

Abstract
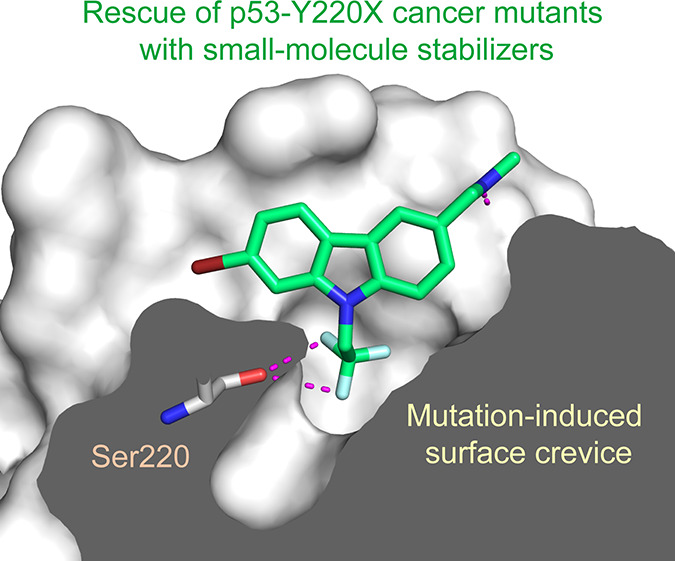
We have previously shown that the thermolabile, cavity-creating p53 cancer mutant Y220C can be reactivated by small-molecule stabilizers. In our ongoing efforts to unearth druggable variants of the p53 mutome, we have now analyzed the effects of other cancer-associated mutations at codon 220 on the structure, stability, and dynamics of the p53 DNA-binding domain (DBD). We found that the oncogenic Y220H, Y220N, and Y220S mutations are also highly destabilizing, suggesting that they are largely unfolded under physiological conditions. A high-resolution crystal structure of the Y220S mutant DBD revealed a mutation-induced surface crevice similar to that of Y220C, whereas the corresponding pocket’s accessibility to small molecules was blocked in the structure of the Y220H mutant. Accordingly, a series of carbazole-based small molecules, designed for stabilizing the Y220C mutant, also bound to and stabilized the folded state of the Y220S mutant, albeit with varying affinities due to structural differences in the binding pocket of the two mutants. Some of the compounds also bound to and stabilized the Y220N mutant, but not the Y220H mutant. Our data validate the Y220S and Y220N mutants as druggable targets and provide a framework for the design of Y220S or Y220N-specific compounds as well as compounds with dual Y220C/Y220S specificity for use in personalized cancer therapy.
Introduction
The tumor suppressor p53 is inactivated by mutation in about half of all tumors, making mutant p53 a prime target for cancer therapy.1−3 Most oncogenic p53 mutations are missense mutations mapping to its DNA-binding domain (DBD), yet the functional spectrum of these mutations is very diverse, depending on the distinct nature of each mutant protein.3−6 Nevertheless, despite this functional heterogeneity, mechanistically, the mutations can be broadly divided into DNA-contact mutations that remove an essential DNA contact, which prevents p53 from binding to its target promoters, and structural mutations. The latter lower the thermostability of the intrinsically unstable p53 protein to varying degrees, causing it to rapidly unfold and aggregate under physiological conditions.7,8 An estimated one-third of common p53 cancer mutants are temperature-sensitive structural mutants that exhibit a loss of function under physiological conditions but retain at least some degree of transcriptional activity when assayed at low (permissive) temperatures, where they adopt a wild-type like conformation.4,7,9 It may, therefore, be possible to restore the function of this subset of cancer mutants with chemical chaperones. The Y220C mutation is an excellent paradigm for this rescue strategy.1 It is the ninth most frequent p53 cancer mutation and accounts for an estimated 100 000 new cancer cases each year. We have previously shown that this highly destabilizing mutation creates an extended surface crevice that can be targeted by stabilizing small molecules (Figure 1).10 Using fragment-based screening and structure-based design, we have developed several chemically diverse classes of small molecules that bind to this pocket, increase the melting temperature, and hence stabilize and slow down the rate of aggregation of the mutant.11−17 Importantly, some of those molecules are biologically active in cancer cells with homozygous Y220C mutation and induce mutant-specific transcription of a series of p53 target genes,17−19 providing the proof-of-concept that pharmacological rescue of thermolabile cancer mutants with molecular chaperones is a viable and promising strategy. In addition, molecular dynamics simulations suggest that it might be possible to target other, transiently open pockets on the DBD surface.20,21 Alternative mutant p53 rescue strategies use thiol modifying alkylating agents, such as PRIMA-1Met (APR-246)22,23 and sulfonylpyrimidines,24 which have been shown to selectively kill cancer cells with compromised p53. In addition, metallochaperones have been developed for the rescue of the zinc-binding deficient R175H mutant by increasing cellular zinc levels.25 These examples illustrate that different reactivation strategies are needed, depending on the underlying mechanism of inactivation. No single mutant is found at a frequency of >6%, and the 10 most frequent and most commonly studied “hotspot” mutants, including the Y220C mutant, together comprise only <25% of all p53 mutations in total.26,27 Although this translates into a staggering 2.5 million new cancer cases per year, the vast majority of cancer patients have nonhotspot mutations with currently largely unknown structural and functional impact.
Figure 1.
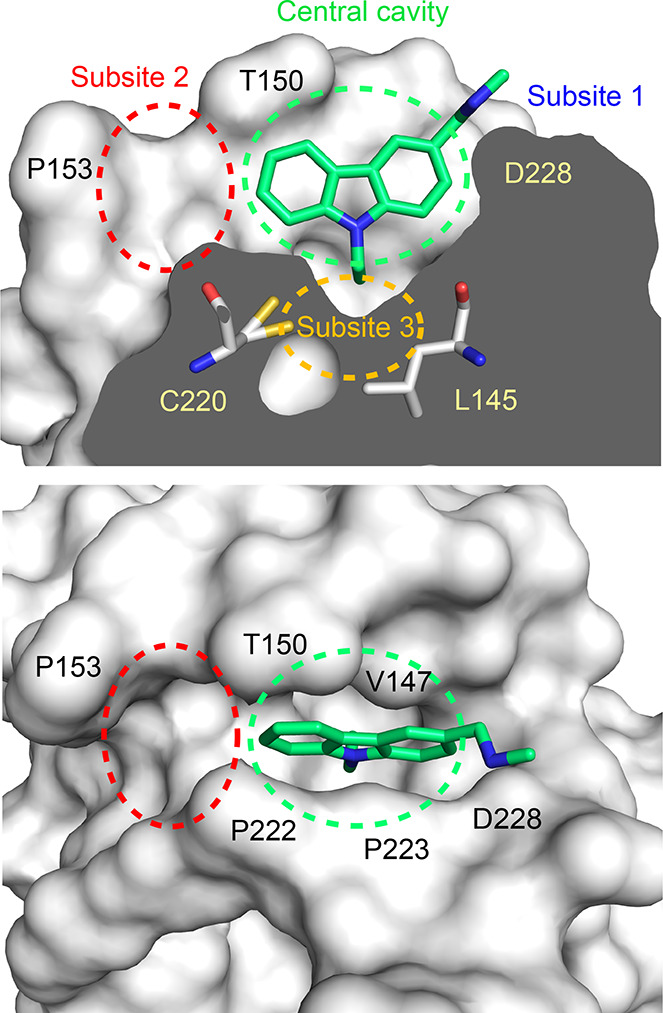
Mutation-induced surface crevice in the p53 cancer mutant Y220C with bound molecule PK083. The mutant protein is shown as a surface representation in two different orientations: cross section of the mutation-induced surface crevice and top view. Bound carbazole-based stabilizer PK083 is shown as a stick model (PDB ID: 2VUK). Specific regions of the pocket that are discussed in this manuscript are highlighted. Adapted with permission from Bauer et al. Copyright 2019 Future Science Ltd.(17)
In our ongoing efforts to structurally characterize the p53 mutome and devise appropriate reactivation strategies, we have now analyzed the effects of other frequently occurring oncogenic mutations at codon 220 on the structure, stability, and dynamics of the p53 DBD. These data show that the oncogenic Y220S and Y220N mutations are also highly destabilizing, even more so than the Y220C mutant, and, like the Y220C mutant, create a unique extended surface crevice that can be specifically targeted by small-molecule stabilizers.
