

Abstract
Identifying additional mitogen-activated protein kinase (MAPK) pathway regulators is invaluable in aiding our understanding of the complex signaling networks that regulate cellular processes, including cell proliferation and survival. Here, using in vitro kinase assays and by expressing WT or kinase-dead MAPK kinase kinase 19 (MAP3K19) in the HEK293T cell line and assessing activation of the extracellular signal–regulated kinase (ERK) and JUN N-terminal kinase (JNK) signaling pathways, we defined MAP3K19 as a novel regulator of MAPK signaling. We also observed that overexpression of WT MAP3K19 activates both the ERK and JNK pathways in a panel of cancer cell lines. Furthermore, MAP3K19 sustained ERK pathway activation in the presence of inhibitors targeting the RAF proto-oncogene Ser/Thr protein kinase (RAF) and MAPK/ERK kinase, indicating that MAP3K19 activates ERK via a RAF-independent mechanism. Findings from in vitro and in-cell kinase assays demonstrate that MAP3K19 is a kinase that directly phosphorylates both MAPK/ERK kinase (MEK) and MAPK kinase 7 (MKK7). Results from an short-hairpin RNA screen indicated that MAP3K19 is essential for maintaining survival in KRAS-mutant cancers; therefore, we depleted or inhibited MAP3K19 in KRAS-mutant cancer cell lines and observed that this reduces viability and decreases ERK and JNK pathway activation. In summary, our results reveal that MAP3K19 directly activates the ERK and JNK cascades and highlight a role for this kinase in maintaining survival of KRAS-mutant lung cancer cells.
Keywords: protein kinase, extracellular-signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), oncogene, GTPase Kras (KRAS), lung cancer, cell proliferation, kinase signaling, kinome, mitogen-activated protein kinase kinase kinase 19 (MAP3K19), YSK4
Introduction
A large portion of the kinome remains poorly characterized, with the functions of ∼25% of kinases yet to be discovered (1). This includes several MAPKs. Elucidating the roles of understudied kinases in cellular processes vital to oncogenesis will provide potential candidates for therapeutic intervention (2–4). It has been well-described that aberrant activation of kinase signaling contributes to cancer initiation and progression. The MAPK signaling pathways represent essential signaling nodes that govern cell cycle progression and cell survival, which underscores the importance of elucidating the function of novel MAPKs (5, 6). A published study using an short-hairpin RNA screen identified mitogen-activated protein kinase kinase kinase 19 (MAP3K19 or YSK4) as a genetic dependence in a panel of KRAS-mutant cancers (7). Consistent with a prosurvival function for MAP3K19, short-hairpin RNA-mediated knockdown of MAP3K19 suppressed HeLa cell proliferation and survival, further indicating that MAP3K19 plays a role in cancer cell survival (8). In addition, MAP3K19 overexpression was detected in patients with chronic obstructive pulmonary disease, a known risk factor for lung cancer (9, 10). MAP3K19 has been reported to regulate transforming growth factor-β–induced SMAD signaling and gene expression, as well as regulate NF-κB to promote the release of various cytokines, although the mechanisms underpinning these activities are unknown (9, 10). Given that these large screening studies have implicated MAP3K19 in promoting cancer cell growth, we aimed to determine the mechanism by which MAP3K19 promotes cancer cell viability. Our data define MAP3K19 as a novel modulator of ERK and JNK signaling cascades and indicate that this kinase may be essential for cell survival in KRAS-mutant cancers.
Results
MAP3K19 promotes ERK pathway activation
To determine the role of MAP3K19 in regulating downstream MAP2Ks, we expressed WT or kinase-dead (KD) MAP3K19 in the HEK 293T cell line and assessed activation of the ERK and JNK signaling pathways. Compared with protein levels in empty vector (EV) control cells and KD-transfected cells, expression of WT MAP3K19 markedly enhanced phosphorylation of MEK, ERK, and JNK proteins (Fig. 1A). We also observed an upward mobility shift in comparing WT to KD MAP3K19, consistent with WT MAP3K19 being post-translationally modified to an active conformation in HEK 293T cells. To verify that MAP3K19 is an ERK pathway activator, we overexpressed MAP3K19 in additional cancer cell lines (LK2, MCF-7, and HCT-116) and monitored ERK pathway activation (Fig. 1, B–D). In all three cell lines, overexpression of WT MAP3K19 led to an increase in both MEK and ERK activation, as assessed by increases in phosphorylation of both MEK and ERK (Fig. 1, B–D; increases in phosphorylation were quantified in bar graphs below representative immunoblots). MAP3K19 WT protein was detected at a higher molecular weight than KD in these cell lines, providing further support that MAP3K19 is post-translationally modified to assume an active conformation. To determine whether the mobility shift between WT and KD MAP3K19 is phosphorylation-dependent, we pretreated HEK 293T and LK2 cell lysates with λ-protein phosphatase (λ-PP). In both cell lines, band migration of WT MAP3K19 notably shifted following λ-PP treatment, minimizing the mobility gap between WT and KD MAP3K19 (Fig. 1, E and F). Together, these results indicate that MAP3K19 can activate both the JNK and ERK pathways upon overexpression.
