

Abstract
A fundamental goal of developmental and stem cell biology is to map the developmental history (ontogeny) of differentiated cell types. Recent advances in high-throughput single-cell sequencing technologies have enabled the construction of comprehensive transcriptional atlases of adult tissues and of developing embryos from measurements of up to millions of individual cells. Parallel advances in sequencing-based lineage-tracing methods now facilitate the mapping of clonal relationships onto these landscapes and enable detailed comparisons between molecular and mitotic histories. Here we review recent progress and challenges, as well as the opportunities that emerge when these two complementary representations of cellular history are synthesized into integrated models of cell differentiation.
Cellular differentiation in composition, organization and function represents one of the major innovations of multicellular life. Determining the molecular mechanisms that govern how cells differentiate in their state is thus a long-standing focus in stem cell and developmental biology1. A comprehensive record of changes in cell states as tissues and organs develop can give insights into the molecular mechanisms and order of events by which cells choose their terminal identities during embryogenesis or regeneration. It can provide clues as to how to manipulate cell fates in vivo, to predict the origins of developmental pathologies and cancer, and to re-create cell differentiation processes in vitro.
Recent advances in single-cell transcriptomics provide a powerful approach to mapping differentiation dynamics by densely sampling cells at different stages. These sampled cells together can be used to construct a continuum of cell states, or a ‘landscape’, a term historically inspired by Waddington’s metaphorical epigenetic landscape2. In this Review, we refer to such depictions as state manifolds, to reflect both their underlying high-dimensional nature and their routine representation as low-dimensional Euclidean surfaces or graphs. State manifolds can provide high-resolution descriptions of cell trajectories as they transition between states during cell differentiation.
While they are powerful, state manifolds and state trajectories offer population-level views of differentiation, without directly revealing the long-term dynamic relationships between individual cells or between cells and their progeny. The gold standard for linking cell states across periods of time is instead through prospective lineage tracing: the practice of labelling an individual cell at an early time point in order to track the state of its clonal progeny at a later time point. Traditionally reliant on microscopy, lineage-tracing approaches have recently evolved to allow the tracking of cell clones via sequencing of inherited DNA sequences, or ‘barcodes’. The migration to sequencing platforms has brought several advantages to lineage-tracing efforts: massive throughput, multiplexing and compatibility with other sequencing-based measurements (for example, RNA sequencing (RNA-seq)).
Recently, we and others have developed approaches to carry out single-cell omic-scale profiling while simultaneously reporting lineage information. These methods offer an opportunity to integrate complementary information about both cell lineage and cell state into synthesized views of differentiation dynamics. In this Review, we survey the currently available strategies for single-cell state manifold reconstruction and lineage barcoding, as well as omics methods for combining lineage and state measurements in the same cells. Both the range of single-cell trajectory construction methods and their assumptions have been reviewed extensively elsewhere3,4, as have foundational molecular strategies for lineage barcoding5,6. Here we aim to draw general lessons from reoccurring conflicts that have emerged between state and fate analyses, and we discuss biological results obtained from first applications of combining the two methods. As this is an emerging field, we also discuss current limitations and potential technical pitfalls in their application. Finally, we speculate on the emerging concepts that might arise.
Inferring cell histories from state manifolds
In measuring the instantaneous state of a cell, one might imagine collecting information on the copy number of every molecular species within a cell, their interactions and spatial organization, the position of the cell in its parent tissue, and its physical and regulatory interactions with other cells. Such a level of information is, of course, impractical. Working definitions of cell state capture only a subset of these attributes and vary dramatically between studies. In the following sections, we describe how cell state designations have evolved from relatively simple observations to quantitative high-dimensional and high-throughput omics measurements. We describe the introduction of cell state manifolds as a relatively recent analytic strategy with important advantages and limitations when inferring cell state relationships.
Defining cell states
A century ago, cells could only be reproducibly defined by simple characteristics: spatial position, morphology, histochemical staining, or basic biochemical or biophysical properties, such as cell density or dye uptake. Accordingly, much of the classical nomenclature associated with cell states (for example, basophilic) reflects these assays. With the advent of molecular biology, cells could be identified more quantitatively by the expression of selected marker genes, through immunocytometry, RNA analysis or the expression of transgenes. The nomenclature of cell state expanded accordingly into marker-based phenotypes (for example, CD34+). The types of measurable determinants of cell identity continue to expand, including epigenetic state (for example, DNA accessibility and conformation, protein–DNA binding, DNA methylation or histone modifications), post-translational protein modifications, protein localization and the metabolic profile of cells.
At present, the most mature technology for genome-scale mapping of cell states is through measurements of the whole transcriptome (single-cell RNA-seq (scRNA-seq)), which can now be carried out rapidly and at low cost, in nanolitre-scale droplets7,8, in microfluidic wells9, or using combinatorial split-pool approaches10. Transcriptomes contain information about multiple aspects of cell identity (for example, cell cycle phase, metabolic state, cell-specific and tissue-specific molecular signatures, and spatially restricted marker genes). These diverse features may or may not be interrelated, but they reinforce a modern view of cell states as multidimensional vectors11,12. Beyond scRNA-seq, recent breakthroughs in single-cell methods capture chromatin accessibility13,14, methylomes15, proteomes16 and metabolic signatures17, as well as multimodal measurements from the same single cells (for example, mRNA and protein18–20 or mRNA and DNA21,22). These measurements incorporate even further dimensions into routine measurements of cell state. Additionally, some highly multiplexed profiling of cell states is now possible in situ, thus complementing cell-intrinsic state information with detailed information on a cell’s local environment and position in tissues23–27. Overall, these innovations set up the coming decade to be an exciting time for stem cell and developmental biology, as well as for tissue physiology in general. These new methods are clarifying the changes that occur in cells during development and, ultimately, the mechanisms governing cell behaviour.
Mapping state manifolds
Large single-cell datasets are now being routinely collected to catalogue the distribution and differentiation of cell states in both embryonic and adult tissues, as well as in disease. Recent examples encompassing entire organ systems include the haematopoietic system28,29, lung30,31, kidney32,33, heart34, gut endoderm35, somitic mesoderm36, nervous system37 and neural crest38. Additionally, whole-organism datasets have been generated for Caenorhabditis elegans39,40, Nematostella vectensis41, Hydra42, annelids43 and planarians44–46. Furthermore, time series data for whole embryos have been mapped for zebrafish47,48, Xenopus laevis49, mouse50,51, Drosophila melanogaster52 and ascidians53. These datasets have revealed novel cell states, and they associate all states with detailed molecular signatures that extend well beyond the previous classifications based on marker genes alone. They also have revealed cells in developmental transitions involving thousands of genes, which change expression at progressive times and between tissues.
Analyses of these and other single-cell data involve several stereotypical steps to predict differentiation dynamics (FIG. 1). First, single-cell datasets noisily sample cells in different states (FIG. 1A). The challenge of data analysis is then to infer the continuum manifold of states from these measurements (FIG. 1B). These manifolds must be constructed, visualized and then used either to predict dynamics directly from cell states or else to represent the measured dynamic information (FIG. 1C). In this section we briefly introduce these steps.
Fig. 1|. inferring cell histories from state manifoids.
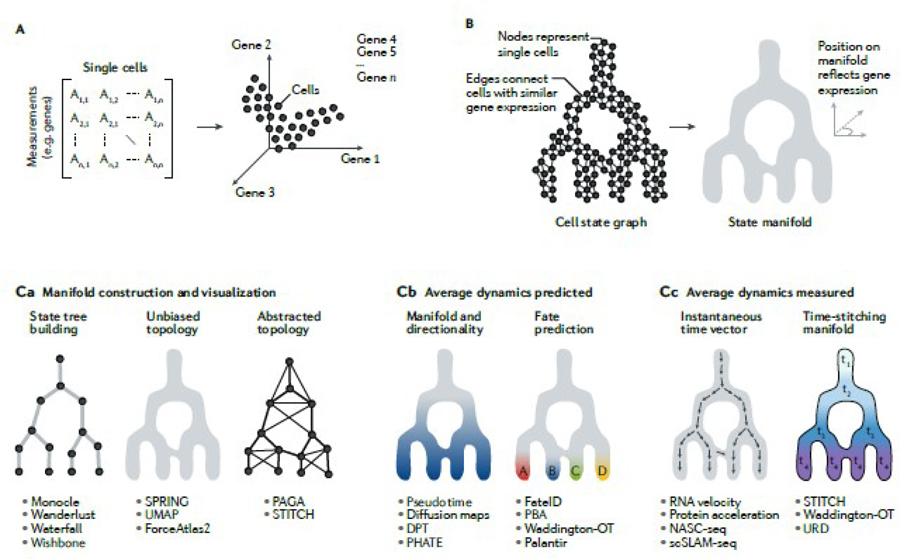
A| Modern omics-based single-cell datasets, conceptuailzed as a measurement xcell count matrix or, alternatively, as cells plotted in a high-dimensional Euciidean space. B | Single-cell graphs, which link cells according to similarity (for example, Euclidean distance) in gene expression space, can be visualized to reveal underlying state manifolds that reflect gene expression dynamics. C| Graph-based tools for constructing and visualizing statement folds (part Ca), computational algorithms for predicting dynamics directly from a state manifold (part Cb) and tools for incorporating independently measured state dynamics into a manifold (part Cc) DPT, diffusion pseudotime: NASC seq, newt ranscriptome aikylation-dependent single-cell RNA sequencing: PAGA, partition-based graph abstraction: PBA. population balance analysis; PHATE, potential of heat diffusion for affinity-based trajectory embedding: scSLAM-seq, single-cell thiol-(SH)}-linked alkylation of RNA for metabolic labelling sequencing; SPRING. a force-layout embedding of single-cell data; STITCH, a method for combining time series of single-cell data; UMAP, uniform manifold state manifolds approximation and projection; URD. a simulated diffusion based computational approach named after the Norse mythological figure: Waddington-OT, Waddington optimal transport.
To infer continuum state manifolds, most methods applied to single-cell data to date have been graph-based: they begin by representing individual cells as nodes, which are then connected by edges that reflect pairwise gene expression similarities (FIG. 1B). Graph-based analyses are useful because they convert a set of isolated measurements (single-cell transcriptomes) into a connected structure (the graph), which can then be analysed using a rich set of pre-existing mathematical methods.
To then visualize state manifolds, several algorithms are used that attempt to preserve the structure of the original cell graph when it is plotted in just two or three dimensions (such as uniform manifold approximation and projection (UMAP)54, SPRING55 and ForceAtlas2 (ref.56)). Two-dimensional representations are popular and do capture meaningful biological trends. However, they can be misleading, as they distort high-dimensional structures upon ‘flattening’ them, and in some cases algorithms force tree-like visual layouts that may further distort the original structure48,57,58. Any 2D and 3D visualizations should serve only as aids for representing the results of more powerful forms of data analysis.
