

Abstract
Most malaria deaths are caused by the protozoan parasite Plasmodium falciparum. Its life cycle is regulated by a cGMP-dependent protein kinase (PfPKG), whose inhibition is a promising antimalaria strategy. Allosteric kinase inhibitors, such as cGMP analogs, offer enhanced selectivity relative to competitive kinase inhibitors. However, the mechanisms underlying allosteric PfPKG inhibition are incompletely understood. Here, we show that 8-NBD-cGMP is an effective PfPKG antagonist. Using comparative NMR analyses of a key regulatory domain, PfD, in its apo, cGMP-bound, and cGMP analog–bound states, we elucidated its inhibition mechanism of action. Using NMR chemical shift analyses, molecular dynamics simulations, and site-directed mutagenesis, we show that 8-NBD-cGMP inhibits PfPKG not simply by reverting a two-state active versus inactive equilibrium, but by sampling also a distinct inactive “mixed” intermediate. Surface plasmon resonance indicates that the ability to stabilize a mixed intermediate provides a means to effectively inhibit PfPKG, without losing affinity for the cGMP analog. Our proposed model may facilitate the rational design of PfPKG-selective inhibitors for improved management of malaria.
Keywords: cyclic GMP (cGMP), malaria, nuclear magnetic resonance (NMR), signaling, plasmodium, protein kinase G (PKG), 8-NBD-cGMP, CNB, cyclic nucleotide–binding domain (CBD), kinase signaling, PfPKG
Introduction
Malaria is a serious health risk for about 40% of the world's population, with more than 200 million diagnoses and 400,000 deaths globally estimated in 2018 (1). Most malarial deaths are caused by Plasmodium falciparum with life cycles in both the human and the mosquito hosts (2, 3). The regulation of such cycles relies on cyclic nucleotide signaling pathways (4). Specifically, the P. falciparum guanosine-3′,5′-cyclic monophosphate (cGMP)2-dependent kinase (PfPKG) plays a major role in both replication and transmission of the Plasmodium parasite, including exflagellation and gametogenesis in the asexual blood stage, invasion of hepatocytes by sporozoites, and gliding motility of ookinetes (5–9). Hence, PfPKG has emerged as a promising target for development of antimalaria therapies complementary to current malaria drugs, whose effectiveness is limited by increasing resistance (2, 10, 11). Furthermore, PfPKG inhibition is an effective and novel strategy not only for malaria treatment, but also for malaria prophylaxis (2, 6, 12–15).
One strategy to inhibit PfPKG is through targeting the active site in the kinase domain. Inhibitors that target such kinase active sites are effective, but poorly selective, because the active sites are highly conserved among eukaryotic kinases (16). One approach to circumvent this is to target allosteric sites, such as the cGMP-binding sites in PfPKG.
PfPKG consists of an N-terminal regulatory region followed by a C-terminal catalytic domain (Fig. 1A), similar to mammalian PKG (17–19). In the absence of cGMP, the regulatory region of PfPKG autoinhibits the catalytic domain. Upon cGMP binding to the regulatory region, the PfPKG autoinhibition is allosterically removed, resulting in activation. The regulatory domain is composed of four cGMP-binding domains (CBDs): A, B, C, and D (Fig. 1A). CBD-C is degenerate and does not bind to cGMP (20). CBD-D (here referred to as “PfD”) is adjacent to the catalytic domain (Fig. 1A) and exhibits the highest affinity and selectivity toward cGMP, with a Kd of 40 nm, compared with Kd ≥ 1 μm for CBD-A and CBD-B (14). In addition, the construct spanning only PfD and the catalytic domain displays low basal activity in the apo state and is efficiently activated by cGMP (21). Hence, PfD serves as the critical controlling unit for both PfPKG autoinhibition and activation.
Figure 1.

Structural architecture of PfCBD-D and hypothetical models of inhibition. A, domain organization of PfPKG, including a regulatory region with four CBDs and a C-terminal catalytic kinase domain. CBD-C is degenerate and does not bind cGMP. B, structures of apo (red) and cGMP-bound (green) CBD-D (PfD). The invariant β-subdomain is gray in both structures. C, cGMP-binding pocket of PfD and selected cGMP-interacting residues. Black dashed lines, hydrogen bonds. Red dashed line, hydrophobic interaction. D, similar to B but with 180º vertical rotation, highlighting the Tyr480–Arg528 interaction. E, four-state thermodynamic equilibrium of the essential regulatory CBD (i.e. PfD). The apo PfD samples an autoinhibitory equilibrium between inactive and active states, which is coupled to the cGMP-binding equilibrium. F and G, hypotheses to explain PfPKG inhibition, as reversal of a two-state equilibrium (F) or stabilization of an intermediate with mixed active and inactive features and diminished activation competency (G). H–K, base-substituted cGMP analogs investigated here. L, maximum PfPKG kinase activities induced by cGMP and the cGMP analogs in I–K. Error bars represent the S.D.
PfD is composed of a contiguous β-subdomain and noncontiguous α-subdomain (Fig. 1B) (14, 22). The β-subdomain forms a β-barrel critical for cGMP recognition and includes the base-binding region (BBR) and the phosphate-binding cassette (PBC) (Fig. 1, B and C), whereas the α-subdomain mediates inhibitory interactions with the catalytic domain and includes three N-terminal and two C-terminal helices, called the N3A motif and αB-αC, respectively (Fig. 1, B and D). In the apo form, the N3A contacts the β-barrel, whereas the C-terminal helices point away from it (Fig. 1B) (14). Upon cGMP binding, the N3A moves away from the β-subdomain, whereas the C-terminal helices are recruited to it by an arginine residue in the PBC, Arg484 (Fig. 1B). Arg484 bridges the PBC to two residues in the αC and defines a unique cGMP-capping triad (Fig. 1B), markedly distinct from human PKG, in which cGMP is capped by a Tyr in the C-terminal helix (23). Disruption of the PfPKG-specific capping triad by mutations blocks proliferation in P. falciparum (14), confirming that PfD is an excellent target for treating malaria. Additionally, Arg528 in the αC plays a pivotal role in regulating PfPKG activity by controlling the αC conformation through a hydrogen bond with Tyr480, which stabilizes the active conformation (Fig. 1D) (14, 21, 24).
The allosteric transition of PfD provides a simple model to rationalize the cGMP-dependent activation of the full-length protein through a four-state allosteric thermodynamic cycle, which arises from the coupling of the PfD inactive-active autoinhibitory equilibrium and the apo-holo cGMP-binding equilibrium (Fig. 1E) (25–27). Of the four states (Fig. 1E), the holo-inactive is particularly relevant for inhibition. This state is selectively stabilized by partial agonists and/or antagonists, which decouple binding from the intramolecular allosteric pathways necessary for activation. We hypothesize that the stabilization of the holo-inactive state arise from a reversal of the two-state inactive/active conformational equilibrium (Fig. 1F) or from a multistate equilibrium sampling a distinct intermediate conformation that exhibits components of both the inactive and the active states of PfD in different regions (Fig. 1G).
To test these hypotheses and understand the mechanism of action of allosteric partial agonists and antagonists that serve as leads for new PfPKG-selective inhibitors, we use comparative NMR to analyze PfD in its apo, cGMP-bound, and cGMP analog–bound forms. The cGMP analogs investigated here include 8-NBD-cGMP, 8-pCPT-cGMP, and PET-cGMP (Fig. 1, I–K). Our results reveal that 8-NBD-cGMP and 8-pCPT-cGMP significantly reduce the kinase activity and induce major perturbations throughout PfD by stabilizing an additional intermediate conformer that is distinct from both the native apo-inactive and holo-active conformations. This “mixed” intermediate state exhibits inhibitory potential comparable with that of the apo-inactive state, as the C-terminal lid is disengaged, but it preserves an affinity similar to that of cGMP, as the key cGMP-binding regions, including the PBC and the adjacent αB, are still engaged. Hence, the stabilization of an allosterically mixed intermediate emerges as a new strategy for selectively inhibiting PfPKG with high potency and efficacy.