Figure 1.

MAP3K19 activates MAPK family members. A–D, HEK293T (A), LK2 (B), MCF7 (C), or HCT-116 (D) cells were transiently transfected with empty vector (EV), WT, or KD-K1089M MAP3K19. Phosphorylation status of MAPK family members was assessed by Western blotting. Band density was quantified by ImageJ software. The data are shown as mean phospho:total protein density normalized to EV control ± S.D. *, p < 0.05; **, p < 0.01. E and F, HEK293T (E) or LK2 (F) cells were transfected with EV, WT, or KD MAP3K19, and lysates were treated with 0 or 1 μl of λ-PP. Western blotting was performed.
We previously characterized the mixed-lineage kinases (MLK1–4) as MEK kinases that can activate the ERK pathway in the presence of RAF inhibitors. To investigate the mechanism by which MAP3K19 stimulates ERK pathway activation, we overexpressed WT MAP3K19 in HEK 293T cells and treated these cells with a pan-RAF inhibitor (L779450), a MEK inhibitor (AZD6244), or both the RAF and MEK inhibitors. Overexpressed MLK1 was used as a control. Expression of MAP3K19 resulted in an increase in MEK phosphorylation that was maintained even in the presence of the RAF inhibitor, indicating that MAP3K19-mediated activation of MEK is independent of RAF, similar to MLK1 (Fig. 2A). Interestingly, MAP3K19 could sustain a lower level of ERK phosphorylation, even in the presence of the MEK inhibitor or the combination of RAF and MEK inhibitors, which raises the possibility that MAP3K19 may be able to activate ERK in a MEK-independent manner (Fig. 2A). MLK1 was not able to maintain ERK phosphorylation in the presence of a MEK inhibitor, concordant with our previous findings (11). To confirm these results and verify that kinase activity is essential for ERK pathway activation, we overexpressed WT or KD MAP3K19 in HEK 293T cells treated with a combination of RAF and MEK inhibitors and monitored ERK phosphorylation. Overexpression of MAP3K19 WT, but not KD, led to an increase in MEK and ERK phosphorylation that was retained even in the presence of the RAF and MEK inhibitor treatment. These data indicate that MAP3K19 catalytic activity is critical for ERK pathway activation and suggest that MAP3K19 is a MEK kinase (Fig. 2B).
Figure 2.

MAP3K19 maintains MEK phosphorylation in the presence of RAF inhibitors. A, 48 h after transfection with EV, MAP3K19-WT, or MLK1-WT HEK293T cells were treated with DMSO vehicle control, 1 μm L779450, 1 μm AZD6244, or a combination of both RAF and MEK inhibitors for 1 h, and Western blotting was performed on cell lysates. B, 48 h after transfection with EV, WT, or KD-MAP3K19, HEK293T cells were treated with DMSO vehicle control or indicated inhibitors: 1 μm vemurafenib (BRAFi), 1 μm selumetinib (S, MEKi), or 500 nm cobimetinib (C, MEKi) for 1 h, and Western blotting was performed on cell lysates. Band density was quantified by ImageJ software. The data are shown as mean phospho:total protein density ± S.D. Dunnett's multiple comparisons test was used for statistical analysis, with EV-DMSO group as control. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
MAP3K19 is a direct MAP2K kinase
To determine whether MAP3K19 is a direct MEK kinase, we performed in vitro kinase assays using purified KD MEK1 as a substrate. Full-length MAP3K19 that was immunoprecipitated from cells phosphorylated MEK1 in a kinase-dependent manner (Fig. 3A). To confirm these results, we used purified MAP3K19 kinase domain in an in vitro kinase assay with KD MEK1. Purified MLK1 kinase domain was used as a control. MAP3K19 directly phosphorylated MEK, indicating that MAP3K19 is a direct MEK kinase, similar to MLK1 (Fig. 3B). To determine whether MAP3K19 can phosphorylate ERK, we performed an in vitro kinase assay using KD ERK2 as a substrate. Purified MEK1, used as a positive control, catalyzed phosphorylation of ERK, but neither MLK1 nor MAP3K19 are ERK kinases (Fig. 3C). To verify that MAP3K19 was not inhibited by RAF or MEK inhibitors, we conducted an in vitro kinase assay using KD MEK1 as a substrate with purified MAP3K19 kinase domain or MLK1 in the presence of MEK and/or RAF inhibitors. MAP3K19-dependent MEK phosphorylation was preserved in the presence of all drugs, confirming that RAF and MEK inhibitors do not inhibit MAP3K19 (Fig. 3D). To assess whether MAP3K19 can phosphorylate MKK7 (MAP2K7), an upstream JNK kinase, we performed an in vitro kinase assay using KD MKK7 as a substrate. MAP3K19 directly phosphorylates MKK7, which leads to activation of JNK (Fig. 3E). To rule out the possibility that MAP3K19 may directly phosphorylate JNK, we used KD JNK1 or JNK2 as substrates in an in vitro kinase assay and purified MKK7 as a control. MAP3K19 did not phosphorylate JNK (Fig. 3F).
