

Abstract
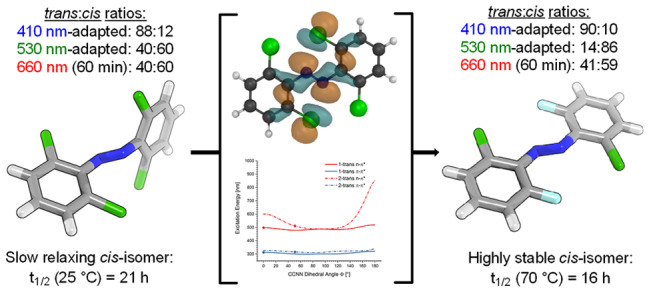
We computationally dissected the electronic and geometrical influences of ortho-chlorinated azobenzenes on their photophysical properties. X-ray analysis provided the insight that trans-tetra-ortho-chloro azobenzene is conformationally flexible and thus subject to molecular motions. This allows the photoswitch to adopt a range of red-shifted geometries, which account for the extended n → π* band tails. On the basis of our results, we designed the di-ortho-fluoro di-ortho-chloro (dfdc) azobenzene and provided computational evidence for the superiority of this substitution pattern to tetra-ortho-chloro azobenzene. Thereafter, we synthesized dfdc azobenzene by ortho-chlorination via 2-fold C–H activation and experimentally confirmed its structural and photophysical properties through UV–vis, NMR, and X-ray analyses. The advantages include near-bistable isomers and an increased separation of the n → π* bands between the trans- and cis-conformations, which allows for the generation of unusually high levels of the cis-isomer by irradiation with green/yellow light as well as red light within the biooptical window.
Introduction
Azobenzenes are versatile photoswitches that can be cycled between their cis- and trans-configurations with light.1,2 Because of their small size, robust photoswitching, synthetic accessibility, and low rate of photobleaching, they serve as excellent building blocks for the generation of elaborate optical tools.3−5 Photopharmaceuticals, for example, contain azobenzene fragments as ON- and OFF-switches, which allow for the control of biological functions with the spatiotemporal precision of light.6−11 To elicit the full potential of these optical tools and to apply them to complex animal tissues, it is crucial to use tissue-penetrating, nonhazardous red and near-infrared (NIR) light within the biooptical window12 (650–950 nm).13−15 In addition, it is highly desirable to use photoswitches with slow thermal relaxation rates for the less stable isomers and near-quantitative photoconversion for each isomer. Fast-relaxing photopharmaceuticals require constant illumination with high intensity light for their photoswitching to outcompete the thermal relaxation. This becomes increasingly difficult in deeper tissue layers.16 The bathochromic shift, the thermal stability, and the photoconversion strongly depend on the substitution pattern of the azobenzene core.7,16,17 Therefore, the design of new photopharmaceuticals that are operated at the biooptical window relies on the availability of azobenzene photoswitches with the appropriate photophysical properties.
A strategy for generating red-shifted photoswitches entails the separation of the n → π* absorption bands, which are typically overlapping for cis- and trans-azobenzene in the visible spectrum. In this context, a significant advancement was made through tetra-ortho-substitution with heteroatoms.18−21 Tetra-ortho-methoxy azobenzenes, for instance, have long-lived cis-isomers (τ ≈ 2 d) and are readily photoswitched to their trans- and cis-configurations with blue and green/red light, respectively.20 Modification of the tetra-ortho-methoxy pattern with electron-donating substituents implements an azonium ion, which can be isomerized with near-infrared light (up to 720 nm) but suffers from a significant decrease in the thermal stability of the cis-isomer (τ = 1 s).22,23 In contrast, the cis-isomer of the tetra-ortho-fluoro azobenzene has a thermal half-life on the order of years (t1/2 = 700 d) and can be reliably photoswitched with blue (trans-configuration) and green (cis-configuration) light.19,20 Significant photoconversion to the cis-isomer can be reached with up to 527 nm light.24 The use of longer wavelengths requires the use of an alternative substitution pattern such as the tetra-ortho-chloro azobenzene.24 It is worth noting that certain photoswitches can be operated with light in the near-infrared region (NIR) by using two-photon excitation.25 Because of the slow photoswitching through two-photon excitation, however, this approach requires the use of photoswitches with long-lived isomers to reach significant photoconversion.26
Remarkably, tetra-ortho-chloro azobenzenes allow for rapid and efficient photoswitching by irradiation of the n → π* band tails. For example, the fastest isomerization rate to the photostationary state (PSS) with the highest content of the cis-isomer for alkyl-substituted derivatives, such as red-AzCA-4 (Figure 1), was reached by using green light (550–560 nm), although the λmax of the trans-isomer (400 nm-adapted) lies close to 450 nm.27 It was further demonstrated that irradiation at far ends of the n → π* band can still produce a PSS with a significant cis-content by using bright light sources and continuous irradiation. For instance, Woolley and co-workers28 reported photoswitching of tetra-ortho-chloro azobenzenes with high intensity red light (625 nm) for 1 min, whereas Feringa and co-workers24 applied longer-wavelength high intensity red light (652 nm) for 2.5 h to reach a PSS with the highest levels of the cis-isomer.
Figure 1.

Structures of red-AzCA-4, azobenzene (1), and tetra-ortho-chloro azobenzene (2).
Intrigued by the potential of the tetra-ortho-chloro substitution pattern (Figure 1) as a basis to develop new photoswitches with long-lived cis-isomers, near-quantitative PSS, and excitation wavelengths within the biooptical window, we focus our study on the evaluation of the influence of azobenzene ortho-chlorination on the photophysical properties. By dissecting the effects of the chlorination into its geometric and electronic subcomponents, we aim to identify the origin of the extended red-shifted n → π* band tails of ortho-chlorinated azobenzenes as well as the separation of the n → π* excitation between the trans- and cis-isomers. Therefore, we employed a combination of quantum-chemical methodologies as well as X-ray analyses. On the basis of our results, the tetra-ortho substitution pattern could be optimized to yield a photoswitch with a highly stable cis-isomer and PSS with high contents for each isomer while retaining the red-shifted transition.29,30
We first computed the geometries of our studied azobenzene substitution patterns as well as their π → π* and n → π* absorption bands. Previous studies have shown that employing a PBE0 hybrid functional provides the closest correlation between the computational and experimental results for determining the λmax of azobenzene-based photoswitches.31 Therefore, we optimized the structures for all studied photoswitches on the PBE0-D3/def2-TZVP32−35 level of theory using the TURBOMOLE program package, to obtain the respective gas-phase ground-state minimum geometry at 0 K followed by the computation of the vertical excitation energies on the TD-PBE0/def2-TZVP level. This gives access to absorption maxima (λmax) of the π → π* (S0 → S2) as well as the n → π* (S0 → S1) transitions for optimized trans- and cis-azobenzene geometries.
