

Abstract
MYC is one of the most important oncogenes and is overexpressed in the majority of cancers. G-quadruplexes are noncanonical four-stranded DNA secondary structures that have emerged as attractive cancer-specific molecular targets for drug development. The G-quadruplex formed in the proximal promoter region of the MYC oncogene (MycG4) has been shown to be a transcriptional silencer that is amenable to small molecule targeting for MYC suppression. Indenoisoquinolines are human topoisomerase I inhibitors in clinical testing with improved physicochemical and biological properties as compared to the clinically used camptothecin anticancer drugs topotecan and irinotecan. However, some indenoisoquinolines with potent anticancer activity do not exhibit strong topoisomerase I inhibition, suggesting a separate mechanism of action. Here, we report that anticancer indenoisoquinolines strongly bind and stabilize MycG4 and lower MYC levels in cancer cells, using various biochemical, biophysical, computer modeling, and cell-based methods. Significantly, a large number of active indenoisoquinolines cause strong MYC downregulation in cancer cells. Structure-activity-relationships of MycG4 recognition by indenoisoquinolines are investigated. In addition, the analysis of indenoisoquinoline analogues for their MYC inhibitory activity, topoisomerase I inhibitory activity, and anticancer activity reveals a synergistic effect of MYC inhibition and topoisomerase I inhibition on anticancer activity. Therefore, this study uncovers a novel mechanism of action of indenoisoquinolines as a new family of drugs targeting the MYC promoter G-quadruplex for MYC suppression. Furthermore, the study suggests that dual targeting of MYC and topoisomerase I may serve as a novel strategy for anticancer drug development.
Keywords: G-quadruplex, MYC, oncogene promoter, indenoisoquinoline, anticancer drug, topoisomerase I
Table of Contents Artwork:
INTRODUCTION
DNA is the target of many important anticancer agents, including human topoisomerase I inhibitors. Recently there has been significant progress in developing molecular-targeted therapies. A therapeutic advantage can be gained from DNA-targeted drugs combined with cancer-specific molecular targeting properties. Indenoisoquinolines are human topoisomerase I inhibitors with improved physicochemical and biological properties as compared to the traditional camptothecin topoisomerase I inhibitors that are clinically used for the treatment of various solid tumors.1–6 Three indenoisoquinolines, indotecan (LMP400), indimitecan (LMP776), and LMP744 (Figure 1A), have entered phase I clinical trials in adults with relapsed solid tumors and lymphomas.7–14 However, some indenoisoquinolines with potent anticancer activity did not show strong topoisomerase I inhibition,3, 15 suggesting additional mechanism of action. Notably, high concentrations of some indenoisoquinoline compounds have been reported to target DNA outside of topoisomerase I action.6–7, 16–17
Figure 1.
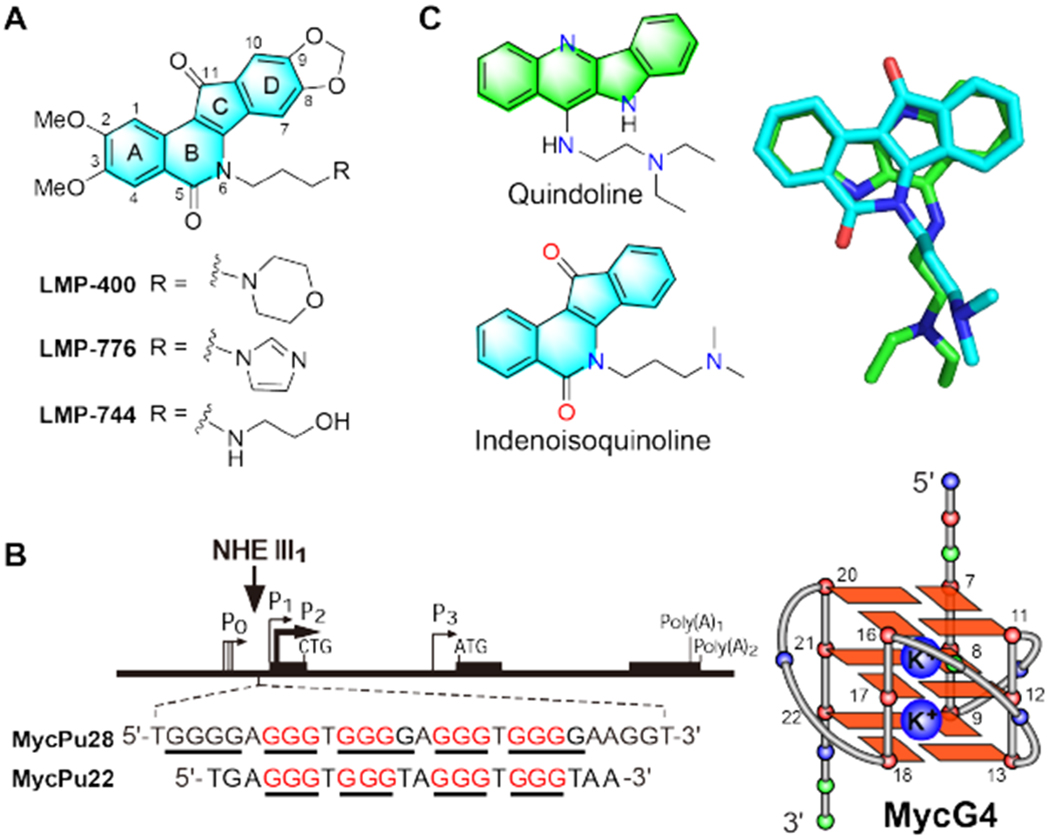
Chemical structures of indenoisoquinoline topoisomerase I inhibitors in Phase I clinical trials and quindoline, as well as the MYC promoter and MYC promoter G-quadruplex. (A) Indenoisoquinoline topoisomerase I inhibitors currently in clinical trials. (B) Left: The structure of the human MYC gene promoter. The G4-forming region NHE III1 sequence is shown, with the guanine runs underlined. The guanine runs involved in the formation of the major MycG4 are highlighted in red. Right: The folding topology of MycG4 adopted by the MycPu22 sequence is a parallel-stranded 3-tetrad G-quadruplex, with the two stabilizing potassium cations shown. Red ball = guanine, green ball = adenine, blue ball = thymine, large blue ball = K+. (C) Left: a MycG4 stabilizer quindoline and a topoisomerase I inhibitor indenoisoquinoline. Right: overlay of the three-dimensional structures of quindoline and an indenoisoquinoline in their energy-minimized states.
MYC is one of the most important oncogenes and is overexpressed in more than 80% of all types of cancer.18–19 The transcription factor MYC protein is involved in cell proliferation, differentiation, and apoptosis, and plays a pivotal role in tumor initiation and progression as well as drug resistance.20–24 MYC is found to be a general transcriptional “amplifier” in cancer cells.25–26 Even a brief inhibition of MYC expression has been shown to permanently stop tumor growth and induce tumor regression in vivo,27 because of the “oncogene addiction” of tumor cells.28 Therefore, MYC is a hotly pursued therapeutic target. However, the MYC protein is not an easy drug target due to its short half-life and lack of a small molecule binding pocket.29–31
The nuclease hypersensitive element (NHE) III1 in the MYC promoter, which controls 85-90% of MYC transcriptional activity, forms a DNA G-quadruplex (G4) under transcription-associated negative supercoiling and functions as a transcriptional silencer (Figure 1B, left).32–36 DNA G-quadruplexes (G4s) are globular four-stranded secondary structures consisting of stacked Hoogsteen hydrogen-bonded G-tetrads stabilized by K+ or Na+.37 DNA G-quadruplexes found in promoter regions of key oncogenes have emerged as a promising new class of cancer-specific molecular targets for drug development.38–40 Using a G4-specific antibody, G4 structures have been visualized in human cells at both telomeric and non-telomeric sites on chromosomes, and G4-loci increase after exposure of live cells to G4 ligands.41 G4s detected in immortalized precancerous cells are at 10 times higher levels than in normal human cells, and G4-sites are found to be specifically enriched in regulatory, transcriptionally active regions of chromatin, particularly the MYC promoter region.42 We previously determined the structures of the MYC promoter G-quadruplexes.43–44 The major MYC promoter G-quadruplex (MycG4) is a parallel-stranded structure with three G-tetrads connected by three propeller loops (Figure 1B, right).32, 43, 45 Significantly, stabilization of the MYC promoter G-quadruplex by small molecules suppresses MYC transcription.32, 36, 46 For example, a quindoline anticancer agent was shown to stabilize the MYC G-quadruplex and downregulate MYC.46–47 We have determined the molecular structure of the 2:1 quindoline-MycG4 complex, which shows specific recognition of the MycG4 by the crescent-shaped quindoline.48 Interestingly, indenoisoquinolines are crescent-shaped and share structural similarity with the quindoline compound (Figure 1C), which is consistent with the report that 6-substituted indenoisoquinolines15 bind the c-Kit promoter G4s.17
Herein, using fluorescence resonance energy transfer (FRET) assays, nuclear magnetic resonance (NMR), fluorescence-based binding assay and competition fluorescence displacement assay, circular dichroism (CD) spectroscopy, and gel electromobility shift assay (EMSA), we demonstrate that a large number of anticancer indenoisoquinolines strongly bind and stabilize MycG4 in vitro. Using cell-based western blotting and quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assays, we show that MycG4-interactive indenoisoquinolines lower MYC mRNA and protein levels in vivo, indicating that targeting the MYC promoter G4 to downregulate MYC is a likely mechanism of action for the anticancer activities of these particular indenoisoquinolines. Furthermore, some active indenoisoquinolines show both MYC downregulation and topoisomerase I inhibition, suggesting that dual-targeting of MycG4 and topoisomerase I could be a potential strategy for anticancer drug development.
