

Abstract
Agonist binding to G protein-coupled receptors (GPCRs) leads to conformational changes in the transmembrane region that activate cytosolic signaling pathways. Although high-resolution structures of different receptor states are available, atomistic details of allosteric signaling across the membrane remain elusive. We calculated free energy landscapes of β2 adrenergic receptor activation using atomistic molecular dynamics simulations in an optimized string of swarms framework, which shed new light on how microswitches govern the equilibrium between conformational states. Contraction of the extracellular binding site in the presence of the agonist BI-167107 is obligatorily coupled to conformational changes in a connector motif located in the core of the transmembrane region. The connector is probabilistically coupled to the conformation of the intracellular region. An active connector promotes desolvation of a buried cavity, a twist of the conserved NPxxY motif, and an interaction between two conserved tyrosines in transmembrane helices 5 and 7 (Y–Y motif), which lead to a larger population of active-like states at the G protein binding site. This coupling is augmented by protonation of the strongly conserved Asp792.50. The agonist binding site hence communicates with the intracellular region via a cascade of locally connected microswitches. Characterization of these can be used to understand how ligands stabilize distinct receptor states and contribute to development drugs with specific signaling properties. The developed simulation protocol can likely be transferred to other class A GPCRs.
G protein-coupled receptors (GPCRs) are membrane proteins that activate cellular signaling pathways in response to extracellular stimuli. There are more than 800 GPCRs in the human genome,1 and these recognize a remarkably large repertoire of ligands such as neurotransmitters, peptides, proteins, and lipids. This large superfamily plays essential roles in numerous physiological processes and has become the most important class of drug targets.2 All GPCRs share a common architecture of seven transmembrane (TM) helices, which recognizes the cognate ligand in the extracellular region and triggers intracellular signals via a more conserved cytosolic domain (Figure 1).3,4 GPCRs are inherently flexible proteins that exist in multiple conformational states, and drug binding alters the relative populations of these. Agonists will shift the equilibrium toward active-like receptor conformations, which promote binding of G proteins leading to initiation of signaling via multiple pathways. In the apo state, GPCRs can still access active-like conformations and thereby exhibit a smaller degree of signaling, which is termed basal activity.
Figure 1.
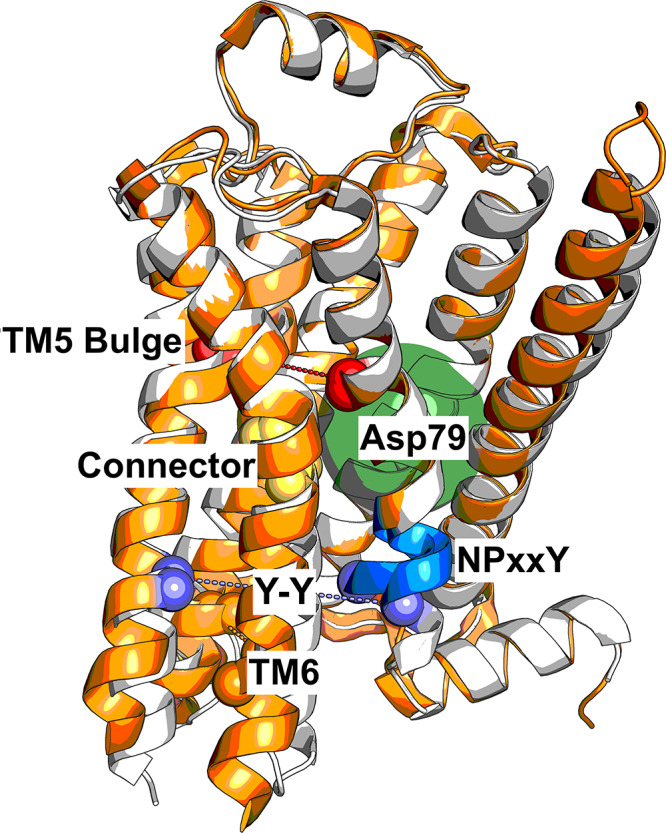
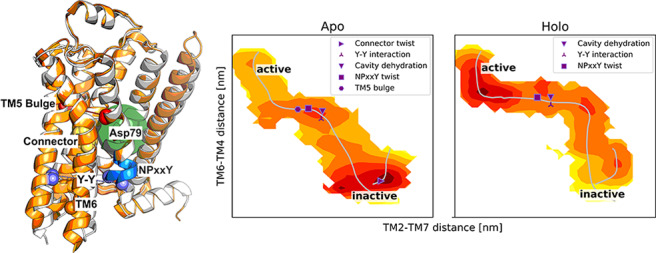
Activation mechanism of GPCRs involving a series of microswitches. Binding of the agonist BI-167107 leads to an inward bulge of TM5 (quantified as the distance between Ser2075.46 and Gly3157.41, red spheres), which leads to a conformational change in the connector region (Ile1213.40 and Phe2826.44, yellow spheres). The transmembrane cavity surrounding Asp792.50 is dehydrated (filled green circle), and the NPxxY motif (blue cartoon) twists upon activation, leading to the Y–Y interaction (Tyr3267.53 and Tyr2195.58, purple spheres). TM6 moves outward to create the G protein binding site. The inactive (PDB entry 2RH1(5)) and active (PDB entry 3P0G(6)) structures are colored white and orange, respectively.
Over the past decade, breakthroughs in GPCR structure determination have provided insights into the process of activation at atomic resolution (Figure 1). In particular, crystal structures of the β2 adrenergic receptor (β2AR) in both active and inactive conformational states5−14 have revealed hallmarks of GPCR activation. The observations made for this prototypical receptor have recently been reinforced by crystal and cryogenic electron microscopy (cryo-EM) structures of other family members.15 The most prominent features of GPCR activation are a large outward movement of TM6 (1.0−1.4 nm) and a slight inward shift of TM7 on the intracellular side (Figure 1), which create a cavity for binding of cytosolic proteins.
Conserved changes in the extracellular part are more difficult to discern due to the lower degree of sequence conservation in this region. In general, the structural changes are relatively subtle and involve only a small contraction of the orthosteric site.5−7 In the case of the β2AR, the catechol group of adrenaline forms hydrogen bonds with Ser2035.42 and Ser2075.46 (superscripts denote Ballesteros–Weinstein numbering16), which leads to an ∼0.2 nm inward bulge of TM5. These structural changes then propagate through the receptor via several conserved motifs. The rearrangement of TM5 influences a connector region (P5.50I3.40F6.44 motif), which is in contact with the highly conserved Asp792.50 and N7.49P7.50xxY7.53 motif via a network of ordered water molecules. A water-filled cavity surrounding Asp792.50 contributes to the stabilization of a sodium ion in several crystal structures of the inactive state, e.g., for the β1AR.17,18 Upon activation, the cavity collapses, leading to dehydration, displacement of the sodium ion and potentially protonation of Asp792.50.19,20 Activation also involves a twist of the NPxxY motif, which reorients Tyr3267.53 to a position where it can form a water-mediated interaction with Tyr2195.58 (Y–Y interaction)21 and together with the displacement of TM6 enable formation of the G protein binding site. Characterization of the role these individual microswitches play in determining the conformational ensemble of structures could guide the design of drugs with tailored signaling profiles.
