

Abstract
While the mechanism of the P450-catalyzed oxidative hydroxylation of organic compounds has been studied in detail for many years, less is known about sulfoxidation. Depending upon the structure of the respective substrate, heme-Fe=O (Cpd I), heme–Fe(III)–OOH (Cpd 0), and heme–Fe(III)–H2O2 (protonated Cpd 0) have been proposed as reactive intermediates. In the present study, we consider the transformation of isosteric substrates via sulfoxidation and oxidative hydroxylation, respectively, catalyzed by regio- and enantioselective mutants of P450-BM3 which were constructed by directed evolution. 1-Thiochromanone and 1-tetralone were used as the isosteric substrates because, unlike previous studies involving fully flexible compounds such as thia-fatty acids and fatty acids, respectively, these compounds are rigid and cannot occur in a multitude of different conformations and binding modes in the large P450-BM3 binding pocket. The experimental results comprising activity and regio- and enantioselectivity, flanked by molecular dynamics computations within a time scale of 300 ns and QM/MM calculations of transition-state energies, unequivocally show that heme-Fe=O (Cpd I) is the common catalytically active intermediate in both sulfoxidation and oxidative hydroxylation.
Introduction
Cytochrome P450 monooxygenases catalyze a remarkable range of oxidative reaction types, including CH-activating hydroxylation, olefin epoxidation, sulfoxidation, and aminoxidation.1−11 This raises the question of whether one and the same catalytically active species is involved or whether the enzyme utilizes a different active species for each type of transformation. Oxidative hydroxylation versus sulfoxidation is a case in point. Thanks to decades of intensive experimental1−7 and theoretical work,8−10 the mechanism of hydroxylation is well understood. All researchers in the field have agreed that it involves high-spin intermediate heme-Fe=O (Cpd I) in a rate-determining radical abstraction process followed by rapid C–O bond formation.1−11 In the most recent QM/MM study, Shaik and co-workers have studied the regio- and enantioselectivity of fatty acid hydroxylation catalyzed by wild type (WT) and mutants of P450-BM3.12 Again, the crucial role of Cpd I was documented, in addition to the dynamics of the flexible carbon chain of fatty acids. Fewer experimental efforts have been devoted to clarifying the mechanism of sulfoxidation, most of which favor Cpd I as the catalytically active species.13−17 In principle, three oxidant candidates (Cpd I, ferric hydroperoxide (Cpd 0), and ferric hydrogen peroxide (Fe(III)–HOOH)) need to be considered (Scheme 1).
Scheme 1. Mechanistic Conjectures Regarding P450-BM3-Catalyzed Oxidations (Sub = Substrate)29.
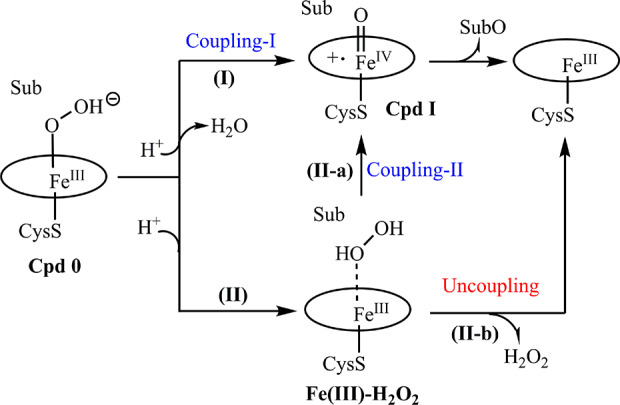
Cpd 0, as a required intermediate in the catalytic cycle of P450, can be either transformed into Cpd I via the coupling-I (pathway I) or heme–Fe(III)–H2O2 intermediate via the protonation of Cpd 0 on the proximal oxygen (pathway II) (Scheme 1). Early experimental and theoretical work on cytochrome P450-cam as the catalyst in the stereoselective sulfoxidation of thioanisole (R/S = 72:28) and p-methylthioanisole (R/S = 48:52) pointed to the role of ferryl form heme–Fe=O as the catalytically active species.18 Indeed, an initial QM-based theoretical study by Shaik and co-workers supported this hypothesis, suggesting that catalysis occurs via Cpd I with a low energy barrier.19 The calculations of the alternative process based on Cpd 0, in which the O atom bound to Fe was assumed to be transferred to the thio-ether in the sulfoxidation reaction, led to a very high energy transition state.19 Thus, it seemed that the common role of Cpd I in hydroxylation and sulfoxidation was well established.
