

Abstract
Despite the success of antiretroviral therapy in suppressing viral load, nearly half of the 37 million people infected with HIV experience cognitive and motor impairments, collectively classified as HIV-associated neurocognitive disorders (HAND). In the CNS, HIV-infected microglia release neurotoxic agents that act indirectly to elicit excitotoxic synaptic injury. HIV trans-activator of transcription (Tat) protein is one such neurotoxin that is thought to play a major role in the neuropathogenesis of HAND. The endocannabinoid (eCB) system provides on-demand neuroprotection against excitotoxicity, and exogenous cannabinoids attenuate neurotoxicity in animal models of HAND. Whether this neuroprotective system is altered in the presence of HIV is unknown. Here, we examined the effects of Tat on the eCB system in rat primary hippocampal cultures. Using whole-cell patch-clamp electrophysiology, we measured changes in retrograde eCB signaling following exposure to Tat. Treatment with Tat significantly reduced the magnitude of depolarization-induced suppression of excitation (DSE) in a graded manner over the course of 48 h. Interestingly, Tat did not alter this form of short-term synaptic plasticity at inhibitory terminals. The Tat-induced decrease in eCB signaling resulted from impaired CB1 receptor (CB1R)-mediated presynaptic inhibition of glutamate release. This novel loss-of-function was particularly dramatic for low-efficacy agonists such as the eCB 2-arachidonoylglycerol (2-AG) and Δ9-tetrahydrocannabinol (Δ9-THC), the main psychoactive ingredient in marijuana. Our observation that HIV Tat decreases CB1R function in vitro suggests that eCB-mediated neuroprotection may be reduced in vivo; this effect of Tat may contribute to synaptodendritic injury in HAND.
Keywords: CB1 receptor, DSE, endocannabinoid, EPSC, HIV Tat, THC
Significance Statement
Activation of the endocannabinoid (eCB) system protects against excitotoxicity. Whether this neuroprotection is altered in the presence of HIV is unknown. We show for the first time in an in vitro model of HIV neurotoxicity that an HIV protein selectively impairs eCB-mediated synaptic plasticity at excitatory but not inhibitory terminals. This selective effect of an HIV protein may unbalance synaptic networks, exacerbating the damage that underlies HIV-associated neurocognitive disorders (HAND). Thus, protecting or enhancing eCB signaling may attenuate the symptoms of HAND.
Introduction
Nearly half of all HIV-positive individuals experience some degree of neurological impairment (Heaton et al., 1995; Tozzi et al., 2005; Bateup et al., 2013; Saylor et al., 2016). While antiretroviral therapy effectively suppresses viral replication, the prevalence of HIV-associated neurocognitive disorders (HAND) remains high and continues to be a significant public health burden. HAND symptoms range from subclinical cognitive impairment to debilitating dementia (Antinori et al., 2007), and their severity correlates with loss of synaptic markers (Ellis et al., 2007). Within the CNS, the hippocampus, prefrontal cortex, and striatum are particularly vulnerable to damage caused by HIV (Masliah et al., 1992; Wiley et al., 1998; Moore et al., 2006). Currently, there are no effective therapeutics to combat the neurological deficits seen in HAND patients.
HIV indirectly affects neurons, where infected microglia and macrophage release viral proteins, inflammatory cytokines, and excitotoxins resulting in synaptodendritic damage and altered network function (Ellis et al., 2007; Green et al., 2019). The HIV protein trans-activator of transcription (Tat) is a potent neurotoxin that evokes the loss of excitatory synapses (Kim et al., 2008; Shin and Thayer, 2013), an increase in the number of inhibitory synapses (Hargus and Thayer, 2013), and ultimately neuronal death (Eugenin et al., 2007). HIV Tat is present in the cerebrospinal fluid (Johnson et al., 2013), brain tissue (Hudson et al., 2000), and sera of HIV-infected individuals at concentrations ranging from 2 to 40 ng/ml (Xiao et al., 2000). Once viral DNA integrates into the host genome, Tat is continuously expressed, even in the presence of combined antiretroviral therapy (Johnson et al., 2013). The titer of antibodies against Tat negatively correlates with HAND symptoms (Bachani et al., 2013), suggesting that Tat accumulation is linked to cognitive deficits.
The endocannabinoid (eCB) system regulates many physiological processes of relevance to HAND including mood, anxiety, appetite, neuroinflammation, motor control, and neuroprotection (Rodríguez de Fonseca et al., 2005). The eCB system protects against excitotoxicity and attenuates epileptiform activity (Shen et al., 1996; Marsicano and Lutz, 1999; Nagayama et al., 1999). Notably, several studies using models of HAND have shown that pharmacological interventions targeted at the eCB system protect against HIV-mediated synaptodendritic damage. Cannabinoid type 2 receptor (CB2R) agonists attenuate neuroinflammation in a murine model of neuroAIDS (Gorantla et al., 2010) and protect human dopaminergic neurons from toxicity elicited by an HIV envelope protein (Hu et al., 2013). Application of the eCBs anandamide and 2-arachidonoylglycerol (2-AG) reduced HIV-induced increases in [Ca2+]i through their actions on cannabinoid receptors in vitro (Xu et al., 2017). Similarly, pharmacological administration of exogenous cannabinoids or inhibitors of eCB metabolism attenuated neuronal damage elicited by HIV proteins (Kim et al., 2011a; Hermes et al., 2018; Zhang and Thayer, 2018). Thus, targeting the eCB system in HAND has therapeutic potential; it is less clear how the presence of HIV in the CNS affects eCB signaling.
Excitotoxic stimuli alter the eCB system. For example, febrile seizures elevate levels of the proinflammatory cytokine interleukin 1β (IL-1β), producing a long-lasting upregulation of eCB-mediated inhibition of GABAergic transmission, resulting in a subsequent increased susceptibility to seizure (Chen et al., 2003; Feng et al., 2016). Interestingly, eCB modulation of glutamatergic transmission was not affected in this model, setting a precedent for differential modulation of eCB signaling at inhibitory versus excitatory synapses under neuroinflammatory conditions. In transgenic mice expressing HIV Tat, CB1Rs are upregulated (Jacobs et al., 2019); whether this increase preferentially affects GABAergic neurons is unclear.
To address this question, we examined the effects of HIV Tat on eCB-mediated retrograde signaling. Treating rat primary hippocampal cultures with Tat reduced the magnitude of depolarization-induced suppression of excitation (DSE) over the course of 48 h. Tat impaired CB1R-mediated presynaptic inhibition of glutamate release, but not GABA release. These results suggest that exposure to the HIV protein Tat may reduce neuroprotection mediated by the eCB system and alter the sensitivity of excitatory synaptic networks to cannabinoids.
Materials and Methods
Materials
Materials were obtained from the following sources: 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), (2R)-amino-5-phosphonovaleric acid (APV), bicuculline methochloride, Win55212-2 (Win-2), and SR141716A were obtained from Tocris Biosciences; 2-AG and JZL184 were obtained from Cayman Chemical; DMEM, fetal bovine serum, and horse serum were obtained from Invitrogen; (S)−3,5-dihydroxyphenylglycine (DHPG) and all other chemicals were obtained from Sigma-Aldrich. A plasmid encoding the tandem C1 domains (C1ab) of PKD fused to GFP (pCMV-PKDC1ab-GFP) was kindly gifted by Tamas Balla (Kim et al., 2011b), National Institute of Child Health and Human Development. HIV-1 Tat1-86 (Clade B, recombinant) was obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases from John Brady. Δ9-tetrahydrocannabinol (Δ9-THC) was obtained from the National Institute on Drug Abuse Drug Supply Program (Research Triangle Institute).
Cell culture
The hippocampus is particularly vulnerable to the neurotoxic effects of HIV (Ellis et al., 2007); thus, we chose rat hippocampal neurons grown in primary culture for this study. Cultures were prepared as previously described (Waataja et al., 2008). Briefly, maternal Sprague Dawley rats were euthanized by CO2 inhalation under a protocol approved by the University of Minnesota Institutional Animal Care and Use Committee in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Male and female fetuses were removed on embryonic day 17, and hippocampi were dissected and placed in Ca2+-free and Mg2+-free HEPES-buffered HBSS. Cells were dissociated by manual trituration using flame-narrowed Pasteur pipettes of decreasing aperture and resuspended in DMEM without glutamine supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 units/ml and 100 μg/ml, respectively). Dissociated cells were then plated at a density of 60,000–80,000 cells per dish on either a 35-mm Petri dish with a 10-mm cover glass bottom (MatTek) or a 25-mm-round cover glass (#1) precoated with Matrigel (150 μl, 0.2 mg/ml). Neurons were grown in a humidified atmosphere of 10% CO2 and 90% air held at 37°C in an incubator. Cells were fed on days 1 and 7 by exchanging 75% of the medium with DMEM supplemented with 10% horse serum and penicillin/streptomycin. Cultures used in this study contained a mixture of 18 ± 2% neurons, 70 ± 3% astrocytes, and 9 ± 3% microglia (Kim et al., 2011a). Cells were grown for 12–13 d in vitro (DIV).