The allosteric control of GPCR activation by extracellular ligands cannot be fully understood from the static structures captured by crystallography or cryo-EM. Mutagenesis and spectroscopy studies22−25 have suggested that the efficacy of a ligand is determined by a complex interplay between different microswitches and that population of distinct states leads to specific functional outcomes. Molecular dynamics (MD) simulations are well suited to study the conformational landscape of GPCRs as this method can provide an atomistic view of the flexible receptor in the presence of a membrane, an aqueous solvent, and ligands.26−34 The seminal paper by Dror et al.31 provided insights into the activation mechanism of the β2AR by monitoring how a crystallized active conformation relaxed to an inactive state upon removal of the intracellular binding partner, a G protein mimicking nanobody, using MD simulations. Although key conformational changes involved in the transition from active to inactive conformations were identified in these simulations, this approach has inherent limitations. Indeed, understanding the roles of different microswitches in activation and the strength of the coupling between these requires mapping of the relevant free energy landscapes, which are still too costly to calculate using brute-force MD simulations. Enhanced sampling methods, on the other hand, provide a means to explore the conformational landscapes of proteins at a relatively low computational cost35 and have been exploited to study the activation mechanism of GPCRs.26−28,30,32−34
In this work, we aimed to identify the most probable path describing the transition between inactive and active states of β2AR and to characterize the full conformational ensemble along this pathway at an atomistic level of resolution. A new version of the string method with swarms of trajectories was first developed for this purpose. The free energy landscapes associated with β2AR activation revealed that whereas agonist binding is only loosely connected to outward movement of TM6, the coupling between microswitch pairs in the transmembrane region ranges from weak to strong. We observed sodium interacting with several binding sites in a state-dependent manner that was consistent with experimental data. Our approach also allowed us to investigate the influence of Asp792.50 protonation and sodium ions on the energy landscape. A protonated (neutral) Asp792.50 favored active-like conformations and led to a distinct conformational state of the NPxxY motif, which closely resembles agonist-bound and active structures of other class A GPCRs. Finally, we demonstrated that the collective variables (CVs) used in this study discriminate active from inactive class A GPCR structures in general. By using the activation path of β2AR as input to simulations of other receptors, our approach can therefore likely be transferred to study activation of other class A GPCRs at a modest computational cost.
Methods
All swarms of trajectories simulations were initiated with the coordinates from the crystal structure of the active β2AR solved in complex with the agonist BI-167107 (hereafter termed the agonist) and an intracellular nanobody (PDB entry 3P0G) (the first two simulation systems in Table S1). The Asn187Glu mutation in 3P0G was reverted, and Glu1223.41 was protonated due to its localization in a hydrophobic pocket, as has been common practice in other simulations of this receptor.36 Residues His1724.64 and His1784.70 were both protonated at the ε position. The systems were parametrized using the CHARMM36m force field37 and the TIP3P water model.38 The protein was inserted in a POPC39 bilayer and solvated in explicit solvent. Na+ and Cl– ions were added at a concentration of 0.15 M. System preparation was performed using CHARMM-GUI.40 MD simulations were run with GROMACS 2016.541 patched with plumed 2.4.142 under a 1 atm pressure and at 310.15 K.
To identify CVs, we performed clustering43 on the frames from an unrestrained MD simulation trajectory of β2AR (condition A in Dror et al.31) (Figure S1). The CVs were selected by training a multilayer perceptron classifier44 using as input all the inter-residue distances and as output the cluster ID, followed by using Deep Taylor decomposition45 to find key distances that could discriminate between clusters.
The end points of the main strings describing the transition between inactive and active states (subscript t) were fixed to the output coordinates of equilibrated structures of each state (PDB entries 2RH1(5) and 3P0G,6 respectively). The initial path for simulations of Holot was guessed using data from Dror et al.. A rough estimate of the free energy landscape was calculated from the probability density landscape estimated using the Scikit-learn44 kernel density estimator with automatic bandwidth detection (Figure S2). Two additional short strings were set up to increase the level of sampling in the active (subscript a) and inactive (subscript i) regions. The average, partially converged path between iterations 20 and 30 from Holot was used as the input path for simulations Apot and HoloAsh79,t. All active (Apoa, Holoa, and HoloAsh79,a) and inactive (Apoi, Holoi, and HoloAsh79,i) substrings were initiated as straight paths between the end points. Even though the system was initiated from the nanobody-bound structure 3P0G, the active state substring sampled conformations closer to the G protein-bound structure 3SN6 (Figure S3).7 The swarms of trajectories simulations with optimizations (Supplementary Methods and Figures S4 and S5) were run for 300 iterations, at which point the strings had not changed on average for many iterations (Figures S6 and S7) and the posterior distribution of free energy profiles given the data was narrow (Figure S8).
By discretizing the system into microstates,
or bins, we were able
to use the short trajectories from the swarms to create a transition
matrix and derive the free energy distribution of the system46 along a given variable (Figure S9). In practice, the transition probabilities Tij of the transition matrix T can be estimated from the normalized number of transitions, Nij, from bin i to bin j: 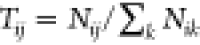 . The transition
matrix of a physical system
at equilibrium is constrained by detailed balance, such that for the
stationary probability distribution, ρ, ρiTij =
ρjTji. A Metropolis Markov chain Monte Carlo (MCMC) method
was used to sample over the posterior distribution of transition matrices,
given the unregularized elements of Tij,47 and thereby obtain
a distribution of free energy profiles for ρ (Figures S8, S10, and S11). All code to run the simulations
and reproduce the results in this paper is available for download.48
. The transition
matrix of a physical system
at equilibrium is constrained by detailed balance, such that for the
stationary probability distribution, ρ, ρiTij =
ρjTji. A Metropolis Markov chain Monte Carlo (MCMC) method
was used to sample over the posterior distribution of transition matrices,
given the unregularized elements of Tij,47 and thereby obtain
a distribution of free energy profiles for ρ (Figures S8, S10, and S11). All code to run the simulations
and reproduce the results in this paper is available for download.48
Results
Optimizing the Enhanced Sampling Protocol for Increased Efficiency
We aimed to compute the most probable transition pathway linking the inactive and active states of β2AR and the relative free energy of the states lining this pathway. For this purpose, we chose the string of swarms method.49 In this framework, the minimum free energy path in an N-dimensional collective variable (CV) space is estimated iteratively from the drift along the local gradient of the free energy landscape (Figure S5). From each point on the string, a number of short trajectories are launched (a swarm), which enables us to calculate the drift. The string is then updated considering this drift and reparametrized to ensure full sampling of the configurational space along the pathway. Convergence is reached when the string diffuses around an equilibrium position. The method allows sampling of a high-dimensional space at a relatively inexpensive computational cost because it samples only along the one-dimensional path of interest.
We characterized the pathway linking equilibrated conformations originating from active and inactive structures of the β2AR (PDB entries 3P0G and 2RH1, respectively), adding two short strings spanning the active and the inactive regions to increase sampling of the end state environments (see Methods and Supplementary Methods). First, we characterized a CV set that embeds receptor activation by automatically inferring the inter-residue distances that are related to activation. On the basis of one of Dror et al.’s simulation trajectories of spontaneous deactivation of the β2AR,31 we identified metastable states by clustering simulation configurations, followed by classification of these by training a fully connected neural network to identify states.50 The most important input features for classification were identified via deep Taylor decomposition45 and taken as CVs (Figure 2 and Figure S1). The set of CVs we inferred was a network of interatomic distances among all seven TM helices (Figure 2). Encouragingly, this five-dimensional CV set captured the main degrees of freedom implicated in β2AR activation, including the large outward movement of TM6 and a smaller inward shift of TM7 at the intracellular face of the receptor.
Figure 2.
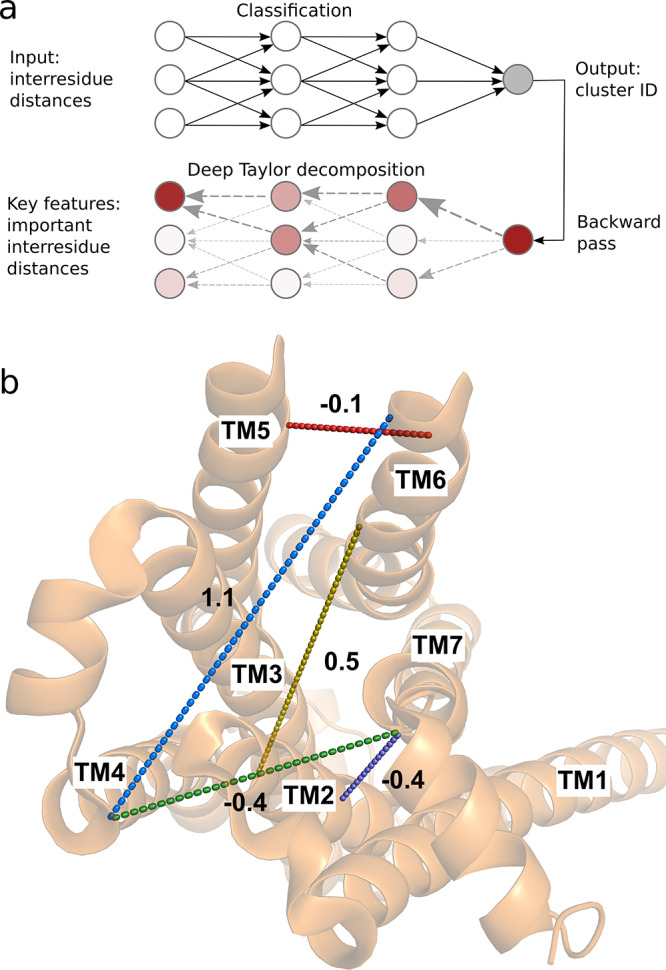
Five-dimensional collective variable (CV) space used to optimize the minimum free energy path identified in a data-driven manner. (a) A fully connected neural network was trained to classify configurations in active, intermediate, and inactive metastable states (clusters). Deep Taylor decomposition was then used to identify the most important input inter-residue distances for the classification decision. The top-ranked distances were used as CVs. (b) The five CVs used in this work projected onto an intracellular view of the active crystal structure (PDB entry 3P0G). The CVs corresponding to TM2–TM7, TM6–TM4, TM7–TM4, TM3–TM6, and TM6–TM5 distances defined in Table S2 are shown as purple, blue, green, yellow, and red dashed lines, respectively. The change of these distance CVs from the inactive to the active state structures is reported in nanometers.