Effects of the Azobenzene ortho-Chlorination
The geometrical changes in the conformation of azobenzenes through isomerization or substitution can be subdivided into three main categories: changes in the N–N bond length, the C(1)–C(2)–N(3)–N(4) dihedral angles Φ, and the C(2)–N(3)–N(4)–C(5) dihedral angle Ψ (Figure 2). The CCNN dihedral angles describe the rotation of the aryl rings out of the plane. This rotation can proceed from two directions and thus can be described by two dihedral angles for each aryl ring: ΦS and ΦL. We focus our discussion on the smaller ΦS angles. The Ψ dihedral angle describes the distortion of the N–N double bond.
Figure 2.

Numerical descriptors for the dihedral angles.
For the evaluation of the influence of tetra-ortho-chlorination on the bathochromic shifts of azobenzene, we computationally optimized structures for trans-1, cis-1, trans-2, and cis-2 and verified that their conformations are true minima on the potential energy surface (PES). We found that trans-azobenzene has a planar structure with an N–N bond length of 1.242 Å and N–N bond geometry (Ψ dihedral angle) of 180.0°. The structure of trans-tetra-ortho-chloro azobenzene is characterized by the rotation of both aryl rings out of the plane by 50.8° (ΦS). In addition, the N–N bond length is shortened to 1.236 Å, and the N–N double bond is slightly distorted to 175.3° (Ψ). Next, we determined their n → π* (S0 → S1) absorption bands (Table 1) and found that the conversion of azobenzene (1) to 2 induces a bathochromic shift of the n → π* absorption band of 14.9 and 4.9 nm for the trans- and cis-isomers, respectively. Because of the higher red-shift for the trans-isomer, the overall difference between the excitation band maxima of both configurations increases from 13.6 nm (1) to 23.6 nm (2). This increased band separation allows for the generation of PSS with higher contents of cis-isomers by irradiation of the trans-2 n → π* excitation band as compared to that of trans-1.
Table 1. Optimized Conformations of 1 and 2: Vertical Excitation Energies (TD-PBE0/def2-TZVP) and Photoswitch Structures (PBE0-D3/def2-TZVP).
| comp. | N=N [Å] | ΦS [deg] | Ψ [deg] | S0 → S1 [nm] |
|---|---|---|---|---|
| trans-1 | 1.242 | 0.0 | 180.0 | 483.6 |
| cis-1 | 1.234 | 52.0 | 8.5 | 470.0 |
| trans-2 | 1.236 | 50.8 | 175.3 | 498.5 |
| cis-2 | 1.229 | 60.5 | 5.1 | 474.9 |
To get a more detailed understanding of the effects of the ortho-chloro substituents, we studied mono-, di-, tri-, and tetra-chlorinated azobenzenes (Figure 3 and Table 2). Substituting one ortho-hydrogen on 1 for chlorine (3) already leads to a significant separation of the n → π* transition band of the trans- and cis-isomers. As compared to 1, trans-3 is red-shifted by 18.3 nm, and cis-3 is blue-shifted by 15.7 nm. Because both structures trans-1 and trans-3 remain planar, the bathochromic shift could be attributed to the electronic contribution of the chlorination. The chlorinated aryl ring of cis-3 has an increased ΦS angle by 11.0°. Because of the asymmetric structure, both aryl rings are rotated at different Φ angles. In this case, the average of both smaller ΦS angles, ΦS(avg), can be used as a descriptor for the overall rotation. For example, changing the substitution pattern from cis-1 to cis-3 results in a difference of 5.4° in the ΦS(avg) (52.0 nm for cis-1 and 57.4 nm for cis-3). Appending a second ortho-chlorine at the opposite aryl ring follows the same trend and provides absorption maxima at 520.6 nm (trans-4) and 438.6 nm (cis-4). To evaluate the origin of the strong shifts in the n → π* excitation wavelengths through ortho-chloro substitution, we investigated the photophysical properties of para-chloro azobenzene (5). If the bathochromic shift is a result of the extended π-system and the concomitant effects on the π* molecular orbital (MO), para-chloro azobenzene 5 should behave in a fashion similar to that of the ortho-substituted derivative 4. The n → π* vertical excitation energies of di-para-chloro azobenzene (5), however, are comparable to those of azobenzene (1). Therefore, the excitation wavelength shifts are an effect of the substitution at the ortho-position. Because the ortho-chlorine atoms of trans-4 are facing toward the nitrogen lone pairs, it is reasonable to assume that their repulsive interaction destabilizes the n molecular orbital and thus decreases the energy gap to the π* molecular orbital.18 In contrast, the bent structure of cis-4 allows the chlorine atoms to face in the opposite directions of the nitrogen lone pairs. We reason that the strong blue-shift of the n → π* excitation band that is observed by transitioning from cis-1 to cis-4 results from a withdrawal of electron density through the electronegative chlorine substituents in proximity to the N–N bond. This effect has previously been reported for tetra-ortho-fluoro azobenzenes and leads to a lowering of the n-orbital energy.19,36
Figure 3.

Mono-, di-, and trichlorinated azobenzenes.
Table 2. Mono-, Di-, Tri-, and Tetrachlorinated Azobenzenes: Vertical Excitation Energies (TD-PBE0/def2-TZVP) and Photoswitch Structures (PBE0-D3/def2-TZVP).
| config. | N=N [Å] | ΦS(Ar1) [deg] | ΦS(Ar2) [deg] | Ψ [deg] | S0 → S1 [nm] | |
|---|---|---|---|---|---|---|
| 1 | trans | 1.242 | 0.0 | 0.0 | 180.0 | 483.6 |
| cis | 1.234 | 52.0 | 52.0 | 8.5 | 470.0 | |
| 3 | trans | 1.242 | 0.2 | 1.4 | 179.9 | 501.9 |
| cis | 1.232 | 51.7 | 63.0 | 9.5 | 454.3 | |
| 4 | trans | 1.243 | 0.7 | 2.7 | 179.8 | 520.6 |
| cis | 1.231 | 63.0 | 63.0 | 10.5 | 438.6 | |
| 5 | trans | 1.243 | 0.1 | 0.1 | 180.0 | 480.0 |
| cis | 1.235 | 50.5 | 50.5 | 9.0 | 478.7 | |
| 6 | trans | 1.239 | 0.6 | 53.6 | 178.1 | 483.6 |
| cis | 1.230 | 50.4 | 74.3 | 6.9 | 444.5 | |
| 7 | trans | 1.239 | 10.6 | 49.8 | 179.1 | 502.8 |
| cis | 1.230 | 51.7 | 71.8 | 7.8 | 453.9 | |
| 2 | trans | 1.236 | 50.8 | 50.8 | 175.3 | 498.5 |
| cis | 1.229 | 60.5 | 60.5 | 5.1 | 474.9 |
In contrast to 4, the introduction of the second ortho-chlorine on the same aryl ring (6) twists the chlorinated aryl group of the trans-isomer out of the plane at a ΦS angle of 53.6° and leads to a hypsochromic shift of the n → π* excitation energy. On the basis of our prior findings, we hypothesize that the hypsochromic shift is a result of a decreased interaction between the chlorine substituent and the nitrogen lone pairs due to the rotation of the aryl ring. Changing the substitution pattern from cis-4 to cis-6 only results in minor changes in the ΦS(avg) angle (63.0 nm for cis-4 and 62.4 nm for cis-6), which is reflected by the subtle n → π* band shift from 438.6 to 444.5 nm. It is worth noting that the rotation around the Φ dihedral angle for trans-6 is accompanied by a contraction of the N–N bond length from 1.243 to 1.239 Å, which is retained in the more chlorinated derivatives 7. Adding a third ortho-chlorine onto 6 induces a red-shift of the n → π* excitation energy of the trans-7 and cis-7 of 19.2 and 9.4 nm, respectively. Finalizing the transition to tetra-ortho-chloro azobenzene (2) from 7 strongly increases the rotational overall twist of the trans structure to 50.8° for both ΦS angles. This distortion leads to a further contraction of the N–N bond length from 1.239 Å (trans-7) to 1.236 Å (trans-2). In addition, a strong contraction of the Ψ dihedral angle to 175.3° occurs, which ranges between 178.1° and 180.0° in the trans-configuration of less chlorinated substrates (1, 3–7). The structural and conformational changes were accompanied by an n → π* hypsochromic shift of 4.3 nm. In contrast to trans-2, cis-2 is subject to a strong n → π* bathochromic shift of 21 nm. We reasoned that the increasing substitution of the ortho-position decreases the rotational flexibility of the individual aryl rings around the Φ dihedral angle. As a consequence, the chlorine atoms of cis-2 are forced into the proximity of the nitrogen lone pairs, which destabilizes the n molecular orbital and decreases the energy gap between the n → π* transition.
