (96% ± 1.2% reduction), which was comparable to treatment during the full course of infection (95% ± 2.6%) (Fig. 2D). There was also a clear reduction in viral infectivity when 5-NT was present prior to (71% ± 2.6%) or during (75% ± 1.2%) virus adsorption, yet this was significantly lower than that under complete treatment conditions. Only a 29% ± 8.7% reduction in infection was seen when 5-NT is added after adsorption of the virus. Collectively, these data suggest that 5-NT predominantly inhibits CHIKV infection during the early stages of the viral replication cycle.
FIG 2.

5-NT inhibits CHIKV infection early in the replication cycle. (A) CHIKV-LR 5′GFP was incubated for 1.5 h at 37°C in U-2 OS medium containing 2% fetal bovine serum (FBS) and 5 μM 5-NT or vehicle control DMSO in a final volume of 300 μl. After incubation, the infectious titer was determined by plaque assay in Vero-WHO cells. Three independent experiments were performed with one replicate per experiment. (B) Growth curve analysis of CHIKV infection. U-2 OS cells were infected with CHIKV-LR 5′GFP at an MOI of 5. Cells and supernatant were collected at 4, 6, 8, 10, and 12 h postinfection (h p.i.) to determine GFP-positive cells using flow cytometry (light gray bars) and infectious virus particle production using a plaque assay on Vero-WHO cells (dark gray bars). Dotted line represents the detection limit of the plaque assay. Two independent experiments were performed, each in duplicate. (C) Schematic representation of the time-of-addition assay. (D) U-2 OS cells were treated during the indicated times with vehicle control DMSO or with 5 μM 5-NT. Virus adsorption was allowed for 1.5 h, after which the inoculum was removed. U-2 OS cells were collected at 10 h p.i. and analyzed for GFP-positive cells using flow cytometry. Three independent experiments were performed with one replicate per experiment. The interpretation of each dot, bar, error bar, and statistic is explained in the legend to Fig. 1.
Cell entry bypass of the viral genome circumvents 5-NT antiviral activity.
To confirm that the serotonin receptor agonist predominantly inhibits CHIKV early in infection, we next evaluated the effect of 5-NT in an infection bypass experiment. To this end, cells pretreated with 5-NT or vehicle control DMSO were harvested and transfected with in vitro transcribed viral RNA by electroporation. Following electroporation, cells were incubated for 12 h in the presence of cell culture medium complemented with the compounds. Alternatively, cells were only exposed to 5-NT or DMSO after electroporation. A later harvesting time point was chosen to allow for cell recovery due to the electroporation procedure. Under these conditions, the infectious virus particle production was 6.6 ± 0.6 log PFU/ml in DMSO control cells. A comparable virus titer, 6.7 ± 0.2 log PFU/ml was detected when cells were solely pretreated with 5-NT. Also, no major effect in infectious virus particle production were seen in cells treated with 5-NT under postelectroporation (6.5 ± 0.6 log PFU/ml) and pre- and postelectroporation (6.3 ± 0.8 log PFU/ml) conditions (Fig. 3A). An inhibitory effect was, however, seen in the percentage of infected cells (Fig. 3B). It is important to note, however, that this result might be slightly biased, since we detected GFP fluorescence at 6 h p.i. under normal infection conditions and thus at 12 h p.i., we may have picked up two rounds of replication. The observed reduction in the number of infected cells may therefore be due to an inhibition in reinfection. Notably, in this experimental setup, we observed high viral titers, which is indicative of a high transfection efficiency. To validate if 5-NT still exhibits potent antiviral activity under these conditions, we next determined the inhibitory capacity of 5-NT following infection at a high MOI. As a control, we also determined the viral titer at 4 h p.i. to ensure that we properly removed the high concentration of virus inoculum and revealed a residual titer of 2.9 log, confirming that we predominantly detected progeny virions at 10 h p.i. Under standard infection conditions at an MOI of 60, a viral titer of 6.0 ± 0.1 log PFU was observed at 10 h p.i. (Fig. 3C). Importantly, under these conditions, we still observed a robust antiviral activity of 5-NT. The viral titer was 5.1 ± 0.3 log PFU, which corresponds to a 0.9-log reduction in infectivity in one round of replication (Fig. 3C). Together, these data confirm that 5-NT predominantly interferes with the early steps of the CHIKV replication cycle, before the viral RNA is released in the cell cytosol.
FIG 3.

The antiviral activity of 5-NT is before viral genome delivery. (A, B) U-2 OS cells were pretreated with 5-NT or vehicle control DMSO for 1 h before in vitro-transcribed viral RNA was transfected by electroporation. Cells were cultured for 12 h with or without 5-NT. (A) Supernatants were harvested, and virus particle production was determined with a plaque assay on Vero-WHO cells. Three independent experiments were performed, each in duplicate. (B) Cells were collected for analysis with flow cytometry for GFP-positive cells. Three independent experiments were performed, each in duplicate. (C) U-2 OS cells were pretreated with 5 μM 5-NT or vehicle control DMSO for 1 h before infection with CHIKV-LR 5′ GFP at an MOI of 60. Supernatants were harvested after 10 h p.i., and virus particle production was determined with a plaque assay on Vero-WHO cells. Four independent experiments were performed, each in duplicate. The interpretation of each dot, bar, error bar, and statistic is explained in the legend to Fig. 1.
5-NT does not affect CHIKV cell binding.
First, we assessed whether the binding capacity and internalization properties of CHIKV in U-2 OS cells is affected in the presence of 5-NT. Initially, we determined the interaction of CHIKV with the host cell surface by use of 35S-labeled CHIKV particles. To mimic the preadsorption and during-adsorption conditions of the time-of-addition experiments, cells were pretreated with 5-NT or vehicle control DMSO for 1 h, after which 1 × 105 dpm 35S-labeled virus (equivalent to ∼1.0 × 109 genome-equivalent copies [GECs] and 2.6 × 108 PFU) was added in cold medium in the presence or absence of 5-NT. Incubation was continued for 3 h at 4°C to maximize virus-cell binding. Under these conditions, internalization of virus particles is inhibited (28). Thereafter, cells were washed thoroughly to remove unbound virions and were harvested by trypsinization. Radioactivity was counted in the total volume by scintillation counting as a measure of virus-cell binding. We measured on average 1.27 × 104 and 1.33 × 104 dpm in the absence or presence of 5-NT, respectively (Fig. 4A). This indicates that 5-NT does not interfere with the virus-cell binding of 35S-labeled CHIKV.
FIG 4.

Binding of CHIKV is not altered in 5-NT treated cells. (A) Binding of CHIKV to U-2 OS cells was tested using 35S-labeled CHIKV particles. 35S-labeled CHIKV was incubated for 3 h at 4°C in the presence and absence of 5 μM 5-NT with U-2 OS cells. The cells were washed to remove unbound virus particles, harvested, and radioactivity was counted. Three independent experiments were performed, each in duplicate. (B) Binding of CHIKV particles in 5 μM 5-NT or vehicle control DMSO-treated U-2 OS cells. Incubation was done for 1.5 h at 37°C. The cells were washed to remove unbound virus particles and collected to determine the total number of viral GECs with specific primers against the viral genome. Three independent experiments were performed, each in duplicate. The interpretation of each dot, bar, error bar, and statistic is explained in the legend to Fig. 1.