To accelerate convergence of the string optimization, we initiated our string simulation from a rough guess of the minimum free energy landscape. The latter was obtained by estimating the density of points from Dror et al.’s trajectory in this CV space (Figure S2). We also introduced algorithmic improvements to the string of swarms method. We adaptively chose the number of trajectories in a swarm, gradually increased the number of points on the string, and introduced a reparametrization algorithm that improves performance and promotes exchanges of configurations between adjacent string points (Supplementary Methods and Figures S4 and S5). We carried out 300 iterations of string optimization for each system, considering a number of points on the string ranging from 20 to 43 and swarms consisting of 16–32 10 ps trajectories. Because we sought to sample only the vicinity of the most probable activation path, derivation of a converged free energy landscape required a mere total of approximately 2–3 μs of simulation time (Figures S6–S8, S10, and S11).
Minimum Free Energy Pathway of β2AR Activation
We derived the most probable transition path (Figure S6) between the active and inactive states of β2AR in the absence and prescence of an agonist ligand. The swarm trajectories allowed us to compute transitions between discrete states in the vicinity of the most probable transition path (Supplementary Methods and Figure S9) and to derive the associated free energy landscape describing the outward movement of TM6 and the inward shift of TM7 upon activation (Figure 3).
Figure 3.

Free energy landscapes are projected onto the first two CVs used to optimize the minimum free energy path. The minimum free energy pathways were optimized (a) in the absence and (b) in the presence of the bound agonist. Characteristic events occurring during activation (see Table S3 for their definition) are marked on the string. The active and inactive labels mark the regions close to the active and inactive structures (PDB entries 3P0G(6) and 2RH1,5 respectively).
For the apo receptor, one distinguishes three minima: one in the active region, one in the inactive one, and an intermediate one between these (Figure 3a). As anticipated, regions close to the inactive end point are stabilized relative to the other two states. Binding of an agonist changes the number of minima to four and shifts the relative stability of states, making regions close to the active conformation of lower free energy (Figure 3b). A number of characteristic variables (defined in Table S2−S3) were calculated for the last iteration of the swarm of trajectories simulations. By localizing sudden shifts in the values of these parameters, we could pinpoint the location of important events in activation on the string (Figure 3). In the absence of a bound agonist, the connector assumes an active conformation in the early stages of activation. The Asp792.50 cavity then dehydrates, followed by the formation of the Y–Y interaction and NPxxY twist. Finally, the TM5 bulge forms before the fully active conformation is reached, confirming an allosteric communication between the orthosteric site and intracellular region.
Binding of the agonist ligand modifies the order in which the helices rearrange, as can be seen when projecting the minimum free energy path along various CVs (Figure 3 and Figure S6). The most probable activation pathway begins with an outward movement of TM6, followed by the inward shift of TM7. The connector and the bulge in TM5 are locked in their active-like states in the presence of the agonist, which enables large outward movements of TM6. As in the apo case, to reach the fully active conformation, the Asp792.50 cavity is dehydrated and the Y–Y interaction forms shortly before the NPxxY twists. In the holo simulation, the binding mode of the ligand remained stable along the activation path (Figure S12a,b,k,l), and it maintained stable interactions with key residues Asp1133.32, Ser2035.43, Ser2075.46, and Asn3127.38 (Figure S12a–j). Occasionally, the shortest heavy atom distance from the ligand to Ser2035.43 was almost 4 Å, although these fluctuations were independent of the receptor state (Figure S12e,f).
Coupling between Orthosteric and G Protein Binding Sites
As the final configurations of the string are at equilibrium in all dimensions, the trajectories from the last iterations can be used to compute the free energy landscape as a function of any variables (Supplementary Methods). This allowed a detailed analysis of how conformational changes induced by agonist binding propagate through the receptor to the G protein binding site. The roles of conserved microswitches were assessed by comparing free energy landscapes in the absence and presence of a bound agonist. We first evaluated how the TM5 bulge, which reflects how the binding site contracts upon activation, influences the intracellular distance between TM6 and TM3 (Figure 4a,b). In the absence of a bound agonist, the receptor accessed both active and inactive conformations of the binding site, with the minimum of the free energy located close to the inactive state distance between TM3 and TM6. The TM3–TM6 distance could be as large as 1.5 nm when the ligand binding site was in the active conformation, an observation compatible with basal activity. Agonist binding led to the stabilization of the TM5 bulge in the orthosteric site (Figure 4b). Although both inactive and active conformations remained accessible in the presence of the ligand, the minimum of the TM3–TM6 distance was shifted toward a more active-like state. A remarkable long-range allosteric coupling (>2 nm) between the orthosteric and G protein binding sites was hence captured by our simulations. The 0.5 nm outward movement of TM6 at the minimum of the landscape (Figure 4a,b) is smaller than that observed in active crystal structures, in agreement with experiments demonstrating that the fully active conformation can be stabilized only in the presence of an intracellular partner.51,23
Figure 4.

Free energy landscapes projected along variables of interest highlight changes in the pairwise coupling of microswitches following binding of an agonist ligand (middle column) and protonation of conserved residue Asp792.50 (right column). The free energy landscapes are projected along (a–c) the TM5 bulge (distance between Ser2075.46 and Gly3157.41, representing the ligand binding site contraction) and the distance between Leu2726.34 and Arg1313.50, representing the outward movement of TM6; (d–f) the TM5 bulge (distance between Ser2075.46 and Gly3157.41) and the difference between the RMSD of Ile1213.40 and Phe2826.44 heavy atoms to the active and inactive crystal structures,5,6 representing the connector region ΔRMSD; (g–i) the connector region ΔRMSD and the number of water molecules within 0.8 nm of Asp792.50, representing the hydration of the Asp792.50 cavity; (j–l) the connector region ΔRMSD and the RMSD of the NPxxY motif relative to the inactive structure 2RH1; (m–o) the connector region ΔRMSD and the distance between the two tyrosines implicated in the Y–Y interaction (Y–Y motif), and (p–r) the Y–Y motif distance and the displacement of TM6. Active and inactive state regions are labeled for each variable pair. Low-free energy regions are colored red, and high-free energy regions light yellow. Free energies are reported in kilocalories per mole. See Table S2 for microswitch definitions. (s–v) Vignettes showing the conformation of the different microswitches in the active and inactive structures 3P0G (orange) and 2RH1 (white): (s) the TM5 bulge shifting Ser2075.46 inward, (t) the connector region containing Ile1213.40 and Phe2826.44, (u) the Asp792.50 cavity, NPxxY motif, and the two tyrosines, Tyr2195.58 and Tyr3267.53, of the Y–Y motif, and (v) the outward movement of TM6 upon activation.
Propagation of Activation through Microswitches
Structural changes in the orthosteric site of the β2AR (Figure 4s) have been proposed to propagate toward the intracellular part via a “connector” centered around Ile1213.40 and Phe2826.44 (Figure 4t).6 Whereas we found the TM5 bulge and outward movement of TM6 to be loosely coupled, the free energy landscapes demonstrated that changes in the orthosteric site have a direct influence on the connector region (Figure 4d,e). In the absence of an agonist, both inactive and active conformations of the connector were populated and the connector region could assume an active-like state even if the TM5 bulge was inactive (Figure 4d). In contrast, agonist binding resulted in a single free energy minimum where both the TM5 bulge and the connector were constrained to their active conformations (Figure 4e).
