

Abstract

The emergence of bacterial resistance against life-saving medicines has forced the scientific community and pharmaceutical industry to take actions in the quest for novel antibacterials. These should not only overcome the existing bacterial resistance but also provide at least interim effective protection against emerging bacterial infections. Research into DNA gyrase and topoisomerase IV inhibitors has become a particular focus, with the description of a new class of bacterial topoisomerase type II inhibitors known as “novel bacterial topoisomerase inhibitors”, NBTIs. Elucidation of the key structural modifications incorporated into these inhibitors and the impact these can have on their general physicochemical properties are detailed in this review. This defines novel bacterial topoisomerase inhibitors with promising antibacterial activities and potencies, which thus represent one potential example of the future “drugs for bad bugs”, as identified by the World Health Organization.
Introduction
Rapidly increasing bacterial resistance is making many antibacterials ineffective, thus threatening the life-saving achievements of modern medicine.1 This includes the therapeutically proven fluoroquinolones, inhibitors of bacterial type II topoisomerases, whose clinical utility for some indications is threatened by resistance. In response to this, the focus of ongoing research has shifted toward not only new antibacterial targets but also the identification of inhibitors against the firmly established bacterial type II topoisomerases, such as DNA gyrase and topoisomerase IV (topo IV) with a completely new mechanism of action. As a result of the strong scientific endeavors in this field, a new class of antibacterials has been developed over the past 2 decades: the novel bacterial type II topoisomerase inhibitors (NBTIs).2,3 While these NBTIs have a somewhat similar intercalating mechanism of action to fluoroquinolones, they differ substantially enough to evade the existing target-mediated bacterial resistance to fluoroquinolones. This is due to their binding to different, nonoverlapping binding pockets on their DNA gyrase and topo IV targets in bacteria, as shown in Figure 1A.4 Furthermore, the antibacterial activities of the NBTIs arise from their well-balanced dual-target inhibition, which is the key for slow development of bacterial resistance due to target mutations.5 As a consequence, the NBTIs should have significant advantages over existing antibacterials.
Figure 1.

(A) Cartoon representations for comparison of the binding modes of the NBTIs (inset, gray, GSK299423) and fluoroquinolones (inset, yellow, clinafloxacin) within Staphylococcus aureus DNA gyrase (PDB code 2XCS).4 For the purpose of comparison of the distinct binding sites between fluoroquinolones and NBTIs, clinafloxacin molecules were artificially inserted after superimposing Streptococcus pneumoniae topo IV (PDB code 3RAD)6 over S. aureus DNA gyrase. The DNA gyrase A subunits are shown in light and dark green, the DNA gyrase B subunits are light and dark violet, and the DNA molecule is orange. (B) Structure of GSK299423 as a representative NBTI, indicating the main important structural fragments: the “left-hand side” (LHS) and the “right-hand side” (RHS) of the molecule (as depicted here) and the central linker.4
Figure 1B shows a representative of these NBTI inhibitors, GSK299423, to illustrate their three essential parts: the DNA-intercalating heteroaromatic “left-hand side” (LHS), the enzyme-bound heteroaromatic “right-hand side” (RHS), and their connection through a cyclic/bicyclic linker.4 This review sheds light on the most successful protocols for optimization of the NBTI-related structure–activity relationships (SARs), with particular emphasis on selection of the appropriate LHS, RHS, and linker motifs to ensure suitable antibacterial activity and spectrum for advanced clinical utility.
How Do the NBTIs Bind to Their Targets?
Limitations of known DNA gyrase inhibitors led to the first published NBTI patent application in 1999.2 The first NBTI-related studies were published in 20057 and 2007, although these provided only a rough insight into their mode of action.8,9 The field was very actively studied during this period by a number of different pharmaceutical R&D groups, which in turn resulted in the discovery of one of the first promising NBTIs, NXL101 (viquidicin).10−13 The mechanism of this NBTI was studied in detail revealing a unique, non-quinolone mode of action, thereby indicating the key differences between NBTIs and quinolones.14 The NBTIs were then more comprehensively studied since 2010, when the very first structure of Staphylococcus aureus DNA gyrase in complex with a potent NBTI (GSK299423) using X-ray crystallography (PDB code 2XCS) became available.4 This allowed the definition of their binding mode and identified the three main structural components, each of which has its own binding pattern. The upper planar LHS moiety illustrated in Figure 1A intercalates between the central DNA base pairs on the 2-fold axis in the middle of each DNA gyrase A (GyrA) active site, helping to stabilize the precleavage enzyme–DNA complex4 and induces DNA single-strand breaks.15 The lower RHS moiety (Figure 1A) interacts through van der Waals forces with the hydrophobic amino acid residues of GyrA (i.e., Ala68, Gly72, Met75, Met121) in the size-restricted binding pocket on the 2-fold axis that is formed upon merging of two GyrA subunits. The LHS and RHS fragments are connected by the central unit (i.e., the linker), which occupies the void space and in principle does not make any contact with the DNA or GyrA, with the exception of the key ionic interaction between the basic amine of the linker and Asp83 of GyrA (Figure 1).16 This original binding mode was independently confirmed by other research groups with their NBTIs in complex with S. aureus DNA gyrase as well (e.g., PDB code 4PLB).17 However, the recently solved crystal structure of the NBTI gepotidacin (GSK2140944) in complex with S. aureus DNA gyrase (PDB code 6QTK) demonstrated that due to the protonated nature of the basic gepotidacin amine, it interacts both directly with Asp83 of one GyrA subunit and indirectly through water-mediated contact with Asp83 of the second GyrA subunit, which significantly strengthens the binding.15 The NBTI interaction with this Asp residue has therefore been recognized as a key element for NBTI binding and activity.4,14 The further role of the linker moiety is not only to provide sufficient rigidity that will ensure the correct spatial arrangement of both LHS and RHS (Figure 1) but also to provide sufficient flexibility to maintain optimal binding as the DNA moves the enzyme.4
Although the binding mechanism of NBTIs to DNA gyrase is being studied intensively, it should be pointed out that topo IV remains an equivalently important antibacterial target. It seems that DNA gyrase and topo IV differ mainly in their sensitivity to NBTIs. While DNA gyrase is mostly preferential as NBTIs antibacterial target in Gram-positive bacteria, it appears that in Gram-negative pathogens NBTIs work more efficiently on topo IV.14 This behavior might be explained by comparing the amino acid sequences constituting NBTIs binding pocket DNA gyrase and topo IV originating from S. aureus and E. coli, respectively (see Figure 2 and Supporting Information, Table S1). The key difference identified is the amino acid residue labeled as Met75 in S. aureus DNA gyrase (Figure 1 and residues in Table S1 highlighted in yellow), which corresponds to Ile74 in E. coli DNA gyrase, Ile71 in S. aureus topo IV, and Leu71 in E. coli topo IV, respectively (Figure 2). Another difference observed is that E. coli topo IV has an extra amino acid residue (Met118) not found in the analyzed sequences of the other three enzymes. We believe that these differences might determine the selectivity of NBTIs. Namely, Ile74 of E. coli DNA gyrase and Ile71 of S. aureus topo IV might sterically hinder the binding of NBTIs through spatial alterations of the volume of the binding pocket. The same may not apply in the case of Leu71 in E. coli topo IV because of that additional amino acid residue. It should be stressed, however, that due to the lack of structural information about NBTIs binding into topo IV binding pocket, one should not get these observations with confidence. Put differently, more attention should be paid to such substantial structural differences between both bacterial topoisomerase II targets in the future design of novel NBTIs with a balanced DNA gyrase/topo IV inhibition profile. Achieving this would be highly beneficial in preventing rapid development of bacterial resistance.