Examining the Effect of the Tetra-ortho-chloro Azobenzene Conformation on Its Excitation Energy
The computational study of the stepwise ortho-chlorination of azobenzene (1) to tetra-ortho-chloro azobenzene (2) highlights that the geometry of the respective photoswitch has a strong influence on the n → π* excitation energies. To determine whether the conformational changes of the same photoswitch structures provide shifts in the excitation energies, we calculated the vertical excitation energies of 1 adopting the optimized conformations of 2 (Table 3, entries A and B) as well as the vertical excitation energies of 2 adopting the optimized conformations of 1 (entries C and D). Because the conformations of cis-1 and cis-2 are closely related, imposing the cis-2 geometry onto cis-1 and vice versa did not provoke any significant alterations of its photophysical properties. In contrast, changing the 3D structure of azobenzene to the tilted geometry of 2 leads to a blue-shift of 39.9 nm for n → π* transitions. Imposing the planar trans-1 geometry onto trans-2 leads to an outstanding n → π* bathochromic shift of 212.8 nm. This result supports the theoretical hypothesis that the photophysical properties of an azobenzene-based photoswitch are determined by both the chemical composition and the conformational geometry.20 It furthermore showcases the profound effect a change in the conformation can have on the n → π* excitation wavelength of tetra-ortho-chloro azobenzenes. By computing the MO energies and visualizing the MOs of the optimized and the planar trans-2 conformations (Figure 4), we show that the strong n → π* bathochromic shift likely originates from the increased interaction between the chlorine and the nitrogen lone pair, which leads to a strong destabilization of the n-MO by 0.60 eV (Figure 4 and Table 3). The planar geometry additionally stabilizes the π*-MO, which decreases the gap of the n → π* transition.
Table 3. Optimized Conformation of 2 Imposed on the Structure of 1 (A,B) and Conformation of 1 Imposed on 2 (C,D): Vertical Excitation Energies (TD-PBE0/def2-TZVP) and Photoswitch Structures (PBE0-D3/def2-TZVP).
| entry | comp. | N=N [Å] | ΦS [deg] | Ψ [deg] | S0 → S1 [nm] |
|---|---|---|---|---|---|
| A | trans-1 (trans-2-adopted) | 1.236 | 50.8 | 175.3 | 443.7 |
| B | cis-1 (cis-2-adopted) | 1.229 | 60.5 | 5.1 | 471.4 |
| C | trans-2 (trans-1-adopted) | 1.242 | 0.0 | 180.0 | 711.3 |
| D | cis-2 (cis-1-adopted) | 1.234 | 52.0 | 8.5 | 463.1 |
Figure 4.

Computed orbital energies and n-MO densities of the optimized 2 conformation (left) and the conformation of 1 imposed on 2 (right).
To gain a more detailed understanding of the influence of the azobenzene conformation, we evaluated each of the main geometrical variables for 1 and 2 separately (Figure 5 and Supporting Information, Chapter 2.8 and Figure S74). Therefore, we simulated the contraction and elongation of the N–N bond lengths (Figure S74A and B), the distortion of the Ψ dihedral angles (Figure S74C), and the selective rotation of one aryl ring around the Φ dihedral angles (Figures 5 and S74E). In each case, elongating the optimized N–N bond lengths (Figure S74A and B) results in a bathochromic shift of the n → π* excitation bands with the strongest shifts for trans-tetra-ortho-chloro azobenzene (2). A distortion of the Ψ dihedral angles (Figure S74C) by contracting the trans-1 and trans-2 structures as well as bending the cis-1 and cis-2 structures evokes a bathochromic shift. Rotating one aryl ring of trans-1 around the Φ dihedral angle (Figure 5) has minor effects on the n → π* transition. The trans-2 n → π* excitation energies, however, were subject to a strong red-shift when the Φ angle approached 0° and 180° (Figure 5). In case of cis-1, rotating one aryl ring (Figure S74E) around the CCNN axis shows that approaching planarity with the N–N double bond in either direction provides a red-shift of n → π* excitation energies. We found that the steric bulk of the ortho-chlorine atoms hinders the full rotation around the CCNN axis of cis-2 (Figure S74E and F). Slight rotations of the optimized cis-2 geometry have minor effects on the n → π* excitation energy, whereas approaching the limit of the rotational axis in each direction strongly decreases the excitation energy barrier.
Figure 5.

Influence of the C–C–N–N dihedral angle ΦS on the excitation energy: optimized structure (PBE0-D3/def2-TZVP) with the corresponding vertical excitation energies (TD-PBE0/def2-SVP) of the photoswitches 1 and 2.
In general, our quantum-chemical calculations have shown that the n → π* vertical excitation energy of trans-2 is strongly dependent on its conformation and that minor structural changes, such as the elongation of the N−N bond length, distortion of the Ψ dihedral angle, and rotations of the Φ dihedral angles can lead to strong bathochromic shifts. To compare our computational findings to experimental conformations of azobenzene (1) and tetra-ortho-chloro azobenzene (2), we recrystallized trans-2 as well as cis-2 and analyzed their structures using X-ray crystallography.