However, in 2006 an experimental investigation was published by Cryle and De Voss in which the regio- and enantioselectivities of fatty acid hydroxylation in comparison to the sulfoxidation of isosteric thia-fatty acids were used as mechanistic probes.20 Since the selectivities proved to be quite different in the two reaction types, the authors speculated that two different active species may be involved, Cpd I in hydroxylation and Cpd 0 in sulfoxidation.20 Interestingly, in two experimental studies featuring nonheme iron catalysts, a species such as Fe(III)–OOH “corresponding” to Cpd 0 was shown to be a sluggish reagent in sulfoxidation.21−23 In yet another experimental study using [FeIII–(TMP)(Cl)] (TMP= meso-tetramesitylporphyrin) as the model for P450 enzymes and m-chloroperbenzoic acid as the oxidant, it was found that [(TMP+•)FeIV=O] is the most active species in sulfoxidation and not the precursor acylperoxoiron(III) porphyrin complex which is orders of magnitude less active.24
In 2007, another QM study by Shaik appeared in which several potential pathways in the sulfoxidation of a different substrate (dimethyl sulfide) were computed using both Cpd I and Cpd 0.25 Cpd I was favored once more as the catalytically active species in both sulfoxidation and hydroxylation,25 as in other theoretical studies.26,27 Following these developments, the Shaik group reported in 2013 computational results which indicate that protonated Cpd 0, i.e., heme–Fe(III)–H2O2, may possibly participate in sulfoxidation via a heterolytic mechanism.28 Though FeIII(H2O2) could be an efficient oxidant in sulfoxidation reactions, the reactivity of H2O2 in the active site of P450 is largely dependent on the persistence of H2O2 and the precise positioning of the substrate. In their most recent studies, the oxidant of Cpd I was favored over heme–Fe(III)–H2O2 in P450.29,30
While Cpd 0 has been amply excluded as an active species in both experiments and theory,1−11,13−19,25−30 the mechanistic dilemma concerning the reactivity of Cpd I and heme–Fe(III)–H2O2 in P450-BM3 has not been fully resolved. This dilemma is augmented by the experimental evidence that H2O2 can be used in the H2O2 shunting of P450-BM3, possibly via coupling-II (II-a in Scheme 1) as demonstrated before.29 To the best of our knowledge, no experimental evidence has been provided to prove or exclude that heme–Fe(III)–H2O2 could function as an oxidant in P450-BM3-catalyzed reactions.
In the present study, we revisit the general mechanistic issue by presenting new empirical data derived from directed evolution6,31−40 studies of P450-BM3 in the regio- and stereoselective sulfoxidation and oxidative hydroxylation of truly isosteric substrates which are structurally rigid. As will be seen, we chose rigid substrates that are structurally quite unlike the flexible fatty acids12 and thia-fatty acids previously used in the De Voss comparative study,20 with our choice allowing for more reliable conclusions. Regio- and enantioselectivities of mutants in sulfoxidation and respective hydroxylation were analyzed by molecular dynamics (MD) computations, flanked by QM/MM computations. Also included are control experiments in the presence of catalase. The combination of experimental and theoretical results allowed us to propose a sound model in which Cpd I plays the common catalytic role.
Results and Discussion: Experimental Results Derived from Directed Evolution
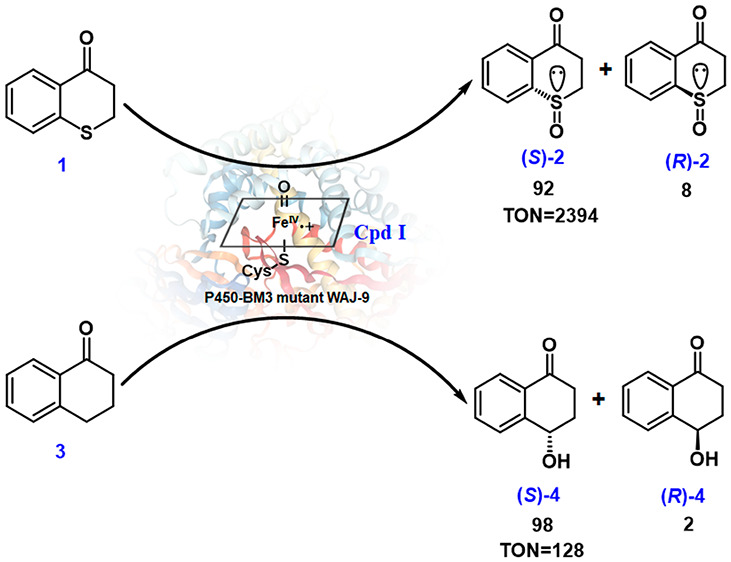
We reasoned that isosteric compounds thiochromanone (1) and 1-tetralone (3) are ideal substrates for the present study (Scheme 2).
Scheme 2. Model Sulfoxidation (a) and Oxidative Hydroxylation (b) of Isosteric Substrates 1 and 3, Respectively.

