

Abstract
The genotype-first approach has been successfully applied and has elucidated several subtypes of autism spectrum disorder (ASD). However, it requires very large cohorts because of the extensive genetic heterogeneity. We investigate the alternate possibility of whether phenotype-specific genes can be identified from a small group of patients with specific phenotype(s). To identify novel genes associated with ASD and abnormal head circumference using a phenotype-to-genotype approach, we performed whole-exome sequencing on 67 families with ASD and abnormal head circumference. Clinically relevant pathogenic or likely pathogenic variants account for 23.9% of patients with microcephaly or macrocephaly, and 81.25% of those variants or genes are head-size associated. Significantly, recurrent pathogenic mutations were identified in two macrocephaly genes (PTEN, CHD8) in this small cohort. De novo mutations in several candidate genes (UBN2, BIRC6, SYNE1, and KCNMA1) were detected, as well as one new candidate gene (TNPO3,) implicated in ASD and related neurodevelopmental disorders. We identify genotype–phenotype correlations for head-size-associated ASD genes and novel candidate genes for further investigation. Our results also suggest a phenotype-to-genotype strategy would accelerate the elucidation of genotype–phenotype relationships for ASD by using phenotype-restricted cohorts.
Keywords: Autism spectrum disorder, whole-exome sequencing, microcephaly, macrocephaly, genotype and phenotype correlations
Graphical Abstract

Introduction
Autism spectrum disorder (ASD) is a group of phenotypic heterogeneous neurodevelopmental disorders (NDDs) with global prevalence around 1%1. Besides the core symptoms, a deficit in social communication and restricted and repetitive behaviors, ASD has a large group of clinical manifestations, such as language disability, intellectual disability (ID), seizure, abnormal head size, gastrointestinal (GI) problems and so on2. To date, de novo and rare gene-disruptive mutations and copy number variants (CNVs) have been found to contribute a significant proportion to the ASD genetic architecture3. However, because of the extremely rare variants of each gene, very large cohorts are needed to prove the significance of an individual gene. We are in the early stages of understanding genotype–phenotype relationships for most risk genes.
ASD genotype–phenotype correlations are important to identify phenotypic subtypes and for early diagnosis and clinical management. The genotype-to-phenotype approach has elucidated several subtypes of ASD, such as CHD84, DYRK1A5 and ADNP6. These studies provide evidence that patients with disruptive mutations at specific genes or CNVs are more likely to share distinctively similar phenotypes. For example, patients with truncating mutations in CHD8 most likely present with macrocephaly, GI disturbance and general overgrowth; patient with truncated mutations in DYRK1A, in contrast, show microcephaly, distinctive facial features and ID. Although the genotype-first approach has been successfully applied, it requires very large cohorts because of the extensive genetic heterogeneity7. Here, we investigate the alternate possibility of whether phenotype-specific genes can be identified from a small group of patients with specific phenotype(s).
Abnormal head size has long been recognized as a co-occurring condition of ASD. Leo Kanner originally described macrocephaly in ASD in 19431. It is estimated that about 20% of ASD patients have increased head size8. Mutations of several ASD genes have been reported causing macrocephaly, such as PTEN9 and CHD84. Meanwhile, microcephaly was also frequently noticed in ASD patients. For example, patients with DYRK1A5 and CDKL510 truncated mutations more likely present with microcephaly. Patients with deletion and duplication of 16p11.2 have opposing head sizes. The deletion is associated with increased head size, whereas the duplication is associated with decreased head size11. Moreover, patients with de novo mutations (DNMs) within genes involved in the β-catenin/Wnt-signaling pathway, which is important in transcription regulation, present reciprocal macrocephaly and microcephaly7.
To test the efficiency of the phenotype-to-genotype approach and identify genes associated with ASD patients and abnormal head size, we sequenced 92 ASD families with abnormal head circumference size. Clinically relevant pathogenic or likely pathogenic events were identified in 17.4% of probands, and most of them are already known to be associated with microcephaly or macrocephaly. Our study implicates new variants and genes for ASD and abnormal head size for future investigation.