Figure 2.
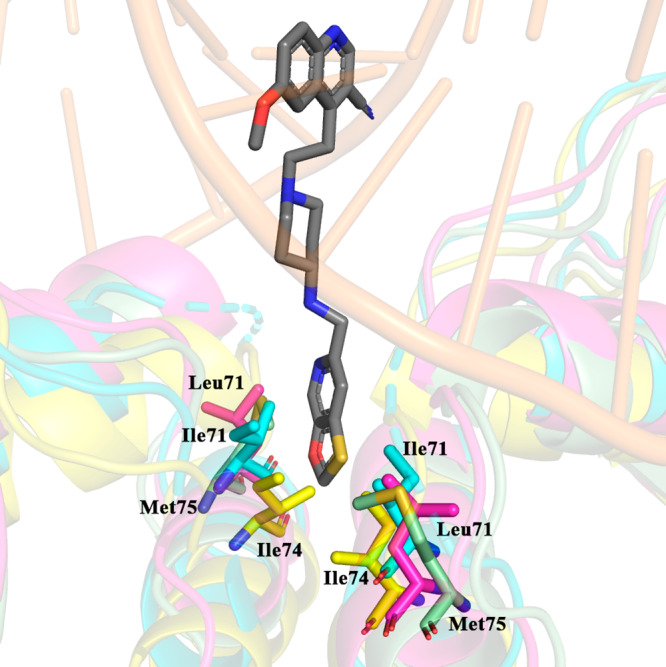
GSK299423 (inset, gray) and Met75 from S. aureus DNA gyrase (inset, green, PDB code 2XCS)4 in comparison to Ile74 in E. coli DNA gyrase (inset, yellow, PDB code 4CKK),18 Ile71 in S. aureus topo IV (inset, cyan, PDB code 2INR), and Leu71 in E. coli topo IV assembled homology model utilizing Klebsiella pneumoniae topo IV structure as a template (inset, magenta, PDB code 5EIX).19 For clarity, the corresponding GyrA and ParC subunits originating from S. aureus and E. coli, respectively, were used for the structural superimposition (see Supporting Information).
Numerous LHS, RHS, and linker moieties have been studied over the years relating to the SARs of the NBTIs. These have generally led to similar conclusions regarding the best fragment selection and substitution pattern to provide the best antibacterial activities (in the low nanomolar range) and spectrum.20,21 In this review, we have compiled and described the optimal fragments that have been reported to date, with a primary focus on NBTIs building blocks that have emerged over the past 2 decades. Due to insufficient structural information related to the binding of NBTIs topo IV, we have primarily focused our SAR on NBTIs of DNA gyrase.
The LHS Fragment Intercalates into DNA to Disturb Its Native Spatial Topology
Two LHS types are appearing among NBTIs, bicyclic and tricyclic heteroaromatics. The optimal bicyclic LHS fragments (Figure 3A) have in general been variants of quinoline, among which substituted quinolines and naphthyridines have been predominantly studied, as these can provide stable intercalation between the DNA base pairs, to enhance the antibacterial activity. Two substitution positions on the LHS fragment have been identified as generally the most suitable for NBTI antibacterial activities and spectrum: position 2, with fluoro, cyano, or hydroxyl substitutions;22−24 position 7, with methoxy or cyano substitutions.23,24 Substitution with fluoro at position 2 also provides improved targeting of topo IV, which thus results in improved dual-target activity.5,25 Alterations to the other positions on the LHS fragment have been reported to have detrimental effects on the bacterial topoisomerase activities and spectrum.23
Figure 3.

(A) Substitution patterns of the LHS fragments identified as most suitable for DNA intercalation and antibacterial activity. (B) 3D representation of a quinoline LHS (inset, gray, stick representation) intercalated between the central DNA base pairs (inset, orange, cartoon representation; PDB code 2XCS).4
Other more polar variants of quinolone have also been investigated for NBTIs. Methoxy-, fluoro-, and cyano-substituted benzoxazinone,26,27 quinoxalinone,27−29 1,8-naphthyridinone,27 and quinolone27,29 have been among the most investigated in the search for the optimal LHS constructs for the desired antibacterial properties. The pivotal reason for the introduction of carbonyl group on the LHS was to restrain the conformational rotation of entire NBTI that may benefit in good target potency. At the same time this LHS fragment provides greater safety profile, due to decreased overall log D.27
The tricyclic LHS compounds were mainly introduced to improve the safety profile of NBTIs by altering their physicochemical properties (e.g., log D and basicity).30 The tricyclic quinolone31 and 1,5-naphthyridinone30,32,33 substituted with fluorine at position 230−33 were also found to be LHS constructs that show good overall antibacterial activities. Hydroxylation of this tricyclic fragment was shown to be promising for NBTI activity as this additionally establishes a favorable H-bond interaction with the DNA,30,31 which in turn strengthens the binding of the LHS fragment itself and thus increases the overall ligand stability. An alternative tricyclic LHS has also been reported to show good inhibitory activity.34 For the LHS fragment, the most common variants that have been reported on to date are shown in Figure 3.
