

Abstract
Introduction:
Phase 2 clinical trials of tuberculosis treatment have shown that once-daily regimens in which rifampin is replaced by high dose rifapentine have potent antimicrobial activity that may be sufficient to shorten overall treatment duration. Herein we describe the design of an ongoing phase 3 clinical trial testing the hypothesis that once-daily regimens containing high dose rifapentine in combination with other anti-tuberculosis drugs administered for four months can achieve cure rates not worse than the conventional six-month treatment regimen.
Methods/Design:
S31/A5349 is a multicenter randomized controlled phase 3 non-inferiority trial that compares two four-month regimens with the standard six-month regimen for treating drug-susceptible pulmonary tuberculosis in HIV-negative and HIV-positive patients. Both of the four-month regimens contain high-dose rifapentine instead of rifampin, with ethambutol replaced by moxifloxacin in one regimen. All drugs are administered seven days per week, and under direct observation at least five days per week. The primary outcome is tuberculosis disease-free survival at twelve months after study treatment assignment. A total of 2500 participants will be randomized; this gives 90% power to show non-inferiority with a 6.6% margin of non-inferiority.
Discussion:
This phase 3 trial formally tests the hypothesis that augmentation of rifamycin exposures can shorten tuberculosis treatment to four months. Trial design and standardized implementation optimize the likelihood of obtaining valid results. Results of this trial may have important implications for clinical management of tuberculosis at both individual and programmatic levels.
Keywords: tuberculosis, TB, multicenter randomized trial, non-inferiority, rifapentine, moxifloxacin
Introduction
Tuberculosis remains an important global health problem. In 2017, 10 million people became ill with tuberculosis and 1.6 million died from the disease, making tuberculosis the leading cause of death from an infectious pathogen.1 For drug-susceptible pulmonary tuberculosis, the current standard treatment lasts six months.2,3 Highly potent treatment regimens of shorter duration could improve treatment completion rates and reduce costs.
Rifamycins remain a cornerstone of tuberculosis treatment based on clinical trials conducted in the 1970s that demonstrated their ability, administered together with other antimicrobial agents, to shorten treatment while maintaining acceptable cure rates.4 Rifampin is the most commonly used rifamycin for tuberculosis treatment. The currently recommended adult dose of rifampin — 10 mg/kg once daily, up to 600 mg once daily — was historically selected based on pharmacokinetic, toxicity, and cost arguments, without detailed analyses of the dosing-exposure at site of infection-response relationships that provide the foundation for contemporary antimicrobial development.5 Animal models of tuberculosis chemotherapy have shown a clear relationship between rifamycin exposure and reduction of bacillary burden, and suggest that the current 600 mg dose of rifampin is at the low end of the dose-response curve.6–9 Phase 1 and 2 clinical trials have confirmed the relationship between rifampin exposure and antimicrobial activity as assessed by early bactericidal activity and time to sputum culture conversion, and rifampin activity does not plateau even at doses as high as 35 mg/kg/day, well above the current standard of 10 mg/kg/day.10,11 No increase in safety signals or change in tolerability has been observed in the phase 2 studies to date for rifampin up to 35 mg/kg/day.10,11
Rifapentine, a cyclopentyl derivative of rifampin, has a longer half-life and a lower minimum inhibitory concentration against M. tuberculosis than rifampin.12 Because of the long half-life, rifapentine’s potential for highly intermittent, e.g. once or twice weekly, administration as a strategy to facilitate, but not shorten the duration of, directly observed therapy was explored in the 1990s.13–15 Rifapentine was approved by the U.S. Food and Drug Administration (FDA) in 1998 for treatment of pulmonary tuberculosis in combination with other antituberculosis drugs, at a rifapentine dosage of 600 mg administered twice weekly during the initial two months of treatment followed by 600 mg administered once weekly for an additional four months. As programmatic emphasis subsequently shifted away from highly intermittent regimens and towards shortening the overall duration of treatment, the potential for daily administration of rifapentine in shortened regimens was explored. Animal studies indicated that rifapentine, administered daily during combination intensive phase treatment, had potent antimycobacterial activity that was associated with the ability to achieve cure without relapse after about three months of total treatment.16 In humans, phase 1 and 2 clinical trials supported the safety and tolerability of rifapentine at daily doses up to 20 mg/kg.17 A phase 2 clinical trial demonstrated a strong drug exposure-response effect for rifapentine using an intermediate marker of time to stable culture conversion,18,19 an indicator of overall efficacy of an anti-tuberculosis regimen.
Moxifloxacin is a fourth-generation fluoroquinolone with potent activity against M. tuberculosis in vitro and in vivo.20–23 Pre-clinical and clinical studies have shown that the single substitution of moxifloxacin for ethambutol increases antimicrobial activity of a multidrug regimen. 21–23 While this single substitution is insufficient to achieve acceptable cure rates after truncation of therapy to four months in the context of standard dosing of rifampin and other companion antituberculosis drugs,24 moxifloxacin nevertheless remains of interest for tuberculosis treatment. Moxifloxacin has good penetration into cellular compartments of tuberculous granulomas in animal models,25,26 is safe and well-tolerated when used for extended periods of time for treatment of tuberculosis in adults,27 based on available data has not been associated with serious arthropathy or other severe toxicity when used for extended periods of time for treatment of tuberculosis in children,28 and is broadly commercially available. Reassuringly, the prevalence of resistance to moxifloxacin at the clinical breakpoint of 2.0 ug/mL was shown to be uniformly low in recent population based surveys from Azerbaijan, Bangladesh, Belarus, Pakistan, and South Africa.29
The aforementioned rifamycin and moxifloxacin results provide rationale for S31/A5349, a phase 3 clinical trial to compare the efficacy of two investigational four-month rifapentine-containing regimens against that of the standard six-month World Health Organization (WHO) and CDC/ATS/IDSA endorsed regimen for treating drug-susceptible pulmonary tuberculosis2,3 using the outcome of relapse-free cure (Figure 1). One investigational regimen enhances antimicrobial activity by the single substitution of high-dose rifapentine for rifampin, administered daily throughout treatment. The other investigational regimen similarly substitutes high-dose rifapentine for rifampin and also replaces ethambutol (which has relatively weak activity) with moxifloxacin, which is administered throughout treatment. This paper describes the S31/A5349 design and the rationale for key aspects of it.
Figure 1.
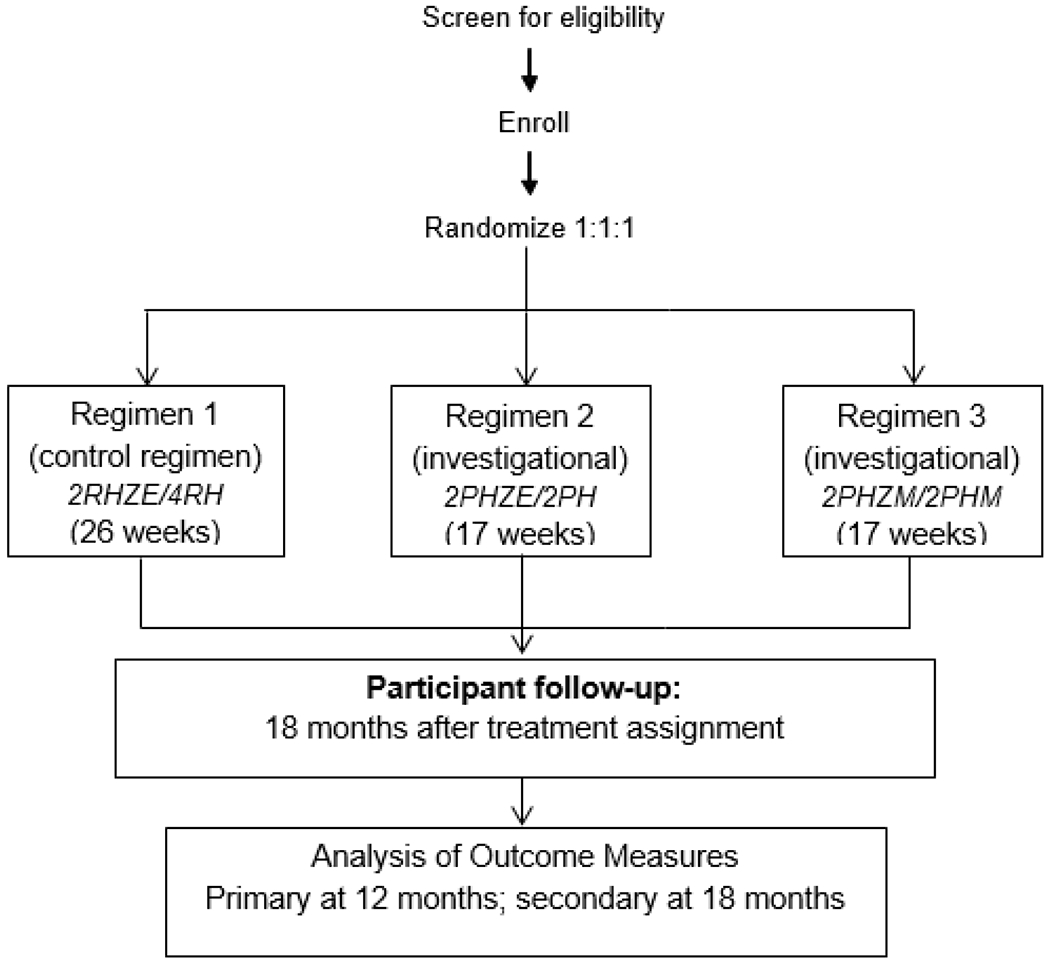
Study schematic for the trial “Rifapentine-containing treatment shortening regimens for pulmonary tuberculosis: a randomized, open-label, controlled phase 3 clinical trial (S31/A5349)”
Note. R=rifampin, H=isoniazid, Z=pyrazinamide, E=ethambutol, P=rifapentine, M=moxifloxacin.