Independently of visualization, a multitude of algorithms propose to predict cell state dynamics and/or differentiation hierarchies directly from a manifold (FIG. 1C). These tools for dynamic inference have been reviewed extensively elsewhere3 and include methods for extracting from the manifold its bare-bones structure, or topology59 ; organizing cells into trajectories57,58,60–63 along an axis (often called pseudotime); and predicting the future fate of cells on the basis of their state28,64–68. To improve these efforts at dynamic inference, some recent studies have succeeded in inferring the instantaneous dynamics of states on the basis of measurements of nascent mRNA abundance, the ratio of spliced to unspliced mRNA (for example, RNA velocity), protein translation or mRNA turnover by metabolite labelling69–73. Temporal information can also be integrated into state manifolds when cells are sampled at time intervals47,48,67 (FIG. 1C). In total, the result of these methods is to order cells along a continuum74,75, which in turn allows for studying changes in the average, variance and correlation of gene expression across the graph, and for inferring tree-like structures from graphs57,58,60,76 that organize cells or cell clusters77,78 into a putative hierarchy.
Limitations of state manifolds for dynamic inference
The representation of cell states as continuous manifolds offers a compelling approach to reconstructing dynamic processes. However, state manifolds average over many individual cells and so lose information on individual dynamics. The missing information includes cell division or death rates, the reversibility of states, and persistent differences between clones, all of which can quantitatively or qualitatively alter the dynamics predicted from snapshot measurements64. The dynamics predicted from cell state snapshots should thus be considered hypotheses. In this respect, the tree-like hierarchies of cell states sharply contrast with those obtained by bona fide lineage analysis (FIG. 2a), in which tree edges link cells with an empirical developmental relationship. On a state manifold, branch-points may be hypothetical: cell division may or may not occur at a branch-point, and sister cells from each division may both progress along one branch of a manifold, rather than exploring all branches. By contrast, in lineage trees, each branch-point strictly corresponds to a division event. State trajectories need not even be strictly tree-like, whereas lineage hierarchies are always strictly branching trees. Therefore, although the population-level structure could trace the dynamic sequence of molecular states experienced by single cells (FIG. 2a,b), several specific reasons could obscure or mislead researchers’ understanding of the underlying dynamics and/or fate relationships (BOX 1; FIG. 2b–h).
Fig. 2 |. Limitations of cell state manifolds.
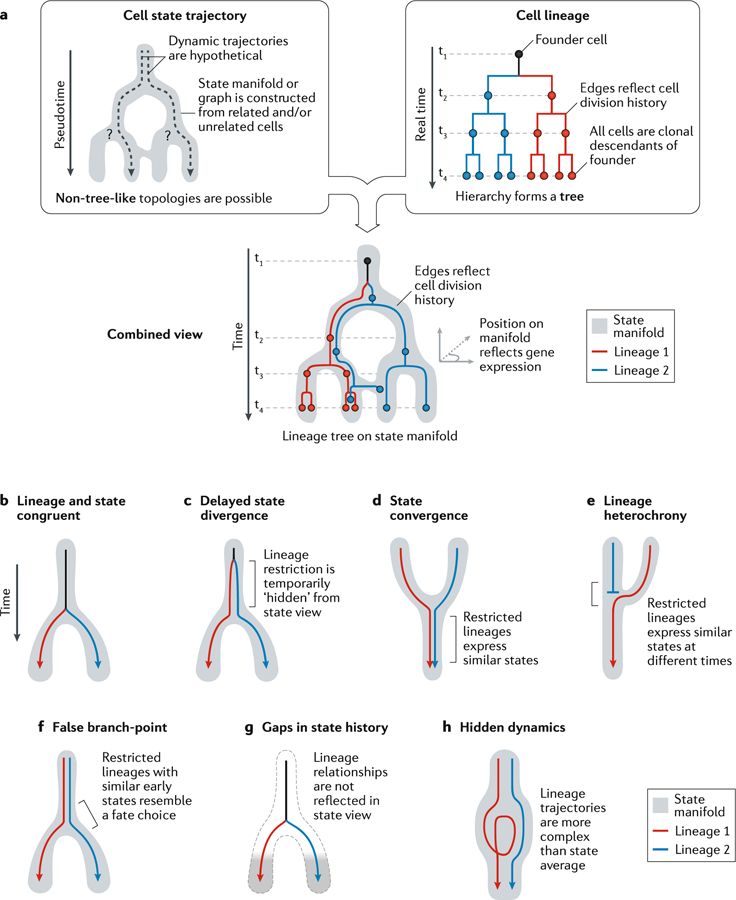
a | Clarification of depictions of cell state manifolds versus cell lineage trees. Trajectory relationships are indirectly inferred from gene expression similarities, whereas lineage relationships reflect measured mitotic histories. Below the boxes, a combined representation highlights a clonal hierarchy of related cells, directly revealing its trajectory along astate manifold. b-h| Hypothetical scenarios of restricted lineage trajectories unfolding ona state manifold. The behaviours of distinct clonal units are presented in simplified form by coloured arrows. Lineage and state congruent (part b): initially all clones share the same fate potential (black); restriction of the clones into distinct trajectories (blue/red) occurs only where the manifold bifurcates. Delayed state divergence (part c): cells become committed to distinct trajectories (blue/red) but continue to occupy similar states for some time. This causes the early state to appear seemingly multipotent despite the cells within each clone being fate-restricted. State convergence (part d): cells with distinct molecular histories converge into similar states, such that the molecular origin of later cells can no longer be inferred. Lineage heterochrony (part e): cells with different origins occupy a sequence of states that implies a false developmental trajectory (blue to red). False branch-point (part f): an extreme case of the situation in part c, in which an apparent branch-point does not represent a decision made by any cell. Instead, it appears artificially when fate-restricted clones overlap in their early state. Gaps in state manifold (part g): disconnected cell states appear when the states of transitional and early progenitors are not represented in the dataset. This occurs when transitional states are very rare or when sampling a developing tissue at a late stage. Hidden dynamics (part h): the extent of stochastic or structured fluctuations in clonal dynamics is not visible from snapshots of cell states.
Box 1 |. How do cells traverse single-cell landscapes?
Single-cell data can be organized into a continuum ‘landscape’ (or ‘manifold’) of cell states, representing cells in progressive states of differentiation. However, these landscapes do not directly clarify how cells or clonal lineages explore these states, or how they choose their trajectory at branch-points. Cells in similar states could show the same dynamic progression and remain uncommitted until they reach a branch-point (FIG. 2b). Dynamic behaviours on a landscape can also be unpredictable (FIG. 2c–h) and can deviate from the global averages inferred from many single-cell measurements. these complications may arise from hidden variables, gaps in manifolds and stochastic dynamics, as detailed here.
Hidden variables
Although single-cell technologies aspire to measure cell state comprehensively, they may still miss important cellular properties that are informative as to fate. such hidden variables may be regulatory molecules that are altogether missing from the state measurement (for example, epigenetic, spatial or post-translational state features that are not captured by single-cell rNa sequencing). they could also be obscured by measurement noise or by ad hoc operational decisions in data processing, such as choices of normalization or dimensionality reduction strategies and of which genes to include in manifold construction. Because cells often participate in multiple dynamic processes, different data-processing choices can emphasize certain biological processes (for example, cell cycle, cell migration or stress) over others and can allow constructing manifolds with qualitatively different structures from the same data. transcriptional signatures of the cell cycle, for example, can overshadow other state differences between unrelated cell types.
Failure to resolve hidden variables can lead to the appearance of ‘delayed state divergence’ on a state manifold (FIG. 2c), obscuring the true point at which fate specification occurs. independent clonal trajectories can also appear to ‘converge’ temporarily on identical or nearly identical states during the differentiation process (FIG. 2d). in these cases, the distinct ontogeny of the cells cannot be deduced from state information alone. Clones or tissue domains in different stages of differentiation may also form a continuum of states that implies a false trajectory (FIG. 2e). additionally, state manifolds may imply ‘false multipotency’ and/or ‘false branch-points’ by superimposing cells with different fate potentials (FIG. 2f). some of these problems can be identified by visualizing the state manifold, but this is only possible if the visualization methods used do not force cells to occupy a tree-like hierarchy. Lineage tracing is essential to identifying and resolving lineage restrictions downstream from a point of state convergence (FIG. 2d,e); not even short-term dynamic information (for example, rNa velocity; see the main text) is informative in these situations.
Gaps
Learning differentiation trajectories works best for systems with a strong flux of cells and coverage of multiple time points. when very few cells differentiate at any moment in time (for example, adult neural stem cells) or transitional time points are missing, analyses become more difficult and are prone to creating artefacts. thus, gaps in the manifold, which may arise from uneven or under-sampling of cell states, can result in apparent discontinuities between clonally related cells (FIG. 2g).
Stochastic dynamics
Even when manifolds faithfully depict the average clonal dynamics, they cannot provide information about distinct dynamic behaviours of cells that appear similar in state, such as stochastic fluctuations about the average or participation in local cycles (FIG. 2h). Collectively resolving the scenarios above would require the ability to track cell state dynamics over both short and long timescales.
Inferring cell histories in lineage tracing
Unlike the state of a cell, the lineage history of a cell can be defined without the operational simplification that comes from reducing the dimensionality of thousands of measurements. By ‘lineage’, we refer to the collective history of cell divisions, as well as the birth, division and death times of a cell’s ancestors and clonal relatives. Lineages can be depicted as detailed trees of mitotic events (FIG. 2a) or, alternatively, as clonal units derived from a common progenitor cell. Lineage measurements, however, do not inherently contain information about the states of the cells they comprise, and as such they are typically combined with other measurements (for example, cell position, morphology or gene expression). In the following sections, we describe classic temporal and clonal analysis paradigms for defining cell lineage, referring the reader elsewhere for more complete reviews5,6. These paradigms have evolved from their roots in imaging-based studies to the recent use of DNA-barcoding-based systems in the post-genomics era.
Lineage-tracing paradigms
Currently there are two major paradigms for defining cell lineages. One major category of approaches, prospective lineage tracing, attempts to establish lineage relationships forwards in time from cells of a defined starting state. Fate mapping, the practice of associating the position of a cell in the early embryo with the ultimate positions and fates of that cell’s descendants, is a form of prospective lineage tracing79. Methods based on CRE or FLIP recombinases, which facilitate permanent genetic labelling of progenitor cells based on the activity of a transgenic promoter, can also be used to learn prospective state relationships80. Prospective lineage tracing requires that some level of state information be known about a starting cell population, and generally the goal is to correlate this state information with future cell states. By contrast, phylogenetic lineage reconstruction methods seek to map the history of lineage relationships with respect to the cell states queried at a single end point in time. With these methods, state and lineage features are generally only measured at the end of the experiment, and lineage relationships are mapped backward in time in order to infer fate decisions that occurred either early or late. An inherent advantage of phylogenetic approaches is that analyses can be performed retrospectively (that is, without the need for experimental labelling) by analysing endogenous, naturally occurring genetic polymorphisms81 — these label-free implementations of phylogenetic lineage analyses are thus also known as retrospective lineage tracing. Such approaches, therefore, can be applied to human patient samples and in other cases in which experimental intervention is not possible. Many additional methods perform similar phylogenetic reconstruction of end states but do so by tracking experimental labels rather than endogenous labels. In practice, when experimental labels are specifically introduced into cells of a particular state, the lineage-tracing experiments combine both prospective and phylogenetic paradigms — for example, by reconstructing lineage phylogenies within a specific tissue.