The cyclic nature of substrates 1 and 3 excludes the possible occurrence of different conformers in the large binding pocket of the enzyme, in contrast to flexible straight-chain compounds such as fatty acids and their thio-analogs. Respective sulfoxide products (R)- and (S)-2 have been prepared previously by other means.41,42
We discovered that wild-type (WT) P450-BM3 catalyzes the sulfoxidation of 1-thiochromanone (1) with >95% chemoselectivity, essentially without any competing oxidative hydroxylation. However, the enantioselectivity is moderate, with the enantiomeric ratio amounting to only er = 75:25 (50% ee) in favor of (S)-2. In the initial work, single codon saturation mutagenesis (SCSM)15 was applied to enhance and invert WT enantioselectivity, with several mutants being created for the asymmetric sulfoxidation of 1. However, the stability of these multimutational variants decreased significantly. Moreover, when too many sites have been mutated, mechanistic investigations by protein crystal structure resolution and/or computer modeling are more difficult. In order to identify the catalytically active species of the reaction of substrate 1 and at the same time to simplify our study of sulfoxidation, we constructed a mutant showing high (S)-selectivity toward substrate 1, this being achieved by the introduction of only three point mutations in two steps. First, seven residues, as in previous work,43 were chosen for NNK-based saturation mutagenesis separately. Seven hundred colonies were screened, and three sites were confirmed to be effective in influencing enantioselectivity positively (L75, F87, and A328). Several variants were found to show enhanced (S)-selectivity (Table S2), while one mutant, F87L, favored (R)-selectivity (er = 53:47), which is different from the WT. In order to evolve further improvement, F87L was used as a template for iterative saturation mutagenesis (ISM)31,44 at L75 and A328. They were mutated simultaneously, likewise using NNK codon degeneracy. After screening a total of about 1200 colonies, the best (S)-selective mutant, WAJ-9 (L75F-F87L-A328F), was evolved, which produces (S)-2 with er = 92:8. As a result of kinetic experiments, WAJ-9 was found to display very high activity for the sulfoxidation of this substrate with a turnover number (TON) of 2394 and a turnover frequency (TOF) of 275 h–1. This mutant plays a crucial role in addressing the basic mechanistic question of our study. Such pronounced enantioselectivity was not found in the (R)-manifold, but this result was of no concern in the present mechanistic work.
Variant WAJ-9 was then tested as a catalyst in the oxidative hydroxylation of isosteric 1-tetralone (3) (Table 1). An enantiomeric ratio amounting to 98:2 in favor of (S)-4 was observed. However, the TON value for the hydroxylation of 3 is only 128, which is similar to that of the WT. A striking result in terms of mechanistic significance is the TOF of the hydroxylation of 3, which decreases to 6 h–1, unlike the sulfoxidation of substrate 1. This mutant can be defined as a sluggish reagent for hydroxylation, although the enantioselectivity is high.
Table 1. Kinetic Data for WT, WAJ-9, and A328F toward the Sulfoxidation of 1 and the Hydroxylation of 3a.
| substrate 1 |
substrate 3 |
|||
|---|---|---|---|---|
| P450-BM3 variants | TON | TOF (h–1) | TON | TOF (h–1) |
| WT | 583 | 134 | 128 | 15 |
| WAJ-9 | 2394 | 275 | 128 | 6 |
| A328F | 2364 | 555 | 442 | 55 |
TON and TOF were obtained by averaging at least three independent experiments. General reaction condition: 0.5 μM P450-BM3 enzyme, 10 mg/mL glucose dehydrogenase, 100 mM glucose, 200 μM NADP+, 2 mM substrate in 100 mM phosphate buffer (pH 8.0), 30 °C, 800 rpm. Experiments were performed for 9 h to calculate TON and for 2 h to calculate TOF.
We then considered the opposite situation by testing variant A328F (Table 1) (the previously evolved hydroxylating variant used in the formation of 4-hydroxy-tetralone 4(45)) as the catalyst for the sulfoxidation of thiochromanone (1). According to the kinetic data of these two reactions, the activity of variant A328F in the sulfoxidation of substrate 1 is higher than in the hydroxylation of 3, and both activities are higher than for WT. However, the enantioselectivity decreased sharply from er = 99:1 to 64:36.
At this stage, a final mechanistic conclusion seemed premature, and further control experiments were planned to test whether heme–Fe(III)–H2O2 could possibly be involved. Due to the lower energy barrier for the dissociation of heme–Fe(III)–H2O2 compared to that of sulfoxidation (2.4 kcal/mol VS 5.2 kcal/mol),28 the rate of dissociation should be faster. Therefore, the presence of a large amount of catalase in the system would be expected to erase most of the H2O2 and induce it to dissociate from the heme–Fe(III)–H2O2 complex (II-b in Scheme 1). Consequently, WAJ-9’s activity toward substrate 1 with the production of 2 should be shut down. To confirm this, we performed all of the kinetic experiments in the presence of catalase (Table 2). The results demonstrate that catalase shows no inhibition because all TONs and TOFs were found to be higher than in the system without any catalase. This result indicates that certainly for the case of P450-BM3, Fe(III)(H2O2) cannot be the active species and that the O2-dependent pathway is more favorable than the H2O2-shunt pathway.29 Moreover, the observation of increased TONs and TOFs for the oxidation of 1 also indicates that some H2O2 is definitely released in the catalase-free system, which to some extent impedes the sulfoxidation reaction. Our results are in line with the presence of bound and unbound hydrogen peroxide: Fe(III)(H2O2) ⇌ Fe(III)–heme + H2O2.