Results and discussion
The apo versus cGMP-bound comparative NMR analyses of PfD reveal pervasive allosteric perturbations
As a first step to probe the allosteric effects of cGMP on the PfD construct (residues 401–542) used in our NMR studies, we acquired NH HSQC spectra of apo and cGMP-bound PfD (Fig. 2A). The cGMP-bound spectrum was assigned through triple-resonance NMR experiments and selective amino acid labeling. However, the apo construct was prone to precipitation during acquisition of 3D NMR data sets. Hence, the apo assignments were obtained by transferring the holo assignments to the apo spectrum through 2D Nz-exchange (Fig. 2, B and D) and 2D difference Nz-exchange experiments acquired in the presence of substoichiometric amounts of cGMP (Fig. 2, C and E). Based on these assignments, the cGMP-induced residue-specific compounded 15N-1H chemical shift changes (ΔCCS) were measured (Fig. 2F).
Figure 2.

The apo versus cGMP-bound comparative NMR analyses of PfD reveal pervasive allosteric perturbations. A, overlay of 15N-1H HSQC spectra of apo (black) and cGMP-bound (green) PfD samples with representative assignments. Nz-exchange (B and D) and Nz-exchange difference experiments (C and E) utilized to transfer the assignments from the cGMP-bound to the apo spectra. F, CCS differences between the apo and cGMP-bound samples. The secondary structure of PfD is depicted at the top of the plot. The cGMP-binding regions BBR and PBC as well as the adjacent β2–3 loop are highlighted in gray background. G, local apo (PDB code 4OFF) versus cGMP-bound (PDB code 4OFG) root mean square deviation for PfD. The secondary structures obtained from the apo and cGMP-bound crystals are reported as red and black dashed lines, respectively. H, Cβ chemical shift values of cGMP-bound PfD obtained from NMR triple-resonance experiments versus the theoretical Cβ chemical shift values predicted form the structure (PDB code 4OFG) using the ShiftX software (54). I, similar to H but for the Cα chemical shifts.
The majority of the residues exhibit significant ΔCCS values upon cGMP binding (Fig. 2F). As expected, significant ppm shifts are observed in the PBC and BBR regions, which directly interact with cGMP, as well as the β2–3 loop, which is adjacent to the PBC (Fig. 2F). Furthermore, major ppm changes are also detected for several α-helical regions, especially the αA, αB, and αC helices (Fig. 2F). The ppm shifts in αB-αC are consistent with the cGMP-dependent structural changes for these regions, as shown by the local root mean square deviation between the apo and cGMP-bound structures (Fig. 2G). The αB-αC structural changes also affect adjacent regions, such as the αA helix and the β8 strand (Fig. 2F), although such regions are not subject to major structural changes upon cGMP binding (Fig. 2G). Hence, the cGMP-dependent ΔCCS values (Fig. 2F) are in full agreement with the structures of apo- and holo-PfD, providing an initial validation of our NMR assignments. Our NMR assignments were further validated by the agreement between the measured and the computed Cα and Cβ chemical shift values based on the apo- and holo-PfD structures (Fig. 2, H and I). Once the NMR assignments were validated, we extended our analyses to evaluate the effects of cGMP analogs.
cGMP analog versus cGMP chemical shift differences report on inhibitory conformational changes of PfD
We focused on three cGMP base analogs with distinct base modifications: 8-NBD-, 8-pCPT-, and PET-cGMP (Fig. 1, I–K), as they act as PfPKG activators to different degrees. Whereas PET-cGMP acts as an agonist, as shown by kinase assays (Fig. 1L), 8-pCPT- and 8-NBD-cGMP elicit progressively increasing potential as antagonists. 8-NBD-cGMP is the most potent inhibitor of PfPKG (Fig. S1), followed by 8-pCPT-cGMP. This range of kinase responses (Fig. 1L) makes this group of cGMP analogs an excellent toolset to investigate the mechanisms underlying the allosteric inhibition of PfPKG. For this purpose, we first measured the cGMP analog versus cGMP differences in compounded chemical shifts (Fig. 3A). The overall distributions of such ppm differences (Fig. 3B) rank in full agreement with the inhibitory potencies of the cGMP analogs (Fig. 1L). Furthermore, the pervasiveness of the effects caused by the cGMP modification (Fig. 3, A and C–E) also reflects the extent of inhibition. The perturbations resulting from the replacement of cGMP with either 8-NBD-cGMP or 8-pCPT-cGMP extend throughout the PfD structure, whereas the least inhibitory analog, PET-cGMP, perturbs only a relatively limited region near the cGMP-binding pocket (Fig. 3, A and C–E).
Figure 3.

cGMP analog versus cGMP chemical shift differences report on inhibitory conformational changes of PfD. A, ΔCCS between cGMP and 8-pCPT-cGMP (orange), 8-NBD-cGMP (red), and PET-cGMP (blue) bound PfD plotted against residue numbers. Dashed line, average ΔCCS + 2 S.D. of PET-cGMP. B, box plot (10–90% percentile) of ΔCCS differences between cGMP-bound and the 8-NBD-cGMP–, 8-pCPT-cGMP–, and PET-cGMP–bound samples. Residues included in the box plot correspond to residues that are assigned in all samples and with at least one of the three ΔCCS > average + 2 S.D. of PET-cGMP (dashed line in A). C–E, residues with CCS changes greater than the dashed line in A mapped onto the active structure of PfD for the 8-NBD-cGMP (C), 8-pCPT-cGMP (D), and PET-cGMP (E) bound samples. Error bars, S.D.
Overall, the data of Fig. 3 suggest that chemical shifts are ideally suited to investigate the dynamical allosteric transitions underlying PfPKG inhibition. This notion is fully consistent with the fast exchange between the inactive and the active states of CBDs (27–33), whereby the observed NMR peak positions are population-weighted averages of the pure inactive and active ppm values. Hence, chemical shifts report on the position of the dynamic inactive-active CBD conformational equilibrium. Chemical shift projection analysis (CHESPA) is an effective means to evaluate the perturbation of such dynamic equilibria caused by ligand modifications (28, 29, 31, 32). In the CHESPA analysis, the NMR peak positions of the cGMP analog–bound sample are evaluated relative to a reference vector connecting the peaks of the apo-inactive and the cGMP-bound active samples (Fig. 4, A and D). Specifically, the angle (θ) arising from the cGMP analog–bound versus cGMP-bound vector relative to the reference vector and the normalized projections onto the reference vector (i.e. fractional shift, X) are quantified through CHESPA (Fig. 4, A and D). The cos(θ) values report on the direction of the cGMP analog–induced shifts, whereas the X values gauge the extent of such shift toward inhibition or activation.
Figure 4.

CHESPA of cGMP analog–bound states reveals a third conformer with a disengaged lid. CHESPA vector scheme, fractional shift (X) and cos(θ) values plotted against residue number for 8-NBD-cGMP (A–C) and for 8-pCPT-cGMP (D–F). The secondary structure of cGMP-bound PfD is depicted at the top of each plot. Pre-lid and lid motifs are highlighted in pink and purple background, respectively. The PBC and BBR are highlighted in gray background. The average 〈X〉 values for residues common in both 8-NBD-cGMP–bound and 8-pCPT-cGMP–bound sample are indicated with a dashed and a solid line for the pre-lid and the lid motifs, respectively. G, distribution of X values for 8-NBD–, 8-pCPT–, and PET-cGMP–bound samples. H and I, HSQC overlay expansions for representative residues in the pre-lid and lid regions of the apo (black) and the 8-NBD-cGMP (red), 8-pCPT-cGMP (orange), PET-cGMP (blue), and cGMP (green) bound samples.