Figure 3.

MAP3K19 directly phosphorylates MAP2Ks. A, MAP3K19 was immunoprecipitated (IP) from HEK293T cells and subjected to a kinase assay with kinase-inactive MEK1. B and C, in vitro kinase assay using recombinant MAP3K19 protein and kinase-inactive MEK1 or ERK2, respectively. D, kinase-inactive MEK1 and purified GST-MAP3K19 or GST-MLK1 kinase domain were subjected to in vitro kinase assay in the presence or absence of inhibitors: 5 μm L779450, 1 μm PLX4032, 5 μm U0126, or 2 μm AZD6244. E and F, in vitro kinase assay using recombinant MAP3K19 protein and kinase-inactive MKK7 or JNK1/2, respectively. The data are shown as mean phospho:total protein density ± S.D. Dunnett's multiple comparisons test was used for statistical analysis, with samples in the first lane as control. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ♢, kinase-inactive.
MAP3K19 does not promote resistance to ERK pathway inhibitors in melanoma
Based on our data showing that MAP3K19 sustains MEK pathway activation in the presence of RAF and MEK inhibitors, we explored the possibility that MAP3K19 may play a role in promoting resistance to ERK pathway inhibitors, similar to the MLKs. We assessed expression of MAP3K19 and observed an increase in MAP3K19 mRNA levels in melanoma cell lines resistant to RAF inhibitors (Fig. 4A). To determine whether MAP3K19 overexpression can promote increased MEK and ERK activation in BRAF-V600E–mutant melanoma cell lines, we expressed doxycycline-inducible MAP3K19 in A375 cells. There was no increase in MEK or ERK phosphorylation, and WT MAP3K19 migrated at the same mobility as KD MAP3K19 in A375 cells, suggesting that overexpressed MAP3K19 is likely to be maintained in an inactive conformation in BRAF-V600E–positive melanoma cells (Fig. 4B). Similar results were observed in the Sk-Mel-28 cell line (Fig. 4C). Lastly, expression of MAP3K19 did not promote ERK pathway activation in the presence of RAF or MEK inhibitors in the A375 cells, indicating that increased expression of MAP3K19 is unlikely to be an acquired mechanism of resistance to ERK pathway inhibitors in melanoma cells (Fig. 4D).
Figure 4.

MAP3K19 expression does not promote resistance to ERK pathway inhibitors in melanoma cell lines. A, RT–quantitative (q) PCR analysis of MAP3K19 expression in vemurafenib-resistant (VR) clones derived from A375 (C2, C4) or MDA-MB-435 (1E, 2D) melanoma cells. B, parental A375 melanoma cell line was used to generate cells with doxycycline (dox)–inducible expression of MAP3K19. Protein expression of MAP3K19 was confirmed, and phosphorylation of MEK1/2 and ERK1/2 was assessed by Western blotting 24 h after doxycycline treatment. The data are shown as mean phospho:total (p:t) protein density ± S.D. C, Sk-Mel-28 cells were transiently transfected with EV, WT, or KD-MAP3K19. Phosphorylation status of MAPK family members was assessed by Western blotting. The data are shown as mean phospho:total protein density ± S.D. D, doxycycline was added to A375-TR cells for 24 h to induce expression of MAP3K19, and then cells were treated with DMSO vehicle control, 500 nm PLX4032 (BRAFi), 500 nm AZD6244 (MEKi), or a combination of both BRAF and MEK inhibitors for 1 h. Western blotting was performed on cell lysates. The data are shown as mean phospho:total protein density ± S.D., with MAP3K19-WT (−dox) DMSO as control. *, p < 0.05; **, p < 0.01.
MAP3K19 enhances KRAS-mediated ERK activation and is required to maintain viability in KRAS-mutant lung cancer cells
MAP3K19 was identified as a genetic dependence in KRAS-mutant cancers. Therefore, we investigated whether MAP3K19 would enhance KRAS-mediated activation of the ERK pathway. Expression of KRAS G12C mutant increased both ERK and MEK activation as expected. Co-expression of MAP3K19 and KRAS G12C led to a marked increase in the expression levels of MAP3K19 and a correlated increase in ERK pathway activation compared with either KRAS or MAP3K19 alone (Fig. 5A). To determine whether MAP3K19 is required to maintain KRAS-mutant lung cancer cell viability, we first assessed a panel of lung cancer cell lines for expression of endogenous MAP3K19 and observed that MAP3K19 is expressed in most lung cancer cells at varying levels. We then depleted MAP3K19 in lung cancer cells with WT KRAS (H3122) or mutant KRAS (H2030 and H2122) by using two unique siRNAs (Fig. 5C). Loss of MAP3K19 resulted in reduced viability in KRAS-mutant lung cancer cells, but not KRAS WT lung cancer cells (Fig. 5C). To determine whether MAP3K19 depletion would alter MAPK pathway activation, we assessed MEK, ERK, and JNK phosphorylation in H2122 cells and observed a decrease in ERK and JNK pathway activation (Fig. 5D). These results indicate that MAP3K19 plays a role in maintaining KRAS-mutant lung cancer cell viability by regulating the ERK and JNK pathways.