Electrophysiology
Synaptic currents were recorded using the whole-cell configuration of the patch-clamp technique. Pipettes were pulled using a horizontal micropipette puller (P-87, Sutter Instruments) from glass capillaries with an outer diameter of 1 mm (Sutter Instrument) and pipette resistances of 3–5 MΩ. Membrane potential was held at −70 mV, and monosynaptic EPSCs/IPSCs were evoked with a bipolar platinum electrode (FHC) placed near a presynaptic neuron. DSE was evoked by a 15-s depolarization to 0 mV, which was previously shown to produce 50% DSE (Roloff et al., 2010). Each cell was exposed to a single stimulus. For DSE recordings pipettes were filled with the following intracellular solution: 120 mm potassium gluconate, 15 mm KCl, 6 mm MgCl2, 0.2 mm EGTA, 10 mm HEPES, and 5 mm Na2ATP, pH 7.3 with KOH, 290 mOsm/kg. Metabotropic suppression of excitation (MSE) was evoked by bath application of the selective group 1 metabotropic glutamate receptor (mGluR) agonist DHPG (1 μm). For MSE recordings, pipettes were filled with the following: 113 mm potassium gluconate, 15 mm KCl, 6 mm MgCl2, 10 mm BAPTA tetrapotassium, 10 mm HEPES, 5 mm Na2ATP, and 6 mm CaCl2, pH 7.2 with KOH, 290 mOsm/kg. Depolarization-induced suppression of inhibition (DSI) was evoked by a 2-, 5-, or 15-s depolarization to 0 mV (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001; Isokawa and Alger, 2005). Most cells were exposed to a single stimulus. Some cells from both control and Tat-treated groups were exposed to up to three different stimulus strengths (2, 5, 15 s); there was a 6-min break between administration of serial depolarizing stimuli. For DSI recordings pipettes were filled with the following: 140 mm KCl, 0.2 mm EGTA, 10 mm HEPES, 10 mm glucose, 5 mm MgATP, and 0.3 mm Na2GTP, pH 7.2 with KOH, 290 mOsm/kg. All recordings were performed at room temperature in an extracellular solution composed of the following: 140 mm NaCl, 5 mm KCl, 9 mm CaCl2, 6 mm MgCl2, 5 mm glucose, and 10 mm HEPES, pH 7.4 with NaOH, 325 mOsm/kg. For EPSC recordings, 10 μm bicuculline methochloride was added to the extracellular recording solution; for IPSC recordings, 10 μm CNQX and 50 μm APV were added. Solutions were applied by a gravity-fed superfusion system. Whole-cell currents were amplified with an AxoPatch 200B (Molecular Devices), low-pass filtered at 2 kHz, and digitized at 10 kHz with a Digidata 1322A (Molecular Devices) digitizer and pClamp v.9.2 software (Molecular Devices). Voltage pulses (0.1 ms) were applied at a fixed rate of 0.5 Hz for all experiments unless otherwise specified using a Grass S44 stimulator with a SIU-5 stimulus isolation unit (Grass Instruments).
Transfection
Rat hippocampal neurons were transfected on DIV10 or DIV11 with plasmid DNA using the calcium phosphate procedure described by Hargus and Thayer (2013). Briefly, hippocampal neurons were incubated for 30 min in DMEM supplemented with 1 mm kynurenic acid, 10 mm MgCl2, and 5 mm HEPES to reduce neurotoxicity. A DNA/calcium phosphate precipitate containing 1 μg of total plasmid DNA per well was prepared, allowed to form for 60 min at room temperature, and added to the culture. After a 60-min incubation period, cells were washed once with DMEM supplemented with MgCl2 and HEPES, then returned to conditioned media that had been saved at the beginning of the procedures.
Confocal microscopy and image processing
Cultures grown in MatTek glass-bottom Petri dishes were transferred to the motorized stage of a Nikon A1 laser-scanning confocal microscope 48 h after transfection with pCMV-PKDC1ab-GFP and viewed through a 60× oil-immersion objective. The objective was focused to the middle of the soma in the z-dimension and held constant using the autofocus feature of the microscope. PKDC1ab-GFP was excited at 488 nm, and emission was collected at 500–550 nm. After recording (1 Hz) baseline images for 60 s, DHPG (1 μm) or vehicle was added directly to the media bathing the cell.
Experimental design and statistical analysis
All data are presented as mean ± SEM. Electrophysiology data were analyzed using pCLAMP v.9.2. Live-cell imaging data were analyzed using NIS Elements and ImageJ. To minimize the effect of variables except the independent variable, experimental controls were run in parallel in all experiments. Only experiments with proper vehicle and/or drug controls were included in statistical analyses. For all experiments, an individual sample (n = 1) is defined as a single cell from a single coverslip/Petri dish. To account for inherent variability across primary cultures from week to week, each experiment was replicated over at least three separate cultures with a sample size of at least six cells/group. Because primary neuronal cultures are derived from a mixture of male and female rat embryos, the culture preparation protocol ensures an unbiased evaluation across both sexes. Hypothesis testing was performed with Prism 8 (GraphPad Software). Data were tested for normality using Bartlett’s test. Experiments with two groups and one factor were analyzed using an unpaired two-tailed Student’s t test. Samples with unequal variance were analyzed with Welch’s two-tailed t test. Experiments with 3+ groups and one factor were analyzed using a one-way ANOVA followed by Tukey’s post hoc test. Experiments with 2+ groups and two factors were analyzed using a two-way ANOVA. Statistical significance was defined as p < 0.05. OriginLab 2019 was used for curve-fitting decay rate and concentration-response data. Unless derived from curve fitting, confidence intervals (CIs) were determined using bootstrap resampling by uploading the same raw data used for hypothesis testing to https://www.estimationstats.com. The results of statistical analyses are presented in the figure legends and Table 1.
Table 1.
Statistics table
| Figure | Group comparison | Datastructure | Type of test | p value | Effectsize | 95% CI (lower toupper bound) | |
|---|---|---|---|---|---|---|---|
| a | 1F | Treatment | Normal | One-way ANOVA | <0.0001 | ||
| Control vs Tat (4 h) | Tukey’s post hoc | 0.5864* | –11.8% DSE | –22.5 to 0.13 | |||
| Control vs Tat (24 h) | Tukey’s post hoc | 0.0002* | –41.8% DSE | –55.6 to –27.4 | |||
| Control vs Tat (48 h) | Tukey’s post hoc | <0.0001* | –46.8% DSE | –60.7 to –32.8 | |||
| Control vs hi-Tat (24 h) | Tukey’s post hoc | 0.9759* | 4.6% DSE | –9.06 to 19.6 | |||
| hi-Tat (24 h) vs Tat (24 h) | Tukey’s post hoc | <0.0001* | –46.5% DSE | –63.2 to –30.2 | |||
| hi-Tat (24 h) vs Tat (48 h) | Tukey’s post hoc | <0.001* | –51.5% DSE | –68.7 to –36.6 | |||
| b | EPSC: control vs Tat (24 h) | Normal | Unpaired t test (two-tailed) | 0.3641 | –35.6 pA | –124.1 to 21.3 | |
| IPSC: control vs Tat (24 h) | Normal# | Welch’s t test (two-tailed) | 0.3406 | –97.5 pA | –329.3 to 68.3 | ||
| c | 2D | Depolarization duration × Tat treatment | Normal | Two-way ANOVA: interaction | 0.9182 | ||
| Depolarization duration | Factor | 0.0007 | |||||
| Tat treatment | Factor | 0.9365 | |||||
| d | 3C | Control vs Tat (24 h) | Normal | Unpaired t test (two-tailed) | 0.0001 | –24.5% MSE | –31.2 to –18.5 |
| e | 4C | Tau: control vs Tat (24 h) | Normal | Unpaired t test (two-tailed) | 0.7611 | –0.4361 s | –3.554 to 2.680§ |
| Peak: control vs Tat (24 h) | Normal | Unpaired t test (two-tailed) | 0.8827 | –0.0411 | –0.646 to 0.564§ | ||
| f | 5C | 2-AG EC50: control vs Tat (24 h) | Normal# | Welch’s t test (two-tailed) | <0.0001 | 1.22 µM | 0.716 to 1.724§ |
| 5D | JZL184 treatment × Tat treatment | Normal | Two-way ANOVA: interaction | 0.8380 | |||
| JZL184 treatment | Factor | 0.3572 | |||||
| Tat treatment | Factor | <0.0001 | |||||
| g | 6D | Win-2 EC50: control vs Tat (24 h) | Normal | Unpaired t test (two-tailed) | 0.93 | –0.16 nM | –3.81 to 4.13§ |
| h | 7D | [THC] × Tat treatment | Normal | Two-way ANOVA: interaction | 0.9165 | ||
| Tat treatment | Factor | <0.0001 | |||||
| [THC] | Factor | 0.0111 | |||||
| i | RAP (24 h) × Tat (24 h) | Normal | Two-way ANOVA: interaction | 0.6978 | |||
| RAP (24 h) treatment | Factor | 0.9528 | |||||
| Tat (24 h) treatment | Factor | <0.0001 | |||||
| j | 8B | CPA: control vs Tat (24 h) | Normal | Unpaired t test (two-tailed) | 0.6862 | 2.65% | –9.05 to 13.9 |
| 8C | CAP: control vs Tat (24 h) | Normal | Unpaired t test (two-tailed) | 0.8557 | –1.83% | –21.9 to 14.7 |
Effect size is calculated as the mean difference.
p values have been corrected for multiple comparisons.
unequal variance.
§CI derived from error associated with curve fit.
Results
HIV Tat inhibits DSE in a time-dependent manner
Depolarization of a postsynaptic hippocampal neuron evokes Ca2+ influx through voltage-gated Ca2+ channels that induces production of the eCB 2-AG (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). 2-AG then traverses the synaptic cleft in a retrograde manner to activate presynaptic CB1Rs, resulting in inhibition of glutamate transmission (Straiker and Mackie, 2005). We studied this process in primary rat hippocampal cultures following exposure to the HIV Tat1-86 (Clade B) protein. We used the whole-cell configuration of the patch-clamp technique to voltage-clamp a hippocampal neuron at −70 mV. ESPCs were evoked by a concentric bipolar stimulating electrode positioned near a presynaptic neuron. After recording a stable baseline, DSE was evoked by depolarizing the postsynaptic cell to 0 mV for 15 s (Fig. 1A; Roloff et al., 2010). Immediately following depolarization, the EPSC amplitude was inhibited by 53 ± 5% (Fig. 1A,F). DSE was completely blocked by pretreatment with the CB1R inverse agonist SR141716A (100 nm, n = 4). To determine whether HIV Tat affects eCB signaling, we assessed DSE in Tat-treated cultures (50 ng/ml). Treatment with Tat for 4 h did not significantly affect DSE (Fig. 1B,F). DSE was significantly reduced following 24 (11 ± 6%; Fig. 1C,F) and 48-h Tat treatment (6 ± 6%; Fig. 1D,F; Table 1a). EPSC amplitude was not affected by 24-h Tat treatment (control = −147 ± 20 pA, Tat = −182 ± 31 pA; t(24) = 0.925, p = 0.36; Table 1b). To confirm that the effect of Tat required structurally intact protein, we treated cells with heat-inactivated Tat (hi-Tat; Fig. 1E) for 24 h and assessed DSE. Exposure to hi-Tat did not affect DSE, which was comparable to control (57 ± 7%; Fig. 1E,F). We conducted a subsequent analysis to determine effect size and precision. Tat treatment for 24 h was estimated to decrease DSE relative to control by 42%. Using bootstrap resampling, we calculated the 95% CI for this effect as 27–56% (Table 1a).