The Central Linker Provides Correct Positioning of LHS and RHS
The central linker is significant not only for the favorable geometrical positioning of LHS and RHS but also for optimizing the overall physicochemical properties of the NBTIs to expand the spectrum of antibacterial activity toward Gram-negative bacteria. These physicochemical properties are particularly important when tackling the “ESKAPE” bacteria for which new antibiotics are urgently needed (i.e., Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.).35 The lipophilic and basic nature of various linkers have been the most studied and modified properties, as they primarily increase the permeability of the NBTI and consequently improve the antibacterial activity. Moreover, the linker also plays a crucial role in improving safety, mainly due to tuned physicochemical properties that affect binding to hERG K+ channels among others.28,29,36
There is little doubt that the suitable spatial orientation and correct positioning of the LHS and RHS fragments are determined by the central core (linker). At the same time, the selection of an appropriate linker can significantly contribute toward the fine-tuning of the physicochemical properties of the NBTIs. Various unsubstituted/substituted central units have therefore been examined with the aim being to optimize the lipophilicity and basicity of the NBTIs, which have included tetrahydroindazole,8,9 piperidinecarboxylic acid,14,37 aminopiperidine,4 oxabicyclooctane,17 tetrahydropyran,5 cyclohexane,22 and 1,3-dioxane36 (Figure 4A). Substitution patterns have been proposed for the two-atom linkage that connects LHS to the central core, where position 2 has been recognized as the more favorable one. Introduction of a hydroxyl group led to improved solubility by over 100-fold, which was associated with better oral efficacy.17 Introduction of a basic −NH2 was shown to increase the polarity (i.e., lowered log D, from 2.0 to 0.9)38 as well as the basicity (i.e., increased pKa, from 7.4 to 8.4). This might also significantly enhance NBTI permeation across the membranes of Gram-negative bacteria, to provide improved whole-cell activity, especially against the hard-to-treat Pseudomonas aeruginosa, as has been demonstrated in various studies.30,38 Replacement of the −OH with −NH2 even improved the P. aeruginosa minimal inhibitory concentration (MIC) by 8-fold (improved MIC, from <8 μg/mL for −OH to 1 μg/mL for −NH2).38 Since passive absorption probably dictates the NBTI membrane permeation, there is a question of whether increasing pKa is an actual improvement and needs further clarification.
Figure 4.

(A) Substitution patterns of the most common NBTI linker fragments. These comprise a representative central unit (linker) and the optimal two-atom linkage. (B) 3D representation of an example aminopiperidine linker moiety (inset, gray, stick representation) and its key ionic interaction (red dots) between the protonated basic amine of the linker and Asp83 of GyrA (inset, green, stick representation; PDB code 2XCS),4 which is required for correct antibacterial activity.
The interconnection between NBTI permeability and basicity was also demonstrated in an independent antibacterial accumulation study, where introduction of a positive charge through a primary amine significantly increased the NBTI permeation for Gram-negative bacterial pathogens.39 As well as this positive impact, the basic nitrogen of the linker moiety was identified as a key structural element that is pivotal for the remarkable antibacterial activities of NBTIs, through its strong ionic interaction with Asp83 of GyrA (Figure 1).4
The RHS Fragment Binds Exclusively within the GyrA binding pocket
With the intention to achieve as high inhibitory potency as possible and consequently better antibacterial activities for NBTIs, a variety of RHS constructs have been examined. These can be classified into two common structural classes: pyridoxazinone (or its thio analog, pyridothiazinone) and pyridodioxane (or oxathiinopyridazine variant). Both structural classes have favorable interactions with the GyrA hydrophobic residues and have been identified as the most beneficial for the NBTI activities and spectrum (Figure 5A). Recently, we demonstrated that the distances between the Cα–Cα atoms of the opposing GyrA α3 helices from S. aureus and Escherichia coli DNA gyrases vary, which led us to conclude that the size-restricted GyrA binding pocket of S. aureus (5.5–7.5 Å) is even narrower in GyrA of E. coli (3.0–4.5 Å).40 Consequently, only few substitutions with small hydrophobic groups (e.g., Cl, F, methyl) at position 3 on the pyridoxazinone RHS moiety have been reported to retain overall NBTI antibacterial activity. In contrast, incorporation of polar groups (e.g., methoxy) can dramatically diminish the antibacterial activity, due to the hydrophobicity of the binding pocket.41
Figure 5.

(A) Substitution patterns of the most common RHS fragments identified as most suitable for appropriate interactions with GyrA, to contribute to improved antibacterial activity. (B) 3D representation of an example bicyclic RHS (inset, gray, stick representation) bound into the GyrA hydrophobic pocket (inset, green) with the key amino acid residues (inset, green, stick representation). The unusual H-bonding between RHS −CH2 and the Ala68 backbone oxygens (inset: red dots; PDB code 2XCS)4 defines the correct positioning and stabilization of RHS within the GyrA binding pocket.
While most studies have focused on these RHS fragments, a different approach was also tried, with the design of a series of NBTIs with the relatively unusual and not frequently occurring cyclobutylphenyl RHS.20,34 Here, 2,5-difuoro substitution was the most suitable for DNA gyrase/topo IV dual-targeting in S. aureus, with the most promising lead compound defined (Table 1, compound 14).23,37 Moreover, it is interesting to note that in comparison to the other NBTIs, these NBTI analogs lack the secondary nitrogen on the linker that serves as H-bond donor, which was commonly identified as a key structural feature for potent antimicrobial activities of NBTIs. Nevertheless, the antibacterial activities of this series of NBTIs were comparable to other NBTIs.
Table 1. Summary of Selected, Most Promising NBTIs for Their Enzyme Inhibition (IC50) and Antibacterial Activities (MIC) against Gram-Negative and Gram-Positive Bacteria, Including Their Cardiotoxicities (hERG)k.

IC50 for enzyme supercoiling (gyrase) and relaxation (topo IV).
MIC of one strain.
MIC90 of more strains.
S. aureus WCUH 29.
S. pneumoniae 1629.
H. influenzae H128.
S. pneumoniae TPS3.
S. aureus ATCC 29213.
E. coli ATCC 25922.
S. pneumoniae ATCC 49619.
MIC = minimum inhibitory concentration. NR = not reported.
The significance of the basic nitrogen of the linker was also investigated by another research group.41 Surprisingly and to some extent contrary to the well-established NBTI SARs, they concluded that alteration of the amine into an amide did not have any particular influence on the NBTI antibacterial activities and spectrum. Unfortunately, the reason for this behavior remains unknown, although they speculated on the possibility of a modified binding mode or altered cell accumulation.
Successful NBTI Stories
Several NBTIs have emerged from the structural optimization studies and have been reported to be promising DNA gyrase/topo IV dual-targeting inhibitors that have broad-spectrum antibacterial activities. Some of these NBTIs are reported in Table 1, along with their MICs against the most widely investigated bacterial pathogens.
