

Abstract
Obstructive sleep apnea (OSA) is a common chronic disease, and is associated with high social and economic costs. OSA is heritable, and there is evidence of both direct genetic contributions to OSA susceptibility and indirect contributions via ‘intermediate’ phenotypes such as obesity, craniofacial structure, neurological control of upper airway muscles, and of sleep and circadian rhythm. Investigation of the genetics of OSA is an important research area, and may lead to improved understanding of disease etiology, pathogenesis, adverse health consequences, and new preventive strategies and treatments. Genetic studies of OSA have lagged behind other chronic diseases, however recent gene discovery efforts have been successful in finding genetic loci contributing to OSA-associated intermediate phenotypes. Nevertheless, many of the seminal questions relating to the genetic epidemiology of OSA and associated factors remain unanswered. This paper reviews the current state of knowledge of the genetics of OSA, with a focus on genomic approaches to understanding sleep apnea.
Keywords: Genetics, obstructive sleep apnea, epidemiology
1. Introduction
Obstructive sleep apnea (OSA) is a genetically complex disease that likely results from multiple interacting genetic and environmental factors1. Obesity is a major risk factor and is present in approximately 70% of patients with OSA2, 3. OSA affects up to 15% of middle-aged adults4-7, with a prevalence that increases rapidly with age and is higher in men. OSA is associated with substantial social and economic costs globally due to its high prevalence in the community, the profound clinical effects on an individual’s cognitive and general functioning, and the increased risk of adverse health complications8.
Concurrent with increased clinical recognition of OSA and importance as a chronic disease, there have been major advances in genetic methodologies to discover genetic susceptibility loci for many conditions. For example, 97 validated loci have been discovered by genome wide association studies (GWAS) for obesity9. Relative to many other common, chronic disorders, the genetic understanding of OSA has lagged behind. In part this is due to a lack of large collections of well-characterized OSA cases with DNA. Much remains unknown regarding the genetic basis of OSA. Understanding the genetic basis of OSA susceptibility, disease progression, and response to therapy are all important goals. In particular, such understanding may prove critical to the development of better ways of targeting efficacious treatments for OSA (i.e., ‘precision medicine’10).
In this review we discuss the definition of adult OSA as a disorder, biomarkers of OSA, what is known about the genetic epidemiology of OSA and different analytic approaches to detect genes for OSA, and the importance of intermediate phenotypes of OSA. Finally, we discuss future genomic approaches in OSA research.
2. Definition of obstructive sleep apnoea
OSA is a syndrome where reduced patency of the upper airway results in repetitive upper airway obstruction, either in the form of reduced airflow (hypopnea) or complete cessation of airflow (apnea), as the soft tissue structures of the upper airway, behind the tongue and soft palate, collapse in upon one another11. Obstructive events lead to hypoxemia and are usually terminated by a cortical arousal from sleep resulting in muscle activation and recovery of airway patency12. The primary measure that is used to define disease in both clinical and research settings is the apnea hypopnea index (AHI). This is calculated from overnight polysomnography (PSG) and is a count of the number of episodes of breathing obstruction per hour of sleep. Male and female subjects are often considered to have OSA if the AHI exceeds a certain level, usually 5-10 events per hour. Other factors enter into the clinical diagnostic decision than AHI, but it remains the primary biomarker of OSA.
3. Biomarkers of obstructive sleep apnea
The use of the AHI to determine disease status in clinical practice and research studies is advantageous because of its simple calculation from PSG and its high night-to-night reproducibility13. Most genetic studies of OSA have thus far used the AHI as the sole disease-defining variable14. The major disadvantages of using the AHI as the only biomarker of disease include: 1) the between-laboratory and within-laboratory variability in measurement and scoring of PSG15; 2) the loss of important information regarding the severity of individual obstructive events (duration, intensity and frequency of hypoxemia and arousal); and 3) the absence of information on the functional impact of the disease on an individual. Additionally, the AHI may include both obstructive and central events, and obstructed breaths may or may not occur in a background of periodic breathing. Since central events and Cheyne-Stokes respiration may occur in association with cardiac failure, central nervous system disease, and with use of certain medications (e.g., opiates)16, the AHI may not accurately reflect the physiological nature of the disease process in an individual.
Other biomarkers used for epidemiologic studies include measures of hypoxemia (e.g., the oxygen desaturation index, which measures the number of episodes of oxygen desaturation of greater than 3 or 4% per hour of sleep or time spent with oxygen saturation below a certain threshold), measures of sleep disruption (e.g., the arousal index), and duration of obstructive events.
Another important biomarker is obesity, as epidemiological, molecular, and physiological studies demonstrate substantial evidence for shared pathogenic pathways for obesity and OSA. Fat located in the intra-abdominal cavity and surrounding vital organs (visceral fat) predicts insulin resistance and other cardiometabolic consequences and is biochemically active. OSA induced hypoxia, sleep fragmentation and sleep loss contributes to further metabolic abnormalities17, which predisposes to obesity. However, total fat (visceral and subcutaneous) around the neck and abdomen is also likely to exert a direct mechanical load on the upper airway. Therefore better characterization of body fat distribution rather than reliance upon the body mass index (BMI) and anthropometry is necessary to define obesity and its impact on OSA. Novel methods to characterize fat deposition such as dual energy X-ray absorptiometry (DEXA) imaging have begun to be used18-20.
New objective biomarkers are necessary for OSA. Future investigations should consider biomarkers of hypoxic stress or arousal, central ventilatory control, and sleep homeostasis. There is a need to utilize other physiological, biochemical and/or anatomic measures to optimally characterize the OSA phenotype, and to couple this work with genetic studies. Recent advances in physiological methodology to measure anatomic (magnetic resonance imaging, CT scans, optical coherence tomography, 3D scanning) and non-anatomic/physiological causes of OSA with simple testing (passive critical closing pressure of the upper airway, arousal threshold, loop gain and muscle responsiveness)21 are likely to play a major role in this area moving forward.