Figure 1.

HIV Tat inhibits DSE in a time-dependent manner. A–E, EPSCs were evoked at 0.5 Hz by stimulation of a presynaptic neuron with an extracellular electrode and recorded from a postsynaptic cell voltage-clamped at −70 mV. DSE was evoked by depolarizing the postsynaptic cell to 0 mV for 15 s at the time indicated by the arrow. Plots show mean EPSC amplitudes as a percentage of baseline (15 responses before depolarization) over time. Cultures were untreated (A, control) or treated with 50 ng/ml Tat for (B) 4, (C) 24, or (D) 48 h. E, Treatment with heat-inactivated Tat (hi-Tat, 90°C for 30 min) for 24 h did not affect DSE. Insets show representative EPSC trace. Black: baseline, average of 15 responses before depolarization. Red: average of two responses after depolarization. Scale bars: 10 ms (horizontal bar) and 50 pA (vertical bar). F, Summary bar graph shows mean magnitude of DSE for control (open bar) and Tat-treated groups (gray bars). Percentage DSE was calculated according to the equation: DSE (%) = 100*(ESPCbaseline – EPSCDSE)/EPSCbaseline; EPSCbaseline: average amplitude of 15 responses immediately before depolarization; EPSCDSE: average amplitude of two responses immediately after depolarization. One-way ANOVA (F(4,25) = 17.9, p < 0.0001; n = 6 cells/group) was followed with Tukey’s post hoc test (***p < 0.001, ****p < 0.0001 relative to control; ####p < 0.0001 relative to hi-Tat). Data are represented as mean ± SEM.
Tat does not affect DSI
Because CB1Rs are also present on inhibitory terminals (Katona et al., 1999), depolarization-induced 2-AG production also inhibits GABAergic transmission (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). To test whether Tat (50 ng/ml, 24 h) similarly affects DSI, we recorded evoked IPSCs, then depolarized the postsynaptic neuron to 0 mV. A 15-s depolarization produced 87 ± 6% inhibition of IPSC amplitude, consistent with the high density of CB1Rs on a subset of hippocampal interneurons (Katona et al., 1999). In cultures treated with Tat, DSI was unaffected (90 ± 5%; Fig. 2A,D). To confirm that we had not saturated DSI, we reduced the stimulus strength by shortening the duration of depolarization (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001; Isokawa and Alger, 2005). Untreated cultures depolarized to 0 mV for 5 s exhibited 75 ± 7% inhibition; Tat treatment did not alter DSI evoked by a 5-s depolarization (72 ± 9%; Fig. 2B,D). A 2-s depolarization in untreated cells induced 57 ± 8% DSI, and Tat did not alter DSI evoked by a 2-s depolarization (58 ± 7%; Fig. 2C,D; Table 1c). IPSC amplitude was not affected by 24-h Tat treatment (control = −541 ± 42 pA, Tat = −638 ± 91 pA; t(25) = 0.97, p = 0.34; Table 1b). Since not all inhibitory synapses contain CB1Rs, not all cells exhibit DSI; we found 29% of control cells responded to depolarization by exhibiting DSI (18 out of 62 cells). All synaptic pairs that failed to exhibit DSI were also insensitive to 1 μm Win-2, a potent and efficacious CB1R agonist, confirming a lack of presynaptic CB1Rs. Tat did not alter the percentage of cells that exhibited DSI, as Tat-treated cultures yielded responses in 32% of the cells (10 out of 31 cells). Overall, Tat does not alter DSI, in contrast to its marked inhibition of DSE, indicating a specificity for eCB signaling at excitatory but not inhibitory synapses.
Figure 2.

HIV Tat does not affect DSI. A–C, IPSCs were recorded from a neuron held at −70 mV and evoked at 0.5 Hz by stimulation of a presynaptic neuron with an extracellular electrode. Cultures were either untreated (control) or treated with 50 ng/ml Tat for 24 h. The postsynaptic cell was depolarized to 0 mV for (A) 15, (B) 5, and (C) 2 s at the time indicated by the arrow. Plots show mean IPSC amplitudes as a percentage of baseline (15 responses before depolarization) over time. Insets show representative IPSC trace. Black: baseline, average of 15 responses before depolarization. Red: average of two responses after depolarization. Scale bars: 25 ms (horizontal bar) and 100 pA (vertical bar). D, Summary bar graph shows the magnitude of DSI for control (open bars) and 24-h Tat-treated groups (gray bars). Percentage DSI was calculated according to the equation: DSI (%) = 100*(ISPCbaseline – IPSCDSI)/IPSCbaseline; IPSCbaseline: average amplitude of 15 responses immediately before depolarization; IPSCDSI: average amplitude of two responses immediately after depolarization. A two-way ANOVA found no interaction (depolarization duration × Tat treatment; F(2,30) = 0.086, p = 0.92, n = 6 cells/group). Depolarization duration had a significant effect on DSI (F(2,30) = 9.4, p = 0.0007); n.s. = no significant difference. Data are represented as mean ± SEM.
Tat inhibits MSE
We next determined whether sensitivity to Tat was dependent on the stimulus used to evoke 2-AG synthesis. MSE is evoked by activating postsynaptic Gq-coupled receptors (Straiker and Mackie, 2007). Activation of group 1 mGluRs stimulates phospholipase Cβ (PLCβ) to produce diacyglycerol (DAG), the rate-limiting precursor for 2-AG synthesis (Murataeva et al., 2014). Bath application of the selective group 1 mGluR agonist DHPG (1 μm) inhibited EPSCs by 53 ± 2% (Fig. 3A). Tat treatment (50 ng/ml, 24 h) significantly reduced MSE to 28 ± 3% (Fig. 3B,C). We conducted a subsequent analysis to determine effect size and precision. Tat treatment for 24 h was estimated to decrease MSE relative to control by 25%. Using bootstrap resampling, we calculated the 95% CI for this effect as 19% to 31% (Table 1d). Thus, Tat impairs two forms of short-term synaptic plasticity mediated by the eCB system, both DSE and MSE.
Figure 3.

Exposure to HIV Tat inhibits MSE. A, B, EPSCs were evoked by 0.5-Hz stimulation of a presynaptic neuron with an extracellular electrode and recorded from a postsynaptic cell voltage-clamped at −70 mV. MSE was evoked by bath application of the selective group 1 mGluR agonist DHPG (1 μm). Plots show mean EPSC amplitudes as a percentage of baseline over time for (A) control and (B) 24-h Tat treatment groups. Insets show representative EPSC trace. Black: baseline, average of 30 responses before DHPG superfusion. Red: average of last 30 responses of DHPG superfusion. Scale bars: 10 ms (horizontal bar) and 50 pA (vertical bar). C, Summary bar graph shows mean magnitude of MSE for control (open bar) and Tat-treated group (gray bar). Tat (50 ng/ml, 24 h) significantly reduced MSE (t(12) = 6.577, ***p < 0.0001; t test, n = 6–8 cells/group). Data are represented as mean ± SEM.
Tat does not alter diacylglycerol (DAG) synthesis
In a previous study, Tat was shown to inhibit PLC (Vitale et al., 2013), the enzyme that produces DAG; DAG is hydrolyzed to produce 2-AG, the eCB that mediates DSE and MSE. To determine whether Tat impairs production of DAG, cells were transfected with an expression vector (pCMV-PKDC1ab-GFP) for a fluorescent DAG bioprobe. Since DAG is a precursor for 2-AG, we used DAG as a proxy to assess changes in 2-AG synthesis. DAG production was stimulated by application of the selective group 1 mGluR agonist DHPG (1 μm). Upon stimulation, the DAG biosensor translocates from the cytosol to the membrane, where it binds DAG (Fig. 4A,B). Tat (50 ng/ml, 24 h) did not significantly alter the translocation kinetics of the DAG biosensor, as indicated by no significant change in the peak membrane-to-cytosol fluorescence intensity ratio (control = 3.9 ± 0.23, Tat = 3.8 ± 0.15) or decay time constant (τ; control = 32 ± 0.6 s, Tat = 32 ± 1.3 s; Fig. 4A,C; Table 1e). While this approach does not directly assess 2-AG production, these data do indicate that the production of DAG, the precursor for 2-AG synthesis, is not altered by Tat.
Figure 4.

HIV Tat does not alter DAG synthesis. Cells expressing a fluorescent DAG bioprobe (pCMV-pKDC1ab-GFP) were stimulated by bath application of DHPG (1 μm). A, Representative images of untreated cells (control, gray) and cells treated with 50 ng/ml Tat for 24 h (red). Scale bars = 5 μm. B, Plot shows intensity values for regions of interest in the cytosol (gray) and membrane (black) monitored over time. C, Ratio of membrane-to-cytosol fluorescence intensity plotted over time. Exponential functions were fitted to plots of peak DHPG response using a nonlinear, least-squares curve-fitting algorithm. The curve was fit by a logistic equation of the form fluorescence intensity ratio = yₒ + Ae-(x-xₒ)/τ. Tat did not significantly alter the translocation kinetics of the DAG biosensor, as indicated by no significant change in the peak response (control = 3.9 ± 0.23, Tat = 3.8 ± 0.15; t(10) = 0.15, p = 0.88, t test; n = 6 cells/group) or decay time constant (τ; t(10) = 0.31, p = 0.76; t test). Data are represented as mean ratio ± SEM.