RESULTS AND DISCUSSION
Indenoisoquinolines can induce and stabilize MycG4
To examine whether the indenoisoquinolines could induce and stabilize the MycG4, we conducted a FRET-quenching assay on indenoisoquinoline compounds. The full-length MYC promoter NHE III1 G4 DNA (MycPu28, Figure 1B) was labeled with FAM (6-fluorescein) on the 3′-end and BHQ-1 (Black Hole-1 quencher) on the 5′-end (Figure 2A left). The MycG4 structure adopted by MycPu22 (Figure 1B) is the major conformation formed by the wild-type MycPu28 in K+ solution. 32, 43, 45 MycPu28 was used for the FRET-quenching screening assay because it has higher FAM-fluorescence than MycPu22 in the unfolded form due to the longer distance between the FAM and BHQ quencher, and thus provided greater range for screening (Figure S2). We confirmed that very similar FRET-quenching effects were observed for MycPu22 and MycPu28 upon compound binding and G4-stabilization (Figure S2). The stable formation of G-quadruplexes requires the presence of K+ or Na+ cations in solution, with a preference of K+ (Figure 1B). In the absence of K+, the MycPu28 is in the extended single-stranded form with its two ends far apart and shows high FAM-fluorescence (Figure 2A left). In the presence of 100 mM K+, the G4 is folded and the FAM-fluorescence is quenched because the quencher and fluorophore at the two ends are in closer proximity (Figure 2A left). Alternatively, the addition of G4-stabilizing ligands can induce G4 formation in the absence of K+ and thereby lead to quenching FAM-fluorescence (Figure 2A left).
Figure 2.
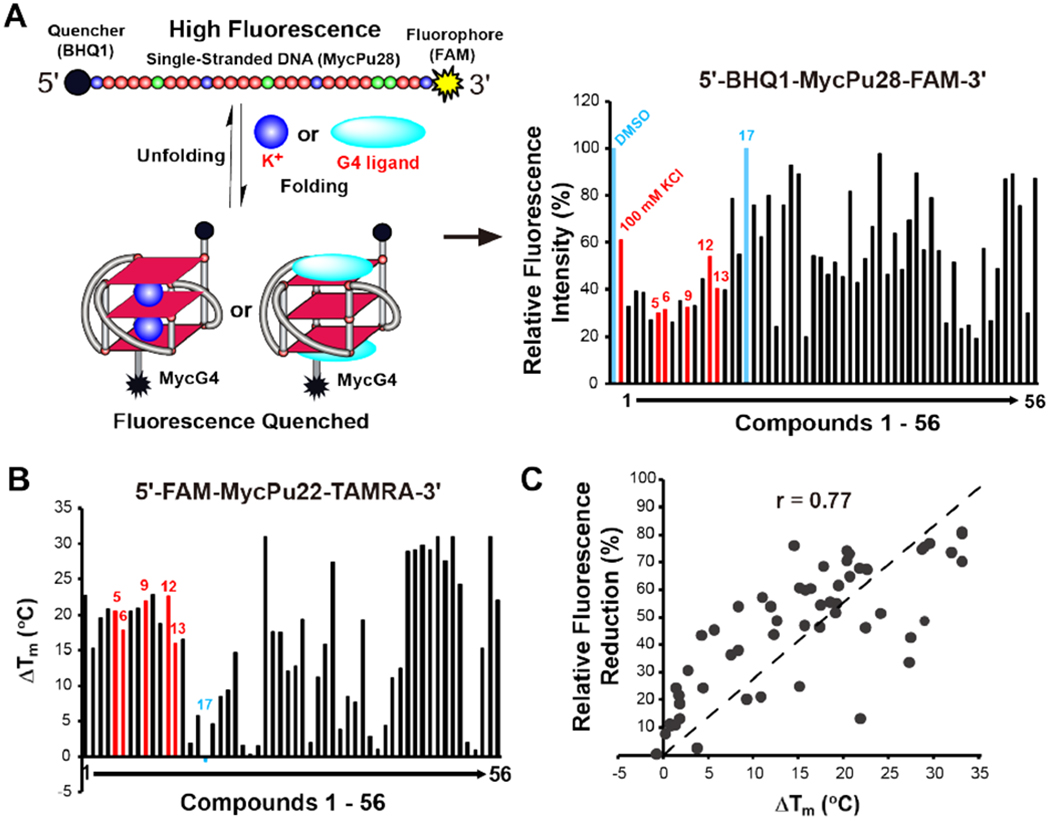
Indenoisoquinolines can induce and stabilize MycG4. (A) Left: schematic of the FRET-quenching assay used for compound screening. The FRET-quenching (shown as fluorophore in black color) caused by MycG4 folding can be induced by K+ or MycG4-inducing compounds. Right: relative fluorescence intensities of the labeled MycG4 in the presence of DMSO, 100 mM K+, and indenoisoquinoline analogs as shown by FRET-quenching assay. Data shown are the average values of the two individual experiments. DMSO (negative control), 100 mM K+ (positive control), and six indenoisoquinolines used for further studies are highlighted and labeled. Conditions: 1 μM labeled DNA, 10 μM compound, 25 °C, 50 mM Tris·acetate, pH 7. (B) Thermal stabilization values (ΔTm) of MycG4 by indenoisoquinoline analogs as shown by FRET-melting assay. Data shown are the average values of the two individual experiments. The six representative indenoisoquinolines used for further studies are highlighted and labeled. Conditions: 150 nM labeled DNA, 1.5 μM compound, 25 °C, pH 7, 10 mM K+. (C) Correlation of FRET-quenching and FRET-melting data. The Pearson correlation coefficient (r) is shown.
We examined 56 indenoisoquinoline compounds (Figure S1) using this FRET-quenching assay (Figure 2A). 100 mM K+ buffer was used as a positive control, which decreased FAM-fluorescence by 39%. We found that 37 compounds decreased the FAM-fluorescence by more than 39%, indicating that these indenoisoquinolines can induce and stabilize MycG4. Some indenoisoquinolines decreased FAM-fluorescence more than 100 mM K+, which is likely due to greater stabilization of MycG4 or its flanking structures. However, it cannot be ruled out that some indenoisoquinolines may interact with the FAM fluorophore directly to quench the FAM-fluorescence.
To confirm the stabilizing effect of indenoisoquinolines on MycG4, the Tm values of MycG4 were measured in the presence of indenoisoquinoline compounds in 10 mM K+ using dual-3′-FAM- and 5′-TAMRA-labeled MycPu22 DNA by FRET-melting experiments. MycPu22 DNA forms a single MycG4 structure and was used for NMR structure determination (Figure 1B, right)43 Therefore, MycPu22 provides the best molecular system for MycG4 and was used in all the subsequent experiments. 10 mM K+ was used in the FRET-melting experiments because the melting temperature of MycG4 at 100 mM K+ is above 90 °C, making it impossible to determine an accurate melting temperature upon compound addition.49 The FRET-melting results showed that forty-four of the fifty-six indenoisoquinolines increased the Tm values of MycG4 by more than 5 °C (Figure 2B). A clear positive correlation was observed between the indenoisoquinolines’ ability to induce MycG4 formation and to increase its thermal stability (Figure 2C).
Some indenoisoquinolines significantly lower MYC levels in cancer cells
G-quadruplex formed in the MYC promoter was found to function as a transcriptional silencer.32–34 To determine the effects of indenoisoquinolines on the MYC protein level, a western blotting experiment was carried out using MCF-7 breast cancer cells treated with 44 indenoisoquinolines that increased the Tm value of MycG4 by more than 5 °C. MCF-7 cells were incubated with each compound at four concentrations (0.5, 1, 2, and 4 μM) for 24 hours, and the MYC protein levels were measured (Figure 3A and Figure S3).
Figure 3.
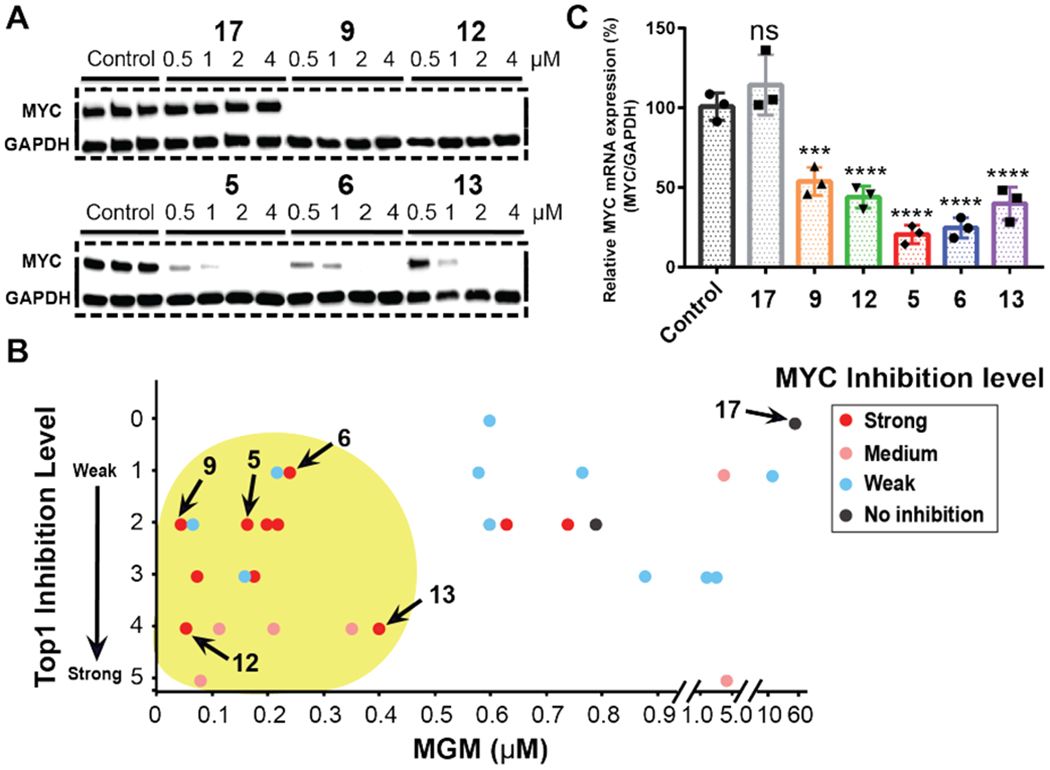
MYC inhibitory activities of indenoisoquinoline analogs. (A) MYC protein expression levels in the absence and presence of various concentrations of indenoisoquinolines (24 hr treatment) were obtained by western blotting experiments in MCF-7 breast cancer cells. GAPDH was used as an internal control. (B) Plot of the topoisomerase I inhibition levels against the MGM values of 31 indenoisoquinolines that were used to determine topoisomerase, MYC, and MGM activities. The yellow shaded area indicated the region of more active indenoisoquinolines. Based on the MYC downregulation shown in the western blotting results (Figures 3A and S3), MYC inhibition levels were classified into four levels: strong inhibition, MYC expression inhibited at 0.5 to 1.0 μM, red dots; medium inhibition, MYC expression inhibited at 2.0 μM or no clear dose-dependent MYC inhibition, pink dots; weak inhibition, MYC expression inhibited at 4.0 μM, blue dots; no inhibition, no MYC expression inhibition up to 4.0 μM, black dots. The relative topoisomerase I (Top1) inhibition levels of the compounds were previously determined and classified into six levels (0 – 5).3, 6–9, 15, 50–52 The MGM values are the average of GI50 values across the entire panel of NCI-60 cancer cell lines; the GI50 values are the concentrations corresponding to 50% growth inhibition which were determined in the NCI-60 cancer cell lines drug screen (Table S3, Figures S8 and S9). (C) MYC transcription levels in the absence and presence of indenoisoquinolines (6 hr treatment) were obtained by qRT-PCR experiments in MCF-7 cancer cells. DMSO was used as the negative control (no inhibition, 100%). The relative MYC mRNA levels were normalized with GAPDH. The experiments were run in triplicate. P values: ***P < 0.0004, ****P < 0.0001.