Results and Discussion
Analysis of Cancer Mutation Frequencies at p53 Codon 220
The TP53 mutation database26 of the International Agency for Research on Cancer lists 402 cancer cases with a somatic Y220C mutation (Table 1). On the basis of the total number of reported p53 cancer mutations in the database, an overall p53 mutation rate in cancer of about 50%, and a total number of 18 million new cancer cases worldwide in 2018,28 this mutation currently accounts for about 125 000 new cancer cases each year, and its tendency is rising. There are also three other mutations at codon 220 that occur with an appreciable frequency in the database: Y220H, Y220N, and Y220S (20, 18, and 17 cases, respectively). As with the Y220C mutation, these mutations are found across different types of cancer, including breast, lung, and liver cancer. Applying the same estimate as for the Y220C mutation, this translates to around 5000–6000 new cancer cases per year worldwide that are caused by each of these mutations. Interestingly, the Y220S mutation is also reported in three cases of familial cancer and has been associated with oncogenic gain of function, including angiogenesis induction and doxorubicin resistance.29 Two other mutations, Y220D and Y220F, are found in tumors, albeit very rarely. The Y220F mutation is reported only once in the database and is most likely a passenger rather than a driver mutation.
Table 1. Frequency of p53 Cancer Mutations at Codon 220.
| mutation | somatic counta | germline counta |
|---|---|---|
| Y220C | 402 | 17 |
| Y220H | 20 | 0 |
| Y220N | 18 | 0 |
| Y220S | 17 | 3 |
| Y220D | 4 | 0 |
| Y220F | 1 | 0 |
Number of cases in release R20 of the TP53 mutation database of the International Agency for Research on Cancer.26N = 28 866 for the somatic and N = 1245 for the germline data set.
Effect of Y220X Cancer Mutations on the Thermostability of the DBD
We determined the effect of the four most frequent Y220X cancer mutations on the thermostability of the DBD by differential scanning fluorimetry (DSF). The mutations were introduced in a superstable variant of the p53 DBD (M133L/V203A/N239Y/N268),30,31 which has been successfully used previously for structural studies of conformationally unstable p53 cancer mutants.10,32 All four mutations were highly destabilizing (Table 2). The Y220H and Y220C mutations lowered the Tm of the DBD by about 6 and 8 °C, respectively. The other two mutations, Y220S and Y220N, were even more destabilizing, lowering the Tm by ∼12 °C. This dramatic stability loss suggests that all four p53-Y220X cancer mutants are largely unfolded under physiological conditions in cancer cells, as shown previously for the Y220C mutant.18 It will, therefore, be interesting to explore whether the more deleterious effect of the Y220S and Y220N mutations on protein stability compared with Y220C and Y220H is also associated with a more severe phenotype.
Table 2. Melting Temperatures of p53-Y220X Mutant DBDs.
Structures of the Y220S, Y220N, and Y220H Mutants
We determined crystal structures of the Y220H and Y220S mutant at a resolution of 1.44 and 1.50 Å, respectively. The crystals used for structure solution were isomorphous to those obtained previously for the Y220C mutant and the stabilized wild-type DBD and contained two molecules in the asymmetric unit. We also collected additional X-ray data for the Y220C mutant, extending the resolution from 1.65 Å (PDB entry 2J1X) to 1.24 Å. The overall structure of the wild-type DBD and its key functional motifs are conserved in the Y220S and Y220H mutants. The Y220S mutant structure, however, also revealed a mutation-induced cavity similar to that observed for the Y220C mutation. In both cases, the large-to-small substitution at residue 220 connects two pre-existing smaller cavities, resulting in an extended surface crevice. A cross-section of the cavity in both mutants is shown in Figure 2, highlighting the conserved water network in both mutant structures. Despite the overall similarity in shape, there are distinct differences. As a result of the smaller, more polar side chain in the Y220S mutant, the subsite-3 region at the bottom of the pocket is slightly deeper and more polar (see Figure 1 for the location and numbering of different subpockets). This is reflected in the incorporation of an additional, weakly bound water molecule that interacts with the hydroxyl group of Ser220 (Water W1 in Figure 2A). We have previously shown that the shape of subsite 3 is modulated by the conformational state of Cys220.13 In some of the Y220C–ligand complexes, but not in the apo structure, the side chain of Cys220 is flipped, with the sulfhydryl group pointing away from the cavity toward a hydrophobic patch formed by the side chains of Leu145, Val157, and Leu257, which increases the depth of the binding pocket. Fluctuation between these alternative states of the Y220C pocket has also been seen in MD simulations.13 In the case of the Y220S mutant, an equivalent flip of the serine side chain would be energetically unfavorable, leaving the hydroxyl group in a hydrophobic environment and impeding H-bond formation with solvent molecules in the pocket. At the upper rim of the binding pocket, the side chains of Thr150 and Pro222 contact each other. This is also observed in some Y220C mutant structures, whereas in other structures, Pro222 is further away and the pocket thereby more open, allowing access of small-molecule binders.13,15
Figure 2.

Structures of the Y220X mutants. Molecular surface representation of a cross-section of the mutation-induced surface crevice in mutants Y220S (A) and Y220C (B). Water molecules bound inside the pocket are shown as small spheres and color-coded according to their crystallographic thermal factors (B factors) in a rainbow gradient from blue (B factor = 10 Å2) to red (B factor = 50 Å2). Water-mediated hydrogen bonds are highlighted with orange dashed lines. Selected side chains in the pocket are highlighted as stick models. (C) The equivalent cross-section of the wild-type structure (PDB ID 1UOL) shows that Tyr220 prevents formation of an extended surface crevice and effectively separates two smaller, pre-existing subpockets. (D) In the structure of the Y220H mutant, the histidine side chain overlays with the position of the Tyr side chain in the wild type, which also blocks the binding pocket. All structures were determined in the same space group, and chain A of the asymmetric unit is shown.