Methods
Objectives
S31/A5349 is an international, randomized, open-label, controlled, three-arm non-inferiority trial among adolescents and adults with smear and culture positive pulmonary tuberculosis. The study objectives are to evaluate the efficacy of a) a rifapentine-containing regimen to determine whether the single substitution of rifapentine for rifampin makes it possible to reduce to four months the duration of treatment for drug-susceptible pulmonary tuberculosis, and b) a rifapentine-containing regimen that additionally substitutes moxifloxacin for ethambutol and continues moxifloxacin throughout treatment, to determine whether the duration of treatment can be reduced.
Study setting
Supported by the United States Centers for Disease Control and Prevention (CDC), the Tuberculosis Trials Consortium (TBTC) collaborates on this trial with the AIDS Clinical Trials Group (ACTG) network, supported by the United States National Institutes of Health. Existing sites from these two networks completed standard startup requirements. Sites submitted detailed descriptions of procedures for direct observation of treatment (DOT) and mycobacteriology laboratory testing. Recruitment occurred in Brazil, Haiti, Hong Kong, India, Kenya, Malawi, Peru, South Africa, Thailand, Uganda, the United States, Vietnam and Zimbabwe (Figure 2); the socio-demographic characteristics of study participants are expected generally to represent those populations most affected by tuberculosis worldwide.
Figure 2.
Geographic distribution of enrolling study sites for the trial “Rifapentine-containing treatment shortening regimens for pulmonary tuberculosis: a randomized, open-label, controlled phase 3 clinical trial (S31/A5349)”
Study population and eligibility
The study enrolled individuals with pulmonary tuberculosis caused by drug-susceptible M. tuberculosis. Inclusion and exclusion criteria are listed in Table 1.
Table 1.
Participant inclusion and exclusion criteria for S31/A5349
| Inclusion criteria |
| Individuals must meet all of the following inclusion criteria in order to participate in this study: |
| A. Suspected pulmonary tuberculosis plus one or both of the following: a) at least one sputum specimen positive for acid-fast bacilli on smear microscopy OR b) at least one sputum specimen positive for M. tuberculosis by Xpert MTB/RIF testing, with semiquantitative result of ‘medium’ or ‘high’ and rifamycin resistance not detected. |
| B. Age twelve years or older |
| C. A verifiable address or residence location that is readily accessible for visiting, and willingness to inform the study team of any change of address during the treatment and follow-up period. |
| D. Women of child-bearing potential who are not surgically sterilized must agree to practice an adequate method of contraception (barrier method or non-hormonal intrauterine device) or abstain from heterosexual intercourse during study drug treatment. |
| E. Documentation of HIV infection status. |
| F. For HIV-positive individuals, CD4 T cell count greater than or equal to 100 cells/mm3 based on testing performed at or within 30 days prior to study entry. HIV-positive individuals will be enrolled in a staged approach: |
| • Group 1 (“EFV1”): receipt of efavirenz-based antiretroviral therapy (ART) for a minimum of 30 days at the time of enrollment AND a documented HIV viral load less than 200 copies/mL at or within 30 days prior to study entry, OR |
| • Group 2 (“EFV2”): for HIV-positive individuals not on ART at enrollment, planned initiation of efavirenz-based ART before or at study week 8 |
| G. Laboratory parameters done at or within 14 days prior to screening: |
| • Serum or plasma alanine aminotransferase (ALT) less than or equal to 3 times the upper limit of normal |
| • Serum or plasma total bilirubin less than or equal to 2.5 times the upper limit of normal |
| • Serum or plasma creatinine level less than or equal to 2 times the upper limit of normal |
| • Serum or plasma potassium level greater than or equal to 3.5 meq/L |
| • Hemoglobin level of 7.0 g/dL or greater |
| • Platelet count of 100,000/mm3 or greater |
| H. For all women who are not surgically sterilized or who do not meet the study definition of postmenopausal, a negative pregnancy test at or within seven days prior to screening |
| I. Karnofsky score greater than or equal to 60 |
| J. Written informed consent. |
| Criteria for exclusion from enrollment |
| An individual meeting any of the following exclusion criteria at the time of enrollment or initiation of study drugs will be excluded from study participation: |
| A. Pregnant or breast-feeding |
| B. Unable to take oral medications |
| C. Previously enrolled in this study |
| D. Received any investigational drug in the past 3 months |
| E. More than five days of treatment directed against active tuberculosis within 6 months preceding initiation of study drugs |
| F. More than five days of systemic treatment with any one or more of the following drugs within 30 days preceding initiation of study drugs: isoniazid, rifampin, rifabutin, rifapentine, ethambutol, pyrazinamide, kanamycin, amikacin, streptomycin, capreomycin, moxifloxacin, levofloxacin, gatifloxacin, ofloxacin, ciprofloxacin, other fluoroquinolones, ethionamide, prothionamide, cycloserine, terizidone, para-aminosalicylic acid, linezolid, clofazimine, delamanid or bedaquiline |
| G. Known history of prolonged QT syndrome |
| H. Suspected or documented tuberculosis involving the central nervous system and/or bones and/or joints, and/or miliary tuberculosis and/or pericardial tuberculosis |
| I. Current or planned use within six months following enrollment of one or more of the following medications: HIV protease inhibitors, HIV integrase inhibitors, HIV entry and fusion inhibitors, HIV non-nucleoside reverse transcriptase inhibitors other than efavirenz; quinidine, procainamide, amiodarone, sotalol, disopyramide, ziprasidone, or terfenadine. |
| J. Weight less than 40.0 kg |
| K. Known allergy or intolerance to any of the study medications |
| L. Individuals will be excluded from enrollment if, at the time of enrollment, their M. tuberculosis isolate is already known to be resistant to any one or more of the following: rifampin, isoniazid, pyrazinamide, ethambutol, or fluoroquinolones. |
| M. Other medical conditions, that, in the investigator’s judgment, make study participation not in the individual’s best interest. |
| N. Current or planned incarceration or other involuntary detention. |
| Criteria for exclusion after enrollment (‘Late exclusion’) |
| Microbiological confirmation of drug-susceptible tuberculosis is not expected always to be available at the time of enrollment. Enrolled individuals who are subsequently determined to meet either of the following criteria will be classified as ‘late exclusions’ and study treatment will be discontinued: |
| A. Screening, baseline, and Week 2 study visit sputum cultures all fail to grow M. tuberculosis. M. tuberculosis cultured or detected through molecular assays (Cepheid Xpert MTB/RIF or Hain MTBDRplus assays) from sputum obtained around the time of study entry is determined subsequently to be resistant to one or more of isoniazid, rifampin, or fluoroquinolones. |
Participants must have had a sputum specimen, collected within two weeks of enrollment, that was either positive for acid-fast bacilli on smear microscopy or positive for M. tuberculosis by GeneXpert MTB/RIF® (“Xpert”, Cepheid Inc., Sunnyvale, CA) testing with semi-quantitative result of ‘medium’ or ‘high’ at the site’s laboratory of record for the trial. Xpert sensitivity is higher than that of smear microscopy, and Xpert semi-quantitative results of medium or high correlate well with smear positivity.30,31 This approach maximized the likelihood that a given participant had culturable M. tuberculosis from sputum, and additionally aligns this study with the pre-Xpert body of clinical trials literature in terms of severity of pulmonary tuberculosis based on sputum bacillary burden.
The lower age limit of eligibility was 12 years. At the time of study initiation there were data showing that the pharmacokinetics of rifapentine were similar in adults and adolescents,32 but insufficient data were available for younger children.