Materials and Methods
Patients recruitment and ethical approval
We selected 67 ASD families (58 trios, 9 simplex quads, 210 individuals) from the Autism Clinical and Genetic Resources in China (ACGC) cohort11 for whole-exome sequencing (WES) (Figure 1). Out of 43 trios and three simplex quads, 46 probands have macrocephaly (> +2 standard deviations (SD)); and 21 probands, from 15 trios and six simplex quads, have microcephaly (< −2SD) (Figure 1b, supplementary table S1). All probands were diagnosed according to DSM-IV or DSM-V by experienced clinicians. In addition, co-occurring conditions including medical problems, such as epilepsy, gastrointestinal issues and sleep disorders; developmental diagnoses, such as intellectual disability and language delay; and mental-health conditions, such as attention deficit hyperactivity disorder (ADHD), obsessive-compulsive disorder and depression, were collected for presenting patients. Peripheral blood of all involved individuals was collected with informed consent. This study is in accordance with the ethical standards of the Institutional Review Board of the School of Life Sciences at Central South University, Changsha, Hunan, China.
Figure 1.

Schematic of phenotype-to-genotype strategy and the performance of whole-exome sequencing (WES). (a). Schematic of phenotype-to-genotype strategy on the identification of genotype-specific variants or genes in this study. P and LP represent the pathogenic and likely pathogenic variants according to ACMG guidelines. (b) Pie plot shows the number distribution for macrocephaly and microcephaly families involved in this study. (c) Histogram shows the mean depth distribution of WES. (d) Histogram shows the mean coverage distribution of WES.
Whole-exome sequencing and single-nucleotide variants (SNVs)/indels calling
Genomic DNA was extracted from the whole blood using a standard proteinase K digestion and the phenol-chloroform method. All samples were captured using SureSelect Human All Exon V6 Library (Agilent) reagents. The pools were then sequenced on the Illumina HiSeq X Ten platform using paired-end 150 bp reads. Clean FASTQ reads were mapped to the reference genome assembly (hg38) using BWA-MEM (v.0.6)12. PCR duplicates were marked using Picard (http://broadinstitute.github.io/picard/) MarkDuplicates (v.2.9.4). Bases were recalibrated using Genome Analysis Toolkit (GATK v.3.7–2) BaseRecalibrator13. Genotypes were then generated with GATK HaplotypeCaller (v.3.7–2) on a per-family basis.
De novo SNV/indel discovery and validation
Family relationships were assessed using the KING program14. De novo SNVs and indels were called using a custom de novo filtering pipeline as previously described15. In brief, the pipeline uses the family-level Variant Call Format (VCF) files; candidate sites were chosen where the parents genotypes are 0/0 and the children’s genotypes are either 0/1 or 1/1. The allele count, read depth, and allele balance were filtered as follows: parental alternate allele count is equal to zero, child allele balance is above 0.25, read depth for all family members is above 10, and child genotype quality was above 20. Merged VCF files were annotated using ANNOVAR16, which annotates different functions of the variants in extensively specific databases, such as population control databases (ExAC, gnomAD, 1000 Genomes Project etc.), functional prediction databases (SIFT, PolyPhen2, MutationTaster, etc.), dbSNP, etc. We applied Sanger sequencing to validate de novo SNVs and indels.
CNV discovery and validation
CNVs were called using XHMM algorithms according to the best-practice guidelines17. In brief, GATK was used to calculate depth of coverage (from BWA-MEM alignments) for each individual, and all individuals were then combined into one composite file. The XHMM-specific steps included hard filtering of samples and targets, PCA on the data, filtering based on the PCA results and discovery of CNVs. Post-discovery CNVs were genotyped by family, and a score cutoff of 10 was ultimately used to determine inheritance in families using SQ and NQ values. Quantitative PCR performed using the Roche LightCycler® 96 System (F. Hoffmann-La Roche AG, Basel, Switzerland), was used to validate the large de novo CNVs called by XHMM. Three pairs of primers were selected from the start, middle, and end of each CNV, separately. The sample was analyzed in triplicate in a 10μl reaction mixture. The values were evaluated using the LightCycler® 96 Application Software Version 1.1 (F. Hoffmann-La Roche AG, Basel, Switzerland). Further data analysis was performed using the qBase method.
Statistical analyses
To identify potential novel candidate genes, we leveraged DNM data from published whole-exome or whole-genome sequencing of 10,842 cases with NDDs, including 5,578 cases with a diagnosis of ASD and 5,264 cases with a diagnosis of ID/developmental delay (DD) collected from five studies18–22 in denovo-db (v.1.5)23. Genes with a significant excess of DNMs from this study and the above reported studies were identified using two statistical models. The first is a probabilistic model that incorporates the overall rate of mutation in coding sequences, estimates of relative locus-specific rates based on chimpanzee–human fixed differences derived from the chimpanzee–human divergence (CH) model24. The second, denovolyzeR, estimates mutation rates based on trinucleotide context and accommodates known mutational biases such as CpG hotspots (denovolyzeR model)25. Both models are well explored for DNM analysis in multiple studies. Default parameters were used for both models. An expected rate of 1.5 DNMs per exome was used for the CH model. P values were corrected for genome-wide multiple testing using the Bonferroni method (n = 18,946 in CH model; n = 19,618 in denovolyzeR).