The human topoisomerase I inhibitory activities of the 44 indenoisoquinolines have been previously determined.3, 6–9, 15, 50–52 Of the 44 compounds tested for their cytotoxicities in the NCI-60 cancer cell lines, the 31 most potent compounds had their mean graph midpoint (MGM) values determined based on the GI50 values obtained from the NCI-60 cancer cell line drug screen (Table S3, Figures S8 and S9).53–55 The topoisomerase I inhibitory activities were plotted against the anticancer activities of these 31 compounds (Figure 3B). Some of the more active compounds (with MGM values < 0.5 μM) showed strong topoisomerase I inhibition. However, many of the active compounds were not strong topoisomerase I inhibitors. The MYC inhibition activities of these compounds were ranked in four groups, i.e., strong, medium, weak, and no inhibition (Figure 3B). Significantly, strong MYC inhibition was concentrated in compounds with potent anticancer activities, including those showing weak topoisomerase I inhibitory activity (Figure 3B). We selected compounds 5, 6, 9, 12 and 13 for further investigation as they showed clear MYC-inhibitory effect (Figures 3B and 4). Compound 17 was used as a negative control (Figures 3B and 4).
Figure 4.
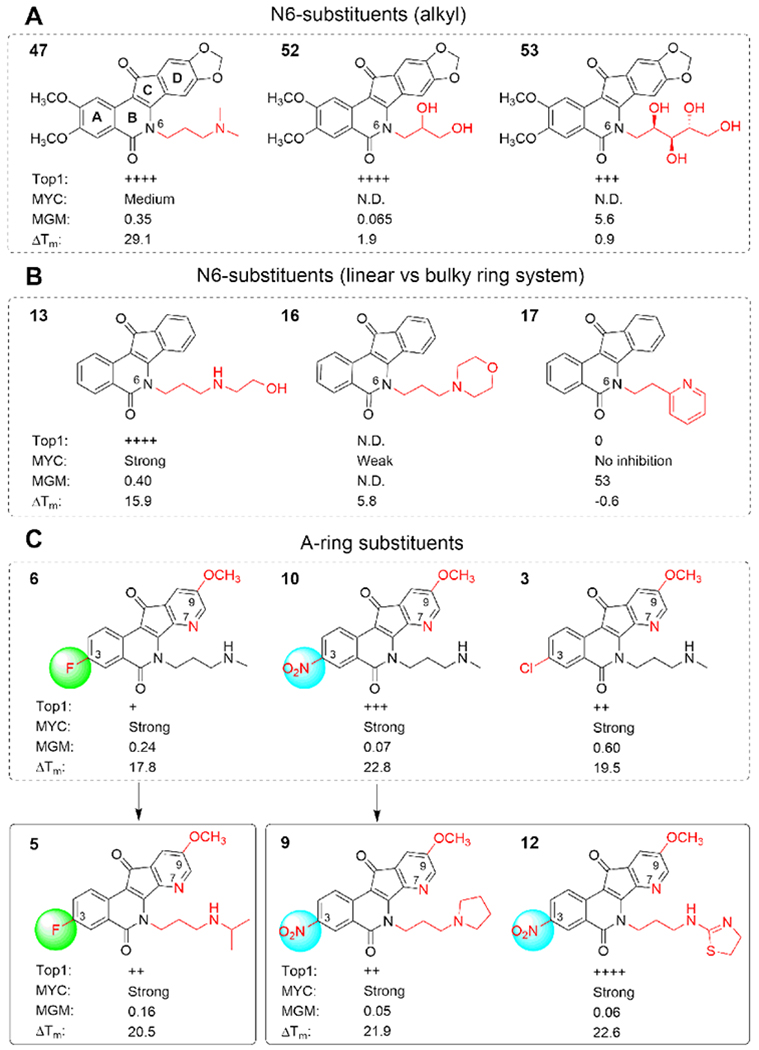
SAR of selected indenoisoquinolines. N.D., not determined.
To confirm the effect on the transcription of the MYC gene in cancer cells by the six selected indenoisoquinoline compounds, the MYC mRNA levels in MCF-7 cancer cells were measured by qRT-PCR. Consistent with the western blotting data, all five MYC-inhibiting compounds significantly lowered MYC mRNA levels at 6 hours post the treatments with 1 μM indenoisoquinolines. The negative control compound 17 showed no reduction of MYC mRNA level (Figure 3C).
MYC-inhibiting indenoisoquinolines are strong MycG4 binding ligands
The binding interactions of six selected indenoisoquinolines with MycG4 were examined using 1H NMR titration experiments in K+-containing solution. The free MycG4 DNA shows 12 imino proton peaks of guanines from the three G-tetrads (Figure 5).43, 48 Upon respective addition of the five MycG4-interactive indenoisoquinolines, clear changes of the tetrad-guanine imino proton signals were observed, confirming the binding of these compounds to MycG4 (Figure 5A–E). The binding appeared to be in the medium-to-fast exchange rate on the NMR time-scale, as shown by the broadening of DNA proton peaks at lower drug equivalence (0.5 and 1) and the sharpening at higher drug equivalence (2 and 3). Indenoisoquinolines appeared to bind at both ends of the MycG4, as shown by the imino proton peaks corresponding to both of the 3′- and 5′- tetrads being significantly shifted upon drug addition. Three MYC-inhibiting compounds, the 7-azaindenoisoquinolines 5 and 6, and the indenoisoquinoline 13, showed more specific binding to MycG4, where a well-defined complex was shown to form at the drug equivalence of 3, with a new set of 12 imino proton peaks. For compound binding at intermediate exchange rate on the NMR time scale, a compound:DNA ratio higher than its binding stoichiometry is needed to push the equilibrium towards the formation of a stable drug-DNA complex, as shown by the sharp, well-resolved proton peaks.48, 56 In contrast, the negative control compound 17 did not show any binding as no change was observed in the 1H NMR spectra upon titration (Figure 5F). The MycG4 complexes of the five MycG4-interactive indenoisoquinolines were monomeric in nature as shown by native EMSA gels (Figure S4).
Figure 5.