CHESPA of the cGMP analog–bound PfDs reveals the presence of a third “mixed” conformer with an engaged pre-lid but a disengaged lid
The CHESPA of 8-NBD-cGMP shows that the majority of residues exhibit negative X values (Fig. 4B), suggestive of most residues shifting toward the inactive state upon binding of 8-NBD-cGMP. However, the magnitudes of the X values are highly residue-dependent, pointing to deviations from the pure inactive-active two-state equilibrium, which predicts similar X values for residues outside the binding site. Another characteristic of a two-state model is the co-linearity of the perturbation and reference vectors (i.e. |cos(θ)| ∼ 1) for residues outside the binding site. However, the CHESPA of 8-NBD-cGMP reveals that several residues not in the binding site exhibit |cos(θ)| values significantly less than 1 (Fig. 4C), suggesting deviations from the two-state model and pointing to the presence of additional state(s) sampled by the PfD:8-NBD-cGMP complex. Similar trends are observed for the 8-pCPT-cGMP–bound sample (Fig. 4, E and F).
The presence of multiple conformational equilibria is further supported by the width of the X value distributions (Fig. 4G). The X distributions measured for 8-NBD-cGMP– and 8-pCPT-cGMP–bound PfD are significantly wider compared with PET-cGMP (Fig. 4G), which is nearly a full agonist and samples mainly the active state (Fig. S2). These observations suggest that the PfD:8-NBD-cGMP and PfD:8-pCPT-cGMP complexes sample a multistate ensemble, including not only the inactive and active conformers represented by the apo and cGMP-bound crystal structures, but also an additional state or states.
To simplify the analysis, we focused on the C-terminal helices that are directly linked to the catalytic domain and are one of the allosteric elements most critical in the inhibition and activation of the kinase (14, 21, 24). The C-terminal helices span two regions with clearly distinct average X values (〈X〉) in the PfD:8-NBD-cGMP complex: residues 515–530, denoted as the pre-lid with 〈X〉 = −0.48, and residues 534–535, denoted as the lid with 〈X〉 = −0.88 (Fig. 4, B, H, and I). This means that for the PfD:8-NBD-cGMP complex, the pre-lid only shifts about halfway toward the inactive state, whereas the lid region shifts almost completely to the inactive state. A similar trend is observed for the PfD:8-pCPT-cGMP complex with average X values of −0.30 and −0.64 in the pre-lid and the lid regions, respectively.
One of the simplest models to explain both the enzymatic and the NMR data in Figs. 1L and 4, respectively, is a three-state equilibrium mechanism involving an inactive, an active, and an intermediate state (Fig. 5A). The inactive state is similar to the apo-PfD structure, where both the pre-lid and the lid are disengaged. The active state is similar to the cGMP-bound structure, where both the pre-lid and the lid are engaged. The intermediate state exhibits a mixed character, as the pre-lid is engaged similarly to the active state, but the lid is disengaged, similarly to the inactive state (Fig. 5A). Next, we checked whether it is possible to utilize this three-state model to back-calculate the relative kinase activities.
Figure 5.
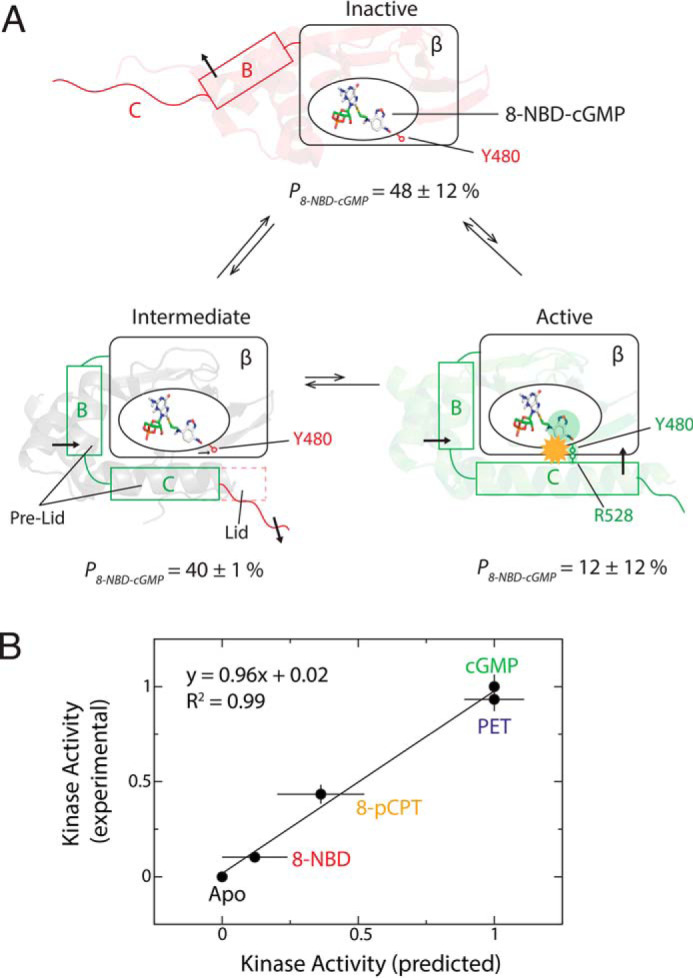
Proposed model for PfD inhibition by 8-NBD-cGMP and comparison of calculated versus measured kinase activities. A, inhibition occurs through a three-state equilibrium, sampling a distinct holo-inactive intermediate state, where the pre-lid and PBC are engaged, similarly to the active state, but the lid is disengaged, similarly to the inactive state. The population of each state (P) is indicated. The structural shifts of the αB and αC helices are indicated with black arrows. The β-barrel is indicated with a rectangular box. The yellow starburst indicates the steric clash between the 8-NBD substituent and the Arg528 side chain. B, correlation plot of experimental versus predicted relative kinase activities. The kinase activity prediction is based on the populations of inactive, intermediate, and active states (Table S1). Error bars for the experimental kinase activity represent the standard deviation of multiple measurements, and for the predicted kinase activity represent the errors propagated from the errors on the populations, which are determined from the standard deviation of the fractional shift (X) values of the residues used to calculate the average X values.
The CHESPA-based three-state model of PfD inhibition explains the experimental kinase activities
Using the average X values for the pre-lid and the lid regions (Fig. 4, B and E) and Fig. S2A), the populations of the inactive, intermediate, and active states were estimated. Because the pre-lid is disengaged only in the inactive state, the average X of the pre-lid reflects the population of the inactive state. Similarly, the lid is engaged only in the active state. Hence, the population of the active state is estimated as 1 − 〈X〉lid. The population of the intermediate is the percentage not accounted for by the other two states. Using this approach, the populations of the inactive, intermediate, and active states for the PfD:8-NBD-cGMP complex are 48 ± 12, 40 ± 12, and 12 ± 1%, respectively (Fig. 5A and Table S1). Similarly, the three state populations can be calculated for the 8-pCPT-cGMP– and PET-cGMP–bound PfD complexes based on the respective CHESPA data (Fig. 4E, Fig. S2, and Table S1). Using these populations and assuming that the inactive and intermediate states are fully inhibitory, given the lid disengagement (14), while the active state is noninhibitory, the relative kinase activity at ligand saturation induced by these cGMP analogs was predicted. The predicted activation values correlate well with the experimental kinase activity values (Fig. 5B).
The ability of our three-state model to recapitulate the enzymatic data has three key implications. First, the measurements on the PfD construct are relevant for the longer PfPKG construct used in the kinase assays to probe the PfPKG function. Second, the NMR chemical shift projection analysis and the resulting fractional shift values are suitable for predicting the populations of different functional states. Third, the mixed intermediate state, where only the lid region is disengaged, is inhibitory to an extent similar to the inactive state, further corroborating the pivotal role of the lid region in activation. Hence, we further probed the inhibitory mixed intermediate stabilized by the 8-NBD-cGMP analog.