Results
Variants discovery and diagnostic yields
We sequenced the protein-coding portion for all individuals with a mean depth of 90 ± 5.3x (Figure 1c). Over 97% of the CCDS region was sequenced over ten times (Figure 1d). All family relationships were confirmed based on variant transmission analysis. We detected 121 de novo SNVs and indels in exomes (supplementary table S2). We attempted to validate all rare (<0.1% in ExAC) de novo non-synonymous SNVs/indels (n = 68). We validated 62 rare SNVs/indels as de novo (five SNVs were validated as false positive, and one located in the segmental duplication region was not resolved by Sanger sequencing) (supplementary table S3). three false positive variants of the five have low alternative allele depth (<5). For the inherited variants, we identified 48 inherited rare likely gene-disruptive (LGD) variants (supplementary table S4). Considering the difficulties associated with exome-based CNV analysis26, we only considered and validated large CNVs with sizes greater than 1 Mbp. Four large CNVs were identified and successfully validated as de novo (supplementary figure S1).
To evaluate the WES diagnostic yield and evaluate the phenotypes corresponding to clinically relevant pathogenic or likely pathogenic variants, we classified the variants described above following the standards and guidelines for the interpretation of sequence variants and CNVs from the American College of Medical Genetics and Genomics (ACMG)27,28. In total, we classified 17 clinically relevant pathogenic or likely pathogenic events from 16 patients, including 13 SNVs/indels (7 de novo LGD, 5 de novo missense and 1 inherited LGD mutation) (Table 1) and 4 de novo CNVs (Table 2). Overall, clinically relevant pathogenic or likely pathogenic variants account for 23.9% patients (16/67). Pathogenic or likely pathogenic variants account for 42.9% patients with microcephaly (9/21, 42.9%) and 15.2% patients with macrocephaly (7/46, 15.2%) (Figure 2a, b). (Table 1, Figure 2b). The diagnostic yield (18%, 12/67) is 2-fold higher than the diagnostic yield (8.9%) among a recent larger unselected ASD cohorts when only considering the SNV/indels29.
Table 1.
Disorder-related SNVs/indels and head-size association.
| Chr | Start(hg38) | End(hg38) | Ref | Alt | Gene | Function1 | NT&AAchange2 | CADD | Sample ID | Inheritance | Ma/Mi3 | HCZ(SD)4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pathogenic variants | ||||||||||||
| 10 | 87961101 | 87961101 | T | - | PTEN | FS | NM_000314:c.1009delT:p.S338Lfs*6 | . | GX0427.p1 | de novo | Ma | +3.23 |
| 14 | 21399998 | 21399998 | T | - | CHD8 | FS | NM_001170629:c.4800delA:p.G1602Vfs*13 | . | HEN0083.p1 | de novo | Ma | +2.00 |
| 14 | 21408388 | 21408388 | T | - | CHD8 | FS | NM_001170629:c.2654delA:p.N885Tfs*14 | . | GX0540.p1 | paternal | Ma | +3.58 |
| 5 | 177191982 | 177191982 | T | A | NSD1 | SG | NM_022455:c.T1026A:p.C342* | 35 | HN0038.p1 | de novo | Ma | +2.38 |
| 2 | 144404880 | 144404880 | - | G | ZEB2 | FS | NM_014795:c.547_548insG:p.L183Rfs*16 | . | GX0430.p1 | de novo | Mi | −3.55 |
| 1 | 224433889 | 224433889 | T | - | WDR26 | FS | NM_025160:c.217delA:p.S73Afs*2 | . | GX0486.p1 | de novo | Mi | −3.92 |