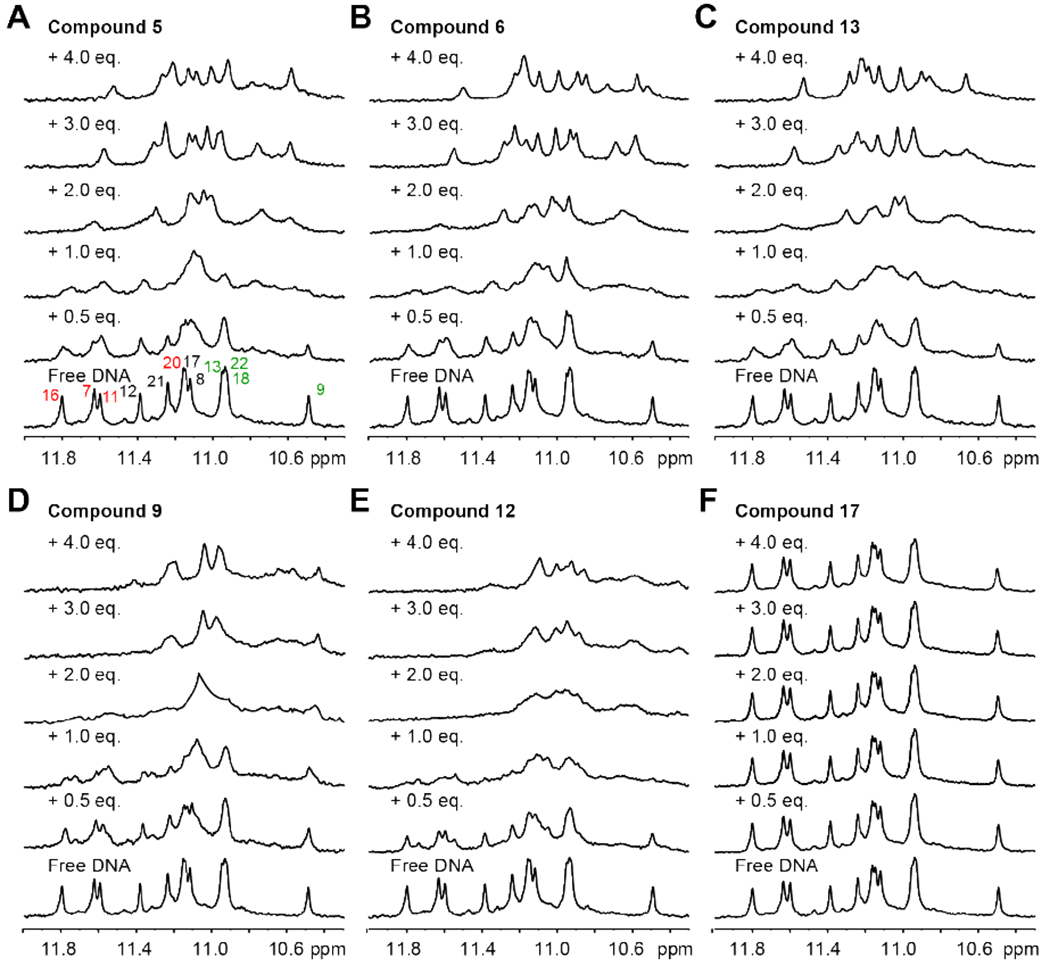
1D 1H NMR titrations of MycPu22 DNA with indenoisoquinolines and 7-azaindenoisoquinolines. Imino proton regions of the titration spectra of MycG4 with compound 5 (A), 6 (B), 13 (C), 9 (D), 12 (E), and 17 (F) are shown. In Figure 5A, the imino proton signals from the 5′ G-tetrad (Figure 1B) are labeled in red, the middle G-tetrad in black, and the 3′ G-tetrad in green. Conditions: 150 μM DNA, 25 °C, pH 7, 100 mM K+.
CD titration experiments with MycG4 were also carried out for the six selected indenoisoquinolines. The free MycPu22 DNA in K+ buffer showed the CD signature of a parallel G-quadruplex, with a positive peak at 264 nm and a negative peak at 242 nm.57 Upon addition of indenoisoquinolines, the CD signature of a parallel G-quadruplex was maintained (Figure S5). The five MycG4-interactive compounds showed a slight decrease in intensity for both the positive peak at 264 nm and the negative peak at 242 nm, likely due to the ligand-induced capping structure formation by the flanking segments. The decrease in intensity in CD spectra of G4 upon ligand binding has been previously reported.58 The negative control compound 17 showed no effect on the CD spectrum.
Binding affinities of these six indenoisoquinolines to MycG4 were measured using a 3′-TAMRA-labeled MycPu22 DNA.59 The five MYC-inhibiting compounds showed strong binding with apparent binding affinity Kd values of 5.6 – 23.9 nM, whereas the negative control compound showed negligible binding (Figure S6). The indenoisoquinolines show negligible fluorescence in either the free or bound state.
Molecular docking study of the binding of indenoisoquinoline 5 to MycG4
NMR titration data showed that 7-azaindenoisoquinoline 5 binds MycG4 to form a well-defined complex at both the 5′- and 3′-ends, as is evident by the significant shifting of the imino proton peaks of the 5′- and 3′-external tetrad guanines (Figure 5A). We have previously determined the NMR structure of the 2:1 quindoline:MycG4 complex in K+ solution (PDB ID 2L7V), in which quindoline binds MycG4 at both ends to form a 5′-complex and 3′-complex.48 As indenoisoquinolines are structurally similar to the quindoline compound (Figure 1C), we performed a molecular docking study to explore the possible binding modes of 7-azaindenoisoquinoline 5 with the MycG4 based on the NMR structure of the 2:1 quindoline:MycG4 complex. The docking program Glide was used in the standard precision (SP) mode: see Methods.60–61 7-Azaindenoisoquinoline 5 was docked to the binding sites at the two ends of the MycG4 using the 2:1 quindoline:MycG4 complex structure (Figure 6). Several similar binding poses were predicted by the docking experiment for both the 5′- and 3′-sites. Docking studies gave docking scores for the 5′- and 3′-complexes at −6.69 and −6.08 kcal/mol, respectively. Figure 6 shows a representative model of the 2:1 7-azaindenoisoquinoline 5:MycG4 complex. The overall binding modes of the indenoisoquinoline resembled those of quindoline in the NMR structure of the 2:1 quindoline:MycG4 complex, in which a flanking DNA base from the 5′- or 3′- flanking segment was recruited to form a ligand-base plane stacking over the external tetrads, except that no H-bond was present in the 3′-complex between the indenoisoquinoline and the recruited base. Notably, the tetracyclic ring scaffold of 7-azaindenoisoquinoline 5 with A- and D-ring substituents stacks very well with both the 5′- and 3′-external tetrads, making extensive stacking interactions. The positively charged amine side chain of indenoisoquinoline 5 resides in the MycG4 groove and forms intermolecular salt bridges with phosphate groups on the nucleotide backbone.
Figure 6.
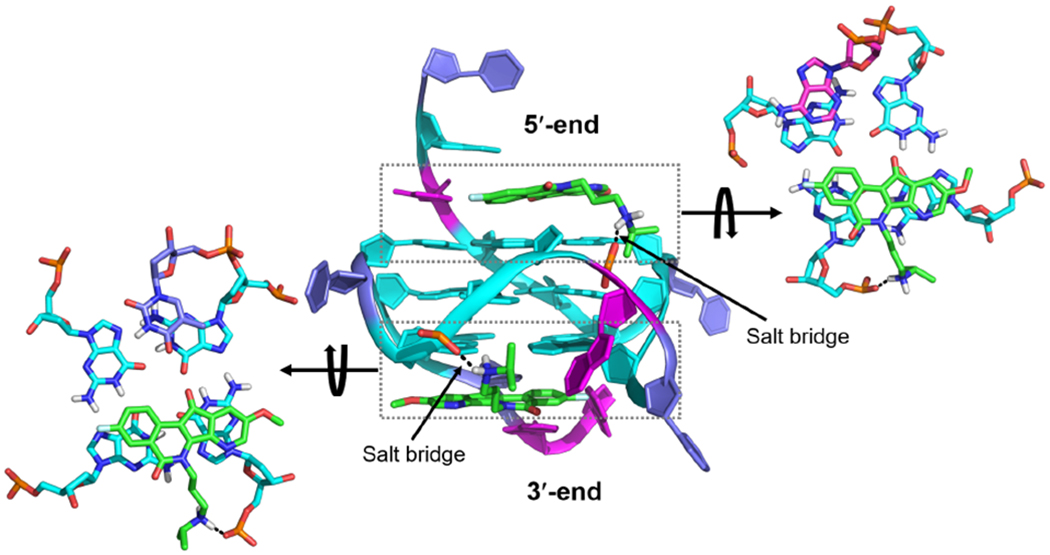
A model of the 2:1 complex of 7-azaindenoisoquinoline 5:MycG4 suggested by Glide docking in different views. 7-azaindenoisoquinoline 5 is shown in green. Intermolecular salt bridges are shown as black dashed lines.
Binding selectivity of MycG4-interactive indenoisoquinolines and 7-azaindenoisoquinolines.
Using a competition fluorescence displacement assay, we determined the binding selectivity of five indenoisoquinolines for MycG4 as compared to a parallel K-Ras promoter G4, a hybrid telomeric G4, and double-stranded (ds) DNA at 1 and 5 equivalents of each compound (Figures 7 and S7). The 3′-TAMRA labeled MycPu22 DNA was used as the fluorescence probe, whose fluorescence was quenched upon the binding of indenoisoquinolines. Upon addition of unlabeled, non-fluorescent competitors (e.g. other DNA G4s and dsDNA), the TAMRA-labeled MycPu22 DNA is displaced by the competitor DNA for indenoisoquinoline binding and the initial high TAMRA-fluorescence is restored. The competition fluorescence displacement assay allows for a straightforward assessment of selective binding towards MycG4 vs. the competitors, i.e. MycG4s (parallel), K-Ras G4 (parallel), telomeric G4 (hybrid), and dsDNA. One and five compound equivalents were used to assess the selectivity of the strongest binding site and other binding sites of each indenoisoquinoline. A quantitative comparison of the competitor affinities (Ki values) of five indenoisoquinolines are summarized in Table S2. As shown in Figure 7 and S7, all five MycG4-interactive indenoisoquinolines showed marked binding selectivity for parallel G4s (MycG4s and K-Ras G4) over dsDNA (Figure 7), and this selectivity became more pronounced at higher compound ratio (Figure S7). Significantly, four 7-azaindenoisoquinolines, 5, 6, 9, and 12, showed remarkable selectivity for DNA G4s over dsDNA (Table S2). However, indenoisoquinoline 13, which has only N6-substitution but no A- and D-ring substituents, showed much less selectivity against dsDNA. This result suggested that substituents on the A- and D-rings are important for selective binding of G4s vs dsDNA. As shown in the modeling study, the substituents on the A- and D-rings of indenoisoquinolines likely contribute to binding MycG4 by more optimal stacking interactions with the external G-tetrads. On the other hand, the increased size of the indenoisoquinoline ring system may hinder intercalation in dsDNA due to possible steric collision with the DNA backbone. Interestingly, the 3-fluoro-substituted 7-azaindenoisoquinolines 5 and 6 showed marked selectivity for parallel G4s over hybrid G4, whereas the 3-nitro-substituted 7-azaindenoisoquinolines 9 and 12 showed much less selectivity, suggesting that the 3-nitro-group may contribute to a less-specific interaction. The less-specific interaction of 7-azaindenoisoquinolines 9 and 12 was also supported by the NMR titration data showing less well-defined MycG4 complexes formed with 9 and 12 (Figure 5). Albeit with low selectivity against dsDNA, 6-substituted indenoisoquinoline 13 showed selectivity for parallel G4s over hybrid G4. 6-Substituted indenoisoquinolines were previously reported to bind to the c-Kit promoter G4s which were also primarily parallel.17
Figure 7.