To verify this finding, we next quantified the number of bound and/or internalized GECs by real-time quantitative reverse-transcription PCR (RT-qPCR). To this end, 5-NT- or control-treated cells were exposed to CHIKV (∼1.5 × 108 GECs, corresponding to an MOI of 5) at 37°C for 1.5 h to allow virus-cell binding and subsequent internalization. We used a shorter incubation time to limit the chance of detecting progeny viral RNA. Furthermore, RT-qPCR is more sensitive compared to the above approach. After 1.5 h, the cells were extensively washed to remove unbound particles and were directly lysed in the cell culture plate for RNA isolation and subsequent RT-qPCR analysis. In agreement with the data shown in Fig. 4A, we found no difference in total GECs bound and/or internalized between samples treated with 5-NT or vehicle control DMSO (Fig. 4B). Collectively, these results indicate that 5-NT does not interfere with CHIKV cell binding at the cell surface.
Virus internalization and membrane hemifusion are not affected upon 5-NT treatment.
Upon internalization, CHIKV traffics through the endosomal pathway toward early endosomes, where membrane fusion occurs (12). To assess whether 5-NT interferes with virus internalization and/or membrane hemifusion, we next applied a microscopic virus internalization/hemifusion assay using 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD)-labeled CHIKV particles (12, 29, 30). Here, membrane hemifusion is evident as an increase in fluorescent activity by dequenching of the DiD probe due to dilution within cellular membranes. Membrane hemifusion is a temporary stage prior to fusion pore formation in which the apposed leaflets of the viral membrane and the endosomal membrane have already merged, yet the inner leaflets of the lipid membranes are still intact (31). In this assay, the total extent of DiD fluorescence is thus taken as a measure of internalization/hemifusion. U-2 OS cells were pretreated with 5-NT or vehicle control DMSO for 1 h and challenged with DiD-labeled CHIKV particles for 20 min in the presence of 5-NT or DMSO. This time point was chosen because our previous studies revealed that 90% of all hemifusion events occur within the first 20 min postinfection (12). Figure 5A shows representative images for all treatment conditions. As a positive control, fusion-inactive diethyl pyrocarbonate (DEPC)-treated CHIKV was used (12). Quantification of the total DiD fluorescence intensity in 15 randomly selected images revealed that there are no differences in the extent of hemifusion between 5-NT and DMSO treatment conditions (Fig. 5B). Taken together, these results suggest that there is no effect of 5-NT on virus-cell entry and the membrane hemifusion capacity of CHIKV.
FIG 5.

5-NT does not interfere with the membrane fusion capacity of CHIKV. U-2 OS cells were pretreated with 5 μM 5-NT or with vehicle control DMSO for 1 h and incubated for 20 min with DiD-labeled CHIKV particles in the presence of 5-NT or DMSO. After washing to remove unbound virus particles, the extent of membrane hemifusion was measured in 15 randomly taken microscopic images per experiment. (A) Representative images showing the DiD signal in DMSO- and 5-NT-treated U-2 OS cells and for DEPC-inactivated CHIKV. White bar, 10 μm. (B) The total extent of fusion is normalized to that of the nontreated positive control (DMSO). Three independent experiments were performed, each in duplicate. The interpretation of each dot, bar, error bar, and statistic is explained in the legend to Fig. 1.
5-NT treatment does not inhibit fusion pore formation and RNA release from endosomes.
To investigate if 5-NT may act at the level of fusion pore formation and nucleocapsid/RNA delivery into the cell cytosol, we separated the cytosol from the endosomal membranes by cell fractionation and analyzed the location of the viral genome by RT-qPCR. To this end, U-2 OS cells were pretreated for 1 h with DMSO, 5-NT, or bafilomycin A1, an inhibitor of the vacuolar H+-ATPase required for membrane fusion. Subsequently, the cells were incubated with CHIKV at 37°C for 1.5 h in the presence and absence of the compound, after which the cells were washed thoroughly. Thereafter, the cells were permeabilized with 50 μM digitonin for 5 min at room temperature (RT), and cells were incubated for 30 min on ice to allow cytoplasmic proteins to diffuse into the supernatant. The supernatant (cytoplasmic fraction; indicative for nucleocapsid/RNA delivery) and extracted cells (endosomal membrane fraction; nonfused particles) were collected for RNA isolation, and the number of GECs was assessed. The results of another study performed by us imply that digitonin treatment does not disrupt the hemifusion intermediate, indicating that the cytosolic fraction only contains RNA from particles that induced complete membrane fusion (32). In addition, we subjected both cellular fractions to SDS-PAGE and Western blotting to verify the efficiency of fractionation (Fig. 6A). Here, we used GAPDH as a marker for the cytoplasmic fraction, and the endosomal markers EEA1 and Rab5 were used for the membrane fraction. Fractionation was very efficient, with 82% ± 1.4% of GAPDH and 77% ± 9.7% of Rab5 ending up in the cytoplasmic and membrane fraction, respectively, based on three independent experiments. Subsequent quantification of the GECs in the membrane and cytoplasmic fraction revealed that bafilomycin A1 treatment abolished RNA delivery into the cytosolic fraction by 0.92 ± 0.04-fold (Fig. 6B). Importantly, 5-NT treatment did not interfere with RNA delivery, as comparable GEC levels were found in the cytosolic fraction of control-treated cells. In conclusion, 5-NT does not inhibit membrane fusion, and the nucleocapsid/RNA is efficiently released from the endosomal membranes into the cytoplasm.
FIG 6.

Genomic RNA delivery to cytoplasm upon 5-NT treatment. Pretreated U-2 OS cells with vehicle control DMSO, 5-NT, or bafilomycin A1 (BafA1) were infected with CHIKV-LR 2006 OPY1 at an MOI of 5 for 1.5 h at 37°C. The cells were washed and permeabilized using 50 μg/ml digitonin for 5 min at RT, followed by 30 min on ice. Separation of the cytoplasmic fraction from the membrane fraction was assessed by Western blot analysis. Subsequently, the total number of GECs was determined in the cytoplasmic and the membrane fraction. (A) Representative Western blot image of the separation. In total, three independent experiments were performed. Shown is GAPDH, which represents the cytoplasmic fraction (C), and EEA1 and Rab5, which represent the membrane fraction (M) from the extracted cells. (B) Total number of viral GECs was assessed by RT-qPCR after Western blot confirmation of successful fractionation. The fold change of cytoplasmic fraction of total gRNA copies is shown. Three independent experiments were performed, each in duplicate. The interpretation of each dot, bar, error bar, and statistic is explained in the legend to Fig. 1.
5-HT receptor antagonist inhibits CHIKV via a different route than that of 5-NT.