Figure 5.

MAP3K19 cooperates with KRAS G12C to enhance ERK activation and plays a role in maintaining cell viability in KRAS-mutant lung cancer cells. A, Western blotting analysis of MAPK phosphorylation changes because of transient expression of MAP3K19-WT, MAP3K19-KD, or KRAS G12C. The data are shown as mean protein density (phospho:total for MEK and ERK) ± S.D. B, Western blotting of endogenous MAP3K19 in lung cancer cell lines. C, top panel, RT–quantitative PCR validation of siRNA-mediated MAP3K19 knockdown in LUAD cell lines 48 h after transfection. Bottom panel, cells were seeded in 96-well plates 24 h after transfection, and cell viability was evaluated after 48 h by crystal violet assay. The data are represented as percentages of cell viability normalized to control (ctrl) ± S.D. D, phosphorylation status of MAPK family members following depletion of MAP3K19 expression in H2122 cells. The data are shown as mean phosphorylated protein density normalized to control (set to 1) ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
A MAP3K19 inhibitor suppresses ERK activation and inhibits viability in RAS-mutant lung cancer
To identify possible pharmacological inhibitors, we evaluated three kinase inhibitors that were previously identified to inhibit MAP3K19 (AT-9283, NVP-TAE226, and GSK-269962A) in an internal pan-kinase inhibitor screen (12, 13). In HEK 293T cells overexpressing MAP3K19, AT-9283 was the only compound that suppressed activation of the ERK pathway and promoted a downward shift in MAP3K19 migration comparable with KD MAP3K19, indicating that this compound inhibits MAP3K19 (Fig. 6A). To validate that AT-9283 targets MAP3K19, we performed an in vitro kinase assay where MAP3K19 was pretreated with AT-9283. We observed a reduction in phosphorylation of inactive MEK at both 1 and 5 μm of drug treatment, which verifies that this compound is a MAP3K19 inhibitor. Treatment of the KRAS-mutant lung cancer cell lines with AT-9283 suppressed viability and promoted a decrease in ERK pathway activation (Fig. 6, C and D). However, there was also a decrease in cell viability in the H3122 cell line (KRAS WT) treated with AT-9283, likely because of off-target effects. Overall, these results suggest that MAP3K19 could serve as a novel therapeutic target in KRAS-mutant lung cancers.
Figure 6.

Analysis of MAP3K19 inhibitor. A, HEK293T cells were transiently transfected with EV or WT MAP3K19. After 48 h, the cells were treated with DMSO vehicle control or indicated doses of inhibitors for 1 h. Western blotting was then performed on cell lysates. The data are shown as mean phospho:total (p:t) protein density ± S.D. with EV vehicle-treated control set to 1. B, kinase-inactive MEK1 and purified GST-MAP3K19 kinase domain were subjected to an in vitro kinase assay in the presence or absence of inhibitors. The data are shown as mean protein density (phospho:total) ± S.D. with vehicle-treated control set to 1. C, Western blotting of MAPK pathway expression and activation in H2122 cells treated with AT-9283 for 24 h. The data are shown as mean protein density (phospho:total for MEK, ERK, and JNK) ± S.D. with vehicle-treated control set to 1. D, viability of lung adenocarcinoma cells was evaluated by crystal violet assay after 72-h treatment with AT-9283. The data are represented as percentages of cell viability normalized to vehicle-treated control ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
MAP3Ks are Ser/Thr kinases that, upon activation, phosphorylate and activate MAP2Ks, ultimately leading to activation of MAPKs (14). The RAFs (MAP3Ks) → MEK1/2 (MAP2Ks) → ERK1/2 (MAPKs) cascade is a classic and cancer-relevant example (15). In addition to the RAF/MEK/ERK cascade, there are others, including those that activate JNKs, p38s, and ERK5 (16). Identifying novel regulators of MAPK signaling cascades is critical to fully understand how dysregulation of these pathways occurs and contributes to cancer and resistance to targeted therapies. Many MAP3Ks are unexplored. Our identification of the MAP3Ks MLK1–4 as kinases that confer resistance to RAF inhibitors in melanoma provides proof that analysis of these understudied kinases has therapeutic implications (11). Our current study describes MAP3K19 as a novel activator of the ERK and JNK pathways (Fig. 7).
Figure 7.