| 20 | 32435608 | 32435611 | GGAG | - | ASXL1 | FS | NM_015338:c.2896_2899del:G966Afs*17 | . | GX0468.p1 | de novo | Mi | −2.54 |
| 19 | 8399883 | 8399883 | G | C | RAB11B | MIS | NM_004218:c.G61C:p.G21R | 35 | GX0152.p1 | de novo | Mi | −3.31 |
| 2 | 1892087 | 1892087 | C | A | MYT1L | SG | NM_001303052:c.G2233T:p.E745* | 45 | GX0391.p1 | de novo | - | −2.54 |
| Likely pathogenic variants | ||||||||||||
| 10 | 87960901 | 87960901 | T | A | PTEN | MIS | NM_000314:c.T809A:p.M270K | 27.8 | HEN0197.p1 | de novo | Ma | +2.31 |
| 15 | 75371990 | 75371990 | T | C | SIN3A | MIS | NM_001145358:c.A3811G:p.K1271E | 17.6 | GX0044.p1 | de novo | Mi | −2.77 |
| X | 53536461 | 53536461 | G | A | HUWE1 | MIS | NM_031407:c.C12344T:p.A4115V | 25.4 | HEN0056.p1 | de novo | Mi | −4.08 |
| 6 | 157181047 | 157181047 | T | C | ARID1B | MIS | NM_017519:c.T3175C:p.W1059R | 27.5 | HN0092.p1 | de novo | - | +2.46 |
| Candidate variants | ||||||||||||
| 7 | 139272328 | 139272328 | C | T | UBN2 | MIS | NM_173569:c.C1603T:p.R535C | 35 | HEN0155.p1 | de novo | - | +2.72 |
| 2 | 32547917 | 32547917 | C | T | BIRC6 | MIS | NM_016252:c.C12878T:p.T4293I | 33 | GX0203.p1 | de novo | - | +2.38 |
| 7 | 128979029 | 128979029 | C | G | TNPO3 | MIS | NM_012470:exon16:c.G2015C:p.C672S | 22.8 | GX0226.p1 | de novo | - | +2.69 |
| 6 | 152211563 | 152211563 | C | T | SYNE1 | MIS | NM_182961:c.G22520A:p.R7507H | 33 | HN0048.p1 | de novo | - | −2.92 |
| 10 | 76891619 | 76891619 | C | T | KCNMA1 | MIS | NM_001161352:c.G3248A:p.R1083K | 22.2 | GX0306.p1 | de novo | - | +2.23 |
Notes:
FS represents frameshift variant, SG represents stop-gain variant, MIS represents missense variants.
The canonical isoforms are listed.
Ma/Mi represent known macrocephaly (Ma) and microcephaly (Mi) association for the respective genes.
HCZ represents head circumference-for-age Z-scores.
Table 2.
Pathogenic CNVs identified in this cohort.
| Interval (hg38) | Size | CNV | Sample | Size | Region | GeneNum | ASD gene | Ma/Mi1 | HCZ (SD)2 |
|---|---|---|---|---|---|---|---|---|---|
| 17:156213–6120665 | 5.96M | DUP | GX0309.p1 | 5.96M | 17p13.2–13.3 | 131 | YWHAE | Micro | −2.31 |
| 11:132310051–134986787 | 2.68M | DEL | GX0309.p1 | 2.68M | 11q25 | 14 | NTM | - | −2.31 |
| 22:47625313–50799120 | 3.17M | DEL | HEN0131.p1 | 3.17M | 22q13.3 | 47 | SHANK3 | Macro | +2.54 |
| 1:733013–2789669 | 2.06M | DEL | GX0418.p1 | 2.06M | 1p36.3 | 75 | GNB1 | - | −2.85 |
Notes:
Ma represents macrocephaly, Mi represents microcephaly.
HCZ represents head circumference-for-age Z-scores.
Figure 2.

The relationship between putative disorder-related variants and head circumference. (a) Shown is the distribution of the head circumference standard deviation for the studied samples. The pie plots show the diagnosed yield (only considering pathogenic and likely pathogenic variants) of WES in microcephaly and macrocephaly families. (b) The dot plot shows the head circumference standard deviation of the corresponding clinically relevant and candidate variants. Red dots represent the LGD mutations or CNVs; blue dots represent the missense variants. P and LP represent the pathogenic and likely pathogenic variants according to ACMG guidelines.