The above data show that 5-NT does not interfere with the initial stages of CHIKV cell entry, which is in contrast to what has been previously described for reovirus (22). In their work, the authors also used methiothepin mesylate (MM), which is a 5-HT receptor antagonist blocking 5HT1, 5HT6, and 5HT7 receptors (33, 34) and showed that MM enhanced reovirus infectivity. In an attempt to better understand the above differences, we next investigated the role of MM in CHIKV infectivity in U-2 OS cells. First, we assessed the cellular cytotoxicity of MM in U-2 OS cells and revealed that MM is nontoxic to the cells at the concentrations used in this study (Fig. 7A). Intriguingly, and in contrast to data described for reovirus, we found a clear dose-dependent reduction in the number of CHIKV-infected cells, with 97% ± 1.0% inhibition at 10 μM MM (Fig. 7B). Due to these contrasting data, we also measured the effect of another 5-HT1A/1B receptor antagonist, isamoltane, on CHIKV infectivity. We chose isamoltane because it has been shown to antagonize signaling pathways downstream of 5-NT stimulation at concentrations ranging from 0.01 to 10μM (35). Our results show that isamoltane at amounts in this concentration range is nontoxic to cells (Fig. 7C) and does not affect CHIKV infectivity (Fig. 7D). This also suggests that the inhibitory effect of MM is independent of 5-HT1A/1B receptor signaling. Importantly, the above data demonstrate that 5-HT agonist and antagonists do not have opposing effects on CHIKV infectivity; rather, 5-NT and MM both appear to act as host-directed antivirals. Given the antiviral role of MM, we next investigated how it interferes with CHIKV infection using similar methods to those described above for 5-NT. Time-of-addition experiments revealed that the strongest inhibitory effect (87% ± 5.9% reduction) on CHIKV infection is seen when MM is present before and during virus adsorption (Fig. 7E). In contrast to results with 5-NT, pretreatment with MM alone barely inhibited CHIKV infection (20% ± 5.5% reduction), indicating that MM needs to be present during CHIKV adsorption to exert its effect. Indeed, MM treatment during CHIKV adsorption resulted in a reduction of 70% ± 3.7%. Lastly, 30% ± 7.6% reduction was seen when MM was added under postadsorption conditions. Collectively, the results show that MM, like 5-NT, predominantly interferes with the early steps in infection. Therefore, we next assessed the capacity of CHIKV to bind cells in the presence of MM. In line with the results obtained for 5-NT, no differences in virus-cell binding were observed in the presence or absence of MM (Fig. 8A and B). Notably, however, the presence of MM did reduce the total extent of hemifusion activity by 54% ± 11% compared to that in the nontreated control, suggesting that MM is likely to interfere with internalization and/or membrane hemifusion activity of the virus (Fig. 8C). The extent of fusion was comparable to that of cells treated with NH4CL (Fig. 8C). In line with these results, subsequent cellular fraction experiments revealed that the levels of cytosolic genomic RNA (gRNA) are reduced 0.38 ± 0.29-fold compared to those of the control (Fig. 8D). Collectively, these data clearly indicate that 5-NT and MM exert different mechanisms for their antiviral activity in CHIKV replication.
FIG 7.

MM inhibits CHIKV infection early in CHIKV infection. (A) MTT assay to determine the cytotoxicity of MM to U-2 OS cells. Cells were treated for 21 h in the presence of increasing concentrations of the inhibitor to mimic conditions during the infectivity assay. Dotted line indicates 75% survival rate. Three independent experiments were performed, each in sextuplicate. (B, D) U-2 OS cells were pretreated for 1 h with 75 mM NH4Cl or with increasing concentrations of (B) MM or (D) isamoltane and subsequently challenged with CHIKV-LR 5′GFP at an MOI of 5 for 20 h. U-2 OS cells were collected and analyzed with flow cytometry for GFP-positive cells. Three independent experiments were performed with one replicate per experiment. (C) ATPlite assay to determine the cytotoxicity of isamoltane to U-2 OS cells. Cells were treated for 21 h in the presence of increasing concentrations of the inhibitor to mimic conditions during the infectivity assay. Dotted line indicates 75% survival rate. Three independent experiments were performed, each in duplicate. (E) U-2 OS cells were treated until the time points indicated in Fig. 2C with or without 10 μM MM. Virus adsorption was allowed for 1.5 h, after which the inoculum was removed. U-2 OS cells were collected at 10 h p.i. and analyzed with flow cytometry for GFP-positive cells. Three independent experiments were performed with one replicate per experiment. The interpretation of each dot, bar, error bar, and statistic is explained in the legend to Fig. 1.
FIG 8.
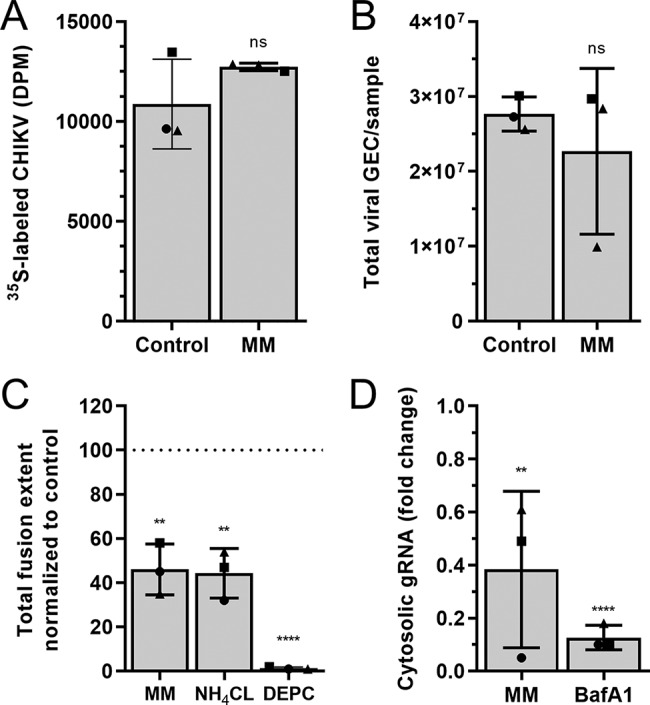
MM inhibits CHIKV infection by restricting membrane fusion activity. (A) Binding of CHIKV to U-2 OS cells was tested using 35S-labeled CHIKV. Radioactive labeled virus was incubated for 3 h at 4°C with U-2 OS cells pretreated for 1 h with or without 10 μM MM. The cells were washed to remove unbound virus particles and harvested by trypsinization. The trypsinized suspension was subjected to scintillation counting. Three independent experiments were performed, each in duplicate. (B) Binding and internalization of CHIKV particles. U-2 OS cells pretreated with or without 10 μM MM for 1 h were infected with CHIKV-LR for 1.5 h at 37°C. The cells were washed, and the total number of viral GECs was assessed with specific primers against the viral genome. Three independent experiments were performed, each in duplicate. (C) U-2 OS cells were pretreated with 10 μM MM or 50 mM NH4Cl for 1 h and incubated for 20 min with DiD-labeled CHIKV particles in the presence of the compounds. DEPC-inactivated CHIKV was used as a negative control. After washing to remove unbound virus particles, the extent of membrane hemifusion was measured in 15 randomly taken microscopic images per experiment. The total extent of fusion was normalized to that of the nontreated control (DMSO). Three independent experiments were performed, each in duplicate. (D) gRNA delivery to the cytoplasm upon MM treatment. Total number of viral GECs was assessed by RT-qPCR. Fold change of cytoplasmic fraction of total gRNA copies is shown. Three independent experiments were performed, each in duplicate. The interpretation of each dot, bar, error bar, and statistic is explained in the legend to Fig. 1.