From the connector region, activation is propagated via several conserved motifs (Figure 4g,h,j,k,m,n,u).3 To investigate the communication between microswitches in the core of the TM region, we analyzed if the connector was coupled to solvation of the cavity surrounding Asp792.50, to the conformation of the NPxxY motif, and to the conformation of the Y–Y motif. In the apo state, an inactive connector region was tightly coupled to a hydrated cavity with approximately 10–17 waters (Figure 4g) and an inactive NPxxY motif (Figure 4j). This result is consistent with a high-resolution crystal structure of an inactive β1 adrenergic receptor (PDB entry 4BVN), in which an ordered solvent network in this region was identified.17 In contrast, the active conformation of the connector was compatible with both the fully hydrated cavity and a desolvated state with only four or five water molecules as well as an inactive and active NPxxY motif. Interestingly, the connector configuration was more tightly coupled to the Y–Y motif. An inactive connector was coupled to a broken Y–Y interaction, whereas it was in the intermediate state or fully formed with an active connector (Figure 4m).
The free energy landscapes suggested that binding of an agonist reduces the number of inactive-like states of the Asp792.50 cavity, NPxxY, and Y–Y motifs (Figure 4h,k,n) as well as the orientation of Met822.53 (Figure S11), a residue located one helix turn above Asp792.50 that has been the focus of nuclear magnetic resonance (NMR) studies.52 For the Asp792.50 cavity and NPxxY motif, both active and inactive conformations were accessible in the presence of an agonist, but the active conformations were more favored energetically (Figure S13). The presence of an agonist resulted in an energy landscape with one active-like and two intermediate distances of the Y–Y motif. The two latter minima were shifted by approximately 0.1 nm toward the active conformation compared to the apo state (Figure 4n).
The final combination of microswitches connected the Y–Y motif to the motion of TM6 (Figure 4p,q). In the apo state, there was a relatively tight coupling between the Y–Y interaction and the TM3–TM6 distance, with several metastable states lining the minimum free energy pathway between inactive and active conformations (Figure 4p). Agonist binding did not alter the coupling between these two microswitches but generally tilted the free energy landscape toward more active states along both of these dimensions (Figure 4q).
Access to the free energy landscapes in the absence and presence of a ligand thus enabled us to characterize the involvement of each microswitch in the transmission of allosteric coupling between the orthosteric ligand and the intracellular partner binding sites.
State-Dependent Sodium Binding
Several structures of class A GPCRs determined in the inactive state have revealed a sodium ion bound to the conserved Asp792.50 (Figure 5a).18 Binding of sodium to this residue has also been studied for several class A GPCRs with simulations.53−55 To investigate potential interactions between Asp792.50 and sodium ions, which were initially randomly added at physiological concentrations to the simulation system, we calculated the free energy landscape as a function of TM6 displacement and the distance between Asp792.50 and the closest sodium ion (Figure 5b,c and Figure S11j–l). In the apo form, five meta-stable states were identified (Figure 5b). In the active-like conformation of TM6, the closest sodium interacted with a specific site in the second extracellular loop. Notably, sodium ions have been confirmed to bind in this pocket in crystal structures of adrenergic receptors (Figure 5a).11 In an intermediate conformation of TM6, the closest sodium was either bound to the second extracellular loop or descended into the binding site and formed a salt bridge to Asp1133.32. Finally, in the completely inactive state of TM6, the closest sodium ion remained bound to Asp1133.32 or was located in the Asp792.50 cavity. Sodium was hence only present in the Asp792.50 cavity when TM6 had completely relaxed to an inactive conformation, and even small increases of the TM3–TM6 distance were incompatible with ion binding to this site. In the agonist-bound receptor with Asp1133.32 ionized, sodium remained strongly bound to the second extracellular loop irrespective of the TM3–TM6 distance (Figure 5c). This difference is likely due to access to the binding site via the extracellular side being blocked by the bound ligand19,53,55 but could also be a result of conformational changes induced by the agonist. Spontaneous Na+ binding to appropriate protein regions further confirms the relevance of the conformational sampling enabled by our computational protocol.
Figure 5.
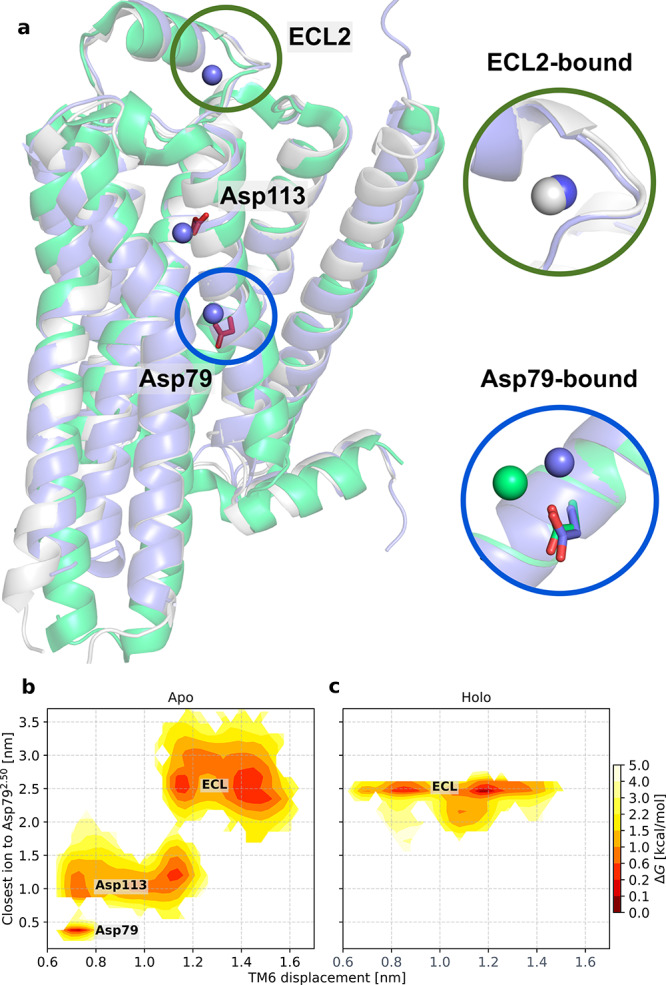
Sodium ions bind to three sites in a state-dependent manner. (a) The inactive β1AR (PDB entry 4BVN(17)) and β2AR (PDB entry 4LDE(11)) structures and a representative inactive state simulation snapshot are shown as green, white, and blue cartoons, respectively. Representative positions of sodium ions in the MD simulations are shown as blue spheres. In the insets, a comparison to the positions of sodium ions found in the crystal structures of active β2AR (white sphere) and inactive β1AR (white spheres) is shown. (b and c) Free energy landscapes along the TM6 displacement and the shortest distance between Asp792.50 and a sodium ion for the apo and holo simulations.
Impact of Asp792.50 Protonation
Protonation of two ionizable residues, Asp792.50 and Asp1303.49, has been proposed to be involved in GPCR activation.20,31,56 In particular, MD simulations have indicated that Asp792.50, the most conserved residue among class A GPCRs, has a pKa value close to physiological pH and that the ionization state of this residue changes upon activation.19,20 As a previous simulation study of the β2AR showed that Asp792.50 (but not Asp1303.49) may alter the activation pathway,31 we repeated the calculations of the minimum free energy pathway of activation with this residue in its protonated (neutral) form (Figure S7).