Among these NBTIs, gepotidacin,42,43 AZD9742,29 GSK945237,31 and NXL10144 have been taken to human clinical trials, although to date only gepotidacin has been reported as promising. NXL101 was discontinued in phase I clinical trials due to QTc prolongation,44 while AZD9742 and GSK945237 were dropped from further investigation. Gepotidacin is composed of a tricyclic LHS and a pyranopyridine RHS that are connected through an aminopiperidine-containing linker. Its antibacterial efficacy has been confirmed in phase 2 clinical trials against Gram-positive acute bacterial skin/skin structure infections, as well as uncomplicated urogenital gonorrhea caused by Neisseria gonorrheae.45,46 Additionally, it is undergoing further studies aimed at identifying further clinical indications caused by Gram-positive and some Gram-negative bacterial pathogens.47,48
The Biggest Challenges
Although the NBTIs have demonstrated outstanding antibacterial activities and potencies against a broad panel of bacterial pathogens, an issue that unfortunately still accompanies these compounds is their relatively high cardiotoxic potential, which has generally prevented their further development toward clinical use. The hERG toxicity has been recognized as a challenge to NBTI development and consequently the main reason for the withdrawing of some of the promising NBTIs from clinical trials. Numerous efforts are being invested in structure optimization toward decreased undesirable cardiotoxic effects while retaining the excellent antibacterial properties of the NBTIs. Unfortunately, it appears that decreased hERG-related cardiotoxicity of these NBTIs is strongly correlated with reduced antibacterial activities and a narrowing of their antibacterial spectrum. A recent review covered the state-of-the-art optimization efforts that have been invested in hERG improvements.50 This indicated that providing the NBTIs with balanced physicochemical properties, based on their log D and pKa, currently appears to be the most reasonable solution toward improving upon the hERG-related issues.
Future Perspectives
The antibacterial activities of the NBTIs have never been disputed, although further improvements appear to be necessary, particularly because of the NBTI-induced cardiotoxicity issues. Nevertheless, if a sufficient range can be provided between their toxic and active concentrations, a safe therapeutic window might be achieved. However, DNA intercalation requires ligand motifs with a planar structural and a size comparable to that of the DNA base pairs to establish suitable π–π stacking interactions. Therefore, the LHS “building blocks” appear not to allow extensive substructural alterations, although some of the more recently designed NBTIs reported below might challenge this notion.
The linker itself is located in a void space with no direct interactions in its vicinity, such as with DNA or the gyrase. However, although the linker has little or no influence on NBTI binding, it might affect the antibacterial activity by improving NBTI permeability, particularly with Gram-negative bacterial strains.
On balance, RHS appears to be the most favorable fragment for further activity optimization at the level of the enzyme target, and we believe that the full spectrum of RHS SARs remains to be fully established, with only a limited aspect uncovered to date. Although bicyclic RHS fragments have demonstrated good activities, it should be stressed that the monocyclic-based RHS building blocks have also been successful. Recently, a series of NBTIs that comprised cyclobutylaryl RHS fragments was studied, and it showed dual-target DNA gyrase/topo IV inhibition as well as good whole-cell activity against S. aureus.25 Moreover, differently substituted monocyclic aryl analogs and in particular chloro-substituted phenyl derivatives were investigated, and they showed antibacterial activities on gyrase comparable to those of the bicyclic NBTI variants.36 In a further study compounds with mono-, di-, and trisubstituted phenyl RHSs were investigated and it was discovered that 4-substituted phenyl seems to be preferred, whereby an appropriate relationship between size of the substituent and its polarity should be considered. While small and more polar substituents decrease potency, large and lipophilic substituents are better tolerated. Examples of substituents identified as more appropriate are chloro, methyl, trifluoromethyl, methoxy, and trifluoromethoxy.51 This finding is strongly supported on the basis that monocyclic RHS fragments can also correctly insert into the GyrA hydrophobic binding pocket and can consequently establish favorable interactions with the amino acid residues that are crucial to the antibacterial activity. Compounds with monocyclic RHSs from these studies were unfortunately unable to exhibit topo IV inhibition.36,51
An important issue to address here is related to the unbalanced Gram-positive and Gram-negative potencies of the NBTIs in general. Put differently, better Gram-positive activities have been reported for the NBTIs, which is also apparent for the small subset of successful NBTIs given in Table 1. The cardinal issue that accompanies the NBTI actions toward Gram-negative pathogens appears to be related to their poor permeability through the bacterial cell wall and the activity of the bacterial efflux pumps.28,38 It seems that the uptake of NBTIs into the bacterial cells depends mainly on their physicochemical properties, a situation that is mandatory when targeting Gram-positive and Gram-negative bacteria. Consequently, the Gram-negative activities might be improved by using more positively charged (i.e., increased basicity) and polar (i.e., lower log D) compounds.38 Such physicochemical properties appear to be more suitable for NBTIs that can target Gram-negative bacteria, compared to those associated with NBTIs that act more on Gram-positive bacteria. Therefore, it is particularly difficult to develop “balanced” NBTIs with broad-spectrum antibacterial activities against bacterial strains from both classes. Some progress toward Gram-negative targeting was achieved by the introduction of −NH2 into the linker moiety.28,38 Furthermore, many patents have indicated that studies in recent years have focused more on achieving better Gram-negative actions through relatively “extreme” transformations of the generic NBTI structure, in terms of LHS and linker replacements, while retaining the established RHS structure. Common bicyclic LHS fragments have been replaced with a biaryl moiety that was substituted with an elongated aliphatic chain designed to provide tighter interactions with the DNA base pairs. In a similarly “unusual” way, the linker was also modified to contain oxazolidinone as its central unit, as shown in Figure 6.52 Interestingly, this compound class lacks the key basic nitrogen that was previously reported to be of crucial significance for antibacterial activity. This once again implies that this basic nitrogen is not necessary and reinforces the theory of a different binding mode or improved cell accumulation. These compounds show good and comparable Gram-negative and Gram-positive antibacterial activities, which indicates that a large structural deviation might be beneficial for the extension of the NBTI antibacterial spectrum.
Figure 6.

Representative of the latest NBTIs that contain modified LHS and linker fragments with an established RHS, in an approach toward improvement of the Gram-negative activity. This compound showed balanced Gram-positive (S. aureus MIC = 0.25 μg/mL) and Gram-negative (E. coli MIC = 0.25 μg/mL) antibacterial activities.52
Although Gram-negative bacteria are indeed of high priority, the development of antibacterials that are effective against Gram-positive bacteria should not be neglected. Additionally, despite the need for broad-spectrum antibacterials, it might be more reasonable to focus on NBTI optimizations toward a narrower antibacterial spectrum, to slow down the rapidly spreading resistance of the existing antibacterials, and to provide long-lasting and effective NBTIs.
