

Abstract
Semiconductor nanoplatelets exhibit spectrally pure, directional fluorescence. To make polarized light emission accessible and the charge transport effective, nanoplatelets have to be collectively oriented in the solid state. We discovered that the collective nanoplatelets orientation in monolayers can be controlled kinetically by exploiting the solvent evaporation rate in self-assembly at liquid interfaces. Our method avoids insulating additives such as surfactants, making it ideally suited for optoelectronics. The monolayer films with controlled nanoplatelets orientation (edge-up or face-down) exhibit long-range ordering of transition dipole moments and macroscopically polarized light emission. Furthermore, we unveil that the substantial in-plane electronic coupling between nanoplatelets enables charge transport through a single nanoplatelets monolayer, with an efficiency that strongly depends on the orientation of the nanoplatelets. The ability to kinetically control the assembly of nanoplatelets into ordered monolayers with tunable optical and electronic properties paves the way for new applications in optoelectronic devices.
Keywords: Semiconductor nanoplatelets, self-assembly, orientation control, angle-dependent photoluminescence spectroscopy, terahertz spectroscopy
Quasi-two-dimensional semiconductor nanoplatelets (NPLs) are atomically flat sheets of semiconductor materials.1−6 Across their thickness, NPLs are much smaller than the Bohr radius, while in their lateral dimensions they are much larger. Their limited thickness induces strong one-dimensional quantum confinement, leading to a highly anisotropic electronic structure.7,8 Since the thickness of NPLs can be controlled with atomic precision, they exhibit outstandingly narrow ensemble absorption and emission bands.4,8,9 Due to their anisotropic shape, they exhibit high absorption cross sections,10,11 enhanced energy-transfer rates,12 boosted optical gain,13 and show strong anisotropic light emission.7,14−16 These properties make NPLs ideal candidates for light-emitting devices,17 field-effect transistors,18 solar cells,12 and lasers.19 Here, the overall device efficiency critically depends on the electronic coupling between nanocrystals and thus on the orientation of the transition dipole moments between adjacent nanocrystals in a solid-state film. For isotropic nanocrystals (e.g., quantum dots), aligning the transition dipole moments is a challenging task, because their isotropic shape leads to a stochastic distribution of transition dipole moment orientations.7 However, in highly anisotropic NPLs, the transition dipole moments are aligned in the NPL plane.7,20 As a consequence, ordered NPL assemblies directly result in ordered transition dipole moment orientations, paving the way for improved optoelectronic and photonic device performances.21 Thus, controlling the orientation of NPLs in solid-state is of utmost importance.
Colloidal self-assembly is a powerful tool to control the orientation of building blocks in highly ordered, functional materials.22−26 In solution, CdSe NPLs can be assembled into ordered superstructures such as long stacks by the addition of polar solvents,14,27 ligand exchange,28 or induced depletion attraction forces.29,30 These approaches make use of additives to tune the thermodynamics, that is, the interaction potentials between NPLs, inducing, for example, preferential interactions between the large, planar faces of NPLs. The formation of films has been investigated as well.7,15 The collective orientation of anisotropic nanorods in films can be controlled31 by controlling the thermodynamics during the self-assembly at liquid–air interfaces.32 The Tisdale group has used this method to prepare highly ordered NPL monolayers by self-assembly at a diethylene glycol–hexane interface,15 where the orientation was controlled thermodynamically by varying the amount of oleic acid in the system to tune the interaction potentials between NPLs and the NPL–diethylene glycol interface. The use of such additives prevents efficient charge transport through the NPL layer. Thus, methods that avoid such insulating additives are highly desirable.
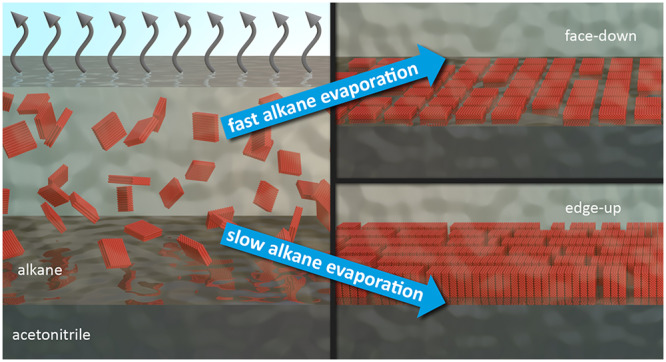
We discovered a mechanism by which the collective NPLs orientation at liquid–liquid interfaces can be controlled kinetically through the evaporation rate of solvents. This allows us to avoid nonvolatile, insulating additives. Both face-down and edge-up NPL assemblies were analyzed by angle-dependent photoluminescence (PL) and terahertz (THz) spectroscopy to study the orientation-dependent collective dipole orientation and the photoconductivity, respectively, of these films. We find that both the edge-up and the face-down assemblies preserve their orientation over an area of at least ∼5 mm2, with an in-plane isotropic dipole distribution for the face-down configuration and an out-of-plane anisotropic dipole distribution for the edge-up configuration, giving rise to macroscopic linearly polarized emission. Our THz spectroscopy experiments reveal that, whereas only excitons are present in isolated NPLs in dispersion, free carriers are observed in the self-assembled films, with limited photoconductivity in the face-down orientation, but remarkably higher photoconductivity in a single layer of NPLs in the edge-up configuration. We thus demonstrate here the ability to collectively orient highly anisotropic nanocrystals at interfaces by an intriguingly simple and effective method, with record long-range order of transition dipole moments and strongly orientation-dependent optical properties and charge mobilities. This previously unknown kinetic self-assembly mechanism of anisotropic nanocrystals opens up new avenues for the orientation of nanocrystals, making their anisotropic properties accessible on the macroscopic scale and thus must be added to our current understanding of collective manipulation of nanoscale building blocks.
Results
Additive-Free Self-Assembly of NPLs
Gao et al. could control the NPL orientation thermodynamically by adding oleic acid to the diethylene glycol subphase.15 Oleic acid acts as a surfactant and alters the interaction potential between the large planar, myristate-passivated facets of the NPLs and the diethylene glycol interface in a concentration-dependent manner.15,33 A large amount of oleic acid increases the interaction potential between the NPL and the diethylene glycol interface leading to face-down assemblies.15 Without or with just a small amount of oleic acid, the inter-NPLs interaction potential is higher, allowing for the edge-up assembly to emerge.15 These observations indicate that the NPLs’ orientation in a dense monolayer film can be controlled by simply tuning the interaction potential between the NPLs and the liquid subphase.