8-NBD-cGMP binds to PfD in a syn conformation and perturbs interactions critical for activation
As a first step toward investigating the mixed intermediate, we examined the base orientation of 8-NBD-cGMP as bound to PfD. Typically, the base of cyclic nucleotides is oriented either in syn or anti relative to the ribose ring (34–38). To determine whether the guanine base of 8-NBD-cGMP is syn or anti, we could not rely on NOE-based assessments, as this cGMP analog lacks nonexchangeable protons in the base. Hence, we focused on the chemical shifts of the Cγ2 methyl of Ile465, which in the PfD:cGMP complex is in direct contact with the guanine base, and of the Cγ methyl of Thr493, whose hydroxyl interacts with the amino group of cGMP (Fig. 1C). The chemical shifts of both Ile465 Cγ2 and Thr493 Cγ show only marginal changes when cGMP is replaced by 8-NBD-cGMP (Fig. 6, A and B), suggesting that the 8-NBD-cGMP guanine base binds in a syn conformation, similar to cGMP. This conclusion was confirmed also by the 3D map of 8-NBD-cGMP versus cGMP NH CCS changes for residues in the β-barrel (Fig. 6C, red surface). Because residues in the β-barrel do not undergo significant structural changes upon cNMP binding, the perturbations sensed by the ΔCCS report mainly on direct binding contacts (33, 39–41). The ΔCCS map is fully consistent with 8-NBD-cGMP binding in syn rather than anti guanine orientation (Fig. 6C, residues within the red surface). Similarly, we confirmed through the β-barrel ΔCCS map that a syn orientation is preserved also for the guanine base of PET-cGMP (Fig. 6D, blue surface). For this analog, the substitution is at the 1- and 2- rather than the 8-position (Fig. 1, I and K), causing the most significant CCS changes to cluster at the opposite side of the β-barrel for PET-cGMP compared with 8-NBD-cGMP, when both analogs maintain a syn guanine orientation (Fig. 6, C and D, blue versus red surfaces).
Figure 6.

Syn conformation of 8-NBD-cGMP bound to PfD and effect of 8-NBD on the Tyr480–Arg528 interaction. A and B, overlaid 13C-1H HSQC spectral expansions of apo (black), cGMP-bound (green), and 8-NBD-cGMP–bound (red) PfD, zoomed into Ile465 (13C2) (A) and Thr493 (13CY) (B), which interact with the guanine base of cGMP. The apo peak for Thr493 (13CY) could not be assigned. C and D, map of residues in the β-barrel that exhibit ΔCCS relative to cGMP-bound PfD greater than the average ΔCCS + 2 S.D. of the PET-cGMP versus cGMP ΔCCS for the 8-NBD-cGMP-bound (C) and PET-cGMP–bound (D) PfD. Circles indicate the cGMP portion of the analog in its syn orientation (PDB code 4OFG). The dashed circles represent the region where the base substituents (i.e. 8-NBD or PET) are expected to be located when the cGMP portion of the analogs is in the syn orientation. E, overlay of the structure of cGMP:PfD (PDB code 4OFG) and representative structures of 8-NBD-cGMP:PfD generated from MD simulations (Table S2). The cGMP (dark gray) aligns with the cGMP portion of the 8-NBD-cGMP (green, carbon; white, hydrogen; blue, nitrogen; red, oxygen; yellow, sulfur; orange, phosphorus). F, aligned representative structures of 4OFG (gray) and 8-NBD-cGMP:PfD (red) generated from MD simulations. The dashed box indicates the portion of the C-terminal αC helix that becomes disordered in the 8-NBD-cGMP:PfD intermediate. G, similar to F, but zoomed into the Tyr480–Arg528 region. The yellow starburst indicates the steric clash of 8-NBD moiety with the Arg528 side chain in the 4OFG structure. The arrow indicates the structural shift of the Arg528 side chain upon binding of 8-NBD-cGMP. H, 1H-15N HSQC spectra of apo (black), cGMP-bound (green), and 8-NBD-cGMP-bound (red) samples overlaid and zoomed in on Tyr480. I, WT versus R528K CCS differences for cGMP-bound (green) and 8-NBD-cGMP–bound (red) samples. Residues assigned in both the cGMP-bound and the 8-NBD-cGMP–bound samples are plotted. The distance (Å) measured from Arg528 using the cGMP-bound structure (PDB code 4OFG) is shown in a gray line. The secondary structure of cGMP-bound PfD is depicted at the top of each plot. The highlighted region indicates residues near Tyr480, where the perturbation induced by the R528K mutation in the cGMP-bound complex is lost when cGMP is replaced by 8-NBD-cGMP. Black arrow, Tyr480; black star, Arg528.
Although 8-NBD-cGMP preserves a syn guanine orientation when bound to PfD, similar to cGMP, the introduction of the 8-NBD moiety has a profound effect on the conformational ensemble accessed by PfD, as shown by the NMR-based three-state model of Fig. 5A. Of the three PfD conformers in Fig. 5A, the mixed intermediate is the least characterized. Hence, we utilized MD simulation to further investigate this intermediate. For this purpose, we built a hybrid structure to resemble the intermediate state proposed based on the CHESPA data (Fig. 5A), relying on the cGMP-bound coordinates (PDB code 4OGF) (14) for residues 401–532 and on the apo coordinates (PDB code 5DYK) (24) for residues 534–542, which span the lid region. The hybrid model provided the initial coordinates for a 1-μs MD trajectory, and Fig. 6 (E and F) shows representative structures selected based on the cluster analysis of such trajectory (Table S2).
In agreement with the data of Fig. 6 (A–C), Fig. 6E shows that the PfD-bound 8-NBD-cGMP preserves a syn guanine orientation. Fig. 6F illustrates that the lid region of the intermediate state is disengaged, whereas the pre-lid is engaged. Fig. 6G shows that when the intermediate conformation is overlaid with the cGMP-bound structure (PDB code 4OFG), the Arg528 side chain of 4OFG clashes with the NBD moiety of 8-NBD-cGMP (Fig. 6G, gray sticks). The steric clash explains why in the simulated intermediate structure, the Arg528 side chain moves away from the 8-NBD-cGMP and rotates outward, breaking the interaction with Tyr480 (Fig. 6G, red sticks). Considering that the Tyr480–Arg528 interaction is critical for activation (14, 21), the perturbation of Arg528 caused by 8-NBD is consistent with the lack of kinase activity observed upon replacement of cGMP with 8-NBD-cGMP.
This perturbation revealed by the MD simulations is consistent with the significant 8-NBD-cGMP versus cGMP chemical shift changes occurring at or near Tyr480 (Fig. 3A). The Tyr480 NH HSQC cross-peak of the cGMP:PfD complex moves completely toward the apo position when cGMP is replaced by 8-NBD-cGMP (Fig. 6H), confirming that the 8-NBD moiety perturbs the Tyr480–Arg528 interaction. To further corroborate this result, we generated the R528K mutant and compared the R528K versus WT chemical shifts for both cGMP– and 8-NBD-cGMP–bound samples (Fig. 6I). For the cGMP:PfD complex, the largest R528K versus WT ΔCCS values cluster primarily close to Tyr480 (Fig. 6I), as expected based on the Arg528–Tyr480 interaction observed in the cGMP:PfD structure (14). However, when cGMP is replaced by 8-NBD-cGMP, the R528K versus WT CCS changes in the vicinity of Tyr480 are markedly suppressed (Fig. 6I), confirming that in the 8-NBD-cGMP:PfD complex, the Arg528–Tyr480 interaction is significantly weakened compared with the cGMP:PfD structure.