4. The genetic epidemiology of OSA and associated traits
Complex genetic diseases such as OSA do not follow Mendelian patterns of inheritance and characteristically involve many genes that interact with many environmental factors.
4.1. Analytic approaches to detect genes for complex diseases such as OSA
Heritability studies measure the covariance of OSA or OSA-related phenotypes among family members and estimate the proportion of phenotypic variance attributable to genetic factors. Candidate gene studies examine associations between selected single nucleotide polymorphisms (SNPs) and OSA phenotypes in case-control or cohort studies. The SNPs are selected based upon a known or presumed contribution of the variant to a functional change in biochemistry, or on the basis of reasonable biological plausibility for a role of the gene in disease etiology. In contrast, the intention of genome-wide linkage studies and GWAS is to find causal genes without having a priori knowledge of functionality or position within the genome – a ‘hypothesis free’ discovery paradigm22. Genome-wide linkage studies use genotyped familial data to identify regions of the genome statistically associated (co-segregating) with the disease or disease-associated phenotype23, 24. GWAS generally genotype a dense set of markers in large samples of unrelated individuals to identify genomic regions containing common genetic variants causally associated with disease phenotypes22, 25. Figure 1 summarizes the GWAS process in more detail for Crohn’s disease (The text and figure reprinted with permission from the American Association for the Advancement of Science).
Figure 1. (Figure and text reprinted with permission from D. Altshuler, M.J. Daly, E.S. Lander Genetic Mapping in Human Disease. Science 2008; 322(5903): 881-888).
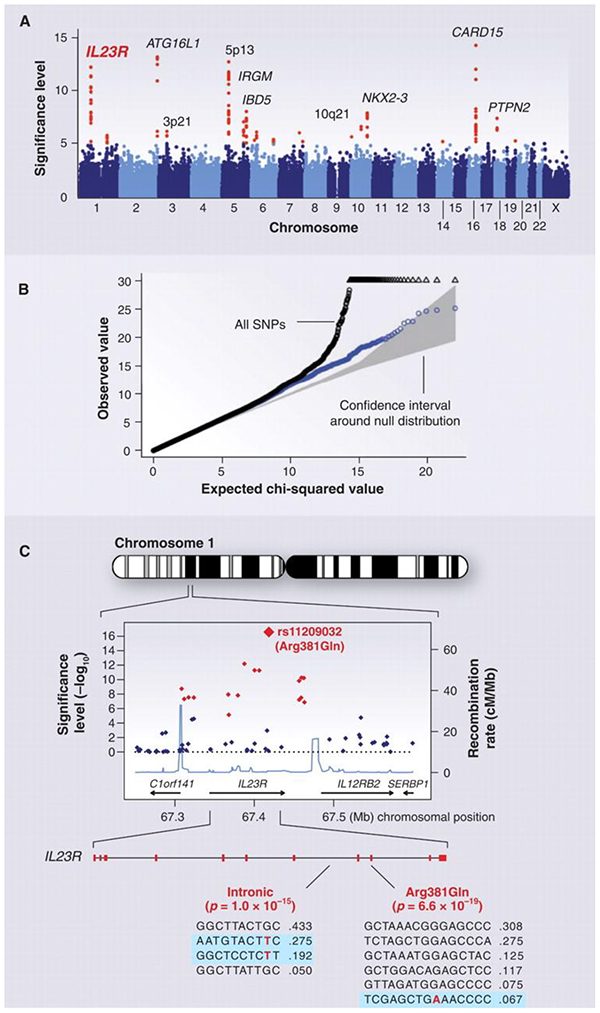
GWAS for Crohn's disease. The panels show data from the study of Crohn's disease by the Wellcome Trust Case Control Consortium. (A) Significance level (P value on log10 scale) for each of the 500,000 SNPs tested across the genome. SNP locations reflect their positions across the 23 human chromosomes. SNPs with significance levels exceeding 10−5(corresponding to 5 on the y axis) are colored red; the remaining SNPs are in blue. Ten regions with multiple significant SNPs are shown, labeled by their location or by the likely disease-related gene (e.g., IL23R on chromosome 1). (B) The fact that the SNPs in red are extreme outliers is made clear from a so-called Q-Q plot. A Q-Q plot is made as follows: The SNPs are ordered (from 1 to n) according to their observed P values; observed and expected P values are plotted for each SNP. Under the null distribution, the expected P value for the ith SNP is i/n. If there are no significant associations, the Q-Q plot will lie along the 45° line; the gray region corresponds to a 95% confidence region around this null expectation. Black points correspond to all 500,000 SNPs studied that passed strict quality control; they diverge strongly from the null expectation. Blue points reflect the P values that remain when the SNPs in the 10 most significant regions are removed; there is still some excess of significant P values, indicating the presence of additional loci of more modest effect. (C) Close-up of the region around the IL23R locus on chromosome 1. The first part shows the significance levels for SNPs in a region of ~400 kb, with colors as in (A). The highest significance level occurs at a SNP in the coding region of the IL23R gene (causing an Arg381 → Gln change). The light blue curve shows the inferred local rate of recombination across the region. There are two clear hotspots of recombination, with SNPs lying between these hotspots being strongly correlated in a few haplotypes. The second part shows that the IL23R locus harbors at least two independent, highly significant disease-associated alleles. The first site is the Arg381 → Gln polymorphism, which has a single disease-associated haplotype (shaded in blue) with frequency of 6.7%. The second site is in the intron between exons 7 and 8; it tags two disease-associated haplotypes with frequencies of 27.5% and 19.2%.
In sharp contrast to linkage or candidate gene studies, GWAS of large, unrelated samples investigating complex diseases have so far discovered >10,000 robust associations with diseases and disease associated phenotypes over the last decade for complex chronic diseases such as type 2 diabetes, autoimmune diseases, and common cancers26.