If this holds, we should be able to obtain NPLs in the face-down configuration by increasing the interaction potential between the NPLs and the subphase at the other liquid–liquid interfaces. Varying the interaction potential does not necessarily require surfactants. The choice of the subphase solvent alone can already change the interaction potential significantly.34 To test this hypothesis, we used a modified design of the Teflon well of Gao et al.15 (Figure S1). We replaced diethylene glycol by acetonitrile as subphase and added 4 monolayers (ML) thick CdSe NPLs (Figure 1) dispersed in hexane on top. We chose acetonitrile for several reasons. First, it is more volatile than diethylene glycol, making it easier to remove completely when the monolayers are deposited on substrates. Second, it has a significantly lower polarity index compared to diethylene glycol (0.460 compared to 0.713).35 This matching of polarities explains the increased solubility of the myristate ligand in acetonitrile compared to diethylene glycol.36,37 Therefore, the interaction potential between the flat, nonpolar, myristate-passivated NPL surfaces and the liquid interface should be significantly greater for acetonitrile than for diethylene glycol, as already demonstrate by Erdem et al.34
Figure 1.
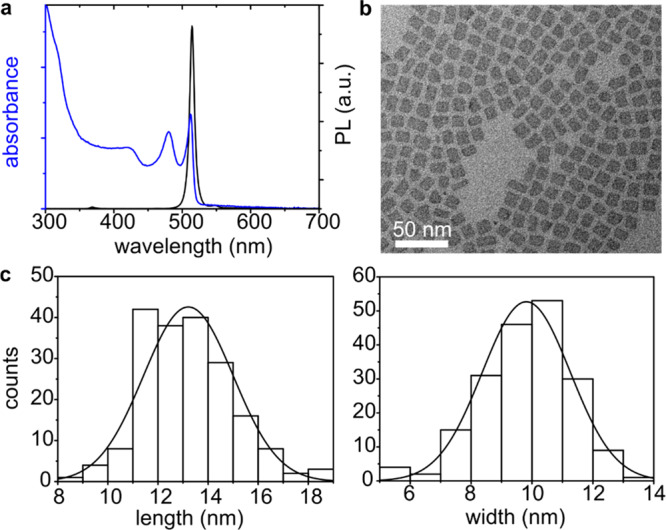
Characterization of the 4 ML CdSe NPLs. (a) Photoluminescence and absorption spectra of 4 ML CdSe NPL dispersed in hexane. (b) TEM micrograph of 4 ML CdSe NPLs facing down. (c) Histograms of the length and width of 4 ML NPLs, ∼200 NPLs were analyzed.
Thus, with acetonitrile as the subphase, we expect to observe the face-down configuration. Indeed, we observed that NPLs assembled face-down when we deposited a hexane NPL dispersion on the acetonitrile subphase and let the hexane evaporate at 20 °C (Figure 2). While this experiment supports our hypothesis that the NPL assembly configuration indeed depends on the interaction potential between the NPLs and the liquid interface, the purity of the NPL dispersion was extremely critical (Figure 2). NPL samples containing octadecene (ODE, the solvent used in NPL syntheses,4Figure S3) in the millimolar regime and of oleic acid in the micromolar regime (used to quench the synthesis; oleic acid could contribute to the ligand shell,38 see Figure S4) resulted in the edge-up configuration (Figure 2) and only painstakingly purified samples (see SI and Figures S3 and S4) that contained no measurable amount of ODE (or oleic acid) reliably deposited in the face-down configuration (Figures 2 and S5). All further experiments were performed using ODE-free samples.
Figure 2.
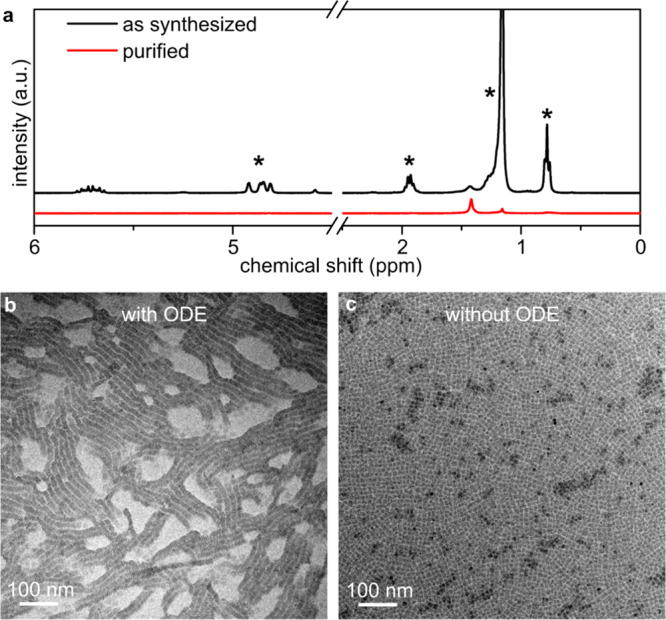
Effect of additional purification on the self-assembly of 4 ML NPLs. (a) NMR spectra of 4 ML NPL before (black line) and after (red line) additional purification. After purification, the peaks assigned to ODE (denoted with *) vanish. (b) TEM micrograph of 4 ML NPLs assembled at the liquid interface in the presence of ODE. The NPLs assembled in the edge-up configuration. (c) TEM micrograph of 4 ML NPLs assembled at the liquid interface in the absence of ODE. The NLPs assembled in the face-down configuration. Clearly, the presence of ODE contamination gave rise to heterogeneity in the assembly process; without ODE, a homogeneous face-down film was produced.
A part of the effect of ODE may be attributable to its activity as depletion agent29,30 in the hexane NPL dispersion, which induces stacking and ultimately leads to discontinuous NPL films in the edge-up configuration (Figure 2). Probably more important, however, is its effect on the kinetics. Kinetic limitations are known to strongly affect particle assembly; they can limit the process and prevail over small differences in interparticle potential.39−41 Here, some time is required for the platelets to form stacks; if evaporation is rapid, the time is insufficient for the stacks to reach a length that stabilizes the edge-up configuration. The low vapor pressure of ODE implies that it accumulates to build up a significant concentration upon the hexane evaporation. Therefore, even a small amount will reduce the evaporation rate of the hexane NPL dispersion. Our experiments indicate that fast evaporation without ODE leads to face-down configurations, while slow evaporation with ODE yields edge-up assembly. If the orientation of NPLs in self-assembled thin films critically depends on the evaporation rate of the solvent, an entirely new and generalizable way to control the collective orientation of NPLs in thin films opens up. However, modifying the evaporation rate by controlling the purity of NPL dispersion precisely is cumbersome. Therefore, we investigate alternative routes that do not rely on impurities such as ODE. Below we show how the collective orientation of NPLs can be kinetically driven and controlled by the solvent vapor pressure in the atmosphere above the self-assembly setup, by choice of the solvent the NPLs are dispersed in, or the temperature at which the assembly is performed.