The free energy landscapes describing changes in the orthosteric site were similar to those obtained in simulations with Asp792.50 ionized (Figure 4c,f). There was a weak coupling between the TM5 bulge and the intracellular region, with two major energy wells describing the conformation of TM6. Compared to the agonist-bound receptor with Asp792.50 ionized, the minima were shifted further toward active-like conformations for the protonated state (Figure 4c). The TM5 bulge remained strongly coupled to conformational changes observed in the connector region irrespective of the ionization state of Asp792.50 (Figure 4e,f). The strongest effects of Asp792.50 protonation were observed for the hydrated cavity surrounding this residue (Figure 4h,i), NPxxY (Figure 4k,l), and Y–Y motifs (Figure 4n,o). Whereas the free energy landscapes showed that both active- and inactive-like conformations of these switches were populated in simulations with ionized Asp792.50, the protonated state resulted only in energy wells close to the active conformation (Figure 4i,l,o). It was also evident that TM6 was stabilized in more active-like conformations by the protonated Asp792.50 (Figure 4r). Interestingly, intermediate states in which the Y–Y motif, as well as the NPxxY, adopted an active-like conformation and a small TM6 displacement were also populated (Figure 4r and Figure S13). Such an intermediate state has been observed, albeit rarely, in simulations considering a protonated Asp792.50.31
Comparison of representative structures from the simulations of ionized and protonated Asp792.50 in active-like states revealed that two distinct conformations of the NPxxY motif were obtained (Figure 6). The simulations carried out with ionized Asp792.50 favored structures that were more similar to the crystal structure of the active β2AR. An alternative conformation of the active NPxxY motif appeared for the protonated Asp792.50, which was not favored energetically in the simulations of the ionized form (Figures S10k,l and S13). Although this conformation of the NPxxY motif did not match any β2AR crystal structure, it was strikingly similar to conformations observed in crystal structures of other class A GPCRs in either agonist-bound (serotonin 5-HT2B and A2A adenosine receptors) or active (angiotensin II type 1) conformations (Figure 6).57,58 Our protocol thus allowed us to sample metastable states not yet resolved by experimental structure determination that may play a role in activation.
Figure 6.
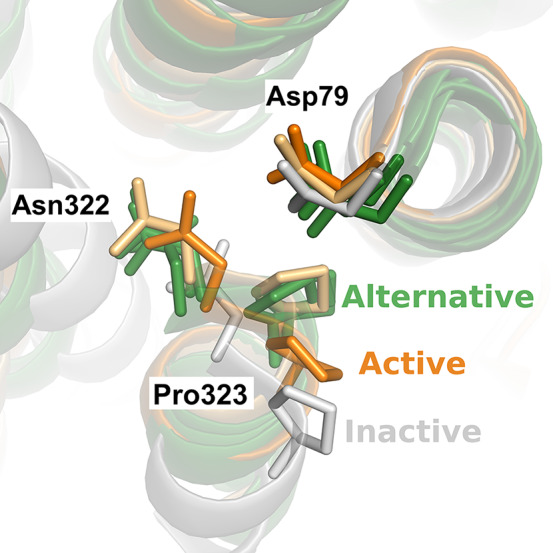
Alternative conformation of the NPxxY identified along the most probable pathway calculated with a protonated Asp792.50. A representative simulation snapshot of active-like states of the β2AR with Asp792.50 ionized and protonated are colored orange and light orange, respectively. A representative simulation snapshot of the inactive-like state of the β2AR is colored white. The structural comparison highlights the resemblance between an alternative conformation of the NPxxY motif (light orange) that is favored by Asp792.50 protonation in the simulations and observed in three crystal structures of other class A GPCRs. Crystal structures of the other GPCRs are colored green (PDB entries 6DRX,57 agonist-bound 5-HT2B serotonin receptor; 3QAK,59 agonist-bound A2A adenosine receptor; and 6DO1,60 angiotensin II type 1 receptor in an active conformation).
Identified CVs Capture Activation of Many Class A GPCRs
Efficient characterization of free energy landscapes with the string method relies on selection of appropriate CVs, which is a nontrivial task. Here, CVs were derived from a conventional MD trajectory of β2AR deactivation in a data-driven fashion. Considering that the conformational changes involved in class A GPCR activation are largely conserved,3 we explored the possibility of transferring the CVs to the conformational sampling of other GPCRs. We mapped the CVs identified for β2AR to 10 other class A GPCRs with active and inactive structures available (Figure 7). Strikingly, the active and inactive structures clearly separate into two distinct clusters. Together with the fact that we could capture relevant receptor conformations that were not observed in the MD trajectory used to identify CVs (Figure 6), this indicates that our approach could potentially be used to characterize the activation of many class A GPCRs.
Figure 7.
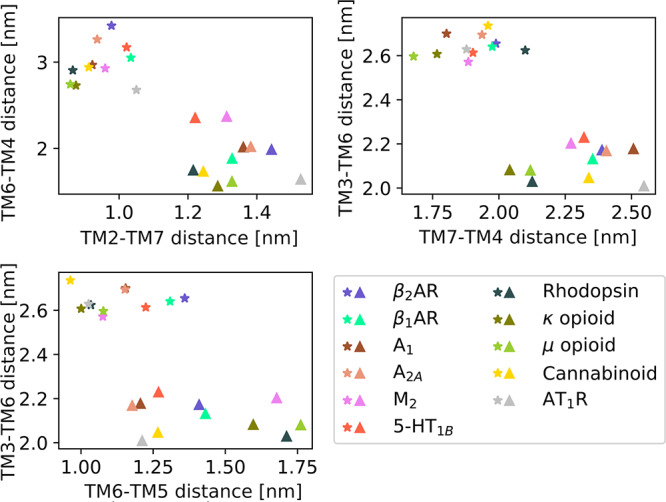
Active and inactive class A GPCR structures cluster into two distinct groups in the five-dimensional CV space used in this work. β2AR and 10 active (stars) and inactive (triangles) structures of other class A GPCRs are projected onto the five original CVs. Details of the mapping can be found in Table S4. Their clustering into two groups highlights that the activation mechanism of all class A GPCRs can likely be described by the CVs identified in this work.
Discussion
Experimental structures of the β2AR in inactive and active conformations provide a basis for the molecular understanding of GPCR signaling. However, it has become increasingly clear that the static structures do not capture all functionally relevant states involved in activation of these molecular machines.26,27,32,33 In a pioneering study, Dror et al. gained insights into the activation pathway of the β2AR from long time scale MD simulations.31 Despite this computational tour de force enabled by the development of hardware specialized for MD, these simulations did not allow quantification of the accessibility of different conformational states. The approach proposed in this work builds on the data generated by Dror et al. and on methodologies suggested in previous molecular simulation studies27,29,30,33 to further allow the assessment of the impact of agonist binding on microswitches involved in activation.
Our work recapitulates a number of key findings of previous enhanced sampling27,29,30,32,33 and long time scale simulations of GPCRs.31 In agreement with previous work, our simulations revealed a complex conformational landscape of the β2AR with only weak coupling between the othosteric site and G protein binding site. Through the analysis of the free energy landscapes, we could quantify the coupling between spatially connected microswitches in the transmembrane region, which ranged from strong to relatively weak and was influenced by protonation of Asp792.50. Our study also further illustrates that the energy landscapes depend on the variables chosen to project the conformational states. This is not an artifact of the protocol but rather an inherent limitation of dimensionality reduction. In particular, it is clear that considering only three states along the activation path (active, intermediate, and inactive states) does not allow us to capture the complexity of the conformational changes induced by ligand binding.26,27,32,33 This work, through the resolution of the complete conformational ensemble lining the most probable activation pathway, can aid in determining the placement of spectroscopic probes to monitor the distribution of specific microswitches under different conditions.
Our work shows agreement with spectroscopy studies, although a quantitative comparison remains challenging. Whereas the β2AR crystal structure solved in complex with an agonist in the absence of an intracellular partner (e.g., G protein or G protein-mimicking nanobody) is similar to those determined with antagonists,36 NMR spectroscopy experiments have demonstrated that agonist binding stabilizes other conformations in certain parts of the TM region, e.g., close to Met2155.54 and Met822.53.52 For example, the region surrounding Met822.53, located one helical turn above Asp792.50, was shown to adopt three conformations in the prescence of a neutral antagonist (with signaling properties similar to those of the apo receptor). A bound agonist, on the other hand, restricted this region to a single active-like state.51,52 Similar to these experiments, our free energy landscapes demonstrate that several conformations of the connector, Asp792.50 cavity, and the orientation of Met822.53 are available under the apo conditions (Figures S10g–i,m–o and S11d–f). In particular, Figure S11d–f shows three conformational Met822.53 states of comparable free energy for the apo receptor, whereas the landscape is significantly shifted to favor a single active state conformation for the agonist-bound receptor. Protonation of Asp792.50 further increases this shift. In the presence of an agonist, the connector is locked in a single state and a desolvated state of the Asp792.50 cavity is stabilized, which creates a more active-like receptor conformation in the vicinity of Met822.53. In agreement with the NMR data, we also find that the agonist cannot stabilize the fully active conformation of the receptor and that TM6 accesses several intermediate conformations that are distinct from those observed in crystal structures.