Clonal versus population tracing
Labelling of cells for prospective lineage tracing can be performed at clonal resolution (such as by delivering complex barcode libraries) or, alternatively, by documenting the collective fate of a population of cells (such as by delivering a common label to the cell population). Population lineage-tracing experiments are generally easier to perform, but they leave open the possibility of internal heterogeneity of the labelled population and/or inclusion of off-target cells. The collective activity of a bulk cell population (for example, unfractionated bone marrow) can easily be misinterpreted as representing the output of a single, multifunctional cell type, even when labelled cells could be restricted in their fate potential82. Such errors can be resolved by increasing the precision of the labelling process to limit any underlying cellular heterogeneity, or by utilizing single-cell lineage methods to track clonal relationships83. Given that even genomics-era state measurements (for example, scRNA-seq) can occasionally fail to fully resolve lineage-restricted groups of cells47,84–86 (FIG. 2c–h), clonal analysis is still the most robust method for establishing the distribution of lineage outputs of a cell population. Once identified, stereotyped clone behaviours can be used to screen for prospective cell state markers that might correlate with and/or predict different lineage outcomes.
Imaging-based methods for lineage tracing
Prospective lineage-tracing experiments date back to the 19th century and initially relied on direct observations via live microscopy to track blastomere divisions in transparent invertebrate embryos, in particular in annelids1 and ascidians87. Ascidian lineage trees were annotated according to the spatial position of each cell, and owing to determinate cleavage patterns and early fate restriction, this relatively simple level of state information was found to be sufficient to predict future cell fates. A similar direct-observation strategy was applied nearly a century later to the nematode C. elegans, again taking advantage of the small size, transparency and determinate embryonic cleavage patterns of this species88.
Embryos of more complex species (for example, vertebrates) often contain many more cells, and cell divisions are generally indeterminate and more difficult to observe directly. Lineage tracing thus expanded to include a wide range of additional approaches, including the injection of tracer dyes, cell transplantation and in vivo genetic recombination methods. The history and applications of these pre-genomic methods have been reviewed extensively elsewhere80. More recent advances in in toto confocal and light-sheet microscopy have reinvigorated modern versions of the direct-observation approach, enabling the tracking of individual cell division patterns in complex vertebrates such as zebrafish and mouse, together with transgenic reporters89,90. One feature common to in toto imaging and nearly all pregenomics methods for live lineage tracing is a reliance on transgenic fluorescent reporters to measure cell state. Thus, these approaches are spectrally limited to relatively few measurements of cell state. Partially countering this limitation, the spatial position of cells and their morphology provide information that may be correlated to molecular state91. Furthermore, recent spatial transcriptomics methods overcome the spectral limit by allowing genome-scale measurements in fixed samples in situ. Using such methods subsequent to live imaging or in combination with lineage tracing allows for combining state information with lineage and position information in one experiment92. However, such experiments remain extremely challenging, and highly multiplexed spatial transcriptomics methods are still generally restricted to the analysis of tissue sections, which may fail to capture all cells in each clone.
Lineage tracing by barcode-sequencing
Recently, high-throughput sequencing has opened up a new generation of lineage-tracing approaches. These new methods use DNA sequence barcodes to encode clonal information (FIG. 3). Although the number of distinct clones that can be simultaneously queried using fluorescent reporters is intrinsically limited, DNA sequence complexity scales exponentially with the length and multiplicity of the engineered barcodes, which is theoretically sufficient to allow a record of every single division event in an organism. The recorded information is read out retrospectively using high-throughput sequencing and can be readily combined with other sequencing-based omics measurements.
Fig. 3 |. Methods and logic for lineage barcoding experiments.
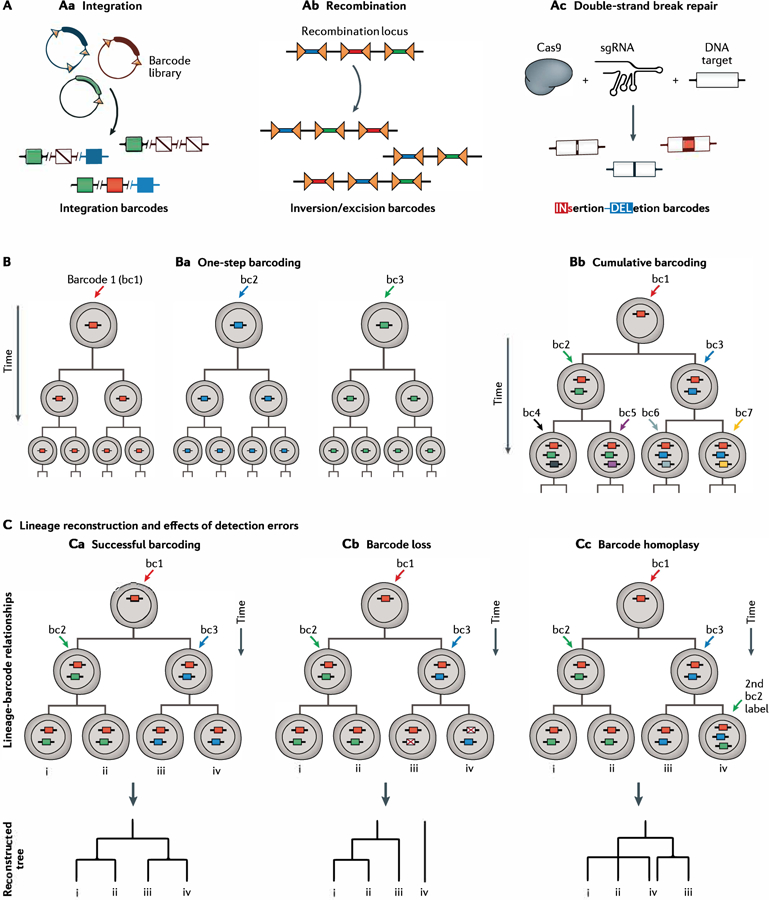
A | Three major paradigms for introducing unique DNA barcodes into cells: by integration of a high-diversity library of DNA barcodes using a transposase (part Aa), by randomrecombination of an array of recombinase target sites (part Ab) and by the accumulation ofrandom errors insertions and dele tions during CRISPR-Cas9 editing of genomic target sites (Part Ac).B | DNA barcoding can be applied in asingle, ins.antaneous pulse, enabling the paraflel tracking of many distinct cell clones (part Ba). When applied cont inuoudy. DNA barcades can repeatedly label a dividing cell clone at sequential levels ofits lineage hierarchy (part Bb) C | Challenges in lineage reconstruction from cumulative barcoding. The upper diagrams depict hypothetical barcode integration events ina cell ineage. Arrows denote the accumulation of novel barcodes, with each colour indicating aunique DNA barcode sequence. Hypothetical lineage correlation heat mapsand trees depict the anticipated results of lineage reconstruction. Lineage phylogenies can be accurately reconstructed from dngle-cell correlations of the detected barcode labels (part Ca), whereby earty versus late clones aredistinguished on the basis of the number of cells that contain theassodated barcode. Errarsin barcoding or barcode det ection can skew the accuracy of phylogenetic inferences (parts Cb and Cc). sgRNA, single-quide RNA.
The use of DNA barcodes to reconstruct lineage relationships initially relied on the identification of unique retroviral integration sites and utilized Southern blot or PCR assays to reveal barcode identity93,94. In the post-genomics sequencing era there has been a burst of innovation in the creation and deployment of far more complex DNA barcodes for lineage tracing (Table 1). A foundational concept for these methods is to use changes in targeted, whole-genome or mitochondrial-genome sequencing data to construct lineage phylogenies 95–97. Targeted barcoding-based methods generally fall into three thematic categories: first, transgenic integration of exogenous DNA sequences (FIG. 3Aa); second, in vivo recombination of transgenic DNA cassettes (FIG. 3Ab); and third, in vivo editing of transgenic DNA targets by CRISPR–Cas9 (FIG. 3Ac). In all of these approaches, a DNA-barcoding event permanently alters the genome of an individual cell, the descendants of which inherit the barcode and can be distinguished as a clonal unit (FIG. 3Ba). Importantly, DNA barcodes can be recorded and measured at high throughput, enabling that interrogation of hundreds or thousands of distinct clonal units in parallel. In addition, these modalities can be adapted for cumulative barcoding, which marks successive/nested clonal units and facilitates phylogenetic reconstruction of cell lineage trees (FIG. 3Bb).
Table 1 |.
Sequencing based technologies for lineage tracing
| Technology | DNA-editing system | Barcode type | Barcode length (bp) | Uniform barcode frequency? | Frequent barcode homoplasy? | Barcode as mRNA | Barcode generation | Species | In vivo? | Refs |
|---|---|---|---|---|---|---|---|---|---|---|
| TracerSeq | Tol2 | Integration | 20 | Yes | No | Yes | Continuous | Zebrafish | Yes | 47 |
| LARRY | Retrovirus | Integration | 28 | Yes | No | Yes | Single-step | Mouse | Yes | 64 |
| CellTag | Retrovirus | Integration | 8 | Yes | No | Yes | Multi-step | Human | No | 114,116 |
| PollyLox | Cre-loxP | Recombination | 2,152 | No | Yes | No | Continuous | Mouse | Yes | 101,108 |
| GESTALT | Cas9 | INDEL | 266 | No | Yes | No | Continuous | Zebrafish | Yes | 95 |
| scGESTALT | Cas9 | INDEL | 363 | No | Yes | Yes | Continuous | Zebrafish | Yes | 101,110 |
| ScarTrace | Cas9 | INDEL | 249 | No | Yes | Yes | Continuous | Zebrafish | Yes | 86 |
| LINNAEUS | Cas9 | INDEL | 75 | No | Yes | Yes | Continuous | Zebrafish | Yes | 102 |
| MARC1 | Cas9 | INDEL + integration | 240 | No | Yes | No | Continuous, evolvable | Mouse | Yes | 92,100 |
| Chen et al. | Cas9 | INDEL + integration | 350 | No | Yes | Yes | Continuous | Mouse | Yes | 85 |
| CHYRON | Cas9+TdT | INDEL(with insertion favoured over deletion) | 100 | No | Minimal | No | Continuous | Human | No | 115 |
A summary of lineage-tracing methods that make use of sequencing DNA barcodes. CHYRON, cell history recording by ordered insertion; GESTALT genome editing of synthetic target arrays for lineage tracing; INDEL, insertion or deletion; LARRY, lineage and RNA recovery; LINNAEUS, lineage tracing by nuclease-activated editing of ubiquitous sequences; MARC1, mouse far actively recording cells 1; scGESTALT, single-cell GESTALT; TdT, terminal deoxynucleotidyl transferase.