4.2. Heritability studies of OSA and related phenotypes
OSA is a heritable disease 27, 28. First degree relatives of an individual with OSA are more likely to snore or have observed apneas, after controlling for obesity, age, and gender28, 29. Around 40% of the variance in AHI has been shown to be explained by genetic factors27. Twin and family studies suggest that ventilatory responsiveness to either hypoxemia or hypercapnia, obesity, and craniofacial morphology are also under a high degree of genetic control, with 30-70% of phenotypic variance being explained by shared familial factors30,31-33. The genetic basis of sleep duration and quality is poorly defined. Heritability has been estimated at ~40% for measures of sleep duration34, 25-45% for insomnia35, and 17% for excessive daytime sleepiness35, but few genetic factors are known. Sleep timing is an essential component of the sleep-wake cycle with twin studies suggesting a genetic component in diurnal types (chronotype), with preference for time to bed and wake time estimated to be about 50% heritable36. Chronotype has a strong genetic basis but is also influenced by light exposure, age and gender37. These observations suggest that genetic factors likely contribute to OSA disease severity.
4.3. Genome-wide linkage studies of OSA
Three genome-wide linkage studies of the Cleveland Family Study (a longitudinal study of cases of OSA, their family members, and control families) were conducted in both Caucasians and African Americans38-40. In common with genome-wide linkage scans for most other common complex diseases, linkage proved to be under-powered for gene discovery in the context of a complex, multifactorial disease, and the results were inconclusive41.
A more recent genome wide linkage analysis from the Cleveland Family study and follow-up association identified a clear link between a polymorphism in the angiopoietin-2 gene (ANGPT2), an endothelial factor which modulates vascular and inflammatory responses, and mean nocturnal oxygen saturation, a surrogate marker of the severity of OSA42. Biological/functional follow-up of the ANGPT2 candidate gene and associated variants is now warranted.
4.4. Candidate gene association studies of OSA
Many candidate gene studies of OSA have examined polymorphisms in genes known to encode biomarkers of inflammatory response and insulin resistance43. Most have been of small sample size and have investigated polymorphic variation in genes with strong biological plausibility in disease risk. As for linkage analysis, it is generally true that most candidate gene studies of complex human diseases and phenotypes have been difficult to replicate and most are likely to be spurious. This failure relates to issues of problematic study design and concomitant low statistical power, and to a failure to replicate findings44. Applied to OSA, the majority of the candidate gene association studies conducted to date have either failed to replicate or replication has yet to attempted. Notably, the majority of candidate genes studied have not been significantly associated with OSA phenotypes in newer GWA studies.
Nevertheless, a few candidate gene association studies of OSA have been independently replicated (Table 1). Some of these suggest a relationship between polymorphic variation in the pro-inflammatory cytokine, tumor necrosis factor-α (TNFα) gene and OSA. The −308G/A polymorphism has been associated with increased levels of TNF-α, which is involved in intermittent hypoxia. Amongst OSA cases, the A allele, in comparison with those homozygous for the G allele, was significantly associated with AHI45, 46, oxygen saturation, and serum TNF-α levels45. However, controls with the A ‘risk’ allele had smaller waists and necks, and lower levels of TNF-α in comparison with cases who had the same genotype, suggesting that factors other than the TNF-α (−308G/A) polymorphism are associated with raised TNF-α levels and obesity in OSA. Meta-analysis of these three studies estimated that the TNF-α (−308G/A) polymorphism is a risk factor for OSA (OR=1.82, 95% CI 1.26-2.61)47.
Table 1:
Replicated candidate gene association studies of OSA
| Gene (publication) |
Abbreviation | Chromosome | Year | Cases/controls | Findings: |
|---|---|---|---|---|---|
| Tumor necrosis factor-α47 | TNF-α | 6 | 2014 | 1369/1064 | TNF-α (−308G/A) polymorphism is a risk factor for OSA |
| Apolipoprotein E Epsilon 449 | APOE ε4 | 19 | 2009 | 1901/4607 | no causal relationship between the APOE ε4 locus and OSA |
| Prostaglandin E2 receptor EP3 subtype50 | PTGER3 | 1 | 2012 | 963/1965 European ancestry | rs1409986 SNP in the PTGER3 gene was significantly associated with OSA |
| Lysophosphatidic acid receptor 150 | LPAR1 | 9 | 2012 | 233/414 African Americans | rs7030789 SNP in the LPAR1 gene was associated with OSA |
Several larger studies have investigated the APOE ε4 locus. The APOE ε4 locus encodes a lipoprotein that influences lipid metabolism, ventilatory stability during sleep, and the development of cardiovascular disease48. Reported associations between OSA and variation at this locus have been conflicting, however a recent meta-analysis of eight studies reviewed data on 1,901 cases of OSA and 4,607 controls and concluded that there was not enough evidence to suggest a causal relationship between the APOE ε4 locus and OSA49 (Table 1).
In 2012, the first multiple cohort candidate gene study of OSA including subjects of both European and African-American ancestry was performed50. European ancestry subjects came from subsets of the Atherosclerosis Risk in Communities study, Framingham Heart Study, and the Cleveland Family Study with a total of 963 cases of OSA and 1,965 controls. Subjects of African American ancestry came from the Cleveland Family Study (233 OSA cases, 414 controls). Unlike previous candidate gene studies of OSA, the CARe study considered a broader spectrum of the genome (>2000 genes) and selected candidate genes implicated in heart, lung, blood, and sleep disorders51. In European ancestry subjects, the rs1409986 SNP in the prostaglandin E2 receptor (PTGER3) gene was significantly associated with OSA. In African-American ancestry subjects, the rs11126184 SNP in the pleckstrin (PLEK) gene was associated with OSA and the rs7030789 SNP in the lysophosphatidic acid receptor 1 (LPAR1) gene was associated with AHI. The authors then sought to replicate these findings in further independent samples. The association of OSA with rs1409986 (PTGER3) was replicated in a sub-sample of European Caucasians (n=1795) from the Western Australian Sleep Health Study52. In a replication study of the findings in African American subjects, 459 clinical cases of OSA from the Case Sleep Apnoea study were compared with 551 controls from the Case Transdisciplinary Research in Energetics and Cancer Colon Polyps Study. The findings with rs7030789 (LPAR1) but not rs11126184 (PLEK) were replicated (Table 1).