Kinetically Driven Self-Assembly of NPLs
A straightforward way to modify the solvent evaporation rates is to control its partial pressure above the solution. This ensures that only the evaporation rate is modified and entropic effects, changes in viscosity or chemical effects (e.g., from impurities), can be neglected. To exploit this for our self-assembly experiments, we constructed a setup allowing us to control the alkane vapor content in the atmosphere of a closed system containing our self-assembly well (Figure S2). With high alkane vapor content in the atmosphere, the evaporation rate of the alkane NPL dispersion is reduced. Under these conditions, the NPLs have more time to assemble and form stacks that are long enough to sustain the edge-up configuration. Indeed, self-assembly experiments with heptane NPL dispersions performed with a high content of heptane vapor in the atmosphere reliably yielded edge-up assemblies (Figure S6). The self-assemblies performed under pure airflow (high evaporation rate) resulted in the face-down configuration instead (Figure S6). This experiment proves unambiguously that the collective orientation of NPLs in self-assemblies can be controlled using solely the solvent evaporation rate. However, to generalize this concept, we investigated ways to systematically change the evaporation rate using a simpler setup.
To do this, we performed a series of NPL self-assembly experiments on acetonitrile interfaces at 20 °C under ambient conditions, where we systematically controlled the vapor pressure (and thus the evaporation rate) by changing the NPL solvent from hexane, to heptane, to octane, keeping all other parameters constant. We observed distinct differences in NPL orientations depending on the type of solvent we used (Figure 3a). For NPLs deposited from hexane, NPLs assemble in the face-down configuration, and when deposited from octane, NPLs assembled in the edge-up configuration. Deposition from heptane resulted in a mix of face-down and edge-up configuration. Since the vapor pressure (at 20 °C) decreases rapidly from hexane (162 hPa), heptane (47 hPa), and octane (14 hPa),35 the measured evaporation rate decreased accordingly (Figure S7), and the time for deposition and organization at the acetonitrile interface systematically increased (Table S2). Therefore, the collective NPL orientation can be controlled kinetically by adjusting the evaporation rate through the choice of solvent. To generalize this further, we varied the evaporation rate by changing the temperature at which the self-assembly is performed.
Figure 3.

Evaporation driven self-assembly of 4 ML thick NPLs. (a) By changing the solvent from hexane to heptane to octane (systematically lowering vapor pressure) at 20 °C, the assembly can be turned from the face-down configuration (left) through a mixture (middle) to the edge-up configuration (right). (b) TEM micrographs of NPL assemblies obtained at 5 °C; the configuration was shifted to the edge-up assembly.
By decreasing the temperature, the evaporation rate of the NPL dispersion was reduced. Thus, the time which the system had to self-assemble was prolonged, and the edge-up configurations should be facilitated. To test this hypothesis, we repeated the experiments shown in Figure 3a at 5 °C. Indeed, we observed that reducing the temperature leads exclusively to the edge-up configuration for both octane and heptane (Figure 3b). For hexane, mixed assemblies with both edge-up and face-down configurations were formed. Since at 5 °C, the alkanes evaporate at a lower rate (Table S3), and the NPLs have more time to arrange, favoring edge-up configurations. Notably, the evaporation rate decreases quickly with temperature due to the exponential relationship between temperature and partial pressure, while the diffusivity only decreases linearly with temperature.42 These data furthermore strengthen our interpretations that the evaporation rate of the solvents, and thus the time the system has to equilibrate, ultimately determines the orientation of NPLs in the self-assemblies.
Mechanism of Kinetically Driven Self-Assemblies
To gain deeper insights into the process, we performed small-angle X-ray scattering (SAXS) experiments on hanging droplets of an evaporating NPL dispersion while concurrently measuring particle concentration.43 Similar to what has been observed for sterically stabilized gold nanocrystals, we found a rapid onset of agglomerative stacking, visible by the evolution of an anisotropic X-ray scattering pattern (Figure S8) when the NPL concentration exceeded a critical concentration of 4.3 μmol/L. This concentration was reached in the liquid film of our self-assembly process when the height of the NPL-containing alkane layer was ∼3 μm thick (∼0.5 μL). Thus, agglomerative stacking can occur before the alkane fully evaporates, but it depends on the evaporation rate whether there is enough time for the particles to arrange (Figure S9, Table S4). The time that the system has until complete evaporation beyond this critical filling height (volume), therefore, determines the orientation of the NPLs in the film (Figure 4a, Table S5).
Figure 4.
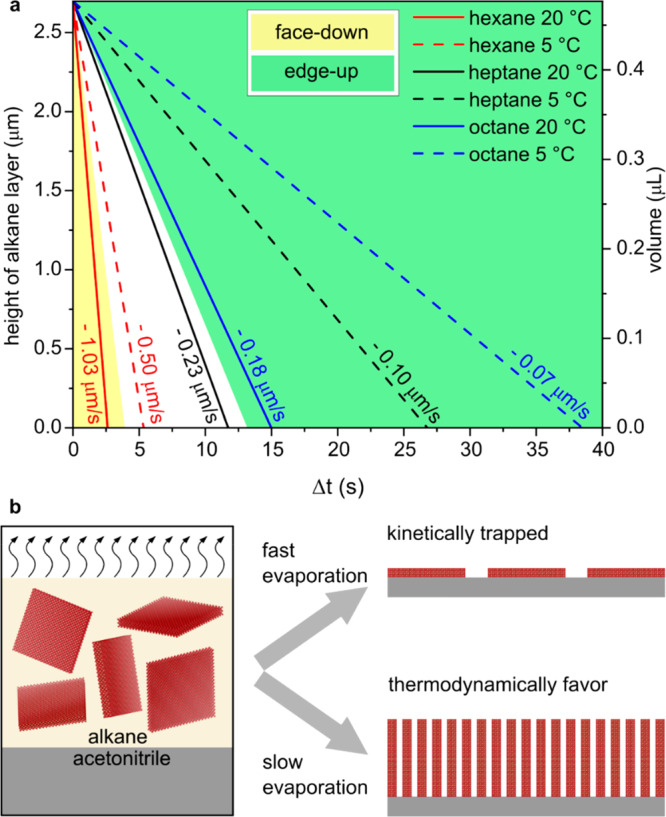
Evaporation rate determines the orientation of NPLs. (a) The height of alkane layer/volume versus the time until complete evaporation after reaching the critical concentration [4.3 μmol/L, reached at alkane layer heights of 2.7 μm (volumes of 0.48 μL)]. Three regions (face-down, intermediate region and edge-up) can be identified. (b) Scheme of kinetically driven self-assembly. Fast evaporation leads to the kinetically trapped product, the face-down assembly. Slow evaporation leads to the thermodynamically favored product, the edge-up assembly.