Clinically relevant variants within known head-circumference-associated genes and corresponding phenotypes
Among the 17 clinically relevant pathogenic or likely pathogenic variants identified in microcephaly or macrocephaly patients, 13 of them are already known to be associated with abnormal head size, which accounts for 81.25% of this clinically resolved patient subset (Figure 2b). Five pathogenic or likely pathogenic variants within three macrocephaly-related genes (PTEN, CHD8, and NSD1) and one pathogenic macrocephaly-related CNV (22q13.3 deletion) were identified in six patients with macrocephaly (Table 1, Table 2). Pathogenic or likely pathogenic variants of PTEN (1 LGD DNM and 1 missense DNM) and CHD8 (1 LGD DNM and 1 inherited LGD mutation) were recurrently identified in two individuals with macrocephaly respectively, and both are well-defined macrocephaly-related genes. Importantly, for the inherited CHD8 LGD mutation (GX0540.p1, p.N885Tfs*14), the carrier father shows increased head size (1.53 SD). In addition, the carrier father also shows autistic traits with high Broad Autism Phenotype Questionnaire score across the domains of autism and NVIQ scores in the borderline range (NVIQ = 79) as we reported in our recently targeted sequencing paper30. NSD1 mutations cause Sotos syndrome, a syndromic ASD with macrocephaly as a recurrent phenotype. Besides ASD and macrocephaly, our patient with the NSD1 LGD DNM also presents with ID, language developmental delay, sleeping problems, and attention deficit hyperactivity disorder (ADHD) (supplementary table S5). Macrocephaly was recurrently reported in patients with Phelan-McDermid syndrome, which is caused by the 22q13.3 deletion. Besides ASD and macrocephaly, this patient also showed ID, motor and language development delay, sleeping problems, ADHD, and obsessive behavior, which is consistent with the phenotypes of Phelan-McDermid syndrome.
Six pathogenic or likely pathogenic variants in six microcephaly-related genes, including ZEB2, ASXL1, WDR26, SIN3A, RAB11B and HUWE1, and one pathogenic microcephaly-related CNV (17p13.2–13.3 duplication) were identified in seven patients with microcephaly (Table 1, Table 2). ZEB2 mutations cause microcephaly-related syndromic ASD (Mowat–Wilson syndrome). Besides autistic phenotypes and microcephaly (−3.55 SD), other phenotypes, including ID, motor and language development delay, abnormal MRI and EEG, sleeping problems, and GI problems, were also present in our patient (supplementary table S5). ASXL1 LGD mutations cause Bohring-Opitz syndrome, which is characterized by severe developmental delay and microcephaly. Loss-of-function mutations of SIN3A are associated with mild ID and often accompanied by ASD and microcephaly. Although the missense mutation we identified here is likely pathogenic, the phenotypes of our patient are consistent with the reported patients: ASD, ID, microcephaly (−2.77 SD), abnormal MRI, and ADHD. Dozens of missense mutations of HUWE1 have been reported in association with ASD and other NDDs. The missense DNM in our patients is located in the HECT domain, where clustered missense mutations have been observed31, and it is very close to the missense mutations co-segregating with the phenotype in multiple-generation families32. Interestingly, most of the patients (6/9) with DNMs in HECT regions present microcephaly31. Chromosome 17p13.3 duplication causes multiple complex NDD phenotypes, including mild to severe developmental phenotypes, ASD, and mild brain malformations including microcephaly2. Our patient presents with ASD, ID, language developmental delay, abnormal MRI, and ADHD. All these phenotypes were recurrently observed in reported patients with chromosome 17p13.3 duplications. Notably, in addition to the 17p13.3 duplication, our patient also carried a 2.7 Mbp pathogenic deletion leading to loss of NTM, an ASD candidate gene. However, no variation in head size was previously reported for this CNV.
Mutations of WDR26 and RAB11B were recently identified for broad NDD phenotypes33,34. Most patients with mutations within these two genes show microcephaly. In addition to ASD and microcephaly (−3.92 SD), our patient with the WDR26 LGD DNM also presents ID, seizure, language developmental delay, abnormal MRI, sleeping problems, and ADHD, which are similar to the patients reported here (supplementary table S5). The missense mutation within RAB11B identified in our patient is located in the reported pathogenic missense cluster within the nucleotide-binding domain, which is important for GTP binding (Figure 3). In addition to ASD and microcephaly (−3.31 SD), the patient also presents with ID, language developmental delay, abnormal EEG and MRI, and ADHD.
Figure 3.