4.5. Genome-wide association studies of OSA
Consistent with genetic research in OSA being an under-explored area, the first GWAS for OSA phenotypes have only recently been conducted.
A recent study reported a GWAS on 12,558 Hispanic American ancestry participants where a number of OSA-associated phenotypic traits were investigated, including AHI, mean oxygen saturation, and mean apnea and hypopnea duration53. AHI was associated with a polymorphism in the G-protein receptor gene (GPR83), which is expressed in several areas of the brain including the hypoglossal nucleus, dorsal motor nucleus of the vagus, and the nucleus of the solitary tract. Measures of sensitivity to hypoxia and respiratory arousability as reflected in the average apnea and hypopnea duration in females were suggestively associated with variants in the β-arrestin 1 (ARRB1) gene, which is an important regulator of hypoxia inducible factor 1 alpha (HIF-1α) and influences the expression of HIF-1α target genes, including vascular endothelial growth factor. HIF-1α plays a role in hypoxic sensitivity in the carotid bodies and therefore the causal variant at this locus may be intimately involved in the etiology of OSA through hypoxic sensitivity and control. The duration of apneas and hypopneas was also associated with variation in several loci associated with an important lipid biosynthesis transcription factor, sterol regulatory element binding protein (SREBP). While independent replication of these findings may be difficult due to the lack of independent cohorts of Hispanic/Latino ancestry with AHI measures and genetic data, future studies should investigate association of independent common or rare variants in these gene regions in other ethnic groups.
The International Sleep Genetic Epidemiology Consortium (ISGEC)54 was formed in 2011 and has recently completed the first of a series of planned GWAS meta-analyses for OSA phenotypes. The first ISGEC study has investigated risk of moderate/severe OSA by conducting a GWAS in case and control samples from 9 independent European ancestry cohorts. Replication was undertaken in 5 mixed ancestry cohorts. In total, 8,336 cases and 76,663 controls were investigated. Analyses were stratified by obesity in order to examine associations with OSA risk in obese and non-obese patients. Results have been presented at international scientific meetings but have yet to be published.
4.6. Genetics of OSA related traits: Intermediate phenotypes
In OSA a number of risk factors interact to increase the likelihood for repetitive upper airway collapse during sleep. In any individual, this is determined by anatomic and non-anatomic factors that influence upper airway size and collapsibility. Recognized risk factors include: obesity, male gender, small upper airway size, ventilatory control mechanisms, and control of sleep and circadian rhythm. Identification of genes that determine ‘intermediate’ phenotypes that are potentially on a causal pathway leading to OSA may be helpful to determine susceptibility genes.
4.6.1. Obesity and Body Fat Distribution.
Obesity increases the risk of OSA 10-14 fold, with the most profound impact observed in middle-age. Obesity, measured as BMI, is expected to explain up to 40% of the genetic variance in AHI55. Regulation of body weight and fat distribution are genetically modulated. Recent GWAS in up to 339,224 individuals have identified 97 loci that contribute to normal variation in BMI, implicating genes and pathways related to synaptic function, glutamate signaling, insulin action and secretion, energy metabolism, lipid biology, and adipogenesis9. In support of the importance of the genetic contribution of BMI to OSA, a recent study tested for association of variation in the BMI-associated FTO/IRX3 locus with >1,000 Electronic Health Record ascertained diagnoses and found significant evidence of association with OSA (2,335 cases and 21,863 controls56). The effect of the FTO/IRX3 region variant was attenuated upon adjustment for BMI. GWAS of body fat distribution in up to 224,459 individuals have identified 49 genetic loci for waist-to-hip ratio adjusted for BMI, with sex-specific associations identified at approximately half of the loci, highlighting the considerable sexual dimorphism in this trait57. Nineteen additional loci for related measures of waist and hip circumference have been identified. Together, these loci highlight adipogenesis, angiogenesis and insulin resistance as important processes contributing to inter-individual differences in body fat distribution9.
4.6.2. Craniofacial Morphology.
Craniofacial morphology, affecting both bony and soft tissues, predisposes to OSA by reducing the size of the upper airway. Commonly identified craniofacial abnormalities in OSA include mandibular deficiency, an inferiorly placed hyoid bone relative to the mandibular plane, a narrow posterior air space, a greater flexion of the cranial base, and elongation of the soft palate. When these abnormalities are coupled with obesity the degree of craniofacial abnormality may determine the extent of obesity required to produce OSA in a given individual58.
Recent GWAS of 20 quantitative traits of normal facial morphology based on 3D surface images have thus far identified and in some cases, replicated, genome-wide significant associations for cranial base width, intercanthal width, nasal width, nasal ala length, and upper facial depth59-61, implicating the genes CACNA2D3, PRDM16, MAFB, PAX9, MIPOL1, ALX3, HDAC8, PAX3, and PAX1. Interestingly, many newly discovered regions harbor genes that are known to play a role in craniofacial development or are mutated in rare syndromes affecting the face, suggesting that larger studies of specific craniofacial abnormalities associated with OSA may be fruitful.
4.6.3. Ventilatory Control
No genetic studies investigating ventilatory control in OSA have been performed. Measurement of loop gain, muscle responsiveness, and passive critical closing pressure of the upper airway will assist in better phenotyping of OSA patients and will be important for future genetic studies21.
4.6.4. Control of sleep and circadian rhythm
Sleep and circadian rhythm are complex, multifactorial traits with sleep being controlled and influenced by many environmental and genetic factors, with the genes regulating sleep-wake control and circadian rhythm62 likely having particular importance. Evidence of a genetic component in the regulation of human sleep is suggested by the heritability of sleep traits, the identification of specific genetic polymorphisms that affect these traits, and known familial sleep disorders63.