The first generation of methods and the logic for sequencing-based lineage tracing have been reviewed extensively elsewhere5,6. It is instructive to review the most recent developments, particularly in CRISPR-editing-based barcoding schemes. This family of methods utilizes a cumulative barcoding strategy to reveal lineage hierarchies that terminate at a single end point in time, typically by introducing three transgenic components: CRISPR–Cas9 DNA endonuclease, an array of DNA target sites, and a panel of single guide RNAs (sgRNAs) or homing guide RNAs (hgRNAs). These components generate high-diversity, ‘evolving’ DNA barcodes within cells by taking advantage of cumulative variability in target sites that results from CRISPR–Cas9 activity. The first methods to demonstrate this principle were genome editing of synthetic target arrays for lineage tracing (GESTALT)98 and homing CRISPR99,100. More recent innovations include the engineering of lineage barcodes into transcribed regions of constitutively expressed or inducible reporter genes, enabling their sequences to be read from mRNA in whole-transcriptome scRNA-seq experiments (FIG. 4A). This innovation was first demonstrated by the single-cell GESTALT (scGESTALT)101, lineage tracing by nuclease-activated editing of ubiquitous sequences (LINNAEUS)102 and ScarTrace86 techniques and has become a standard feature in subsequent methods. Other common innovations include the use of barcode arrays, which increase the number of barcoding possibilities, as well as the use of inducible promoters and integrated fluorescent reporters to both control and monitor the barcoding process in real time (FIG. 4B).
Fig. 4 |. Reading and writing transgenic DNA barcodes.
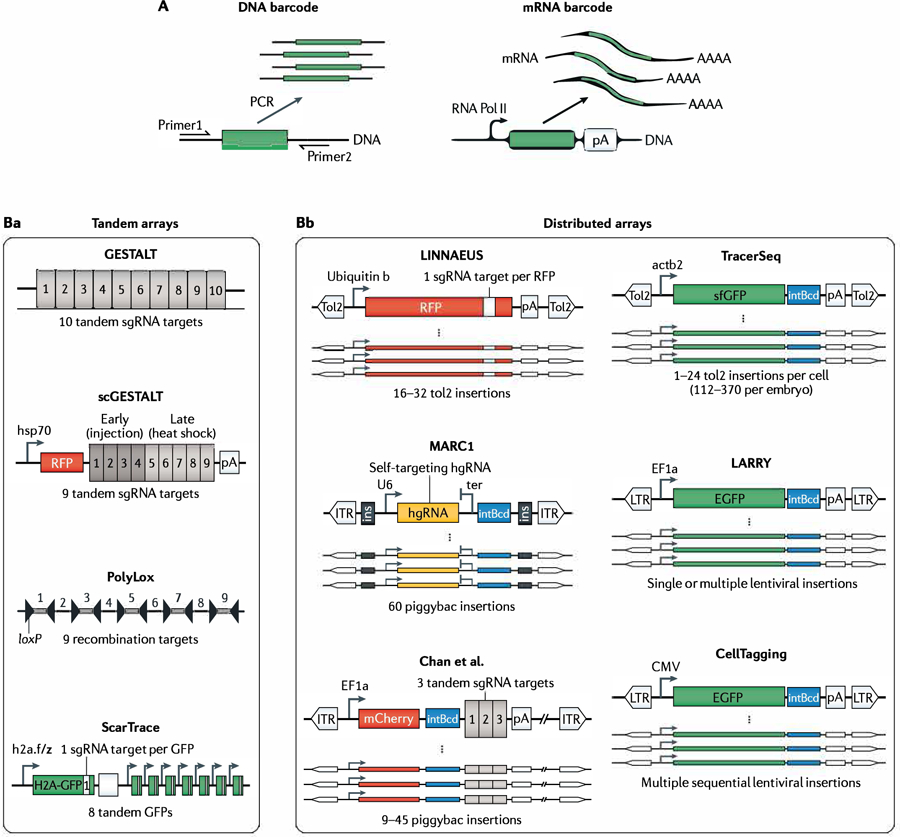
A | DNA barcodes can be encoded exclusively in genomic DNA (left) or expressed as mRNA, to allow detection concurrent with single-cell RNA sequencing. Reliable detection of barcode sequences requires amplification. For DNA barcodes this is achieved by P CR or in vitro transcription, whereas mRNA-based barcodes are endogenously amplified via RNA polymerase II (Pol II) transcription and can be detected as part of each single-cell transcriptome. B | Transgenic strategies for storing and transcribing DNA barcodes. The schematics show the diversity of DNA arrays used to store lineage information for each method. The arrays can be grouped according to whether they store lineage information at a single genomic locus using a tandem array (part Ba) or whether they store lineage information at multiple genomic loci using distributed arrays (part Bb). Right-angled black arrows indicate promoters used to drive barcode expression for detection by RNA sequencing in a subset of methods. The methods differ in whether they utilize recombination (PolyLox), barcode library integration using a lentivirus or transposase (TracerSeq, LARRY. CellTagging) or CRISP R-Cas9 targeting of single guide RNA (sgRNA) arrays (all remaining methods). GESTALT, genome editing of synthetic target arrays for lineage tracing; hgRNA, homing guide RNA; LARRY. lineage and RNA recovery; LINNAEUS, lineage tracing by nuclease-activated editing of ubiquitous sequences; MARCl, mouse for actively recording cells 1; scGESTALT, single-cell GESTALT.
Performance, trade-offs and further innovations
DNA-barcoding technologies show considerable potential as future tools for lineage tracing. As this is a rapidly evolving field, the published methods are likely to be revised substantially in the coming years. For this reason, we do not recommend any single published method at present over others. It is helpful instead to appreciate the limitations that are likely to be resolved, as well as some methodological improvements that are already emerging.
DNA-damage-induced toxicity
Most CRISPR–Cas9 barcoding methods rely on random insertions and deletions introduced during the process of double-strand break repair by non-homologous end joining (NHEJ). Recently, CRISPR–Cas9 activity has been shown to cause cell death in human induced pluripotent stem cells (iPSCs)103 and cell lines104, and also it can result in developmental delay in mouse embryos85, raising potential concerns about maintaining continuous endonuclease activity. The extent and effect of potential off-target double-strand breaks also remains generally unaddressed. Going forward, it will therefore be important to validate that these systems do not perturb the developmental dynamics that they are being used to interrogate.
The alternatives to CRISPR–Cas9-based methods may not face the same concern of excessive DNA damage. One alternative is TracerSeq47, a method for clonal barcoding demonstrated in zebrafish. TracerSeq makes use of ongoing transposase activity to successively integrate a pool of predefined barcodes, delivered as an injected plasmid library into embryos. The progressive integration of plasmids into the genome provides a heritable label of clones and sub-clones without inducing unrepaired double-strand breaks, yet it does require injection or electroporation. Other alternatives that similarly avoid double-strand breaks use genetic recombination105,106 (for example, PolyLox), CRISPR-associated transposase systems (CAST and Vibrio cholerae Tn6677)107,108 and base-editing enzymes109,110. Base-editing enzymes, however, can have substantial off-target effects that could perturb biological function111–113.
Barcode detection
Failure to detect edited barcode sequences (for example, due to measurement drop-outs) can skew inferred lineage relationships (FIG. 3Cb). Such errors arise, for example, from low or noisy levels of barcode reporter expression or from endogenous silencing of integrated transgenes or lentiviral constructs114. We do not at present know the precise barcode detection rates of existing methods, but the extent of such errors for any barcoding method can be estimated in principle through control experiments in which lineage relationships can be independently verified115. At a minimum, studies using DNA barcodes should assess the per-cell barcode detection rate, and may need to consider taking steps to improve experimental detection (for example, introducing strong RNA polymerase II promoters that drive the transcription of mRNA-based barcodes).
Assay calibration
Because lineage tracing and single-cell omics assays can take weeks to analyse and are expensive, it is desirable to be able to assess the efficiency of barcoding before detection. In integration-based systems, the expression of barcode-linked fluorescent proteins can report on the level and specificity of barcoding activity in live specimens. Some CRISPR–Cas9 systems (for example, LINNAEUS and ScarTrace) target DNA editing to the coding region of a fluorescent transgene, such that loss of fluorescence can be used to monitor the barcoding process. Such live-reporting schemes for barcode generation provide a simple means for sample validation before sequencing.
Barcode diversity.
Failures to resolve unique clones (that is, barcode homoplasy or type I errors) occur when cells inherit identical barcode sequences despite having no true lineage relationship (FIG. 3Cc). To avoid such errors, lineage-tracing methods should generate far more barcodes than the number of clones to be analysed. In CRISPR–Cas9 systems, the barcode diversities generated by Cas9 that are quoted in different studies have varied considerably. The true barcode diversity obtained in such systems, however, is likely to be overestimated, in part, because certain errors in double-strand break repair re-occur frequently102. In addition, the generation of multiple DNA double-strand breaks in close proximity leads to the excision of intervening sequences, resulting in the loss of previously generated edits101. Finally, the activity of DNA repair machinery may differ between organisms, tissues and/or species.
To minimize the negative effects of barcode homoplasy, it is possible to utilize biological replicates to empirically identify high-frequency barcodes and exclude them from downstream analyses85,102. However, the presence of species-specific and tissue-specific differences in barcode diversity argues that diversity should be evaluated in each experimental system to which the methods are applied. An additional innovation for increasing Cas9-edit barcode diversity includes the use of terminal deoxynucleotidyl transferase (TdT) as an additional transgenic component expressed at the time of barcoding115. In the presence of double-strand breaks, TdT was demonstrated to catalyse the random incorporation of nucleotides at the DNA cut site, resulting in an increased frequency of insertion-based edits over deletion-based edits.
It is also possible to expand barcode diversity by increasing the number of barcoding events per cell, although this strategy can carry experimental trade-offs. In CRISPR–Cas9 systems, barcode diversity can be increased through the parallel editing of several transgenic DNA target sites arranged in tandem or distributed throughout the genome (FIG. 4B). Tandem barcode arrays (FIG. 4Ba) face a practical limit, as they form repeat-rich sequences that are problematic substrates for both molecular cloning and most modern single-cell sequencing pipelines. Most of the recent methods therefore use distributed barcode arrays (FIG. 4Bb), which greatly reduce the number of nucleotides that must be sequenced in order to recover the barcode identity and also provide the advantage of being far less susceptible to internal deletions and information loss47,84,85,100,116. Distributed arrays can resolve otherwise identical DNA target sites through the use of an additional layer of integration barcodes that are specific to each transgenic insertion site. Distributed arrays are thus inherently scalable and can increase the barcode-space complexity while avoiding the need for long sequencing reads. However, they too face a limitation, in that a failure to detect some barcodes (type II errors) may lead to partial barcode recovery for many cells. Additionally, distributed arrays may be lost during outbreeding of transgenic animals and/or through endogenous silencing of transgenic or lentiviral constructs.
Barcode diversity may be less of a challenge for integration-based or recombination systems. Integration-based systems use high-diversity, uniform barcode libraries that are both simple to recover by sequencing and straightforward to interpret. TracerSeq barcodes, for example, are sampled evenly from a large sequence space (20-nucleotide sequences, yielding ~1012 possible variants), greatly simplifying computational analyses and the assignment of cells to clones47. Furthermore, increasing the integration rate expands combinatorial diversity by allowing more than one barcode to label each cell114,116. A drawback to the use of defined barcode libraries is that they require the introduction of exogenous transgenic DNA libraries into cells through injection, viral transduction or electroporation/lipofection, which limits their experimental possibilities47,114. In recombination systems, the number of barcode possibilities increases with the number of recombination sites. In the PolyLox system, 9 loxP sites yields >1.8 million Cre recombination possibilities105; this diversity could be further increased by adding more sites.