Tat impairs CB1R function
Because the effects of Tat on eCB signaling were independent of postsynaptic stimulus and Tat did not appear to affect 2-AG synthesis, we next examined presynaptic components of the eCB system. We assessed changes in presynaptic CB1R function by measuring EPSC amplitude in the presence of various CB1R agonists. EPSCs were evoked at 0.5 Hz by stimulating the presynaptic neuron with an extracellular electrode. Exogenous 2-AG was bath applied at varying concentrations and found to inhibit EPSCs in a concentration-dependent manner (Fig. 5A,C). Following 24 h treatment with 50 ng/ml Tat, the concentration-response relationship for 2-AG-mediated inhibition of EPSCs was right-shifted (Fig. 5B,C), and the EC50 was significantly increased from 0.39 ± 0.07 μm (control) to 1.6 ± 0.24 μm, indicating reduced potency of 2-AG (Table 1f). The maximal inhibition produced by 2-AG trended lower in Tat-treated cells, as indicated by a shift in the asymptote of the sigmoidal curve fit from 87% in control to 70% after Tat treatment. We examined whether this Tat-induced shift in the 2-AG concentration-response relationship was the result of an increase in 2-AG metabolism by monoacylglycerol lipase (MGL) by treating the culture with the selective, irreversible MGL inhibitor JZL184 (1 μm; Long et al., 2009) for 24 h. We first confirmed that JZL184 effectively inhibited MGL. JZL184 treatment prolonged the duration of DSE, as indicated by a markedly slowed recovery from depolarization-induced inhibition (n = 2). However, pretreating control or Tat-treated (50 ng/ml, 24 h) cultures with JZL184 did not affect 2-AG-mediated inhibition of EPSCs (Fig. 5D; Table 1f). These data suggest that breakdown of bath-applied 2-AG by MGL was not enhanced by Tat.
Figure 5.

Exposure to HIV Tat reduces the potency of 2-AG-induced inhibition of EPSCs. A, B, Representative traces show EPSCs during baseline and in the presence of varying [2-AG] for (A) control and (B) Tat-treated groups (50 ng/ml, 24 h). Scale bars: 10 ms (horizontal bar) and 50 pA (vertical bar). C, The concentration-response curve for 2-AG-mediated inhibition of EPSCs shows a rightward shift following Tat treatment. The curves were fit with a logistic equation of the form percentage inhibition = A1 + [(A2 – A1)/(1 + 10(logxₒ-x)p)], where xₒ = EC50, × = log[2-AG], A1 = % inhibition in the absence of drug, A2 = % inhibition at a maximally effective drug concentration, and p = slope factor. The following values were calculated using a nonlinear, least-squares curve fitting program: A1 = 13.1 for control, 10.4 for Tat; A2 = 87.1 for control, 70.3 for Tat; EC50 = 0.39 ± 0.07 μm for control, 1.6 ± 0.2 μm for Tat; p = 1.6 ± 0.04 for control, 1.1 ± 0.1 for Tat. The EC50 was significantly increased relative to control by treatment with Tat (t test; t(37) = 4.91, p < 0.0001), indicating reduced 2-AG potency. D, Bar graph shows the mean magnitude of 0.2 μm 2-AG-mediated inhibition of EPSCs in control (open bars) and Tat-treated groups (gray bars) in cultures treated with vehicle or 1 μm JZL184 for 24 h. A two-way ANOVA found no interaction (JZL184 treatment × Tat treatment; F(1,20) = 0.043, p = 0.838, n = 6 cells/group). Tat treatment had a significant effect on 2-AG-induced inhibition of EPSC amplitude (F(1,20) = 59.01, p < 0.0001). Data are represented as mean ± SEM.
Interestingly, Tat did not alter the concentration-response relationship for Win-2-mediated inhibition of EPSCs (Fig. 6). The EC50 for Win-2 inhibition of EPSC amplitude was 7.8 ± 1.6 nm for control and 8.0 ± 1.1 nm after 24 h exposure to Tat (Table 1g). Win-2 is a highly efficacious agonist for cannabinoid receptors (Shen et al., 1996; Luk et al., 2004) and has been shown to elicit near maximal effect while occupying only 7.5% of CB1Rs (Gifford et al., 1999). Thus, the presence of spare receptors may explain the discrepancy. If spare receptors account for the insensitivity of Win-2-mediated inhibition of EPSCs to Tat treatment relative to 2-AG-mediated presynaptic inhibition, then we would predict that the inhibition produced by Δ9-tetrahydrocannabinol (Δ9-THC), the psychoactive ingredient in marijuana and a drug that acts as a partial agonist for CB1Rs (Shen and Thayer, 1999; Roloff and Thayer, 2009), to be especially sensitive to Tat exposure. Presynaptic inhibition mediated by Gi-coupled GPCRs is attenuated at high stimulus frequencies (Bean, 1989), which is more pronounced for low-efficacy agonists such as Δ9-THC (Roloff and Thayer, 2009); thus, to study the effect of Δ9-THC we reduced the stimulus frequency to 0.1 Hz. Following Tat treatment, presynaptic inhibition produced by Δ9-THC was markedly reduced (Fig. 7; Table 1h). The inhibition of EPSC amplitude at a maximally effective concentration of Δ9-THC was reduced from 55% in control to 15% after Tat treatment. Overall, Tat impairs CB1R function and this loss-of-function is most evident by the attenuation of the responses to low-efficacy agonists such as Δ9-THC.
Figure 6.

HIV Tat does not affect Win-2-mediated inhibition of EPSCs. Representative traces show EPSCs during baseline and in the presence of varying [Win-2] for (A) control and (B) Tat-treated groups (50 ng/ml, 24 h). Scale bars: 10 ms (horizontal bar) and 50 pA (vertical bar). C, The concentration-response curve for Win-2-mediated inhibition of EPSCs was not affected by Tat treatment. The curves were fit with a logistic equation of the form percentage inhibition = A1 + [(A2 – A1)/(1 + 10(logxₒ-x)p)], where xₒ = EC50, × = log[Win-2], A1 = % inhibition in the absence of drug, A2 = % inhibition at a maximally effective drug concentration, and p = slope factor. The following values were calculated using a nonlinear, least-squares curve fitting program: A1 = 21.7 for control, 19.9 for Tat; A2 = 81.1 for control, 77.0 for Tat; EC50 = 7.8 ± 1.6 nm for control, 8.0 ± 1.1 nm for Tat; p = 2.5 ± 1.7 for control, 2.5 ± 1.2 for Tat. Tat treatment did not significantly affect the EC50 for Win-2-mediated inhibition of EPSCs (t test; t(39) = 0.08, p = 0.93).
Figure 7.

Exposure to HIV Tat reduces Δ9-THC-mediated inhibition of EPSCs. A, B, Representative traces show EPSCs during baseline and in the presence of varying [Δ9-THC] for (A) control and (B) Tat-treated groups (50 ng/ml, 24 h). Scale bars: 10 ms (horizontal bar) and 50 pA (vertical bar). C, The concentration-response curve for Δ9-THC-mediated inhibition of EPSCs shows a decrease in potency and efficacy following exposure to Tat. The curves were fit with a logistic equation of the form percentage inhibition = A1 + [(A2 – A1)/(1 + 10(logxₒ-x)p)], where xₒ = EC50, × = log[Δ9-THC], A1 = % inhibition in the absence of drug, A2 = % inhibition at a maximally effective drug concentration, and p = slope factor. The following values were calculated using a nonlinear, least-squares curve fitting program: A1 = −4.4 for control, −0.8 for Tat; A2 = 54.6 for control, 15.3 for Tat; EC50 = 0.04 ± 0.03 μm for control, 0.27 μm for Tat; p = 1.3 ± 0.8 for control, 3.4 for Tat. The marked reduction in Δ9-THC-mediated inhibition following Tat exposure limited the quality of the curve fit precluding determination of error associated with the EC50. To determine the significance of the Tat effect, we performed a two-way ANOVA over the 0.1–1 μm THC concentrations that were tested in both control and Tat-treated cultures and found no interaction ([Δ9-THC] × Tat treatment; F(2,19) = 0.088, p = 0.92, n = 4–5 cells/group). Tat treatment had a significant effect on Δ9-THC-induced inhibition of EPSC amplitude (F(1,19) = 53.71, p < 0.0001). Data are represented as mean ± SEM.
Several reports have shown that Tat produces synaptodendritic injury and cell death via a pathway initiated by Tat binding to low-density lipoprotein receptor-related protein (LRP; Eugenin et al., 2007; Kim et al., 2008; Shin and Thayer, 2013). Thus, we examined whether binding to LRP is necessary for Tat-mediated impairment of eCB signaling. We found that blocking LRP by pretreating with the selective antagonist receptor-associated protein (RAP) did not prevent Tat-mediated reduction of DSE. In untreated cells DSE was 51 ± 4% and was similar in the presence of 50 nm RAP (55 ± 6%). Tat treatment (50 ng/ml, 24 h) reduced DSE to 14 ± 12% and was not affected by RAP (12 ± 7%). A two-way ANOVA found no interaction (RAP treatment × Tat treatment; F(1,16) = 0.156, p = 0.698, n = 4–6 cells/group) while Tat treatment had a significant effect on DSE (F(1,16) = 30.94, p < 0.0001; Table 1i), indicating that Tat acts via a mechanism independent of LRP.