Many candidate gene studies have targeted circadian genes, based on the detailed understanding of the molecular nature of the circadian clock and the connection with sleep regulation in humans64. Thirty years after the seminal work on Period gene mutants in Drosophila65, it was discovered that familial advanced sleep phase syndrome is caused by mutations in analogous human clock-related genes. A mutation in Period 2 results in individuals having a 4-6 hour phase advance in sleep and wake times, on a background of normal sleep architecture66. Subsequently, several associations between clock genes and circadian rhythm disorders have been reported. The core set of clock genes involved in the generation of circadian rhythmicity are known67. A Mendelian short sleep mutation in BHLHE41 (P385R) has been identified, and confirmed in a mouse model68.
Three recent GWAS of self-reported morningness/eveningness preference in large-scale populations of European ancestry with >100,000 individuals (the UK Biobank resource and 23andMe company database) identified over 20 genome-wide significant associations, implicating known circadian clock genes and providing new opportunities for enhanced molecular understanding of chronotype and its impact on health outcomes69-71. Future studies require GWAS in large samples with better measures of chronotype, and objective measures of sleep timing to fully characterize the genetic architecture of chronotype.
GWAS for sleep duration have been reported70 , 72, 73 but only associations at the thyroid expressed PAX8 locus and schizophrenia linked VRK2 locus have reached genome-wide significance in well-powered studies with >40,000 individuals70, 72, 73. Suggestive association is seen for dopamine D2 receptor gene (DRD2) variation in a multi-ethnic study74. In UK Biobank (n=112,586), genetic associations with insomnia symptoms (near MEIS1, TMEM132E, CYCL1, TGFBI in females and WDR27 in males) and excessive daytime sleepiness (near AR/OPHN1) have been identified72, 75 that should be useful for understanding of the contribution of sleep quantity and quality to the etiology of OSA. Further studies in this important area are needed.
5. Genetic links between sleep apnea and adverse health outcomes
OSA has an established independent association with CVD, metabolic syndrome, and hypertension. More severe OSA is more frequently associated with adverse cardiovascular, metabolic, and neuropsychiatric outcomes76, 77. Observational studies have consistently shown significantly fewer cardiovascular events, reduced vascular endothelial dysfunction, inflammation, and oxidative stress, and reduced HbA1c levels in patients adherent to CPAP therapy compared to those who are not adherent77-80. OSA is associated with cognitive dysfunction, which can be at least partially reversed with CPAP therapy, and with accelerated cognitive decline and dementia81-88.
All of these traits associated with OSA are themselves genetically determined, and therefore, a key question for future studies is to examine if genetic factors are shared between OSA and these adverse health outcomes. If genetic variants are found to associate both with OSA and a co-morbid disorder, Mendelian Randomization89, 90, a technique that uses inherited genetic effects as instrumental variables to elucidate if a trait is causally linked to another trait, can be used to determine whether pathways disrupted in the pathogenesis of OSA causally contribute to these outcomes, or whether genetic effects are pleiotropic, i.e. the genetic variants contribute simultaneously to both phenotypes through different mechanistic effects.
6. New in silico and genomic approaches to understanding OSA
Online databases containing sequencing and functional data have improved dramatically over the last decade, and such ‘in silico’ resources greatly aid identification of the causal variant and biological interpretation of findings from GWAS discovery efforts26, 91. For instance, the ongoing accrual of data on coding variants (via exome sequencing and/or exome array genotyping) has been extremely useful in assigning correct causal coding variants to GWAS peaks92, 93. Additionally, given that GWAS findings have largely implicated non-coding variation, sequence annotation of tissue-specific chromatin states and protein binding (e.g., from the Roadmap Epigenomics94 and ENCODE projects95), promoter, enhancer, and transcriptomic atlases that assess combinatorial gene regulation (e.g., the FANTOM project96), and databases that enable assessment of the effect of SNPs on tissue-specific gene expression (e.g., the gTEX consortium97) have been particularly useful to identify functional variants and to generate hypotheses about their mechanism of action.
Current technologies such as exome or whole genome sequencing92, epigenetic analysis (that measures DNA methylation and chromatin modification98), and transcriptomics, proteomics, and metabolomics (measuring changes in levels of gene expression, protein or metabolite levels)99 have only recently begun to be applied to OSA. Such technologies are promising, and have accelerated the understanding of other diseases100, 101. Rare variants may play an important role in chronic disease102. Indeed, discovery of rare, protective variants for cardiovascular disease has already resulted in successful development of cholesterol-lowering drugs that mimic human mutations103. Analytically, genome sequencing simply results in a very dense GWAS, but novel methods that aggregate the effect of putatively functional rare variants across genes or genomic windows increase power98. Epigenome-wide association studies (EWAS) assess DNA methylation across diseased and normal individuals to identify differentially methylated sites that could be environmental or genetic causal contributors to OSA or may be a consequence of the disease process. Initial studies have reported altered transcriptomic signatures in OSA patients versus controls in blood leukocytes104, 105 and visceral fat106, and suppression of cancer-associated gene expression signatures in cases after treatment with CPAP106, but future research in transcriptomics, proteomics and metabolomics and integrative genomics analysis will be important to derive biological insights into the causes and consequences of sleep apnea.
7. Conclusions
OSA is a complex disease genetically with many potential genetic and environmental factors combining to produce the disease. The future for gene discovery in complex diseases such as OSA will likely involve hundreds of thousands of samples with whole-genome sequencing data. New approaches involving in silico research and advanced sequencing technologies together with ongoing efforts in GWAS and international cooperation and collaboration54 are causes for optimism that the genetic basis of OSA will be defined over the coming decade. Such knowledge will lead to better understanding of the pathogenesis of the disease and potentially to better therapies and to ‘precision medicine’. Ongoing work on model organisms for OSA will also be important to fully understand causative pathways.