Table 2. Kinetic Tests in the Presence of Catalasea.
| substrate 1 |
substrate 3 |
|||
|---|---|---|---|---|
| P450-BM3 variants | TON | TOF (h–1) | TON | TOF (h–1) |
| WT | 737 | 255 | 142 | 36 |
| WAJ-9 | 2796 | 382 | 135 | 7 |
| A328F | 2668 | 677 | 716 | 86 |
Reactions were performed under general conditions but with >1200 U/mL catalase.
At this point, the extensive experimental data excludes Fe(III)(H2O2) as a catalyst in sulfoxidation. However, some questions remained to be answered. What are the poses of isosteric substrates 1 and 3 in the large binding pocket of mutant WAJ-9?
In order to gain insight into the origin of enantioselectivity diversity in the mutants and to answer the question of whether Cpd I functions as the catalytically active species in sulfoxidation, MD simulations of substrates 1 and 3 were carried out in the presence of Cpd I species for three P450-BM3 variants.
For WT, our MD simulations predict an enantiomeric ratio of 68:32 for the sulfoxidation of substrate 1 (Table 3), which is in good agreement with the experimental value of 75:25. In particular, the predicted enantiomeric ratio is slightly sensitive to the time scale of the MD run, but the trend is constant even on the extended MD time scale. For instance, the calculated enantiomeric ratio in WT is 69:31 within 50 ns, 68:32 within 200 ns, and 79:21 within a time scale of 300 ns, respectively, all in good agreement with the experimental result. As indicated by the structures from the MD trajectory (Figure 1), substrate 1 displays mobility in the binding pocket of WT P450-BM3, in which it adopts either a “left” position or a “right” position in two different binding processes (Figure 1), which correspond to (R)- and (S)-selectivity, respectively. The behavior of 1 in A328F is quite similar to that of WT (Figure S7), and the A328F mutation has a minor effect on the predicted enantioselectivity (71:29). In both WT and variant A328F, 1 forms a π–π stacking interaction with the Phe87 residue below (Figure S7). However, due to the large cavity above 1, the binding of this substrate reveals mobility in both WT and A328F, which results in suboptimal enantioselectivity.
Table 3. Enantioselectivity of Sulfoxidation in the Reaction of Substrate 1, Derived from Experiment and 200 ns MD Simulations, and the Computed Average Ox–S Distance for Cpd I of WT, WAJ-9, and A328F in the Presence of Substrate 1.
| P450-BM3 | (R)/(S) |
Ox–S distance (Å) | |
|---|---|---|---|
| substrate 1 | experiment | MD | MD |
| WT | 25:75 | 32:68 | 4.3 |
| WAJ-9 | 8:92 | 16:84 | 3.6 |
| A328F | 36:64 | 29:71 | 4.1 |
Figure 1.
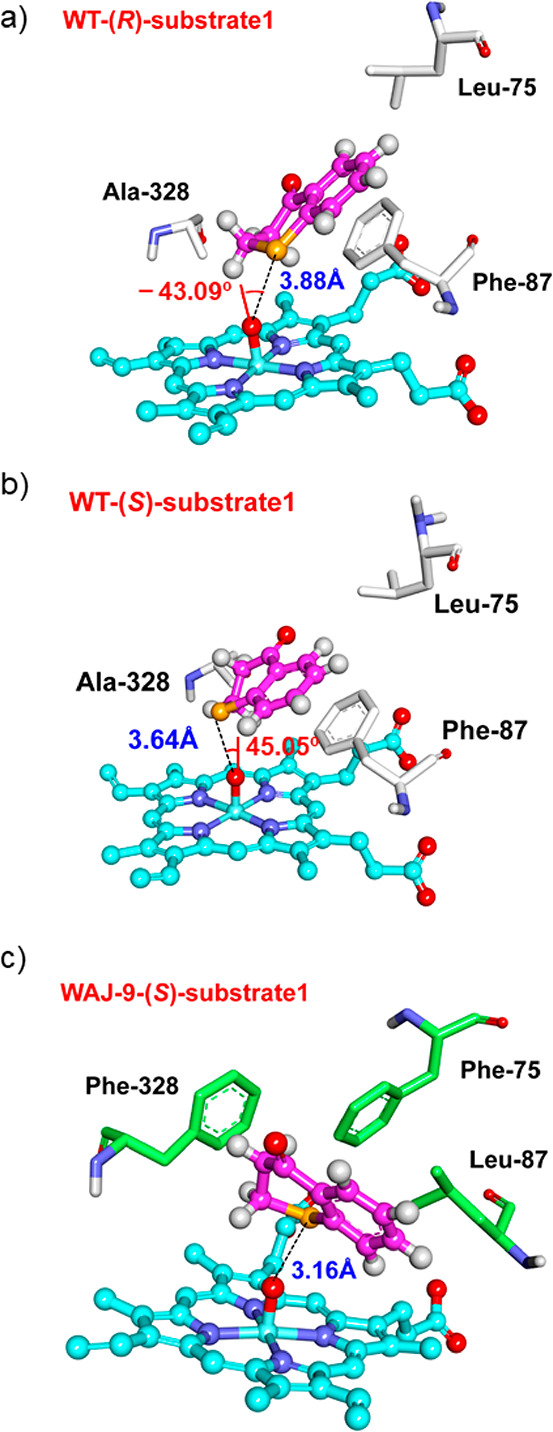
Representative binding modes of (R)-substrate 1 (a), (S)-substrate 1 (b) in WT, and (S)-substrate 1 (c) in WAJ-9.