In the crystal structure of the Y220H mutant, the imidazole ring of the His220 side chain roughly superimposes with the position of the tyrosine aromatic ring in the wild-type structure (Figure 2C,D), thereby effectively blocking the binding pocket seen in the other Y220X cancer mutant structures. Both imidazole nitrogen atoms form a hydrogen bond with a structural water molecule. The partial retention of Tyr220-mediated interactions in the wild type is reflected in the less severe stability loss compared with the other Y220X mutants tested.
For the Y220N mutant, only poorly diffracting crystals were obtained; hence the structure of this mutant was modeled with the SWISS-MODEL33 server using the Y220C mutant structure as a template. According to this model, the Y220N mutation also creates an extended surface crevice, but the center of the pocket is not as deep as in the Y220C and Y220S mutants and more polar (Supporting Information Figure S1).
Dynamics of the Y220X Surface Crevice
We performed molecular dynamics (MD) simulations on the different p53 DBD variants to provide insights into the structural plasticity of the mutation-induced binding pocket. The stabilized pseudo wild type and the four Y220X mutants of this scaffold were simulated using atomistic MD, each for a total of 800 ns (see Methods for details) under physiological conditions and in the absence of any stabilizing ligand (only the zinc atom was retained). During the simulated time, none of the mutants underwent unfolding, although specific fluctuations around the X-ray conformation were observed, revealing the dynamics of the systems. The fluctuations of the proteins around their average structure, measured as Cα root-mean-square fluctuations (RMSF) of residues 97–289, ranked them according to their experimentally determined stability, as seen for other model systems,34 i.e., the lower the thermostability of the mutant, the higher the average RMSF in the simulations (Table S1). The profile of the RMSF (Supporting Information Figure S2) shows enhanced fluctuations of all the Y220X mutants around the binding pocket (residue 145–157 and 218–232) compared with the wild type, which was expected, given that they lack the Tyr side chain that holds the S3/S4 and S7/S8 loops together. The distributions of the distances between the Cδ atoms of Pro151 and Pro223 (d1) and between the Cα atom of Pro153 and the Cγ atom of Pro222 (d2), which line the pocket, are bimodal for the mutants (but not for the wild type), hinting at the presence of open and closed conformations of the pocket in the Y220X mutants (Figure 3). The first distance is a measure for the opening/collapse of the central part of the pocket, whereas the second distance describes the expanse of subsite 2. The time series of these distances shows that open and collapsed conformations are visited repeatedly during the simulations (Supporting Information Figures S3 and S4). Cluster analysis of the side chains of the residues around the pocket (109, 145–157, 218–232, 257) was used to extract representative conformations of the open and collapsed state of the central cavity. In the closed state, as shown for the Y220S mutant (Figure 3A), Pro153 collapses into the pocket and packs against the side chain of Val147 and Pro151 on the other side of the pocket. Y220H, the most thermostable of the four Y220X mutants, which incidentally also has the smallest pocket, had the smallest population of this state. In the Y220H simulations, collapse of Pro223 into the pocket is linked to a displacement of the His220 side chain, which swings out of the pocket, thereby facilitating its collapse (Figure 3B). Subsite 2 was generally less prone to collapse than the central pocket in the simulations. There was also an interesting difference in the coupling between the two subsites in the various mutants. An open state of the central pocket in the Y220S and Y220C mutants was generally linked to an open state of subsite 2, but collapse of the central pocket did not necessitate the collapse of the subsite-2 region. In contrast, the state of both subpockets is coupled in the Y220H mutant, where displacement of His220 favors the coordinated collapse of both subsites (Figure 3B and Supporting Information Figure S5).
Figure 3.

MD simulations of Y220X mutants showing open and closed states of the pocket region. (A) Superposition of Cα traces of the crystal structure of the Y220S mutant (green) and three representative structures of open and closed states along the trajectory. Selected side chains are shown as stick models. Selected distances across the central cavity (d1) and subsite 2 (d2) are highlighted with magenta dashed lines. (B) Superposition of Cα traces of the crystal structure of the Y220H mutant (green) and three representative structures of open and closed states along the trajectory. The view is the same as in panel A. Concerted collapse of the pocket region is associated with His220 swinging out of the pocket. (C) Distribution of representative distances shows that the Y220X mutants visit open and closed states during the simulations, whereas the wild type stays close to the conformation of the crystal structure. The distance between the Cδ atoms of Pro151 and Pro223 (left) is a measure for the width of the central cavity, whereas the distance between the Cα atom of Pro153 and the Cγ of Pro222 spans subsite 2. The green line indicates the distance in the corresponding crystal structures (chain A) or the starting model in case of the Y220N mutant.
Carbazole-Based Y220C Binders Also Bind to the Y220S and Y220N Mutants
Given the similarity of the mutation-induced surface crevice in the structures of the Y220C and Y220S mutants, we speculated that the molecules we had previously designed for rescuing the function of the Y220C mutant might also bind to the Y220S mutant and stabilize it. We therefore tested a selection of our carbazole-based lead structures by DSF thermal shift assays and determined the binding constant, Kd, of a few selected compounds by isothermal calorimetry (ITC; Table 3). All Y220C binders tested also stabilized the Y220S mutant, albeit to a different extent. Generally, compounds with a larger subsite-2 moiety had a much smaller stabilizing effect on the Y220S than on the Y220C mutant at a given concentration, with a notable exception being PK9322 and PK9323, which stabilized both mutants equally well. Both compounds contain a thiazole moiety that can potentially form a hydrogen bond with Ser220. In contrast, compounds with a trifluoroethyl anchor and lacking extensive subsite-2 extensions had a more stabilizing effect on Y220S than on Y220C. The highest thermostabilization among all compounds tested was observed for PK9301, which increased the Tm of the Y220S mutant by more than 5 °C, higher than any stability increase observed for the Y220C mutant to date. PK9301 bound to Y220S with a Kd of 4 μM (Supporting Information Figure S6), a slightly higher affinity than determined previously for the Y220C mutant (Kd = 6 μM). The preference of Y220S for a trifluorylated ethyl anchor was even more pronounced in the absence of a subsite-2 targeting moiety. PK9255 bound about 2 times more tightly to the Y220S mutant (Kd = 16 μM vs Kd = 28 μM for Y220C). When comparing the compound-induced thermal shifts for the two mutants, one has to take into consideration that the Y220S mutation has a higher destabilizing effect than the Y220C mutation (ΔTm of 12 vs 8 °C). The potential stability gains from optimally targeting the Y220S (and also Y220N) pocket are therefore expected to be higher than from targeting the Y220C pocket.
Table 3. Thermostabilization and Dissociation Constants of Carbazole-Based Y220S and Y220C Binders.
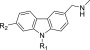

We also tested a few representative carbazoles against the Y220N and Y220H mutants (Table 4). Intriguingly, PK9255 also stabilized the Y220N mutant by 1.4 °C and bound with a Kd of 40 μM as measured by ITC (Supporting Information Figure S7). Larger compounds with a subsite-2 extension had no additional stabilizing effects and were in one case actually less effective in stabilizing Y220N than PK9255, clearly indicating significant differences in the binding mode and/or the architecture of the Y220N pocket. PK9255 had no significant stabilizing effect on the Y220H mutant, consistent with the crystal structure of the mutant showing that the histidine side chain effectively blocks the center of the binding pocket. These data establish that the mutation-induced pocket in the Y220N mutant is also druggable but requires a different design strategy or an alternative scaffold to optimally target it.