Similar analyses were carried out to probe the effect of 8-NBD-cGMP on the capping triad interactions between Arg484, Gln532, and Asp533, which are also critical for activation (14). We found that the Arg484–Asp533 interaction is perturbed in the simulated intermediate structure of the 8-NBD-cGMP:PfD complex (Fig. S3A). To confirm such perturbation, we measured the CCS changes between WT and the R484A mutant for both the cGMP– and 8-NBD-cGMP–bound samples (Fig. S3B). Fig. S3B reveals that the R484A versus WT CCS changes in the C-terminal helices, especially in the vicinity of Gln532–Asp533, are suppressed when cGMP is replaced with 8-NBD-cGMP, confirming that 8-NBD-cGMP perturbs the capping interaction between Arg484 and the C-terminal helices. These results are in full agreement with the MD simulations, suggesting that the 8-NBD substituent destabilizes also the capping triad.
Hence, we conclude that upon introduction of the NBD group at the 8-guanine position, the syn base orientation is preserved but the Tyr480–Arg528 contact and the capping triad interactions are significantly perturbed, thus compromising the full engagement of the C-helix and explaining why 8-NBD-cGMP functions as an effective allosteric antagonist of the PfPKG kinase.
Sampling a mixed intermediate state is beneficial for maximizing kinase inhibition while minimizing affinity losses
The three-state model with the mixed intermediate state featuring an engaged pre-lid and a disengaged lid (Fig. 5A) provides an excellent strategy for PfPKG kinase inhibition without significantly compromising affinity. Reconciling high inhibitory efficacy and potency would be more challenging for a simple two-state model. In this case, the disengagement of the lid necessary for inhibition also implies the concurrent disengagement of other PfD elements necessary for high-affinity binding, such as the PBC in its entirety. On the contrary, in the mixed intermediate state (Fig. 5A), which exhibits an inhibitory effect similar to that of the inactive state, it maintains the pre-lid and the adjacent PBC engaged. For example, whereas the capping carboxylate of Asp533 is displaced away from Arg484 in the PBC, the side chain of this arginine in the mixed intermediate remains in an orientation similar to the active conformation (Fig. S3A), thus minimizing perturbations of the interactions with the cyclic nucleotide. Hence, based on the three-state model of Fig. 5A, we hypothesize that the 8-NBD-cGMP preserves an affinity comparable with the endogenous allosteric effector, cGMP.
Our hypothesis is confirmed through surface plasmon resonance (SPR) measurements (Fig. 7) showing that the affinities of 8-NBD-cGMP and cGMP are comparable (i.e. Kd = 59 ± 4 nm versus 51 ± 7 nm; Table S3), despite their markedly different efficacies in terms of kinase activation (Fig. 1L and Fig. S1A). This conservation of affinity is especially important, because 8-NBD-cGMP acts as a competitive antagonist (Fig. S1B). Hence, 8-NBD-cGMP serves as an excellent example for how sampling a mixed intermediate state enables significant inhibition without major affinity losses by selectively disengaging only structural units strictly necessary for kinase activation. Similar inhibition strategies are anticipated to inform the design of future PfPKG allosteric inhibitors, which may serve as anti-malaria drug leads.
Figure 7.

Surface plasmon resonance binding studies of cGMP and 8-NBD-cGMP to PfPKG 401–853 (PfD-catalytic domain) demonstrate similar binding kinetics. Direct binding assays were performed by capturing polyhistidine-tagged PfPKG 401–853 on a Ni-NTA sensor chip and injecting a dilution series of cGMP for 45 s (A) and 8-NBD-cGMP for 70 s (B), respectively. Rate and equilibrium binding constants are reported in Table S3. A Langmuir 1:1 model was applied for fitting.
Experimental procedures
Expression and purification
The PfPKG CBD-D construct (residues 401–542) (i.e. PfD) within a His-tagged expression vector pQTEV (14) was expressed in the Escherichia coli strain BL21 (DE3). The transformed E. coli was grown in a minimal medium enriched with 15NH4Cl and [13C]glucose and supplemented with d-biotin, thiamine HCl, MgSO4, and CaCl2. The cells were grown at 37 °C to an optical density of 0.7 (at λ = 600 nm), induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and then incubated for an additional 18 h at 18 °C. Cells were harvested, resuspended in the lysis buffer (50 mm Tris, pH 7.4, 500 mm NaCl, 1 mm BME), and lysed using a cell disruptor. The cell debris was removed through centrifugation, and then the supernatant was loaded onto an Ni2+-Sepharose resin. The cell lysate was allowed to flow through, and the column was rinsed with 50 mm Tris, pH 7.4, 500 mm NaCl, 1 mm BME, and 20 mm imidazole. After thorough rinsing, the protein was eluted with 50 mm Tris, pH 7.4, 500 mm NaCl, 1 mm BME, and 300 mm imidazole. The eluted protein was then dialyzed in 50 mm Tris, pH 7.4, 100 mm NaCl, 1 mm BME with tobacco etch virus protease for 24 h at 4 °C. The cleaved products were loaded on the Ni2+-Sepharose resin. The protein without the His-tag was obtained through collecting the flow-through and was further purified with a HiLoad 16/60 Superdex 75 gel filtration column (GE Healthcare) equilibrated with NMR buffer (50 mm Tris, pH 7, 100 mm NaCl, 10 mm MgCl2, 1 mm DTT). The His-tagged PfPKG 401–853 construct including the catalytic domain was expressed in E. coli TP2000 and purified using Ni-NTA and size-exclusion chromatography, as described previously (21).
NMR spectroscopy
NMR experiments were performed on a Bruker Avance 700-MHz spectrometer equipped with a 5-mm TCI cryoprobe or a Bruker Avance 850-MHz spectrometer equipped with a triple-resonance TXI probe. All experiments were acquired at 306 K in NMR buffer (50 mm Tris, pH 7, 100 mm NaCl, 10 mm MgCl2, 1 mm DTT) with 5% D2O. All NMR spectra were processed with NMRPipe (42) and analyzed using Sparky (43). Three-dimensional triple-resonance NMR experiments (i.e. HNCACB, CBCA(CO)NH, HNCA, HN(CO)CA, HNCO, HCCH-TOCSY, and (H)CC(CO)NH) were acquired and used to assign the two-dimensional 1H-15N HSQC and 1H-13C HSQC NMR spectra of cGMP-bound PfD. Because the apo sample was prone to precipitation, 2D Nz-exchange, 2D difference Nz-exchange, and 2D Cz-exchange experiments with mixing time of 250 ms were used to transfer the chemical shift assignments from the cGMP-bound PfD to the apo-PfD spectra. The cGMP analog–bound spectra were assigned through spectral comparison with cGMP-bound spectra, if no ambiguities were present, and through 2D Nz exchange and 2D difference Nz exchange with the apo state. Chemical shift assignments of R528K:cGMP and R528K:8-NBD-cGMP were obtained through spectral comparison with WT spectra if no ambiguities were present.
Chemical shift analyses
All spectra utilized for chemical shift analyses were acquired with 100 μm PfD and saturating amounts of cGMP or cGMP analogs (2 mm) or without any cNMP addition for apo spectra. For accurate chemical shift comparisons, an internal reference (i.e. 15N-labeled N-acetylglycine) was used to align the spectra in Sparky, and the resulting chemical shift values were used to compute the compounded chemical shift differences and perform CHESPA following protocols described previously (29, 30, 34, 44, 45). The fractional shift (X) values and cos(θ) values were computed for residues with cGMP analog–bound versus cGMP-bound CCS difference >0.05 ppm. The average 〈X〉 values for the pre-lid and the lid were calculated using residues that were assigned in all five samples (i.e. apo, cGMP-bound, 8-NBD-cGMP-bound, 8-pCPT-cGMP-bound, and PET-cGMP-bound PfD), had X values between 0 and −1, and had ΔCCS (cGMP analog–bound versus cGMP-bound) values greater than 0.05 ppm, ensuring reliable X and cos(θ) values. The distribution of X values for cGMP analogs was computed using the residues that exhibited |cos(θ)| > 0.75 to minimize biases from nearest-neighbor effects.