In the case of triple mutant WAJ-9 (L75F-F87L-A328F), the F87L mutation removes the π–π stacking interactions with 1 (Figure 1c). Accordingly, a cavity is created underneath 1, resulting in a proximal conformation closer to that of Fe-porphyrin. As evidenced by the nonbonding interaction analysis, substrate 1 is significantly stabilized by “enclosing” residues Phe75, Leu87, and Phe328 (Figure S9), leading to firm binding, restrained mobility, and essentially complete (S)-enantioselectivity in WAJ-9 (Table 3). Thus, our MD simulations provide a detailed mechanistic picture of the effect of mutations in variant WAJ-9 on the enantioselectivity of sulfoxidation.
As indicated in Table 3, the predicted order of average Ox–S distances WAJ-9 < A328F < WT is in accord with the experimentally determined reactivity order (Table 2). To identify if the angle defined by ∠Fe–O–S could play some role in the reactivity of Cpd I-mediated sulfoxidation, four randomly selected snapshots from MD simulations (two snapshots from WT, one from WAJ-9, and one from A328F) were used for QM/MM calculations. It was found that the sulfoxidation reactivities (Table S5) correlate well with the Ox–S distance, while they have little dependence on the Fe–Ox–S angle. Taken together, our combined MD simulations and QM/MM calculations suggest that the average Ox–S distance is a good indicator of the reactivity of Cpd I-mediated sulfoxidation.
When considering the hydroxylation of substrate 3, the MD simulations point to a much higher mobility and long distance to Cpd I than in the case of substrate 1 in WT (Figure S11).
This dynamic phenomenon explains the low activity observed experimentally in the hydroxylation of 3 (Table 1). In both WAJ-9 and A328F, 3 adopts an “upright” binding juxtaposition with its pro-(S) H-atom being closest to Cpd I (Figure S11), which is in line with the observed experimental enantioselectivity (Table S4). Moreover, our MD simulations predict an order of A328F < WT < WAJ-9 for the average distance between Ox and the pro-(S) H-atom (Table S4), which is in agreement with the order of observed activity (Table 1). To confirm that the MD-predicted structure is reactive toward the hydroxylation of substrate 3, a random snapshot from MD of substrate 3 was used for QM/MM calculations (Figure S17). The calculated energy barrier is ∼23 kcal/mol, which is in excellent agreement with the experimental value of ∼20 kcal/mol. This indicates that in spite of the relatively long computed average distance (>5 Å), substrate 3 has a position from which it can still get close to the active center for hydroxylation.
The present combination of laboratory experiments and MD simulations clearly leads to a model according to which Cpd I is the catalytically active species in sulfoxidation and hydroxylation, supporting the most recent prediction made by Shaik and co-workers based on a combined MD and QM/MM study.30
Conclusions
In order to shed light on the question of a single or two different catalytically active species in P450-BM3-catalyzed sulfoxidation versus oxidative hydroxylation, we have applied experimental and theoretical techniques. On the experimental side, two isosteric compounds were chosen as substrates, thiochromanone (1) for sulfoxidation and 1-tetralone (3) for hydroxylation. In contrast to a previous study employing inherently flexible fatty acids and thia-fatty acids in hydroxylation and sulfoxidation, respectively,20 which makes definitive interpretations difficult, the model compounds used in our study are rigid due to their cyclic nature and therefore do not allow a multitude of possible conformations to be formed in the large P450-BM3 binding pocket. Indeed, the already-mentioned earlier theoretical study of P450-BM3 harboring N-palmitoylglycine demonstrated the flexibility of the respective carbon chain of this fatty acid-derived substrate.12
Active and selective mutants evolved for the sulfoxidation of substrate 1 proved to be somewhat sluggish catalysts in the hydroxylation of substrate 3 (very low activity) but with high enantioselectivity. In contrast, a mutant evolved earlier for the hydroxylation of substrate 3 was found to retain high activity in the sulfoxidation of substrate 1, although with low enantioselectivity. Such catalytic profiles could lead to premature conclusions regarding the occurrence of two different catalytically active species, e.g., heme–Fe(III)–H2O2 in sulfoxidation and Cpd I in oxidative hydroxylation. However, the possibility that heme–Fe(III)–H2O2 may be involved as the catalytically active species was excluded for the first time by performing the enzyme assays in the presence of excess catalase. The results of applying this mechanistic probe showed that Cpd I is the only catalytically active species catalyzing both sulfoxidation and hydroxylation. This general conclusion is in line with MD computations within a time scale of 300 ns and QM/MM calculations of transition-state energies.