DISCUSSION
In this study, we aimed to understand the efficacy and mode of action of serotonergic drugs in CHIKV infection. We focused on the 5-HT receptor agonist 5-NT and 5-HT antagonists, MM, and isamoltane. Intriguingly, we observed a strong antiviral effect for both 5-NT and MM on CHIKV infection, whereas no effect was seen for isamoltane. We showed that 5-NT and MM interfere with distinct steps in the replication cycle of CHIKV.
Addition of 5-NT to cells led to a stark reduction in the number of infected cells and lowered the secretion of progeny virions. Detailed analysis of steps of the replication cycle revealed that 5-NT did not interfere with CHIKV attachment, internalization, hemifusion activity, and gRNA delivery to the cell cytosol. Interestingly, however, we also observed that upon transfection of RNA transcripts in 5-NT-treated cells, the antiviral activity of 5-NT is almost completely diminished. We have two possible explanations for these intriguing findings. First, even though we did not notice an effect on gRNA delivery to the cell cytosol, we do not know whether the gRNA is still part of the nucleocapsid or not. Thus, based on our findings we hypothesize that 5-NT interferes with nucleocapsid uncoating, thereby reducing the chance to productively infect a cell. The process of nucleocapsid uncoating is currently ill understood, and early data suggest that ribosomes are involved in this process (36). However, it has been speculated that as-yet-unknown host factors might further contribute to nucleocapsid uncoating (36, 37). Indeed, more recent evidence suggest that ubiquitination and cytoskeleton-associated motor proteins are important for nucleocapsid disassembly in dengue virus, HIV-1, and influenza type A virus infections (38–42). Alternatively, 5-NT stimulation of 5-HT receptors may affect the transport/cellular location of CHIKV-containing endosomes, thereby releasing the nucleocapsid at sites that do not support efficient translation and replication of the genome. Indeed, Mainou and colleagues showed that 5-NT treatment did alter the distribution of Rab5 endosomes in CCL2 HeLa cells (24).
In this study, we also demonstrate that MM behaves as a strong antiviral compound and predominantly controls infectivity after virus-cell binding but prior to fusion pore formation and gRNA delivery. Although MM is mainly reported as an antagonist of the 5-HT1b receptor, it also has nonselective properties and can bind to several other receptor subtypes, including 5-HT6/7 receptors (33, 43, 44). For example, MM has been described to function as an inverse agonist inducing desensitization of forskolin-stimulated cAMP formation in cells overexpressing the 5-HT7 receptor (43–45). The lack of antiviral activity of isamoltane strengthens the notion that CHIKV infectivity is not controlled by antagonizing the 5-HT1B receptor. MM and isamoltane are both described as 5-HT1B receptor antagonists, yet have distinct alternative effects. For example, isamoltane and MM have been shown to act differentially from the forskolin-induced cAMP formation in renal epithelial cells (46). Future research is required to delineate the precise functions of MM in cells and how MM controls CHIKV internalization and/or the process of membrane hemifusion.
Many chemical compound library screen studies have revealed that agonist and antagonist serotonergic drugs can interfere with viral infections (22, 23, 47, 48). For many of these compounds, the mechanism of action remains unclear, but many seem to act on cell entry of viruses. For example, in hepatitis C virus infection, 5-HT2 receptor antagonists inhibit cell entry at a late endocytic stage. This has been linked to alterations in the protein kinase A (PKA) pathway that interfere with claudin 1, an important receptor for postbinding steps of hepatitis C virus cell entry (17, 47). For JC polyomavirus, 5-HT2 receptor antagonists inhibit infection due to interference with binding of β-arrestin to the 5-HT2A receptors, which is required for internalization of the virus via clathrin-coated vesicles (20, 49, 50). During reovirus infection, 5-NT strongly inhibits the cell entry of reovirus, whereas MM enhances reovirus infectivity. This is contradictory to what we observed for CHIKV, and this is likely related to differences in the virus cell entry process between both viruses. Reovirus particles traffic toward late endosomes, where cathepsin-mediated proteolysis is required for efficient infection, whereas CHIKV fusion is solely dependent on low pH and is triggered from within early endosomes (51). Thus, these serotonergic drugs may regulate a host factor that is beneficial for one virus and inhibitory for the other.
The widespread abundance of serotonin receptors in the periphery and the potent effect of serotonergic drugs on CHIKV infectivity described in this study suggest that targeting 5-HT receptors might be an interesting approach to alleviate CHIKV disease. Pharmacological targeting of specific 5-HT receptors is, however, challenging due to the various roles of these receptors in multiple parts of the body. To minimize the chance of side effects, it is probably best to use combination treatments with low concentrations of multiple serotonergic drugs acting on different stages of infection. This will also further reduce the chance of developing resistance to the treatment. Future studies should be performed to investigate the in vivo efficacy of single and combination serotonergic drug treatments on CHIKV infection in mice.
MATERIALS AND METHODS
Cells, compounds, and cell viability.
Human bone osteosarcoma epithelial U-2 OS cells (a gift from the Department of Cell Biology, University Medical Center Groningen, Groningen, The Netherlands) were maintained in high-glucose Dulbecco’s modified Eagle medium (DMEM; Gibco, the Netherlands) with GlutaMax supplemented with 10% fetal bovine serum (FBS) (Life Science Production, Barnet, UK). Green monkey kidney Vero-WHO cells (European Collection of Cell Cultures accession no. 88020401) were cultured in DMEM supplemented with 5% FBS. Baby hamster kidney cells (BHK-21 cells; ATCC CCL-10) were cultured in RPMI medium (Gibco) supplemented with 10% FBS. All media were supplemented with penicillin (100 U/ml) and streptomycin (100 U/ml) (Gibco). All cells were tested as Mycoplasma negative and were maintained at 37°C under 5% CO2.
Ammonium chloride (NH4Cl; Merck, Darmstadt, Germany) was diluted to a 1 M stock concentration in H2O. Bafilomycin A1 was diluted to a 200 mM stock in dimethyl sulfoxide (DMSO). 5-Nonyloxytryptamine oxalate (5-NT; Tocris, Bristol, United Kingdom) was diluted to a 5 mM stock concentration in DMSO (Merck). Methiothepin mesylate (MM) (Tocris) was diluted to a 10 mM stock concentration in H2O. All chemicals were stored according to the respective manufacturer’s instructions.