Acknowledgments
The financial support of this work from the Slovenian Research Agency (Grants P1-0017 and P1-0208) is gratefully acknowledged. The authors thank Christopher Berrie for proofreading the manuscript and a lot of useful advice.
Glossary
Abbreviations Used
- NBTI
novel bacterial topoisomerase inhibitor
- LHS
left-hand side
- RHS
right-hand side
- GyrA
gyrase A subunit
- R&D
research and development
- ParC
topoisomerase IV C subunit
- ESKAPE
Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.
- hERG
human ether-a-go-go-related gene
Biographies
Anja Kolarič obtained her Master’s degree in Pharmacy at the Faculty of Pharmacy, University of Ljubljana, Slovenia. Currently she is employed at the National Institute of Chemistry in Ljubljana, Slovenia, and is doing her Ph.D., with her research focused on the identification of novel antibacterial agents in the class of novel bacterial topoisomerase inhibitors.
Marko Anderluh received his Ph.D. at the Faculty of Pharmacy, University of Ljubljana, Slovenia, in 2004. In 2002 he worked at the University of Karlsruhe, Germany, within a bilateral collaboration with the group of Prof. Dr. Athanassios Giannis. From June 2007 to early 2008, he was a postdoctoral researcher in the group of Prof. Dr. Anna Bernardi at the University of Milan, Italy. In 2017, he became Full Professor, at the Faculty of Pharmacy, University of Ljubljana. His research is devoted to the design, synthesis, and the evaluation of drugs and molecular probes. He focuses on novel antibacterial agents (novel bacterial topoisomerase inhibitors, gyrase B inhibitors, bacterial autolysin inhibitors), CCK2R ligands as PET probes, and glycomimetics and glyco-based molecular probes.
Nikola Minovski graduated from Ss. Cyril and Methodius University, Macedonia, with a Master’s in Pharmacy in 2005. In 2007, he moved to Slovenia and joined the Ph.D. Program in Biomedicine at the University of Ljubljana, Slovenia. In 2011, he completed his Ph.D. studies that were focused on the integration of in silico methods for the design of novel 6-fluoroquinolones. Since 2007, he has been Senior Researcher in the Laboratory for Cheminformatics at the National Institute of Chemistry in Ljubljana, Slovenia. His main research interests include the design and optimization of novel antibacterial agents (DNA gyrase/topo IV inhibitors such as NBTIs), ligand–protein interactions, enzyme inhibition, molecular dynamics simulations, and development of efficient in silico platforms for the design and optimization of novel hit compounds.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01738.
Sequence comparison results (PDF)
Author Contributions
The manuscript was written through contributions of all of the authors. All of the authors have given their approval of the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization. Antimicrobial Resistance: Global Report On Surveillance 2014. http://www.who.int/drugresistance/documents/surveillancereport/en (accessed Aug 7, 2019).
- Coates W. J.; Gwynn M. N.; Hatton I. K.; Masters P. J.; Pearson N. D.; Rahman S. S.; Slocombe B.; Warrack J. D.. Preparation Of Piperidinylalkylquinolines As Antibacterials. Patent WO 1999037635, 1999.
- Malleron J.-L.; Tabart M.; Carry J. C.; Evers M.; El Ahmad Y.; Mignani S.; Viviani F.. Quinolyl Propyl Piperidine Derivatives And Theiruse As Antibacterial Agents. Patent WO 200125227, 2001.
- Bax B. D.; Chan P. F.; Eggleston D. S.; Fosberry A.; Gentry D. R.; Gorrec F.; Giordano I.; Hann M. M.; Hennessy A.; Hibbs M.; Huang J.; Jones E.; Jones J.; Brown K. K.; Lewis C. J.; May E. W.; Saunders M. R.; Singh O.; Spitzfaden C. E.; Shen C.; Shillings A.; Theobald A. J.; Wohlkonig A.; Pearson N. D.; Gwynn M. N. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- Surivet J. P.; Zumbrunn C.; Rueedi G.; Hubschwerlen C.; Bur D.; Bruyère T.; Locher H.; Ritz D.; Keck W.; Seiler P.; Kohl C.; Gauvin J. C.; Mirre A.; Kaegi V.; Dos Santos M.; Gaertner M.; Delers J.; Enderlin-Paput M.; Boehme M. Design, synthesis, and characterization of novel tetrahydropyran-based bacterial topoisomerase inhibitors with potent anti-Gram-positive activity. J. Med. Chem. 2013, 56, 7396–7415. 10.1021/jm400963y. [DOI] [PubMed] [Google Scholar]
- Laponogov I.; Pan X. S.; Veselkov D. A.; Cirz R. T.; Wagman A.; Moser H. E.; Fisher L. M.; Sanderson M. R. Exploring the active site of the Streptococcus pneumoniae topoisomerase IV–DNA cleavage complex with novel 7,8-bridged fluoroquinolones. Open Biol. 2016, 6, 160157. 10.1098/rsob.160157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur P.; Delachaume C.; Lowther J.; Hodgson J.. Minimum Inhibitory Concentrations (MIC) And Mutation Prevention Concentrations (MPC) Of NXL101, A Novel Topoisomerase IV Inhibitor, Against Staphylococcus aureus Including Multi-resistant Strains. Abstracts, 45th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; American Society for Microbiology, 2005; Abstract F-507, p 184 [Google Scholar]
- Gomez L.; Hack M. D.; Wu J.; Wiener J. J. M.; Venkatesan H.; Santillán A. Jr; Pippel D. J.; Mani N.; Morrow B. J.; Motley S. T.; Shaw K. J.; Wolin R.; Grice C. A.; Jones T. K. Novel pyrazolederivates as potent inhibitors of type II topoisomerases. Part 1: synthesis and preliminary SAR analysis. Bioorg. Med. Chem. Lett. 2007, 17, 2723–2727. 10.1016/j.bmcl.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Wiener J. J. M.; Gomez L.; Venkatesan H.; Santillán A. Jr; Allison B. D.; Schwarz K. L.; Shinde S.; Tang L.; Hack M. D.; Morrow B. J.; Motley S. T.; Goldschmidt R. M.; Shaw K. J.; Jones T. K.; Grice C. A. Tetrahydroindazole inhibitors of bacterial type II topoisomerases. Part 2: SAR development and potency against multidrug resistant strains. Bioorg. Med. Chem. Lett. 2007, 17, 2718–2722. 10.1016/j.bmcl.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Andes D.; Lowther J.; Craig W. A.. In vivo determination of the magnitude of the pharmacodynamic parameter associated with efficacy for the topoisomeraseinhibitor, AVE 6971, against multiple bacteria in a murine infection model. Presented at the 45th Interscience Conference on Antimicrobial Agents and Chemotherapy, Washington, DC, U S., December 16–19, 2005.