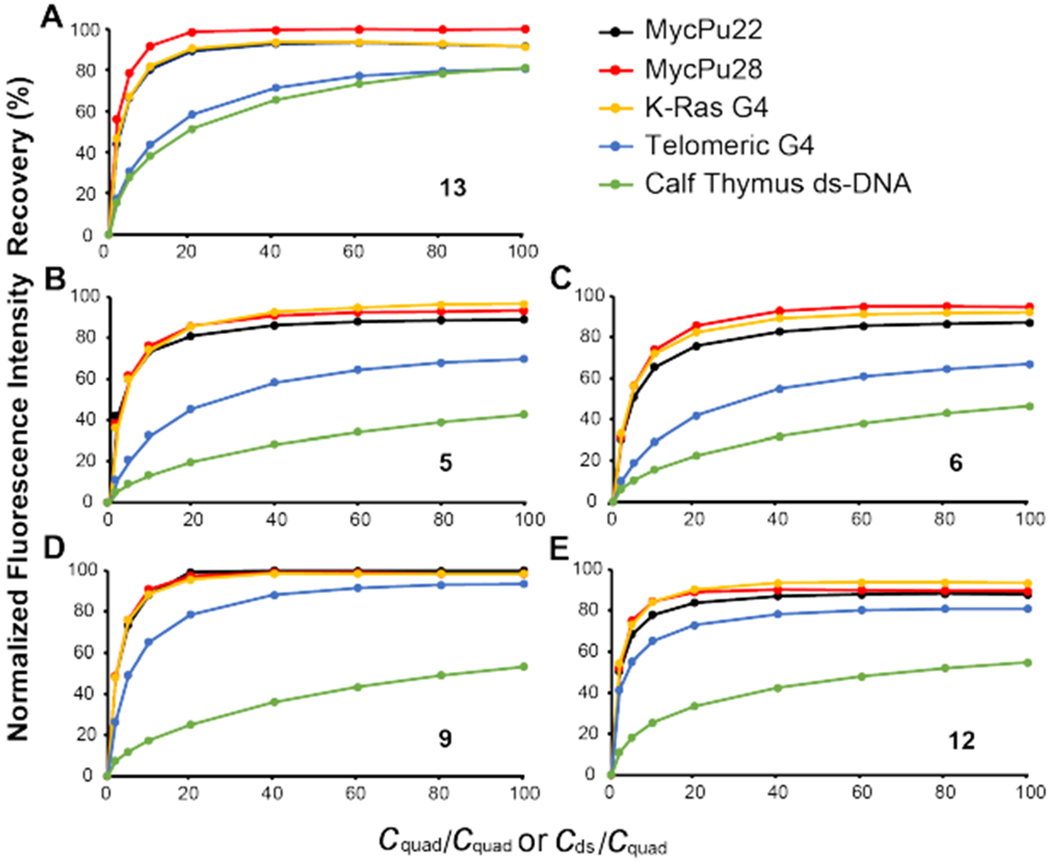
Binding selectivities of MycG4-interactive indenoisoquinolines. Competition fluorescence displacement experiments with increasing concentrations of unlabeled G4s and dsDNA added to 3′-TAMRA-labeled MycPu22 (20 nM) mixed with 1 equivalent of compound 13 (A), 5 (B), 6 (C), 9 (D), and 12 (E). The normalized TAMRA fluorescence intensities at 580 nm were plotted as a function of molar ratio of added G4 DNA (in 3 G-tetrads) or calf thymus dsDNA (in 11 bp) to labeled MycPu22 DNA. The fluorescence intensity of free 3′-TAMRA labeled MycPu22 was defined as 100%, and 1:1 mixture of 3′-TAMRA labeled MycPu22 and indenoisoquinoline was defined as 0%. Conditions: 20 °C, pH 7, 100 mM K+.
Structure-activity relationship of MycG4 binding by indenoisoquinolines.
To understand the factors that govern indenoisoquinoline recognition for MycG4, indenoisoquinoline analogues were analyzed for their MycG4 interactions and MYC inhibitory activity. Clear trends could be established to generate structure-activity relationships for MycG4 binding (Figure 4). It was shown that N6-substituents play a critical role in MycG4 binding and stabilization (Figures 4A–B). For example, indenoisoquinoline 47 with an N6-dimethylaminopropyl moiety, showed medium MycG4 stabilizing activity, whereas indenoisoquinolines 52 and 53, which lack the aminopropyl side chain structure, were found to be poor MycG4 binders and stabilizers. These results suggested that an alkyl amine-containing side chain at N6 of ring B is important for MycG4 binding (Figure 4A), likely due to the favorable electrostatic interactions between the positively charged N-containing side chain and the negatively charged phosphate backbone in the groove of MycG4 at physiological pH 7.4. However, this favorable interaction (compound 13) appeared to be weakened by a more bulky N-containing ring-system (compound 16), and abolished by an aromatic N-containing ring-system (compound 17, reduced positive charge for N) (Figure 4B), suggesting that the bulky nitrogen-containing group may sterically hinder the binding.
Significantly, the newer generation 9-methoxy-7-azaindenoisoquinolines, which were developed with improved water solubility and increased charge-transfer properties,62–63 appeared to bind MycG4 well and show potent MYC-inhibitory activity (Figure 4C). 7-Azaindenoisoquinolines with small substituents, such as 3-fluoro-, 3-nitro-, and 3-chloro, on the A-ring were found to be strong MycG4 binders and stabilizers and showed potent MYC-inhibitory activity.
CONCLUSION
This study demonstrates that anticancer indenoisoquinolines and 7-azaindenoisoquinolines strongly bind and stabilize MycG4 and lower MYC levels in cancer cells as revealed by various biophysical, biochemical, computer modeling, and cell-based experiments. Significantly, a large number of active indenoisoquinolines and 7-azaindenoisoquinolines caused strong MYC downregulation. Indenoisoquinoline analogs are clinically useful anticancer drugs and present a promising scaffold for MycG4-targeting anticancer drug development (Figure 8A). The insights into structure-activity-relationships of MycG4 recognition by indenoisoquinolines were also elucidated. In addition, some active indenoisoquinolines and 7-azaindenoisoquinolines were shown to cause both MYC downregulation and topoisomerase I inhibition. The analysis of indenoisoquinoline analogues for their MYC inhibitory activity, topoisomerase I inhibitory activity, and anticancer activity led to the discovery of a synergistic effect of MYC inhibition and topoisomerase I inhibition on anticancer activity (Figures 8B and S10). Notably, topoisomerase I specifically relaxes transcription-induced negative supercoiling,4 which is the key to the formation of the MYC promoter G4 (Figure 8A), thus dual targeting of MycG4 and topoisomerase I could be an effective mechanism of action for cancer intervention. Collectively, the results uncover a novel mechanism of action of the clinically useful indenoisoquinoline scaffold as a new family of drugs targeting MycG4 for MYC downregulation. Furthermore, the study suggests that dual targeting of MYC and topoisomerase I may serve as a novel strategy for anticancer drug development.
Figure 8.
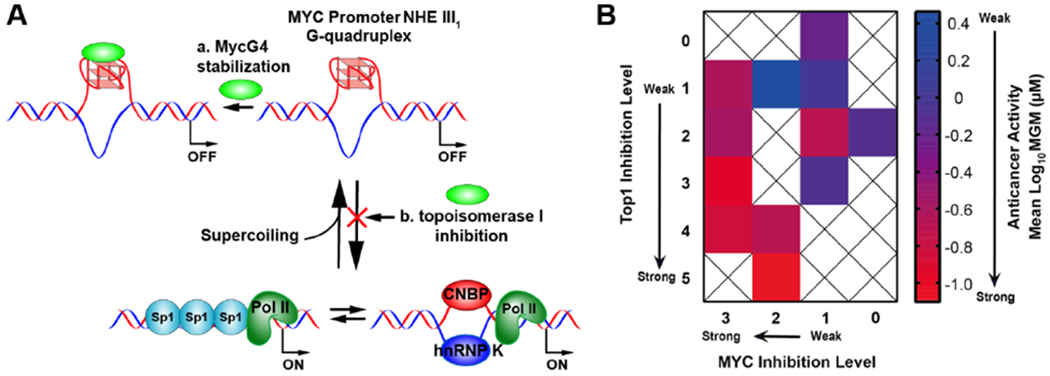
(A) A schematic model showing the potential mechanisms of MYC suppression by indenoisoquinolines by (a) stabilization of MycG4 in the MYC promoter to inhibit transcription, and (b) inhibition of topoisomerase I to maintain negative supercoiling for G4 formation. (B) A heat map showing the synergistic effect of MYC inhibition and topoisomerase I inhibition on the anticancer activities of 29 indenoisoquinolines. The 29 indenoisoquinolines are grouped by their MYC inhibition levels and topoisomerase I inhibition levels. The anticancer activity for each group is determined by the mean(log10MGM) value of the grouped compounds (Table S4), which is displayed as color gradient in the heat map. The MGM values are the approximate average of GI50 values across the entire panel of NCI-60 cancer cell lines for each compound (Table S3). The synergistic effect of MYC inhibition and topoisomerase I inhibition is reflected by the increased anticancer activities (redder color) towards the bottom left comer with strong MYC and topoisomerase I inhibitory activities.
MATERIALS AND METHODS
Sample Preparation.