Tat does not affect adenosine A1 receptor (A1AR)-mediated presynaptic inhibition
To determine whether Tat acts selectively on CB1R, or whether it similarly affects other presynaptic Gi/o-coupled receptors, we examined the effects of Tat on A1ARs. These receptors are localized on presynaptic terminals and modulate the release of neurotransmitters, including glutamate and GABA (Dunwiddie and Masino, 2001). We assessed whether Tat impairs A1AR function by monitoring EPSC amplitude in the presence of a selective A1AR agonist N6-cyclopentyladenosine (CPA; 10 nm; Hargus et al., 2009; Gonçalves et al., 2015) and partial agonist capadenoson (30 nm; Albrecht-Kupper et al., 2012). CPA inhibited EPSC amplitude by 74 ± 5% in control cultures and by 77 ± 5% in cultures treated with 50 ng/ml Tat for 24 h (Fig. 8B). The partial agonist capadenoson inhibited EPSC amplitude by 43 ± 8% in control cultures and by 41 ± 6% in Tat-treated cultures (Fig. 8C; Table 1j). Thus, Tat treatment does not affect inhibition of glutamate release initiated by selective A1AR agonists, indicating that A1AR function is not altered by Tat. These results suggest that Tat acts selectively to inhibit CB1R function and provides evidence that Tat exposure does not broadly suppress the function of all presynaptic Gi/o-coupled receptors.
Figure 8.
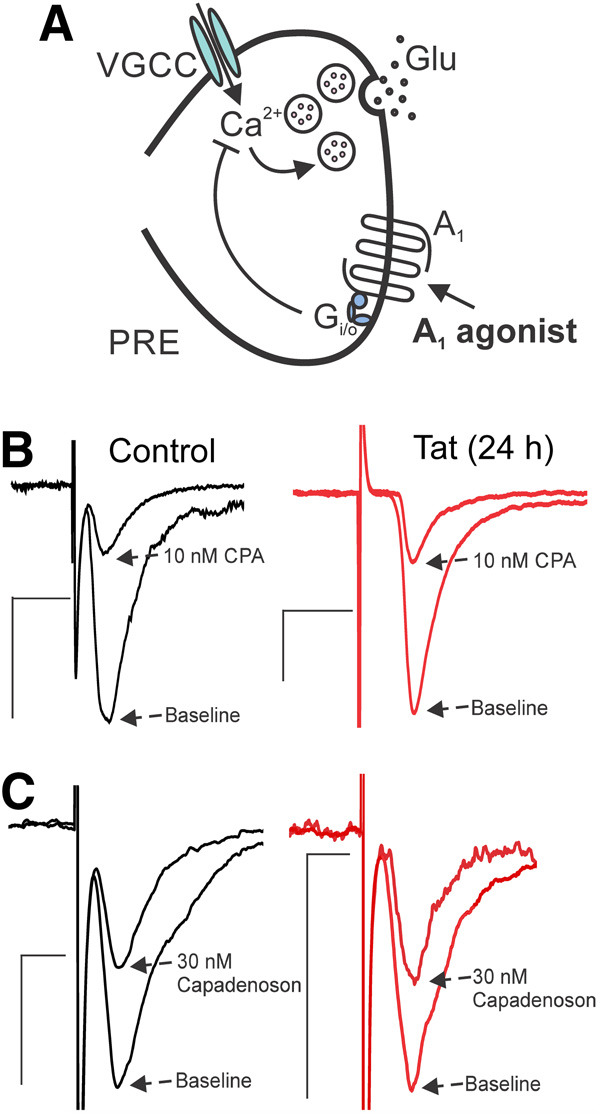
HIV Tat does not affect A1AR-mediated presynaptic inhibition. A, Presynaptic terminal illustrating localization of A1 adenosine receptors. When these receptors are activated, glutamate transmission is reduced. B, Representative traces show EPSCs during baseline and in the presence of CPA (10 nm, selective A1AR agonist) for control and Tat-treated group (50 ng/ml, 24 h). Tat did not affect CPA-mediated inhibition of glutamate release (t(10) = 0.416, p = 0.686; t test). C, Representative traces showing EPSCs during baseline and in the presence of the partial agonist capadenoson (30 nm) for control and Tat-treated group. Tat did not affect capadenoson-mediated inhibition of glutamate release (t(10) = 0.187, p = 0.856; t test). Scale bars: 10 ms (horizontal bar) and 50 pA (vertical bar).
Discussion
Treating rat hippocampal cultures with the HIV protein Tat inhibited DSE and MSE, two forms of short-term plasticity mediated by the eCB system. While Tat is a neurotoxin known to contribute to synaptodendritic damage in HAND (Li et al., 2009), this is the first report to show that Tat disrupts function of the eCB system. Our principal finding is that Tat impairs CB1R-mediated presynaptic inhibition at excitatory but not inhibitory terminals. This selective attenuation of eCB signaling may unbalance network excitability, with potentially significant effects on symptoms associated with HAND, the progression of neurological disease in people living with HIV, and the sensitivity of HIV-positive individuals to exogenous cannabinoids. Furthermore, the selective loss of eCB signaling at excitatory synapses has implications for the effects of neuroinflammatory stimuli on the eCB system.
Primary neuronal cultures provide an experimentally accessible system that exhibits synaptic changes that are a hallmark of HAND (Ellis et al., 2007; Eugenin et al., 2007; Kim et al., 2008, 2011a; Shin and Thayer, 2013; Raybuck et al., 2017; Hermes et al., 2018). Advantages of primary hippocampal cultures for this study include robust DSE and DSI, the ability to administer HIV Tat at specific concentrations with accurate timing, and the ease with which the culture can be treated with highly lipophilic cannabinoid receptor ligands. However, there are caveats to cell culture models. The rat primary cultures used in this study were derived from fetal neurons, and while they mature in culture, we cannot precisely match their stage of development to in vivo models. Furthermore, dissociated cultures lack the synaptic architecture found in vivo, precluding our ability to identify the specific synapses affected by HIV Tat in our model. Finally, the effects of chronic exposure to Tat produced in transgenic models cannot be replicated with short-lived primary cultures. Thus, the novel changes in the eCB system described here provide compelling evidence to extend these findings to chronic in vivo models to explore regional and developmental effects on synaptic networks.
Tat-induced attenuation of CB1R-mediated inhibition of glutamatergic, but not GABAergic neurotransmission, might result from several mechanisms. CB1Rs at inhibitory synapses are expressed at a much higher density relative to the density at excitatory terminals (Katona et al., 1999). Thus, Tat-induced loss of CB1R-mediated signaling at GABAergic terminals may not be sufficient to significantly impair eCB-mediated inhibition of GABA release. The importance of spare receptors in modulating eCB signaling is illustrated by comparing the failure of Tat to affect inhibition of glutamate release by the highly efficacious agonist Win-2, which elicits maximal effects while occupying only 7.5% of CB1Rs (Gifford et al., 1999), to the marked attenuation of the effects of the partial agonist Δ9-THC. Even when the duration of the depolarizing stimulus to evoke 2-AG production was reduced, Tat failed to affect DSI. Thus, following exposure to Tat, physiologically relevant eCB-mediated retrograde signaling remains functional at inhibitory terminals, but is impaired at excitatory synapses.
The rightward shift in the 2-AG concentration-response relationship produced by Tat treatment is reminiscent of CB1R desensitization (Kouznetsova et al., 2002; Sim-Selley, 2003; Lundberg et al., 2005; Daigle et al., 2008; Wu et al., 2008). Tat is a powerful excitotoxin; perhaps Tat-induced excitatory drive chronically activates the eCB system, desensitizing CB1Rs. The development of Tat-induced attenuation of CB1R function over the span of 4–48 h is consistent with gradual receptor downregulation. Alternatively, Tat can be taken up by glutamatergic neurons where it activates protein kinase C (Haughey et al., 2001), which is known to produce heterologous desensitization of CB1Rs (Garcia et al., 1998; Chu et al., 2010). The time course of the Tat effect also suggests that Tat does not simply bind to and occlude CB1Rs; direct receptor interaction would be expected to equilibrate more rapidly and also affect receptors on GABAergic terminals similarly to those on glutamatergic terminals. Tat can be internalized into neurons by binding to LRP, leading to synaptodendritic injury and cell death (Liu et al., 2000; Eugenin et al., 2007; Kim et al., 2008; Shin and Thayer, 2013). We found that Tat-mediated impairment of eCB signaling is independent of LRP. Tat elicits a range of toxic effects on neuronal networks, some of which are mediated by neuroinflammation (Lu et al., 2011; Chivero et al., 2017; Gonek et al., 2018; Thangaraj et al., 2018), leading us to speculate that Tat may impair eCB signaling through an indirect mechanism involving non-neuronal cells, possibly via the release of inflammatory cytokines (Kim et al., 2018). The degree to which this damage results from direct or adaptive responses to the presence of Tat is unclear.
In contrast to the loss of CB1R function described here, CB1R protein increased in infralimbic cortex from transgenic mice expressing Tat (Jacobs et al., 2019). The mPFC and hippocampus display differential sensitivity to Tat (Cirino et al., 2020), suggesting the effects of Tat on the eCB system may also exhibit regional differences. Xu et al. (2016) found that Tat expression occluded the effects of CB1R agonists. We found DSE to be an extremely sensitive assay for Tat-induced modulation of the eCB system. Thus, the failure of Tat to occlude DSI provides strong evidence that Tat does not affect CB1R-mediated presynaptic inhibition at GABAergic synapses in the hippocampal culture model. Tat failed to affect DSI evoked by three different stimulus strengths; this physiological method of graded CB1R activation essentially produces a 2-AG concentration-response. It is possible that Tat upregulates CB1Rs on GABAergic terminals while also decreasing CB1R levels on glutamatergic terminals. Tat elicits a neuroinflammatory response (Nath et al., 1999; Pu et al., 2003), and neuroinflammation selectively upregulates CB1Rs on GABAergic terminals (Chen et al., 2003; Feng et al., 2016), providing a precedent for this type of change in the eCB system. Alternatively, the loss of CB1R function at excitatory synapses described here may be an early event initiated by Tat, akin to the initial stages of exposure, and the gain-of-function at inhibitory terminals (Xu et al., 2016) may result from adaptations in the eCB system that occur during prolonged Tat expression. Thus, unique changes in the synaptic network may predominate at different disease stages.