The rapidly increasing number of crystal structures provides insights into how agonist binding to GPCRs, despite recognizing disparate molecules, can result in very similar conformational changes. The bulge of TM5, which we found to be strongly coupled to the connector region, is also present in active state structures of other GPCRs.61,62 Whereas this conformational change is the result of a direct interaction with TM5 in the case of the β2AR, interactions of the agonist with other helices appear to indirectly influence the same region in the case of the μ opioid and A2A adenosine receptor.61,62 In other cases, the connector region was found to have a similar arrangement in the active and inactive states (e.g., the M2 muscarinic receptor).63 In such cases, receptor activation may be controlled by direct modulation of the Asp792.50 cavity, e.g., via the conserved Trp2866.48. Several recent experimental studies have demonstrated that Asp792.50 and residues forming the hydrated TM cavity play an important role in signaling and can even steer activation via G protein-dependent and G protein-independent pathways.25,64,65 One mechanism by which Asp792.50 could control the receptor conformation is via its protonation state. Agonist binding destabilizes the water network in the solvated TM cavity, which may lead to a larger population of protonated Asp792.50 and disrupt binding of sodium to this pocket.19,20 In turn, the protonated Asp792.50 stabilizes a structure of the NPxxY that has been observed for other class A GPCRs crystallized in active and active-like states, suggesting that this alternative conformation of TM7 may be relevant for function. NMR experiments have shown that different ligands stabilize different active states.66 In particular, agonists that preferentially signal via arrestin mainly affect the conformation of TM7, whereas a neutral agonist stabilizes both the G protein and the arrestin signaling state. While two distinct active state conformations of the NPxxY motif were identified in our simulations for a neutral agonist, a ligand with different biased signaling properties may preferentially stabilize one of these states. Interestingly, the ionized and protonated forms of Asp792.50 also stabilize different TM6 conformations, which could change the intracellular interface that interacts with G proteins and arrestins.9 These results, combined with the fact that Asp792.50 is the most conserved residue in the class A receptor family, support the idea that this region is a central hub for controlling receptor activation.
With the protocol developed herein, we sampled enough transitions along the activation path to obtain free energy profiles of GPCR activation by accumulating a few microseconds of total simulation time. Compared to regular MD simulations, the optimized string of swarms method can thus provide reliable energetic insights using 1–2 orders magnitude less simulation time than plain MD simulations31,32 and comparable to other enhanced sampling methods such as GaMD.26 From a practical point of view, the short trajectories in the swarms of trajectories method are easy to run in parallel with minimal communication overhead even in a heterogeneous computational environment. An important consideration that has guided our choice of enhanced sampling methodology is that the method has the advantage of functioning well in high-dimensional space, i.e., with many CVs. This is because we only optimize the one-dimensional string instead of opting to sample the entire landscape spanned by the CVs. This means we can utilize a high-dimensional CV space, thus alleviating the need to reduce the dimensionality of the conformational landscape to one or two dimensions, as is done in most CV-based methods such as umbrella sampling or metadynamics.67
On the other hand, a well-known limitation of the string with swarms method is that it is guaranteed to converge to the most probable path closest only to the initial guess of the path, and not necessarily the globally most probable path. The naive assumption of a straight initial path is not guaranteed to converge to the latter. Here we have proposed to alleviate this shortcoming by exploiting previous knowledge of the activation pathway and deriving an initial guess of the pathway likely to be close to the globally most probable path. An initial pathway can also be transferred from a similar system (as revealed in Figure 7) or inferred from available experimental data. If multiple pathways are nevertheless expected, the protocol presented herein provides the tools necessary to compare them: the swarms from separate string simulations can be included in the same transition matrix and used to compute a single free energy landscape. It is also worth noting that the relative free energies of the end point conformations (evaluated by integration over the end point basins) do not depend on the transition pathway and should be estimated correctly. The protocol is also applicable to complex transitions involving many intermediate states: in such a case, one may launch multiple strings to explore different parts of the activation path, and let every substring converge separately, eventually combining the transitions derived from them to yield a single free energy landscape. Finally, we note that the string of swarms method can also be used as a complementary method to instantiate Markov state model (MSM) simulations.68
Conclusion
Despite the major progress in structural biology for GPCRs, many aspects of receptor function remain poorly understood. Here, we derived the most probable activation path of β2AR using an improved variant of the string method with swarms of trajectories. The associated free energy landscapes revealed that agonist binding is only loosely connected to outward movement of TM6, while the coupling between microswitch pairs in the transmembrane region, in particular the TM5 bulge and the connector conformation, was strongly coupled to ligand binding. We also investigated the influence of sodium binding to known sodium interaction sites and found that a sodium ion travels along a distinct pathway from the Asp792.50 cavity via the orthosteric site as the apo receptor is activated. We then studied the effect of Asp792.50 protonation on the agonist-bound receptor and found that a protonated Asp792.50 did not shift the free energy landscape to favor active-like conformations; it also led to a distinct conformational state of the NPxxY motif. This protonated active state was reminiscent of active structures of other class A GPCRs, suggesting that protonation of this conserved residue is a shared feature across several GPCRs. We expect that insights from atomistic MD simulations will continue to be valuable tools for interpreting experimental data. In particular, we anticipate that our methodology will enable further insights into how binding of different ligands influences the conformational landscape, potentially making it possible to design ligands with biased signaling properties.25 The method is equally well suited to study the effect of allosteric modulators and the influence of different lipidic environments.69 As the same approach can likely be transferred to other class A GPCRs, future applications will shed light on the common principles of activation as well as on the details that give each receptor a unique signaling profile, paving the way to the design of more effective drugs.
Acknowledgments
The simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the PDC Centre for High Performance Computing (PDC-HPC). The authors thank D. E. Shaw Research for providing access to their MD trajectories.
Glossary
Abbreviations
- MD
molecular dynamics
- GPCR
G protein-coupled receptor
- CV
collective variable
- β2AR
β2 adrenergic receptor
- PDB
Protein Data Bank.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.9b00842.
Tables S1–S4, Supplementary Methods (system preparation, MD simulation details, CV selection, swarms of trajectories simulation setup, algorithmic improvements to the swarms of trajectories method, and free energy computation), and Figures S1–S13 (PDF)
This work was supported by grants from the Göran Gustafsson Foundation and Science for Life Laboratory to J.C. and L.D. This work was also supported by grants from the Swedish Research Council (2017-4676) and the Swedish strategic research program eSSENCE to J.C.
The authors declare no competing financial interest.