Our analysis of the distances between adjacent NPLs in the self-assembled films gave us some hints about the origin of the kinetically driven self-assembly mechanism. The distance between the NPLs in the assemblies (Figure S10 a-b) was significantly shorter in the edge-up configuration (2.5 ± 0.45 nm) than in the face-down configuration (2.9 ± 0.71 nm). Hence, in the edge-up configuration, the inter-NPL distance was significantly shorter than the expected distance of 3.4 nm between adjacent myristate-passivated NPLs. Thus, the NPLs ligand shells experienced a significant amount of interpenetration. This requires the rearrangement and intercalation of a large number of myristate molecules on the NPL surfaces. Adopting this energetically favorable conformation requires a certain amount of time.44 Our experiments conclusively show that slow evaporation of the NPL dispersion gives the system enough time to adopt the thermodynamically favored edge-up configuration. In contrast, upon fast evaporation, the NPLs become kinetically trapped by the quickly moving alkane solvent front45 and deposit in the face-down configuration at the acetonitrile interface. Thus, we are able to trap the transition from the face-down to the edge-up configuration at different states (fully face-down, fully edge-up, and anything in between). This means that we are not limited to one trapped state, but we can actually tune which fraction is in the face-down and edge-up configuration (Figure 3). Notably, NPLs adapted a well-defined collective orientation even when kinetically trapped by fast evaporation, which is in contrast to the glassy states obtained in previous works under similar conditions with nanorods.31 Thus, our self-assembly method yields a well-defined monolayer under kinetic and under thermodynamic control. Both the edge-up and the face-down configurations were maintained even if the NPL concentration or the NPL dispersion volume was varied (Table S6 and Figures S11–S13), that is, even if the maximum possible surface coverage of NPLs was much lower than the total acetonitrile surface area. This further validates our proposed mechanism (Figure 4b).
Angle-Dependent PL of NPL Self-Assemblies
To assess the long-range order of our assemblies, we studied net transition dipole moment orientations by angle-dependent PL spectroscopy (spot size ∼5 mm2, see SI Scheme S1). Figure 5a shows the s and p radial intensity of the face-down assembly at detection angles ranging from −80° to 80° with respect to the substrate normal, and their respective fit curves obtained by optical simulations (see SI). The fitting indicates that on the face-down sample, the dipole orientation θ is preferentially parallel to the substrate plane (θ = 0.16).
Figure 5.

Angle-resolved PL, polarized emission and ultrafast THz conductivity dynamics for NPL self-assemblies. (a, b) Angle-resolved PL of a face-down (a) and a edge-up (b) assembly. The s (green) and p (blue/purple) radial intensity with their fits (solid green and blue/purple line) are displayed. For comparison also the p-polarized curve corresponding to an isotropic orientation of the dipoles (θ = 1/3, gray curve) is displayed. (c) Polarization dependence of the PL for face-down and edge-up assemblies at a fixed detection angle of 0° with respect to the substrate normal. (d–f) Ultrafast THz conductivities for NPLs dispersed in (d) hexane and assembled in (e) face-down and (f) edge-up configurations. The solid (dashed) lines represent the real (imaginary) part of the complex conductivity.
This proves that the in-plane arrangement of the platelets observed under the TEM is maintained at least over the area of the entire spot size (∼5 mm2), highlighting the uniformity of the self-assembly. Note that the fit model assumes an in-plane isotropic orientation of the dipoles. This is the case for face-down samples as evidenced by the TEM Fourier transform analysis (Figure S14). On the other hand, the shape of the p-polarized curve of the edge-up assemblies (Figure 5b) differs from the face-down characteristics (Figure 5a), reflecting a different arrangement of the dipoles with respect to the face-down configuration. We can derive a dipole orientation of θ = 0.56, indicating that dipoles are preferentially oriented perpendicular to the substrate normal. The discrepancy between experimental data and simulations for the edge-up assemblies stems from the preferential orientation of NPLs along a specific in-plane direction (direction of stacking), in line with our TEM Fourier analysis (Figure S14). This should give rise to macroscopically observable linear light polarization. We investigated this by collecting the PL signals of face-down and edge-up assemblies at a fixed detection angle (0° with respect to the normal) and varying the in-plane polarization angle of the detector (Figure 5c). For the face-down assembly, no periodical variation of the signal was observed, as expected for isotopically distributed dipoles. In striking contrast, a periodical oscillation of the PL signal was found for the edge-up sample. Clearly, the preferential NPL stack orientation is maintained over the spot size area of ∼5 mm2, resulting in the anisotropic in-plane PL signal. Thus, edge-up NPL films could have interesting applications in optoelectronics, such as the emerging field of polarized light-emitting diodes.46 To integrate confined nanomaterials into optoelectronic devices requires efficient electronic coupling between the building blocks, so that charge carriers can be efficiently extracted from (or injected into) the nanomaterials. Given the anisotropy of NPLs and their assemblies, we investigate the orientation-dependent photoconductivity of face-down and edge up-assemblies.
Photoconductivity of NPL Self-Assemblies
In principle, the coupling strength in NPL solids depends strongly on the degree of wave function overlap between neighboring NPLs: the shorter the NPL-NPL distance, the higher the electronic coupling.47 The inter-NPLs distance in the edge-up (2.5 ± 0.45 nm) was shorter than in the face-down configuration (2.9 ± 0.71 nm, Figures S10 and S14). Along with the edge-to-edge distance, other parameters including the total NPL–NPL interaction area (larger for edge-up than for face-down) and the dipole–dipole interactions between NPLs play also critical roles on the inter-NPL coupling strength and thus charge transport dynamics in NPL samples. To investigate and compare the coupling strength between samples, we measured PL of NPLs in dispersion and in assemblies (on borosilicate glass substrates, self-assembly performed with octane at 5 °C, overnight temperature equilibration of equipment). The edge-up and face-down assemblies both displayed broader PL spectra than NPLs in dispersion (Figure S15), illustrating the enhanced coupling strength in NPL assemblies. The edge-up samples showed slightly broader and red-shifted emission compared to the face-down case (Figure S15 and Table S7). This redshift was larger than that expected from the different dielectric environments in dispersion and particle assemblies on borosilicate and thus indicates stronger coupling between NPLs for the edge-up configuration.