Barcoding precision.
A critical requirement for any lineage-barcoding experiment is the need to capture a minimum of two cells (ideally, many more) per clone. This requirement argues strongly for the need to label small numbers of cells in a defined tissue of interest, in order to ensure adequate sampling of their resulting progeny. In addition, the interpretation of clonal-tracing experiments depends strongly on precisely controlling the time interval in which cells are labelled. To date, published methods have not yet been optimized to achieve both tissue and temporal specificity in barcoding. Targeting clonal labelling to specific tissues can be facilitated by expressing components of the barcoding machinery under the control of tissue-specific promoters. Achieving temporal specificity is a more complex challenge. For CRISPR–Cas9-based methods, an open problem is that of target site ‘exhaustion’, in which all editing is completed early in the developmental period of interest. We expect the practical challenges of targeting clonal labelling to be resolved in the coming years.
Applications of lineage tracing on state manifolds
Lineage-tracing methods can now integrate high-dimensional state information with clonal and phylogenetic barcoding. In doing so, they greatly increase the number of clones that can be tracked, and they establish clonal composition without requiring prior knowledge of the marker genes. Both of these advantages should greatly reduce transgene-centric observation biases. However, omics lineage-tracing experiments demand novel experimental designs and controls, as compared with traditional methods. These methods also demand far more computational support than do traditional methods, owing to the high-dimensional nature of omics measurements, the difficulty of studying thousands of lineage trees that may be heterogeneous, and the unique nature of noise in these methods compared with previous approaches. We next survey three general experimental strategies that recently have been utilized to map how lineages unfold on state manifolds. We review outstanding challenges encountered in the computational analysis of state–lineage relationships, as well as potential pitfalls in experimental design.
Prospective lineage tracing on state manifolds
Prospective lineage tracing is still most commonly deployed by marking cells of a defined state at an early time point and establishing their collective cellular products at a later time. This approach has its roots in classical fate-mapping and genetic-labelling methods, which are implemented at either bulk or single-cell resolution. A modern version of this approach combines a sequencing-readable DNA recombination event as the genetic label, cell sorting and downstream analysis by scRNA-seq or some other high-resolution genome-scale measurement (FIG. 5Aa). Such approaches can provide detailed state resolution for the resulting cell populations, but prospective lineage labels are still comparatively low-resolution when they rely on the promoter activity of a single gene.
Fig. 5 |. Applications and pitfalls of lineage tracing on state manifolds.
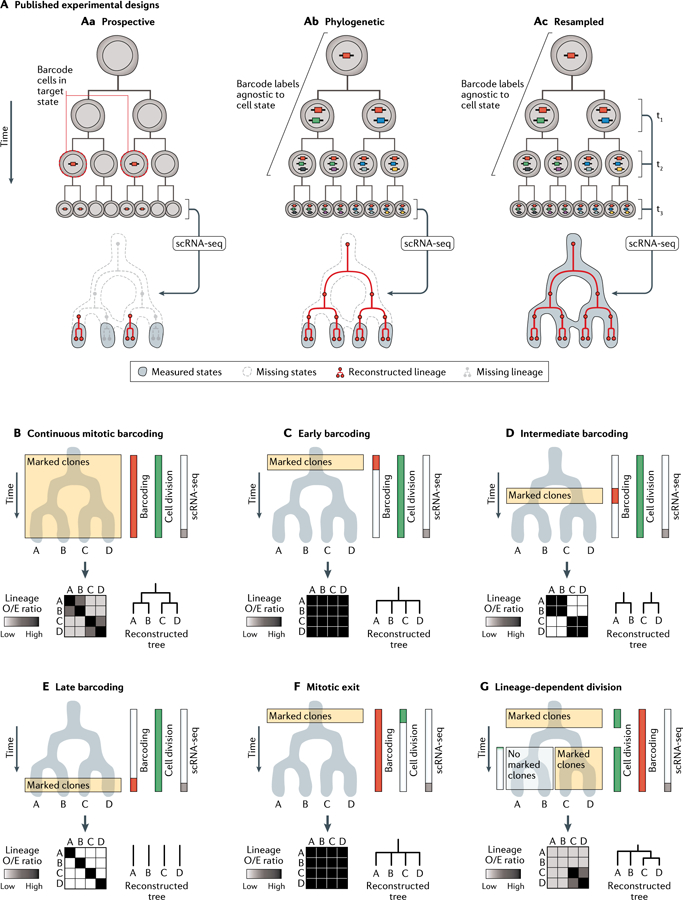
A | Recent studies have highlighted three experimental designs for combining lineage and state measurements. For simplicity, the panels depict largely congruent state-lineage hierarchies. Prospective (part Aa): a bulk genetic label is applied to cells of a particular state; labelled cells are subsequently captured and sequenced to reveal the gene expression states and lineage barcodes for each cell. Phylogenetic (part Ab): gene expression states and lineage barcodes are measured at a defined end point with respect to a biological process. Prior lineage relationships can be reconstructed retrospectively from the lineage barcodes, whereas state information is limited to the final time point. Resampled (part Ac): gene expression states and lineage barcodes are repeatedly sub-sampled over time, enabling the mapping of lineage trends directly on the state manifold. B-G | Phylogenetic reconstruction of fate hierarchies from end-point state and lineage measurements. The results of hypothetical lineage-state reconstruction analyses are displayed for each scenario; they vary dramatically, depending on the timing of both cell division and lineage barcoding. Heat maps depict the number of shared barcodes observed between each pair of states, normalized by the expected number of barcodes under a null hypothesis in which barcodes are distributed at random (‘Lineage 0/E ratio’). For a thorough definition of this statistic, see Weinreb et al. (2020)84. Lineage relationships can only be inferred at the time points when marked clones are generated and expanded. Given constant cell division rates and identical state manifolds, different time windows of barcode induction will lead to different inferences about lineage relationships. B | Continuous lineage barcoding in an actively dividing cell population enables all major lineage restriction events to be well-represented in a lineage-state reconstruction analysis. C-E | Lineage relationships can only be inferred at time points when marked clones are generated and expanded. Given constant cell division rates and identical state manifolds, different time windows of barcode induction will lead to distinct inferences about lineage-state relationships. F | In postmitotic differentiation hierarchies, despite continuous DNA barcoding, an absence of cell division precludes the formation of marked clones containing> 1 cell. Barcodes are no longer enriched across the state manifold and cannot be used to reconstruct fate restriction hierarchies. G | Lineage inferences require well-sampled barcode data from marked clones. Variable rates of cell division on a state manifold skew clone sizes and, hence, the statistical power to detect lineage-barcode correlations. scRNA-seq, single-cell RNA sequencing.
In an instructive example, Rajagopal and colleagues30 made use of the classical genetic recombination-based lineage-tracing method to label cells, followed by scRNA-seq to analyse the fates of the labelled cells. This ‘pulse-seq’ method was applied to study the fate of basal cells in airway epithelium labelled at a defined time using a conditional CreER recombinase expressed from the Krt5 locus, which activates permanent and heritable expression of a fluorescent reporter gene. At later time points, scRNA-seq established that Krt5–CreER-marked basal cells regenerated all epithelial cell types of the airway. Crucially, this approach did not require knowledge of the markers for any of the derivative cell types and thus could be used to establish the basal origin of a novel cell population. In this study, scRNA-seq also revealed that Krt5-expressing basal cells are not homogeneous in their transcriptomes. Thus, while pulse-seq could collectively mark all basal cells, it could not distinguish which individual clonal behaviours are stem cell-like, nor could it correlate such clonal behaviours to a particular subset of the Krt5-expressing population.
A more refined approach to prospective lineage tracing would make use of DNA barcodes to uniquely label each cell in the initial cell population. Although resolution into the labelled cell state would still be limited to the expression of a single marker gene, the end point measurements would provide information on heterogeneity in the clonal output of the labelled cells. From such data, one could assess whether the entire labelled cell population was equal in its fate potential or whether further fractionation of the labelled cells might be needed to resolve distinct cellular subsets. Such experiments could use scRNA-seq to analyse the clonal progeny. Evolving barcode approaches could also be adapted as a variant of such prospective lineage tracing, by requiring that barcode evolution be conditional on the expression of a state-prognostic transgenic promoter. To our knowledge, however, such applications have not yet been demonstrated.
Lineage phylogenies on state manifolds
Although there are now multiple methods for phylogenetic lineage barcoding (FIG. 4), all share a common goal: to determine the shared division history of the cells collected at a single end point in time. A common innovation in recent lineage-barcoding studies has been the engineering of lineage barcode cassettes into expressed sequences (FIG. 4A, right), enabling the simultaneous measurement of lineage information and whole-transcriptome state measurements for each cell. While these approaches succeed in revealing detailed states for the end-point-sequenced cells (FIG. 5Ab), they fail to capture the transcriptional states of progenitor cells that existed at time points before sequencing. Thus far, early applications of phylogenetic state–lineage approaches have largely recapitulated known developmental hierarchies in proof-of-concept studies. They have, however, revealed a recurring insight: namely, that similar cell states can arise (or ‘converge’) from qualitatively different developmental origins. Lineage construction using the tools reviewed above (Figs 3,4) can therefore be useful to identify converging developmental trajectories (FIG. 2d) and to distinguish other trajectories (FIG. 2b–h; BOX 1) that are not immediately highlighted by state manifold approaches alone. They also can be used to identify measured features of cells (for example, novel marker transcripts) that reflect their cell ontogeny.
Three recent studies spanning different embryonic tissues illustrate the recurring observation of state convergence, although these examples are far from exhaustive. In a first example, integration-based barcodes were detected in scRNA-seq data (by TracerSeq; FIG. 3) in zebrafish embryos. From these data, collected at a single time point 24 hours after embryo fertilization, it was possible to determine the shared lineage history of tens of transcriptionally defined cell states. Notably, one set of structures in the embryo, known as the pharyngeal arches, could be seen to arise from different clonal origins, despite appearing transcriptionally similar. These structures arise from either neural crest or lateral plate mesoderm47. Once the origin of the cells was established, it became possible to identify genes whose expression in the pharyngeal arches was specific to the crest-derived cells47. In a second example, CRISPR–Cas9 barcoding using the ScarTrace system revealed that the zebrafish fin harbours resident immune cells (RICs) with an ontogeny distinct from that of other immune cells86. These experiments could reveal precisely which cells were RICs amongst all immune cells in the fin, and they defined Epcam as a putative marker for this population. In yet a third example, Chan et al.85 used cumulative Cas9 editing to study the ontogeny of endodermal tissues in the mouse embryo. These tissues are known to comprise a mixture of visceral and epiblast-derived cells117. Chan et al. could resolve between the visceral and epiblast lineages, despite their converging onto similar endodermal gene expression programs. The researchers could then identify differences between the two endodermal lineages in the expression of two genes: Rhox5 and Trap1a. The ubiquity of converging trajectories has been further supported by complementary observations in the mouse extra-embryonic endoderm35,51, in C. elegans embryogenesis40 and in the parallel progression of excitatory and inhibitory neuronal states in the mouse central nervous system50. Collectively, these findings highlight a recurring phenomenon that these methods are particularly suited to address: they resolve different clonal origins among identical or nearly identical cell states (FIG. 2c–e), and they can reveal features of a cellular transcriptome (however subtle) that correlate with lineage behaviour and could be used to label or isolate cell subsets for further study.