Altered eCB signaling could affect many physiological processes relevant to HAND (Rodríguez de Fonseca et al., 2005). Using brain tissue from Alzheimer’s patients, autoradiography, in situ hybridization, and GTPγS binding studies have demonstrated reduced CB1R agonist binding, regionally discrete losses of CB1R mRNA expression, and less efficient coupling to G-proteins (Westlake et al., 1994; Ramírez et al., 2005). The selective loss of functional eCB signaling at excitatory terminals described here emphasizes the need to understand synapse-specific changes in the eCB system over the course of neurodegenerative disease. Antagonism of CB1R function elicits depression and anxiety-like behaviors (Moreira et al., 2009). If the loss of CB1R function on excitatory terminals we describe for hippocampal cultures also occurs in the prefrontal cortex (Rigucci et al., 2010), it might contribute to the production of these symptoms in HAND. The motor impairment produced by cannabinoids results from CB1R activation on both excitatory and inhibitory terminals in basal ganglia and cerebellum (Sañudo-Peña et al., 1999; Kishimoto and Kano, 2006), suggesting Tat-mediated motor effects could result from unbalanced eCB signaling (Kim et al., 2003). CB1Rs on glutamatergic terminals in the nucleus accumbens regulate the hedonic impact of food, while CB1Rs in the hypothalamus regulate the release of hormones that regulate appetite and energy expenditure (Lau et al., 2017). Thus, their loss could contribute to the loss of appetite and wasting observed in AIDS patients.
The eCB system protects against excitotoxicity (Shen et al., 1996; Shen and Thayer, 1998; Marsicano and Lutz, 1999; Nagayama et al., 1999; Chen et al., 2003; Li et al., 2012; Feng et al., 2016), including HIV-induced neurotoxicity (Kim et al., 2011a; Xu et al., 2017; Hermes et al., 2018; Zhang and Thayer, 2018). Thus, Tat-induced loss of neuroprotection may accelerate the development of neurological complications in HAND. There is precedent for changes in eCB signaling contributing to epileptogenesis following neuroinflammatory insult (Chen et al., 2003; Feng et al., 2016; Sugaya and Kano, 2018). Furthermore, in the Fmr1-knockout mouse model of fragile X syndrome, impaired eCB signaling contributes to synaptic plasticity defects that underlie impairments in episodic memory (Wang et al., 2018). Superfusion of amyloid-β1-42 onto brain slices from wild-type mice prolonged DSI, thus altering GABAergic transmission and possibly contributing to synaptic dysfunction in Alzheimer’s disease (Mulder et al., 2011).
While evidence linking impaired eCB signaling to disease progression is limited, there is considerable evidence demonstrating that enhanced CB1R signaling protects neurological function (Aymerich et al., 2018). In HAND models, cannabinoids protect dopaminergic neurons against damage from the HIV envelope glycoprotein gp120 (Hu et al., 2013), reduce Tat-induced release of nitric oxide (Esposito et al., 2002), and diminish Tat-induced increases in neuronal [Ca2+]i (Xu et al., 2017). Boosting eCB tone by inhibiting eCB metabolism also attenuates HIV neurotoxicity; inhibition of MGL with the selective inhibitor JZL184 protects against excitatory synapse loss evoked by gp120 (Zhang and Thayer, 2018). Similarly, pharmacological inhibition of fatty acid amide hydrolase, the enzyme responsible for degrading the eCB anandamide, protects against Tat-mediated increases in [Ca2+]i and dendritic damage (Hermes et al., 2018). Thus, cannabimimetics and drugs that inhibit eCB metabolism may slow the course of HIV neurotoxicity (Wu et al., 2019).
This study suggests that the converse may also be true; in the presence of HIV, impaired CB1R function at excitatory terminals may alter the response to exogenous cannabinoids. Because efforts to legalize medicinal and recreational marijuana are increasing access (Charilaou et al., 2017; Jones et al., 2018) and cannabis use is prevalent among people living with HIV (Hartzler et al., 2017), altered response to cannabinoids could impact many patients. We found a dramatic loss of Δ9-THC efficacy at excitatory synapses in the presence of Tat. However, CB1R signaling at inhibitory synapses was normal in the presence of Tat. Thus, the network effects of Δ9-THC could shift to an excitatory response due to CB1R-mediated suppression of GABA release without a corresponding decrease in glutamate release. This prediction is consistent with the increased miniature EPSC frequency observed in mice expressing Tat (Jacobs et al., 2019).
In conclusion, we have shown in an in vitro model of HIV neurotoxicity that the HIV protein Tat, an established contributor to HIV neurotoxicity, inhibits short-term eCB-mediated plasticity selectively at excitatory synapses. We speculate that this novel loss of CB1R function might contribute to excitotoxicity under neuroinflammatory conditions. Thus, our conclusions from this in vitro work provide a framework for future work assessing the status of the eCB system in vivo in neurodegenerative disease noting the duration of disease, the specific brain regions affected and, as shown here, the specific neurotransmitters involved.
Acknowledgments
Acknowledgements: We thank Dr. Tamas Balla (National Institute of Child Health and Human Development, National Institutes of Health) for the DAG biosensor.
Synthesis
Reviewing Editor: Lindsay De Biase, University of California, Los Angeles
Decisions are customarily a result of the Reviewing Editor and the peer reviewers coming together and discussing their recommendations until a consensus is reached. When revisions are invited, a fact-based synthesis statement explaining their decision and outlining what is needed to prepare a revision will be listed below. The following reviewer(s) agreed to reveal their identity: NONE. Note: If this manuscript was transferred from JNeurosci and a decision was made to accept the manuscript without peer review, a brief statement to this effect will instead be what is listed below.
The previous reviews of this manuscript acknowledge that this is a well-executed in vitro study with important observations regarding the impact of TAT on the endocannabinoid system. The experiments added at suggestion of reviewers have strengthened the manuscript. However, the abstract and discussion need to be further modified to acknowledge the limitations of the in vitro approach and to discuss additional experiments that could clarify if TAT-induced changes in endocannabinoid DSE and MSE contribute to neurological symptoms in vivo.
Please address the following concerns:
1) In the abstract: the final sentence should be modified to explicitly state that these in vitro findings suggest that similar effects of TAT could contribute to HAND in vivo.
2) The discussion needs to devote an entire paragraph to acknowledging possible differences between in vitro and in vivo neuronal and eCB function and point to critical future experiments that could reveal whether similar phenomena are observed in vivo. This would include acknowledging that cultured neurons generally exhibit more immature cellular phenotypes and may not fully reflect how mature neurons in vivo will respond to TAT insults. This should also include a discussion of any reported differences in the expression levels of CB1Rs in neurons in vitro and in vivo as well as any reported differences in the magnitude of endocannabinoid mediated DSE in vitro and in vivo.
3) It is not clear from the methods whether the authors used a range of stimulation intensities for each recorded neuron before settling on a stimulus intensity that evoked 50% max or 80% max EPSC or IPSC currents. If this was carried out, this should be described in the methods. In addition, the authors should use this data to comment on whether there were any baseline differences in EPSC or IPSC amplitude or the relationship between stimulus intensity and amplitude following TAT treatment but before inducing DSE or DSI.
4) Include in the introduction or results a description of the brain regions most affected by HAND as part of the rationale for using hippocampal cultures to investigate TAT-mediated effects on the eCB system.
5) Page 14 line 8 and Page 16 lines 11-14 discuss the implications of the LRP experiments and suggest that TAT acts through an “indirect” mechanism, potentially via glial cells. In the absence of experimental evidence showing that glial cells are involved, it seems more accurate to simply say that the effect of TAT on CB1R function is occurring through an “LRP-independent mechanism.”