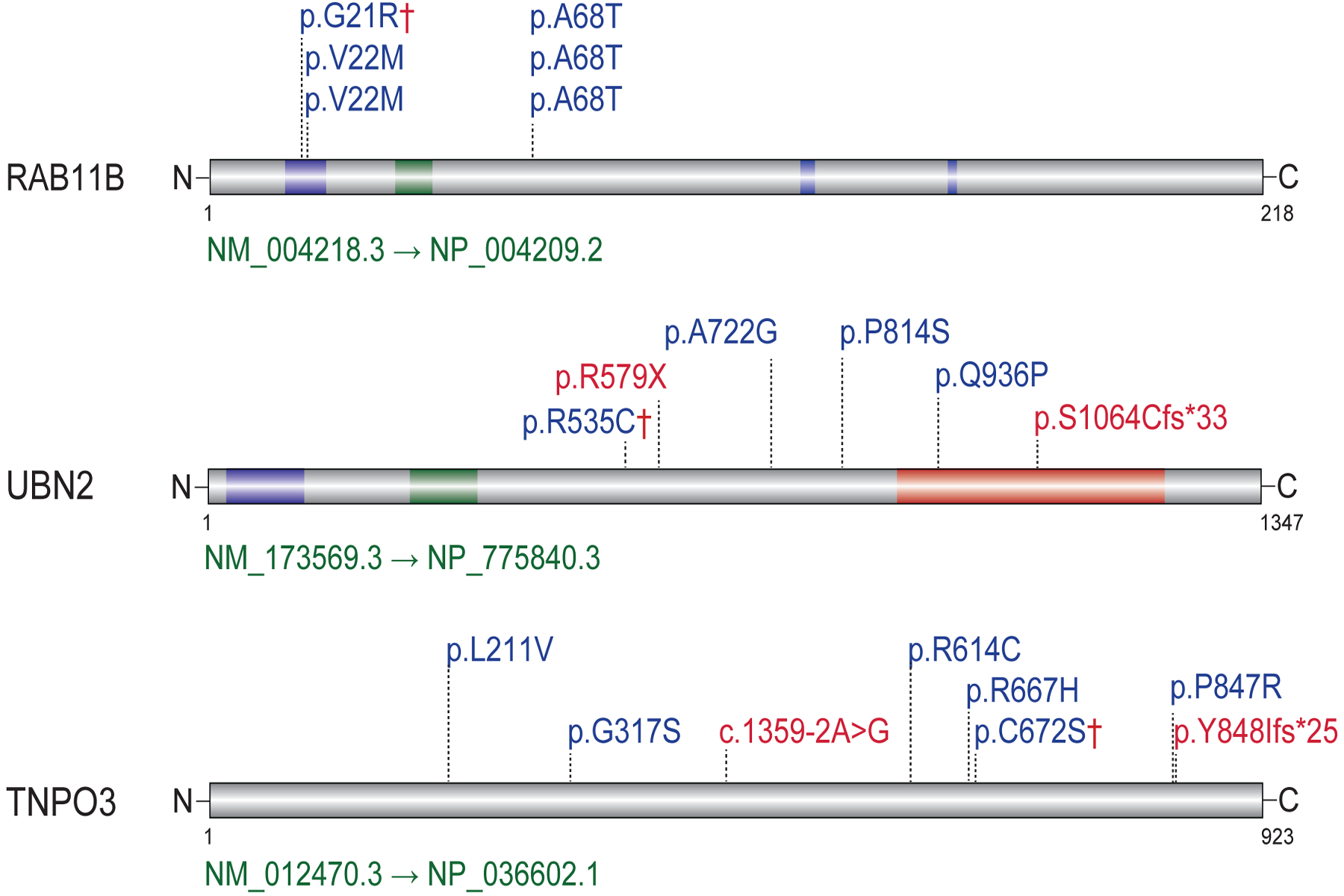
The Spectrum of de novo mutations (DNMs) within one known (RAB11B) and two candidate genes (UBN2, TNPO3). Diagrams of the canonical isoform with DNMs from this study indicated with a dagger (†) and from the published whole-exome or whole-genome sequencing studies of large NDD cohorts are displayed for each gene. Red indicates LGD, and blue indicates missense.
De novo variants in ASD candidate genes
In addition to clinically relevant pathogenic or likely pathogenic variants, four missense DNMs within four ASD candidate genes (SFARI: https://gene.sfari.org/), including UBN2 (p.R535C), BIRC6 (p.T4293I), KCNMA1 (p.R1083K) and SYNE1 (p.R7507H), were identified in three patients with macrocephaly and one patient with microcephaly (Table 1, Figure 2). We applied the Combined Annotation Dependent Depletion (CADD) score as well as other functional prediction tools to predict the damaging effect of these missense DNMs in gene function (supplementary table S2). The missense DNM within UBN2, previously associated with ASD19, has a CADD score of 35 and is predicted as damaging by both SIFT and Polyphen2 (supplementary table S2). Two LGD DNMs and three missense DNMs were reported in NDDs (Figure 3, supplementary table S6). Two LGD DNMs and one missense DNM are from ASD, and two missense DNMs are from DD. The number of DNMs among individuals with NDD is nominally significant (P = 4.7 × 10−5, CH model; P = 4.5 × 10−4, denovolyzeR model) although neither survives genome-wide multiple comparisons.