- Robbins M. J.; Bryskier A.; Felmingham D.. Comparative in vitro activity of AVE 6971, a novel inhibitor of DNA topoisomerase IV, against Staphylococcus aureus (SA). Presented at the 44th Interscience Conference on Antimicrobial Agents and Chemotherapy, Washington, DC, U.S., October 30–Nowember 2, 2004.
- Salvador A.; Gautier J. Y.; Pasquier O.; Merdjan H. Liquid chromatography–tandem mass spectrometric determination of a new antibacterial agent (AVE6971) in human white blood cells. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2007, 855, 173–179. 10.1016/j.jchromb.2007.04.034. [DOI] [PubMed] [Google Scholar]
- Lesuisse D.; Tabart M.. Importance of chirality in the field of anti-infective agents. In Comprehensive Chirality, 1st ed.; Carreira E. M., Yamamoto H., Eds.; Elsevier: Amsterdam, 2012; pp 8–29. [Google Scholar]
- Black M. T.; Stachyra T.; Platel D.; Girard A.-M.; Claudon M.; Bruneau J.-M.; Miossec C. Mechanism of action of the antibiotic NXL101, a novel non-fluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 2008, 52, 3339–3349. 10.1128/AAC.00496-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson E. G.; Bax B.; Chan P. F.; Osheroff N. Mechanistic and structural basis for the actions of the antibacterial gepotidacin against Staphylococcus aureus gyrase. ACS Infect. Dis. 2019, 5, 570–581. 10.1021/acsinfecdis.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax B. D.; Murshudov G.; Maxwell A.; Germe T. DNA topoisomerase inhibitors: trapping a DNA-cleaving machine in motion. J. Mol. Biol. 2019, 431, 3427–3449. 10.1016/j.jmb.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. B.; Kaelin D. E.; Wu J.; Miesel L.; Tan C. M.; Meinke P. T.; Olsen D.; Lagrutta A.; Bradley P.; Lu J.; Patel S.; Rickert K. W.; Smith R. F.; Soisson S.; Wei C.; Fukuda H.; Kishii R.; Takei M.; Fukuda Y. Oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad spectrum antibacterial agents. ACS Med. Chem. Lett. 2014, 5, 609–614. 10.1021/ml500069w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearnshaw S. J.; Edwards M. J.; Stevenson C. E.; Lawson D. M.; Maxwell A. A new crystal structure of the bifunctional antibiotic simocyclinone D8 bound to DNA gyrase gives fresh insight into the mechanism of inhibition. J. Mol. Biol. 2014, 426, 2023–2033. 10.1016/j.jmb.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veselkov D. A.; Laponogov I.; Pan X.-S.; Selvarajah J.; Skamrova G. B.; Branstrom A.; Narasimhan J.; Prasad J. V.; Fisher L. M.; Sanderson M. R. Structure of a quinolone-stabilized cleavage complex of topoisomerase IV from Klebsiella pneumonia and comparison with a related Streptococcus pneumonia complex. ActaCrystallogr. D Struct. Biol. 2016, 72, 488–496. 10.1107/S2059798316001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C.; Janin Y. L. Non-quinolone inhibitors of bacterial type IIA topoisomerases: a feat of bioisosterism. Chem. Rev. 2014, 114, 2313–2342. 10.1021/cr4003984. [DOI] [PubMed] [Google Scholar]
- Mitton-Fry M. J. Novelbacterial type II topoisomerase inhibitors. Med. Chem. Rev. 2017, 52, 281–302. 10.29200/acsmedchemrev-v52.ch15. [DOI] [Google Scholar]
- Miles T. J.; Barfoot C.; Brooks G.; Brown P.; Chen D.; Dabbs S.; Davies D. T.; Downie D. L.; Eyrisch S.; Giordano I.; Gwynn M. N.; Hennessy A.; Hoover J.; Huang J.; Jones G.; Markwell R.; Rittenhouse S.; Xiang H.; Pearson N. Novel cyclohexyl-amides as potent antibacterials targeting bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2011, 21, 7483–7488. 10.1016/j.bmcl.2011.09.114. [DOI] [PubMed] [Google Scholar]
- Singh S. B.; Kaelin D. E.; Wu J.; Miesel L.; Tan C. M.; Meinke P. T.; Olsen D. B.; Lagrutta A.; Wei C.; Peng X.; Wang X.; Fukuda H.; Kishii R.; Takei M.; Shibata T.; Ohata K.; Takano H.; Kurasaki H.; Takeuchi T.; Nishimura A.; Fukuda Y. Structure activity relationship of substituted 1, 5-naphthyridine analogs of oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents (Part 4). Bioorg. Med. Chem. Lett. 2015, 25, 2409–2415. 10.1016/j.bmcl.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Singh S. B.; Kaelin D. E.; Meinke P. T.; Wu J.; Miesel L.; Tan C. M.; Olsen D. B.; Lagrutta A.; Fukuda H.; Kishii R.; Takei M.; Takeuchi T.; Takano H.; Ohata K.; Kurasaki H.; Nishimura A.; Shibata T.; Fukuda Y. Structure activity relationship of C-2 ether substituted 1,5-naphthyridine analogs of oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents (Part-5). Bioorg. Med. Chem. Lett. 2015, 25, 3630–3635. 10.1016/j.bmcl.2015.06.061. [DOI] [PubMed] [Google Scholar]