To further investigate the role of coupling strength for charge transport properties, we measured the photoconductivity on well-defined NPL assemblies (for AFM micrographs with height profile see Figure S16), employing time-resolved, contact-free optical pump-THz probe (OPTP) spectroscopy: A well-established tool for characterizing the nature (exciton or free carrier) and transport properties of photogenerated charge carriers in bulk or low dimensional, quantum-confined semiconductors48 (see methods section and SI). Here, we compare the THz photoconductivity in three different samples: dispersed (isolated) NPLs in hexane and NPLs in the face-down and edge-up assemblies (Figure 5d–f). The signals are normalized to the absorbed photodensity (i.e., photogenerated electron–hole pairs) and can, therefore, be compared quantitatively. For isolated NPLs dispersed in hexane, the THz response is purely excitonic (Figure 5d), as evident from the purely imaginary THz photoconductivity. This observation of stable exciton states at room temperature in our NPLs indicates an intrinsically strong Coulomb interaction between photogenerated electrons and holes, leading to a large exciton binding energy Eb beyond the thermal fluctuation (∼ 26 meV), in line with previous spectroscopic results (with Eb∼ 170 meV).49 For NPL assemblies, the electronic coupling between NPLs allows for charge transport to occur: Charge carriers in the assemblies are sufficiently mobile to give rise to a decent THz conductivity (Figure 5e,f). Here we propose two plausible scenarios accounting for the free charge carrier generation in the assemblies: (1) in a more “compact” configuration, the charge carriers are screened better, resulting in a reduction of the exciton binding energy50 and thus an increase in the fraction of free charges. Note that a decrease in exciton binding energy is not necessarily evident from the PL.51 (2) With enhancing inter-NPL coupling strength (donor/acceptor wave function overlap), the inter-NPL charge transfer should increase. As a result, the efficiency of free charge generation by exciton dissociation is expected to increase, and free charge carriers can become delocalized. The THz photoconductivities in NPL samples (Figure 5d–f) agree well with these considerations: We observe a clear transition from pure excitonic response for isolated NPLs in dispersion to free charge carrier response in the edge-up sample as a result of enhanced NPL coupling (with the face-down as the intermediate situation). To support our experimental observations, we have further measured the frequency-resolved THz conductivity for NPLs in dispersion and in the edge-up case. For NPLs dispersed in hexane, in line with pure excitonic picture based on the dynamics shown in Figure 5d, we observe that the complex conductivity is dominated by its imaginary component (see Figure S17a). More importantly, the complex conductivity can be well fitted by a Lorentzian resonance, which has been well-established for describing intraexcitonic transitions (e.g., 1S-2P) in the excitonic material systems.52,53 For NPLs in the edge-up configuration, on the other hand, we observed a free carrier dominant conductivity which can be described and fitted by a modified Drude model (see Figure S16b and associated discussion related to Drude–Smith model). Finally, in the normalized THz conductivity dynamics, the decay time for the face-down assembly is found to be much shorter than that of the edge-up (Figure S18). It is worth pointing out that in our THz measurements, we probe the photoconductivity, which is proportional to the product of photogenerated carrier density and carrier mobility (both of which can be time-dependent). In our study, the UV–vis absorption spectra of the two NPL solids was nearly identical, and the pump fluence was set to be the same. As such, we made sure that the initial photogenerated carrier densities were the same, and therefore the fast decay in the face-down sample can be attributed to a transition of some (if not all) of the free charges into the exciton states. During the formation of an “insulating” exciton gas, the absolute value for both the real and imaginary photoconductivity is expected to go down (which is in line with experimental result). The exciton formation picture is further supported by observation of a long-lived imaginary and zero real conductivity (Figure 5e) following the fast decays in the dynamics, which has been reported for other excitonic systems including semiconducting polymer and graphene nanoribbons.54−56 The long-lived (>10 ps) lifetime of the free conductivity in the edge-up NPLs indicates the effective separation of electrons and holes in the assembly.
Discussions and Conclusion
We discovered a method allowing for kinetic control over the collective orientation of NPLs in self-assembled monolayers. This simple and effective strategy opens up new ways to control the collective orientation of anisotropic, nanoscale building blocks in solid-state superstructures, making their anisotropic and orientation-dependent properties accessible. By adjusting the evaporation rate of NPL dispersion, either by choice of solvent, temperature, or solvent partial pressure, NPL films in the face-down or edge-up configuration can be obtained. These assemblies show long-range order of the individual NPL transition dipole moments over at least ∼5 mm2, with the edge-up assemblies possessing linearly polarized light emission due to the hierarchical stacking in the self-assembled monolayers. Therefore, these NPL assemblies could be useful to create novel optoelectronic devices, such as polarized light-emitting diodes by bottom-up approaches. Since our method does not rely on nonvolatile additives that could influence charge carrier transport, it is ideally suited for optoelectronic applications. Employing THz spectroscopy, we demonstrate a clear orientation-dependent photoconductivity in self-assembled NPL solids. We unveil that such dependence originates from the different inter-NPL electronic coupling strength. We observed a transition from the excitonic response in dispersed NPLs (with the lowest inter-NPL coupling) to quasi-free carriers in the edge-up assembly (with the highest inter-NPL coupling among our samples) by enhancing the coupling strength between the NPLs.
Acknowledgments
The authors are grateful for the financial support of the Max Planck Center for Complex Fluid Dynamics. H.H. was supported by a fellowship from the Max Planck Graduate Center (MPGC), Mainz, Germany. We thank Ingo Lieberwirth and Gunnar Glasser for help with electron microscopy and Shuai Fu for his contributions to the THz measurements. We thank Hans-Jürgen Butt and Michael Kappl for discussions and for their help with the partial pressure control setup. We thank Katharina Landfester, Markus Mezger, Enrique Canovas, Maksim Grechko, and Kaloian Koynov for the fruitful discussions.
Glossary
Abbreviations
- NPLs
quasi-two-dimensional semiconductor nanoplatelets
- THz
terahertz
- ML
monolayer
- ODE
octadecene
- SAXS
small-angle X-ray scattering
- PL
photoluminescence
- TEM
transmission electron microscope
- OPTP
optical pump-THz probe
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.nanolett.9b05270.