By leveraging DNMs identified from the published NDD cohorts, we identified a potential new candidate gene in ASD and related NDDs. We identified one missense DNM (p.C672S) in TNPO3 (CADD = 22) and seven DNMs (2 LGD, 5 missense) from previously published large-scale sequencing studies (Figure 3, supplementary table S6). We calculated the probability of detecting eight or more DNMs (including LGD and missense) in the combined NDD cohorts (n = 10842) and find an excess of TNPO3 DNMs in NDD patients (P = 5.9 × 10−8, Padj = 0.001, CH model; P = 1.3 × 10−6, Padj = 0.026, denovolyzeR model; Bonferroni correction).
Discussion
We have applied WES to an ASD cohort with abnormal head circumferences to investigate the efficiency of restricting phenotypes to increase gene discovery and to better resolve ASD genotype–phenotype correlations. Clinically relevant pathogenic and likely pathogenic variants account for 23.9% patients. Specifically, clinically relevant variants account for 19.4% patients with microcephaly or macrocephaly. When we consider the DNMs in candidate genes together, WES diagnostic yields arrive 22.4%. Of note, these genes of which 76.5% clinically relevant pathogenic or likely pathogenic variants are already known to be associated with macrocephaly or microcephaly. In addition, we could miss some potential clinically relevant variants with postzygotic de novo mutations since we only consider variants with AB above 0.25 which is not very sensitive for mosaic mutations. These data provide evidence that the phenotype-to-genotype approach is useful to detect variants or genes associated with specific phenotype.
We analyzed the detailed clinical information for patients with clinically relevant pathogenic and likely pathogenic mutations. Our study expanded the phenotype spectrum of several ASD and related NDD genes recently identified, including RAB11B and WDR26. RAB11B missense DNMs are also extremely rare among NDDs. Only five patients with RAB11B missense DNMs have been reported33, and four of them have microcephaly. The phenotype of our patient is consistent with those reported patients, including ID, absent speech, and abnormal MRI. Specifically, recurrent missense DNMs of RAB11A, an important homolog of RAB11B, were identified in patients with epilepsy and/or DD35. WDR26 was recently identified causing a recognizable NDD syndrome, and more than half of the patients showed autistic behaviors and share microcephaly34. WDR26 involves in Wnt-signaling and PI3K-AKT pathway and directly interacts with GNB1, a newly identified NDD gene36. The function of WDR26 as it relates to neurodevelopment remains to be investigated.
Our study also provides further genetic evidence for several ASD candidate risk genes, especially within the Chinese cohort, including UBN2, BIRC6, SYNE1 and KCNMA1. UBN2 was recently indicated in ASD risk19, but the function of this gene is completely unknown. Gene ontology annotations related this gene to DNA binding transcription factor activity. Dozens of BIRC6 missense DNMs have been reported in patients with NDDs (denovo-db), although de novo burden has not yet achieved statistical significance. Homozygous SYNE1 missense mutations have been reported to cause ASD while homozygous truncated mutations cause cerebellar ataxia37 and a recessive form of arthrogryposis multiplex congenital38. Although no other mutation within this gene was identified in the same patient and the pathogenesis of the missense DNM identified in this study is still uncertain, several SYNE1 missense DNMs have been reported in NDD patients (denovo-db)23. KCNMA1 encodes the α-subunit of the large conductance Ca2+-activated K+ channel (BK channels), which plays an essential role in neuronal excitability. Disruption of KCNMA1 was reported in a single ASD case39, and several missense DNMs have been reported (denovo-db). Recently, Liang et al. reported nine patients with de novo KCNMA1 missense variants and a broad spectrum of neurodevelopmental and neurological phenotypes including autistic features40.
In addition to the variants within known ASD candidate genes, our results also implicate a novel candidate gene, TNPO3. TNPO3 encodes a nuclear import receptor for serine/arginine-rich (SR) proteins. Unfortunately, the function of this gene in neurodevelopment has not been investigated. Further genetic study and functional assays are needed to investigate the role of this gene in ASD risk and neurodevelopment.
Supplementary Material
Acknowledgements
We thank Tonia Brown for assistance in editing this manuscript. We are grateful to all of the families at the participating this study. This work was supported by the following grants: the National Natural Science Foundation of China (81330027, 81525007, 81671122, 31400919, 31671114) to K.X., Z.H. and H.G.; the Natural Science Foundation of Hunan Province (2016RS2001, 2016JC2055, 2018SK1030, 2018DK2016) to K.X. and Z.H.; the U.S. National Institutes of Health (NIH) (R01MH10221) to E.E.E. H.G. was also supported by the China Hunan Provincial Science & Technology Department (2019RS2005). E.E.E. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc. All other authors declare no competing financial interests.