X-ray Analysis of Tetra-ortho-chloro Azobenzene
To gain insight into the experimental 3D structures of tetra-ortho-chloro azobenzenes, we investigated trans-2 and cis-2 using X-ray crystallography (Figure 6 and Table 4). In accordance with our computational results, we found that the steric bulk of the ortho-chlorine atoms twists the two aryl rings of trans-2 out of the plane. Remarkably, the extended cell of the crystal structure contains three conformations for trans-2 with strong variations between the conformations. For example, the N–N bond length ranges from 1.182 to 1.248 Å, the ΦS dihedral angles reach from 35.2° to 79.7°, and the Ψ dihedral angle is distorted from 180.0° to 176.3°. This result is an essential cornerstone for the development of new photoswitches, because it provides a new perspective on tetra-ortho-substituted azobenzenes: the twisted trans-isomers are not stiff structures but are highly flexible and subject to molecular motions. In contrast, the trans-azobenzene (1) X-ray structure contains one conformation, which is near-planar with a N–N bond length of 1.249, an ideal nondistorted Ψ angle, and a ΦS dihedral of 9.7°.37 The X-ray structures cis-138 and cis-2 also contain a single conformation, which suggests that these structures have a more restricted conformation as compared to that of trans-2. Conformational NMR studies by Brittain and co-workers,39 however, have shown that cis-tetra-ortho-substituted azobenzenes are subject to a certain degree of structural flexibility. Comparing the structures of the cis-isomers shows that the relative changes in the conformation are accurately portrayed by our quantum-chemical calculations. For instance, the average distortion of the ΦS dihedral angle is increased from 51.6° (cis-1) to 60.3° (cis-2), and the Ψ dihedral angle is decreased from 7.7° (cis-1) to 3.5° (trans-2).
Figure 6.
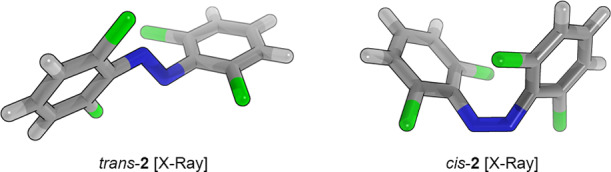
X-ray structures of trans-2 and cis-2.
Table 4. X-ray Analysis of the Structural Flexibility.
| config. | N=N [Å] | ΦS(Ar1) [deg] | ΦS(Ar2) [deg] | Ψ [deg] | |
|---|---|---|---|---|---|
| 1 | trans | 1.249 | 9.7 | 9.7 | 180.0 |
| cis | 1.251 | 51.6 | 51.6 | 7.7 | |
| 2 | trans1 | 1.182 | 35.2 | 79.7 | 180.0 |
| trans2 | 1.245 | 51.6 | 65.4 | 178.1 | |
| trans3 | 1.248 | 53.1 | 59.3 | 176.3 | |
| cis | 1.251 | 57.0 | 63.6 | 3.5 |
We hypothesize that the broad n → π* excitation bands and the concomitant extended band tail that allow for the photoswitching to PSS with a high cis-content with red light are a result of the conformational flexibility of trans-2. We have provided computational evidence that azobenzene-based photoswitches are subject to strong changes in their excitation energies through the slightest alterations in their conformation. Consequentially, we deduce that the excitation of the n → π* bands at the red end of the spectrum is affected by highly red-shifted conformations. The low absorption intensity of the tail regions is a result of the low abundance of the conformations that are shifted into the red region. According to our calculations, highly red-shifted conformations are characterized by long N–N bonds, small ΦS angles, and/or distorted Ψ angles. In contrast, the strong absorption intensity of the band maxima is a result of the high abundance of conformations that are closely related to the optimized structures. Irradiation at the band tail thus allows for the photoswitching of red-shifted conformations to the cis-isomer. On the basis of our hypothesis, trans-2 continuously cycles through a variety of 3D structures, which include these red-shifted conformations. Therefore, a PSS with a high cis-content can be reached with red light by the gradual accumulation of the long-lived cis-isomer, while the flexible trans-2 molecules continuously adopt red-shifted conformations and are photoswitched.
It is reasonable to assume that a large fraction of the highly red-shifted conformations is characterized by small average ΦS angles, due to the outstanding n → π* band shifts that result from the interaction between the chlorine atoms and the nitrogen lone pairs. Therefore, we envisage that modifying the substitution pattern to allow for a more planar structure yields a photoswitch that could be isomerized using red light within the biooptical window. Substituting one ortho-chlorine atom on each aryl ring of 2 for the smaller fluorine atoms, in this context, may lead to smaller ΦS dihedral angles and thereby enable a better overlap between the two retained chlorine atoms and the nitrogen lone pairs. In addition, incorporation of fluorine atoms leads to a stronger withdrawal of electron density in proximity to the diazene unit. This effect is known to produce a hypsochromic shift of the n → π* band of the cis-isomer, which increases the separation of the excitation energies between both isomers.19,36 Irradiation of the individual bands close to the absorption maxima could consequently result in a PSS with higher isomer levels using shorter light pulses.
Computational Optimization of the Tetra-ortho Substitution Pattern
A computational investigation of the substitution of two ortho-chlorines of 2 for the more electronegative fluorine (8, Figure 7) shows that, in accordance with our hypothesis, the ΦS dihedral angles decrease from 50.8° to 37.3°. In addition, the N–N bond is lengthened from 1.236 to 1.243 Å, and the distortion of the Ψ angle is nearly retained. The combination of these structural changes induces a bathochromic shift of 9.1 nm by transitioning from trans-2 to trans-8 (Table 5). Comparing cis-2 to cis-8 showed a stronger distortion of the Ψ angle and an increased N–N bond length from 1.229 to 1.232 Å, without any significant alteration of the ΦS dihedral angle. Although these geometric adjustments have been shown to contribute to a bathochromic shift, the electronic contribution to the photophysical properties that stem from the withdrawing effect of the fluorine atoms results in an overall hypsochromic shift of 22.2 nm. To exclude the possibility that the di-ortho-fluoro di-ortho-chloro (dfdc) azobenzene preferentially adopts a conformation, where the chlorines are arranged on the same face of the molecule (8s), we computed the total energies of both structures. The optimized geometries trans-8 and cis-8 are more stable by 4.0 kJ/mol than are trans-8s and cis-8s, respectively (Table 5). To evaluate the differences between the newly designed tetra-ortho-fluoro/tetra-ortho-chloro hybrid (8) and the literature-known tetra-ortho-fluoro structure (9),19,36 we investigated 9 with the same quantum-chemical methodology. As expected, transitioning from cis-8 to cis-9 does not significantly alter the conformation, but the increased withdrawal of electron density in proximity to the diazene unit leads to a hypsochromic shift of 12.2 nm. In comparison to trans-8, trans-9 is approaching a planar conformation. The ΦS angle decreased to 27.7°, the Ψ angle increased to 176.7°, and the N–N double bond is elongated to 1.246 Å. These combined structural changes lead to a positioning of the n → π* excitation maxima (trans-9: λmax = 505.5 nm) similar to that of trans-8. On the basis of our previous findings, however, we hypothesize that the geometric flexibility of trans-8 enables an overlap between the bulky chlorine atoms with the nitrogen lone pairs through molecular motions. This interaction strongly contributes to the extended n → π* band tails and allows for photoswitching with red light. The small fluorine atoms of trans-9, in comparison, are unable to overlap with the n molecular orbitals to the same extent, which leads to narrower n → π* absorption bands and photoswitching with wavelengths close to the λmax.