Site-directed mutagenesis
The R528K mutation was created through site-directed mutagenesis using a KOD Hot Start Master mix (Novagen) and primer pairs (synthesized by IDT): forward, CTGGCCCACTTGGAAGAGAAGATTAAGATGCAGGATAC; reverse, GTATCCTGCATCTTAATCTTCTCTTCCAAGTGGGCCAG. The reaction protocol provided by the KOD Hot Start kit was followed. Template DNA was degraded with DpnI for 2 h at 37 °C, and the reaction product was used for transformation of E. coli top10 cells. The transformed colony was grown in lysogeny broth or Luria-Bertani medium medium for 18 h at 37 °C. Plasmids were isolated from the E. coli using the GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific). The purified plasmids were sequenced to verify the mutation. The expression and purification of PfD R528K were carried out using the same protocol as for the WT PfD.
Kinase assays
Specific kinase activities were determined using an enzyme-coupled spectrophotometric assay (46). In a quartz cuvette, purified PfPKG 401–853 was mixed with the reaction buffer (100 mm MOPS, pH 7.0, 10 mm MgCl2, 5 mm β-mercaptoethanol, 1 mm ATP, 1 mm phosphoenolpyruvate, 230 μm NADH, 15 units/ml lactate dehydrogenase, 8.4 units/ml pyruvate kinase). The reactions were started by adding 1 mm PKStide (GRTGRRNSI; GeneCust, Luxembourg) and monitored in the absence and presence of a 100 μm concentration of each cyclic nucleotide (cGMP, 8-pCPT-cGMP, and PET-cGMP) for at least 1 min. Specific kinase activity was calculated according to the Lambert–Beer law.
Kinase activity induced by 8-NBD-cGMP was measured by microfluidic mobility shift assay using a Caliper DeskTop Profiler (PerkinElmer Life Sciences). Here, 100 μm cGMP or 8-NBD-cGMP was mixed with assay buffer (20 mm MOPS (pH 7), 300 mm NaCl, 10 mm MgCl2, 1 mm ATP, 1 mm DTT, 990 μm PKStide, 10 μm FITC-PKStide (FITC-GRTGRRNSI; GeneCust, Luxembourg, 0.05% L-31) and a fixed protein concentration and continued as described previously (21). The assay was also conducted with multiple concentrations of 8-NBD-cGMP or cGMP, ranging from 0.1 nm to 300 μm. For the competition assay, the protein was preincubated with 350 nm cGMP, varying concentrations of 8-NBD-cGMP were added to the reaction, and substrate conversion was monitored over 2 h. The combined kinase inhibition data were further analyzed and normalized using GraphPad Prism 8.3.0 (GraphPad Software).
SPR
Direct binding data were obtained via SPR using a Biacore T200 (GE Healthcare Life Sciences). Briefly, 200–300 RU of Ni2+ (5 mm NiCl2 in running buffer) were immobilized on a poly-NTA–derivatized sensor chip (NIHC 1000M; XanTec Bioanalytics GmbH). Subsequently, His-tagged PfPKG 401–853 in running buffer (20 mm HEPES, 150 mm NaCl, 50 μm EDTA, 0.01% P20, pH 7.3) was captured at a flow rate of 10 μl/min to an immobilization level of 500–2500 RU. Next, association was measured by injection of a dilution series of the respective cyclic nucleotide for 45–75 s. Then running buffer was injected for 60–180 s to initiate dissociation. Regeneration of the sensor chip surface was achieved by consecutively injecting 0.5 m EDTA (pH 8) for 600 s, followed by injections of 3 m guanidinium HCl, 0.5% SDS, and 3 m guanidinium HCl for 240 s each. Unless stated otherwise, all kinetic measurements were performed at 25 °C in running buffer and a flow rate of 50 μl/min. Data were evaluated with the Biacore T200 Evaluation Software 3.0 using a global fit and assuming a 1:1 Langmuir binding model. Further data processing was performed using GraphPad Prism 8.3.0 (GraphPad Software).
Molecular dynamic simulations: initial model preparation
The structure of 8-NBD-cGMP in a syn conformation was generated using RDKit (47). A model of the mixed intermediate state sampled by the PfD:8-NBD-cGMP complex was then built using residues 401–533 from the active PfD:cGMP crystal structure (PDB code 4OFG) (14) and lid region residues 534–542 from the inactive apo-PfD crystal structure (PDB code 5DYK) (24). The 8-NBD-cGMP ligand was docked into the resulting mixed structure of PfD using AutoDock4 with default parameters generated by AutoDockTools (48). Residues 463–494, where the PBC and BBR regions are located, were assigned as the binding pocket for AutoDock. The structure in which the ribose-phosphate and guanine moieties of the docked ligand were best aligned with the position of cGMP in the cGMP-bound active structure (PDB code 4OFG) (14) was selected as the initial coordinates for MD simulations.
Molecular dynamics simulation protocol
All simulations were performed using the AMBER 16 software PMEMD.CUDA (49) on the Shared Hierarchical Academic Research Computing Network (SHARCNET). The Amber ff99SBnmr force field was employed for the protein, whereas RESP charge and the general Amber force field were applied to generate the parameters for 8-NBD-cGMP using the Gaussian09 program (50) and the antechamber module implemented in AMBER 16, as described previously (51). The TIP3P water model was applied in the form of a rectangular MD solvent box surrounding the PfD:8-NBD-cGMP structure, with a minimum distance of 12 Å between the PfD:8-NBD-cGMP structure and the box edge. To mimic the experimental conditions, the charged residues and N/C termini were set to the protonation states expected at pH 7, following a protocol similar to that implemented previously (52), and Na+ and Cl− ions were added to mimic a 100 mm salt concentration.
The simulations started with an energy minimization performed with harmonic restraints on the protein backbone (with a force constant of 5 kcal/mol·Å2). The system was then gradually heated from 0 to 100 K at constant volume and then heated from 100 to 306 K in the NPT ensemble, and all of the heating process employed harmonic restraints on protein backbone (with a force constant of 3 kcal/mol·Å2) for a total of 1 ns followed by an equilibration period of 1.2 ns with main-chain atoms restrained with a force constant of 1 kcal/mol·Å2. Then a 1-μs production MD trajectory was generated in the NPT ensemble at 306 K and 1 atm with a weak-coupling algorithm, saving structures every 10 ps for subsequent analysis. A cluster analysis for 20,000 frames recorded from the MD trajectory (i.e. the structures recorded at 50-ps intervals) was performed, with β-core alignment, using the CPPTRAJ protocol (53).
Data availability
The NMR chemical shift assignment for cGMP-bound PfD has been deposited in the Biological Magnetic Resonance Data Bank (BMRB) with the code 50203. All remaining data are contained within the article.
Author contributions
J. A. B. and G. M. conceptualization; J. A. B., K. V., J. H., P. H., and E. F. data curation; J. A. B., K. V., J. H., P. H., F. W. H., and G. M. formal analysis; J. A. B., F. W. H., and G. M. validation; J. A. B. and G. M. visualization; J. A. B., M. A., and G. M. methodology; J. A. B. and G. M. writing-original draft; J. A. B. and G. M. writing-review and editing; F. W. H. and G. M. supervision; F. W. H. and G. M. funding acquisition; C. K. resources; G. M. investigation; G. M. project administration.
Supplementary Material
Acknowledgments
We thank Dr. B. VanSchouwen, Dr. S. Boulton, H. Shao, R. Ahmed, N. Jafari, and Dr. S. S. Taylor for helpful discussions. Furthermore, we thank A. Morell and K. Zupfer for help with protein purification and kinase activity assays.
This work was supported by Canadian Institutes of Health Research Grant 389522 (to G. M.), Natural Sciences and Engineering Research Council of Canada Grant RGPIN-2019-05990 (to G. M.), Deutsche Forschungsgemeinschaft (DFG) Grant He1818/10 (to F. W. H.), and the funding line Future “PhosMOrg” and Graduate School “Clocks” of the University of Kassel (to F. W. H.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains Tables S1–S3 and Figs. S1–S3.