All MD-predicted enantioselectivity and reactivity values are consistent with the experimental results, strongly supporting the notion that Cpd I is the competent active species for both sulfoxidation and hydroxylation reactions. They also provide a detailed mechanistic picture for explaining the origin of enantioselectivity in sulfoxidations catalyzed by various P450-BM3 mutants. The relatively large cavity above bound substrate 1 in both WT and mutant A328F allows dynamic movements and different binding modes of this compound, thus resulting in suboptimal enantioselectivity. By contrast, variant WAJ-9 (L75F-F87L-A328F) and substrate-enclosing residues Phe75, Leu87, and Phe328 significantly stabilize the binding mode, thus leading to high enantioselectivity and activity for sulfoxidation. We expect that the mechanistic insights from our study will aid future protein engineering of cytochrome P450 enzymes.
Acknowledgments
B.W. is grateful for financial support from NSFC (nos. 21933009 and 21907082). M.T.R. acknowledges support from the Max-Planck-Society and LOEWE Research Cluster SynChemBio. J.-b.W. thanks Hunan Normal University for the start-up funding.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b13061.
All chemicals, materials for molecular biology, and experimental details (PDF)
Author Contributions
# These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Modi A. R.; Dawson J. H.. Oxidizing Intermediates in P450 Catalysis: A Case for Multiple Oxidants. In Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450; Hrycay E. G., Bandiera S. M., Eds.; Springer International Publishing: Cham, 2015; pp 63–81. [Google Scholar]
- Whitehouse C. J. C.; Bell S. G.; Wong L.-L. P450BM3 (CYP102A1): Connecting the Dots. Chem. Soc. Rev. 2012, 41, 1218–1260. 10.1039/C1CS15192D. [DOI] [PubMed] [Google Scholar]
- Isin E. M.; Guengerich F. P. Complex Reactions Catalyzed by Cytochrome P450 Enzymes. Biochim. Biophys. Acta, Gen. Subj. 2007, 1770, 314–329. 10.1016/j.bbagen.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Munro A. W.; Girvan H. M.; McLean K. J. Variations on a (t) heme-novel Mechanisms, Redox Partners and Catalytic Functions in the Cytochrome P450 Superfamily. Nat. Prod. Rep. 2007, 24, 585–609. 10.1039/B604190F. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R. Hydrocarbon Hydroxylation by Cy-tochrome P450 Enzymes. Chem. Rev. 2010, 110, 932–948. 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urlacher V. B.; Girhard M. Cytochrome p450 Monooxygenases in Biotechnology and Synthetic Biology. Trends Biotechnol. 2019, 37, 882–897. 10.1016/j.tibtech.2019.01.001. [DOI] [PubMed] [Google Scholar]
- Holtmann D.; Fraaije M. W.; Arends I. W. C. E.; Opperman D. J.; Hollmann F. The Taming of Oxygen: Biocatalytic Oxyfunctionalisations. Chem. Commun. 2014, 50, 13180–13200. 10.1039/C3CC49747J. [DOI] [PubMed] [Google Scholar]
- Meunier B.; de Visser S. P.; Shaik S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Kumar D.; de Visser S. P.; Altun A.; Thiel W. Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 2005, 105, 2279–2328. 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Cohen S.; Wang Y.; Chen H.; Kumar D.; Thiel W. Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 2010, 110, 949–1017. 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- Dong J.-J.; Fernandez-Fueyo E.; Hollmann F.; Paul C.; Pesic M.; Schmidt S.; Wang Y.; Younnes S.; Zhang W. Biocatalytic Oxidation Reactions: A Chemist’s Perspective. Angew. Chem., Int. Ed. 2018, 57, 9238–9261. 10.1002/anie.201800343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey K. D.; Wang B.; Shaik S. Molecular Dynamics and QM/MM Calculations Predict the Substrate-induced Gating of Cytochrome P450-BM3 and the Regio-and Stereoselectivity of Fatty Acid Hydroxylation. J. Am. Chem. Soc. 2016, 138, 837–845. 10.1021/jacs.5b08737. [DOI] [PubMed] [Google Scholar]