Table 4. Differential Stabilization of Y220X Mutants.

Measured at a compound concentration of 250 μM. Mean values of triplicate measurements ± SEM are shown.
Binding Modes of Carbazole-Based Y220S Mutant Stabilizers
To provide a rationale for the observed binding affinities, we determined crystal structures of the Y220S mutant in complex with the best stabilizers, PK9301 and PK9323, as well as the structure of the Y220C mutant with PK9323. The resolution of the structures ranged from 1.4 to 1.8 Å, and there was clear electron density for the bound ligand in each case. The overall binding mode of PK9323 in the Y220S mutant resembles closely that observed for other carbazoles bound to the Y220C mutant (Figure 4A,B). The carbazole ring system is flanked on one side by Val147, Thr150, and Pro151 and on the other side by Pro222, Pro223, and Thr230. The secondary amine forms a hydrogen bond with the backbone oxygen of Asp228, and the ethyl anchor forms hydrophobic interactions in the subsite-3 region at the bottom of the binding pocket. The subsite-2 thiazole moiety shows good shape complementarity with this subpocket, with the sulfur atom facing Pro153. Importantly, the thiazole nitrogen forms a hydrogen bond with the Ser220 hydroxyl (2.9 Å distance), which explains the preference of the Y220S mutant for thiazole-substituted carbazoles over other subsite-2 targeting moieties (Table 3). Compared with the structure of the Y220C–PK9323 complex, the overall scaffold is shifted slightly toward subsite 2 in the Y220S mutant, and the thiazole ring shows a stronger deviation from coplanarity with the carbazole ring system to accommodate the binding pocket (average dihedral angle of 14° vs 5°).
Figure 4.

Structures of the p53-Y220S mutant in complex with PK9301 and PK9323. (A) Molecular surface representation of a cross-section of the mutation-induced surface crevice in the Y220S mutant with bound PK9323, showing that the thiazole moiety of the ligand forms a hydrogen bond with Ser220. (B) Structure of the Y220C–PK9323 complex; same view as for the Y220S–PK9323 complex in A. (C) Structure of the Y220S–PK9301 complex. Key residues in the binding pocket are shown as stick models. A structural water molecule interacting with the aminomethyl substituent is shown as a red sphere. Ligand-mediated hydrogen bonds are shown as orange dashed lines. (D) Superposition of the binding modes of PK9323 in Y220S and Y220C as well as PK9301 in Y220S. The ligands and the mutated side chain are shown as stick models. Polar interactions with Ser220 are shown as dashed lines in the same color as the corresponding structure.
Relative to its orientation in most Y220C complexes, the PK9301 carbazole scaffold is shifted slightly toward subsite 2, with the bromine substituent reaching into this subsite, where it forms extensive hydrophobic interactions (Figure 4C,D). In the subsite-3 region at the bottom of the binding pocket, two fluorine atoms of the trifluoroethyl anchor form a weak hydrogen bond with the Ser220 hydroxyl group each (distance of 3.0 and 3.1 Å, respectively), and one strongly polarized proton next to the trifluoromethyl group interacts with the hydroxyl oxygen atom via weak hydrogen bonding (C···O distance = 3.3 Å). These interactions rationalize why a trifluoroethyl anchor is strongly favored over an ethyl anchor (cf. Table 3). The third fluorine atom points toward the backbone carbonyls of Leu145 and Trp146, as seen for the Y220C mutant, where it forms orthogonal multipolar interactions. The F···O=C distance is 3.9 and 4.2 Å, respectively, which is slightly larger than the typical interaction distance (3.0–3.7 Å),35 indicating that the trifluoroethyl group prefers to interact with the Ser220 side chain. Instead of directly interacting with the Asp228 backbone, as seen for PK9323, the secondary methylamine group forms a hydrogen bond with the backbone oxygen of Val147 and a number of water-mediated contacts with the protein, for example, with the Thr150 side chain at the entrance of the pocket.
Taken together, the analysis of the ligand binding modes in the Y220S and Y220C mutant and the SAR (Table 3) provide a framework for the design of binders with dual Y220C/Y220S specificity.
Reduced Dynamics of the Y220C and Y220S Mutant Proteins upon Ligand Binding
We performed additional MD simulations on the Y220C/S-ligand structures described above (Y220S–PK9301, Y220S–PK9323, and Y220C–PK9323) to gain insights into the effect of ligand binding on the dynamics of the mutant proteins. After aligning the protein backbone of all trajectory frames to the initial conformation, the all-atom RMSD of the ligand with respect to its initial conformation was calculated. These values fluctuated around an average of 2.1 Å for the two PK9323 complexes and 1.8 Å for the Y220S–PK9301 complex, with a standard deviation of 0.5 Å in all three cases. The small fluctuations around the initial structure indicate that the protein–ligand complexes were very stable during the time scale of the simulations. Ligand binding stabilized the protein in all three simulations, resulting in drastically reduced average RMSFs compared with the unbound mutant structure (Table S1). For the Y220C–PK9323 complex, the overall RMSF was actually lower than that of the wild-type DBD. For the Y220S–ligand complexes studied, PK9323 and PK9301, reduction of the average RMSF correlated with the affinity and effect on thermostability of the compound, i.e., the more potent binder and stabilizer PK9301 induced a stronger decrease of the average RMSF than PK9323 (cf. Tables 3 and S1). Analysis of the RMSF for individual residues also provided interesting information (Supporting Information Figure S8). The elevated RMSFs for residues on both sides of the mutation-induced binding pocket in the MD simulations of the native Y220C and Y220S mutant, i.e., residues 147–153 and around residue 220, went back to wild-type levels upon ligand binding, indicating stabilization of the local structure of the pocket region. This local stabilization was also reflected in the distances of specific pocket parameters describing the state of the central cavity (d1) and subsite 2 (d2), i.e., the distances between the Cδ atoms of Pro151 and Pro223 (d1) and between the Cα atom of Pro153 and the Cγ atom of Pro222 (d2; see Figure 3). In the MD simulations of the native mutants, there was a bimodal distribution of both pocket parameters, corresponding to open and collapsed states of the pocket. In contrast, in both the Y220C and Y220S ligand complexes, there was a reversal to a unimodal distribution, as seen for the wild type, and the distances stayed close to the values in the starting structure throughout the simulation (Figures S3 and S4). Comparison of the subsite-2 parameter, d2, in the two Y220S–ligand complex simulations also shows that there is a slight narrowing of subsite 2 with PK9301, which has a smaller subsite-2 targeting moiety than PK9323 (bromine vs thiazole). Especially in the Y220S–ligand complexes, a number of long-range effects were also observed, resulting in reduced structural fluctuations in the distant L2 and L3 loops in the ligand-bound mutant (Figure S8).
Overall, the MD simulations support the notion that our molecules act as a molecular glue that stabilizes both the local and global structure, reducing conformational sampling and increasing the thermostability of the Y220X mutant proteins.