With regard to HIV status, we attempted to balance the general importance of broad eligibility with guide lines-based antiretroviral therapy at the time of study initiation and then-existing uncertainties about the magnitude of the drug-drug interaction between high-dose daily rifapentine and efavirenz. At the time of protocol development, efavirenz-based ART was the global preference. To be eligible, HIV-positive individuals must have had a CD4 T cell count ≥100 cells/mm3 obtained prior to enrollment. The bases for this CD4 threshold were: (1) the need to initiate concomitant antiretroviral therapy (ART) within a very short period of time in individuals with very low CD4 counts (as recommended by both WHO and U.S. guidelines)2,3; (2) the potential for severe immune reconstitution events in that context; and (3) the high rates of mortality and serious adverse events in individuals with very low CD4 counts.33–37. Initially enrollment was open only to those HIV-positive patients who were already on efavirenz based ART, with documented viral load less than 200 copies/mL at or within 30 days prior to study entry (“EFV Group 1”). For this initial cohort of participants, blood efavirenz concentrations and HIV viral load were measured on study treatment and those data were analyzed early during the trial in order to determine whether or not the standard dose of efavirenz resulted in adequate efavirenz exposure and viral suppression in the presence of daily 1200 mg rifapentine treatment. Demonstration of acceptable efavirenz concentrations in the initial cohort of EFV Group 1 participants was required before enrollment was opened to HIV-positive individuals not already on ART at enrollment (“EFV Group 2”).38,39
Recruitment process
Potentially eligible patients (and their parent/legal guardian, in the case of adolescents) were given information about the study by their routine tuberculosis care provider. Interested individuals were referred to study staff, who provided detailed information about the study, including risks and potential benefits of all study procedures. If site staff were satisfied that the patient understood the information and was interested in study participation, then the potential participant was asked to provide written informed consent. For adolescents, written parental permission was obtained from the parent/legal guardian, and written assent was obtained from the patient. Study-specific procedures were initiated only after obtaining informed consent. The schedule of study procedures and assessments is in Table 2.
Table 2.
Schedule of Procedures/Evaluations
| Visit Window | Up to 7 days after screen | +/− three (3) days | +/− seven (7) days | Treatment Response | Post Early Termination Visitg | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Visit | Screen | Baseline | WK 2 | WK 4 | WK 8 | WK 12 | WK 17 | WK 22 | WK 26 | MO 9 | MO 12 | MO 15 | MO 18 | ||
| Informed Consent | X | ||||||||||||||
| Inclusion/Exclusion | X | X | |||||||||||||
| Demographics, medical history | X | ||||||||||||||
| Contact information | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Symptoms | X | X | X | X | X | X | X | X | X | X | X | X | X | X | |
| Concomitant medications | X | X | X | X | X | X | X | X | X | X | X | X | X | X | |
| Adverse events | X | X | X | X | X | X | X | X | |||||||
| Interval medical history | X | X | X | X | X | ||||||||||
| Height | X | ||||||||||||||
| Weight | X | X | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Chest radiograph | X | Xf | Xf | X | |||||||||||
| Visual tests | X | X | |||||||||||||
| HIV test | X | ||||||||||||||
| CD4, HIV viral load (if HIV-pos) | Xa | ||||||||||||||
| Pregnancy testing | X | ||||||||||||||
| Randomization | X | ||||||||||||||
| Sputum for smear and cultureb | X | X | X | X | X | X | XX | XX | XX | XX | XXc | XX | XXc | XXX | |
| Sputum for rapid molecular test, if available at site | X | ||||||||||||||
| Storage of Mtb bacterial isolate | X | X | X | X | X | X | X | X | X | X | |||||
| Diabetes screend | X | ||||||||||||||
| ALT, bilirubin, creatinine, hemoglobin, WBC with differential, platelets | X | X | X | X | X | X | X | X | |||||||
| Serum albumin, potassium | X | ||||||||||||||
| PK sampling for TB drugs | within this interval | ||||||||||||||
| Blood sample for pharmacogenomics | Obtain anytime after enrollment | ||||||||||||||
| EFV1: Plasma for EFV PK | At screening or baseline | X | X | X | |||||||||||
| EFV1: HIV viral load | At screening or baseline | X | X | ||||||||||||
| EFV2: Plasma for EFV PK | Obtain at about 4 weeks after starting EFV AND at about 8 weeks after starting EFV | X | |||||||||||||
| EFV2: HIV viral load | Required per eligibility criteria | Obtain once at about 8 weeks after starting EFV | X | ||||||||||||
| Sputum, blood, urine for research | X | X | X | X | Xe | Xe | X | ||||||||
| Contact central study clinician | X | ||||||||||||||
NOTES
Unless results of a test performed at or within 30 days prior to screening are available.
All sputa should be sent to the designated study laboratory with the exception of the screening specimen, which may be evaluated at any locally acceptable laboratory. If a screening specimen has been found to be smear or culture positive at a non-study laboratory, then either store the isolate if culture positive from the non-study laboratory or get an additional specimen for culture and storage of isolate. Two specimens (i.e. one at screening and one at baseline) are required prior to initiation of study treatment. At least two (2) sputa should be obtained at each of weeks 17, 22, 26 and at each of months 9, 12, 15, and 18.
If both of the month 12 sputa or both of the month 18 sputa are contaminated, then the participant should be asked to provide at least two (2) additional sputa as soon as possible after contamination is recognized.
Hemoglobin A1C is the preferred test. If such testing is not available, then fasting or random blood glucose can be measured.
To be obtained at the end-of-study treatment visit (i.e. either week 17 or week 26).
This visit occurs approximately 14 days after stopping study drugs.
Treatment allocation
Randomization and treatment arm assignment were computer-generated centrally at the TBTC Data and Coordinating Center (DCC) in Atlanta, GA. Eligible persons who met all of the inclusion criteria and none of the exclusion criteria were randomly assigned in a 1:1:1 ratio to the study arms (Figure 1). Random assignments were based on a Big Stick Design with a maximum imbalance of 2.40 This design allowed random assignments to be generated in a way that limits the imbalance between arms within strata at sites, while ensuring that the sequence is not predictable based on previous assignments. Randomization was stratified by site, and by lung cavitation (present vs. absent based on baseline chest radiograph) or both cavitation and HIV status (HIV-negative vs. HIV-positive), based on the expected number of enrollments of participants with HIV infection at the site. Cavitation was defined as a gas-containing lucent space at least 1 cm in diameter, located within the lung parenchyma and surrounded by an infiltrate or fibrotic wall greater than 1 mm thick on chest radiograph. Cavitation seen only on chest tomography (e.g. CT) did not satisfy this definition. Cavitation and HIV infection are both associated with a decreased rate of microbiological response to tuberculosis treatment and an increased risk for relapse after treatment completion, providing the rationale for stratifying enrollment based on these variables.13,14
Rifamycin dosing strategies
This trial used a fixed dose of rifapentine, 1200 mg, administered once daily with food, irrespective of participant weight. This strategy was based on a) demonstration of the safety of rifapentine at 1200 mg in phase 1 and 2 trials, b) demonstration that body weight does not significantly affect rifapentine clearance, c) the known effect of food in substantially increasing rifapentine absorption, and d) modeling predictions that the target rifapentine exposure (area under the concentration-time curve [AUC] of approximately 500 to 600 mcg*h/L) was achievable using this strategy.17–19,41–43 In a priorphase 2 rifapentine dose-ranging study, all participants received study drugs along with a very high fat meal (intended to augment rifapentine exposure) and the target rifapentine AUC was achieved at daily doses of 900 to 1200 mg.18,19 We reasoned that a very high fat meal might be poorly feasible under usual care conditions, and therefore for S31/A5349 we selected the higher daily dose (i.e. 1200 mg) and a more modest food requirement (i.e. “Participants receiving a rifapentine-containing investigational regimen should take study drugs within one hour after ingesting food”).
For rifampin, administration with food slows the rate of absorption and decreases the maximal concentration (Cmax) by about 36% in healthy adults but to a lesser extent (5 to 15%) in patients with tuberculosis, with little to no effect on AUC.44,45 The clinical consequences of these PK effects are unclear. Therefore, rifampin doses were administered without food in S31/A5349.
Study regimens
A web-based application was used to randomly assign each participant to receive one of the three regimens. Regimen 1(control regimen) was eight weeks of daily treatment with rifampin (R), isoniazid (H), pyrazinamide (Z), and ethambutol (E), followed by eighteen weeks of daily treatment with rifampin and isoniazid (2RHZE/4RH).2,3 Regimen 2 (investigational) was eight weeks of daily rifapentine (P), isoniazid, pyrazinamide, and ethambutol, followed by nine weeks of daily rifapentine and isoniazid (2PHZE/2PH). Regimen 3 (investigational) was eight weeks of daily rifapentine, isoniazid, pyrazinamide, and moxifloxacin (M), followed by nine weeks of daily rifapentine, isoniazid, and moxifloxacin (2PHZM/2PHM). Doses of study drugs are shown in Table 3. Weight-based doses used to initiate treatment were automatically calculated by the web-based application at the time of randomization. Assigned doses for pyrazinamide and ethambutol were based on weight at enrollment. During intensive phase treatment, doses for pyrazinamide and ethambutol were adjusted for the participant’s actual weight.