Methods, syntheses, purification protocols, additional analyses, and supporting data (PDF)
Author Contributions
R.M. and A.R. conceived the project. Synthesis, self-assembly, optical characterization, evaporation rate determination and microscopies were performed by R.M., H.H., E.J., and S.Y. SAXS experiments and data analysis were done by D.D. and T.K. Angle-dependent PL measurements and fitting of the data were performed by D.B. and B.B. THz measurements and data analysis were done by H.Z., S.C., M.B., and H.I.W. R.M., B.B., H.I.W, and A.R. wrote the manuscript. All authors contributed to the discussion of the results and to the revision of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Liu Y.-H.; Wang F.; Wang Y.; Gibbons P. C.; Buhro W. E. Lamellar assembly of cadmium selenide nanoclusters into quantum belts. J. Am. Chem. Soc. 2011, 133 (42), 17005–17013. 10.1021/ja206776g. [DOI] [PubMed] [Google Scholar]
- Schliehe C.; Juarez B. H.; Pelletier M.; Jander S.; Greshnykh D.; Nagel M.; Meyer A.; Foerster S.; Kornowski A.; Klinke C.; Weller H. Ultrathin PbS sheets by two-dimensional oriented attachment. Science 2010, 329 (5991), 550–553. 10.1126/science.1188035. [DOI] [PubMed] [Google Scholar]
- Joo J.; Son J. S.; Kwon S. G.; Yu J. H.; Hyeon T. Low-temperature solution-phase synthesis of quantum well structured CdSe nanoribbons. J. Am. Chem. Soc. 2006, 128 (17), 5632–5633. 10.1021/ja0601686. [DOI] [PubMed] [Google Scholar]
- Ithurria S.; Dubertret B. Quasi 2D Colloidal CdSe Platelets with Thicknesses Controlled at the Atomic Level. J. Am. Chem. Soc. 2008, 130 (49), 16504–16505. 10.1021/ja807724e. [DOI] [PubMed] [Google Scholar]
- Ott F. D.; Riedinger A.; Ochsenbein D. R.; Knüsel P. N.; Erwin S. C.; Mazzotti M.; Norris D. J. Ripening of Semiconductor Nanoplatelets. Nano Lett. 2017, 17 (11), 6870–6877. 10.1021/acs.nanolett.7b03191. [DOI] [PubMed] [Google Scholar]
- Riedinger A.; Ott F. D.; Mule A.; Mazzotti S.; Knüsel P. N.; Kress S. J. P.; Prins F.; Erwin S. C.; Norris D. J. An intrinsic growth instability in isotropic materials leads to quasi-two-dimensional nanoplatelets. Nat. Mater. 2017, 16, 743. 10.1038/nmat4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R.; Heckmann J.; Prudnikau A. V.; Antanovich A.; Mikhailov A.; Owschimikow N.; Artemyev M.; Climente J. I.; Woggon U.; Grosse N. B.; Achtstein A. W. Directed emission of CdSe nanoplatelets originating from strongly anisotropic 2D electronic structure. Nat. Nanotechnol. 2017, 12, 1155. 10.1038/nnano.2017.177. [DOI] [PubMed] [Google Scholar]
- Ithurria S.; Tessier M. D.; Mahler B.; Lobo R. P. S. M.; Dubertret B.; Efros A. L. Colloidal nanoplatelets with two-dimensional electronic structure. Nat. Mater. 2011, 10, 936. 10.1038/nmat3145. [DOI] [PubMed] [Google Scholar]
- Tessier M. D.; Mahler B.; Nadal B.; Heuclin H.; Pedetti S.; Dubertret B. Spectroscopy of Colloidal Semiconductor Core/Shell Nanoplatelets with High Quantum Yield. Nano Lett. 2013, 13 (7), 3321–3328. 10.1021/nl401538n. [DOI] [PubMed] [Google Scholar]
- Yeltik A.; Delikanli S.; Olutas M.; Kelestemur Y.; Guzelturk B.; Demir H. V. Experimental Determination of the Absorption Cross-Section and Molar Extinction Coefficient of Colloidal CdSe Nanoplatelets. J. Phys. Chem. C 2015, 119 (47), 26768–26775. 10.1021/acs.jpcc.5b09275. [DOI] [Google Scholar]
- Achtstein A. W.; Antanovich A.; Prudnikau A.; Scott R.; Woggon U.; Artemyev M. Linear Absorption in CdSe Nanoplates: The Thickness and Lateral Size Dependency of the Intrinsic Absorption. J. Phys. Chem. C 2015, 119 (34), 20156–20161. 10.1021/acs.jpcc.5b06208. [DOI] [Google Scholar]
- Rowland C. E.; Fedin I.; Zhang H.; Gray S. K.; Govorov A. O.; Talapin D. V.; Schaller R. D. Picosecond energy transfer and multiexciton transfer outpaces Auger recombination in binary CdSe nanoplatelet solids. Nat. Mater. 2015, 14 (5), 484–489. 10.1038/nmat4231. [DOI] [PubMed] [Google Scholar]
- She C.; Fedin I.; Dolzhnikov D. S.; Demortière A.; Schaller R. D.; Pelton M.; Talapin D. V. Low-Threshold Stimulated Emission Using Colloidal Quantum Wells. Nano Lett. 2014, 14 (5), 2772–2777. 10.1021/nl500775p. [DOI] [PubMed] [Google Scholar]
- Abécassis B.; Tessier M. D.; Davidson P.; Dubertret B. Self-Assembly of CdSe Nanoplatelets into Giant Micrometer-Scale Needles Emitting Polarized Light. Nano Lett. 2014, 14 (2), 710–715. 10.1021/nl4039746. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Weidman M. C.; Tisdale W. A. CdSe Nanoplatelet Films with Controlled Orientation of their Transition Dipole Moment. Nano Lett. 2017, 17 (6), 3837–3843. 10.1021/acs.nanolett.7b01237. [DOI] [PubMed] [Google Scholar]
- Ma X.; Diroll B. T.; Cho W.; Fedin I.; Schaller R. D.; Talapin D. V.; Wiederrecht G. P. Anisotropic Photoluminescence from Isotropic Optical Transition Dipoles in Semiconductor Nanoplatelets. Nano Lett. 2018, 18 (8), 4647–4652. 10.1021/acs.nanolett.8b00347. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Nadal B.; Mahler B.; Aubin H.; Dubertret B. Quasi-2D Colloidal Semiconductor Nanoplatelets for Narrow Electroluminescence. Adv. Funct. Mater. 2014, 24 (3), 295–302. 10.1002/adfm.201301711. [DOI] [Google Scholar]