Supplementary Material
References
- Fredriksson R.; Lagerström M. C.; Lundin L.-G.; Schiöth H. B. (2003) The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63, 1256–1272. 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Hauser A. S.; Attwood M. M.; Rask-Andersen M.; Schiöth H. B.; Gloriam D. E. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery 16, 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis W. I.; Kobilka B. K. (2018) The Molecular basis of G protein–coupled receptor activation. Annu. Rev. Biochem. 87, 897–919. 10.1146/annurev-biochem-060614-033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C. (2017) Structural Basis for G Protein-Coupled Receptor Activation. Biochemistry 56, 5628–5634. 10.1021/acs.biochem.7b00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V.; Rosenbaum D. M.; Hanson M. A.; Rasmussen S. G. F.; Thian F. S.; Kobilka T. S.; Choi H.-J.; Kuhn P.; Weis W. I.; Kobilka B. K.; Stevens R. C. (2007) High-Resolution Crystal Structure of an Engineered Human 2-Adrenergic G Protein-Coupled Receptor. Science 318, 1258–1265. 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G.; Choi H.-J.; Fung J. J.; Pardon E.; Casarosa P.; Chae P. S.; DeVree B. T.; Rosenbaum D. M.; Thian F. S.; Kobilka T. S.; et al. (2011) Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469, 175–180. 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G.; DeVree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D.; Mathiesen J. M.; Shah S. T. A.; Lyons J. A.; Caffrey M.; Gellman S. H.; Steyaert J.; Skiniotis G.; Weis W. I.; Sunahara R. K.; Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 477, 549–555. 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masureel M.; Zou Y.; Picard L.-P.; van der Westhuizen E.; Mahoney J. P.; Rodrigues J. P. G. L. M.; Mildorf T. J.; Dror R. O.; Shaw D. E.; Bouvier M.; Pardon E.; Steyaert J.; Sunahara R. K.; Weis W. I.; Zhang C.; Kobilka B. K. (2018) Structural insights into binding specificity, efficacy and bias of a β2AR partial agonist. Nat. Chem. Biol. 14, 1059–1066. 10.1038/s41589-018-0145-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus D. P.; Strachan R. T.; Manglik A.; Pani B.; Kahsai A. W.; Kim T. H.; Wingler L. M.; Ahn S.; Chatterjee A.; Masoudi A.; Kruse A. C.; Pardon E.; Steyaert J.; Weis W. I.; Prosser R. S.; Kobilka B. K.; Costa T.; Lefkowitz R. J. (2016) Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 535, 448–452. 10.1038/nature18636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Ahn S.; Kahsai A. W.; Meng K.-C.; Latorraca N. R.; Pani B.; Venkatakrishnan A. J.; Masoudi A.; Weis W. I.; Dror R. O.; Chen X.; Lefkowitz R. J.; Kobilka B. K. (2017) Mechanism of intracellular allosteric β2AR antagonist revealed by X-ray crystal structure. Nature 548, 480–484. 10.1038/nature23652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring A. M.; Manglik A.; Kruse A. C.; Enos M. D.; Weis W. I.; Garcia K. C.; Kobilka B. K. (2013) Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579. 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson M. A.; Cherezov V.; Griffith M. T.; Roth C. B.; Jaakola V.-P.; Chien E. Y.; Velasquez J.; Kuhn P.; Stevens R. C. (2008) A Specific Cholesterol Binding Site Is Established by the 2.8 Å Structure of the Human β2-Adrenergic Receptor. Structure 16, 897–905. 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Fenalti G.; Brown M. A.; Katritch V.; Abagyan R.; Cherezov V.; Stevens R. C. (2010) Conserved Binding Mode of Human β2Adrenergic Receptor Inverse Agonists and Antagonist Revealed by X-ray Crystallography. J. Am. Chem. Soc. 132, 11443–11445. 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G. F.; DeVree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D.; Mathiesen J. M.; Shah S. T. A.; Lyons J. A.; Caffrey M.; Gellman S. H.; Steyaert J.; Skiniotis G.; Weis W. I.; Sunahara R. K.; Kobilka B. K. (2007) Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 450, 383–387. 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- García-Nafría J.; Tate C. G. (2019) Cryo-EM structures of GPCRs coupled to Gs, Gi and Go. Mol. Cell. Endocrinol. 488, 1–13. 10.1016/j.mce.2019.02.006. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A., and Weinstein H. (1995) Methods in Neurosciences, pp 366–428, Elsevier. [Google Scholar]
- Miller-Gallacher J. L.; Nehme R.; Warne T.; Edwards P. C.; Schertler G. F.; Leslie A. G.; Tate C. G. (2014) The 2.1 Å resolution structure of cyanopindolol-bound β1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor. PLoS One 9, e92727. 10.1371/journal.pone.0092727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V.; Fenalti G.; Abola E. E.; Roth B. L.; Cherezov V.; Stevens R. C. (2014) Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 39, 233–244. 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickery O. N.; Carvalheda C. A.; Zaidi S. A.; Pisliakov A. V.; Katritch V.; Zachariae U. (2018) Intracellular Transfer of Na+ in an Active-State G-Protein-Coupled Receptor. Structure 26, 171–180.e2. 10.1016/j.str.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan A.; Dror R. O.; Carlsson J. (2014) Insights into the Role of Asp792.50 in β2 Adrenergic Receptor Activation from Molecular Dynamics Simulations. Biochemistry 53, 7283–7296. 10.1021/bi5008723. [DOI] [PubMed] [Google Scholar]
- Latorraca N. R.; Venkatakrishnan A.; Dror R. O. (2017) GPCR dynamics: structures in motion. Chem. Rev. 117, 139–155. 10.1021/acs.chemrev.6b00177. [DOI] [PubMed] [Google Scholar]
- Lamichhane R.; Liu J. J.; Pljevaljcic G.; White K. L.; van der Schans E.; Katritch V.; Stevens R. C.; Wüthrich K.; Millar D. P. (2015) Single-molecule view of basal activity and activation mechanisms of the G protein-coupled receptor β2AR. Proc. Natl. Acad. Sci. U. S. A. 112, 14254–14259. 10.1073/pnas.1519626112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kim T. H.; Masureel M.; Altenbach C.; Yang Z.; Hilger D.; Lerch M. T.; Kobilka T. S.; Thian F. S.; Hubbell W. L.; Prosser R. S.; Kobilka B. K. (2015) Structural Insights into the Dynamic Process of β 2 -Adrenergic Receptor Signaling. Cell 161, 1101–1111. 10.1016/j.cell.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio G. G.; Masureel M.; Hilger D.; Terry D. S.; Juette M.; Zhao H.; Zhou Z.; Perez-Aguilar J. M.; Hauge M.; Mathiasen S.; et al. (2017) Single-molecule analysis of ligand efficacy in β2AR–G-protein activation. Nature 547, 68–73. 10.1038/nature22354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard L.-P.; Schonegge A.-M.; Bouvier M. (2019) Structural Insight into G Protein-Coupled Receptor Signaling Efficacy and Bias between Gs and β-Arrestin. ACS Pharmacol Transl Sci 2, 148–154. 10.1021/acsptsci.9b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; McCammon J. A. (2016) Graded activation and free energy landscapes of a muscarinic G-protein–coupled receptor. Proc. Natl. Acad. Sci. U. S. A. 113, 12162–12167. 10.1073/pnas.1614538113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S.; Vaidehi N. (2010) Computational Mapping of the Conformational Transitions in Agonist Selective Pathways of a G-Protein Coupled Receptor. J. Am. Chem. Soc. 132, 5205–5214. 10.1021/ja910700y. [DOI] [PubMed] [Google Scholar]
- Niesen M. J. M.; Bhattacharya S.; Vaidehi N. (2011) The Role of Conformational Ensembles in Ligand Recognition in G-Protein Coupled Receptors. J. Am. Chem. Soc. 133, 13197–13204. 10.1021/ja205313h. [DOI] [PubMed] [Google Scholar]
- Shan J.; Khelashvili G.; Mondal S.; Mehler E. L.; Weinstein H. (2012) Ligand-Dependent Conformations and Dynamics of the Serotonin 5-HT2A Receptor Determine Its Activation and Membrane-Driven Oligomerization Properties. PLoS Comput. Biol. 8, e1002473. 10.1371/journal.pcbi.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jonsson A. L.; Beuming T.; Shelley J. C.; Voth G. A. (2013) Ligand-Dependent Activation and Deactivation of the Human Adenosine A2A Receptor. J. Am. Chem. Soc. 135, 8749–8759. 10.1021/ja404391q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror R. O.; Arlow D. H.; Maragakis P.; Mildorf T. J.; Pan A. C.; Xu H.; Borhani D. W.; Shaw D. E. (2011) Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A. 108, 18684–18689. 10.1073/pnas.1110499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhoff K. J.; Shukla D.; Lawrenz M.; Bowman G. R.; Konerding D. E.; Belov D.; Altman R. B.; Pande V. S. (2014) Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 6, 15–21. 10.1038/nchem.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provasi D.; Artacho M. C.; Negri A.; Mobarec J. C.; Filizola M. (2011) Ligand-induced modulation of the free-energy landscape of G protein-coupled receptors explored by adaptive biasing techniques. PLoS Comput. Biol. 7, e1002193. 10.1371/journal.pcbi.1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova I. G.; Selvam B.; Ivetac A.; Wereszczynski J.; McCammon J. A. (2013) Simulations of biased agonists in the β2 adrenergic receptor with accelerated molecular dynamics. Biochemistry 52, 5593–5603. 10.1021/bi400499n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpole T. J.; Delemotte L. (2018) Conformational landscapes of membrane proteins delineated by enhanced sampling molecular dynamics simulations. Biochim. Biophys. Acta, Biomembr. 1860, 909–926. 10.1016/j.bbamem.2017.10.033. [DOI] [PubMed] [Google Scholar]