Figure 7.

Structures of di-ortho-fluoro di-ortho-chloro (dfdc, 8), tetra-ortho-fluoro (9) azobenzene, as well as the dfdc derivatives 10 and 11.
Table 5. Exchange of Two Chlorine Atoms for Fluorine on Tetra-ortho-chloro Azobenzene: Vertical Excitation Energies (TD-PBE0/def2-TZVP) and Photoswitch Structures (PBE0-D3/def2-TZVP).
| config. | N=N [Å] | ΦS(Ar1) [deg] | ΦS(Ar2) [deg] | Ψ [deg] | S0 → S1 [nm] | |
|---|---|---|---|---|---|---|
| 2 | trans | 1.236 | 50.8 | 50.8 | 175.3 | 498.5 |
| cis | 1.229 | 60.5 | 60.5 | 5.1 | 474.9 | |
| 8 | trans | 1.243 | 37.3 | 37.3 | 175.7 | 507.6 |
| cis | 1.232 | 58.6 | 58.6 | 8.4 | 452.7 | |
| 8s | trans | 1.241 | 37.6 | 42.8 | 175.5 | 508.0 |
| cis | 1.231 | 56.4 | 66.8 | 6.8 | 452.4 | |
| 9 | trans | 1.246 | 27.7 | 27.7 | 176.7 | 505.5 |
| cis | 1.233 | 59.5 | 59.5 | 8.9 | 440.5 | |
| 10 | trans | 1.242 | 40.5 | 40.5 | 175.1 | 526.9 |
| cis | 1.233 | 60.9 | 60.9 | 6.6 | 450.6 | |
| 11 | trans | 1.248 | 27.0 | 27.0 | 176.7 | 523.2 |
| cis | 1.234 | 57.1 | 57.1 | 9.8 | 463.8 |
To account for the influence of electronically active substituents on the conformation and photophysical properties of the dfdc azobenzene, we computationally investigated the addition of ethyl benzoate (10) and acetamide (11) groups to its para-positions. Transitioning from trans-8 to trans-10 and from cis-8 to cis-10 does not show any significant geometrical changes, respectively. The electron-withdrawing effect of the ethyl benzoate substituents (10), however, leads to a strong bathochromic shift of 19.3 nm for trans-10 and a slight hypsochromic shift of 2.1 nm of cis-10. Implementing an electron-rich substitution pattern (11) on the dfdc azobenzene backbone leads to an elongation of the N–N bond from 1.243 Å (trans-8) to 1.248 Å for trans-11, which is accompanied by a relaxation of the twisted geometry by decreasing the ΦS dihedral angle from 37.3° to 27.0°, whereas no significant alterations are observed for the cis-11 conformation. For the combination of these structural properties with the electron-donating effect of the acetamide substituents, we observe a bathochromic shift of 15.6 nm for trans-11 and of 11.1 nm for cis-11.
Synthesis and Photophysical Characterization of Di-ortho-fluoro Di-ortho-chloro Azobenzene
To confirm our theoretical results and to evaluate whether the designed hybrid combines the red-shift of the tetra-ortho-chloro azobenzene trans-isomer with the blue-shift of the tetra-ortho-fluoro azobenzene cis-isomer, we synthesized 8. In addition, we were intrigued by the strong computed bathochromic shift that occurred by substituting trans-8 with two ethyl benzoate substituents (trans-10) and synthesized 10 to experimentally validate the effect of an electron-withdrawing para-substituent on the photophysical properties of dfdc azobenzenes. For these syntheses, we leveraged our previously established C–H chlorination methodology.27 By optimizing this methodology for the dichlorination of the di-ortho-fluoro azobenzenes 12 and 13, we could increase its substrate scope to mixed ortho-substituted azobenzenes and provide our desired products 8 and 10 in ample quantities (Figure 8).
Figure 8.

Synthesis of the tetra-ortho-hydrids 8 and 10 using a palladium-catalyzed C–H chlorination.
In general, the positions of the π → π* and n → π* excitation bands were determined through UV–vis analysis using a 50 μM solution in DMSO. Because of the low absorption of the n → π* transition, the visualization of the full extent of the band tail required a measurement at higher concentrations (500 μM solution in DMSO).40 After the position of the excitation bands was determined, the extent of the n → π* band tail was verified through photoswitching by illuminating the compounds with light at the red end of the visible spectrum (Figure 9). The light-dependent PSS were then analyzed by NMR spectroscopy using a 500 μM solution in DMSO-d6 to allow for a direct comparison with the UV–vis spectra. For example, the n → π* of trans-azobenzene (trans-1) absorbs light up to 550 nm (Figure 9c). This was experimentally confirmed by irradiation of the dark-adapted state with green/yellow light (see the Supporting Information). Illumination with 590 nm does not effect a change in the UV–vis spectrum, whereas 530 nm light produces an altered PSS of the molecule (1). However, due to the substantial overlap of the n → π* excitation bands of the trans- and cis-isomers, the application of 530 nm light only leads to minor photoconversion in comparison to irradiation of the π → π* band maximum with UV-A light. UV–vis analysis of tetra-ortho-chloro azobenzene (2, Figure 9) shows slightly separated n → π* band tails with a more red-shifted absorption spectrum for the dark-adapted state (100% trans-isomer). This slight separation of the n → π* band tails allows one to reach significant photoconversion by irradiating with green light (530 nm, trans:cis = 40:60). The extent of the tail was determined by applying 660 nm red light with an intensity of 24.1 mW/cm2 to the blue-adapted (410 nm) compound, which produces a trans:cis ratio of 40:60 after 60 min. Both azobenzene (1) and tetra-ortho-chloro azobenzene (2) have long-lived cis-isomers with thermal half-lives of 75.5 and 20.7 h in DMSO at 25 °C, respectively.
Figure 9.

(a) Photoswitching of tetra-ortho-chloro azobenzene (2). UV–vis spectra of 1 and 2: (b) 50 μM in DMSO is used to show the full spectrum and (c) 500 μM in DMSO is used to visualize the extent of the n → π* band tails.