- Mitton-Fry M. J.; Brickner S. J.; Hamel J. C.; Barham R.; Brennan L.; Casavant J. M.; Ding X.; Finegan S.; Hardink J.; Hoang T.; Huband M. D.; Maloney M.; Marfat A.; McCurdy S. P.; McLeod D.; Subramanyam C.; Plotkin M.; Reilly U.; Schafer J.; Stone G. G.; Uccello D. P.; Wisialowski T.; Yoon K.; Zaniewski R.; Zook C. Novel 3-fluoro-6-methoxyquinoline derivatives as inhibitors of bacterial DNA gyrase and topoisomerase IV. Bioorg. Med. Chem. Lett. 2017, 27, 3353–3358. 10.1016/j.bmcl.2017.06.009. [DOI] [PubMed] [Google Scholar]
- Geng B.; Comita-Prevoir J.; Eyermann C. J.; Reck F.; Fisher S. Exploring left-hand side substitutions in the benzoxazinone series of 4-amino-piperidine bacterial type IIa topoisomerase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 5432–5435. 10.1016/j.bmcl.2011.06.126. [DOI] [PubMed] [Google Scholar]
- Reck F.; Alm R.; Brassil P.; Newman J.; DeJonge B.; Eyermann C. J.; Breault G.; Breen J.; Comita-Prevoir J.; Cronin M.; Davis H.; Ehmann D.; Galullo V.; Geng B.; Grebe T.; Morningstar M.; Walker P.; Hayter B.; Fisher S. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II: broad-spectrum antibacterial agents with reduced hERG activity. J. Med. Chem. 2011, 54, 7834–7847. 10.1021/jm2008826. [DOI] [PubMed] [Google Scholar]
- Reck F.; Ehmann D. E.; Dougherty T. J.; Newman J. V.; Hopkins S.; Stone G.; Agrawal N.; Ciaccio P.; McNulty J.; Barthlow H.; O’Donnell J.; Goteti K.; Breen J.; Comita-Prevoir J.; Cornebise M.; Cronin M.; Eyermann C. J.; Geng B.; Carr G. R.; Pandarinathan L.; Tang X.; Cottone A.; Zhao L.; Bezdenejnih-Snyder N. Optimization of psysicochemical properties and safety profile of novel bacterial topoisomerase type II inhibitors (NBTIs) with activity against Pseudomonas aeruginosa. Bioorg. Med. Chem. 2014, 22, 5392–5409. 10.1016/j.bmc.2014.07.040. [DOI] [PubMed] [Google Scholar]
- Reck F.; Alm R. A.; Brassil P.; Newman J. V.; Ciaccio P.; McNulty J.; Barthlow H.; Goteti K.; Breen J.; Comita-Prevoir J.; Cronin M.; Ehmann D. E.; Geng B.; Aydon Godfrey A.; Fisher S. L. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II with reduced pKa: antibacterial agents with an improved safety profile. J. Med. Chem. 2012, 55, 6916–6933. 10.1021/jm300690s. [DOI] [PubMed] [Google Scholar]
- Singh S. B.; Kaelin D. E.; Wu J.; Miesel L.; Tan C. M.; Black T.; Nargund R.; Meinke P. T.; Olsen D.; Lagrutta A.; Lu J.; Patel S.; Rickert K. W.; Smith R. F.; Soisson S.; Sherer E.; Joyce L. A.; Wei C.; Peng X.; Wang X.; Fukuda H.; Kishii R.; Takei M.; Takano H.; Shibasaki M.; Yajima M.; Nishimura A.; Shibata T.; Fukuda Y. Tricyclic 1,5-naphthyridinone oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents-SAR of left-hand-side moiety (Part 2). Bioorg. Med. Chem. Lett. 2015, 25, 1831–1835. 10.1016/j.bmcl.2015.03.044. [DOI] [PubMed] [Google Scholar]
- Miles T. J.; Hennessy A. J.; Bax B.; Brooks G.; Brown B. S.; Brown P.; Cailleau N.; Chen D.; Dabbs S.; Davies D. T.; Esken J. M.; Giordano I.; Hoover J. L.; Jones G. E.; Sukmar S. K. K.; Markwell R. E.; Minthorn E. A.; Rittenhouse S.; Gwynn M. N.; Pearson N. D. Novel tricyclics (e.g. GSK945237) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2016, 26, 2464–2469. 10.1016/j.bmcl.2016.03.106. [DOI] [PubMed] [Google Scholar]
- Miles T. J.; Hennessy A. J.; Bax B.; Brooks G.; Brown B. S.; Brown P.; Cailleau N.; Chen D.; Dabbs S.; Davies D. T.; Esken J. M.; Giordano I.; Hoover J. L.; Huang J.; Jones G. E.; Sukmar S. K. K.; Spitzfaden C.; Markwell R. E.; Minthorn E. A.; Rittenhouse S.; Gwynn M. N.; Pearson N. D. Novel hydroxyl tricyclics (e.g., GSK966587) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2013, 23, 5437–5441. 10.1016/j.bmcl.2013.07.013. [DOI] [PubMed] [Google Scholar]
- Singh S. B.; Kaelin D. E.; Wu J.; Miesel L.; Tan C. M.; Gill C.; Black T.; Nargund R.; Meinke P. T.; Olsen D.; Lagrutta A.; Wei C.; Peng X.; Wang X.; Fukuda H.; Kishii R.; Takei M.; Takeuchi T.; Shibue T.; Ohata K.; Takano H.; Ban S.; Nishimura A.; Fukuda Y. Hydroxy tricyclic 1,5-naphthyridinone oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents-SAR of RHS moiety (Part 3). Bioorg. Med. Chem. Lett. 2015, 25, 2473–2478. 10.1016/j.bmcl.2015.04.063. [DOI] [PubMed] [Google Scholar]
- Charrier C.; Salisbury A.-M.; Savage V. J.; Duffy T.; Moyo E.; Chaffer-Malam N.; Ooi N.; Newman R.; Cheung J.; Metzger R.; McGarry D.; Pichowicz M.; Sigerson R.; Cooper I. R.; Nelson G.; Butler H. S.; Craighead M.; Ratcliffe A. J.; Best S. A.; Stokes N. R. Novel bacterial topoisomerase inhibitors with potent broad-spectrum activity against drug-resistant bacteria. Antimicrob. Agents Chemother. 2017, 61, e02100-16 10.1128/AAC.02100-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed Aug 10, 2019).