Table 3.
Doses of study medications by body weight
| Drug | Dose |
|---|---|
| Rifapentine | 1200 mg |
| Moxifloxacin | 400 mg |
| Rifampin | 600 mg |
| Isoniazid | 300 mg |
| Pyrazinamide | |
| < 55 kg | 1000 mg |
| ≥ 55-75 kg | 1500 mg |
| > 75 kg | 2000 mg |
| Ethambutol | |
| < 55 kg | 800 mg |
| ≥ 55-75 kg | 1200 mg |
| > 75 kg | 1600 mg |
| Vitamin B6 | 25 or 50 mg (based on local site norms) |
All study drugs were administered orally, seven days per week, throughout treatment. Five of seven doses per week were given under directly observed therapy by study personnel, or by a healthcare worker or lay treatment supervisor designated by the site investigator and trained regarding the study protocol. Doses on weekends and on holidays (up to three consecutive days) were either DOT or self-administered. Guidance on timing of food intake and administration of study drugs was specified in the protocol and differed based on treatment assignment, in keeping with the differential food effect on rifampin vs. rifapentine. As mentioned above, participants assigned to a rifapentine-containing regimen took study drugs within one hour after ingesting food. Neither the type nor the amount of food was prescribed in order to enhance operational flexibility and generalizability of study findings. Participants assigned to the rifampin control regimen took study drugs on an empty stomach. Individuals with gastrointestinal upset after taking the rifampin control regimen on an empty stomach were allowed to take subsequent doses with food.
Blinding to treatment assignment
Neither participants nor clinical site staff were blinded to individual participant treatment assignment, for two main reasons. First, blinding through use of placebos would have required the already substantial pill burden in the investigational arms to increase to approximately 20 pills per day. We feared that this would affect tolerability, which in turn could impact adherence and efficacy, and diminish validity of study findings. From a physiologic perspective, the dissolution and absorption of pills in the gastrointestinal tract might be reduced with this high pill burden, resulting in diminished treatment efficacy and increased risk for acquisition of drug resistance. Second, as noted above, there is a differential effect of food on rifampin and rifapentine absorption, such that different food guidance is required to optimize regimen pharmacokinetics. It would not have been feasible to implement blinding of food vs. no food with treatment administration.
Two key aspects of the study were intended to minimize ascertainment bias in the face of the open-label design. First was the use of frequent scheduled sputum collections along with use of objective laboratory measures and blinding of laboratory technicians to treatment assignment and study week. Second was the uniform application of study visits and procedures regardless of treatment assignment, including a pre-specified set of triggers and processes for evaluating individuals who might not be responding well to treatment (so called “possible poor treatment response” [PPTR] procedures, Table 4).
Table 4.
Triggers and Procedures for Participants with Possible Poor Treatment Response (PPTR)
| A PPTR evaluation is triggered by any one or more of the following, and applies to all regimens: |
| • A positive culture confirmed as M. tuberculosis from a sputum specimen collected at or after week 17 |
| • A positive smear from a sputum specimen collected at or after week 17 |
| • Worsening signs and/or symptoms consistent with tuberculosis at or after week 17 |
| • Radiographic worsening consistent with tuberculosis at or after week 17 |
| • The site investigator is considering an extension of tuberculosis treatment beyond that of the participant’s assigned regimen |
| • The site investigator is considering re-treatment with any tuberculosis therapy after the participant has completed their assigned regimen |
| • For a participant on assigned study treatment, the site investigator is considering a change in treatment for efficacy reasons (this does not apply to changes in treatment for pregnancy, temporary drug challenge, or toxicity) |
| Study procedures implemented for all participants who have a PPTR trigger: |
| • Symptom assessment |
| • Review interval medical history |
| • Concomitant medical assessment |
| • Measure weight |
| • Obtain at least 3 sputum specimens and send them to the study laboratory for smear and culture. At least two of these specimens should be obtained prior to changing or re-starting tuberculosis treatment (if change or re-start are being considered), and at least 4 hours apart. At least one of the specimens should be a first morning specimen, if feasible. If M. tuberculosis is isolated in culture, then DST should be performed, and the isolate stored frozen. |
| • Chest radiograph |
| • For participants consenting to collection of specimens for biomarker research, sputum, urine, and blood should be obtained |
| • Review participant’s contact information |
| • Complete the appropriate case report form |
| • Contact the Study Clinician to review PPTR procedures and ensure that all procedures are completed |
| • Participant should continue to be followed in the study per the Protocol/Manual of Operating Procedures unless participant withdraws consent |
Assessment of study outcomes and duration of follow-up
The primary efficacy outcome is tuberculosis disease-free survival at twelve months after randomization. This time point was selected based on historical trial data showing that over 75% of relapses occur within 6 months of stopping treatment,46 and is consistent with other recent and ongoing trials.47–49 Total duration of participant follow-up is 18 months; a secondary efficacy outcome will consider tuberculosis disease-free survival at eighteen months after randomization.
Sputum specimens are collected at each study visit according to the Schedule of Procedures/Evaluations (Table 2) and sent to the designated site laboratory for smear microscopy and mycobacteriology culture on both liquid and solid media. Phenotypic drug susceptibility testing (DST) for at least isoniazid, rifampin, and fluoroquinolones is performed on the first study isolate of M. tuberculosis and on the first of any M. tuberculosis isolates obtained at or after week 17 (time of completion of the four-month regimens). Mycobacteriology laboratory procedures are harmonized across study site laboratories for standardized key elements, and test results are collected on a study-specific case report form. Laboratorians handling study specimens are blinded to treatment assignment and time point of collection. For each participant, the baseline. M. tuberculosis isolate as well as the first of any M. tuberculosis isolates from sputum obtained at or after week 17 are shipped to the mycobacteriology laboratory at the CDC, where whole genome sequencing is performed for paired isolates to determine if recurrent tuberculosis is due to relapse of the same strain or re-infection with a new strain.
For each participant, a primary outcome status of “Absence of cure”, “Cure”, or “Not Assessable” is assigned based on definitions in Table 5. The Not Assessable group is defined by exclusions that are unlikely to introduce selection bias, and are aligned with definitions used in other recent phase 3 trials for treatment of drug-sensitive pulmonary tuberculosis.24,50,51
Table 5.
Definitions for primary outcome status.
| Each participant will be classified into one of the following 3 outcome categories of Absence of Cure (Unfavorable Outcome), Cure (Favorable Outcome), or Not Assessable. The primary efficacy outcome will be assessed at 12 months after treatment assignment; a secondary efficacy outcome will consider the follow-up period to be 18 months after treatment assignment. |
| Absence of cure (unfavorable outcome) is assigned to a participant who meets any one or more of the following criteria: |
| • Absence of bacteriological cure. A participant will be considered to have absence of bacteriological cure if he/she has a sputum sample, obtained at or after Week 17, that is culture positive for an M. tuberculosis strain that is indistinguishable from the initial isolate, and this is confirmed by a second sample that is culture positive for M. tuberculosis. A second confirmatory sample is required as a single positive sputum culture in isolation will not be considered absence of bacteriological cure. |
| • Participants who die from any cause during study treatment, except from violent or accidental cause (e.g. road traffic accident). |
| • Participants failing to complete treatment and not assessable at the end of the follow-up period. |
| • Participants who had a positive culture for M. tuberculosis when last seen, whether confirmed by a second sample or not, unless determined to have been re-infected |
| • Participants receiving any one or more of the following: a) extension of treatment beyond that permitted by the protocol; b) a re-start of treatment; c) a change in treatment for any reason except re-infection, pregnancy, or temporary drug challenge |
| Cure (favorable outcome) is assigned to a participant who meets any one of the following criteria and has not already been classified as having an unfavorable outcome: |
| • Participants with negative cultures at the end of the follow-up period |
| • Participants who at the end of the follow-up period are clinically without symptoms/signs of ongoing active TB and are unable to produce a sputum specimen |
| • Participants who at the end of the follow-up period are clinically without symptoms/signs of ongoing active TB and produce a sputum specimen that is contaminated without evidence of M. tuberculosis |
| An outcome of Not Assessable is assigned to a participant meeting any one or more of the following criteria and not already classified as having an unfavorable outcome: |
| • Participants who completed assigned treatment and then default from follow-up, with their last culture being negative for M. tuberculosis |
| • Women who become pregnant during their assigned active treatment and stop their assigned treatment |
| • Participants who die during the follow-up phase (≥ 15 days after completion of study treatment) |
| • Participants who die from a violent (e.g. homicide) or accidental (e.g. road traffic) cause during their assigned active treatment. As above, suicide will be considered an unfavorable outcome |
| • Participants re-infected with a new strain of M. tuberculosis, demonstrated to be different from that identified at study entry through genotyping methods |
The main safety outcome is proportion of participants with grade 3 or higher adverse events during study drug treatment. Detected adverse events are graded by the site investigator according to the Common Terminology Criteria for Adverse Events version 4.03 published on June 14, 2010.52
Assessment of efficacy outcomes at fixed times post randomization has some inherent bias against the treatment shortening regimens because relatively more time is spent off treatment, and therefore participants in the shortened regimen arms have relatively more time both for relapse of the original disease and for re-infection due to a different M. tuberculosis strain. Relapse and re-infection are handled differently in the trial, since relapse is considered an absence of cure and is thus associated with the original disease and intervention, whereas re-infection is driven mainly by local tuberculosis epidemiology and HIV status. With regard to relapse, the trial analysis plan does not include adjustments or corrections for the two additional months of post-treatment follow-up for participants assigned to investigational four-month regimens. A major factor in this trial design decision was the observation of others that the risk for relapse is not constant throughout follow-up (most relapses occur early after treatment discontinuation),46 such that the time available for follow-up for both the control and intervention arms should be sufficient to detect the majority of relapses in our trial. In our trial, the threat of bias related to re-infection is mitigated through design approaches (randomization by study site and HIV-status) and analysis approaches that take bacterial genotype into consideration.