Unlabeled DNA sequences used for NMR and competition fluorescence displacement assays were synthesized and purified using commercially available reagents as previously described.48, 64 The sequences are listed in Table S1. 3′-6-Carboxytetramethylrhodamine (3′-TAMRA)-labeled MycPu22 and 3′-TAMRA, 5′-6-carboxyfluorescein (5′-FAM) dual-labeled MycPu22 DNA sequences were obtained from Sigma-Aldrich. 3′-FAM, 5′-BlackHole Quencher-1 (5′-BHQ1) dual-labeled MycPu28 DNA sequence was synthesized using an Expedite 8909 DNA Synthesizer, with 3′-(6-FAM) CPG (20-2961-xx) and BHQ-1 phosphoramidite (10-5931-xx) obtained from Glen Research Corporation. The synthesized 5′-BHQl-MycPu28-FAM-3′ DNA sequence was purified using MicroPure II columns and dialyzed against water before lyophilization. DNA concentrations were quantified by UV/Vis absorption at 260 nm using their extinction coefficients. Calf thymus DNA was purchased from Sigma-Aldrich. Indenoisoquinoline stock solutions were dissolved in DMSO at 40 mM by quantifying the mass. For all experiments, indenoisoquinoline stock solutions were further diluted with DMSO or desired buffers.
Fluorescence Resonance Energy Transfer (FRET) Experiments.
FRET-quenching experiments.
The stock solution containing 100 μM 3′-FAM (Ex. 490 nm/Em. 520 nm), 5′-BHQ1 (Abs. 480-580 nm) dual-labeled MycPu28 DNA sequence was first diluted to 2 μM using 50 mM Tris·acetate buffer, pH 7.0. The 2 μM probe solution was equilibrated for 1 h at room temperature. Subsequently, the FRET probe (1 μM) was incubated with the indenoisoquinolines (10 μM) or KC1 (100 mM) in 50 mM Tris-acetate buffer at pH 7.0 for another 1 h, using a black 96-well plate (ThermoFisher Scientific) with a total volume of 100 μL in each well. Fluorescence measurements were then recorded by a Synergy Neo2 plater reader (Bio Tek) at 25 °C with 10 nm bandwidth. The excitation and emission wavelengths were set to 490 and 520 nm, respectively. The final fluorescence intensity was plotted as the average relative fluorescence intensity of two individual experiments after correction for background. Relative fluorescence intensity (%) = FCompound/FDMSO × 100%. Relative fluorescence reduction (%) = (1 –FCompound/FDMSO) × 100%.
FRET-melting experiments.
The stock solution containing 100 μM 3′-TAMRA (Ex. 555 nm/Em. 580 nm), 5′-FAM (Ex. 490 nm/Em. 520 nm) dual-labeled MycPu22 DNA sequence was first diluted to 2 μM using 7.5 mM KCl, 2.5 mM phosphate buffer, pH 7.0. The 2 μM probe solution was heated to 95 °C for 1 min then cooled down slowly to room temperature for G4 formation. Subsequently, the FRET probe (150 nM) was incubated with the indenoisoquinolines (1.5 μM) in 7.5 mM KCl, 2.5 mM phosphate buffer at pH 7.0 for 1 h, using a blank 96-well plate (ThermoFisher Scientific) with a total volume of 100 μL for each well. In the presence of 10 mM K+, the labeled MycPu22 is mainly present in a G4 form where the FAM is in close proximity to the TAMRA, which shows a low FAM fluorescence due to the FRET effect. With gradually increasing temperature, the MycPu22 DNA is unfolded from the G4 form to a single-stranded conformation where the FAM is far apart to the TAMRA, which results in a high FAM fluorescence. Melting curves for the determination of Tm were then obtained by recording FAM fluorescence with increasing temperatures from 25 to 95 °C at a rate of 0.9 °C/min using a QuantStudio 6 Flex Real-Time PCR System. The Tm values were determined by the maximum of the first derivative plot of the melting curves. The final Tm values were plotted as the average Tm values of two individual experiments.
Cell Culture.
MCF-7 (Michigan Cancer Foundation-7) cancer cell lines were originally obtained from the Arizona Cancer Center and grown in RMPI 1640 (10-040-CV, Corning) supplemented with 10% fetal bovine serum (35-010-CV, Coming). Cells were incubated at 37 °C with 5% CO2.
Western Blotting.
After collecting cells from 6-well plates, the cell pellets were re-suspended in 150 μL of 1X RIPA buffer supplemented with 1X Protease Inhibitor Cocktail (11836153001, Roche) and 1X NuPAGE LDS Sample Buffer (NP0007, Invitrogen) and then proteins were immediately denatured at 80 °C for 10 min. After sonication, 7 μL of each sample was analyzed using 4-15% Mini-PROTEAN TGX Gels (456-1086, Bio-Rad). The gels were cut into strips that contained the proteins of interest and transferred to nitrocellulose membrane (IB23002, Invitrogen) using an iBlot 2 Dry Transfer Device (Invitrogen). Immunoblotting was carried out according to standard procedures using the ECL detection method. The membrane was hybridized with the following antibodies: monoclonal anti-MYC (1:1000 dilution; rabbit, Cell Signaling Technology), monoclonal anti-GAPDH (1: 2000 dilution; rabbit, Cell Signaling Technology).
NCI-60 Cancer Cell Line Drug Screen.
The antiproliferative activities of the indenoisquinoline compounds were determined in the NCI-60 cancer cell lines of the National Cancer Institute Developmental Therapeutics Program (NCI-DTP) (Table S3, Figures S8 and S9),53–55 Compounds showed sufficient cytotoxicity during the pre-screen were subjected to the five-dose assay to determine the 50% growth inhibition (GI50) values. Cancer cells were incubated with the test compounds at five concentrations ranging from 100 μM to 10 nM for 48 h. After the treated cancer cells had been stained with sulforhodamine B dye, the percentage growth was plotted as a function of the common logarithm of the tested compound concentration. The GI50 values were determined by interpolation between the points located above and below the 50% cell growth. GI50 values above and below the tested range (10−4 to 10−8 M) were taken as the maximum (10−4 M) and minimum (10−8 M) drug concentrations, respectively, used in the screening test. The approximate average of GI50 values across the entire panel of NCI-60 cancer cell lines for each compound was recorded as the MGM value.
Quantitative Reverse Transcription PCR (qRT-PCR).
Total RNA was isolated using TRIzol reagent (Invitrogen). To remove phenol contamination, purified RNA was dissolved in DEPC-treated water and re-precipitated with 75% ethanol. RNA (1 μg) was subjected to cDNA synthesis using the qScript cDNA Synthesis kit (Quanta Biosciences) according to manufacturer’s instructions. Real-time PCR was performed in triplicate reactions. For each reaction, a mix of the following reaction components was prepared to the indicated end-concentration: 3 μl water, 1 μl cDNA synthesis products, 0.25 μM of each primer for MYC or GAPDH and 5 μl of SYBR Green PCR Master Mix. Cycling conditions were 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 15 s and 72 °C for 15 s. Relative gene expression was calculated by using the 2−ΔΔCT, in which the amount of MYC mRNA was normalized to an endogenous reference (GAPDH). Melting curve analysis or agarose gel electrophoresis was carried out to confirm correct PCR products.
Nuclear Magnetic Resonance (NMR) Spectroscopy Experiments.
All NMR experiments were conducted using a Bruker AV-500 spectrometer equipped with a Prodigy cryoprobe at 25 °C. Watergate water suppression technique was used to suppress water signals. Briefly, each DNA sample was prepared to a final concentration of 150 μM oligonucleotide in 75 mM KCl, 25 mM phosphate buffer at pH 7.0, and containing 90/10% H2O/D2O. DNA samples were heated to 95 °C for 5 min then cooled slowly to room temperature for G4 formation. 1H-NMR titrations were performed by adding increasing amounts of the compound (0.5 to 4 equivalents) to the oligonucleotide solution.
Native Gel Electrophoretic Mobility Shift Assay (EMSA).
Native PAGE experiments were performed with a 1.5 mm thick 10 × 7 cm native gel containing 15% acrylamide (acrylamide:bisacrylamide 29:1) in 1X TBE buffer, pH 8.0, supplemented with 12.5 mM KCl. MycG4 DNA samples were the samples from NMR titration experiments in the absence and presence of indenoisoquinolines. Each sample contains 4 μL of 150 μM DNA. DNA bands were visualized using ultraviolet (UV) light absorption at 260 nm.
Circular Dichroism (CD) Spectroscopy Experiments.
Circular dichroism spectra were recorded using a Jasco-1100 spectropolarimeter (Jasco Inc.) equipped with a temperature controller. Samples were prepared in 3.8 mM KCl, 1.2 mM phosphate buffer at a DNA concentration of 15 μM in the absence and presence of the indenoisoquinolines. CD measurements were taken through a quartz cell with a 1 mm path length, 1 nm bandwidth, and 1 s response time for spectra at 25 °C. Spectra were obtained using three averaged scans between 230 and 330 nm. The baseline was corrected by subtracting the buffer spectrum.
Fluorescence-based Binding Assay.
The fluorescence-based binding assay was performed on a Jasco FP-8300 spectrofluorometer equipped with a temperature controller at 20 °C. The stock solution containing 2 μM 3′-TAMRA labelled MycPu22 oligonucleotide was diluted to 0.5 nM using 75 mM KC1, 25 mM phosphate buffer, pH 7.0. To check the binding affinity of each indenoisoquinoline to MycG4 DNA, the compound was gradually added to the DNA solution in a volume of 1.6 mL using a quartz cell with a 10 mm path length. After each addition of the compound, the solution was allowed to equilibrate for at least 2 min. The fluorescence spectrum was recorded at a range from 570 to 600 nm with an excitation wavelength of 555 nm, 10 nm bandwidths, 100 nm/min scan speed, and 1 s response time. The fluorescence intensity at the emission maximum (λmax = 580 nm) was used in all calculations. The apparent binding affinity Kd values were determined by fitting the data to a one site-specific binding model using GraphPad Prism software, with a simplified equation of , where ΔF represents the fluorescence intensity change of the indenoisoquinolines bound to MycPu22 DNA and [L]T represents the total ligand concentration that is the independent variable, varying with each measurement.