Remarkably, by exchanging two chlorine atoms of 2 for fluorine (8), the separation between the n → π* excitation bands of the trans-isomer and cis-isomer of 8 strongly improved, which allows for the generation of near-quantitative PSS (Figure 10). Irradiation with green light (530 nm), for example, leads to an excellent PSS with significantly increased cis-isomer content (trans:cis = 14:86) as compared to 2 (530 nm, trans:cis = 40:60). In addition, high levels of trans-8 (trans:cis = 90:10) could be produced with blue light (410 nm). The extent of the n → π* band tail was determined by applying 660 nm light (24.1 mW/cm2) to the blue-adapted compound, which produces a trans:cis ratio of 41:59 after 60 min, 20:80 after 120 min, and even reaches 3:97 after 300 min. By employing a deeply red LED with higher intensity (660 nm, 32.3 mW/cm2) under the same setting, photoswitching could be accelerated to provide trans:cis ratios of 30:70 and 10:90 after 60 and 120 min, respectively. To further increase the rate of photoswitching, a three-membered LED array could be used (660 nm; 24.1, 32.3, and 44.3 mW/cm2), which leads to a trans:cis ratio of 24:76 after 60 min, 13:87 after 90 min, and 5:95 after 120 min. Next, we investigated the thermal half-lives of 8 and found that it is near-bistable. Full relaxation of green-irradiated 8 (530 nm) to its trans-configuration requires heating to 90 °C for 7 h (t1/2 = 2 h, DMSO). The thermal half-life at 70 °C was determined to be 16 h in DMSO. X-ray analysis of trans-8 provides the conformation trans-8x where both aryl rings are oriented on parallel, slightly displaced planes. In comparison to trans-2, trans-8x has a decreased ΦS angle (ΦS = 35.9°), a diminished distortion of the Ψ angle (Ψ = 180°), and an elongated N–N bond length (N–N = 1.254 Å), which is in accordance with our calculations. By subjecting trans-8x to our quantum-chemical methodology, we determined that the n → π* absorption maximum of the X-ray conformation lies at 478.3 nm, which is a hypsochromic shift of 29.3 nm as compared to trans-8. On the basis of this result, it is likely that the highly red-shifted conformations are adopted in the process of transitioning between the twisted trans-8 and trans-8x, where both aryl rings are oriented on parallel, slightly displaced planes.
Figure 10.

(a) Photoswitching of di-ortho-fluoro di-ortho-chloro azobenzene (8). UV–vis spectra of 8: (b) 50 μM in DMSO is used to show the full spectrum; (c) 500 μM in DMSO and 9:1 DMSO:H2O are used to visualize the extent of the n → π* band tails; and (d) X-ray structure of trans-8x and its calculated vertical excitation energy (TD-PBE0/def2-TZVP).
We assessed the utility of the dfdc azobenzene as a photoswitch for the development of photopharmaceuticals by analyzing its photophysical properties in an aqueous solution (9:1 DMSO:H2O). Overall, we did not observe any significant changes in the PSS and thermal stability by transitioning from DMSO to a mixture of DMSO and water. The PSS that is reached with green light (530 nm, trans:cis = 14:86) remains unchanged, and irradiation with blue light (410 nm, trans:cis = 85:15) only decreases the content of the trans-isomer by a small fraction. Prolonged irradiation (60 min) of the blue-adapted photoswitch (410 nm) with deep red light (660 nm, 24.1 mW/cm2, trans:cis = 40:60) produces a PSS similar to that from irradiation in DMSO. The thermal half-life of cis-8 at 70 °C is 12 h in 9:1 DMSO:H2O.
To confirm that the dfdc azobenzene (8) is subject to a stronger bathochromic shift than are both of its parent azobenzenes, we examined the extent of the n → π* excitation band of tetra-ortho-fluoro azobenzene (9). UV–vis analysis shows that the n → π* band tail of 9 does not exceed 590 nm (see the Supporting Information for experimental details), which was subsequently confirmed by NMR analysis. Irradiating the blue-adapted tetra-ortho-fluoro azobenzene (420 nm, trans:cis = 83:17) with yellow light (595 nm, 10.1 mW/cm2) for 60 min provided a slight change in the PSS (trans:cis = 76:14). However, subjecting 9 to a 60 min pulse of red light (625 nm, 26.7 mW/cm2) or deep red light (660 nm, 24.1 mW/cm2) did not provide a significant alteration of the trans:cis ratio.
Next, we examined the effect of electron-withdrawing para-substituents on the photophysical properties of the dfdc azobenzene by implementing ethyl benzoate groups (10, Figure 11). As compared to trans-8 and cis-8, the λmax(n → π*) values of both isomers, trans-10 and cis-10, are subject to a bathochromic shift (Figure 11c). This provides an excellent separation of the n → π* excitation bands between the trans- and cis-isomers in the blue as well as orange/red regions of the visible spectrum and a stronger overlap in the green region. Therefore, green light (530 nm) produces a trans:cis ratio of 41:59, whereas photoswitching with 410 and 440 nm blue light results in outstanding PSS with trans-isomer contents of 96% and 95%, respectively. This effect is further demonstrated by irradiating the 410 nm-adapted 10 with deep red light (660 nm, 24.1 mW/cm2), which shows accelerated photoswitching kinetics as compared to 8 and produces a trans:cis ratio of 17:83 after 60 min. The only drawback of the electron-poor 10 as compared to the unsubstituted dfdc azobenzene is the diminished thermal stability of the cis-isomer, which has a half-life of 5 h at 25 °C in DMSO.
Figure 11.

(a) Photoswitching of the electron-poor di-ortho-fluoro di-ortho-chloro azobenzene 10. UV–vis spectra of 8 and 10: (b) 50 μM in DMSO is used to show the full spectrum; and (c) 500 μM in DMSO is used to visualize the extent of the n → π* band tails.
Significance of the Di-ortho-fluoro Di-ortho-chloro Azobenzene
Azobenzenes are the most commonly used photoswitches to develop advanced optical tools such as photopharmaceuticals, due to their small size, robust photoswitching, and low rate of photobleaching.1−5 To fine-tune the photophysical properties of these optical devices, it is highly desirable to have access to an arsenal of azobenzene core structures with defined photophysical properties. In this regard, the dfdc azobenzene (8) provides an unprecedented solution to the long-standing problem to identify a substitution pattern that combines near-bistable isomers, PSS with high levels of trans- and cis-isomers, and photoswitching within the biooptical window. Although photoswitching with deep red light (660 nm) requires long irradiation times (trans:cis = 41:59 after 60 min with 24.1 mW/cm2), we show that by increasing the light intensity (trans:cis = 30:70 after 60 min with 32.3 mW/cm2) and/or using arrays of multiple LEDs (trans:cis = 24:76 after 60 min with 24.1, 32.3, and 44.3 mW/cm2), the photoswitching rate can be significantly accelerated. In addition, the installation of electron-poor substituents on the 8para-position (10) is accompanied by a bathochromic shift, which allows for faster isomerization with deep red light. Given that photodynamic therapy is carried out with light sources that provide an intensity of up to 200 mW/cm2 which can, in theory, be incorporated into an array, we reason that the dfdc azobenzene could be photoswitched within a photopharmaceutically relevant time frame in a therapeutic setting.41 The decreased steric bulk of the ortho-substitution of 8 as compared to 2 allows the relaxation of the twisted structure to a near-planar geometry, which leads to a closer 3D relationship of dfdc azobenzene to azobenzene. This factor plays a crucial role in the process of red-shifting photopharmaceuticals.42 The ability to add a para-substituent to 8 increases the utility of this pattern by allowing its anchoring to optical devices through an electron-active substituent (10). This substitution pattern leads to a shift of the λmax(n → π*) to the red end of the spectrum, increases the trans-content of the blue-irradiated photoswitch, and only decreases the thermal stability of the cis-isomer to an extent that 10 is still considered a slow-relaxing azobenzene. These excellent photophysical properties that were demonstrated by evaluating two dfdc derivatives leave a great margin for improvement with regard to the substitution of the meta- and para-positions. Therefore, 8 serves an ideal basis for the development of a photoswitch that operates with near-infrared light while retaining highly bistable isomers by performing an in-depth investigation into the effects of alternative substitution patterns on its photophysical properties.