Cytotoxicity of the compounds were tested by use of an MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay (Merck) at a final MTT concentration of 0.45 mg/ml or by use of a ATPlite luminescence assay system (PerkinElmer, Waltham, MA) according to the manufacturer’s instructions.
RT-qPCR of serotonin receptors.
RNA was isolated from U-2 OS cells with the RNeasy minikit (Qiagen, Hilden, Germany). RNA (0.5 μg) was reverse transcribed into cDNA using the PrimeScript RT reagent kit (TaKaRa, Kusatsu, Japan). Real-time qPCR was conducted on a StepOnePlus real-time PCR system (Applied Biosystems) using specific primers (Table 1) (Eurogentec, Seraing, Belgium), SYBR green reagents, and ROX reference dye (Thermo Scientific, Waltham, MA). The cDNA was diluted 1:10 for the amplification with GAPDH-specific primers. Data were analyzed using StepOne v2.3 software.
TABLE 1.
List of primers used for 5-HT receptor genes
| Target gene | Product size (bp) | Forward primer | Reverse primer |
|---|---|---|---|
| 5-HTR1A | 132 | GGTAACCTGCGACCTGTTCA | GGCGTCCTCTTGTTCACGTA |
| 5-HTR1B | 101 | GGTAACCTGCGACCTGTTCA | GCATCACCAGGATGGACACA |
| 5-HTR1D | 135 | GGGCCTTCTACATTCCCTCG | CAGAGCCTGTGATGAGGTGG |
| 5-HTR2A | 155 | ACTGTGAGAGATGCAGCGAG | TTCTCACCAAACCGAGGACAAA |
| 5-HTR2B | 85 | ACAGCAGCAAGCAAGTCTAGT | CATGCCAGAGAGTTCCCCCT |
| 5-HTR2C | 162 | GGCCAGCACTTTCAATCGTC | GGGGCATGACAAGTAGTCCC |
| 5-HTR3 | 110 | CCCTGGTTATGCTCTGGTCC | GGGGCCTAAGCAGAAATCCT |
| 5-HTR4 | 125 | ATGGTGCTGGCCTATTACCG | TGAGTGCTATGCTGGTCTGC |
| 5-HTR6 | 131 | CAGATTCGGACTCAGACGCA | GAAGAAATTGACGGCGGCAG |
| 5-HTR7 | 149 | TGGTGATCTCCGTGTGCTTC | TCCAAAGATCCACTTGCCCC |
| GAPDH | 75 | TTGCCCTCAACGACCACTTT | CCACCACCCTGTTGCTGTAG |
Virus production, purification, and quantification.
The infectious clone based on CHIKV strain La Reunion (CHIKV-LR) 2006 OPY1 was kindly provided by Andres Merits (University of Tartu, Tartu, Estonia). CHIKV-LR 5′GFP was kindly provided by the European Virus Archive (EVA; Marseille, France). GFP was cloned after a second subgenomic promoter 5′ to the structural genes (52). Virus production was done as described previously (12, 53). Briefly, BHK-21 cells were transfected with in vitro-transcribed RNA transcripts by electroporation with a Gene Pulser Xcell system (1.5 kV, 25 μF, and 200 Ω; Bio-Rad, Hercules, CA). At 22 h posttransfection, the supernatant was harvested (p0) and used to inoculate Vero-WHO cells at a multiplicity of infection (MOI) of 0.01 (p1) to generate working stocks.
Purified virus was prepared by inoculating monolayers of BHK-21 cells with CHIKV-LR (p0) at an MOI of 4. At 25 h postinfection (h p.i.), the supernatant was harvested and cleared from cell debris by low-speed centrifugation. Subsequently, the virus particles were pelleted by ultracentrifugation in a Beckman type 19 rotor (Beckman Coulter, Brea, CA) at 54,000 × g for 2.5 h at 4°C. The pellet was resuspended overnight in HNE buffer (5 mM HEPES [Gibco], 150 mM NaCl [Merck], and 0.1 mM EDTA [pH 7.4] [Merck]) before it was purified by ultracentrifugation on a sucrose density gradient (20% to 50% [wt/vol] sucrose in HNE) in a Beckman SW41 rotor at 75,000 × g for 18 h at 4°C. Upon centrifugation, the virus particles were in the 40% to 45% sucrose layer, which was harvested and aliquoted before storage at −80°C.
L-[35S]methionine/L-[35S]cysteine-labeled CHIKV was produced by inoculation of a confluent monolayer of BHK-21 cells with CHIKV-LR (p0) at an MOI of 10. At 2.5 h p.i., the BHK-21 cells were starved for 1.5 h with DMEM without cysteine/methionine (Gibco) at 37°C. Next, [35S]-EasyTag Express protein labeling mix (PerkinElmer) was added, and the cells were incubated overnight at 37°C. The medium was harvested, and cell debris were removed with low-speed centrifugation. Purification was done by ultracentrifugation for 2 h at 154,000 × g at 4°C in an SW41 rotor (Beckman) using a two-step sucrose gradient (20%/50% [wt/vol] in HNE). Radioactive virus was collected at the 20%/50% sucrose interface, and radioactivity was counted by liquid scintillation analysis. Fractions were pooled based on radioactivity counts and stored at −80°C.
The infectious virus titers of all virus preparations were determined with a plaque assay in Vero-WHO cells. Additionally, the number of genome equivalent copies (GECs) was determined by RT-qPCR, as described previously (11).
Flow cytometry analysis.
Flow cytometry analysis was used to determine the number of infected cells. U-2 OS cells were washed and preincubated for 1 h with or without compounds diluted in U-2 OS medium containing 2% FBS. Thereafter, CHIKV-LR 5′GFP was added to the cells at the indicated MOI. At 1.5 h p.i., inoculum was removed, and fresh U-2 OS medium containing 10% FBS was added in the presence or absence of the compound and incubated for a specified time point at 37°C under 5% CO2. Upon collection, cells were washed and fixed with 4% paraformaldehyde (PFA; Alfa Aesar, Haverhill, MA) and analyzed by flow cytometry. Flow cytometry was performed with a FACSVerse instrument (BD Biosciences, Franklin Lakes, NJ) and analyzed with FlowJo vX.0.7.
Viricidal assay.
CHIKV-LR 5′GFP was incubated for 1.5 h at 37°C in U-2 OS medium containing 2% FBS and 5 μM 5-NT or DMSO in a final volume of 300 μl. After incubation, the infectious titer was determined by plaque assay on Vero-WHO cells.
Binding assay with 35S-labeled CHIKV.
U-2 OS cells were seeded to 80% confluence in a 12-well plate and washed twice with HNE supplemented with 0.5 mM CaCl2 (Merck), 0.5 mM MgCl2 (Merck), and 1% FBS (HNE+). Cells were incubated with HNE+ supplemented with the compounds of interest or with vehicle control for 45 min at 37°C and subsequently for 15 min at 4°C. Next, 1 × 105 dpm 35S-labeled CHIKV (2.6 × 108 PFU, corresponding to an MOI of 500) diluted in HNE+ was added to the cells and incubated for 3 h at 4°C to allow virus-cell binding. Unbound virus was removed by washing two times with HNE+. The cells were harvested by trypsinization, and the total volume was subjected to liquid scintillation analysis to count radioactivity.