Analysis groups
The four pre-specified analysis groups are shown in Table 6. For analyses of the Microbiologically Eligible group, participants with an outcome of “Not Assessable” are considered to have an unfavorable outcome.
Table 6.
Analysis Groups
| Intention-to-Treat (ITT) |
| Includes all enrolled participants who receive a treatment assignment. |
| Microbiologically Eligible |
| Includes the subset of Intention-to-Treat participants who, in addition, have culture confirmation of drug-susceptible tuberculosis at study entry. Participants classified as ‘not assessable’ will be considered to have an unfavorable outcome. |
| Assessable |
| Includes the subset of Microbiologically Eligible participants who, in addition, are not classified as ‘not assessable’. |
| Adherent Per-Protocol |
| Includes the subset of Assessable participants who, in addition, complete assigned study treatment and follow-up. |
Sample size assumptions
The primary objective of the trial is to evaluate whether four-month rifapentine-containing regimens can produce outcomes that are non-inferior to (i.e., “not worse than”) standard six-month therapy. Therefore, the trial is designed as a non-inferiority study. The non-inferiority margin is 6.6%.
We assumed a proportion of 15% unfavorable outcomes for the standard regimen arm (Microbiologically Eligible population). This rate is based on observed results for the control arm (modified intention to treat [MITT] analysis group) in recently completed phase 3 clinical trials that used similar outcome definitions as S31/A5349 -- 14.4% unfavorable in the RIFAQUIN trial,50 13.2% in the Oflotub trial,51 and 16% in the ReMOX trial.24 Based on TBTC phase 2 trial experience18,42 and considering the anticipated increasing use of rapid molecular resistance testing during S31/A5349, the proportion of enrolled participants who would be found to be late exclusions due to microbiological ineligibility was estimated at 12%. The proportion of enrolled patients who would be found to be “not assessable” was estimated at 12%.24,50
Total required sample size was calculated to be 2500, with approximately 612 assessable per arm. With the expected 15% unfavorable outcomes among those who are assessable, a 6.6% non-inferiority margin and a two-sided 5% type 1 error rate, the study will have 90% power to test the primary hypothesis among the Assessable subgroup.
Justification for non-inferiority margin
A 6% margin of non-inferiority has been used in other recent trials of single-drug substitution treatment shortening trials (REMoxTB, OFLOTUB, RIFAQUIN)24,50,51. The justification of the 6% margin is published in the online supplements with those papers and is based on a meta-analysis of historical trials including six-month or four-month rifampin-based regimens.53–57 The extension from 6% to the 6.6% as used in our trial is based on the following statistical and clinical considerations.
From a statistical perspective, the justification for a 4.8% margin in the FDA Guidance for Industry for Pulmonary Tuberculosis Trials is based on historical trials under ‘per protocol’ type analyses with many post-randomization exclusions, relapse as the primary outcome, and largely hospitalized populations.58 Contemporary trials, with limited post-randomization exclusions, composite outcomes, and in which the majority of participants are treated in the community, have had larger proportions of unfavorable outcomes (e.g. 16% in REMoxTB MITT) in the six-month control arm than was seen in the historical relapse-only analyses from previous trials.24,50,51 This provides justification for use of a margin that is larger than 4.8% and also larger than the 6% that was justified for the REMoxTB trial.
Additionally, the rationale for a 4.8% margin is based on the situation where a single drug that has an unknown contribution to the regimen is replaced by a new drug (the replacement of ethambutol, for example). In the current trial, rifampin is replaced by rifapentine (in addition to the substitution of moxifloxacin for ethambutol in one arm). As rifampin is the most important sterilizing drug in the current regimen, for estimation of the effect of the active control compared with placebo (“M1”) it is therefore appropriate to consider not just the removal of the final two months of therapy (following the argument in lines 829-832 in the FDA Guidance),58 but also the removal of rifampin from the regimen. As a basis for the margin of non-inferiority, the FDA guidance document identifies two studies that compare four and six months of tuberculosis therapy and there by provide data to estimate M1. We are not aware of any trials that evaluated a four-month regimen without rifampin. However, one of these studies also included two four-month regimens without a rifamycin in the continuation phase, 2HRZ+streptomycin (S)/2HZ and 2HRZS/2H56, and the combined relapse rate in these two arms was 31% (63/203). Using the figures quoted in the FDA guidance document for the 2HRZS/4HR regimen from this study (4.7%, 8/172), the treatment effect (four-month regimen minus six-month regimen) is 26.4%, 95% CI (19.3%, 33.5%) for the unstratified risk difference. This lower bound of 19.3% provides an estimate of M1 for the removal of the final 2 months of HR therapy, and the removal of R in months 3 and 4. For the current study, the 6.6% margin of non-inferiority preserves more than 50% of the above 19.3% M1 estimate.
Considerng the clinical argument,58,59the investigators for the current study, in broader consultation within the two large publicly-funded international consortia of TB stakeholders that participate in the current study (CDCTB Trials Consortium and NIH AIDS Clinical Trials Group), consider the benefits of a shortened rifapentine-based regimen to justify the margin of 6.6%. Study investigators consider 600 patients per arm sufficiently large to provide adequate precision on the difference in efficacy between the regimens to determine whether an intervention regimen might be considered not inferior to the control regimen.
Analysis of the primary outcomes
There will be two co-primary efficacy analyses. One will consider the Microbiologically Eligible analysis population, and the other will consider the Assessable population. For each, the comparison of Regimen 1 (control regimen) versus Regimen 3 (2PHZM/2PHM) will be considered first, and, if non-inferiority criteria are met then the comparison of Regimen 1 versus Regimen 2 (2PHZE/2PH) will be considered (in this approach there is no adjustment for multiple comparisons). In each comparison, non-inferiority will be assessed by comparing the upper bound of the 95%, 2-sided confidence interval for the difference between the proportions of participants who are classified as having an unfavorable outcome on the intervention regimen vs. the control regimen (Regimen 1) to the predefined non-inferiority margin of 6.6%. Non-inferiority must be demonstrated in both analysis populations in order to declare non-inferiority for an intervention regimen. The Microbiologically Eligible population and its subset, the Assessable population, are modified intention-to-treat populations. The Assessable population excludes those participants who experience an event (e.g. death from violent or accidental cause at any time during participation) that is unlikely to be related to the disease or to the intervention, while the Microbiologically Eligible population classifies those non-assessable participants as unfavorable and thus is closer to an intention-to-treat population.
The primary safety analysis population will include all randomized participants that took at least one dose of study medication. The primary safety analysis will include two comparisons, namely Regimen 1 (control regimen) vs. Regimen 2 (2PHZE/2PH) and Regimen 1 vs. Regimen 3 (2PHZM/2PHM).
Pharmacokinetic sampling
Sampling for pharmacokinetic (PK) analysis of study tuberculosis drugs is performed for all study participants. An intensive sampling scheme (used at a few sites with capacity to perform this activity) is described in a separate protocol with an additional informed consent process. All other participants, comprising the large majority of study participants, undergo sparse PK sampling as a component of the main S31/A5349 protocol. The recommended time of sparse PK sampling is at the week 2, 4 or 8 visit (when sputum is also collected), but sparse sampling is permitted any time between the 14th study drug dose and the week 8 study visit. All PK blood samples are collected in reference to an observed dose of study drugs -- the reference dose of study drugs must have been preceded by three directly observed study drug doses given approximately 24 hours, 48 hours, and 72 hours prior. Two or three blood samples, 10 ml each, are obtained over a 9-hour period. Blood specimens are shipped to a designated laboratory in Europe, where concentrations of tuberculosis drugs and their metabolites are measured.