References
- Albrecht-Kupper BE, Leineweber K, Nell PG (2012) Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signal 8:91–99. 10.1007/s11302-011-9274-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, et al. (2007) Updated research nosology for HIV-associated neurocognitive disorders. Neurology 69:1789–1799. 10.1212/01.WNL.0000287431.88658.8b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymerich MS, Aso E, Abellanas MA, Tolon RM, Ramos JA, Ferrer I, Romero J, Fernández-Ruiz J (2018) Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem Pharmacol 157:67–84. 10.1016/j.bcp.2018.08.016 [DOI] [PubMed] [Google Scholar]
- Bachani M, Sacktor N, McArthur JC, Nath A, Rumbaugh J (2013) Detection of anti-tat antibodies in CSF of individuals with HIV-associated neurocognitive disorders. J Neurovirol 19:82–88. 10.1007/s13365-012-0144-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Denefrio CL, Johnson CA, Saulnier JL, Sabatini BL (2013) Temporal dynamics of a homeostatic pathway controlling neural network activity. Front Mol Neurosci 6:28. 10.3389/fnmol.2013.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP (1989) Neurotransmitter inhibition on neuronal calcium currents by changes in channel voltage dependence. Nature 340:153–156. 10.1038/340153a0 [DOI] [PubMed] [Google Scholar]
- Charilaou P, Agnihotri K, Garcia P, Badheka A, Frenia D, Yegneswaran B (2017) Trends of cannabis use disorder in the inpatient: 2002 to 2011. Am J Med 130:678–687.e7. 10.1016/j.amjmed.2016.12.035 [DOI] [PubMed] [Google Scholar]
- Chen K, Ratzliff A, Hilgenberg L, Gulyás A, Freund TF, Smith M, Dinh TP, Piomelli D, Mackie K, Soltesz I (2003) Long-term plasticity of endocannabinoid signaling induced by developmental febrile seizures. Neuron 39:599–611. 10.1016/s0896-6273(03)00499-9 [DOI] [PubMed] [Google Scholar]
- Chivero ET, Guo ML, Periyasamy P, Liao K, Callen SE, Buch S (2017) HIV-1 Tat primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation. J Neurosci 37:3599–3609. 10.1523/JNEUROSCI.3045-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Zheng H, Zhang Y, Loh HH, Law PY (2010) Agonist-dependent mu-opioid receptor signaling can lead to heterologous desensitization. Cell Signal 22:684–696. 10.1016/j.cellsig.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirino TJ, Harden SW, McLaughlin JP, Frazier CJ (2020) Region-specific effects of HIV-1 Tat on intrinsic electrophysiological properties of pyramidal neurons in mouse prefrontal cortex and hippocampus. J Neurophysiol 123:1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Kearn CS, Mackie K (2008) Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacol 54:36–44. 10.1016/j.neuropharm.2007.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24:31–55. 10.1146/annurev.neuro.24.1.31 [DOI] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E (2007) HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci 8:33–44. 10.1038/nrn2040 [DOI] [PubMed] [Google Scholar]
- Esposito G, Ligresti A, Izzo AA, Bisogno T, Ruvo M, Di Rosa M, Di Marzo V, Iuvone T (2002) The endocannabinoid system protects rat glioma cells against HIV-1 Tat protein-induced cytotoxicity - Mechanism and regulation. J Biol Chem 277:50348–50354. 10.1074/jbc.M207170200 [DOI] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW (2007) HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci USA 104:3438–3443. 10.1073/pnas.0611699104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, Tang Y, Chen B, Xu C, Wang Y, Dai Y, Wu D, Zhu J, Wang S, Zhou Y, Shi L, Hu W, Zhang X, Chen Z (2016) Transient increase of interleukin-1β after prolonged febrile seizures promotes adult epileptogenesis through long-lasting upregulating endocannabinoid signaling. Sci Rep 6:21931. 10.1038/srep21931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DE, Brown S, Hille B, Mackie K (1998) Protein kinase C disrupts cannabinoid actions by phosphorylation of the CB1 cannabinoid receptor. J Neurosci 18:2834–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford AN, Bruneus M, Gatley SJ, Lan RX, Makriyannis A, Volkow ND (1999) Large receptor reserve for cannabinoid actions in the central nervous system. J Pharmacol Exp Ther 288:478–483. [PubMed] [Google Scholar]
- Gonçalves FQ, Pires J, Pliassova A, Beleza R, Lemos C, Marques JM, Rodrigues RJ, Canas PM, Köfalvi A, Cunha RA, Rial D (2015) Adenosine A2b receptors control A1 receptor-mediated inhibition of synaptic transmission in the mouse hippocampus. Eur J Neurosci 41:878–888. 10.1111/ejn.12851 [DOI] [PubMed] [Google Scholar]
- Gonek M, McLane VD, Stevens DL, Lippold K, Akbarali HI, Knapp PE, Dewey WL, Hauser KF, Paris JJ (2018) CCR5 mediates HIV-1 Tat-induced neuroinflammation and influences morphine tolerance, dependence, and reward. Brain Behav Immun 69:124–138. 10.1016/j.bbi.2017.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorantla S, Makarov E, Roy D, Finke-Dwyer J, Murrin LC, Gendelman HE, Poluektova L (2010) Immunoregulation of a CB2 receptor agonist in a murine model of neuroAIDS. J Neuroimmune Pharmacol 5:456–468. 10.1007/s11481-010-9225-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MV, Raybuck JD, Zhang X, Wu MM, Thayer SA (2019) Scaling synapses in the presence of HIV. Neurochem Res 44:234–246. 10.1007/s11064-018-2502-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Thayer SA (2013) Human immunodeficiency virus-1 Tat protein increases the number of inhibitory synapses between hippocampal neurons in culture. J Neurosci 33:17908–17920. 10.1523/JNEUROSCI.1312-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Bertram EH, Patel MK (2009) Adenosine A1 receptors presynaptically modulate excitatory synaptic input onto subiculum neurons. Brain Res 1280:60–68. 10.1016/j.brainres.2009.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzler B, Carlini BH, Newville H, Crane HM, Eron JJ, Geng EH, Mathews WC, Mayer KH, Moore RD, Mugavero MJ, Napravnik S, Rodriguez B, Donovan DM (2017) Identifying HIV care enrollees at-risk for cannabis use disorder. AIDS Care 29:846–850. 10.1080/09540121.2016.1271393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD (2001) HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem 78:457–467. 10.1046/j.1471-4159.2001.00396.x [DOI] [PubMed] [Google Scholar]
- Heaton RK, Grant I, Butters N, White DA, Kirson D, Atkinson JH, McCutchan JA, Taylor MJ, Kelly MD, Ellis RJ (1995) The HNRC 500–neuropsychology of HIV infection at different disease stages. HIV Neurobehavioral Research Center. J Int Neuropsychol Soc 1:231–251. 10.1017/s1355617700000230 [DOI] [PubMed] [Google Scholar]
- Hermes DJ, Xu C, Poklis JL, Niphakis MJ, Cravatt BF, Mackie K, Lichtman AH, Ignatowska-Jankowska BM, Fitting S (2018) Neuroprotective effects of fatty acid amide hydrolase catabolic enzyme inhibition in a HIV-1 Tat model of neuroAIDS. Neuropharmacol 141:55–65. 10.1016/j.neuropharm.2018.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Sheng WS, Rock RB (2013) CB2 receptor agonists protect human dopaminergic neurons against damage from HIV-1 gp120. PLoS One 8:e77577. 10.1371/journal.pone.0077577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I (2000) Detection of the human immunodeficiency virus regulatory protein Tat in CNS tissues. J Neurovirol 6:145–155. 10.3109/13550280009013158 [DOI] [PubMed] [Google Scholar]
- Isokawa M, Alger BE (2005) Retrograde endocannabinoid regulation of GABAergic inhibition in the rat dentate gyrus granule cell. J Physiol 567:1001–1010. 10.1113/jphysiol.2005.094219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs IR, Xu C, Hermes DJ, League AF, Xu C, Nath B, Jiang W, Niphakis MJ, Cravatt BF, Mackie K, Mukhopadhyay S, Lichtman AH, Ignatowska-Jankowska BM, Fitting S (2019) Inhibitory control deficits associated with upregulation of CB1R in the HIV-1 Tat transgenic mouse model of hand. J Neuroimmune Pharmacol 14:661–678. 10.1007/s11481-019-09867-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R, Nath A (2013) Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc Natl Acad Sci USA 110:13588–13593. 10.1073/pnas.1308673110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J, Nicole Jones K, Peil J (2018) The impact of the legalization of recreational marijuana on college students. Addict Behav 77:255–259. 10.1016/j.addbeh.2017.08.015 [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF (1999) Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19:4544–4558. 10.1523/JNEUROSCI.19-11-04544.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ (2003) Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol 162:1693–1707. 10.1016/S0002-9440(10)64304-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Martemyanov KA, Thayer SA (2008) Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci 28:12604–12613. 10.1523/JNEUROSCI.2958-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Shin AH, Thayer SA (2011a) Activation of cannabinoid type 2 receptors inhibits HIV-1 envelope glycoprotein gp120-induced synapse loss. Mol Pharmacol 80:357–366. 10.1124/mol.111.071647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Guzman-Hernandez ML, Balla T (2011b) A highly dynamic ER-derived phosphatidylinositol-synthesizing organelle supplies phosphoinositides to cellular membranes. Dev Cell 21:813–824. 10.1016/j.devcel.2011.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Hahn YK, Podhaizer EM, McLane VD, Zou S, Hauser KF, Knapp PE (2018) A central role for glial CCR5 in directing the neuropathological interactions of HIV-1 Tat and opiates. J Neuroinflammation 15:285. 10.1186/s12974-018-1320-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto Y, Kano M (2006) Endogenous cannabinoid signaling through the CB1 receptor is essential for cerebellum-dependent discrete motor learning. J Neurosci 26:8829–8837. 10.1523/JNEUROSCI.1236-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouznetsova M, Kelley B, Shen M, Thayer SA (2002) Desensitization of cannabinoid-mediated presynaptic inhibition of neurotransmission between rat hippocampal neurons in culture. Mol Pharmacol 61:477–485. 10.1124/mol.61.3.477 [DOI] [PubMed] [Google Scholar]
- Lau BK, Cota D, Cristino L, Borgland SL (2017) Endocannabinoid modulation of homeostatic and non-homeostatic feeding circuits. Neuropharmacol 124:38–51. 10.1016/j.neuropharm.2017.05.033 [DOI] [PubMed] [Google Scholar]
- Li W, Li G, Steiner J, Nath A (2009) Role of Tat protein in HIV neuropathogenesis. Neurotox Res 16:205–220. 10.1007/s12640-009-9047-8 [DOI] [PubMed] [Google Scholar]