- Lhuillier E.; Pedetti S.; Ithurria S.; Heuclin H.; Nadal B.; Robin A.; Patriarche G.; Lequeux N.; Dubertret B. Electrolyte-Gated Field Effect Transistor to Probe the Surface Defects and Morphology in Films of Thick CdSe Colloidal Nanoplatelets. ACS Nano 2014, 8 (4), 3813–3820. 10.1021/nn500538n. [DOI] [PubMed] [Google Scholar]
- Grim J. Q.; Christodoulou S.; Di Stasio F.; Krahne R.; Cingolani R.; Manna L.; Moreels I. Continuous-wave biexciton lasing at room temperature using solution-processed quantum wells. Nat. Nanotechnol. 2014, 9 (11), 891–895. 10.1038/nnano.2014.213. [DOI] [PubMed] [Google Scholar]
- Yoon D.-E.; Kim W. D.; Kim D.; Lee D.; Koh S.; Bae W. K.; Lee D. C. Origin of Shape-Dependent Fluorescence Polarization from CdSe Nanoplatelets. J. Phys. Chem. C 2017, 121 (44), 24837–24844. 10.1021/acs.jpcc.7b07216. [DOI] [Google Scholar]
- Antanovich A.; Prudnikau A.; Artemyev M. Anisotropy of Structure and Optical Properties of Self-Assembled and Oriented Colloidal CdSe Nanoplatelets. Z. Phys. Chem. 2018, 232 (9–11), 1619. 10.1515/zpch-2018-1131. [DOI] [Google Scholar]
- Boles M. A.; Engel M.; Talapin D. V. Self-Assembly of Colloidal Nanocrystals: From Intricate Structures to Functional Materials. Chem. Rev. 2016, 116 (18), 11220–11289. 10.1021/acs.chemrev.6b00196. [DOI] [PubMed] [Google Scholar]
- Thorkelsson K.; Bai P.; Xu T. Self-assembly and applications of anisotropic nanomaterials: A review. Nano Today 2015, 10 (1), 48–66. 10.1016/j.nantod.2014.12.005. [DOI] [Google Scholar]
- Reichhelm A.; Haubold D.; Eychmuller A. Ligand Versatility in Supercrystal Formation. Adv. Funct. Mater. 2017, 27 (39), 1700361–1700361. 10.1002/adfm.201700361. [DOI] [Google Scholar]
- Boneschanscher M. P.; Evers W. H.; Geuchies J. J.; Altantzis T.; Goris B.; Rabouw F. T.; van Rossum S. A. P.; van der Zant H. S. J.; Siebbeles L. D. A.; Van Tendeloo G.; Swart I.; Hilhorst J.; Petukhov A. V.; Bals S.; Vanmaekelbergh D. Long-range orientation and atomic attachment of nanocrystals in 2D honeycomb superlattices. Science 2014, 344 (6190), 1377–1380. 10.1126/science.1252642. [DOI] [PubMed] [Google Scholar]
- Barrow S. J.; Funston A. M.; Wei X. Z.; Mulvaney P. DNA-directed self-assembly and optical properties of discrete 1D, 2D and 3D plasmonic structures. Nano Today 2013, 8 (2), 138–167. 10.1016/j.nantod.2013.02.005. [DOI] [Google Scholar]
- Guzelturk B.; Erdem O.; Olutas M.; Kelestemur Y.; Demir H. V. Stacking in Colloidal Nanoplatelets: Tuning Excitonic Properties. ACS Nano 2014, 8 (12), 12524–12533. 10.1021/nn5053734. [DOI] [PubMed] [Google Scholar]
- Antanovich A.; Prudnikau A.; Matsukovich A.; Achtstein A.; Artemyev M. Self-Assembly of CdSe Nanoplatelets into Stacks of Controlled Size Induced by Ligand Exchange. J. Phys. Chem. C 2016, 120 (10), 5764–5775. 10.1021/acs.jpcc.5b12139. [DOI] [Google Scholar]
- Jana S.; Davidson P.; Abécassis B. CdSe Nanoplatelets: Living Polymers. Angew. Chem., Int. Ed. 2016, 55 (32), 9371–9374. 10.1002/anie.201603880. [DOI] [PubMed] [Google Scholar]
- Jana S.; Phan T. N. T.; Bouet C.; Tessier M. D.; Davidson P.; Dubertret B.; Abécassis B. Stacking and Colloidal Stability of CdSe Nanoplatelets. Langmuir 2015, 31 (38), 10532–10539. 10.1021/acs.langmuir.5b02152. [DOI] [PubMed] [Google Scholar]
- Diroll B. T.; Greybush N. J.; Kagan C. R.; Murray C. B. Smectic Nanorod Superlattices Assembled on Liquid Subphases: Structure, Orientation, Defects, and Optical Polarization. Chem. Mater. 2015, 27 (8), 2998–3008. 10.1021/acs.chemmater.5b00355. [DOI] [Google Scholar]
- Dong A.; Chen J.; Vora P. M.; Kikkawa J. M.; Murray C. B. Binary nanocrystal superlattice membranes self-assembled at the liquid–air interface. Nature 2010, 466, 474. 10.1038/nature09188. [DOI] [PubMed] [Google Scholar]
- Paik T.; Ko D.-K.; Gordon T. R.; Doan-Nguyen V.; Murray C. B. Studies of liquid crystalline self-assembly of GdF3 nanoplates by in-plane, out-of-plane SAXS. ACS Nano 2011, 5 (10), 8322–8330. 10.1021/nn203049t. [DOI] [PubMed] [Google Scholar]
- Erdem O.; Gungor K.; Guzelturk B.; Tanriover I.; Sak M.; Olutas M.; Dede D.; Kelestemur Y.; Demir H. V. Orientation-Controlled Nonradiative Energy Transfer to Colloidal Nanoplatelets: Engineering Dipole Orientation Factor. Nano Lett. 2019, 19 (7), 4297–4305. 10.1021/acs.nanolett.9b00681. [DOI] [PubMed] [Google Scholar]
- Reichert C.; Welton T.. Solvents and solvent effects in organic chemistry, 3rd ed.; Wiley-VCH Publishers: Weinheim, 2003; Vol. 15, p 2014. [Google Scholar]
- Sedgwick R. S.; Hoerr C. W.; Harwood H. J. Solubilities of Saturated Fatty Acid Ester. J. Org. Chem. 1952, 17 (2), 327–337. 10.1021/jo01136a023. [DOI] [Google Scholar]
- Calvo B.; Collado I.; Cepeda E. A. Solubilities of Palmitic Acid in Pure Solvents and Its Mixtures. J. Chem. Eng. Data 2009, 54 (1), 64–68. 10.1021/je8005979. [DOI] [Google Scholar]