Binding and internalization assay by RT-qPCR.
U-2 OS cells were seeded to 80% confluence in a 24-well plate and washed three times with HNE+ before incubation with HNE+ supplemented with the compounds of interest or with vehicle control DMSO for 1 h at 37°C. Next, CHIKV-LR was added to the cells at an MOI of 5 and incubated at 37°C for 1.5 h. Thereafter, unbound virus was removed by washing three times with PBS (Life Technologies, Carlsbad, CA). Next, cells were lysed with the RNAeasy minikit (Qiagen) according to the manufacturer’s instructions, and the number of GECs was determined as described before (11). In addition to this protocol, single-stranded RNA was degraded after cDNA synthesis by RNase A (Thermo Scientific).
Microscopic fusion assay.
For the microscopic fusion assay, purified CHIKV particles were labeled with the lipophilic fluorescent probe 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD) (Life Technologies), as described before (12). U-2 OS cells were cultured to 80% confluence in Nunc 8-well Lab-Tek II Chambered Coverglass slides (Thermo Scientific). Upon infection, the cells were washed three times with serum-free, phenol red-free minimal essential medium (MEM; Gibco) medium and incubated with phenol red-free MEM supplemented with 1% glucose (Merck) and the compounds of interest. After 1 h of treatment, DiD-labeled CHIKV (MOI, ∼10) was added to the cells and incubated at 37°C for 20 min to allow virus cell entry and membrane fusion. Subsequently, unbound particles were removed by washing three times with serum-free, phenol red-free MEM, after which fresh phenol-red free MEM supplemented with 1% glucose was added. Image fields were randomly selected using differential interference contrast (DIC), and 15 snapshots were taken per experiment in both the DIC and DiD channels with a 6000B instrument (Leica Biosystems, Amsterdam, The Netherlands). All snapshots were analyzed for the total area of fluorescent spots, which was quantified using the ParticleAnalyzer plugin of ImageJ. Total fluorescent area was averaged per experiment and normalized to the total fluorescent area of the vehicle control DMSO.
Cell entry bypass assay.
In vitro-transcribed RNA derived from the infectious clone CHIKV-LR was electroporated in 1 × 107 U-2 OS cells treated beforehand with 5 μM 5-NT or DMSO for 1 h, using a Gene Pulser Xcell system (250V, 95 μF, and 186 Ω). After electroporation, the cells were seeded into a 12-well plate and incubated in medium containing either 5-NT at an end concentration of 5 μM or the vehicle control DMSO for 12 h at 37°C. Cell supernatants were harvested and analyzed for infectious particle production with a plaque assay on Vero-WHO cells. Additionally, the transfected cells were harvested and prepared for flow cytometry analysis. To this end, cells were fixed with 4% PFA, permeabilized, and stained with a rabbit anti-E2-stem antibody (1:1,000; obtained from G. Pijlman, Wageningen University, Wageningen, The Netherlands) and Alexa Fluor 647-conjugated chicken anti-rabbit antibody (1:300; Life Technologies).
Digitonin-based cell fractionation.
Cell fractionation of U-2 OS cells was performed as described previously (54). Briefly, the cells were seeded to 80% confluence in a 12-well plate, washed 3 times with HNE+, and incubated with HNE+ supplemented with the compounds of interest or with vehicle control DMSO for 1 h at 370C. CHIKV-LR was added to the cells at an MOI of 5 and incubated at 37°C for 1.5 h, after which the inoculum was removed. Cells were first washed with PBS, treated for 2 min with a high-salt, high-pH buffer, i.e., 1 M NaCl in H2O (pH 9.5), and then washed another three times with PBS. Next, cells were permeabilized by incubation with 50 μg/ml digitonin dissolved in PBS (Sigma-Aldrich, St. Louis, MS) for 5 min at RT and subsequently for 30 min on ice. Directly after this incubation step, the supernatant was carefully collected to obtain the cytosolic fraction. Thereafter, the adherent but permeabilized cells, representing the membrane fraction, were collected. RNA was isolated using a viral RNA kit and the RNAeasy minikit for the cytosolic and the membrane fraction, respectively, according to the manufacturer’s instructions. The number of GECs was determined as described before (11). In addition to this protocol, single-stranded RNA was degraded after cDNA synthesis by RNase A. Additionally, Western blot analysis was performed to verify the fractionation step. To this end, the fractions were diluted in 4× SDS sample buffer (Merck) and heated at 95°C for 5 min prior to fractionation by SDS-PAGE. The antibodies used were mouse-anti-EEA1 (1:5,000; BD Biosciences), mouse-anti-GAPDH (1:10,0000; Abcam, Cambridge, UK), and rabbit-anti-Rab5 (1:1,000; Abcam). Secondary horseradish peroxidase (HRP)-conjugated antibodies, either anti-mouse or anti-rabbit (Thermo Fisher Scientific), were used as recommended by manufacturer. Quantification was done in ImageQuant TL.
Statistical analysis.
All data were analyzed in GraphPad Prism software. Data are presented as mean ± standard deviation (SD) unless otherwise indicated. Student’s t test was used to evaluate statistical differences. A P value of ≤0.05 was considered significant; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ***, P ≤ 0.0001. EC50, the concentration at which 5-NT reduces virus particle production by 50%, was determined by a dose-response curve that was fitted by nonlinear regression analysis employing a sigmoidal model.