References cited
- 1.Palmer LJ, Redline S. Genomic approaches to understanding obstructive sleep apnea. Respir Physiol Neurobiol. 2003; 135: 187–205. [DOI] [PubMed] [Google Scholar]
- 2.Elmasry A, Lindberg E, Berne C, et al. Sleep-disordered breathing and glucose metabolism in hypertensive men: a population-based study. Journal of Internal Medicine. 2001; 249: 153–61. [DOI] [PubMed] [Google Scholar]
- 3.Grunstein RR, Ho KY, Berthon-Jones M, et al. Central sleep apnea is associated with increased ventilatory response to carbon dioxide and hypersecretion of growth hormone in patients with acromegaly. American Journal of Respiratory & Critical Care Medicine. 1994; 150: 496–502. [DOI] [PubMed] [Google Scholar]
- 4.Young T, Palta M, Dempsey J, et al. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993; 328: 1230–5. [DOI] [PubMed] [Google Scholar]
- 5.Bearpark H, Elliott L, Grunstein R, et al. Snoring and sleep apnea. A population study in Australian men. Am J Respir Crit Care Med. 1995; 151: 1459–65. [DOI] [PubMed] [Google Scholar]
- 6.Ferini-Strambi L, Fantini ML, Castronovo C. Epidemiology of obstructive sleep apnea syndrome. Minerva Med. 2004; 95: 187–202. [PubMed] [Google Scholar]
- 7.Simpson L, Hillman DR, Cooper MN, et al. High prevalence of undiagnosed obstructive sleep apnoea in the general population and methods for screening for representative controls. Sleep Breath. 2013; 17: 967–73. [DOI] [PubMed] [Google Scholar]
- 8.Tarasiuk A, Reuveni H. The economic impact of obstructive sleep apnea. Curr Opin Pulm Med. 2013; 19: 639–44. [DOI] [PubMed] [Google Scholar]
- 9.Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015; 518: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015; 372: 793–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fogel RB, Malhotra A, White DP. Sleep. 2: pathophysiology of obstructive sleep apnoea/hypopnoea syndrome. Thorax. 2004; 59: 159–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012; 8: 597–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Redline S, Tosteson T, Boucher MA, et al. Measurement of sleep-related breathing disturbances in epidemiologic studies. Assessment of the validity and reproducibility of a portable monitoring device. Chest. 1991; 100: 1281–6. [DOI] [PubMed] [Google Scholar]
- 14.Sleiman P, Hakonarson H. Genetic underpinnings of obstructive sleep apnea: are we making progress? Sleep. 2011; 34: 1449–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manser RL, Rochford P, Naughton MT, et al. Measurement variability in sleep disorders medicine: the Victorian experience. Intern Med J. 2002; 32: 386–93. [DOI] [PubMed] [Google Scholar]
- 16.Farney RJ, McDonald AM, Boyle K, et al. Sleep disordered breathing (SDB) in patients receiving therapy with buprenorphine/naloxone. Eur Respir J. 2012. [DOI] [PubMed] [Google Scholar]
- 17.Van Cauter E, Spiegel K, Tasali E, et al. Metabolic consequences of sleep and sleep loss. Sleep Med. 2008; 9 Suppl 1: S23–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpson L, Mukherjee S, Cooper MN, et al. Sex differences in the association of regional fat distribution with the severity of obstructive sleep apnea. Sleep. 2010; 33: 467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sforza E, Chouchou F, Collet P, et al. Sex differences in obstructive sleep apnoea in an elderly French population. Eur Respir J. 2011; 37: 1137–43. [DOI] [PubMed] [Google Scholar]
- 20.Saint Martin M, Roche F, Thomas T, et al. Association of body fat composition and obstructive sleep apnea in the elderly: A longitudinal study. Obesity (Silver Spring). 2015; 23: 1511–6. [DOI] [PubMed] [Google Scholar]
- 21.Eckert DJ, White DP, Jordan AS, et al. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med. 2013; 188: 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.WTCCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007; 447: 661–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morton NE. Sequential tests for the detection of linkage. Am J Hum Genet. 1956; 7: 277–318. [PMC free article] [PubMed] [Google Scholar]
- 24.Haseman JK, Elston RC. The investigation of linkage between a quantitative trait and a marker locus. Behav Genet. 1972; 2: 3–19. [DOI] [PubMed] [Google Scholar]
- 25.Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008; 118: 1590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Visscher PM, Wray NR, Zhang Q, et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet. 2017; 101: 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev. 2000; 4: 583–602. [DOI] [PubMed] [Google Scholar]
- 28.Redline S, Tosteson T, Tishler PV, et al. Studies in the genetics of obstructive sleep apnea. Familial aggregation of symptoms associated with sleep-related breathing disturbances. Am Rev Respir Dis. 1992; 145: 440–4. [DOI] [PubMed] [Google Scholar]
- 29.Redline S, Tosteson T, Tishler PV, et al. Studies in the genetics of obstructive sleep apnea: familial aggregation of symptoms associated with sleep related breathing disturbances. Am Rev Respir Dis. 1992; 145: 440–4. [DOI] [PubMed] [Google Scholar]
- 30.Villaneuva AT, Buchanan PR, Yee BJ, et al. Ethnicity and obstructive sleep apnoea. Sleep Med Rev. 2005; 9: 419–36. [DOI] [PubMed] [Google Scholar]
- 31.Patel SR, Frame JM, Larkin EK, et al. Heritability of upper airway dimensions derived using acoustic pharyngometry. Eur Respir J. 2008; 32: 1304–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herrera BM, Lindgren CM. The genetics of obesity. Curr Diab Rep. 2010; 10: 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roosenboom J, Hens G, Mattern BC, et al. Exploring the Underlying Genetics of Craniofacial Morphology through Various Sources of Knowledge. Biomed Res Int. 2016; 2016: 3054578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Castro JM. The influence of heredity on self-reported sleep patterns in free-living humans. Physiol Behav. 2002; 76: 479–86. [DOI] [PubMed] [Google Scholar]
- 35.Wing YK, Zhang J, Lam SP, et al. Familial aggregation and heritability of insomnia in a community-based study. Sleep Med. 2012; 13: 985–90. [DOI] [PubMed] [Google Scholar]
- 36.Koskenvuo M, Hublin C, Partinen M, et al. Heritability of diurnal type: a nationwide study of 8753 adult twin pairs. J Sleep Res. 2007; 16: 156–62. [DOI] [PubMed] [Google Scholar]