- Li L.; Okumu A.; Dellos-Nolan S.; Li Z.; Karmahapatra S.; English A.; Yalowich J. C.; Wozniak D. J.; Mitton-Fry M. J. Synthesis and anti-staphylococcal activity of novel bacterial topoisomerase inhibitors with a 5-amino-1, 3-dioxane linker moiety. Bioorg. Med. Chem. Lett. 2018, 28, 2477–2480. 10.1016/j.bmcl.2018.06.003. [DOI] [PubMed] [Google Scholar]
- Mitton-Fry M. J.; Brickner S. J.; Hamel J. C.; Brennan L.; Casavant J. M.; Chen M.; Chen T.; Ding X.; Driscoll J.; Hardink J.; Hoang T.; Hua E.; Huband M. D.; Maloney M.; Marfat A.; McCurdy S. P.; McLeod D.; Plotkin M.; Reilly U.; Robinson S.; Schafer J.; Shepard R. M.; Smith R. F.; Stone G. G.; Subramanyam C.; Yoon K.; Yuan W.; Zaniewski R. P.; Zook C. Novel quinoline derivatives as inhibitors of bacterial DNA gyrase and topoisomerase IV. Bioorg. Med. Chem. Lett. 2013, 23, 2955–2961. 10.1016/j.bmcl.2013.03.047. [DOI] [PubMed] [Google Scholar]
- Surivet J. P.; Zumbrunn C.; Bruyère T.; Bur D.; Kohl C.; Locher H. H.; Seiler P.; Ertel E. A.; Hess P.; Enderlin-Paput M.; Enderlin-Paput S.; Gauvin J. C.; Mirre A.; Hubschwerlen C.; Ritz D.; Rueedi G. Synthesis and characterization of tetrahydropyran-based bacterial topoisomerase inhibitors with antibacterial activity against Gram-negative bacteria. J. Med. Chem. 2017, 60, 3776–3794. 10.1021/acs.jmedchem.6b01831. [DOI] [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarič A.; Novak D.; Weiss M.; Hrast M.; Zdovc I.; Anderluh M.; Minovski N. Cyclohexyl amide-based novel bacterial topoisomerase inhibitors with prospective GyrA-binding fragments. Future Med. Chem. 2019, 11, 935–945. 10.4155/fmc-2018-0472. [DOI] [PubMed] [Google Scholar]
- Singh S. B.; Kaelin D. E.; Wu J.; Miesel L.; Tan C. M.; Meinke P. T.; Olsen D.; Lagrutta A.; Wei C.; Liao Y.; Peng X.; Wang X.; Fukuda H.; Kishii R.; Takei M.; Yajima M.; Shibue T.; Shibata T.; Ohata K.; Nishimura A.; Fukuda Y. Structure activity relationships of pyridoxazinone substituted RHS analogs of oxabicyclooctane-linked 1,5-naphthyridinyl novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents (Part 6). Bioorg. Med. Chem. Lett. 2015, 25, 3636–3643. 10.1016/j.bmcl.2015.06.057. [DOI] [PubMed] [Google Scholar]
- Biedenbach D. J.; Bouchillon S. K.; Hackel M.; Miller L. A.; Scangarella-Oman N. E.; Jakielaszek C.; Sahm D. F. In-vitro activity of gepotidacin, a novel triazaacenaphthylene bacterial topoisomerase inhibitor, against a broad spectrum of bacterial pathogens. Antimicrob. Agents Chemother. 2016, 60, 1918–1923. 10.1128/AAC.02820-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell D. J.; Sader H. S.; Rhomberg P. R.; Scangarella-Oman N. E.; Flamm R. K. In vitro activity of gepotidacin (GSK2140944) against Neisseria gonorrhoeae. Antimicrob. Agents Chemother. 2017, 61, e02047-16 10.1128/AAC.02047-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black M. T.; Coleman K. New inhibitors of bacterial topoisomerase GyrA/ParC subunits. Curr. Opin. Invest. Drugs. 2009, 10, 804–810. [PubMed] [Google Scholar]
- O’Riordan W.; Tiffany C.; Scangarella-Oman N.; Perry C.; Hossain M.; Ashton T.; Dumont E. Efficacy, safety, and tolerability of gepotidacin (GSK2140944) in the treatment of patients with suspected or confirmed Gram-positive acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 2017, 61, e02095-16 10.1128/AAC.02095-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S. N.; Morris D. H.; Avery A. K.; Workowski K. A.; Batteiger B. E.; Tiffany C. A.; Perry C. R.; Raychaudhuri A.; Scangarella-Oman N. E.; Hossain M.; Dumont E. F. Gepotidacin for the treatment of uncomplicated urogenital gonorrhea: A phase 2, randomized, dose-ranging, single-oral dose evaluation. Clin. Infect. Dis. 2018, 67, 504–512. 10.1093/cid/ciy145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waites K. B.; Crabb D. M.; Xiao L.; Duffy L. B. In vitro activities of gepotidacin (GSK2140944) and other antimicrobial agents against human mycoplasmas and ureaplasmas. Antimicrob. Agents Chemother. 2017, 61, e01064-17. 10.1128/AAC.01064-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamm R. K.; Farrell D. J.; Rhomberg P. R.; Scangarella-Oman N. E.; Sader H. S. Gepotidacin (GSK2140944) in vitro activity against Gram-positive and Gram-negative bacteria. Antimicrob. Agents Chemother. 2017, 61, 00468-17. 10.1128/AAC.00468-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surivet J. P.; Zumbrunn C.; Rueedi G.; Bur D.; Bruyère T.; Locher H.; Ritz D.; Seiler P.; Kohl C.; Ertel E. A.; Hess P.; Gauvin J. C.; Mirre A.; Kaegi V.; Dos Santos M.; Kraemer S.; Gaertner M.; Delers J.; Enderlin-Paput M.; Weiss M.; Sube R.; Hadana H.; Keck W.; Hubschwerlen C. Novel tetrahydropyran-based bacterial topoisomerase inhibitors with potent anti-gram positive activity and improved safety profile. J. Med. Chem. 2015, 58, 927–942. 10.1021/jm501590q. [DOI] [PubMed] [Google Scholar]
- Kolarič A.; Minovski N. Novel bacterial topoisomerase inhibitors: challenges and perspectives in reducing hERG toxicity. Future Med. Chem. 2018, 10, 2241–2244. 10.4155/fmc-2018-0272. [DOI] [PubMed] [Google Scholar]
- Li L.; Okumu A. A.; Nolan S.; English A.; Vibhute S.; Lu Y.; Hervert-Thomas K.; Seffernick J. T.; Azap L.; Cole S. L.; Shinabarger D.; Koeth L. M.; Lindert S.; Yalowich J. C.; Wozniak D. J.; Mitton-Fry M. J. 1,3-Dioxane-linked bacterial topoisomerase inhibitors with enhanced antibacterial activity and reduced hERGinhibition. ACS Infect. Dis. 2019, 5, 1115–1128. 10.1021/acsinfecdis.8b00375. [DOI] [PubMed] [Google Scholar]
- Cren S.; Friedli A.; Rueedi G.; Zumbrunn C.. Antibacterial Basic Biaromatic Derivatives With Aminoalkoxy Substitution. Patent WO 2016/059097 A1, 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.