- cGMP
- guanosine-3′,5′-cyclic monophosphate
- PfPKG
- P. falciparum cGMP-dependent kinase
- 8-NBD-cGMP
- 8-(2-[7-nitro-4-benzofurazanyl]aminoethylthio)guanosine-3′,5′-cyclic monophosphate
- 8-pCPT-cGMP
- 8-(4-chlorophenylthio)guanosine-3′,5′-cyclic monophosphorothioate
- PET-cGMP
- β-phenyl-1,N2-ethenoguanosine-3′,5′-cyclic monophosphate
- CBD
- cyclic nucleotide–binding domain
- BBR
- base-binding region
- PBC
- phosphate-binding cassette
- 2D
- two-dimensional
- CHESPA
- chemical shift projection analysis
- PDB
- Protein Data Bank
- HSQC
- heteronuclear single quantum coherence
- SPR
- surface plasmon resonance
- BME
- β-mercaptoethanol
- CCS
- compounded 15N-1H chemical shift
- NTA
- nitrilotriacetic acid.
References
- 1. World Health Organization (2019) World Malaria Report 2019, World Health Organization, Geneva [Google Scholar]
- 2. Cowman A. F., Healer J., Marapana D., and Marsh K. (2016) Malaria: biology and disease. Cell 167, 610–624 10.1016/j.cell.2016.07.055 [DOI] [PubMed] [Google Scholar]
- 3. Doerig C., Billker O., Pratt D., and Endicott J. (2005) Protein kinases as targets for antimalarial intervention: kinomics, structure-based design, transmission-blockade, and targeting host cell enzymes. Biochim. Biophys. Acta 1754, 132–150 10.1016/j.bbapap.2005.08.027 [DOI] [PubMed] [Google Scholar]
- 4. Brochet M., and Billker O. (2016) Calcium signalling in malaria parasites. Mol. Microbiol. 100, 397–408 10.1111/mmi.13324 [DOI] [PubMed] [Google Scholar]
- 5. Hopp C. S., Bowyer P. W., and Baker D. A. (2012) The role of cGMP signalling in regulating life cycle progression of Plasmodium. Microbes Infect. 14, 831–837 10.1016/j.micinf.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Taylor H. M., McRobert L., Grainger M., Sicard A., Dluzewski A. R., Hopp C. S., Holder A. A., and Baker D. A. (2010) The malaria parasite cyclic GMP-dependent protein kinase plays a central role in blood-stage schizogony. Eukaryot. Cell 9, 37–45 10.1128/EC.00186-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moon R. W., Taylor C. J., Bex C., Schepers R., Goulding D., Janse C. J., Waters A. P., Baker D. A., and Billker O. (2009) A cyclic GMP signalling module that regulates gliding motility in a malaria parasite. PLoS Pathog. 5, e1000599 10.1371/journal.ppat.1000599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McRobert L., Taylor C. J., Deng W., Fivelman Q. L., Cummings R. M., Polley S. D., Billker O., and Baker D. A. (2008) Gametogenesis in malaria parasites is mediated by the cGMP-dependent protein kinase. PLoS Biol. 6, e139 10.1371/journal.pbio.0060139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Govindasamy K., Jebiwott S., Jaijyan D. K., Davidow A., Ojo K. K., Van Voorhis W. C., Brochet M., Billker O., and Bhanot P. (2016) Invasion of hepatocytes by Plasmodium sporozoites requires cGMP-dependent protein kinase and calcium dependent protein kinase 4. Mol. Microbiol. 102, 349–363 10.1111/mmi.13466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miller L. H., Ackerman H. C., Su X. Z., and Wellems T. E. (2013) Malaria biology and disease pathogenesis: insights for new treatments. Nat. Med. 19, 156–167 10.1038/nm.3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fairhurst R. M. (2015) Understanding artemisinin-resistant malaria. Curr. Opin. Infect. Dis. 28, 417–425 10.1097/QCO.0000000000000199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alam M. M., Solyakov L., Bottrill A. R., Flueck C., Siddiqui F. A., Singh S., Mistry S., Viskaduraki M., Lee K., Hopp C. S., Chitnis C. E., Doerig C., Moon R. W., Green J. L., Holder A. A., et al. (2015) Phosphoproteomics reveals malaria parasite protein kinase G as a signalling hub regulating egress and invasion. Nat. Commun. 6, 7285 10.1038/ncomms8285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diaz C. A., Allocco J., Powles M. A., Yeung L., Donald R. G. K., Anderson J. W., and Liberator P. A. (2006) Characterization of Plasmodium falciparum cGMP-dependent protein kinase (PfPKG): antiparasitic activity of a PKG inhibitor. Mol. Biochem. Parasitol. 146, 78–88 10.1016/j.molbiopara.2005.10.020 [DOI] [PubMed] [Google Scholar]
- 14. Kim J. J., Flueck C., Franz E., Sanabria-Figueroa E., Thompson E., Lorenz R., Bertinetti D., Baker D. A., Herberg F. W., and Kim C. (2015) Crystal structures of the carboxyl cGMP binding domain of the Plasmodium falciparum cGMP-dependent protein kinase reveal a novel capping triad crucial for merozoite egress. PLOS Pathog. 11, e1004639 10.1371/journal.ppat.1004639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collins C. R., Hackett F., Strath M., Penzo M., Withers-Martinez C., Baker D. A., and Blackman M. J. (2013) Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLoS Pathog. 9, e1003344 10.1371/journal.ppat.1003344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cohen P. (2002) Protein kinases—the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1, 309–315 10.1038/nrd773 [DOI] [PubMed] [Google Scholar]
- 17. Baker D. A., and Deng W. (2005) Cyclic GMP-dependent protein kinases in protozoa. Front. Biosci. 10, 1229–1238 10.2741/1615 [DOI] [PubMed] [Google Scholar]
- 18. Kim J. J., Lorenz R., Arold S. T., Reger A. S., Sankaran B., Casteel D. E., Herberg F. W., and Kim C. (2016) Crystal structure of PKG I:cGMP complex reveals a cGMP-mediated dimeric interface that facilitates cGMP-induced activation. Structure 24, 710–720 10.1016/j.str.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moon T. M., Sheehe J. L., Nukareddy P., Nausch L. W., Wohlfahrt J., Matthews D. E., Blumenthal D. K., and Dostmann W. R. (2018) An N-terminally truncated form of cyclic GMP–dependent protein kinase Iα (PKG Iα) is monomeric and autoinhibited and provides a model for activation. J. Biol. Chem. 293, 7916–7929 10.1074/jbc.RA117.000647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deng W., Parbhu-Patel A., Meyer D. J., and Baker D. A. (2003) The role of two novel regulatory sites in the activation of the cGMP-dependent protein kinase from Plasmodium falciparum. Biochem. J. 374, 559–565 10.1042/bj20030474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Franz E., Knape M. J., and Herberg F. W. (2018) cGMP binding domain D mediates a unique activation mechanism in Plasmodium falciparum PKG. ACS Infect. Dis. 4, 415–423 10.1021/acsinfecdis.7b00222 [DOI] [PubMed] [Google Scholar]
- 22. Berman H. M., Ten Eyck L. F., Goodsell D. S., Haste N. M., Kornev A., and Taylor S. S. (2005) The cAMP binding domain: an ancient signaling module. Proc. Natl. Acad. Sci. U.S.A. 102, 45–50 10.1073/pnas.0408579102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang G. Y., Kim J. J., Reger A. S., Lorenz R., Moon E. W., Zhao C., Casteel D. E., Bertinetti D., Vanschouwen B., Selvaratnam R., Pflugrath J. W., Sankaran B., Melacini G., Herberg F. W., and Kim C. (2014) Structural basis for cyclic-nucleotide selectivity and cGMP-selective activation of PKGi. Structure 22, 116–124 10.1016/j.str.2013.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El Bakkouri M., Kouidmi I., Wernimont A. K., Amani M., Hutchinson A., Loppnau P., Kim J. J., Flueck C., Walker J. R., Seitova A., Senisterra G., Kakihara Y., Kim C., Blackman M. J., Calmettes C., et al. (2019) Structures of the cGMP-dependent protein kinase in malaria parasites reveal a unique structural relay mechanism for activation. Proc. Natl. Acad. Sci. U.S.A. 116, 14164–14173 10.1073/pnas.1905558116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Monod J., Wyman J., and Changeux J.-P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 10.1016/S0022-2836(65)80285-6 [DOI] [PubMed] [Google Scholar]