HIV-positive participants randomized to either of the rifapentine containing regimens undergo efavirenz PK measurements as described above. Refer to Table 2 for schedule of PK sampling for EFV1 and EFV2 groups.
Biorepository development
At participating sites, sputum, urine, and blood were collected from participants who provided explicit consent for use of their specimens for future research to identify potential biomarkers of tuberculosis treatment response. Consenting participants are asked to provide sputum, blood (approximately 8 ml blood per blood draw), and urine samples at 5 visits (baseline, week 2, week 4, week 8, and at end of treatment). Additionally, samples are also collected if a) treatment failure or relapse is suspected and evaluated at an unscheduled visit or a visit other than one of the 5 visits indicated above; and/or b) the participant voluntarily withdraws from the study. Biorepository samples are stored in the United Kingdom as part of the Consortium for TB Biomarkers initiative.60 Priority areas of investigation include identification of host blood and sputum transcriptional biomarkers of treatment response as well as host and bacterial macromolecule signatures potentially present in blood or urine.61–63 An additional initiative seeks to expand on recent work by Colangeli et al showing that, among tuberculosis patients with M. tuberculosis strains having sub-breakpoint minimum inhibitory concentrations (MICs) of isoniazid and rifampin (i.e. “susceptible”), higher MICs were associated with a greater risk of relapse than lower MICs.64 In the context of S31/A5349 this initiative aims to develop a highly predictive algorithm that identifies tuberculosis patients who are cured by shortened treatment regimens in a model that includes bacterial measures, PK data, and measured host characteristics.
Ethical approvals
The trial is approved by the CDC Institutional Review Board (IRB). Each participating institution provides for the review and approval of this protocol and its informed consent documents by a local IRB or ethics committee, or relies formally on the CDC IRB approval.
Dissemination of trial findings
Following completion of the study, the investigators will publish the results in peer-reviewed journals in accordance with the Consolidated Standards of Reporting Trials (CONSORT) statement.65 Additional dissemination of results will be through the press, national meetings, and international conferences. Aggregate study results will be shared with study participants through mechanisms and materials approved by the TBTC Community Research Advisors Group (CRAG).
Discussion
Study S31/A5349 is a phase 3 clinical trial that is the culmination of over 15 years of work to determine if optimized rifamycin dosing–in the form of rifapentine administered daily at a dose of 1200 mg — can meaningfully shorten the required duration of treatment for pulmonary tuberculosis. A finding of non-inferiority of one or both rifapentine regimens, with good safety and tolerability, would open up a new shorter course treatment option for a broad group of tuberculosis patients. If the rifapentine regimens do not meet the non-inferiority threshold, this would not exclude the possibility that optimized high doses of rifampin could shorten treatment, including through stratified medicine approaches,66 but our findings would underscore the need for additional trials that include newly developed drugs.
This trial has several notable features. As a mainly explanatory trial, it includes rigorous DOT implementation that, while perhaps not achievable in all tuberculosis program settings, will nevertheless allow a detailed understanding of regimen efficacy under pre-specified conditions. Pooled analysis of individual participant data from other recent phase 3 tuberculosis treatment-shortening trials has shown that even minimal non-adherence or missed doses increases the risk of unfavorable outcomes.66 The schemes of DOT implementation are setting-specific and vary from fully centralized (all DOT provided at the clinical research site) to semi-decentralized (DOT at tuberculosis clinics or primary healthcare clinics closer to participants’ residences) to fully decentralized (DOT done at participants’ homes or another participant-preferred setting). Second, the trial includes frequent, standardized, and comprehensive microbiological testing that serves as the main component of outcome assessment; results of this testing will also provide useful longitudinal information about potential differences in antimicrobial activity across the regimens. Whole genome sequencing (WGS) is used to assess reinfection versus relapse. These features, along with embedded PK measurements for all participants, should provide nuanced information about factors associated with treatment failure, relapse and cure, thereby potentially contributing to development of new strategies for individualized management of patients with tuberculosis. Study eligibility criteria were deliberately kept as broad as possible based on data available prior to study start. Inclusion of Xpert or smear positivity as eligibility criteria added flexibility and anticipated changes in tuberculosis diagnostics algorithms. Children less than 12 years of age, pregnant women, and HIV-positive patients with low CD4 cell counts are important populations that were excluded from this trial, and if one or both rifapentine regimens is shown to be non-inferior to the control regimen in S31/A5349, we advocate for future evaluation of rifapentine-based regimens in these populations. Further, S31/A5349 does not provide information on high-dose rifapentine regimens administered together with now-common ART agents such as integrase inhibitors and tenofovir alafenamide fumarate.
Contemporary trials build on tuberculosis clinical trials conducted between the 1950s and 1980s that established the current six-month standard regimen. That said, there are important differences between these “historical” and modern clinical trials that we considered during S31/A5349 planning. The first is an observed difference in proportions of participants assessed as having cure after treatment with the six-month standard regimen of 2HRZE/4HR. Higher proportions of unfavorable outcomes in contemporary trials may be driven by several factors including treatment setting (formerly mostly hospital-based and now almost all outpatient/ambulatory based), less stringent application of DOT, minimization of post-randomization exclusions, and the HIV epidemic. This has implications for a non-inferiority trial in terms of selection of the non-inferiority margin, because, for a fixed design, the maximum difference consistent with a non-inferiority conclusion decreases as the proportion of unfavorable outcomes in the control arm increases. The non-inferiority margin selected for S31/A5349 considers these issues as well as expert opinion, and is slightly higher than the 6% margin of non-inferiority used in other recent 1- or 2-drug substitution phase 3 trials of drug-susceptible tuberculosis. Second, in keeping with contemporary practice, for this trial mycobacteriology cultures are performed using both solid and liquid media. Trials done during the 20th century mainly or exclusively used solid media for cultures. Liquid media supports mycobacterial growth better and perhaps differently from solid media, which could increase the overall proportion of unfavorable responses in the current trial compared to earlier trials.
This trial purposefully incorporates a number of features of other recent phase 3 clinical trials for treatment of drug-susceptible pulmonary tuberculosis. This aided in estimating parameters critical for sample size calculations and will facilitate cross-trial analyses66. An important example is our use of diagnostic eligibility criteria that aimed to enroll a cohort of patients with pulmonary tuberculosis of approximately the same severity, at least based on sputum bacillary burden, as enrolled in recent and historical phase 3 clinical trials. S31/A5349 was performed in the context of increasing availability of Xpert, a relatively new rapid nucleic acid amplification test that has inherent sensitivity that is appreciably higher than that of sputum smear microscopy. We allowed Xpert testing for assessing study eligibility but required that the bacillary burden, as assessed by Xpert semiquantitative readout, meet or exceed the minimum burden detectable by smear microscopy.
S31/A5349 data collection and management for S31/A5349 use methods that optimize harmonization with data from other recent clinical trials, both to enable multi-trial analyses and to satisfy the requirements of stringent regulatory authorities. The trial database was designed to be compliant with standards of the Clinical Data Interchange Standards Consortium (CDISC) Clinical Data Acquisition Standards Harmonization (CDASH),67 in order to assure clarity and facilitate regulatory review.
Additional implementation details that strengthen the study include 1) a custom-built, secure, and highly responsive web-based data-management system, 2) provision and distribution of all study drugs in pre-packaged individual-participant kits by the manufacturer of rifapentine, Sanofi, and 3) strong collaboration and excellent communication between two large clinical trial networks.
Blinding of participants and investigators to treatment assignment is commonly used as a strategy to reduce bias. In S31/A5349 blinding of participants and investigators through using placebo pills was not feasible based on pill-burden, and was inappropriate based on differential food effects on pharmacokinetics of rifampin vs. rifapentine. Nevertheless, several trial design and implementation features should minimize bias around ascertainment of outcomes. Outcomes are based mainly on mycobacteriology data, and laboratorians are blinded to treatment assignment and study visit at which sputum was collected. Sputum specimens are collected at pre-specified time points that are uniform across the three study treatment regimens. The trial incorporates a novel “PPTR” process to ensure that, regard less of study treatment assignment, a uniform set of data and specimens are collected whenever a participant experiences any of a set of pre-specified triggers. Data and Coordinating Center staff that provide study implementation guidance to site staff are blinded to participant treatment assignment.
Despite long-standing evidence that rifamycin exposures are suboptimal using currently recommended doses of rifampin, this is one of the first studies to formally test the hypothesis that augmentation of rifamycin exposures can shorten tuberculosis treatment to four months. Results of this trial will have important implications for clinical management of tuberculosis at the individual and programmatic levels, and for future tuberculosis therapeutics trials.