ACKNOWLEDGMENTS
This work was supported by the Graduate School of Medical Sciences of the University of Groningen and by a research grant from De Cock-Hadders Stichting of the University of Groningen (grant to E.M.B.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Weaver SC, Lecuit M. 2015. Chikungunya virus and the global spread of a mosquito-borne disease. N Engl J Med 372:1231–1239. doi: 10.1056/NEJMra1406035. [DOI] [PubMed] [Google Scholar]
- 2.Burt FJ, Rolph MS, Rulli NE, Mahalingam S, Heise MT. 2012. Chikungunya: a re-emerging virus. Lancet 379:662–671. doi: 10.1016/S0140-6736(11)60281-X. [DOI] [PubMed] [Google Scholar]
- 3.Suhrbier A, Jaffar-Bandjee M-C, Gasque P. 2012. Arthritogenic alphaviruses—an overview. Nat Rev Rheumatol 8:420–429. doi: 10.1038/nrrheum.2012.64. [DOI] [PubMed] [Google Scholar]
- 4.Rodríguez-Morales AJ, Cardona-Ospina JA, Fernanda Urbano-Garzón S, Sebastian Hurtado-Zapata J. 2016. Prevalence of post-chikungunya infection chronic inflammatory arthritis: a systematic review and meta-analysis. Arthritis Care Res (Hoboken) 68:1849–1858. doi: 10.1002/acr.22900. [DOI] [PubMed] [Google Scholar]
- 5.Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. 2018. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discov 17:35–56. doi: 10.1038/nrd.2017.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka A, Tumkosit U, Nakamura S, Motooka D, Kishishita N, Priengprom T, Sa-ngasang A, Kinoshita T, Takeda N, Maeda Y. 2017. Genome-wide screening uncovers the significance of N-sulfation of heparan sulfate as a host cell factor for chikungunya virus infection. J Virol 91:1–22. doi: 10.1128/JVI.00432-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karlas A, Berre S, Couderc T, Varjak M, Braun P, Meyer M, Gangneux N, Karo-Astover L, Weege F, Raftery M, Schönrich G, Klemm U, Wurzlbauer A, Bracher F, Merits A, Meyer TF, Lecuit M. 2016. A human genome-wide loss-of-function screen identifies effective chikungunya antiviral drugs. Nat Commun 7:11320. doi: 10.1038/ncomms11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Clercq E, Li G. 2016. Approved antiviral drugs over the past 50 years. Clin Microbiol Rev 29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ching K-C, F P Ng L, Chai C. 2017. A compendium of small molecule direct-acting and host-targeting inhibitors as therapies against alphaviruses. J Antimicrob Chemother 72:2973–2989. doi: 10.1093/jac/dkx224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang R, Kim AS, Fox JM, Nair S, Basore K, Klimstra WB, Rimkunas R, Fong RH, Lin H, Poddar S, Crowe JE, Doranz BJ, Fremont DH, Diamond MS. 2018. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557:570–574. doi: 10.1038/s41586-018-0121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Duijl-Richter MKS, Blijleven JS, van Oijen AM, Smit JM. 2015. Chikungunya virus fusion properties elucidated by single-particle and bulk approaches. J Gen Virol 96:2122–2132. doi: 10.1099/vir.0.000144. [DOI] [PubMed] [Google Scholar]
- 12.Hoornweg TE, van Duijl-Richter MKS, Ayala Nuñez NV, Albulescu IC, van Hemert MJ, Smit JM. 2016. Dynamics of chikungunya virus cell entry unraveled by single-virus tracking in living cells. J Virol 90:4745–4756. doi: 10.1128/JVI.03184-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solignat M, Gay B, Higgs S, Briant L, Devaux C. 2009. Replication cycle of chikungunya: a re-emerging arbovirus. Virology 393:183–197. doi: 10.1016/j.virol.2009.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silva LA, Dermody TS. 2017. Chikungunya virus: epidemiology, replication, disease mechanisms, and prospective intervention strategies. J Clin Invest. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sodhi A, Montaner S, Gutkind JS. 2004. Viral hijacking of G-protein-coupled-receptor signalling networks. Nat Rev Mol Cell Biol 5:998–1012. doi: 10.1038/nrm1529. [DOI] [PubMed] [Google Scholar]
- 16.Zhang N, Huang H, Tan B, Wei Y, Xiong Q, Yan Y, Hou L, Wu N, Siwko S, Cimarelli A, Xu J, Han H, Qian M, Liu M, Du B. 2017. Leucine-rich repeat-containing G protein-coupled receptor 4 facilitates vesicular stomatitis virus infection by binding vesicular stomatitis virus glycoprotein. J Biol Chem 292:16527–16538. doi: 10.1074/jbc.M117.802090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao L, Chen J, Wang Y, Yang Y, Qing J, Rao Z, Chen X, Lou Z. 2019. Identification of serotonin 2A receptor as a novel HCV entry factor by a chemical biology strategy. Protein Cell 10:178–195. doi: 10.1007/s13238-018-0521-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duman RS. 1998. Novel therapeutic approaches beyond the serotonin receptor. Biol Psychiatry 44:324–335. doi: 10.1016/s0006-3223(98)00031-6. [DOI] [PubMed] [Google Scholar]
- 19.Hannon J, Hoyer D. 2008. Molecular biology of 5-HT receptors. Behav Brain Res 195:198–213. doi: 10.1016/j.bbr.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 20.Assetta B, Maginnis MS, Gracia Ahufinger I, Haley SA, Gee GV, Nelson CDS, O’Hara BA, Allen Ramdial SA, Atwood WJ. 2013. 5-HT2 Receptors Facilitate JC Polyomavirus Entry. J Virol 87:13490–13498. doi: 10.1128/JVI.02252-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manéglier B, Guillemin GJ, Clayette P, Rogez-Kreuz C, Brew BJ, Dormont D, Advenier C, Therond P, Spreux-Varoquaux O. 2008. Serotonin decreases HIV-1 replication in primary cultures of human macrophages through 5-HT(1A) receptors. Br J Pharmacol 154:174–182. doi: 10.1038/bjp.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng H, Lear-Rooney CM, Johansen L, Varhegyi E, Chen ZW, Olinger GG, Rong L. 2015. Inhibition of Ebola and Marburg virus entry by G protein-coupled receptor antagonists. J Virol 89:9932–9938. doi: 10.1128/JVI.01337-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashbrook AW, Lentscher AJ, Zamora PF, Silva LA, May NA, Bauer JA, Morrison TE, Dermody TS. 2016. Antagonism of the sodium-potassium ATPase impairs chikungunya virus infection. mBio 7:1–14. doi: 10.1128/mBio.00693-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mainou BA, Ashbrook AW, Smith EC, Dorset DC, Denison MR, Dermody TS. 2015. Serotonin receptor agonist 5-nonyloxytryptamine alters the kinetics of reovirus cell entry. J Virol 89:e00739-15. doi: 10.1128/JVI.00739-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glennon RA, Hong SS, Dukat M, Teitler M, Davis K. 1994. 5-(Nonyloxy)tryptamine: a novel high-affinity 5-HT1D beta serotonin receptor agonist. J Med Chem 37:2828–2830. doi: 10.1021/jm00044a001. [DOI] [PubMed] [Google Scholar]