Conclusion and Outlook
We have shown that the four most common oncogenic Y220X mutations drastically impair the thermostability of the p53 DBD. Crystal structures and MD simulations further revealed that three of them result in the formation of an extended surface crevice with unique properties. Importantly, our data establish that both the Y220S and Y220N mutants, in addition to the Y220C mutant, are druggable targets for restoring the function of those mutants in tumors with small-molecule stabilizers. The structural and biophysical data presented here provide a framework for the development of both specific and dual Y220S/Y220C stabilizers, whereas the Y220N mutant most likely requires a different design strategy for achieving the required potency. In addition, MD simulations provided complementary structural and dynamical information that can inform the rational design and optimization of Y220X ligands. Systematic structural studies on other p53 cancer mutants, combined with virtual and fragment screening approaches, are likely to unearth more oncogenic variants with druggable pockets. These studies should further expand the druggable p53 mutome for cancer therapy and aid the development of new personalized cancer drugs.
Shortly before submission of this manuscript, a docking study with our initial Y220C stabilizer PK083 was published, suggesting that it may preferentially bind to the Y220S mutant, although no experimental verification was provided.36
Materials and Methods
Compound Synthesis
Synthesis routes of chemical compounds, except PK9301, have been described elsewhere.17 1-(7-Bromo-9-(2,2,2-trifluoroethyl)-9H-carbazol-3-yl)-N-methylmethanamine (PK9301). To a solution of 7-bromo-9H-carbazole-3-carbaldehyde (200 mg, 0.72 mmol), in anhydrous DMF (3 mL), was added cesium carbonate (475 mg, 1.45 mmol) and 2,2,2-trifluoroethyl-p-toluenesulfonate (356 mg, 1.45 mmol). The reaction mixture was irradiated in the microwave at 150 °C for 30 min. After cooling, the crude product was purified by chromatography on silica gel using EtOAc/hexane 1/4 to 1/1 over 20 min. 7-Bromo-9-(2,2,2-trifluoroethyl)-9H-carbazole-3-carbaldehyde was isolated as a white solid, yield 52% (134 mg, 0.37 mmol). 1H NMR (500 MHz, DMSO-d6): δ 10.06 (s, 1H, CHO), 8.78 (s, 1H, ar CH), 8.25 (d, J = 8.5 Hz, 1H, ar CH), 8.14 (s, 1H, ar CH), 8.05 (d, J = 8.5 Hz, 1H, ar CH), 7.91 (d, J = 8.5 Hz, 1H, ar CH), 7.49 (d, J = 8.5 Hz, 1H, ar CH), 5.55 (q, 3JFH = 9.2 Hz, 2H, CH2) ppm. 13C NMR (126 MHz, DMSO-d6): δ 192.3, 144.3, 142.3, 130.2, 127.9, 125.3 (q, 1JFC = 281.7 Hz), 124.5, 124.2, 123.0, 122.7, 122.1, 120.4, 113.9, 11.2, 44.1 (q, 2JFC = 33.0 Hz) ppm. MS-EI (m/z) found: 355 [M]+. Calcd.: 355.
To a solution of the latter compound (85 mg, 0.24 mmol), used as such, in anhydrous EtOH/DCM (2 mL) was added methylamine hydrochloride (32 mg, 0.48 mmol), triethylamine (49 μL, 0.35 mmol), and titanium(IV) isopropoxide (141 μL, 0.48 mmol). The resulting solution was stirred at RT for 18 h before the addition of sodium borohydride (18 mg, 0.48 mmol). The solution was stirred at RT for 8 h before pouring into 2 M aqueous ammonia solution (10 mL). The suspension was filtered through Celite, and to the filtrate was added H2O. Crude product was extracted with DCM, dried over anhydrous K2CO3. The suspension was filtered, and the filtrate was concentrated in vacuo to yield crude product that was purified by chromatography on silica gel using 9:1 DCM/MeOH as an eluent to yield the product as a colorless solid, yield: 36% (32 mg, 0.08 mmol). 1H NMR (500 MHz, DMSO-d6): δ 8.32 (s, 1H, ar CH), 8.08 (s, 1H, ar CH), 8.05 (d, J = 8.5 Hz, 1H, ar CH), 7.80 (d, J = 8.5 Hz, 1H, ar CH), 7.69 (d, J = 8.5 Hz, 1H, ar CH), 7.44 (d, J = 8.5 Hz, 1H, ar CH), 5.49 (q, 3JFH = 9.0 Hz, 2H, CH2), 4.23 (s, 2H, CH2), 2.53 (s, 3H, CH3) ppm. 13C NMR (126 MHz, DMSO-d6): δ 141.9, 140.9, 128.9, 124.5, 123.7, 123.4 (q, 1JFC = 285.3 Hz), 122.8, 122.4, 122.3, 121.8, 119.8, 113.6, 110.8, 51.9, 43.9 (q, 2JFC = 33.2 Hz), 32.2 ppm. HRMS-ESI (m/z) found: 339.9470 [M]+. Calcd.: 339.9943 for [C15H10BrF3N]+ (loss of NHMe). LC-MS purity = 97% (UV), ret. time = 12.55 min.
Differential Scanning Fluorimetry
Melting temperatures of the p53 mutants were determined by differential scanning fluorimetry (DSF) using the dye SYPRO Orange (Invitrogen), which quantitatively binds to hydrophobic protein patches that become exposed upon thermal unfolding. Real-time melt analyses were performed using either an Agilent MX3005P (heating rate of 270 K/h excitation/emission filters = 492/610 nm; Tables 2 and 4) or a Corbett Rotor-Gene 6000 real-time qPCR thermocycler (heating rate of 180 K/h, excitation/emission filters = 460/510 nm; Table 3). Tm measurements were performed with 8 μM p53 protein and 5–10 × SYPRO Orange (Life Technologies) in a 25 mM KPi (pH 7.2), 150 mM NaCl, and 1 mM TCEP assay buffer. For measurements of compound-induced Tm shifts (ΔTm), the assay buffer additionally contained 5% DMSO (v/v), with either 250 μM ligand or no ligand (control). ΔTm values were calculated as ΔTm = Tm (protein + compound) – Tm (protein). Native melting curves were measured in quadruplicate and compound-induced thermal shifts in triplicate or higher. Mean values ± SEM are given.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) experiments for the Y220C and Y220S mutants were performed using a MicroCal (Amherst) iTC200 calorimeter as described.14 Briefly, the cell unit contained 30–50 μM protein in a 25 mM KPi (pH 7.2), 150 mM NaCl, 1 mM TCEP, and 5% (v/v) DMSO assay buffer. The syringe contained 0.5–2 mM compound in the same buffer, using injection steps of 2 μL at a rate of 0.5 μL/s (initial injection: 0.5 μL) and 120 s spacing. For the poorly soluble PK9223, a reverse titration was performed. Data analysis was performed using the MicroCal Origin software. ITC measurements for the Y220N mutant were carried out with the same buffers as above but using a Nano ITC microcalorimeter (TA Instruments) and injection steps of 8 μL, at a rate of 0.5 μL/s, and 200 s spacing. Data were processed using the NanoAnalyze software (Version 3.5.0) supplied with the instrument. All measurements were performed at 20 °C.