- 26. Akimoto M., Moleschi K., Boulton S., VanSchouwen B., Selvaratnam R., Taylor S. S., and Melacini G. (2014) Allosteric linkers in cAMP signalling. Biochem. Soc. Trans. 42, 139–144 10.1042/BST20130257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akimoto M., Selvaratnam R., McNicholl E. T., Verma G., Taylor S. S., and Melacini G. (2013) Signaling through dynamic linkers as revealed by PKA. Proc. Natl. Acad. Sci. U.S.A. 110, 14231–14236 10.1073/pnas.1312644110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. VanSchouwen B., Selvaratnam R., Giri R., Lorenz R., Herberg F. W., Kim C., and Melacini G. (2015) Mechanism of cAMP partial agonism in protein kinase G (PKG). J. Biol. Chem. 290, 28631–28641 10.1074/jbc.M115.685305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Selvaratnam R., VanSchouwen B., Fogolari F., Mazhab-Jafari M. T., Das R., and Melacini G. (2012) The projection analysis of NMR chemical shifts reveals extended EPAC autoinhibition determinants. Biophys. J. 102, 630–639 10.1016/j.bpj.2011.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Selvaratnam R., Chowdhury S., VanSchouwen B., and Melacini G. (2011) Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. U.S.A. 108, 6133–6138 10.1073/pnas.1017311108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Akimoto M., McNicholl E. T., Ramkissoon A., Moleschi K., Taylor S. S., and Melacini G. (2015) Mapping the free energy landscape of PKA inhibition and activation: a double-conformational selection model for the tandem cAMP-binding domains of PKA RIα. PLoS Biol. 13, e1002305 10.1371/journal.pbio.1002305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boulton S., Akimoto M., Akbarizadeh S., and Melacini G. (2017) Free energy landscape remodeling of the cardiac pacemaker channel explains the molecular basis of familial sinus bradycardia. J. Biol. Chem. 292, 6414–6428 10.1074/jbc.M116.773697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boulton S., Selvaratnam R., Blondeau J.-P., Lezoualc'h F., and Melacini G. (2018) Mechanism of selective enzyme inhibition through uncompetitive regulation of an allosteric agonist. J. Am. Chem. Soc. 140, 9624–9637 10.1021/jacs.8b05044 [DOI] [PubMed] [Google Scholar]
- 34. Das R., Chowdhury S., Mazhab-Jafari M. T., Sildas S., Selvaratnam R., and Melacini G. (2009) Dynamically driven ligand selectivity in cyclic nucleotide binding domains. J. Biol. Chem. 284, 23682–23696 10.1074/jbc.M109.011700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rehmann H., Arias-Palomo E., Hadders M. A., Schwede F., Llorca O., and Bos J. L. (2008) Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature 455, 124–127 10.1038/nature07187 [DOI] [PubMed] [Google Scholar]
- 36. Zagotta W. N., Olivier N. B., Black K. D., Young E. C., Olson R., and Gouaux E. (2003) Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 425, 200–205 10.1038/nature01922 [DOI] [PubMed] [Google Scholar]
- 37. Su Y., Dostmann W. R., Herberg F. W., Durick K., Xuong N. H., Ten Eyck L., Taylor S. S., and Varughese K. I. (1995) Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science 269, 807–813 10.1126/science.7638597 [DOI] [PubMed] [Google Scholar]
- 38. Clayton G. M., Silverman W. R., Heginbotham L., and Morais-Cabral J. H. (2004) Structural basis of ligand activation in a cyclic nucleotide regulated potassium channel. Cell 119, 615–627 10.1016/j.cell.2004.10.030 [DOI] [PubMed] [Google Scholar]
- 39. Dolgova N. V., Yu C., Cvitkovic J. P., Hodak M., Nienaber K. H., Summers K. L., Cotelesage J. J. H., Bernholc J., Kaminski G. A., Pickering I. J., George G. N., and Dmitriev O. Y. (2017) Binding of copper and cisplatin to Atox1 is mediated by glutathione through the formation of metal–sulfur clusters. Biochemistry 56, 3129–3141 10.1021/acs.biochem.7b00293 [DOI] [PubMed] [Google Scholar]
- 40. McNicholl E. T., Das R., SilDas S., Taylor S. S., and Melacini G. (2010) Communication between tandem cAMP binding domains in the regulatory subunit of protein kinase A-Iα as revealed by domain-silencing mutations. J. Biol. Chem. 285, 15523–15537 10.1074/jbc.M110.105783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fayos R., Melacini G., Newlon M. G., Burns L., Scott J. D., and Jennings P. A. (2003) Induction of flexibility through protein-protein interactions. J. Biol. Chem. 278, 18581–18587 10.1074/jbc.M300866200 [DOI] [PubMed] [Google Scholar]
- 42. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 10.1007/bf00197809 [DOI] [PubMed] [Google Scholar]
- 43. Goddard T. D., and Kneller D. G. (2006) SPARKY, NMR Assignment and Integration Software, version 3, University of California, San Francisco [Google Scholar]
- 44. Boulton S., Akimoto M., Selvaratnam R., Bashiri A., and Melacini G. (2014) A tool set to map allosteric networks through the NMR chemical shift covariance analysis. Sci. Rep. 4, 7306 10.1038/srep07306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Selvaratnam R., Mazhab-Jafari M. T., Das R., and Melacini G. (2012) The auto-inhibitory role of the EPAC hinge helix as mapped by NMR. PLoS One 7, e48707 10.1371/journal.pone.0048707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cook P. F., Neville M. E. Jr., Vrana K. E., Hartl F. T., and Roskoski R. Jr. (1982) Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 21, 5794–5799 10.1021/bi00266a011 [DOI] [PubMed] [Google Scholar]
- 47. Riniker S., and Landrum G. A. (2015) Better informed distance geometry: using what we know to improve conformation generation. J. Chem. Inf. Model. 55, 2562–2574 10.1021/acs.jcim.5b00654 [DOI] [PubMed] [Google Scholar]
- 48. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Case D. A., Betz R. M., Cerutti D. S., Cheatham T. E., Darden T., and Duke R. E. (2016) AMBER16, University of California, San Francisco [Google Scholar]
- 50. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., et al. (2016) Gaussian 09, revision E01, Gaussian, Inc., Wallingford, CT [Google Scholar]
- 51. Huang J., Sun B., Yao Y., and Liu J. (2017) Fast and reliable thermodynamic approach for determining the protonation state of the Asp dyad. J. Chem. Inf. Model. 57, 2273–2280 10.1021/acs.jcim.7b00207 [DOI] [PubMed] [Google Scholar]
- 52. VanSchouwen B., Selvaratnam R., Fogolari F., and Melacini G. (2011) Role of dynamics in the autoinhibition and activation of the exchange protein directly activated by cyclic AMP (EPAC). J. Biol. Chem. 286, 42655–42669 10.1074/jbc.M111.277723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roe D. R., and Cheatham T. E. 3rd (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
- 54. Han B., Liu Y., Ginzinger S. W., and Wishart D. S. (2011) SHIFTX2: significantly improved protein chemical shift prediction. J. Biomol. NMR 50, 43–57 10.1007/s10858-011-9478-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The NMR chemical shift assignment for cGMP-bound PfD has been deposited in the Biological Magnetic Resonance Data Bank (BMRB) with the code 50203. All remaining data are contained within the article.