- Li Y, Krogh KA, Thayer SA (2012) Epileptic stimulus increases Homer 1a expression to modulate endocannabinoid signaling in cultured hippocampal neurons. Neuropharmacol 63:1140–1149. 10.1016/j.neuropharm.2012.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ (2000) Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med 6:1380–1387. 10.1038/82199 [DOI] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5:37–44. 10.1038/nchembio.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SM, Tremblay ME, King IL, Qi J, Reynolds HM, Marker DF, Varrone JJ, Majewska AK, Dewhurst S, Gelbard HA (2011) HIV-1 Tat-induced microgliosis and synaptic damage via interactions between peripheral and central myeloid cells. PLoS One 6:e23915. 10.1371/journal.pone.0023915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk T, Jin WZ, Zvonok A, Lu D, Lin XZ, Chavkin C, Makriyannis A, Mackie K (2004) Identification of a potent and highly efficacious, yet slowly desensitizing CB1 cannabinoid receptor agonist. Br J Pharmacol 142:495–500. 10.1038/sj.bjp.0705792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg DJ, Daniel AR, Thayer SA (2005) Delta9-tetrahydrocannabinol-induced desensitization of cannabinoid-mediated inhibition of synaptic transmission between hippocampal neurons in culture. Neuropharmacol 49:1170–1177. 10.1016/j.neuropharm.2005.07.012 [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B (1999) Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci 11:4213–4225. 10.1046/j.1460-9568.1999.00847.x [DOI] [PubMed] [Google Scholar]
- Masliah E, Ge N, Achim CL, Hansen LA, Wiley CA (1992) Selective neuronal vulnerability in HIV encephalitis. J Neuropathol Exp Neurol 51:585–593. 10.1097/00005072-199211000-00003 [DOI] [PubMed] [Google Scholar]
- Moore DJ, Masliah E, Rippeth JD, Gonzalez R, Carey CL, Cherner M, Ellis RJ, Achim CL, Marcotte TD, Heaton RK, Grant I (2006) Cortical and subcortical neurodegeneration is associated with HIV neurocognitive impairment. AIDS 20:879–887. [DOI] [PubMed] [Google Scholar]
- Moreira FA, Grieb M, Lutz B (2009) Central side-effects of therapies based on CB1 cannabinoid receptor agonists and antagonists: focus on anxiety and depression. Best Pract Res Clin Endocrinol Metab 23:133–144. 10.1016/j.beem.2008.09.003 [DOI] [PubMed] [Google Scholar]
- Mulder J, Zilberter M, Pasquaré SJ, Alpár A, Schulte G, Ferreira SG, Köfalvi A, Martín-Moreno AM, Keimpema E, Tanila H, Watanabe M, Mackie K, Hortobágyi T, de Ceballos ML, Harkany T (2011) Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 134:1041–1060. 10.1093/brain/awr046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murataeva N, Straiker A, Mackie K (2014) Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br J Pharmacol 171:1379–1391. 10.1111/bph.12411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA (1999) Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci 19:2987–2995. 10.1523/JNEUROSCI.19-08-02987.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO (1999) Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem 274:17098–17102. 10.1074/jbc.274.24.17098 [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29:729–738. 10.1016/s0896-6273(01)00247-1 [DOI] [PubMed] [Google Scholar]
- Pu H, Tian J, Flora G, Lee YW, Nath A, Hennig B, Toborek M (2003) HIV-1 Tat protein upregulates inflammatory mediators and induces monocyte invasion into the brain. Mol Cell Neurosci 24:224–237. 10.1016/s1044-7431(03)00171-4 [DOI] [PubMed] [Google Scholar]
- Ramírez BG, Blázquez C, Gómez del Pulgar T, Guzmán M, de Ceballos ML (2005) Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci 25:1904–1913. 10.1523/JNEUROSCI.4540-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybuck JD, Hargus NJ, Thayer SA (2017) A GluN2B-selective NMDAR antagonist reverses synapse loss and cognitive impairment produced by the HIV-1 protein Tat. J Neurosci 37:7837–7847. 10.1523/JNEUROSCI.0226-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigucci S, Serafini G, Pompili M, Kotzalidis GD, Tatarelli R (2010) Anatomical and functional correlates in major depressive disorder: the contribution of neuroimaging studies. World J Biol Psychiatry 11:165–180. 10.3109/15622970903131571 [DOI] [PubMed] [Google Scholar]
- Rodríguez de Fonseca F, Del Arco I, Bermudez-Silva FJ, Bilbao A, Cippitelli A, Navarro M (2005) The endocannabinoid system: physiology and pharmacology. Alcohol Alcohol 40:2–14. 10.1093/alcalc/agh110 [DOI] [PubMed] [Google Scholar]
- Roloff AM, Thayer SA (2009) Modulation of excitatory synaptic transmission by Delta 9-tetrahydrocannabinol switches from agonist to antagonist depending on firing rate. Mol Pharmacol 75:892–900. 10.1124/mol.108.051482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roloff AM, Anderson GR, Martemyanov KA, Thayer SA (2010) Homer 1a gates the induction mechanism for endocannabinoid-mediated synaptic plasticity. J Neurosci 30:3072–3081. 10.1523/JNEUROSCI.4603-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sañudo-Peña MC, Tsou K, Walker JM (1999) Motor actions of cannabinoids in the basal ganglia output nuclei. Life Sci 65:703–713. 10.1016/s0024-3205(99)00293-3 [DOI] [PubMed] [Google Scholar]
- Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, Mankowski JL, Brown A, Volsky DJ, McArthur JC (2016) HIV-associated neurocognitive disorder - pathogenesis and prospects for treatment. Nat Rev Neurol 12:234–248. 10.1038/nrneurol.2016.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M, Thayer SA (1998) Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol 54:459–462. 10.1124/mol.54.3.459 [DOI] [PubMed] [Google Scholar]
- Shen M, Thayer SA (1999) Delta9-tetrahydrocannabinol acts as a partial agonist to modulate glutamatergic synaptic transmission between rat hippocampal neurons in culture. Mol Pharmacol 55:8–13. 10.1124/mol.55.1.8 [DOI] [PubMed] [Google Scholar]
- Shen M, Piser TM, Seybold VS, Thayer SA (1996) Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci 16:4322–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin AH, Thayer SA (2013) Human immunodeficiency virus-1 protein Tat induces excitotoxic loss of presynaptic terminals in hippocampal cultures. Mol Cell Neurosci 54:22–29. 10.1016/j.mcn.2012.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim-Selley LJ (2003) Regulation of cannabinoid CB1 receptors in the central nervous system by chronic cannabinoids. Crit Rev Neurobiol 15:91–119. 10.1615/critrevneurobiol.v15.i2.10 [DOI] [PubMed] [Google Scholar]
- Straiker A, Mackie K (2005) Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol 569:501–517. 10.1113/jphysiol.2005.091918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mackie K (2007) Metabotropic suppression of excitation in murine autaptic hippocampal neurons. J Physiol 578:773–785. 10.1113/jphysiol.2006.117499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugaya Y, Kano M (2018) Control of excessive neural circuit excitability and prevention of epileptic seizures by endocannabinoid signaling. Cell Mol Life Sci 75:2793–2811. 10.1007/s00018-018-2834-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangaraj A, Periyasamy P, Liao K, Bendi VS, Callen S, Pendyala G, Buch S (2018) HIV-1 TAT-mediated microglial activation: role of mitochondrial dysfunction and defective mitophagy. Autophagy 14:1596–1619. 10.1080/15548627.2018.1476810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi V, Balestra P, Lorenzini P, Bellagamba R, Galgani S, Corpolongo A, Vlassi C, Larussa D, Zaccarelli M, Noto P, Visco-Comandini U, Giulianelli M, Ippolito G, Antinori A, Narciso P (2005) Prevalence and risk factors for human immunodeficiency virus–associated neurocognitive impairment, 1996 to 2002: results from an urban observational cohort. J Neurovirol 11:265–273. 10.1080/13550280590952790 [DOI] [PubMed] [Google Scholar]
- Vitale N, Beaumelle B, Bader MF, Tryoen-Tóth P (2013) HIV-1 Tat protein perturbs diacylglycerol production at the plasma membrane of neurosecretory cells during exocytosis. Commun Integr Biol 6:e25145. 10.4161/cib.25145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waataja JJ, Kim HJ, Roloff AM, Thayer SA (2008) Excitotoxic loss of post-synaptic sites is distinct temporally and mechanistically from neuronal death. J Neurochem 104:364–375. 10.1111/j.1471-4159.2007.04973.x [DOI] [PubMed] [Google Scholar]
- Wang W, Cox BM, Jia Y, Le AA, Cox CD, Jung KM, Hou B, Piomelli D, Gall CM, Lynch G (2018) Treating a novel plasticity defect rescues episodic memory in fragile X model mice. Mol Psychiatry 23:1798–1806. 10.1038/mp.2017.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlake TM, Howlett AC, Bonner TI, Matsuda LA, Herkenham M (1994) Cannabinoid receptor binding and messenger RNA expression in human brain: an in vitro receptor autoradiography and in situ hybridization histochemistry study of normal aged and Alzheimer’s brains. Neuroscience 63:637–652. 10.1016/0306-4522(94)90511-8 [DOI] [PubMed] [Google Scholar]
- Wiley CA, Soontornniyomkij V, Radhakrishnan L, Masliah E, Mellors J, Hermann SA, Dailey P, Achim CL (1998) Distribution of brain HIV load in AIDS. Brain Pathol 8:277–284. 10.1111/j.1750-3639.1998.tb00153.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588–592. 10.1038/35069076 [DOI] [PubMed] [Google Scholar]
- Wu DF, Yang LQ, Goschke A, Stumm R, Brandenburg LO, Liang YJ, Höllt V, Koch T (2008) Role of receptor internalization in the agonist-induced desensitization of cannabinoid type 1 receptors. J Neurochem 104:1132–1143. 10.1111/j.1471-4159.2007.05063.x [DOI] [PubMed] [Google Scholar]
- Wu MM, Zhang X, Asher MJ, Thayer SA (2019) Druggable targets of the endocannabinoid system: implications for the treatment of HIV-associated neurocognitive disorder. Brain Res 1724:146467. 10.1016/j.brainres.2019.146467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT (2000) Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci USA 97:11466–11471. 10.1073/pnas.97.21.11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Hermes DJ, Mackie K, Lichtman AH, Ignatowska-Jankowska BM, Fitting S (2016) Cannabinoids occlude the HIV-1 Tat-induced decrease in GABAergic neurotransmission in prefrontal cortex slices. J Neuroimmune Pharmacol 11:316–331. 10.1007/s11481-016-9664-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Hermes DJ, Nwanguma B, Jacobs IR, Mackie K, Mukhopadhyay S, Lichtman AH, Ignatowska-Jankowska B, Fitting S (2017) Endocannabinoids exert CB1 receptor-mediated neuroprotective effects in models of neuronal damage induced by HIV-1 Tat protein. Mol Cell Neurosci 83:92–102. 10.1016/j.mcn.2017.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Thayer SA (2018) Monoacylglycerol lipase inhibitor JZL184 prevents HIV-1 gp120-induced synapse loss by altering endocannabinoid signaling. Neuropharmacol 128:269–281. 10.1016/j.neuropharm.2017.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]