Acknowledgments:
Funding and Role of the Funder/ Sponsor
Funding support for this trial was provided by the U.S. Centers for Disease Control and Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, Division of Tuberculosis Elimination; and by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Numbers UM1 AI068634, UM1 AI068636 and UM1 AI106701.
Sanofi (Paris, France, and Bridgewater, New Jersey) donated rifapentine and all other study drugs, supported shipping of study drugs to all sites, and provided funding support for pharmacokinetic testing. From 2007 until 2016, Sanofi donated a total of $2.9 million to the CDC Foundation to supplement CDC funding for rifapentine research; these funds supported prior TBTC studies of rifapentine, but were not part of the support for this trial.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Trial registration: This trial was registered with ClinicalTrials.gov (Identifier: NCT02410772) on April 8, 2015.
Trial status
Recruitment began in January 2016, and the last participant was enrolled in October 2018. Participant follow-up is expected to be completed in May 2020.
Ethics approval and consent to participate
This study is approved by the institutional review board of the US CDC. Additionally, each site follows local review policies and procedures. All adult study participants provide written informed consent. For adolescents, written parental permission is obtained from the parent/legal guardian, and written assent is obtained from the adolescent.
Competing interests
The authorship team members have declared (below or attached) any potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Sanofi commercial interests did not influence the study design; the collection, analysis, or interpretation of data; the preparation of this manuscript; or the decision to submit this manuscript for publication. A Sanofi technical expert served on the protocol team.
Consortium Identifiers: Tuberculosis Trials Consortium Study 31. AIDS Clinical Trials Group A5349
Disclaimer
The contents of this report are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention (CDC), the National Institute of Allergy and Infectious Diseases (NIAID/NIH), or the U.S. Department of Health and Human Services.
IND Number: 46,954.
IND Sponsor: U.S. Centers for Disease Control and Prevention.
References
- 1.World Health Organization. Global tuberculosis report 2018. Geneva, Switzerland: World Health Organization; 2018. Report No.: 978-92-4-156564-6. [Google Scholar]
- 2.Nahid P, Dorman S, Alipanah N, et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clinical infectious diseases 2016;63:e147–e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. Guidelines for treatment of drug-susceptible tuberculosis and patient care, 2017 update. Geneva, Switzerland: World Health Organization; 2017. [Google Scholar]
- 4.Fox W, Ellard GA, Mitchison DA. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946-1986, with relevant subsequent publications. The international journal of tuberculosis and lung disease : the official journal of the International Union against Tuberculosis and Lung Disease 1999;3:S231–79. [PubMed] [Google Scholar]
- 5.van Ingen J, Aarnoutse RE, Donald PR, et al. Why Do We Use 600 mg of Rifampicin in Tuberculosis Treatment? Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2011;52:e194–9. [DOI] [PubMed] [Google Scholar]
- 6.de Steenwinkel JE, Aarnoutse RE, de Knegt GJ, et al. Optimization of the rifampin dosage to improve the therapeutic efficacy in tuberculosis treatment using a murine model. American journal of respiratory and critical care medicine 2013;187:1127–34. [DOI] [PubMed] [Google Scholar]
- 7.Hu Y, Liu A, Ortega Muro F, Alameda Martin L, Mitchison D, Coates A. High-dose rifampicin kills persisters, shortens treatment duration, and reduces relapse rate in vitro and in vivo. Frontiers in microbiology 2015;6:641-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jayaram R, Gaonkar S, Kaur P, et al. Pharmacokinetics-pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrobial agents and chemotherapy 2003;47:2118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenthal I, Tasneen R, Peloquin C, et al. Dose-ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrobial agents and chemotherapy 2012;56:4331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diacon AH, Patientia RF, Venter A et al. Early bactericidal activity of high-dose rifampin in patients with pulmonary tuberculosis evidenced by positive sputum smears. Antimicrob Agents Chemother 2007;51:2994–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Svensson EM, Svensson RJ, Te Brake LHM et al. The potential for treatment shortening with higher rifampicin doses: relating drug exposure to treatment response in patients with pulmonary tuberculosis. Clin Infect Dis 2018;67:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bemer-Melchior P, Bryskier A, Drugeon HB. Comparison of the in vitro activities of rifapentine and rifampicin against Mycobacterium tuberculosis complex. The Journal of antimicrobial chemotherapy 2000;46:571–6. [DOI] [PubMed] [Google Scholar]
- 13.Benator D, Bhattacharya M, Bozeman L, et al. Rifapentine and isoniazid once a week versus rifampicin and isoniazid twice a week for treatment of drug-susceptible pulmonary tuberculosis in HIV-negative patients: a randomised clinical trial. Lancet (British edition) 2002;360:528–34. [DOI] [PubMed] [Google Scholar]
- 14.Vernon A, Burman W, Benator D, Khan A, Bozeman L. Acquired rifamycin monoresistance in patients with HIV-related tuberculosis treated with once-weekly rifapentine and isoniazid. Tuberculosis Trials Consortium. Lancet (British edition) 1999;353:1843–7. [DOI] [PubMed] [Google Scholar]
- 15.Pritin package insert. Accessed 1 January 2020 at http://products.sanofi.us/priftin/Priftin.pdf
- 16.Rosenthal IM, Zhang M, Almeida D, Grosset JH, Nuermberger EL. Isoniazid or moxifloxacin in rifapentine-based regimens for experimental tuberculosis? American journal of respiratory and critical care medicine 2008;178:989–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dooley KE, Bliven-Sizemore EE, Weiner M, et al. Safety and pharmacokinetics of escalating daily doses of the antituberculosis drug rifapentine in healthy volunteers. Clinical pharmacology and therapeutics 2012;91:881–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dorman S, Savic R, Goldberg S, et al. Daily rifapentine for treatment of pulmonary tuberculosis. A randomized, dose-ranging trial. American Journal of Respiratory & Critical Care Medicine 2015;191:333–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savic RM, Weiner M, MacKenzie WR, et al. Defining the optimal dose of rifapentine for pulmonary tuberculosis: Exposure-response relations from two phase II clinical trials. Clinical Pharmacology & Therapeutics 2017;102:321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji B, Lounis N, Maslo C, Truffot Pernot C, Bonnafous P, Grosset J. In vitro and in vivo activities of moxifloxacin and clinafloxacin against Mycobacterium tuberculosis. Antimicrobial agents and chemotherapy 1998;42:2066–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li S-Y, Irwin S, Converse P, Mdluli K, Lenaerts A, Nuermberger E. Evaluation of moxifloxacin-containing regimens in pathologically distinct murine tuberculosis models. Antimicrobial agents and chemotherapy 2015;59:4026–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burman WJ, Goldberg S, Johnson JL, et al. Moxifloxacin versus ethambutol in the first 2 months of treatment for pulmonary tuberculosis. American journal of respiratory and critical care medicine 2006;174:331–8. [DOI] [PubMed] [Google Scholar]
- 23.Conde MB, Efron A, Loredo C, et al. Moxifloxacin versus ethambutol in the initial treatment of tuberculosis: a double-blind, randomised, controlled phase II trial. Lancet 2009;373:1183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillespie SH, Crook AM, McHugh TD, et al. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. The New England journal of medicine 2014;371:1577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kjellsson MC, Via LE, Goh A, et al. Pharmacokinetic evaluation of the penetration of antituberculosis agents in rabbit pulmonary lesions. Antimicrobial agents and chemotherapy 2012;56:446–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prideaux B, Dartois V, Staab D, et al. High-sensitivity MALDI-MRM-MS imaging of moxifloxacin distribution in tuberculosis-infected rabbit lungs and granulomatous lesions. Analytical chemistry 2011;83:2112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu P, Chen H, Xu J et al. Moxifloxacin is an effective and safe candidate agent for tuberculosis treatment: a meta-analysis. Int J Infect Dis 2017;60:35–41. [DOI] [PubMed] [Google Scholar]
- 28.Thee S, Garcia-Prats AJ, Donald PR, Hesseling AC, Schaaf HS. Fluoroquinolones for the treatment of tuberculosis in children. Tuberculosis (Edinb) 2015;95:229–245. [DOI] [PubMed] [Google Scholar]
- 29.Zignol M, Dean AS, Alikhanova N, et al. Population-based resistance of Mycobacterium tuberculosis isolates to pyrazinamide and fluoroquinolones: results from a multicountry surveillance project. Lancet Inf Dis 2016;16:1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blakemore R, Nabeta P, Davidow AL et al. A multisite assessment of the quantitative capabilities of the Xpert MTB/RIF assay. Am J Respir Crit Care Med 2011;184:1076–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boehme CC, Nabeta P, Hillemann D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med 2010;363:1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall JD, Abdel-Rahman S, Johnson K, Kauffman RE, Kearns GL. Rifapentine pharmacokinetics in adolescents. The Pediatric infectious disease journal 1999;18:882–8. [DOI] [PubMed] [Google Scholar]
- 33.Abdool Karim S, Naidoo K, Grobler A, et al. Timing of initiation of antiretroviral drugs during tuberculosis therapy. The New England journal of medicine 2010;362:697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breen RAM, Smith CJ, Bettinson H, et al. Paradoxical reactions during tuberculosis treatment in patients with and without HIV co-infection. Thorax 2004;59:704–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burman W, Weis S, Vernon A, et al. Frequency, severity and duration of immune reconstitution events in HIV-related tuberculosis. The international journal of tuberculosis and lung disease 2007;11:1282–9. [PubMed] [Google Scholar]
- 36.Havlir D, Kendall M, Ive P, et al. Timing of antiretroviral therapy for HIV-1 infection and tuberculosis. The New England journal of medicine 2011;365:1482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guidelines for the prevention and treatment of opportunistic infections in adults and adolescents with HIV: recommendations from the Centers for Disease Control and Prevention, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. (Accessed 11 July, 2019, at http://aidsinfo.nih.gov/contentfiles/lvguidelines/adult_oi.pdf.)