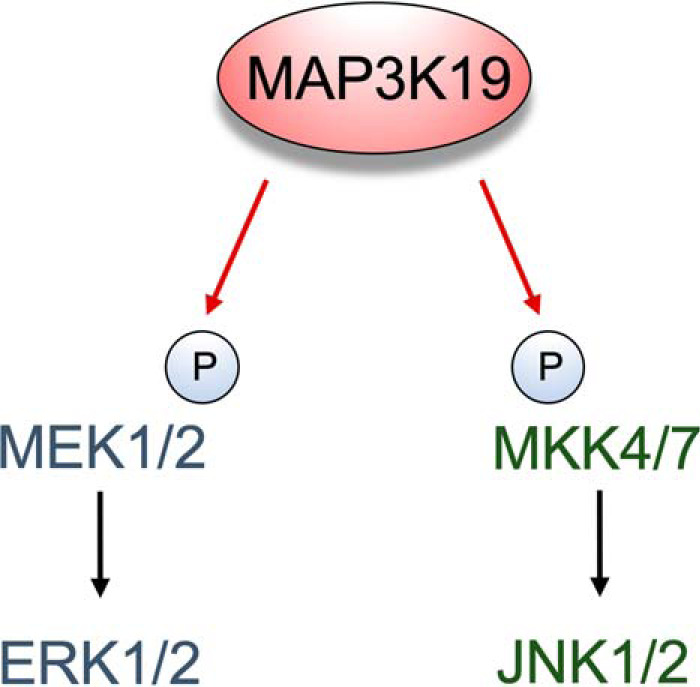
Schematic representation of MAP3K19 regulating MAPK pathways. MAP3K19 can directly phosphorylate MEK1/2 and MKK7, leading to ERK and JNK activation.
Mutations that activate the RAF/MEK/ERK pathway occur in RAF proteins, such as the BRAF V600E mutation, and in the RAS family of proteins, which are upstream activators of RAF. Although KRAS gain-of-function mutations are detected in ∼25% of lung adenocarcinoma (LUAD) patients, direct inhibition of all KRAS mutants, aside from G12C/G13C, has not been therapeutically possible, and resistance often develops to inhibitors of the RAF/MEK/ERK pathway. An alternative approach for targeting KRAS-mutant tumors is identification of targetable vulnerabilities through synthetic lethal screens (7). Our interest in MAP3K19 arises from two studies indicating that this MAP3K may be involved in lung cancer. One study identified MAP3K19 as a genetic dependence in a panel of KRAS-mutant cancers, including LUAD samples (7). The authors of the second study observed that MAP3K19 is more abundant in patients with chronic obstructive pulmonary disease, a known risk factor for lung cancer (9, 10). Consistent with these observations, our data demonstrate that siRNA-mediated depletion or pharmacological inhibition of MAP3K19 significantly reduces cell viability and decreases ERK and JNK pathway activation in KRAS-mutant lung cancer cells. However, the AT-9283 inhibitor also decreased the viability of KRAS WT LUAD cells, indicating the need for additional studies using more specific MAP3K19 inhibitors to further validate MAP3K19 as a genetic vulnerability and potential therapeutic target in KRAS-mutant lung cancer cells.
Reactivation of MAPK signaling is a prominent mechanism of resistance to BRAF inhibitors (17–23). From our observation that MAP3K19 maintained ERK activation in the presence of RAF and MEK inhibitors, we explored the role of MAP3K19 in regulating resistance to MAPK-targeted therapy. Although we observed that expression of MAP3K19 was elevated in vemurafenib-resistant BRAF-mutant melanoma cell lines, induced expression of MAP3K19 could not sustain ERK pathway activation in the presence of RAF or MEK inhibitors in melanoma cells. Thus, MAP3K19 likely does not play a role in promoting resistance to ERK pathway inhibitors in these cells. Our findings establishing MAP3K19 as a novel direct kinase of MEK may be relevant in other cancer models. Response to BRAF inhibitor therapy has been limited in patients with BRAF-V600E-mutant metastatic colorectal cancer, and it is possible that MAP3K19 plays a role in resistance in tumors of different origins (24). In addition, MEK inhibitor therapy has limited efficacy against NRAS-mutant melanoma in the clinic (25). Future studies examining MAP3K19 activity in the context of BRAF-mutant colorectal cancer and NRAS-mutant melanoma may shed light on MAP3K19's potential role in promoting resistance to ERK pathway inhibitors in these cancers. Overall, our findings indicate that MAP3K19 is an upstream regulator of the ERK and JNK cascades and is required to maintain lung cancer cell survival in KRAS-mutant cancers.
Experimental procedures
Cell lines and reagents
Lung cancer cell lines NCI-H2122 and NCI-H2030 were acquired from American Type Culture Collection. NCI-H3122 cells were obtained from the National Cancer Institute Repository. All LUAD cell lines were maintained in RPMI 1640 medium (Quality Biological) supplemented with 10% fetal bovine serum (Atlanta Biologicals), 1% penicillin/streptomycin (Invitrogen), and 1% GlutaMAX (Invitrogen). HEK 293T cells were cultured in DMEM supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% GlutaMAX. Inhibitors were purchased as follows: PLX4032 (vemurafenib), U0126, AZD6244 (selumetinib), and GDC-0973 (cobimetinib) from Selleck Chemicals; L779450 from Abcam; and AT-9283 from Cayman Chemical. All inhibitors were dissolved in DMSO, and aliquots were stored at −20 °C.