Clonal resampling on a state manifold
Recently, several groups have utilized an alternative approach for linking detailed cell states across time. The approach relies on ‘clonal resampling’: experimentally isolating part of a clone for single-cell transcriptomic analysis recurrently, as the clone differentiates. When scaled to large numbers of cells, this method facilitates the construction of state manifolds on which the trajectories of individual clones may be revealed (FIG. 5Ac). This method requires both that cells be sampled over time without excessively disrupting the behaviour of the surviving cells and that cells divide symmetrically, such that all cells within a clone initially possess similar states. Due to these requirements, this method is best applied to either in vitro systems or regenerative systems in which cells or tissues may be serially removed or transplanted. Early realizations of this approach have been applied in culture, in which individual clones of related cells can be physically split, grown and sampled independently. For example, Tian et al.118 recently applied this approach to analysing dendritic cell clones derived from single haematopoietic stem cells cultured and assayed in vitro. By physically splitting small clones of cells into separate culture wells, they were able to perform two distinct types of measurement on the clonal ‘sister’ cells: scRNA-seq at an early time point, to establish the transcriptional features of each clone before differentiation, and in vitro assays, to establish the ability of the same clone to generate three distinct types of differentiated dendritic cell population. This approach, which the researchers termed ‘SIS-seq’, was able to reveal rich transcriptional features of early progenitor cells that were predictive of the later fate outcomes.
More recent applications of this approach have relied on DNA barcoding, rather than physical isolation, to simultaneously track large numbers of cell clones. In an instructive example, Biddy et al.114 developed a method, ‘CellTagging’, to trace the state of cells undergoing direct reprogramming from fibroblast to endoderm progenitors in vitro during serial rounds of passaging. The researchers made use of a lentiviral library to genetically barcode cells by integration of a constitutively expressed GFP-encoding gene with random barcodes engineered into its 3ʹ untranslated region sequence. During serial passaging, they applied additional rounds of lentiviral barcoding to mark successive lineage restriction events and simultaneously sampled subsets of the growing culture for scRNA-seq analysis. From this analysis, they identified that successful lineage conversion observed late in the reprogramming process correlated with a distinct expression profile of clonally related cells at an earlier time point. Such correlative analyses raise hypotheses for genes whose early expression influences future cell behaviours. In the study, Biddy et al. found that incorporating one such predictor gene, Mettl7a1, into the reprogramming procedure increased the efficiency of generating endodermal progenitors. Crucially, in this study neither the initial, transient nor end state of the dynamics had to be resolved in advance, and no marker genes were required to label cells for lineage tracing. A similar logic was applied by Weinreb et al.84, who also used a lentiviral DNA-barcoding approach to demarcate fate boundaries in haematopoietic progenitor cell differentiation in order to link early biases in gene expression to later fate potential.
Clonal resampling thus offers a powerful approach to fully integrate state manifolds with lineage tracing and can be used to identify prospective fate markers. This approach has been most thoroughly applied in vitro, but it can also be used to interrogate in vivo systems that permit physical resampling, such as the haematopoietic system84 and the regenerative zebrafish fin86. A persisting challenge in studying in vivo systems in this way is the need to obtain sufficient statistical sampling of each clone of interest, which can be difficult when isolating and sequencing cells from large endogenous populations.
Computational tools for state–lineage mapping
Computational approaches to analyse combined lineage and state datasets are still in their infancy. They are likely to evolve considerably and to require steps that are sensitive to the choice of experimental platform. As choices in data analysis could affect the conclusions drawn from such methods, we briefly review the key steps here.
The first step for DNA-barcoding pipelines is to assign a unique DNA barcode sequence to each cell clone. In doing so, pipelines must eliminate putative sequencing errors, remove cell doublets that could lead to two clonal barcodes appearing in one cell, and correct platform-specific artefacts. For the CRISPR-based and PolyLox methods, some barcodes can be formed with high probability, leading to frequent barcode homoplasy. Computational pipelines must therefore decide which barcodes are informative and which must be discarded from the analysis. Current methods are naive to recombination or error preferences of DNA-modifying enzymes; future methods could learn and incorporate editing biases to correct for observed barcode frequencies.
Computational pipelines then face decisions about how to reconstruct lineage phylogenies from large sets of clonal barcodes. In some cases, tree-building methods established for evolutionary phylogenetics have been applied directly to lineage reconstruction efforts; for example, GESTALT studies have utilized maximum parsimony98,101,119, whereas homing CRISPR and TracerSeq have utilized neighbour-joining methods47,100. However, previously established tree-building methods are not necessarily robust to the frequent detection errors encountered in single-cell measurements102. LINNAEUS102 and Chan et al.85 have therefore developed custom tree-building algorithms to minimize the influence of drop-outs and have also incorporated empirical likelihood estimates for each barcode in order to minimize the influence of barcode homoplasy on the final inferred tree topology. Inference of lineage relationships from DNA-barcoding data is an active area of research, with several additional groups now favouring maximum-likelihood approaches and ground-truth benchmarking of algorithm performance against empirical120–122 or simulated123 datasets.
At present, no universal computational tools exist for end-to-end lineage tree inference, starting from raw single-cell DNA barcode sequences. Given the wide diversity of DNA modification strategies, barcode lengths and barcode probability distributions, the development of a single universal tool might be unlikely. However, for CRISPR–Cas9 editing systems, in particular, community benchmarking efforts such as the DREAM challenge124 are now providing opportunities to directly compare the performance of dozens to hundreds of independent algorithms. Standardization of metrics and input data types could further enable meta-approaches that draw results from the consensus of multiple different tools. Because all lineage-barcoding methods — including non-CRISPR –Cas9 methods — face similar downstream analysis challenges (for example, tree building, the analysis of large tree ensembles and increasing dataset sizes), the field as a whole will undoubtedly benefit from these and other computational innovations.
In addition to tree construction, there are also other — perhaps simpler — data representations that can reveal intuitive lineage–state relationships. Rather than focusing on the structure of individual lineage trees, one can instead integrate information from multiple trees so as to infer the average lineage relationships between cell states. For many organisms and tissues, such approaches may be crucial, because individual lineage trees can be highly variable. Various metrics can be used for establishing lineage coupling between states, including the covariance of barcode abundances between states or the ratio of the barcodes observed to be shared between two transcriptional states to that expected after data randomization47,84 (FIG. 5B). Maximum-likelihood frameworks can similarly be leveraged so as to combine individual lineage trees into ‘consensus’ lineage trees by integrating gene expression and lineage data122. This approach permits the integration of information across biological specimens, to separate core systematic trends from chance relationships that occur in just a single lineage tree.
Pitfalls in lineage barcoding on a state manifold
New biological assays can generate unforeseen artefacts that often become appreciated only after technologies mature. In the case of sequencing-based lineage tracing, the details of an experimental design can profoundly affect the relationships encoded in sequencing data. In FIG. 5 we have detailed two parameters that can strongly influence the observed clonal overlaps between states. These include the effects of the timing of barcode induction (FIG. 5B–E) and of changes in endogenous cell division rates (FIG. 5F,G). Altering these parameters can lead to strong differences in apparent lineage structure by affecting both the presence and size — that is, the detect-ability — of the marked clones. Other barcode detection errors, including both type I and type II errors (FIG. 3C), can similarly interfere with lineage reconstruction efforts. Although the negative effects of detection errors can be minimized by means of certain tree reconstruction algorithms (for example, maximum parsimony), the frequency of such errors for a particular method should be quantified, and minimized wherever possible. Good experimental practices should further ensure that biological conclusions are robust to such errors, including through performing adequate biological/technical replicates and the use of multiple data analysis strategies.
Emerging concepts
State trajectories and lineage codify two distinctive yet complementary aspects of a cell’s developmental history, and each type of analysis can provide insights into ontogeny and gene regulation. In this Review we have outlined some important limitations of state manifolds, and we have described the motivation and tools for integrating bona fide lineage measurements with single-cell omics. From the early application of these methods, we propose to highlight three emerging concepts: first, state manifolds as models; second, the modes of coupling of cell state bifurcation with cell division; and third, the validity of trees as descriptions of cell differentiation hierarchies.
State manifolds as models
In this Review we have raised the contradictions that can appear between lineage and state representations (FIG. 2) and discussed how clonal information could be used to clarify such developmental relationships. These contradictions demonstrate that representations of state manifolds are not infallible — rather, they are data-driven models that follow from particular sets of assumptions and data-processing criteria. Currently, most state manifolds are constructed in an unbiased fashion from the most prominent sources of covariation in the original state measurement. Under this practice, the defining features of an scRNA-seq manifold will reflect robust, variable transcriptional signatures and thus are not guaranteed to emphasize cell fate decisions, which might correlate with small sets of regulatory genes expressed at low levels at the time that fate restrictions occur. State manifolds have until now been constructed without incorporating information from clonal data. However, state and lineage relationships need not remain in conflict: once information on lineage is established, it can be used to improve our methods for representing state manifolds. An immediate and simple use of lineage information, for example, is in identifying molecular markers of lineage-biased progenitor cell states. Indeed, novel fate markers have been inferred both from combined lineage and state phylogenetic experiments85 and from clonal-resampling studies84,114. Lineage information could also be used to train algorithms in the construction of state manifolds in a way that avoids errors such as those in FIG. 2. Such actions demand a conceptual shift towards treating state manifolds as models of a particular set of high-dimensional gene expression features, rather than as absolute or universal references on which to overlay cell differentiation trajectories.
Variability of individual lineage trees
Both state manifolds and mitotic lineage trees can define hierarchies. What is the nature of the relationship between these two hierarchies? Drawing on lessons from imaging-based clonal analysis125, we propose two potential relationships: one of mitotic coupling, and another of population coupling. Mitotic coupling will occur in cases in which a branch-point identified on the cell state manifold closely corresponds to a cell division event (FIG. 6a, left). Determinate lineage trees of ascidians87 and C. elegans88 stand as instructive examples. Population coupling, by contrast, will occur in cases in which the clonal and division histories do not influence the progression of any individual cell along the manifold or its fate choice. Instead, cell behaviours are indeterminate and can be described by a set of transition probabilities for moving down a particular trajectory (FIG. 6a, right). Accordingly, population coupling can lead to highly variable lineage trees that resemble those from a stochastic branching process and that will not be precisely reproducible within or between organisms (FIG. 6b, right). In such cases, efforts towards high-resolution reconstruction of fate hierarchies may fail to produce a single representative lineage tree of development, but the distribution of state–lineage couplings across multiple observed lineage trees should nonetheless prove highly informative (FIG. 6c).
Fig. 6 |. Developmental paradigms that shape state-lineage relationships.