Protein Expression and Purification
Protein expression and purification were performed following published protocols.11 Briefly, we used a pET24a-based expression vector encoding a fusion protein with an N-terminal 6xHis tag, followed by the lipoyl domain of the dihydrolipoamide acetyltransferase from Bacillus stearothermophilus, a TEV protease cleavage site, and the mutant p53 DBD (residues 94–312) of interest. Point mutations in the p53 DBD were introduced using the QuickChange site-directed mutagenesis kit (Agilent). All mutations were introduced into a stabilized quadruple mutant variant of the human DBD (M133L/V203A/N239Y/N268D) that we used successfully for studying the effects of destabilizing p53 mutations in the past.10,37 The recombinant proteins were expressed in E. coli C41. Cells harboring the expression vector were grown in 2TY medium at 37 °C up to an OD600 = 0.6–0.8. At this point, the temperature was lowered to 20 °C, and protein expression was induced by adding IPTG (0.5 mM). The cells were further supplemented with 0.1 mM zinc chloride. Cells were grown overnight and then broken by either sonication or French press. The p53 mutants were purified using a nickel column, followed by TEV protease cleavage overnight and a second purification step on a heparin column. After an additional gel filtration chromatography step, the mutants were concentrated to 6 mg mL–1, flash frozen in liquid nitrogen, and stored at −80 °C.
Protein Crystallization and Structure Determination
Crystals of the Y220C, Y220S, and Y220H mutant were grown at 20 °C using the sitting drop vapor diffusion technique by mixing equal amounts of protein solution (6 mg mL–1 mutant protein in 25 mM phosphate buffer, pH 7.2, 150 mM NaCl, and 5 mM DTT) and reservoir buffer (19% (w/v) polyethylene glycol 4000, 100 mM HEPES, pH 7.2, and 5 mM DTT). Rod shaped crystals grew within a few days. Crystals of the Y220S–PK9323 complex were grown under the same conditions but with the crystallization buffer supplemented with 2 mM PK9323. All crystals were cryoprotected with mother liquor supplemented with 20% glycerol and flash frozen in liquid nitrogen. For additional soaking experiments, crystals of the Y220S mutant were soaked for 60 min in a saturated solution of PK9301 in cryoprotection buffer prior to flash freezing, and crystals of the Y220C mutant were soaked for 3.5 h with a saturated solution of PK9323 in the same cryoprotection buffer as above prior to flash freezing. X-ray data sets were collected at 100 K at beamline I03 of the Diamond Light Source, Oxford, UK, and beamline X06SA of the Swiss Light Source, Villigen, Switzerland. The diffraction data were integrated with the program XDS38 and scaled with either SCALA39 or AIMLESS,40 which are both implemented in the CCP4 package.41 The structures were solved by difference Fourier analysis in PHENIX42 using PDB entry 2J1X as a starting model with initial rigid body refinement. Structure refinement was performed using iterative cycles of manual model building in COOT43 and refinement in PHENIX. Dictionary files for the PK9301 and PK9323 ligands were generated using the Grade Web Server (http://grade.globalphasing.org). In the structure of the Y220S complex with the brominated ligand PK9301, there was significant negative difference electron density at the bromine after refinement, as observed previously for a similar compound with the Y220C mutant,17 suggesting a partial, radiation-induced loss of the bromine atom. Data collection and refinement statistics are listed in Table 5. Structural figures were prepared using PyMOL (www.pymol.org).
Table 5. X-ray Data Collection and Refinement Statistics.
| structure | Y220C | Y220C-9323 | Y220H | Y220S | Y220S-9301 | Y220S-9323 |
|---|---|---|---|---|---|---|
| data collection | ||||||
| space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| a (Å) | 65.18 | 64.88 | 64.76 | 65.06 | 64.98 | 65.22 |
| b (Å) | 71.15 | 71.14 | 70.77 | 71.22 | 71.30 | 71.19 |
| c (Å) | 105.04 | 104.74 | 104.88 | 105.40 | 105.72 | 105.16 |
| molecules/AU | 2 | 2 | 2 | 2 | 2 | 2 |
| resolution (Å)a | 29.5–1.24 (1.31–1.24) | 47.9–1.53 (1.56–1.53) | 47.8–1.44 (1.47–1.44) | 29.6–1.50 (1.58–1.50) | 29.6–1.40 (1.48–1.40) | 48.1–1.80 (1.84–1.80) |
| unique reflections | 135,992 | 72,947 | 86,307 | 78,532 | 96,281 | 45,643 |
| completeness (%)a | 98.0 (96.2) | 99.0 (97.7) | 98.5 (97.7) | 99.4 (99.4) | 99.3 (99.4) | 99.1 (99.9) |
| multiplicitya | 5.5 (5.5) | 4.1 (3.9) | 5.5 (5.6) | 4.7 (4.6) | 4.7 (4.7) | 4.3 (4.4) |
| Rmerge (%)a | 5.3 (58.2) | 5.3 (63.6) | 8.6 (63.5) | 6.0 (52.9) | 5.3 (48.4) | 8.9 (54.1) |
| CC(1/2)a | 0.999 (0.887) | 0.999 (0.798) | 0.995 (0.888) | 0.999 (0.875) | 0.999 (0.895) | 0.996 (0.873) |
| mean I/σ(I)a | 15.6 (3.1) | 14.1 (2.5) | 9.3 (2.4) | 14.5 (2.9) | 13.1 (3.0) | 8.9 (2.5) |
| refinement | ||||||
| Rwork (%)b | 15.1 | 14.3 | 15.2 | 14.7 | 15.1 | 19.0 |
| Rfree (%)b | 17.2 | 17.3 | 17.8 | 17.7 | 17.6 | 22.4 |
| no. of atoms | ||||||
| proteinc | 3171 | 3100 | 3131 | 3082 | 3117 | 3064 |
| zinc | 2 | 2 | 2 | 2 | 2 | 2 |
| water | 513 | 474 | 516 | 474 | 445 | 392 |
| ligands | 14 | 71 | 31 | 18 | 62 | 52 |
| RMSD bonds (Å) | 0.005 | 0.006 | 0.006 | 0.005 | 0.005 | 0.006 |
| RMSD angles (deg) | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 |
| mean B (Å2) | 18.5 | 21.0 | 18.8 | 22.6 | 21.5 | 22.6 |
| PDB entry | 6SHZ | 6SI0 | 6SI1 | 6SI2 | 6SI3 | 6SI4 |
Values in parentheses are for the highest resolution shell.
Rwork and Rfree = ∑||Fobs| – |Fcalc||/∑|Fobs|, where Rfree was calculated with 5% of the reflections chosen at random and not used in the refinement.
Number includes alternative conformations.