Competition Fluorescence Displacement Experiments.
The competition fluorescence displacement experiments were performed on a Jasco FP-8300 Spectrofluorometer equipped with a temperature controller at 20 °C. The stock solution containing 2 μM 3′-TAMRA-labelled MycPu22 oligonucleotide was diluted to 20 nM using 75 mM KCl, 25 mM phosphate buffer at pH 7.0. To check the binding selectivity of each compound to MycG4 DNA, 20 nM or 100 nM of the indenoisoquinoline was added to the DNA solution in a total volume of 1.6 mL in a quartz cell with a 10 mm path length. Subsequently, various unlabeled MycG4s (MycPu22 and MycPu28), K-Ras G4, telomeric G4 DNAs, or calf thymus dsDNA were gradually added to the complex solution. For each addition of the DNA, the sample was equilibrated at least 2 min. The fluorescence spectrum was recorded between 570 and 600 nm with an excitation wavelength of 555 nm, 10 nm bandwidths, 100 nm/min of scan speed, and 1 s of response time. The fluorescence intensities at the emission maximum (λmax = 580 nm) were plotted for figures. The competitor binding affinities (Ki values) were calculated by , using data from 20 nM compound. The C50 value was the concentration of the unlabeled competing DNA that recovers the fluorescence of the labelled DNA by 50%. [L] represents the ligand concentration that is a constant value of 20 nM. Kd,app values were obtained by fluorescence-based binding assay.
Molecular Modeling
The binding sites at the two ends of the MycG4 were defined by using the NMR structure of the 2:1 quindoline:MycG4 complex (PDB ID 2L7V).48 The docking program Glide (Schrodinger Inc.) was used in the standard precision (SP).60–61 During docking, the DNA was fixed while the ligand was flexible. Before running docking, the 3D energy-minimized structure of the ligand was generated using the LigPrep from Maestro (Schrödinger Inc.). The protonation state of the ligand was assigned at pH 7.0 using the program Epik (Schrödinger Inc.).65 The following default settings in the Glide protocol were used for docking: the OPLS3 force field was used to describe the DNA-ligand complex and a distance-dependent dielectric constant ε = 2.0 was used to mimic the solvent effect.66 A maximum of 5000 poses passed through the initial phase of docking, and a maximum of 400 best poses were kept for energy minimization. The maximum number of the minimization steps was set to be 100.
Supplementary Material
ACKNOWLEDGEMENT
This research was supported by the National Institutes of Health R01CA177585 (DY) and P30CA023168 (Purdue Center for Cancer Research). We thank Drs. Clement Lin and Jonathan Dickerhoff for insightful comments and proofreading the manuscript.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: https://doi.org/10.1021/jacs.9b02679 DNA sequences, Ki values of indenoisoquinolines, complete GI50 values and log10MGM data, indenoisoquinoline chemical structures, fluorescence emission spectra of labeled MycPu28 and MycPu22 in the presence or absence of indenoisoquinoline 5, MYC protein expression levels in the absence and presence of indenoisoquinolines in MCF-7 breast cancer cells, EMSA and CD of MycPu22 in the presence and absence of indenoisoquinolines, fluorescence binding affinities of the indenoisoquinolines, competition fluorescence displacement experiments, example dose response curves and Mean Graphs from NCI-60 cancer cell lines drug screen, GI50 bar graphs of selected indenoi soquinolines.
Publisher's Disclaimer: Just Accepted
Publisher's Disclaimer: “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
REFERENCES
- 1.Kohlhagen G; Pauli KD; Cushman M; Nagafuji P; Pommier Y, Protein-linked DNA strand breaks induced by NSC 314622, a novel noncamptothecin topoisomerase I poison. Mol. Pharmacol 1998, 54(1), 50–58. [DOI] [PubMed] [Google Scholar]
- 2.Strumberg D; Pommier Y; Pauli K; Jayaraman M; Nagafuji P; Cushman M, Synthesis of cytotoxic indenoisoquinoline topoisomerase I poisons. Synthesis of cytotoxic indenoisoquinoline topoisomerase I poisons. J. Med. Chem 1999, 42 (3), 446–457. [DOI] [PubMed] [Google Scholar]
- 3.Cushman M; Jayaraman M; Vroman JA; Fukunaga AK; Fox BM; Kohlhagen G; Strumberg D; Pommier Y, Synthesis of new indeno [1, 2-c] isoquinolines: cytotoxic noncamptothecin topoisomerase I inhibitors. J. Med. Chem 2000, 43 (20), 3688–3698. [DOI] [PubMed] [Google Scholar]
- 4.Pommier Y; Sun Y; Shar-yin NFL; Nitiss JL, Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell. Biol 2016, 17 (11), 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pommier Y, DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem. Rev 2009, 109 (7), 2894–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang P; Elsayed MSA; Plescia CB; Ravji A; Redon CE; Kiselev E; Marchand C; Zeleznik O; Agama K; Pommier Y; Cushman M, Synthesis and biological evaluation of the first triple inhibitors of human topoisomerase I, tyrosyl-DNA phosphodiesterase 1 (Tdp1), and tyrosyl-DNA phosphodiesterase 2 (Tdp2). J. Med. Chem 2017, 60 (8), 3275–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elsayed MSA; Su Y; Wang P; Sethi T; Agama K; Ravji A; Redon CE; Kiselev E; Horzmann KA; Freeman JL; Pommier Y; Cushman M, Design and synthesis of chlorinated and fluorinated 7-azaindenoisoquinolines as potent cytotoxic anticancer agents that inhibit topoisomerase I. J. Med. Chem 2017, 60 (13), 5364–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cinelli MA; Reddy PV; Lv PC; Liang JH; Chen L; Agama K; Pommier Y; van Breemen RB; Cushman M, Identification, synthesis, and biological evaluation of metabolites of the experimental cancer treatment drugs indotecan (LMP400) and indimitecan (LMP776) and investigation of isomerically hydroxylated indenoisoquinoline analogues as topoisomerase I poisons. J. Med. Chem 2012, 55 (24), 10844–10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagarajan M; Morrell A; Ioanoviciu A; Antony S; Kohlhagen G; Agama K; Hollingshead M; Pommier Y; Cushman M, Synthesis and evaluation of indenoisoquinoline topoisomerase I inhibitors substituted with nitrogen heterocycles. J. Med. Chem 2006, 49 (21), 6283–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doroshow JH; Ji JJ; Chen A; Allen D; Zhang Y; Lawrence SM; Pfister TD; Wang L; Redon CE; Bonner W; Speranza G; Weil MK; Eiseman J; Holleran JL; Kinders RJ; Beumer JH; Parchment RE; Pommier Y; Tomaszewski JE; Kummar S, Proof of mechanism (POM) in the first-in-human trial of two novel indenoisoquinoline, non-camptothecin topoisomerase I (TOPI) inhibitors. J. Clin. Oncol 2012, 30 (15_suppl), 3031–3031.22802319 [Google Scholar]
- 11.Kummar S; Chen A; Gutierrez M; Pfister TD; Wang L; Redon C; Bonner WM; Yutzy W; Zhang Y; Kinders RJ, Clinical and pharmacologic evaluation of two dosing schedules of indotecan (LMP400), a novel indenoisoquinoline, in patients with advanced solid tumors. Cancer Chemother. Pharmacol 2016, 78 (1), 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coyne GHOS; Kummar S; Meehan RS; Streicher H; Takebe N; Sharon E; Conley BA; Harris L; Collins JM; Moore N; Juwara L; Rubinstein L; Quinn MF; Pommier Y; Cushman M; Doroshow JH; Chen AP, Phase I study of indenoisoquinolines LMP776 in adults with relapsed solid tumors and lymphomas. J. Clin. Oncol 2017, 35 (15_suppl), 2558–2558.28574779 [Google Scholar]
- 13.A phase I study of indenoisoquinolines LMP400 and LMP776 in adults with relapsed solid tumors and lymphomas. https://clinicaltrials.gov/ct2/show/NCT01051635 (Accessed Apr 20, 2019).
- 14.Indenoisoquinoline LMP744 in adults with relapsed solid tumors and lymphomas. https://clinicaltrials.gov/ct2/show/NCT03030417 (accessed Apr 20, 2019).