Conclusion and Outlook
We have used a combination of quantum-chemical calculations and X-ray analyses to dissect the effects of the ortho-chlorination in its electronic and structural subcomponents and identified the origin of the extended red-shifted n → π* band tails of tetra-ortho-chloro azobenzenes. On the basis of our findings, we designed the dfdc azobenzene substitution pattern and synthesized 8 and 10 by employing a C–H dichlorination methodology. UV–vis and NMR studies showed significant improvements of the photophysical properties of 8 over the tetra-ortho-chloro and tetra-ortho-fluoro substitution pattern. Photoswitching can be achieved with red light within the biooptical window and provides a PSS with up to 97% of the highly stable cis-isomer. These outstanding properties make the dfdc azobenzene an ideal photoswitch for the development of photopharmaceuticals that target complex animal tissues. In addition, the dfdc azobenzene scaffold is an excellent basis for the development of bistable photoswitches that can be isomerized with near-infrared light. Implementing an electron-poor substitution pattern by adding an ethyl benzoate group to both para-positions (10), for example, leads to a significant bathochromic shift of the n → π* excitation bands of both isomers, which results in an accelerated photoconversion by applying deep red light as compared to 8. To evaluate the full potential of the dfdc azobenzene, studies on the effect of meta- and para-substituents are currently underway.
Acknowledgments
This work was supported by the SFB749 to D.T. and C.O. as well as a Georg Forster Research Fellowship (Alexander von Humboldt Foundation) to A.M.A. C.O. also acknowledges financial support by the Innovative Training Network “Computational Spectroscopy in Natural Sciences and Engineering” (ITN-COSINE). D.B.K. received funding from the European Union’s Framework Program for Research and Innovation Horizon 2020 (2014-2020) under the Marie Skłodowska-Curie Grant agreement no. 754388 (LMUResearchFellows) and from LMU Munich’s Institutional Strategy LMUexcellent within the framework of the German Excellence Initiative (no. ZUK22). A.M.A. is grateful to Prof. Dr. Klaus Wanner for hosting the second year of his AvH fellowship. We thank Prof. Ivan Huc for hosting revision experiments in his laboratory, Dr. Oliver Thorn-Seshold and Li Gao for their help with establishing a LED setup, Dr. Peter Mayer for X-ray analyses, as well as Marvin Thielert for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b10430.
Author Contributions
∇ D.B.K. and G.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Bandara H. M. D.; Burdette S. C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. 10.1039/C1CS15179G. [DOI] [PubMed] [Google Scholar]
- Hartley G. S. The Cis-form of Azobenzene. Nature 1937, 140, 281. 10.1038/140281a0. [DOI] [Google Scholar]
- Merino E. Synthesis of azobenzenes: the coloured pieces of molecular materials. Chem. Soc. Rev. 2011, 40, 3835–3853. 10.1039/c0cs00183j. [DOI] [PubMed] [Google Scholar]
- Clara B.; Falk R.; Alexander G.; Günter M.; Alexander H. Light-Controlled Tools. Angew. Chem., Int. Ed. 2012, 51, 8446–8476. 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]
- Dong L.; Feng Y.; Wang L.; Feng W. Azobenzene-based solar thermal fuels: design, properties, and applications. Chem. Soc. Rev. 2018, 47, 7339–7368. 10.1039/C8CS00470F. [DOI] [PubMed] [Google Scholar]
- Timm F.; Schönberger M.; Dirk T. Optochemical Genetics. Angew. Chem., Int. Ed. 2011, 50, 12156–12182. 10.1002/anie.201103236. [DOI] [PubMed] [Google Scholar]
- Beharry A. A.; Woolley G. A. Azobenzene photoswitches for biomolecules. Chem. Soc. Rev. 2011, 40, 4422–4437. 10.1039/c1cs15023e. [DOI] [PubMed] [Google Scholar]
- Broichhagen J.; Frank J. A.; Trauner D. A Roadmap to Success in Photopharmacology. Acc. Chem. Res. 2015, 48, 1947–1960. 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]
- Lerch M. M.; Hansen M. J.; van Dam G. M.; Szymanski W.; Feringa B. L. Emerging Targets in Photopharmacology. Angew. Chem., Int. Ed. 2016, 55, 10978–10999. 10.1002/anie.201601931. [DOI] [PubMed] [Google Scholar]
- Szymański W.; Beierle J. M.; Kistemaker H. A. V.; Velema W. A.; Feringa B. L. Reversible Photocontrol of Biological Systems by the Incorporation of Molecular Photoswitches. Chem. Rev. 2013, 113, 6114–6178. 10.1021/cr300179f. [DOI] [PubMed] [Google Scholar]
- Velema W. A.; Szymanski W.; Feringa B. L. Photopharmacology: Beyond Proof of Principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. 10.1021/ja413063e. [DOI] [PubMed] [Google Scholar]
- Hammerich M.; Schütt C.; Stähler C.; Lentes P.; Röhricht F.; Höppner R.; Herges R. Heterodiazocines: Synthesis and Photochromic Properties, Trans to Cis Switching within the Bio-optical Window. J. Am. Chem. Soc. 2016, 138, 13111–13114. 10.1021/jacs.6b05846. [DOI] [PubMed] [Google Scholar]
- Frangioni J. V. In vivo near-infrared fluorescence imaging. Curr. Opin. Chem. Biol. 2003, 7, 626–634. 10.1016/j.cbpa.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Smith A. M.; Mancini M. C.; Nie S. Second window for in vivo imaging. Nat. Nanotechnol. 2009, 4, 710. 10.1038/nnano.2009.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissleder R. A clearer vision for in vivo imaging. Nat. Biotechnol. 2001, 19, 316. 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- Dong M.; Babalhavaeji A.; Samanta S.; Beharry A. A.; Woolley G. A. Red-Shifting Azobenzene Photoswitches for in Vivo Use. Acc. Chem. Res. 2015, 48, 2662–2670. 10.1021/acs.accounts.5b00270. [DOI] [PubMed] [Google Scholar]