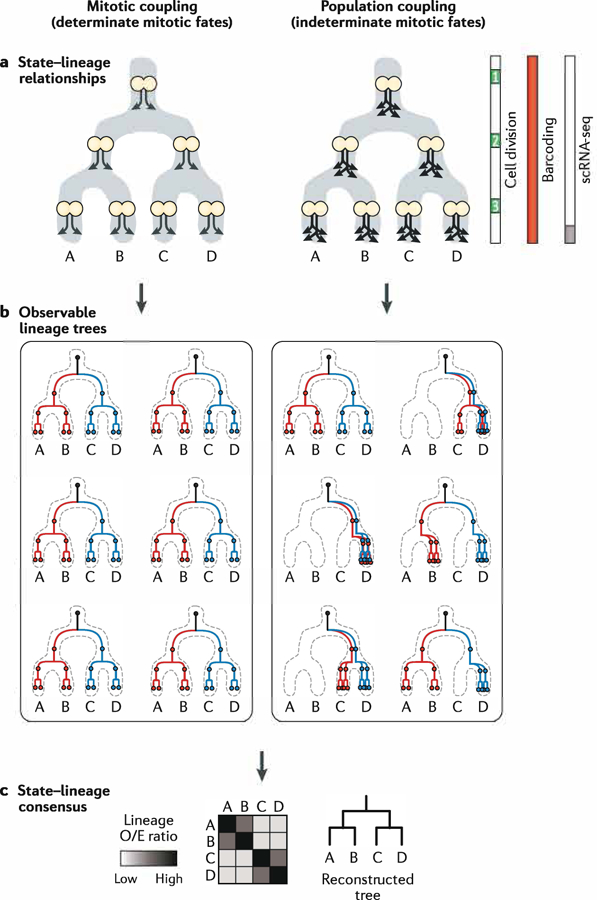
a | State manifold diagrams depicting the timing and fates of mitotic daughter cells. In cases of mitotic coupling (left), cells divide asymmetrically and give rise to distinct daughter states. In cases of population coupling (right), the average flux of cells down branches of the state manifold is maintained, but the fates of individual daughter cells are largely unpredictable. b | Examples of observable lineage trees that result from mitotic or population coupling. Mitotic coupling (left) leads to invariant, determinant lineage trees. Population coupling (right) permits a large number of observable lineage tree possibilities (six shown). c | Consensus relationships derived from a large number of individual tree observations. Despite the varied possibilities for the individual lineage trees in part b, the lineage relationships between states will be similar for both mitotic- and population-coupling scenarios. The heat map plots lineage observed/expected (0/E) ratios (see the FIG. 5 legend and Weinreb et al. (2020)84 for the definition). scRNA-seq, single-cell RNA sequencing.
Is development a tree?
What is the structure of a differentiation hierarchy? Answers to this question depend first on whether one is considering a state manifold or a mitotic lineage. In the absence of cell fusion, lineages can generally be treated as bifurcating trees, with each branch-point representing a mitotic event. State manifolds can be tree-like but, depending on the biology of the system, they need not be. State manifolds therefore represent an opportunity to discover the structure (that is, the topology) of a cell differentiation process. When state manifolds are integrated with lineage measurements, one has an opportunity to independently reject or confirm specific hypotheses regarding these structures. As we described above, several recent studies have shown evidence for state convergence, in which two or more distinct fate trajectories converge onto the same final position on a state manifold. This end point state thus comprises cells of mixed origins, which may or may not retain distinct functions or potentials. We reviewed examples of state convergence among immune cells86, neural crest lineages47 and endodermal populations35,85. The reverse scenario (state divergence) has also been observed, in which mitotic sister cells (highly related in lineage) rapidly adopt discontinuous states40. State divergence can occur as a result of asymmetric cell division, particularly in cases in which partitioned cytoplasmic components are delivered to only one of the two mitotic daughter cells. Such cases may produce state transitions that lack intermediate states and that thus would not appear as a bifurcation event on a state manifold, at any sampling depth. Both of these scenarios — convergence and divergence — will cause a state manifold to depart from a strict tree structure and can result from well-described biological scenarios. Mapping novel examples of such scenarios from single-cell datasets will therefore require integrated state and lineage measurements.
Conclusions
With the emergence of genome-scale single-cell analyses, representations of differentiation dynamics have shifted in the span of a few years from cartoons of discrete state transitions to data-driven views of dynamic state manifolds. Such representations provide not just predictions for the differentiation dynamics of thousands of genes but also hypotheses for the structure of differentiation hierarchies, including novel transitional and terminal cell states, interactions with cell cycle and the appearance of convergent differentiation that takes the form of ‘loops’ between cell states. In this Review we described the errors and ambiguities that can arise in inferring dynamics directly from single-cell state measurements, and we argued that the integration of lineage-barcoding data can improve state manifold representations by facilitating a faithful reconstruction of dynamics. Such integrative measurements can identify prospective fate markers, localize fate boundaries on state manifolds, allow the inference of tree-like and non-tree-like differentiation hierarchies, and should allow for resolving consensus fate relationships even when the individual lineage trees are highly variable. We thus anticipate that integrated measurements of cell state and lineage will greatly clarify the key events in cellular differentiation and become an important tool in the arsenal of stem cell, tissue and developmental biologists.
Cell differentiation
The process by which uncommitted progenitor cells are specified and transform into functional (and typically postmitotic) cells that carry out the specialized tasks of a particular tissue or organ.
Landscape
An informal term for a state manifold, typically used in developmental biology to represent the ensemble of cell states during their differentiation.
State manifolds
Approximate representations of high-dimensional cell states (for example, the whole-animal embryonic cell state atlas Tabula Muris) as lower-dimensional shapes.
State trajectories
The paths taken by individual cells or clones of cells through a state manifold.
Prospective lineage tracing
A lineage-tracing experiment that introduces a label for marking cells in a specified state.
Barcodes
Units of DNA with a large number of sequence possibilities, such as those used to uniquely label cells and their progeny.
Cell lineage
A representation of a series of mitotic events that trace back to a single founder cell.
Cell state
A designation of cell identity (defined with respect to a particular measurement) that can be used to classify or quantify physical or molecular differences between cells (for example, ‘basophilic’, ‘KRT4+’, ‘columnar’, ‘RNA-Seq cluster 4’).
RNA velocity
The rate of change in mRNA transcript abundance — more specifically, a set of computational techniques for calculating these rates across all genes from measurements of spliced and unspliced transcript abundances.
Clonal analysis
A lineage-tracing experiment that involves marking an individual cell, followed by state analysis of that founder cell’s clonal descendants.
Retrospective lineage tracing
A lineage-tracing experiment based on phylogenetic reconstruction of endogenous genetic polymorphisms (that is, no experimental intervention).
Hidden variables
Molecular or environmental properties of a cell that correlate with — or could be used to predict — a cell fate decision, which are obscured from a state manifold.
Direct observations
Lineage-tracing experiments that rely on in vivo live imaging of cells as they divide.
Determinate
In the context of developmental processes, when the relationship between lineage and molecular state is tightly controlled at each cell division event and is invariant between individuals.
Indeterminate
In the context of developmental processes, when the relationship between lineage and molecular state can vary greatly between individuals and between cell clones.
Lineage phylogenies
Trees of lineage relationships constructed from end point measurements.
Drop-outs
Type II errors that are common in single-cell omics experiments in which transcripts, lineage barcodes or other features present in cells fail to be detected.
Barcode homoplasy
A type I error in which identical DNA sequence barcodes are randomly recovered from cells with no close lineage relationship.
Cell ontogeny
The developmental history of a cell.
State convergence
A differentiation scenario in which cells with distinct origins converge onto the same end point on a state manifold.
Mitotic coupling
A class of developmental fate regulation mechanisms that specify states to the daughter cells of a mitotic division, either symmetrically or asymmetrically.
Population coupling
A class of developmental fate regulation mechanisms in which the cell state specification is uncoupled from cell division but the proportion of cells specified to each state is controlled.
State divergence
A scenario in which the asymmetric partitioning of cellular components between two daughters of a single cell division differentiates them rapidly or instantaneously into distinct states.
Acknowledgements
The authors thank R. Ward and S. Mekhoubad for critical reading of the manuscript. D.E.W. is supported by grant R00GM121852.
Footnotes
Competing interests
A.M.K. is a founder of 1CellBio, Inc. D.E.W. declares no competing interests.
Peer review information
Nature Reviews Genetics thanks J. P. Junker and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Whitman CO Memoirs: the embryology of clepsine. J. Cell Sci s2-18, 215–315 (1878). [Google Scholar]
- 2.Waddington CH The strategy of the genes A discussion of some aspects of theoretical biology. (George Allen & Unwin, Ltd., London, 1957). [Google Scholar]
- 3.Saelens W, Cannoodt R, Todorov H & Saeys Y A comparison of single-cell trajectory inference methods. Nat. Biotechnol 37, 547–554 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Tritschler S et al. Concepts and limitations for learning developmental trajectories from single cell genomics. Development 146, dev170506 (2019). [DOI] [PubMed] [Google Scholar]
- 5.McKenna A & Gagnon JA Recording development with single cell dynamic lineage tracing. Development 146, dev169730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kester L & van Oudenaarden A Single-cell transcriptomics meets lineage tracing. Cell Stem Cell 23, 166–179 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macosko EZ et al. Highly parallel genome-wide expression profiling of individual cells using nanolite droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gierahn TM et al. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 14, 395–398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner A, Regev A & Yosef N Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol 34, 1145–1160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotliar D et al. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. eLife 8, e43803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lareau CA et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol 37, 916–924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mezger A et al. High-throughput chromatin accessibility profiling at single-cell resolution. Nat. Commun 9, 3647 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karemaker ID & Vermeulen M Single-cell DNA methylation profiling: technologies and biological applications. Trends Biotechnol 36, 952–965 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Budnik B, Levy E, Harmange G & Slavov N SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol 19, 161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duncan KD, Fyrestam J & Lanekoff I Advances in mass spectrometry based single-cell metabolomics. Analyst 144, 782–793 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mimitou EP et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 16, 409–412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson VM et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol 35, 936–939 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Dey SS, Kester L, Spanjaard B, Bienko M & van Oudenaarden A Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol 33, 285–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han KY et al. SIDR: simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells. Genome Res 28, 75–87 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M & Cai L Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eng CL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriques SG et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JH et al. Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tusi BK et al. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 555, 54–60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tikhonova AN et al. The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montoro DT et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plasschaert LW et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park J et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young MD et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 361, 594–599 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Soysa TY et al. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 572, 120–124 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nowotschin S et al. The emergent landscape of the mouse gut endoderm at single-cell resolution. Nature 569, 361–367 (2019).The authors analyse >100,000 single-cell transcriptomes from developing mouse endoderm and describe the convergence of visceral and definitive lineages into spatially defined transcriptional states.
- 36.Diaz-Cuadros M et al. In vitro characterization of the human segmentation clock. Nature 10.1038/s41586-019-1885-9 (2020). [DOI] [PMC free article] [PubMed]
- 37.Zeisel A et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e1022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soldatov R et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 364, eaas9536 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Cao J et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Packer JS et al. A lineage-resolved molecular atlas of C. elegans embryogenesis at single-cell resolution. Science 365, eaax1971 (2019).The authors interrogate the temporal dynamics of lineage–state relationships, as well as transcriptional convergence and divergence, in the invariant C. elegans embryonic lineage.