- Rosenbaum D. M.; Zhang C.; Lyons J. A.; Holl R.; Aragao D.; Arlow D. H.; Rasmussen S. G. F.; Choi H.-J.; DeVree B. T.; Sunahara R. K.; Chae P. S.; Gellman S. H.; Dror R. O.; Shaw D. E.; Weis W. I.; Caffrey M.; Gmeiner P.; Kobilka B. K. (2011) Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature 469, 236–240. 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmüller H.; MacKerell A. D. Jr (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O'Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D. Jr; Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Cheng X.; Swails J. M.; Yeom M. S.; Eastman P. K.; Lemkul J. A.; Wei S.; Buckner J.; Jeong J. C.; Qi Y.; et al. (2016) CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. (2014) PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 185, 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Westerlund A. M.; Delemotte L. (2018) Effect of Ca2+ on the promiscuous target-protein binding of calmodulin. PLoS Comput. Biol. 14, e1006072. 10.1371/journal.pcbi.1006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V.; Vanderplas J.; Passos A.; Cournapeau D.; Brucher M.; Perrot M.; Duchesnay E. (2011) Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830. [Google Scholar]
- Montavon G.; Lapuschkin S.; Binder A.; Samek W.; Müller K.-R. (2017) Explaining nonlinear classification decisions with deep taylor decomposition. Pattern Recognit 65, 211–222. 10.1016/j.patcog.2016.11.008. [DOI] [Google Scholar]
- Lev B.; Murail S.; Poitevin F.; Cromer B. A.; Baaden M.; Delarue M.; Allen T. W. (2017) String method solution of the gating pathways for a pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. U. S. A. 114, E4158–E4167. 10.1073/pnas.1617567114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp K. A.; Bowman G. R.; Lane T. J.; Maibaum L.; Haque I. S.; Pande V. S. (2011) MSMBuilder2: modeling conformational dynamics on the picosecond to millisecond scale. J. Chem. Theory Comput. 7, 3412–3419. 10.1021/ct200463m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleetwood O.Code to study GPCRs using the String Method with swarms of trajectories. 2019. https://github.com/delemottelab/gpcr-string-method-2019 10.5281/zenodo.2662201. [DOI]
- Pan A. C.; Sezer D.; Roux B. (2008) Finding transition pathways using the string method with swarms of trajectories. J. Phys. Chem. B 112, 3432–3440. 10.1021/jp0777059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleetwood O.; Kasimova M. A.; Westerlund A. M.; Delemotte L. (2019) Molecular insights from conformational ensembles via machine learning. Biophys. J. 10.1016/j.bpj.2019.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R.; Zou Y.; Dror R. O.; Mildorf T. J.; Arlow D. H.; Manglik A.; Pan A. C.; Liu C. W.; Fung J. J.; Bokoch M. P.; Thian F. S.; Kobilka T. S.; Shaw D. E.; Mueller L.; Prosser R. S.; Kobilka B. K. (2013) The Dynamic Process of β2-Adrenergic Receptor Activation. Cell 152, 532–542. 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuku Y.; Ueda T.; Okude J.; Shiraishi Y.; Kondo K.; Maeda M.; Tsujishita H.; Shimada I. (2012) Efficacy of the β2-adrenergic receptor is determined by conformational equilibrium in the transmembrane region. Nat. Commun. 3, 1045–1053–9. 10.1038/ncomms2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selent J.; Sanz F.; Pastor M.; De Fabritiis G. (2010) Induced Effects of Sodium Ions on Dopaminergic G-Protein Coupled Receptors. PLoS Comput. Biol. 6, e1000884. 10.1371/journal.pcbi.1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Wang Y.; Hunkele A.; Provasi D.; Pasternak G. W.; Filizola M. (2019) Kinetic and thermodynamic insights into sodium ion translocation through the μ-opioid receptor from molecular dynamics and machine learning analysis. PLoS Comput. Biol. 15, e1006689. 10.1371/journal.pcbi.1006689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvam B.; Shamsi Z.; Shukla D. (2018) Universality of the Sodium Ion Binding Mechanism in Class A G-Protein-Coupled Receptors. Angew. Chem. 130, 3102–3107. 10.1002/ange.201708889. [DOI] [PubMed] [Google Scholar]
- Arnis S.; Fahmy K.; Hofmann K. P.; Sakmar T. P. (1994) A conserved carboxylic acid group mediates light-dependent proton uptake and signaling by rhodopsin. J. Biol. Chem. 269, 23879–23881. [PubMed] [Google Scholar]
- McCorvy J. D.; Wacker D.; Wang S.; Agegnehu B.; Liu J.; Lansu K.; Tribo A. R.; Olsen R. H. J.; Che T.; Jin J.; Roth B. L. (2018) Structural determinants of 5-HT2B receptor activation and biased agonism. Nat. Struct. Mol. Biol. 25, 787–796. 10.1038/s41594-018-0116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler L. M.; McMahon C.; Staus D. P.; Lefkowitz R. J.; Kruse A. C. (2019) Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell 176, 479–490.e12. 10.1016/j.cell.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F.; Wu H.; Katritch V.; Han G. W.; Jacobson K. A.; Gao Z.-G.; Cherezov V.; Stevens R. C. (2011) Structure of an Agonist-Bound Human A2A Adenosine Receptor. Science 332, 322–327. 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler L. M.; McMahon C.; Staus D. P.; Lefkowitz R. J.; Kruse A. C. (2019) Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell 176, 479–490.e12. 10.1016/j.cell.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Manglik A.; Venkatakrishnan A. J.; Laeremans T.; Feinberg E. N.; Sanborn A. L.; Kato H. E.; Livingston K. E.; Thorsen T. S.; Kling R. C.; Granier S.; Gmeiner P.; Husbands S. M.; Traynor J. R.; Weis W. I.; Steyaert J.; Dror R. O.; Kobilka B. K. (2015) Structural insights into μ-opioid receptor activation. Nature 524, 315–321. 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G.; Warne T.; Edwards P. C.; Bennett K.; Langmead C. J.; Leslie A. G. W.; Tate C. G. (2011) Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521–525. 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse A. C.; Ring A. M.; Manglik A.; Hu J.; Hu K.; Eitel K.; Hübner H.; Pardon E.; Valant C.; Sexton P. M.; Christopoulos A.; Felder C. C.; Gmeiner P.; Steyaert J.; Weis W. I.; Garcia K. C.; Wess J.; Kobilka B. K. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106. 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G.; Giguere P. M.; Katritch V.; Huang X.-P.; Thompson A. A.; Cherezov V.; Roth B. L.; Stevens R. C. (2014) Molecular control of δ-opioid receptor signalling. Nature 506, 191–196. 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy M. T.; Lee M.-Y.; Gao Z.-G.; White K. L.; Didenko T.; Horst R.; Audet M.; Stanczak P.; McClary K. M.; Han G. W.; Jacobson K. A.; Stevens R. C.; Wüthrich K. (2018) Allosteric Coupling of Drug Binding and Intracellular Signaling in the A2A Adenosine Receptor. Cell 172, 68–80.e12. 10.1016/j.cell.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. J.; Horst R.; Katritch V.; Stevens R. C.; Wuthrich K. (2012) Biased Signaling Pathways in β2-Adrenergic Receptor Characterized by 19F-NMR. Science 335, 1106–1110. 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrucci F. (2017) Strategies for the exploration of free energy landscapes: Unity in diversity and challenges ahead. Rev. Phys. 2, 32–45. 10.1016/j.revip.2017.05.001. [DOI] [Google Scholar]
- Pan A. C.; Roux B. (2008) Building Markov state models along pathways to determine free energies and rates of transitions. J. Chem. Phys. 129, 064107–064107–8. 10.1063/1.2959573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Yen H.-Y.; Robinson C. V.; Sansom M. S. (2019) State-dependent Lipid Interactions with the A2a Receptor Revealed by MD Simulations Using In Vivo-Mimetic Membranes. Structure 27, 392–403.e3. 10.1016/j.str.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.