- Bléger D.; Hecht S. Visible-Light-Activated Molecular Switches. Angew. Chem., Int. Ed. 2015, 54, 11338–11349. 10.1002/anie.201500628. [DOI] [PubMed] [Google Scholar]
- Beharry A. A.; Sadovski O.; Woolley G. A. Azobenzene Photoswitching without Ultraviolet Light. J. Am. Chem. Soc. 2011, 133, 19684–19687. 10.1021/ja209239m. [DOI] [PubMed] [Google Scholar]
- Knie C.; Utecht M.; Zhao F.; Kulla H.; Kovalenko S.; Brouwer A. M.; Saalfrank P.; Hecht S.; Bléger D. ortho-Fluoroazobenzenes: Visible Light Switches with Very Long-Lived Z Isomers. Chem. - Eur. J. 2014, 20, 16492–16501. 10.1002/chem.201404649. [DOI] [PubMed] [Google Scholar]
- Samanta S.; Beharry A. A.; Sadovski O.; McCormick T. M.; Babalhavaeji A.; Tropepe V.; Woolley G. A. Photoswitching Azo Compounds in Vivo with Red Light. J. Am. Chem. Soc. 2013, 135, 9777–9784. 10.1021/ja402220t. [DOI] [PubMed] [Google Scholar]
- Samanta S.; McCormick T. M.; Schmidt S. K.; Seferos D. S.; Woolley G. A. Robust visible light photoswitching with ortho-thiol substituted azobenzenes. Chem. Commun. 2013, 49, 10314–10316. 10.1039/c3cc46045b. [DOI] [PubMed] [Google Scholar]
- Dong M.; Babalhavaeji A.; Collins C. V.; Jarrah K.; Sadovski O.; Dai Q.; Woolley G. A. Near-Infrared Photoswitching of Azobenzenes under Physiological Conditions. J. Am. Chem. Soc. 2017, 139, 13483–13486. 10.1021/jacs.7b06471. [DOI] [PubMed] [Google Scholar]
- Dong M.; Babalhavaeji A.; Hansen M. J.; Kalman L.; Woolley G. A. Red, far-red, and near infrared photoswitches based on azonium ions. Chem. Commun. 2015, 51, 12981–12984. 10.1039/C5CC02804C. [DOI] [PubMed] [Google Scholar]
- Wegener M.; Hansen M. J.; Driessen A. J. M.; Szymanski W.; Feringa B. L. Photocontrol of Antibacterial Activity: Shifting from UV to Red Light Activation. J. Am. Chem. Soc. 2017, 139, 17979–17986. 10.1021/jacs.7b09281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner S.; Berlin S. Two-Photon Excitation of Azobenzene Photoswitches for Synthetic Optogenetics. Appl. Sci. 2020, 10, 805. 10.3390/app10030805. [DOI] [Google Scholar]
- Cabré G.; Garrido-Charles A.; Moreno M.; Bosch M.; Porta-De-La-Riva M.; Krieg M.; Gascón-Moya M.; Camarero N.; Gelabert R.; Lluch J. M.; Busqué F.; Hernando J.; Gorostiza P.; Alibés R. Rationally designed azobenzene photoswitches for efficient two-photon neuronal excitation. Nat. Commun. 2019, 10, 907. 10.1038/s41467-019-08796-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad D. B.; Frank J. A.; Trauner D. Synthesis of Redshifted Azobenzene Photoswitches by Late-Stage Functionalization. Chem. - Eur. J. 2016, 22, 4364–4368. 10.1002/chem.201505061. [DOI] [PubMed] [Google Scholar]
- Rullo A.; Reiner A.; Reiter A.; Trauner D.; Isacoff E. Y.; Woolley G. A. Long wavelength optical control of glutamate receptor ion channels using a tetra-ortho-substituted azobenzene derivative. Chem. Commun. 2014, 50, 14613–14615. 10.1039/C4CC06612J. [DOI] [PubMed] [Google Scholar]
- Calbo J.; Weston C. E.; White A. J. P.; Rzepa H. S.; Contreras-Garcéa J.; Fuchter M. J. Tuning Azoheteroarene Photoswitch Performance through Heteroaryl Design. J. Am. Chem. Soc. 2017, 139, 1261–1274. 10.1021/jacs.6b11626. [DOI] [PubMed] [Google Scholar]
- Weston C. E.; Richardson R. D.; Haycock P. R.; White A. J. P.; Fuchter M. J. Arylazopyrazoles: Azoheteroarene Photoswitches Offering Quantitative Isomerization and Long Thermal Half-Lives. J. Am. Chem. Soc. 2014, 136, 11878–11881. 10.1021/ja505444d. [DOI] [PubMed] [Google Scholar]
- Simeth N. A.; Bellisario A.; Crespi S.; Fagnoni M.; König B. Substituent Effects on 3-Arylazoindole Photoswitches. J. Org. Chem. 2019, 84, 6565–6575. 10.1021/acs.joc.8b02973. [DOI] [PubMed] [Google Scholar]
- Ahlrichs R.; Bär M.; Häser M.; Horn H.; Kölmel C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Weigend F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Häser M.; Patzelt H.; Ahlrichs R. RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. 10.1016/S0009-2614(98)00862-8. [DOI] [Google Scholar]
- Bléger D.; Schwarz J.; Brouwer A. M.; Hecht S. o-Fluoroazobenzenes as Readily Synthesized Photoswitches Offering Nearly Quantitative Two-Way Isomerization with Visible Light. J. Am. Chem. Soc. 2012, 134, 20597–20600. 10.1021/ja310323y. [DOI] [PubMed] [Google Scholar]
- Harada J.; Ogawa K. X-ray Diffraction Analysis of Nonequilibrium States in Crystals: Observation of an Unstable Conformer in Flash-Cooled Crystals. J. Am. Chem. Soc. 2004, 126, 3539–3544. 10.1021/ja038203l. [DOI] [PubMed] [Google Scholar]
- Mostad A.; Rømming C. A. Refinement of the Crystal Structure of cis-Azobenzene. Acta Chem. Scand. 1971, 25, 3561–3568. 10.3891/acta.chem.scand.25-3561. [DOI] [Google Scholar]
- Rastogi S. K.; Rogers R. A.; Shi J.; Gao C.; Rinaldi P. L.; Brittain W. J. Conformational Dynamics of o-Fluoro-Substituted Z-Azobenzene. J. Org. Chem. 2015, 80, 11485–11490. 10.1021/acs.joc.5b02202. [DOI] [PubMed] [Google Scholar]
- Wang D.; Wagner M.; Butt H.-J.; Wu S. Supramolecular hydrogels constructed by red-light-responsive host-guest interactions for photo-controlled protein release in deep tissue. Soft Matter 2015, 11, 7656–7662. 10.1039/C5SM01888A. [DOI] [PubMed] [Google Scholar]
- Bäumler W.Light sources for photodynamic therapy and fluorescence diagnosis in dermatology. In Comprehensive Series in Photosciences; Calzavara-Pinton P., Szeimies R.-M., Ortel B., Eds.; Elsevier: New York, 2001; Vol. 2, Chapter 6, pp 83–98. [Google Scholar]
- Trads J. B.; Burgstaller J.; Laprell L.; Konrad D. B.; de la Osa de la Rosa L.; Weaver C. D.; Baier H.; Trauner D.; Barber D. M. Optical control of GIRK channels using visible light. Org. Biomol. Chem. 2017, 15, 76–81. 10.1039/C6OB02153K. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.