- 26.van Duijl-Richter MKS, Hoornweg TE, Rodenhuis-Zybert IA, Smit JM. 2015. Early events in chikungunya virus infection—from virus cell binding to membrane fusion. Viruses 7:3647–3674. doi: 10.3390/v7072792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen W, Foo S-S, Rulli NE, Taylor A, Sheng K-C, Herrero LJ, Herring BL, Lidbury BA, Li RW, Walsh NC, Sims NA, Smith PN, Mahalingam S. 2014. Arthritogenic alphaviral infection perturbs osteoblast function and triggers pathologic bone loss. Proc Natl Acad Sci U S A 111:6040–6045. doi: 10.1073/pnas.1318859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang G, Hernandez R, Weninger K, Brown DT. 2007. Infection of cells by Sindbis virus at low temperature. Virology 362:461–467. doi: 10.1016/j.virol.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 29.Ayala-Nunez NV, Hoornweg TE, van de Pol DPI, Sjollema KA, Flipse J, van der Schaar HM, Smit JM. 2016. How antibodies alter the cell entry pathway of dengue virus particles in macrophages. Sci Rep 6:28768. doi: 10.1038/srep28768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ayala-Nuñez NV, Wilschut J, Smit JM. 2011. Monitoring virus entry into living cells using DiD-labeled dengue virus particles. Methods 55:137–143. doi: 10.1016/j.ymeth.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Harrison SC. 2015. Viral membrane fusion. Virology 479–480:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoornweg TE, Bouma EM, van de Pol DPI, Rodenhuis-Zybert IA, Smit JM. 2020. Chikungunya virus requires an intact microtubule network for efficient viral genome delivery. bioRxiv 10.1101/2020.03.24.004820. [DOI] [PMC free article] [PubMed]
- 33.Jin H, Oksenberg D, Ashkenazi A, Peroutka SJ, Duncan AMV, Rozmahel R, Yang Y, Mengod G, Palacios JM, O’Dowd BF. 1992. Characterization of the human 5-hydroxytryptamine(1B) receptor. J Biol Chem 267:5735–5738. [PubMed] [Google Scholar]
- 34.Bard JA, Zgombick J, Adham N, Vaysse P, Branchek TA, Weinshank RL. 1993. Cloning of a novel human serotonin receptor (5-HT 7) positively linked to adenylate cyclase. J Biol Chem 268:23422–23426. [PubMed] [Google Scholar]
- 35.McDuffie JE, Motley ED, Limbird EL, Maleque MA. 2000. 5-Hydroxytryptamine stimulates phosphorylation of p44/p42 mitogen-activated protein kinase activation in bovine aortic endothelial cell cultures. J Cardiovasc Pharmacol 35:398–402. doi: 10.1097/00005344-200003000-00008. [DOI] [PubMed] [Google Scholar]
- 36.Singh I, Helenius A. 1992. Role of ribosomes in Semliki Forest virus nucleocapsid uncoating. J Virol 66:7049–7058. doi: 10.1128/JVI.66.12.7049-7058.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wengler G. 2009. The regulation of disassembly of alphavirus cores. Arch Virol 154:381–390. doi: 10.1007/s00705-009-0333-9. [DOI] [PubMed] [Google Scholar]
- 38.Yamauchi Y, Greber UF. 2016. Principles of virus uncoating: cues and the snooker ball. Traffic 17:569–592. doi: 10.1111/tra.12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banerjee I, Miyake Y, Philip Nobs S, Schneider C, Horvath P, Kopf M, Matthias P, Helenius A, Yamauchi Y. 2014. Influenza A virus uses the aggresome processing machinery for host cell entry. Science -) 346:473–477. doi: 10.1126/science.1257037. [DOI] [PubMed] [Google Scholar]
- 40.Francis AC, Melikyan GB. 2018. Live-cell imaging of early steps of single HIV-1 infection. Viruses 10:275. doi: 10.3390/v10050275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lukic Z, Dharan A, Fricke T, Diaz-Griffero F, Campbell EM. 2014. HIV-1 uncoating is facilitated by dynein and kinesin 1. J Virol 88:13613–13625. doi: 10.1128/JVI.02219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malikov V, Da Silva ES, Jovasevic V, Bennett G, De Souza Aranha Vieira DA, Schulte B, Diaz-Griffero F, Walsh D, Naghavi MH. 2015. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat Commun 6:6660. doi: 10.1038/ncomms7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLoughlin DJ, Strange PG. 2000. Mechanisms of agonism and inverse agonism at serotonin 5-HT(1A) receptors. J Neurochem 74:347–357. doi: 10.1046/j.1471-4159.2000.0740347.x. [DOI] [PubMed] [Google Scholar]
- 44.Schoeffter P, Ullmer C, Bobirnac I, Gabbiani G, Lübbert H. 1996. Functional, endogenously expressed 5-hydroxytryptamine 5-ht7 receptors in human vascular smooth muscle cells. Br J Pharmacol 117:993–994. doi: 10.1111/j.1476-5381.1996.tb16687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krobert KA, Andressen KW, Levy FO. 2006. Heterologous desensitization is evoked by both agonist and antagonist stimulation of the human 5-HT7 serotonin receptor. Eur J Pharmacol 532:1–10. doi: 10.1016/j.ejphar.2005.11.039. [DOI] [PubMed] [Google Scholar]
- 46.Pauwels PJ, Palmier C. 1994. Inhibition by 5-HT of forskolin-induced cAMP formation in the renal opossum epithelial cell line OK: mediation by a 5-HT1B like receptor and antagonism by methiothepin. Neuropharmacology 33:67–75. doi: 10.1016/0028-3908(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 47.Riva L, Song O-r, Prentoe J, Helle F, L'homme L, Gattolliat C-H, Vandeputte A, Fénéant L, Belouzard S, Baumert TF, Asselah T, Bukh J, Brodin P, Cocquerel L, Rouillé Y, Dubuisson J. 2018. Identification of piperazinylbenzenesulfonamides as new inhibitors of claudin-1 trafficking and hepatitis C virus entry. J Virol 92:1–19. doi: 10.1128/JVI.01982-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barrows NJ, Campos RK, Powell ST, Prasanth KR, Schott-Lerner G, Soto-Acosta R, Galarza-Muñoz G, McGrath EL, Urrabaz-Garza R, Gao J, Wu P, Menon R, Saade G, Fernandez-Salas I, Rossi SL, Vasilakis N, Routh A, Bradrick SS, Garcia-Blanco MA. 2016. A screen of FDA-approved drugs for inhibitors of Zika virus infection. Cell Host Microbe 20:259–270. doi: 10.1016/j.chom.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Assetta B, Morris-Love J, Gee GV, Atkinson AL, O’Hara BA, Maginnis MS, Haley SA, Atwood WJ. 2019. Genetic and functional dissection of the role of individual 5-HT 2 receptors as entry receptors for JC polyomavirus. Cell Rep 27:1960–1966.e6. doi: 10.1016/j.celrep.2019.04.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mayberry CL, Soucy AN, Lajoie CR, DuShane JK, Maginnis MS. 2019. JC polyomavirus entry by clathrin-mediated endocytosis is driven by β-arrestin. J Virol 93:e01948-18. doi: 10.1128/JVI.01948-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mainou BA, Dermody TS. 2012. Transport to late endosomes is required for efficient reovirus infection. J Virol 86:8346–8358. doi: 10.1128/JVI.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsetsarkin K, Higgs S, McGee CE, De Lamballerie X, Charrel RN, Vanlandingham DL. 2006. Infectious clones of chikungunya virus (La Réunion isolate) for vector competence studies. Vector Borne Zoonotic Dis 6:325–337. [DOI] [PubMed] [Google Scholar]
- 53.Scholte FEM, Tas A, Martina BEE, Cordioli P, Narayanan K, Makino S, Snijder EJ, van Hemert MJ. 2013. Characterization of synthetic chikungunya viruses based on the consensus sequence of recent E1-226V isolates. PLoS One 8:e71047. doi: 10.1371/journal.pone.0071047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fahrer J, Rausch J, Barth H. 2013. A cell-permeable fusion protein based on Clostridium botulinum C2 toxin for delivery of p53 tumorsuppressor into cancer cells. PLoS One 8:e72455. doi: 10.1371/journal.pone.0072455. [DOI] [PMC free article] [PubMed] [Google Scholar]