Western blotting
48 h after transient transfection, the cells were treated with inhibitors or DMSO vehicle control for 1 h before lysis with M-PER mammalian protein extraction reagent (Thermo Scientific) supplemented with 1% protease inhibitor and 1% phosphatase inhibitors (I/II) (Invitrogen). The following primary antibodies were used: MAP3K19 (antibody was generated at the Dundee antibody production facility in collaboration with Dr. James Hastie); MLK1, MEK1/2, p-MEK1/2 (S217/221), ERK1/2, p-ERK1/2 (T202/Y204), p-MKK7 (S271/T275), JNK, p-JNK (T183/Y185), GST, Tubulin (Cell Signaling Technology), and V5 (Bio-Rad). Tubulin antibody was used at 1:5,000 dilution. All other antibodies were used at 1:1000 dilution. Sheep horseradish peroxidase–conjugated antibody (Cell Signaling Technology) was used as a secondary antibody at 1:5000 dilution in 5% BSA for MAP3K19 detection. After incubation with anti-rabbit DyLightTM 680 conjugate or anti-mouse DyLightTM 800 conjugate (Cell Signaling Technology) at 1:10,000 dilution in Odyssey Blocking Buffer (LI-COR Biosciences), bands were detected using the ChemiDoc MP imaging system (Bio-Rad). All Western blots are representative of at least three independent experiments.
λ-PP treatment
HEK 293T or LK2 cells were lysed in radioimmune precipitation assay buffer and centrifuged at 4 °C. After MnCl2 (2 mm) was added to the lysates, the samples were treated with 0 or 1 μl of λ phosphatase (New England Biolabs) for 1.5 h at 30 °C. Laemmli buffer was added to terminate the reaction, and samples were boiled for 5 min.
Quantitative RT-PCR
The Cells were collected in cold phosphate-buffered saline and pelleted by centrifugation at 4 °C. Total RNA was extracted using the RNeasy mini kit in accordance with the manufacturer's protocol (Qiagen). The quality and concentration of RNA were determined using the NanoDrop One spectrophotometer (Thermo Scientific). Total RNA (1 μg) was reverse-transcribed using the iScript kit (Bio-Rad), and qPCR was performed using SYBR Green (Bio-Rad). The following primer sequences were used: MAP3K19 forward, GCATCAGCAGAAGTGAGGAG; MAP3K19 reverse, TCAACACCTTCTGTCCTGGG; Actin forward, GGCACCCAGCACAATGAAGA; and Actin reverse, ACTCCTGCTTGCTGATCCAC. The cycle numbers were normalized to β-actin, and control conditions were scaled to one. The experiments were conducted in triplicate with internal duplicates.
Cell viability assays
The cells were treated with inhibitor at the indicated concentrations and time points or were reverse-transfected with siRNA for 72 h in a 96-well plate format. The cells were fixed with glutaraldehyde for 20 min and then stained with 1% crystal violet in 10% methanol solution for 15 min. The wells were washed thoroughly and allowed to air dry. For quantification, 100 μl of 33% acetic acid was added to each well and incubated for at least 1 h with shaking. The absorbance values were read at 595 nm. The data are represented as mean cell viability normalized to control ± S.E. of triplicate experiments, each consisting of three technical replicates.
Transient transfections
For ectopic expression of MAP3K19, HEK 293T cells were seeded at 50% confluency and allowed to adhere overnight. The following day, the cells were transfected with the designated amount of plasmid DNA using jetPRIME transfection reagent (Polyplus Transfection) for the indicated time points. MAP3K19 and MLK1 WT constructs were purchased as entry vector (OriGene). The KD mutation was introduced by site-directed mutagenesis (QuikChange II kit, Stratagene) in the critical lysine required to bind ATP (within the VAIK motif) (K1089M-MAP3K19). The following primer sequences were used: K1089M forward, GGTATCCAAAGCCACCTGCATTACAGCTATTAGCTGTCCT; and K1089M reverse, AGGACAGCTAATAGCTGTAATGCAGGTGGCTTTGGATACC. MAP3K19 constructs were cloned into pLenti6.3/TO/V5-DEST vector (Thermo Scientific) with the Gateway system according to the manufacturer's instructions. KRAS G12C plasmid (pCDH-CMV-HA backbone) was kindly provided by Dr. Deborah Morrison (National Cancer Institute).
For RNAi experiments, cells were reverse-transfected using Lipofectamine RNAiMAX (Thermo Scientific) and 40 nm of siRNA oligonucleotides (OriGene) diluted with Opti-MEM I reduced serum medium (Thermo Scientific) in 10-cm-plate format. Knockdown of mRNA was assessed by quantitative RT-PCR 48 h after transfection.