- Fukushima D.; Kim Y. H.; Yanagi T.; Oae S. Enzymatic Oxidation of Disulfides and Thiolsulfinates by Both Rabbit Liver Microsomes and a Reconstituted System with Purified Cytochrome P450. J. Biochem. 1978, 83, 1019–1027. 10.1093/oxfordjournals.jbchem.a131990. [DOI] [PubMed] [Google Scholar]
- Toshikazu T.; Ken F.; Hae K. Y.; Takashi I.; Shigeru O. Enzymatic Oxygenation of Sulfides with Cytochrome P450 from Rabbit Liver. Stereochemistry of Sulfoxide Formation. Bull. Chem. Soc. Jpn. 1983, 56, 2300–2310. 10.1246/bcsj.56.2300. [DOI] [Google Scholar]
- Filipovic D.; Paulsen M. D.; Loida P. J.; Sligar S. G.; Ornstein R. L. Ethylbenzene Hydroxylation by Cytochrome P450cam. Biochem. Biophys. Res. Commun. 1992, 189, 488–495. 10.1016/0006-291X(92)91584-D. [DOI] [PubMed] [Google Scholar]
- Miao E.; Joardar J.; Zuo C.; Clouthier N. J.; Nagahisa A.; Byon C.; Wilson S. R. Cytochrome P-450scc-mediated Oxidation of (20S)-22-thiacholesterol: Characterization of Mechanism-based Inhibition. Biochemistry 1995, 34, 8415–8421. 10.1021/bi00026a024. [DOI] [PubMed] [Google Scholar]
- Volz T. J.; Rock D. A.; Jones J. P. Evidence for Two different Active Oxygen Species in Cytochrome P450-BM3Mediated Sulfoxidation and N-dealkylation Reactions. J. Am. Chem. Soc. 2002, 124, 9724–9725. 10.1021/ja026699l. [DOI] [PubMed] [Google Scholar]
- Fruetel J.; Chang Y. T.; Collins J.; Loew G.; Montellano P. R. Thioanisole Sulfoxidation by Cytochrome P450cam (CYP101): Experimental and Calculated Absolute Stereochemistries. J. Am. Chem. Soc. 1994, 116, 11643–11648. 10.1021/ja00105a003. [DOI] [Google Scholar]
- Sharma P. K.; de Visser S. P.; Sason S. Can a Single Oxidant with Two Spin States Masquerade as Two Different Oxidants? A Study of the Sulfoxidation Mechanism by Cytochrome P450. J. Am. Chem. Soc. 2003, 125, 8698–8699. 10.1021/ja035135u. [DOI] [PubMed] [Google Scholar]
- Cryle M. J.; De Voss J. J. Is the Ferric Hydroperoxy Species Responsible for Sulfur Oxidation in Cytochrome P450s?. Angew. Chem., Int. Ed. 2006, 45, 8221–8223. 10.1002/anie.200603411. [DOI] [PubMed] [Google Scholar]
- Park M. J.; Lee J.; Suh Y.; Kim J.; Nam W. Reactivities of Mononuclear Non-heme Iron Intermediates Including Evidence that Iron (III)- hydroperoxo Species is a Sluggish Oxidant. J. Am. Chem. Soc. 2006, 128, 2630–2634. 10.1021/ja055709q. [DOI] [PubMed] [Google Scholar]
- Seo M. S.; Kamachi T.; Kouno T.; Murata K.; Park M. J.; Yoshizawa K.; Nam W. Experimental and Theoretical Evidence for Nonheme Iron (III) Alkylperoxo Species as Sluggish Oxidants in Oxygenation Reactions. Angew. Chem., Int. Ed. 2007, 46, 2291–2294. 10.1002/anie.200604219. [DOI] [PubMed] [Google Scholar]
- Nam W. Synthetic mononuclear nonheme iron–oxygen intermediates. Acc. Chem. Res. 2015, 48, 2415–2423. 10.1021/acs.accounts.5b00218. [DOI] [PubMed] [Google Scholar]
- Franke A.; Fertinger C.; van Eldik R. Which Oxidant is Really Responsible for P450 Model Oxygenation Reactions? A Kinetic Approach. Angew. Chem., Int. Ed. 2008, 47, 5238–5242. 10.1002/anie.200800907. [DOI] [PubMed] [Google Scholar]
- Li C.; Zhang L.; Zhang C.; Hirao H.; Wu W.; Shaik S. Which Oxidants is Really Responsible for Sulfur Oxidation by Cytchrome P450?. Angew. Chem., Int. Ed. 2007, 46, 8168–8170. 10.1002/anie.200702867. [DOI] [PubMed] [Google Scholar]
- Rydberg P.; Ryde U.; Olsen L. Sulfoxide, Sulfur, and Nitrogen Oxidation and Dealkylation by Cytochrome P450. J. Chem. Theory Comput. 2008, 4, 1369–1377. 10.1021/ct800101v. [DOI] [PubMed] [Google Scholar]
- Porro C. S.; Sutcliffe M. J.; de Visser S. P. Quantum Mechanics/Molecular Mechanics Studies on the Sulfoxidation of Dimethyl Sulfide by Compound I and Compound 0 of Cytochrome P450: Which is the Better Oxidant?. J. Phys. Chem. A 2009, 113, 11635–11642. 10.1021/jp9023926. [DOI] [PubMed] [Google Scholar]