Molecular Dynamics Simulations
Molecular dynamics (MD) simulations were set up for the DBD of stabilized pseudo-wild-type p53 and the four Y220X mutants of this scaffold. Starting models were PDB entries 1UOL and the mutant crystal structures determined in this study (chain A). For the Y220N mutant, a model using SWISS-MODEL33 with the Y220C mutant structure (PDB ID: 2J1X) as a template was used. Ligands (e.g., buffer molecules, glycerol from the crystallization conditions) were removed from the calculations. The structures of the mutants were solvated in truncated octahedron simulation boxes with walls at least 1.2 nm away from the protein. The TIP3P44 model was used for the explicit simulation of water molecules. The system was neutralized and taken to physiological ion concentration (0.15 M) by changing some water molecules into sodium and chlorine ions. Simulations were carried out with the CHARMM36m force field45 and the program GROMACS.2018.46 The zinc ion and the coordinating cysteine/histidine residues were modeled according to Budiman et al. and Godwin et al.(47,48) This resulted in systems comprising between 35 570 and 42 173 atoms. The temperature of the system was kept constant at 300 K using a Nosé–Hoover thermostat49,50 with τt = 1 ps, and the pressure was kept constant at 1 bar with the Parrinello–Rahman algorithm51 with τp = 1 ps. Direct nonbonded interactions were cut off at 1.2 nm with a switch function on Van-der-Waals interactions starting at 1.0 nm. Long-range electrostatic interactions were computed using sPME.52 The covalent bonds with hydrogen atoms were constrained with the LINCS algorithm,53 and the integration time step was set to 2 fs. The systems were minimized for a max 50 000 steps, then equilibrated in the NVT ensemble for 1 ns with positional restraints on the positions of the heavy atoms of the protein. Then, they were further equilibrated for 1 ns in the NPT ensemble with no restraints. Four production runs were started for each mutant. Each run was 200 ns long. The resulting trajectories were analyzed using VMD54 and WORDOM.55
PK9301 and PK9323 ligands were modeled using the CHARMM general force field (CGenFF56 4.0, program version 2.2.0, https://cgenff.umaryland.edu) based on their similarity to previously parametrized compounds. The very small deviation observed along the MD simulations (maximal average RMSD of 2.1 Å over all atoms including hydrogens) of the ligands from their position in the crystallographic structure (after addition and minimization of the hydrogens) confirmed that the parametrization was appropriate.
Acknowledgments
This work was funded by German Research Foundation (DFG) grant JO 1473/1-1 (A.C.J.) and Worldwide Cancer Research (grants 14-1002, 18-0043). G.S. is funded by the DFG grant SFB1066 project Q1 and acknowledges support from the Max-Planck Graduate Center with the University of Mainz. We gratefully acknowledge support with computing time from the HPC facility Hazelhen at the High Performance Computing Center Stuttgart (project Flexadfg) and the HPC facility Mogon at the University of Mainz. We are grateful for support by the SGC, a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute, Innovative Medicines Initiative (EU/EFPIA; ULTRA-DD grant no. 115766), Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust. We also thank the staff at beamline I03 of the Diamond Light Source, Oxford, UK and beamline X06SA of the Swiss Light Source, Villigen, Switzerland for their assistance during data collection. We thank L. Holtkamp and S. Truong for help with protein purification and setting up crystallization trials.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.9b00748.
Model of the mutation-induced cavity in the p53-Y220N mutant; RMSFs of Cα atoms in MD simulations of p53-Y220X mutant DBDs and correlation between average RMSFs and protein thermostability; selected pocket parameters as a function of time over all MD simulations of Y220X mutant DBDs; correlation between subsite 2 and the central cavity in the MD simulations of the Y220S, Y220C, and Y220H mutant; ITC curve for binding of PK9301 to mutant Y220S and binding of PK9255 to mutant Y220N (PDF)
Accession Codes
Coordinates and structure factors of the p53-Y220X mutant structures have been deposited in the Protein Data Bank (PDB). Accession codes: 6SHZ, 6SI0, 6SI1, 6SI2, 6SI3, and 6SI4.
Author Contributions
# These authors contributed equally
The authors declare no competing financial interest.
Supplementary Material
References
- Joerger A. C.; Fersht A. R. (2016) The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 85, 375–404. 10.1146/annurev-biochem-060815-014710. [DOI] [PubMed] [Google Scholar]
- Khoo K. H.; Verma C. S.; Lane D. P. (2014) Drugging the p53 pathway: understanding the route to clinical efficacy. Nat. Rev. Drug Discovery 13, 217–236. 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- Stiewe T.; Haran T. E. (2018) How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist. Updates 38, 27–43. 10.1016/j.drup.2018.05.001. [DOI] [PubMed] [Google Scholar]
- Joerger A. C.; Fersht A. R. (2007) Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 26, 2226–2242. 10.1038/sj.onc.1210291. [DOI] [PubMed] [Google Scholar]
- Freed-Pastor W. A.; Prives C. (2012) Mutant p53: one name, many proteins. Genes Dev. 26, 1268–1286. 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainaut P.; Pfeifer G. P. (2016) Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harbor Perspect. Med. 6, a026179. 10.1101/cshperspect.a026179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock A. N.; Henckel J.; Fersht A. R. (2000) Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19, 1245–1256. 10.1038/sj.onc.1203434. [DOI] [PubMed] [Google Scholar]
- Wang G.; Fersht A. R. (2017) Multisite aggregation of p53 and implications for drug rescue. Proc. Natl. Acad. Sci. U. S. A. 114, E2634–E2643. 10.1073/pnas.1700308114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearth L. R.; Qian H.; Wang T.; Baroni T. E.; Zeng J.; Chen S. W.; Yi S. Y.; Brachmann R. K. (2007) Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis 28, 289–298. 10.1093/carcin/bgl132. [DOI] [PubMed] [Google Scholar]
- Joerger A. C.; Ang H. C.; Fersht A. R. (2006) Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. U. S. A. 103, 15056–15061. 10.1073/pnas.0607286103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckler F. M.; Joerger A. C.; Jaggi G.; Rutherford T. J.; Veprintsev D. B.; Fersht A. R. (2008) Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. U. S. A. 105, 10360–10365. 10.1073/pnas.0805326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken R.; Liu X.; Zimmermann M. O.; Rutherford T. J.; Fersht A. R.; Joerger A. C.; Boeckler F. M. (2012) Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J. Am. Chem. Soc. 134, 6810–6818. 10.1021/ja301056a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger A. C.; Bauer M. R.; Wilcken R.; Baud M. G.; Harbrecht H.; Exner T. E.; Boeckler F. M.; Spencer J.; Fersht A. R. (2015) Exploiting Transient Protein States for the Design of Small-Molecule Stabilizers of Mutant p53. Structure 23, 2246–2255. 10.1016/j.str.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M. R.; Jones R. N.; Baud M. G.; Wilcken R.; Boeckler F. M.; Fersht A. R.; Joerger A. C.; Spencer J. (2016) Harnessing Fluorine-Sulfur Contacts and Multipolar Interactions for the Design of p53 Mutant Y220C Rescue Drugs. ACS Chem. Biol. 11, 2265–2274. 