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
References
- 1.Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet (London, England). 2014;383(9920):896–910. [DOI] [PubMed] [Google Scholar]
- 2.Blazejewski SM, Bennison SA, Smith TH, Toyo-Oka K. Neurodevelopmental Genetic Diseases Associated With Microdeletions and Microduplications of Chromosome 17p13.3. Frontiers in genetics. 2018;9:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nature medicine. 2016;22(4):345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernier R, Golzio C, Xiong B, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158(2):263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Bon BW, Coe BP, Bernier R, et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol Psychiatry. 2016;21(1):126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helsmoortel C, Vulto-van Silfhout AT, Coe BP, et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nature genetics. 2014;46(4):380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stessman HA, Bernier R, Eichler EE. A genotype-first approach to defining the subtypes of a complex disease. Cell. 2014;156(5):872–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanner L Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- 9.Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nature genetics. 1997;16(1):64–67. [DOI] [PubMed] [Google Scholar]
- 10.Scala E, Ariani F, Mari F, et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. Journal of medical genetics. 2005;42(2):103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qureshi AY, Mueller S, Snyder AZ, et al. Opposing brain differences in 16p11.2 deletion and duplication carriers. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34(34):11199–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 2010;26(5):589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics (Oxford, England). 2010;26(22):2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turner TN, Coe BP, Dickel DE, et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell. 2017;171(3):710–722.e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fromer M, Moran JL, Chambert K, et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. American journal of human genetics. 2012;91(4):597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lelieveld SH, Reijnders MR, Pfundt R, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nature neuroscience. 2016;19(9):1194–1196. [DOI] [PubMed] [Google Scholar]
- 19.RK CY, Merico D, Bookman M, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nature neuroscience. 2017;20(4):602–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Rubeis S, He X, Goldberg AP, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deciphering Developmental Disorders S Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542(7642):433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iossifov I, O’Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner TN, Yi Q, Krumm N, et al. denovo-db: a compendium of human de novo variants. Nucleic acids research. 2017;45(D1):D804–d811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Roak BJ, Vives L, Fu W, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science (New York, NY). 2012;338(6114):1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samocha KE, Robinson EB, Sanders SJ, et al. A framework for the interpretation of de novo mutation in human disease. Nature genetics. 2014;46(9):944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Retterer K, Scuffins J, Schmidt D, et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(8):623–629. [DOI] [PubMed] [Google Scholar]
- 27.Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genetics in medicine : official journal of the American College of Medical Genetics. 2011;13(7):680–685. [DOI] [PubMed] [Google Scholar]
- 28.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo H, Duyzend MH, Coe BP, et al. Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Genetics in medicine : official journal of the American College of Medical Genetics. 2019;21(7):1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo H, Wang T, Wu H, et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Molecular autism. 2018;9:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moortgat S, Berland S, Aukrust I, et al. HUWE1 variants cause dominant X-linked intellectual disability: a clinical study of 21 patients. European journal of human genetics : EJHG. 2018;26(1):64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Froyen G, Corbett M, Vandewalle J, et al. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. American journal of human genetics. 2008;82(2):432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamers IJC, Reijnders MRF, Venselaar H, et al. Recurrent De Novo Mutations Disturbing the GTP/GDP Binding Pocket of RAB11B Cause Intellectual Disability and a Distinctive Brain Phenotype. American journal of human genetics. 2017;101(5):824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skraban CM, Wells CF, Markose P, et al. WDR26 Haploinsufficiency Causes a Recognizable Syndrome of Intellectual Disability, Seizures, Abnormal Gait, and Distinctive Facial Features. American journal of human genetics. 2017;101(1):139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamdan FF, Myers CT, Cossette P, et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. American journal of human genetics. 2017;101(5):664–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petrovski S, Kury S, Myers CT, et al. Germline De Novo Mutations in GNB1 Cause Severe Neurodevelopmental Disability, Hypotonia, and Seizures. American journal of human genetics. 2016;98(5):1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gros-Louis F, Dupre N, Dion P, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nature genetics. 2007;39(1):80–85. [DOI] [PubMed] [Google Scholar]
- 38.Attali R, Warwar N, Israel A, et al. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Human molecular genetics. 2009;18(18):3462–3469. [DOI] [PubMed] [Google Scholar]
- 39.Laumonnier F, Roger S, Guerin P, et al. Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. The American journal of psychiatry. 2006;163(9):1622–1629. [DOI] [PubMed] [Google Scholar]
- 40.Liang L, Li X, Moutton S, et al. De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Human molecular genetics. 2019;28(17):2937–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.