- 38.Podany ATSE, Samaneka W, Mohapi L, Johnson J, Mayanja H, Lalloo U, Badal-Faesen S, Campbell K, Pham M, Miyahara S, Bryant K, Vernon A, Hafner R, Kurbatova E, Chaisson RE, Dorman S, Nahid P, Dooley K, Swindells S, and the AIDS Clinical Trials Group and the Tuberculosis Trials Consortium. Efavirenz pharmacokinetics in HIV/TB coinfected persons initiating ART while receiving high dose rifapentine. 20th International Workshop on Clinical Pharmacology of HIV, Hepatitis & Other Antiviral Drugs 2019; Netherlands. [Google Scholar]
- 39.Podany ASE, Chen M, Martinson NA, Dawson R, Badal-Faesen S, Miyahara S, Kurbatova E, Whitworth WC, Chaisson RE, Dorman SE, Nahid P, Dooley K, Swindells S, for the AIDS Clinical Trials Group & Tuberculosis Trials Consortium A5349 / Study 31 Team. Efavirenz Pharmacokinetics in HIV/TB Coinfected Persons Receiving Rifapentine. Conference on Retroviruses and Opportunistic Infections (CROI) 2018; Boston, MA. [Google Scholar]
- 40.Soares JF, Jeff Wu CF. Some Restricted randomization rules in sequential designs. Communications in Statistics - Theory and Methods Communications in Statistics - Theory and Methods 1983;12:2017–34.. [Google Scholar]
- 41.Chan SL, Yew WW, Porter JH, et al. Comparison of Chinese and Western rifapentines and improvement of bioavailability by prior taking of various meals. International journal of antimicrobial agents 1994;3:267–74. [DOI] [PubMed] [Google Scholar]
- 42.Dorman SE, Goldberg S, Stout JE, et al. Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: study 29 of the tuberculosis trials consortium. The Journal of infectious diseases 2012; 206:1030–40. [DOI] [PubMed] [Google Scholar]
- 43.Zvada SP, Van Der Walt JS, Smith PJ, et al. Effects of four different meal types on the population pharmacokinetics of single-dose rifapentine in healthy male volunteers. Antimicrobial agents and chemotherapy 2010;54:3390–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peloquin CA, Namdar R, Singleton MD, Nix DE. Pharmacokinetics of rifampin under fasting conditions, with food, and with antacids. Chest 1999;115:12–8. [DOI] [PubMed] [Google Scholar]
- 45.Zent C, Smith P. Study of the effect of concomitant food on the bioavailability of rifampicin, isoniazid and pyrazinamide. Tubercle and lung disease : the official journal of the International Union against Tuberculosis and Lung Disease 1995;76:109–13. [DOI] [PubMed] [Google Scholar]
- 46.Nunn AJ, Phillips PP, Mitchison DA. Timing of relapse in short-course chemotherapy trials for tuberculosis. Int J Tuberc Lung Dis 2010;14:241–242. [PubMed] [Google Scholar]
- 47.Nunn AJ, Phillips PPJ, Meredith SK et al. A trial of a shorter regimen for rifampin-resistant tuberculosis. N Engl J Med 2019;380:1201–1213. [DOI] [PubMed] [Google Scholar]
- 48.A phase 3 study assessing the safety and efficacy of bedaquiline plus PA-824 plus linezolid in subjects with drug resistant pulmonary tuberculosis. NCT02333799. Accessed 13 January 2020 at https://clinicaltrials.gov/ct2/show/NCT02333799
- 49.Trial to evaluate the efficacy, safety and tolerability of BPaMZ in drug-sensitive (DS-TB) adult patients and drug-resistant (DR-TB) adult patients. NCT03338621. Acessed 13 January 2020 at https://clinicaltrials.gov/ct2/show/NCT03338621
- 50.Jindani A, Harrison TS, Nunn AJ, et al. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. The New England journal of medicine 2014;371:1599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Merle CS, Fielding K, Sow OB, et al. A four-month gatifloxacin-containing regimen for treating tuberculosis. The New England journal of medicine 2014;371:1588–98. [DOI] [PubMed] [Google Scholar]
- 52.The Common Terminology Criteria for Adverse Events version 4.03 published on June 14, 2010 (Accessed 11 July, 2019, at https://www.eortc.be/services/doc/ctc/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf.)
- 53.East African/British Medical Research Council. Controlled clinical trial of four 6-month regimens of chemotherapy for pulmonary tuberculosis. Second report. Second East African/British Medical Research Council Study. The American review of respiratory disease 1976;114:471–5. [DOI] [PubMed] [Google Scholar]
- 54.East African/British Medical Research Council. Results at 5 years of a controlled comparison of a 6-month and a standard 18-month regimen of chemotherapy for pulmonary tuberculosis. The American review of respiratory disease 1977; 116:3–8. [DOI] [PubMed] [Google Scholar]
- 55.East African/British Medical Research Councils Study. Controlled clinical trial of five short-course (4-month) chemotherapy regimens in pulmonary tuberculosis. Second report of the 4th study. East African/British Medical Research Councils Study. The American review of respiratory disease 1981;123:165–70. [DOI] [PubMed] [Google Scholar]
- 56.East and Central Africa/British Medical Research Council. Controlled clinical trial of 4 short-course regimens of chemotherapy (three 6-month and one 8-month) for pulmonary tuberculosis: final report. East and Central African/British Medical Research Council Fifth Collaborative Study. Tubercle 1986;67:5–15. [DOI] [PubMed] [Google Scholar]
- 57.Singapore Tuberculosis Service/British Medical Research Council. Long-term follow-up of a clinical trial of six-month and four-month regimens of chemotherapy in the treatment of pulmonary tuberculosis. Singapore Tuberculosis Service/British Medical Research Council. The American review of respiratory disease 1986; 133:779–83. [PubMed] [Google Scholar]
- 58.Guidance for Industry Pulmonary Tuberculosis: Developing Drugs for Treatment. 2013. (Accessed 11 July, 2019, at https://www.fda.gov/media/87194/download.)
- 59.Nunn AJ, Phillips PPJ, Gillespie SH. Design issues in pivotal drug trials for drug sensitive tuberculosis (TB). Tuberculosis Tuberculosis 2008;88:S85–S92. [DOI] [PubMed] [Google Scholar]
- 60.The Consortium for TB Biomarkers (CTB2) Biorepository (Accessed 11 July, 2019, at https://www.tbbiorepository.org/.)
- 61.Nahid P, Saukkonen J, Mac Kenzie WR et al. CDC/NIH Workshop. Tuberculosis biomarker and surrogate endpoint research roadmap. Am J Respir Crit Care Med 2011;184:972–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walter ND, de Jong BC, Garcia BJ et al. Adaptation of Mycobacterium tuberculosis to impaired host immunity in HIV-infected patients. J Infect Dis 2016;214:1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walter ND, Dolganov GM, Garcia BJ et al. Transcriptional adaptation of drug-tolerant Mycobacterium tuberculosis during treatment of human tuberculosis. J Infect Dis 2015;212:990–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Colangeli R, Jedrey H, Kim S et al. Bacterial factors that predict relapse after tuberculosis therapy. New Engl J Med 2018;379:823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schulz KF, Altman DG, Moher D, Schulz K. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. bmjbritmedj BMJ: British Medical Journal 2010;340:698–702. [Google Scholar]
- 66.Imperial MZ, Nahid P, Phillips PPJ et al. A patient-level pooled analysis of treatment shortening regimens for drug-susceptible pulmonary tuberculosis. Nat Med 2018. 24:1708–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clinical Data Interchange Standards Consortium (CDISC) Clinical Data Acquisition Standards Harmonization (CDASH). (Accessed 11 July, 2019, at https://www.cdisc.org/standards/foundational/cdash.)