- 37.Roenneberg T, Kuehnle T, Juda M, et al. Epidemiology of the human circadian clock. Sleep Med Rev. 2007; 11: 429–38. [DOI] [PubMed] [Google Scholar]
- 38.Palmer LJ, Buxbaum SG, Larkin EK, et al. Whole genome scan for obstructive sleep apnea and obesity in African-American families. Am J Respir Crit Care Med. 2004; 169: 1314–21. [DOI] [PubMed] [Google Scholar]
- 39.Palmer LJ, Buxbaum SG, Larkin EK, et al. A whole-genome scan for obstructive sleep apnea and obesity. Am J Hum Genet. 2003; 72: 340–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larkin EK, Patel SR, Elston RC, et al. Using linkage analysis to identify quantitative trait loci for sleep apnea in relationship to body mass index. Ann Hum Genet. 2008; 72: 762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altmuller J, Palmer LJ, Fischer G, et al. Genomewide scans of complex human diseases: true linkage is hard to find. Am J Hum Genet. 2001; 69: 936–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Cade BE, Chen H, et al. Variants in angiopoietin-2 (ANGPT2) contribute to variation in nocturnal oxyhaemoglobin saturation level. Hum Mol Genet. 2016; 25: 5244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varvarigou V, Dahabreh IJ, Malhotra A, et al. A review of genetic association studies of obstructive sleep apnea: field synopsis and meta-analysis. Sleep. 2011; 34: 1461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmer LJ, Cardon LR. Shaking the tree: mapping complex disease genes with linkage disequilibrium. Lancet. 2005; 366: 1223–34. [DOI] [PubMed] [Google Scholar]
- 45.Bhushan B, Guleria R, Misra A, et al. TNF-alpha gene polymorphism and TNF-alpha levels in obese Asian Indians with obstructive sleep apnea. Respir Med. 2009; 103: 386–92. [DOI] [PubMed] [Google Scholar]
- 46.Riha RL, Brander P, Vennelle M, et al. Tumour necrosis factor-alpha (−308) gene polymorphism in obstructive sleep apnoea-hypopnoea syndrome. Eur Respir J. 2005; 26: 673–8. [DOI] [PubMed] [Google Scholar]
- 47.Zhong A, Xiong X, Xu H, et al. An updated meta-analysis of the association between tumor necrosis factor-alpha −308G/A polymorphism and obstructive sleep apnea-hypopnea syndrome. PLoS One. 2014; 9: e106270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gottlieb DJ, DeStefano AL, Foley DJ, et al. APOE epsilon4 is associated with obstructive sleep apnea/hypopnea: the Sleep Heart Health Study. Neurology. 2004; 63: 664–8. [DOI] [PubMed] [Google Scholar]
- 49.Thakre TP, Mamtani MR, Kulkarni H. Lack of association of the APOE epsilon 4 allele with the risk of obstructive sleep apnea: meta-analysis and meta-regression. Sleep. 2009; 32: 1507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel SR, Goodloe R, De G, et al. Association of genetic loci with sleep apnea in European Americans and African-Americans: the Candidate Gene Association Resource (CARe). PLoS One. 2012; 7: e48836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keating BJ, Tischfield S, Murray SS, et al. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS One. 2008; 3: e3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mukherjee S, Hillman D, Lee J, et al. Cohort profile: the Western Australian Sleep Health Study. Sleep Breath 2011. [DOI] [PubMed]
- 53.Cade BE, Chen H, Stilp AM, et al. Genetic Associations with Obstructive Sleep Apnea Traits in Hispanic/Latino Americans. Am J Respir Crit Care Med. 2016; 194: 886–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.2017. International Sleep Genetic Epidemiology Consortium (ISGEC) Website. https://sleepgenetics.org.
- 55.Patel SR, Larkin EK, Redline S. Shared genetic basis for obstructive sleep apnea and adiposity measures. Int J Obes (Lond). 2008; 32: 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cronin RM, Field JR, Bradford Y, et al. Phenome-wide association studies demonstrating pleiotropy of genetic variants within FTO with and without adjustment for body mass index. Front Genet. 2014; 5: 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shungin D, Winkler TW, Croteau-Chonka DC, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015; 518: 187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cistulli PA. Craniofacial abnormalities in obstructive sleep apnoea: implications for treatment. Respirology. 1996; 1: 167–74. [DOI] [PubMed] [Google Scholar]
- 59.Shaffer JR, Orlova E, Lee MK, et al. Genome-Wide Association Study Reveals Multiple Loci Influencing Normal Human Facial Morphology. PLoS Genet. 2016; 12: e1006149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paternoster L, Zhurov AI, Toma AM, et al. Genome-wide association study of three-dimensional facial morphology identifies a variant in PAX3 associated with nasion position. Am J Hum Genet. 2012; 90: 478–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu F, van der Lijn F, Schurmann C, et al. A genome-wide association study identifies five loci influencing facial morphology in Europeans. PLoS Genet. 2012; 8: e1002932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franken P, Chollet D, Tafti M. The homeostatic regulation of sleep need is under genetic control. J Neurosci. 2001; 21: 2610–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sehgal A, Mignot E. Genetics of sleep and sleep disorders. Cell. 2011; 146: 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Allada R, Emery P, Takahashi JS, et al. Stopping time: the genetics of fly and mouse circadian clocks. Annu Rev Neurosci. 2001; 24: 1091–119. [DOI] [PubMed] [Google Scholar]
- 65.Dubowy C, Sehgal A. Circadian Rhythms and Sleep in Drosophila melanogaster. Genetics. 2017; 205: 1373–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Toh KL, Jones CR, He Y, et al. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001; 291: 1040–3. [DOI] [PubMed] [Google Scholar]