- 41.Sebé-Pedrós A et al. Cnidarian cell type diversity and regulation revealed by whole-organism single-cell RNA-seq. Cell 173, 1520–1534.e1520 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Siebert S et al. Stem cell differentiation trajectories in Hydra resolved at single-cell resolution. Science 365, eaav9314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Achim K et al. Whole-body single-cell sequencing reveals transcriptional domains in the annelid larval body. Mol. Biol. Evol 35, 1047–1062 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng A et al. Prospectively isolated tetraspanin(+) neoblasts are adult pluripotent stem cells underlying planaria regeneration. Cell 173, 1593–1608.e1520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fincher CT, Wurtzel O, de Hoog T, Kravarik KM & Reddien PW Cell type transcriptome atlas for the planarian Schmidtea mediterranea. Science 360, eaaq1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plass M et al. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science 360, eaaq1723 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Wagner DE et al. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 360, 981–987 (2018).The authors describe non-tree-like cell state trajectories using combined lineage barcoding and single-cell transcriptomics in zebrafish embryos.
- 48.Farrell JA et al. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 360, eaar3131 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Briggs JA et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science 360, eaar5780 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao J et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).This study presents the largest single-cell transcriptome atlas for mouse embryogenesis to date, spanning >2 million cells and 56 cell state trajectories.
- 51.Pijuan-Sala B et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karaiskos N et al. The Drosophila embryo at single-cell transcriptome resolution. Science 358, 194–199 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Cao C et al. Comprehensive single-cell transcriptome lineages of a proto-vertebrate. Nature 571, 349–354 (2019).This study performs comprehensive single-cell profiling of ascidian embryos from the early gastrula to larval stages and maps the transcriptomic signatures onto a virtual map of the determinate embryonic lineage tree.
- 54.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 10.1038/nbt.4314 (2018). [DOI] [PubMed]
- 55.Weinreb C, Wolock S & Klein AM SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246–1248 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jacomy M, Venturini T, Heymann S & Bastian M ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS One 9, e98679 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qiu X et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolf FA et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59 (2019).This study presents ‘PAGA’, a graph-based computational approach for mapping non-tree-like topologies in single-cell state landscapes.
- 60.Setty M et al. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol 34, 637–645 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shin J et al. Single-Cell RNA-Seq with Waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell 17, 360–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haghverdi L, Buttner M, Wolf FA, Buettner F & Theis FJ Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Bendall SC et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinreb C, Wolock S, Tusi BK, Socolovsky M & Klein AM Fundamental limits on dynamic inference from single-cell snapshots. Proc. Natl Acad. Sci. USA 115, E2467–E2476 (2018).This is one of several studies to provide a framework for predicting fate trajectories from single-cell state manifolds.
- 65.Herman JS, Sagar & Grün D FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nat. Methods 15, 379–386 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Setty M et al. Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol 37, 451–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schiebinger G et al. Optimal-transport analysis of single-cell gene expression identifies developmental trajectories in reprogramming. Cell 176, 1517 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Furchtgott LA, Melton S, Menon V & Ramanathan S Discovering sparse transcription factor codes for cell states and state transitions during development. eLife 6, e20488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.La Manno G et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hendriks G-J et al. NASC-seq monitors RNA synthesis in single cells. Nat. Commun 10, 3138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Erhard F et al. scSLAM-seq reveals core features of transcription dynamics in single cells. Nature 571, 419–423 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Gorin G, Svensson V & Pachter L Protein velocity and acceleration from single-cell multiomics experiments. Genome Biol 21, 39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qiu X et al. Mapping vector field of single cells Preprint at 10.1101/696724 (2019). [DOI]
- 74.Haghverdi L, Buettner F & Theis FJ Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 31, 2989–2998 (2015). [DOI] [PubMed] [Google Scholar]
- 75.Coifman RR et al. Geometric diffusions as a tool for harmonic analysis and structure definition of data: diffusion maps. Proc. Natl Acad. Sci. USA 102, 7426–7431 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen H et al. Single-cell trajectories reconstruction, exploration and mapping of omics data with STREAM. Nat. Commun 10, 1903 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Traag VA, Waltman L & van Eck NJ From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blondel VD, Guillaume J-L, Lambiotte R & Lefebvre E Fast unfolding of communities in large networks. J. Stat. Mech 10.1088/1742-5468/2008/10/P10008 (2008). [DOI]
- 79.Kimmel CB, Warga RM & Schilling TF Origin and organization of the zebrafish fate map. Development 108, 581–594 (1990). [DOI] [PubMed] [Google Scholar]
- 80.Kretzschmar K & Watt FM Lineage tracing. Cell 148, 33–45 (2012). [DOI] [PubMed] [Google Scholar]
- 81.Lodato MA et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wagers AJ & Weissman IL Plasticity of adult stem cells. Cell 116, 639–648 (2004). [DOI] [PubMed] [Google Scholar]
- 83.Wagers AJ, Sherwood RI, Christensen JL & Weissman IL Little evidence for developmental plasticity of adult hematopoietic stem cells. Science 297, 2256–2259 (2002). [DOI] [PubMed] [Google Scholar]
- 84.Weinreb C, Rodriguez-Fraticelli A, Camargo FD & Klein AM Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367, eaaw3381 (2020).The authors implement the ‘LARRY’ clonal resampling approach to map single-cell transcriptomes and lineage relationships in differentiating cells in the mouse haematopoietic system.
- 85.Chan MM et al. Molecular recording of mammalian embryogenesis. Nature 570, 77–82 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alemany A, Florescu M, Baron CS, Peterson-Maduro J & van Oudenaarden A Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112 (2018).This study describes the Cas9-editing-based ‘ScarTrace’ method for simultaneous measurement of single-cell transcriptomes and lineage relationships in the zebrafish embryo and the regenerating fin of its adult form.
- 87.Conklin EG The organization and cell lineage of the ascidian egg. J. Acad. Nat. Sci. Phila 13, 1–119 (1905). [Google Scholar]
- 88.Sulston JE, Schierenberg E, White JG & Thomson JN The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol 100, 64– 119 (1983). [DOI] [PubMed] [Google Scholar]
- 89.Keller PJ, Schmidt AD, Wittbrodt J & Stelzer EH Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science 322, 1065–1069 (2008). [DOI] [PubMed] [Google Scholar]
- 90.McDole K et al. In toto imaging and reconstruction of post-implantation mouse development at the single-cell level. Cell 175, 859–876.e833 (2018). [DOI] [PubMed] [Google Scholar]
- 91.Satija R, Farrell JA, Gennert D, Schier AF & Regev A Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Frieda KL et al. Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Keller G, Paige C, Gilboa E & Wagner EF Expression of a foreign gene in myeloid and lymphoid cells derived from multipotent haematopoietic precursors. Nature 318, 149–154 (1985). [DOI] [PubMed] [Google Scholar]
- 94.Lemischka IR, Raulet DH & Mulligan RC Developmental potential and dynamic behavior of hematopoietic stem cells. Cell 45, 917–927 (1986). [DOI] [PubMed] [Google Scholar]
- 95.Ludwig LS et al. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell 176, 1325–1339.e1322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Woodworth MB, Girskis KM & Walsh CA Building a lineage from single cells: genetic techniques for cell lineage tracking. Nat. Rev. Genet 18, 230–244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xu J et al. Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. eLife 8, e45105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McKenna A et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kalhor R, Mali P & Church GM Rapidly evolving homing CRISPR barcodes. Nat. Methods 14, 195–200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kalhor R et al. Developmental barcoding of whole mouse via homing CRISPR. Science 361, eaat9804 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raj B et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol 36, 442–450 (2018).This study combines the previously established Cas9-editing GESTALT approach for lineage barcoding with inDrops-based single-cell transcriptome analysis to reconstruct developmental trajectories in the zebrafish brain.
- 102.Spanjaard B et al. Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat. Biotechnol 36, 469–473 (2018).This study introduces ‘LINNAEUS’ and a network algorithm for reconstructing Cas9-editing-based lineage phylogenies between cell states of the 5-day-old zebrafish embryo.
- 103.Ihry RJ et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med 24, 939–946 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Haapaniemi E, Botla S, Persson J, Schmierer B & Taipale J CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med 24, 927–930 (2018). [DOI] [PubMed] [Google Scholar]
- 105.Pei W et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 548, 456–460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pei W et al. Using Cre-recombinase-driven Polylox barcoding for in vivo fate mapping in mice. Nat. Protoc 14, 1820–1840 (2019). [DOI] [PubMed] [Google Scholar]
- 107.Klompe SE, Vo PLH, Halpin-Healy TS & Sternberg SH Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature 571, 219–225 (2019). [DOI] [PubMed] [Google Scholar]
- 108.Strecker J et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hwang B et al. Lineage tracing using a Cas9-deaminase barcoding system targeting endogenous L1 elements. Nat. Commun 10, 1234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hess GT et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13, 1036–1042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Grunewald J et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jin S et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 364, 292–295 (2019). [DOI] [PubMed] [Google Scholar]
- 113.Zuo E et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364, 289–292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Biddy BA et al. Single-cell mapping of lineage and identity in direct reprogramming. Nature 564, 219–224 (2018).This study introduces the ‘CellTag’ clonal resampling method for retroviral barcoding of cell lineages with a combined single-cell transcriptomic readout.
- 115.Loveless TB et al. Ordered insertional mutagenesis at a single genomic site enables lineage tracing and analog recording in mammalian cells Preprint at 10.1101/639120 (2019). [DOI]
- 116.Guo C et al. CellTag Indexing: genetic barcode-based sample multiplexing for single-cell genomics. Genome Biol 20, 90 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kwon GS, Viotti M & Hadjantonakis A-K The endoderm of the mouse embryo arises by dynamic widespread intercalation of embryonic and extraembryonic lineages. Dev. Cell 15, 509–520 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tian L et al. SIS-seq, a molecular ‘time machine’, connects single cell fate with gene programs Preprint at 10.1101/403113 (2018). [DOI]
- 119.Raj B, Gagnon JA & Schier AF Large-scale reconstruction of cell lineages using single-cell readout of transcriptomes and CRISPR–Cas9 barcodes by scGESTALT. Nat. Protoc 13, 2685–2713 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jones MG et al. Inference of single-cell phylogenies from lineage tracing data Preprint at 10.1101/800078 (2019). [DOI] [PMC free article] [PubMed]
- 121.Feng J et al. Estimation of cell lineage trees by maximum-likelihood phylogenetics Preprint at 10.1101/595215 (2019). [DOI] [PMC free article] [PubMed]
- 122.Zafar H, Lin C & Bar-Joseph Z Single-cell lineage tracing by integrating CRISPR–Cas9 mutations with transcriptomic data Preprint at 10.1101/630814 (2019). [DOI] [PMC free article] [PubMed]
- 123.Salvador-Martínez I, Grillo M, Averof M & Telford MJ Is it possible to reconstruct an accurate cell lineage using CRISPR recorders? eLife 8, e40292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Synapse. Allen Institute Cell Lineage Reconstruction DREAM Challenge. Sage Bionetworks https://www.synapse.org/#!Synapse:syn20692755/wiki/595096 (2019).
- 125.Klein AM & Simons BD Universal patterns of stem cell fate in cycling adult tissues. Development 138, 3103–3111 (2011). [DOI] [PubMed] [Google Scholar]