- Wang B.; Li C.; Cho K. B.; Nam W.; Shaik S. The Fe(III)(H2O2) Complex as a Highly Efficient Oxidant in Sulfoxidation Reactions: Revival of an Underrated Oxidant in Cytochrome P450. J. Chem. Theory Comput. 2013, 9, 2519–2525. 10.1021/ct400190f. [DOI] [PubMed] [Google Scholar]
- Wang B.; Li C.; Dubey K. D.; Shaik S. Quantum Mechanical/Molecular Mechanical Calculated Reactivity Networks Reveal How cytochrome P450cam and Its T252A Mutant Select Their Oxidation Pathways. J. Am. Chem. Soc. 2015, 137, 7379–7390. 10.1021/jacs.5b02800. [DOI] [PubMed] [Google Scholar]
- Dubey K. D.; Wang B.; Vajpai M.; Shaik S. MD Simulations and QM/MM Calculations Show that Single-site Mutations of Cytochrome P450 BM3 Alter the Active Site’s Complexity and the Chemoselectivity of Oxidation Without Changing the Active Species. Chem. Sci. 2017, 8, 5335–5344. 10.1039/C7SC01932G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu G., Li A., Sun Z., Acevedo-Rocha C. G., Reetz M. T.. The Crucial Role of Methodology Development in Directed Evolution of Selective Enzymes. Angew. Chem., Int. Ed. 2019, 58, 10.1002/anie.201901491. [DOI] [PubMed] [Google Scholar]
- Arnold F. H. Innovation by Evolution: Bringing New Chemistry to Life (Nobel Lecture). Angew. Chem., Int. Ed. 2019, 58, 14420–14426. 10.1002/anie.201907729. [DOI] [PubMed] [Google Scholar]
- Zeymer C.; Hilvert D. Directed Evolution of Protein Catalysts. Annu. Rev. Biochem. 2018, 87, 131–157. 10.1146/annurev-biochem-062917-012034. [DOI] [PubMed] [Google Scholar]
- Sun H.; Zhang H.; Ang E. L.; Zhao H. Biocatalysis for the Synthesis of Pharmaceuticals and Pharmaceutical Intermediates. Bioorg. Med. Chem. 2018, 26, 1275–1284. 10.1016/j.bmc.2017.06.043. [DOI] [PubMed] [Google Scholar]
- Badenhorst C. P. S.; Bornscheuer U. T. Getting Momentum: from Biocatalysis to Advanced Synthetic Biology. Trends Biochem. Sci. 2018, 43, 180–198. 10.1016/j.tibs.2018.01.003. [DOI] [PubMed] [Google Scholar]
- Alcalde M.Directed Enzyme Evolution: Advances and Applications; Springer International Publishing: Stuttgart, 2017. [Google Scholar]
- Ebert M. C.; Pelletier J. N. Computational Tools for Enzyme Improvement: Why Everyone Can–and Should–use Them. Curr. Opin. Chem. Biol. 2017, 37, 89–96. 10.1016/j.cbpa.2017.01.021. [DOI] [PubMed] [Google Scholar]
- Reetz M. T.Directed Evolution of Selective Enzymes: Catalysts for Organic Chemistry and Biotechnology; Wiley VCH: Weinheim, 2016. [Google Scholar]
- Bommarius A. S. Biocatalysis: a Status Report. Annu. Rev. Chem. Biomol. Eng. 2015, 6, 319–345. 10.1146/annurev-chembioeng-061114-123415. [DOI] [PubMed] [Google Scholar]
- Widersten M. Protein Engineering for Development of New Hydrolytic Biocatalysts. Curr. Opin. Chem. Biol. 2014, 21, 42–47. 10.1016/j.cbpa.2014.03.015. [DOI] [PubMed] [Google Scholar]
- Devlin F. J.; Stephens P. J.; Scafato P.; Superchi S.; Rosini C. Determination of Absolute Configuration Using Vibrational Circular Dichroism Spectroscopy: The Chiral sulfoxide 1-Thiochromanone S-oxide. Chirality 2002, 14, 400–406. 10.1002/chir.10103. [DOI] [PubMed] [Google Scholar]
- Matsumoto K.; Yamaguchi T.; Katsuki T. Asymmetric Oxidation of Cyclic Sulfides Catalyzed by an Aluminum (salalen) Complex as the Catalyst. Heterocycles 2008, 76, 191–196. 10.3987/COM-08-S(N)37. [DOI] [Google Scholar]
- Wang J.-B.; Ilie A.; Reetz M. T. Chemo-and Stereoselective Cytochrome P450-BM3-Catalyzed Sulfoxidation of 1-Thiochroman-4- ones Enabled by Directed Evolution. Adv. Synth. Catal. 2017, 359, 2056–2060. 10.1002/adsc.201700414. [DOI] [Google Scholar]
- Reetz M. T. Laboratory Evolution of Stereoselective Enzymes: a Prolific Source of Catalysts for Asymmetric Reactions. Angew. Chem., Int. Ed. 2011, 50, 138–174. 10.1002/anie.201000826. [DOI] [PubMed] [Google Scholar]
- Roiban G. D.; Agudo R.; Ilie A.; Lonsdale R.; Reetz M. T. CH-activating Oxidative Hydroxylation of 1-Tetralones and Related Compounds with High Regio-and Stereoselectivity. Chem. Commun. 2014, 50, 14310–14313. 10.1039/C4CC04925J. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.