Immunoprecipitation and kinase assays
48 h after transfection, the cells were treated with inhibitors or DMSO and lysed with Triton X-100 lysis buffer (Cell Signaling Technology) supplemented with protease inhibitors (Roche) dissolved in sterile water. Cell lysates were incubated overnight (4 °C) with anti-V5 antibody (1:100) and then incubated with 50 μl of a 50% slurry of Protein G Dynabeads (Thermo Scientific) for 1 h. The beads were washed three times, first with lysis buffer and then with kinase buffer (Cell Signaling Technology). The kinase assay was performed in the presence of ATP (50 μm) and 100 ng of kinase-inactive MEK1 as substrate (Carna Biosciences) at 30 °C for 30 min. The kinase assay was terminated after adding SDS sample buffer and boiling for 15 min. The proteins were resolved by SDS-PAGE and analyzed by Western blotting.
In vitro kinase assay
Recombinant human GST-tagged MAP3K19 (YSK4) kinase domain (Thermo Fisher Scientific) or human MLK1 kinase domain (Carna Biosciences) was incubated at room temperature with kinase-inactive MEK1, ERK2, MKK7, JNK1, or JNK2 (Carna Biosciences) in the absence or presence of MAPK pathway inhibitors for 5 min. The kinase reaction was initiated with the addition of ATP (50 μm) at 30 °C for 30 min, and the reaction was stopped by adding 4× SDS sample buffer. After the samples were boiled for 15 min, the proteins were resolved by SDS-PAGE and analyzed by Western blotting.
Generation of tetracycline-inducible cell lines
WT or KD MAP3K19 plasmids cloned into pLenti6.3/TO/V5-DEST and pLenti3.3/TR vector (for tetracycline repressor expression) were transfected into HEK 293FT cells using Lipofectamine 2000, and lentiviral supernatant was collected. Parental A375 melanoma cells were transduced with lentiviral stocks and treated with antibiotics for selection (blasticidin (Invitrogen) and Geneticin (Gibco)). Doxycycline (Sigma) was used to induce expression of MAP3K19.
Generation of vemurafenib-resistant cell lines
Melanoma cell lines A375 and MDA-MB-435 were generated by chronic treatment with 1 μm vemurafenib, as described previously (11). The cells were cultured in medium with fresh vemurafenib added 1.
Statistical analysis
Statistical analyses were performed using unpaired Student's t test or one-way analysis of variance (Dunnett's multiple comparisons test) on GraphPad Prism software (GraphPad Software Inc.). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Data availability
All data reported are contained within the manuscript.
Acknowledgments
We thank Dr. Deborah Morrison and her lab (National Cancer Institute) for providing reagents as well as advice for our studies and Roger Liang for analysis of MAP3K19 constructs. We are also grateful to Dr. Craig Thomas (National Cancer Institute) for helpful suggestions regarding MAP3K19 inhibitors.
Author contributions—V. T. H., K. N., and J. B. data curation; V. T. H. and J. B. formal analysis; V. T. H., K. N., and J. B. validation; V. T. H. and J. B. investigation; V. T. H., K. N., P. T.-A., and J. B. methodology; V. T. H. and J. B. writing-original draft; V. T. H. and J. B. project administration; V. T. H., K. N., P. T.-A., and J. B. writing-review and editing; J. B. conceptualization; J. B. supervision; J. B. funding acquisition; J. B. visualization.
Funding and additional information—This research was supported by the Intramural Research Program of NCI, National Institutes of Health Grant 1ZIABC011691. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- MAPK
- mitogen-activated protein kinase
- KD
- kinase-dead
- MAP3K19
- MAPK kinase kinase 19
- ERK
- extracellular signal–regulated kinase
- JNK
- JUN N-terminal kinase
- RAF
- RAF proto-oncogene Ser/Thr protein kinase
- MEK
- MAPK/ERK kinase
- MKK
- MAPK kinase
- λ-PP
- λ-protein phosphatase
- MLK
- mixed-lineage kinase
- LUAD
- lung adenocarcinoma
- GST
- glutathione S-transferase
- EV
- empty vector.