- Singh S.; Tomar R.; ten Brinck S.; De Roo J.; Geiregat P.; Martins J. C.; Infante I.; Hens Z. Colloidal CdSe Nanoplatelets, A Model for Surface Chemistry/Optoelectronic Property Relations in Semiconductor Nanocrystals. J. Am. Chem. Soc. 2018, 140 (41), 13292–13300. 10.1021/jacs.8b07566. [DOI] [PubMed] [Google Scholar]
- Mann S. Self-assembly and transformation of hybrid nano-objects and nanostructures under equilibrium and non-equilibrium conditions. Nat. Mater. 2009, 8 (10), 781–792. 10.1038/nmat2496. [DOI] [PubMed] [Google Scholar]
- Xia Y. S.; Nguyen T. D.; Yang M.; Lee B.; Santos A.; Podsiadlo P.; Tang Z. Y.; Glotzer S. C.; Kotov N. A. Self-assembly of self-limiting monodisperse supraparticles from polydisperse nanoparticles. Nat. Nanotechnol. 2011, 6 (9), 580–587. 10.1038/nnano.2011.121. [DOI] [PubMed] [Google Scholar]
- Ramsden J. J. The Stability of Superspheres. Proc. R. Soc. London, Ser. A 1987, 413 (1845), 407–414. 10.1098/rspa.1987.0122. [DOI] [Google Scholar]
- Born P.; Munoz A.; Cavelius C.; Kraus T. Crystallization Mechanisms in Convective Particle Assembly. Langmuir 2012, 28 (22), 8300–8308. 10.1021/la2048618. [DOI] [PubMed] [Google Scholar]
- Doblas D.; Kister T.; Cano-Bonilla M.; Gonzalez-Gariia L.; Kraus T. Colloidal Solubility and Agglomeration of Apolar Nanoparticles in Different Solvents. Nano Lett. 2019, 19 (8), 5246–5252. 10.1021/acs.nanolett.9b01688. [DOI] [PubMed] [Google Scholar]
- Napper D.; Steric H. stabilization. J. Colloid Interface Sci. 1977, 58 (2), 390–407. 10.1016/0021-9797(77)90150-3. [DOI] [Google Scholar]
- Bigioni T. P.; Lin X.-M.; Nguyen T. T.; Corwin E. I.; Witten T. A.; Jaeger H. M. Kinetically driven self assembly of highly ordered nanoparticle monolayers. Nat. Mater. 2006, 5, 265. 10.1038/nmat1611. [DOI] [PubMed] [Google Scholar]
- Matioli E.; Brinkley S.; Kelchner K. M.; Hu Y. L.; Nakamura S.; DenBaars S.; Speck J.; Weisbuch C. High-brightness polarized light-emitting diodes. Light: Sci. Appl. 2012, 1, e22 10.1038/lsa.2012.22. [DOI] [Google Scholar]
- Tisdale W. A.; Zhu X. Y. Artificial atoms on semiconductor surfaces. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (3), 965. 10.1073/pnas.1006665107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbricht R.; Hendry E.; Shan J.; Heinz T. F.; Bonn M. Carrier dynamics in semiconductors studied with time-resolved terahertz spectroscopy. Rev. Mod. Phys. 2011, 83 (2), 543–586. 10.1103/RevModPhys.83.543. [DOI] [Google Scholar]
- Scott R.; Achtstein A. W.; Prudnikau A. V.; Antanovich A.; Siebbeles L. D. A.; Artemyev M.; Woggon U. Time-Resolved Stark Spectroscopy in CdSe Nanoplatelets: Exciton Binding Energy, Polarizability, and Field-Dependent Radiative Rates. Nano Lett. 2016, 16 (10), 6576–6583. 10.1021/acs.nanolett.6b03244. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Ling X.; Yu L.; Huang S.; Hsu A. L.; Lee Y.-H.; Kong J.; Dresselhaus M. S.; Palacios T. Dielectric Screening of Excitons and Trions in Single-Layer MoS2. Nano Lett. 2014, 14 (10), 5569–5576. 10.1021/nl501988y. [DOI] [PubMed] [Google Scholar]
- Komsa H.-P.; Krasheninnikov A. V. Effects of confinement and environment on the electronic structure and exciton binding energy of MoS${}_{2}$ from first principles. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 86 (24), 241201. 10.1103/PhysRevB.86.241201. [DOI] [Google Scholar]
- Tries A.; Osella S.; Zhang P.; Xu F.; Kläui M.; Mai Y.; Beljonne D.; Wang H. I.. Gigantic Binding Energy and Ultrafast Formation of Excitons in Graphene Nanoribbons. arXiv (Mesoscale and Nanoscale Physics), https://arxiv.org/abs/1911.04431 (accessed 2019-11-21).
- Kaindl R. A.; Hägele D.; Carnahan M. A.; Chemla D. S. Transient terahertz spectroscopy of excitons and unbound carriers in quasi-two-dimensional electron-hole gases. Phys. Rev. B: Condens. Matter Mater. Phys. 2009, 79 (4), 045320 10.1103/PhysRevB.79.045320. [DOI] [Google Scholar]
- Chen Z.; Wang H. I.; Teyssandier J.; Mali K. S.; Dumslaff T.; Ivanov I.; Zhang W.; Ruffieux P.; Fasel R.; Räder H. J.; Turchinovich D.; De Feyter S.; Feng X.; Kläui M.; Narita A.; Bonn M.; Müllen K. Chemical Vapor Deposition Synthesis and Terahertz Photoconductivity of Low-Band-Gap N = 9 Armchair Graphene Nanoribbons. J. Am. Chem. Soc. 2017, 139 (10), 3635–3638. 10.1021/jacs.7b00776. [DOI] [PubMed] [Google Scholar]
- Hendry E.; Schins J. M.; Candeias L. P.; Siebbeles L. D. A.; Bonn M. Efficiency of Exciton and Charge Carrier Photogeneration in a Semiconducting Polymer. Phys. Rev. Lett. 2004, 92 (19), 196601. 10.1103/PhysRevLett.92.196601. [DOI] [PubMed] [Google Scholar]
- Ivanov I.; Hu Y.; Osella S.; Beser U.; Wang H. I.; Beljonne D.; Narita A.; Müllen K.; Turchinovich D.; Bonn M. Role of Edge Engineering in Photoconductivity of Graphene Nanoribbons. J. Am. Chem. Soc. 2017, 139 (23), 7982–7988. 10.1021/jacs.7b03467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.