10.1021/acschembio.6b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basse N.; Kaar J. L.; Settanni G.; Joerger A. C.; Rutherford T. J.; Fersht A. R. (2010) Toward the rational design of p53-stabilizing drugs: probing the surface of the oncogenic Y220C mutant. Chem. Biol. 17, 46–56. 10.1016/j.chembiol.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Wang G.; Boeckler F. M.; Fersht A. R. (2012) Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl. Acad. Sci. U. S. A. 109, 13584–13589. 10.1073/pnas.1211550109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M. R.; Jones R. N.; Tareque R. K.; Springett B.; Dingler F. A.; Verduci L.; Patel K. J.; Fersht A. R.; Joerger A. C.; Spencer J. (2019) A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med. Chem. 11, 2491–2504. 10.4155/fmc-2019-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Wilcken R.; Joerger A. C.; Chuckowree I. S.; Amin J.; Spencer J.; Fersht A. R. (2013) Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 41, 6034–6044. 10.1093/nar/gkt305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud M. G. J.; Bauer M. R.; Verduci L.; Dingler F. A.; Patel K. J.; Horil Roy D.; Joerger A. C.; Fersht A. R. (2018) Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 152, 101–114. 10.1016/j.ejmech.2018.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassman C. D.; Baronio R.; Demir O.; Wallentine B. D.; Chen C.-K.; Hall L. V.; Salehi F.; Lin D.-W.; Chung B. P.; Wesley Hatfield G.; Richard Chamberlin A.; Luecke H.; Lathrop R. H.; Kaiser P.; Amaro R. E. (2013) Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 4, 1407. 10.1038/ncomms2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan M. R.; Siau J. W.; Kannan S.; Nguyen M. N.; Ouaray Z.; Kwoh C. K.; Lane D. P.; Ghadessy F.; Verma C. S. (2019) Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic Acids Res. 47, 1637–1652. 10.1093/nar/gky1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J. M.; Gorzov P.; Veprintsev D. B.; Soderqvist M.; Segerback D.; Bergman J.; Fersht A. R.; Hainaut P.; Wiman K. G.; Bykov V. J. (2009) PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15, 376–388. 10.1016/j.ccr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Bykov V. J.; Zhang Q.; Zhang M.; Ceder S.; Abrahmsen L.; Wiman K. G. (2016) Targeting of Mutant p53 and the Cellular Redox Balance by APR-246 as a Strategy for Efficient Cancer Therapy. Front. Oncol. 6, 21. 10.3389/fonc.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M. R.; Joerger A. C.; Fersht A. R. (2016) 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. U. S. A. 113, E5271–5280. 10.1073/pnas.1610421113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanden A. R.; Yu X.; Wolfe A. J.; Gilleran J. A.; Augeri D. J.; O’Dell R. S.; Olson E. C.; Kimball S. D.; Emge T. J.; Movileanu L.; Carpizo D. R.; Loh S. N. (2015) Synthetic Metallochaperone ZMC1 Rescues Mutant p53 Conformation by Transporting Zinc into Cells as an Ionophore. Mol. Pharmacol. 87, 825–831. 10.1124/mol.114.097550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaoun L.; Sonkin D.; Ardin M.; Hollstein M.; Byrnes G.; Zavadil J.; Olivier M. (2016) TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 37, 865–876. 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- Leroy B.; Fournier J. L.; Ishioka C.; Monti P.; Inga A.; Fronza G.; Soussi T. (2013) The TP53 website: an integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 41, D962–969. 10.1093/nar/gks1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F.; Ferlay J.; Soerjomataram I.; Siegel R. L.; Torre L. A.; Jemal A. (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-Cancer J. Clin. 68, 394–424. 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Capponcelli S.; Pedrini E.; Cerone M. A.; Corti V.; Fontanesi S.; Alessio M.; Bachi A.; Soddu S.; Ribatti D.; Picci P.; Helman L. J.; Cantelli-Forti G.; Sangiorgi L. (2005) Evaluation of the molecular mechanisms involved in the gain of function of a Li-Fraumeni TP53 mutation. Hum. Mutat. 26, 94–103. 10.1002/humu.20192. [DOI] [PubMed] [Google Scholar]
- Nikolova P. V.; Henckel J.; Lane D. P.; Fersht A. R. (1998) Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. U. S. A. 95, 14675–14680. 10.1073/pnas.95.25.14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger A. C.; Allen M. D.; Fersht A. R. (2004) Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J. Biol. Chem. 279, 1291–1296. 10.1074/jbc.M309732200. [DOI] [PubMed] [Google Scholar]
- Ang H. C.; Joerger A. C.; Mayer S.; Fersht A. R. (2006) Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 281, 21934–21941. 10.1074/jbc.M604209200. [DOI] [PubMed] [Google Scholar]
- Bienert S.; Waterhouse A.; de Beer T. A.; Tauriello G.; Studer G.; Bordoli L.; Schwede T. (2017) The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 45, D313–D319. 10.1093/nar/gkw1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Interlandi G.; Wetzel S. K.; Settanni G.; Pluckthun A.; Caflisch A. (2008) Characterization and further stabilization of designed ankyrin repeat proteins by combining molecular dynamics simulations and experiments. J. Mol. Biol. 375, 837–854. 10.1016/j.jmb.2007.09.042. [DOI] [PubMed] [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. (2010) A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 53, 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan V.; Agrahari M.; Gowda D. K. (2019) Virtual screening of p53 mutants reveals Y220S as an additional rescue drug target for PhiKan083 with higher binding characteristics. Comput. Biol. Chem. 80, 398–408. 10.1016/j.compbiolchem.2019.05.005. [DOI] [PubMed] [Google Scholar]
- Joerger A. C.; Ang H. C.; Veprintsev D. B.; Blair C. M.; Fersht A. R. (2005) Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J. Biol. Chem. 280, 16030–16037. 10.1074/jbc.M500179200. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmuller H.; MacKerell A. D. Jr. (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Budiman M. E.; Knaggs M. H.; Fetrow J. S.; Alexander R. W. (2007) Using molecular dynamics to map interaction networks in an aminoacyl-tRNA synthetase. Proteins: Struct., Funct., Genet. 68, 670–689. 10.1002/prot.21426. [DOI] [PubMed] [Google Scholar]
- Godwin R. C.; Melvin R. L.; Gmeiner W. H.; Salsbury F. R. Jr. (2017) Binding Site Configurations Probe the Structure and Dynamics of the Zinc Finger of NEMO (NF-kappaB Essential Modulator). Biochemistry 56, 623–633. 10.1021/acs.biochem.6b00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosé S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519. 10.1063/1.447334. [DOI] [Google Scholar]
- Hoover W. G. (1985) Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A: At., Mol., Opt. Phys. 31, 1695–1697. 10.1103/PhysRevA.31.1695. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. (1980) Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 45, 1196–1199. 10.1103/PhysRevLett.45.1196. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8592. 10.1063/1.470117. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472. 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Seeber M.; Cecchini M.; Rao F.; Settanni G.; Caflisch A. (2007) Wordom: a program for efficient analysis of molecular dynamics simulations. Bioinformatics 23, 2625–2627. 10.1093/bioinformatics/btm378. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; Mackerell A. D. Jr. (2009) CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt T.; Kaar J. L.; Fersht A. R.; Veprintsev D. B. (2012) Stability of p53 homologs. PLoS One 7, e47889. 10.1371/journal.pone.0047889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.