- 15.Nagarajan M; Morrell A; Fort BC; Meckley MR; Antony S; Kohlhagen G; Pommier Y; Cushman M, Synthesis and anticancer activity of simplified indenoisoquinoline topoisomerase I inhibitors lacking substituents on the aromatic rings. J. Med. Chem 2004, 47 (23), 5651–5661. [DOI] [PubMed] [Google Scholar]
- 16.Pommier Y; Cushman M, The indenoisoquinoline noncamptothecin topoisomerase I inhibitors: update and perspectives. Mol. Cancer. Ther 2009, 8 (5), 1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bejugam M; Gunaratnam M; Muller S; Sanders DA; Sewitz S; Fletcher JA; Neidle S; Balasubramanian S, Targeting the c-Kit promoter G-quadruplexes with 6-substituted indenoisoquinolines. ACS Med. Chem. Lett 2010, 1 (7), 306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slack GW; Gascoyne RD, MYC and aggressive B-cell lymphomas. Adv. Anat. Pathol 2011, 18 (3), 219–228. [DOI] [PubMed] [Google Scholar]
- 19.Nesbit CE; Tersak JM; Prochownik EV, MYC oncogenes and human neoplastic disease. Oncogene 1999, 18 (19), 3004–3016. [DOI] [PubMed] [Google Scholar]
- 20.Dang CV, MYC on the path to cancer. Cell 2012, 149 (1), 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouvard C; Lim SM; Ludka J; Yazdani N; Woods AK; Chatteijee AK; Schultz PG; Zhu S, Small molecule selectively suppresses MYC transcription in cancer cells. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (13), 3497–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adhikary S; Eilers M, Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell. Biol 2005, 6 (8), 635–645. [DOI] [PubMed] [Google Scholar]
- 23.Meyer N; Penn LZ, Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8 (12), 976–990. [DOI] [PubMed] [Google Scholar]
- 24.Dang CV, c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol 1999, 79(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin CY; Loven J; Rahl PB; Paranal RM; Burge CB; Bradner JE; Lee TL; Young RA, Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012,151 (1), 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nie Z; Hu G; Wei G; Cui K; Yamane A; Resch W; Wang R; Green DR; Tessarollo L; Casellas R; Zhao K; Levens D, c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151 (1), 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jain M; Arvanitis C; Chu K; Dewey W; Leonhardt E; Trinh M; Sundberg CD; Bishop JM; Felsher DW, Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002, 297 (5578), 102–104. [DOI] [PubMed] [Google Scholar]
- 28.Weinstein IB; Joe AK, Mechanisms of disease: Oncogene addiction—a rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol 2006, 3 (8), 448–457. [DOI] [PubMed] [Google Scholar]
- 29.Dang CV; Reddy EP; Shokat KM; Soucek L, Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 77(8), 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Felsenstein KM; Saunders LB; Simmons JK; Leon E; Calabrese DR; Zhang S; Michalowski A; Gareiss P; Mock BA; Schneekloth JS Jr., Small molecule microarrays enable the identification of a selective, quadruplex-binding inhibitor of MYC expression. ACS Chem. Biol 2016, 77 (1), 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitfield JR; Beaulieu ME; Soucek L, Strategies to inhibit Myc and their clinical applicability. Front. Cell Dev. Biol 2017, 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siddiqui-Jain A; Grand CL; Bearss DJ; Hurley LH, Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U. S. A 2002, 99 (18), 11593–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brooks TA; Hurley LH, The role of supercoiling in transcriptional control of MYC and its importance in molecular therapeutics. Nat. Rev. Cancer 2009, 9 (12), 849–861. [DOI] [PubMed] [Google Scholar]
- 34.Brown RV; Danford FL; Gokhale V; Hurley LH; Brooks TA, Demonstration that drug-targeted downregulation of MYC in non-hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J. Biol. Chem 2011, 286 (47), 41018–41027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kouzine F; Sanford S; Elisha-Feil Z; Levens D, The functional response of upstream DNA to dynamic supercoiling in vivo. Nat. Struct. Mol. Biol 2008, 15 (2), 146–154. [DOI] [PubMed] [Google Scholar]
- 36.Kang HJ; Park HJ, Novel molecular mechanism for Actinomycin D activity as an oncogenic promoter G-quadruplex binder. Biochemistry 2009, 48 (31), 7392–7398. [DOI] [PubMed] [Google Scholar]
- 37.Yang D; Okamoto K, Structural insights into G-quadruplexes: towards new anticancer drugs. Future Med. Chem 2010, 2 (4), 619–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin Y; Hurley LH, Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie 2008, 90 (8), 1149–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y; Yang D, Sequence, stability, and structure of G-quadruplexes and their interactions with drugs. Curr. Protoc. Nucleic Acid Chem 2012, Chapter 17, Unit17 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balasubramanian S; Hurley LH; Neidle S, Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat. Rev. Drug Discov 2011, 10 (4), 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biffi G; Tannahill D; McCafferty J; Balasubramanian S, Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem 2013, 5 (3), 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hansel-Hertsch R; Beraldi D; Lensing SV; Marsico G; Zyner K; Parry A; Di Antonio M; Pike J; Kimura H; Narita M; Tannahill D; Balasubramanian S, G-quadruplex structures mark human regulatory chromatin. Nat. Genet 2016, 48 (10), 1267–1275. [DOI] [PubMed] [Google Scholar]
- 43.Ambrus A; Chen D; Dai J; Jones RA; Yang D, Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Biochemistry 2005, 44 (6), 2048–2058. [DOI] [PubMed] [Google Scholar]
- 44.Mathad RI; Hatzakis E; Dai J; Yang D, c-MYC promoter G-quadruplex formed at the 5’-end of NHE III1 element: insights into biological relevance and parallel-stranded G-quadruplex stability. Nucleic Acids Res 2011, 39 (20), 9023–9033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seenisamy J; Rezler EM; Powell TJ; Tye D; Gokhale V; Joshi CS; Siddiqui-Jain A; Hurley LH, The dynamic character of the G-quadruplex element in the c-MYC promoter and modification by TMPyP4. J. Am. Chem. Soc 2004, 126 (28), 8702–8709. [DOI] [PubMed] [Google Scholar]
- 46.Ou TM; Lu YJ; Zhang C; Huang ZS; Wang XD; Tan JH; Chen Y; Ma DL; Wong KY; Tang JC; Chan AS; Gu LQ, Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J. Med. Chem 2007, 50 (7), 1465–1474. [DOI] [PubMed] [Google Scholar]
- 47.Ou TM; Lin I; Lu YJ; Hou JQ; Tan JH; Chen SH; Li Z; Li YP; Li D; Gu LQ, Inhibition of cell proliferation by quindoline derivative (SYUIQ-05) through its preferential interaction with c-myc promoter G-quadruplex. J. Med. Chem 2011, 54 (16), 5671–5679. [DOI] [PubMed] [Google Scholar]
- 48.Dai J; Carver M; Hurley LH; Yang D, Solution structure of a 2:1 quindoline-c-MYC G-quadruplex: insights into G-quadruplex-interactive small molecule drug design. J. Am. Chem. Soc 2011, 133 (44), 17673–17680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hatzakis E; Okamoto K; Yang D, Thermodynamic stability and folding kinetics of the major G-quadruplex and its loop isomers formed in the nuclease hypersensitive element in the human c-Myc promoter: effect of loops and flanking segments on the stability of parallel-stranded intramolecular G-quadruplexes. Biochemistry 2010, 49 (43), 9152–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morrell A; Placzek M; Parmley S; Antony S; Dexheimer TS; Pommier Y; Cushman M, Nitrated indenoisoquinolines as topoisomerase I inhibitors: a systematic study and optimization. J. Med. Chem 2007, 50 (18), 4419–4430. [DOI] [PubMed] [Google Scholar]
- 51.Conda-Sheridan M; Reddy PN; Morrell A; Cobb BT; Marchand C; Agama K; Chergui A; Renaud A; Stephen AG; Bindu LK, Synthesis and biological evaluation of indenoisoquinolines that inhibit both tyrosyl-DNA phosphodiesterase I (Tdp1) and topoisomerase I (Top1). J. Med. Chem 2012, 56(1), 182–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beck DE; Agama K; Marchand C; Chergui A; Pommier Y; Cushman M, Synthesis and biological evaluation of new carbohydrate-substituted indenoisoquinoline topoisomerase I inhibitors and improved syntheses of the experimental anticancer agents indotecan (LMP400) and indimitecan (LMP776). J. Med. Chem 2014, 57(4), 1495–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.NCI-60 human tumor cell lines screen, https://dtp.cancer.gov/discovery_development/nci-60/ (accessed Apr 20, 2019).
- 54.Kiselev E; Sooryakumar D; Agama K; Cushman M; Pommier Y, Optimization of the lactam side chain of 7-azaindenoisoquinoline topoisomerase I inhibitors and mechanism of action studies in cancer cells. J. Med. Chem 2014, 57 (4), 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shoemaker RH, The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6 (10), 813. [DOI] [PubMed] [Google Scholar]
- 56.Kotar A; Wang B; Shivalingam A; Gonzalez-Garcia J; Vilar R; Plavec J, NMR structure of a triangulenium-based long-lived fluorescence probe bound to a G-quadruplex. Angew. Chem. Int. Ed 2016, 55 (40), 12508–12511. [DOI] [PubMed] [Google Scholar]
- 57.Del Villar-Guerra R; Trent JO; Chaires JB, G-quadruplex secondary structure obtained from circular dichroism spectroscopy. Angew. Chem. Int. Ed 2018, 130 (24), 7289–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brown RV; Wang T; Chappeta VR; Wu G; Onel B; Chawla R; Quijada H; Camp SM; Chiang ET; Lassiter QR, The consequences of overlapping G-Quadruplexes and i-Motifs in the platelet-derived growth factor receptor β core promoter nuclease hypersensitive element can explain the unexpected effects of mutations and provide opportunities for selective targeting of both structures by small molecules to downregulate gene expression. J. Am. Chem. Soc 2017, 139 (22), 7456–7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le DD; Di Antonio M; Chan L; Balasubramanian S, G-quadruplex ligands exhibit differential G-tetrad selectivity. Chem. Commun 2015, 51 (38), 8048–8050. [DOI] [PubMed] [Google Scholar]
- 60.Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK, Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47 (7), 1739–1749. [DOI] [PubMed] [Google Scholar]
- 61.Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL, Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem 2004, 47 (7), 1750–1759. [DOI] [PubMed] [Google Scholar]
- 62.Kiselev E; DeGuire S; Morrell A; Agama K; Dexheimer TS; Pommier Y; Cushman M, 7-azaindenoisoquinolines as topoisomerase I inhibitors and potential anticancer agents. J. Med. Chem 2011, 54 (17), 6106–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kiselev E; Agama K; Pommier Y; Cushman M, Azaindenoisoquinolines as Topoisomerase I Inhibitors and Potential Anticancer Agents: A Systematic Study of Structure-Activity Relationships. J. Med. Chem 2012, 55 (4), 1682–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Onel B; Carver M; Wu G; Timonina D; Kalarn S; Larriva M; Yang D, A new G-quadruplex with hairpin loop immediately upstream of the human BCL2 PI promoter modulates transcription. J. Am. Chem. Soc 2016, 138 (8), 2563–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shelley JC; Cholleti A; Frye LL; Greenwood JR; Timlin MR; Uchimaya M, Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J. Computer Aid. Mol. Des 2007, 21 (12), 681–691. [DOI] [PubMed] [Google Scholar]
- 66.Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL, OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput 2015, 72 (1), 281–296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.