References
- 1. Fedorov O., Müller S., and Knapp S. (2010) The (un)targeted cancer kinome. Nat. Chem. Biol. 6, 166–169 10.1038/nchembio.297 [DOI] [PubMed] [Google Scholar]
- 2. Knapp S. (2018) New opportunities for kinase drug repurposing and target discovery. Br. J. Cancer 118, 936–937 10.1038/s41416-018-0045-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhullar K. S., Lagarón N. O., McGowan E. M., Parmar I., Jha A., Hubbard B. P., and Rupasinghe H. P. V. (2018) Kinase-targeted cancer therapies: progress, challenges and future directions. Mol. Cancer 17, 48 10.1186/s12943-018-0804-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cohen P., and Alessi D. R. (2013) Kinase drug discovery: what's next in the field? ACS Chem. Biol. 8, 96–104 10.1021/cb300610s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Atay O., and Skotheim J. M. (2017) Spatial and temporal signal processing and decision making by MAPK pathways. J. Cell Biol. 216, 317–330 10.1083/jcb.201609124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang L., and Karin M. (2001) Mammalian MAP kinase signalling cascades. Nature 410, 37–40 10.1038/35065000 [DOI] [PubMed] [Google Scholar]
- 7. Barbie D. A., Tamayo P., Boehm J. S., Kim S. Y., Moody S. E., Dunn I. F., Schinzel A. C., Sandy P., Meylan E., Scholl C., Fröhling S., Chan E. M., Sos M. L., Michel K., Mermel C., et al. (2009) Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 10.1038/nature08460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grueneberg D. A., Degot S., Pearlberg J., Li W., Davies J. E., Baldwin A., Endege W., Doench J., Sawyer J., Hu Y., Boyce F., Xian J., Munger K., and Harlow E. (2008) Kinase requirements in human cells: I. Comparing kinase requirements across various cell types. Proc. Natl. Acad. Sci. U.S.A. 105, 16472–16477 10.1073/pnas.0808019105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boehme S. A., Franz-Bacon K., Ludka J., DiTirro D. N., Ly T. W., and Bacon K. B. (2016) MAP3K19 is overexpressed in COPD and is a central mediator of cigarette smoke-induced pulmonary inflammation and lower airway destruction. PLoS One 11, e0167169 10.1371/journal.pone.0167169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Durham A. L., and Adcock I. M. (2015) The relationship between COPD and lung cancer. Lung Cancer 90, 121–127 10.1016/j.lungcan.2015.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marusiak A. A., Edwards Z. C., Hugo W., Trotter E. W., Girotti M. R., Stephenson N. L., Kong X., Gartside M. G., Fawdar S., Hudson A., Breitwieser W., Hayward N. K., Marais R., Lo R. S., and Brognard J. (2014) Mixed lineage kinases activate MEK independently of RAF to mediate resistance to RAF inhibitors. Nat. Commun. 5, 3901 10.1038/ncomms4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anastassiadis T., Deacon S. W., Devarajan K., Ma H., and Peterson J. R. (2011) Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045 10.1038/nbt.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davis M. I., Hunt J. P., Herrgard S., Ciceri P., Wodicka L. M., Pallares G., Hocker M., Treiber D. K., and Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 10.1038/nbt.1990 [DOI] [PubMed] [Google Scholar]
- 14. Gallo K. A., and Johnson G. L. (2002) Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat. Rev. Mol. Cell Biol. 3, 663–672 10.1038/nrm906 [DOI] [PubMed] [Google Scholar]
- 15. Morrison D. K. (2012) MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 4, a011254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoang V. T., Yan T. J., Cavanaugh J. E., Flaherty P. T., Beckman B. S., and Burow M. E. (2017) Oncogenic signaling of MEK5-ERK5. Cancer Lett. 392, 51–59 10.1016/j.canlet.2017.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arozarena I., and Wellbrock C. (2017) Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 5, 387 10.21037/atm.2017.06.09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nazarian R., Shi H., Wang Q., Kong X., Koya R. C., Lee H., Chen Z., Lee M. K., Attar N., Sazegar H., Chodon T., Nelson S. F., McArthur G., Sosman J. A., Ribas A., and Lo R. S. (2010) Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977 10.1038/nature09626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi H., Moriceau G., Kong X., Lee M. K., Lee H., Koya R. C., Ng C., Chodon T., Scolyer R. A., Dahlman K. B., Sosman J. A., Kefford R. F., Long G. V., Nelson S. F., Ribas A., and Lo R. S. (2012) Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 3, 724 10.1038/ncomms1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Su F., Viros A., Milagre C., Trunzer K., Bollag G., Spleiss O., Reis-Filho J. S., Kong X., Koya R. C., Flaherty K. T., Chapman P. B., Kim M. J., Hayward R., Martin M., Yang H., et al. (2012) RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 366, 207–215 10.1056/NEJMoa1105358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Villanueva J., Vultur A., Lee J. T., Somasundaram R., Fukunaga-Kalabis M., Cipolla A. K., Wubbenhorst B., Xu X., Gimotty P. A., Kee D., Santiago-Walker A. E., Letrero R., D'Andrea K., Pushparajan A., Hayden J. E., et al. (2010) Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18, 683–695 10.1016/j.ccr.2010.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilson T. R., Fridlyand J., Yan Y., Penuel E., Burton L., Chan E., Peng J., Lin E., Wang Y., Sosman J., Ribas A., Li J., Moffat J., Sutherlin D. P., Koeppen H., et al. (2012) Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487, 505–509 10.1038/nature11249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun C., Wang L., Huang S., Heynen G. J., Prahallad A., Robert C., Haanen J., Blank C., Wesseling J., Willems S. M., Zecchin D., Hobor S., Bajpe P. K., Lieftink C., Mateus C., et al. (2014) Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122 10.1038/nature13121 [DOI] [PubMed] [Google Scholar]
- 24. Prahallad A., Sun C., Huang S., Di Nicolantonio F., Salazar R., Zecchin D., Beijersbergen R. L., Bardelli A., and Bernards R. (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103 10.1038/nature10868 [DOI] [PubMed] [Google Scholar]
- 25. Thumar J., Shahbazian D., Aziz S. A., Jilaveanu L. B., and Kluger H. M. (2014) MEK targeting in N-RAS mutated metastatic melanoma. Mol. Cancer 13, 45 10.1186/1476-4598-13-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data reported are contained within the manuscript.