- 67.Buhr ED, Takahashi JS. Molecular components of the Mammalian circadian clock. Handb Exp Pharmacol. 2013: 3–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He Y, Jones CR, Fujiki N, et al. The transcriptional repressor DEC2 regulates sleep length in mammals. Science. 2009; 325: 866–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu Y, Shmygelska A, Tran D, et al. GWAS of 89,283 individuals identifies genetic variants associated with self-reporting of being a morning person. Nat Commun. 2016; 7: 10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jones SE, Tyrrell J, Wood AR, et al. Genome-Wide Association Analyses in 128,266 Individuals Identifies New Morningness and Sleep Duration Loci. PLoS Genet. 2016; 12: e1006125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lane JM, Vlasac I, Anderson SG, et al. Genome-wide association analysis identifies novel loci for chronotype in 100,420 individuals from the UK Biobank. Nat Commun. 2016; 7: 10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lane JM, Liang J, Vlasac I, et al. Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nat Genet. 2017; 49: 274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gottlieb DJ, Hek K, Chen TH, et al. Novel loci associated with usual sleep duration: the CHARGE Consortium Genome-Wide Association Study. Mol Psychiatry. 2015; 20: 1232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cade BE, Gottlieb DJ, Lauderdale DS, et al. Common variants in DRD2 are associated with sleep duration: the CARe consortium. Hum Mol Genet. 2016; 25: 167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hammerschlag AR, Stringer S, de Leeuw CA, et al. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nat Genet. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Macey PM, Woo MA, Kumar R, et al. Relationship between obstructive sleep apnea severity and sleep, depression and anxiety symptoms in newly-diagnosed patients. PLoS One. 2010; 5: e10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Malik JA, Masoodi SR, Shoib S. Obstructive sleep apnea in Type 2 diabetes and impact of continuous positive airway pressure therapy on glycemic control. Indian J Endocrinol Metab. 2017; 21: 106–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marin JM, Carrizo SJ, Vicente E, et al. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet. 2005; 365: 1046–53. [DOI] [PubMed] [Google Scholar]
- 79.Jelic S, Lederer DJ, Adams T, et al. Vascular inflammation in obesity and sleep apnea. Circulation. 2010; 121: 1014–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Campos-Rodriguez F, Martinez-Garcia MA, de la Cruz-Moron I, et al. Cardiovascular mortality in women with obstructive sleep apnea with or without continuous positive airway pressure treatment: a cohort study. Ann Intern Med. 2012; 156: 115–22. [DOI] [PubMed] [Google Scholar]
- 81.Yaffe K, Laffan AM, Harrison SL, et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA. 2011; 306: 613–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yaffe K, Nettiksimmons J, Yesavage J, et al. Sleep Quality and Risk of Dementia Among Older Male Veterans. Am J Geriatr Psychiatry. 2015; 23: 651–4. [DOI] [PubMed] [Google Scholar]
- 83.Findley LJ, Barth JT, Powers DC, et al. Cognitive impairment in patients with obstructive sleep apnea and associated hypoxemia. Chest. 1986; 90: 686–90. [DOI] [PubMed] [Google Scholar]
- 84.Ferini-Strambi L, Baietto C, Di Gioia M, et al. Cognitive dysfunction in patients with obstructive sleep apnea (OSA): partial reversibility after continuous positive airway pressure (CPAP). Brain research bulletin. 2003; 61: 87–92. [DOI] [PubMed] [Google Scholar]
- 85.Engleman H, Douglas N. Sleep· 4: Sleepiness, cognitive function, and quality of life in obstructive sleep apnoea/hypopnoea syndrome. Thorax. 2004; 59: 618–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dealberto MJ, Pajot N, Courbon D, et al. Breathing disorders during sleep and cognitive performance in an older community sample: the EVA Study. Journal of the American Geriatrics Society. 1996; 44: 1287–94. [DOI] [PubMed] [Google Scholar]
- 87.Alchanatis M, Zias N, Deligiorgis N, et al. Comparison of cognitive performance among different age groups in patients with obstructive sleep apnea. Sleep and breathing. 2008; 12: 17–24. [DOI] [PubMed] [Google Scholar]
- 88.Ayalon L, Ancoli-Israel S, Drummond SP. Obstructive sleep apnea and age: a double insult to brain function? Am J Respir Crit Care Med. 2010; 182: 413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smith GD, Ebrahim S. Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol. 2004; 33: 30–42. [DOI] [PubMed] [Google Scholar]
- 90.Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003; 32: 1–22. [DOI] [PubMed] [Google Scholar]
- 91.McClelland KS, Yao HH. Leveraging Online Resources to Prioritize Candidate Genes for Functional Analyses: Using the Fetal Testis as a Test Case. Sex Dev. 2017; 11: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536: 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang H, Fang M, Jostins L, et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature. 2017; 547: 173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518: 317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lizio M, Harshbarger J, Shimoji H, et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015; 16: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Consortium GTEx. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348: 648–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee S, Abecasis GR, Boehnke M, et al. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014; 95: 5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017; 18: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cellerino A, Ori A. What have we learned on aging from omics studies? Semin Cell Dev Biol. 2017. [DOI] [PubMed] [Google Scholar]
- 101.Kan M, Shumyatcher M, Himes BE. Using omics approaches to understand pulmonary diseases. Respir Res. 2017; 18: 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bomba L, Walter K, Soranzo N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017; 18: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stitziel NO, Kathiresan S. Leveraging human genetics to guide drug target discovery. Trends Cardiovasc Med. 2017; 27: 352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lin SW, Tsai CN, Lee YS, et al. Gene expression profiles in peripheral blood mononuclear cells of Asian obstructive sleep apnea patients. Biomed J. 2014; 37: 60–70. [DOI] [PubMed] [Google Scholar]
- 105.Hoffmann MS, Singh P, Wolk R, et al. Microarray studies of genomic oxidative stress and cell cycle responses in obstructive sleep apnea. Antioxid Redox Signal. 2007; 9: 661–9. [DOI] [PubMed] [Google Scholar]
- 106.Gharib SA, Seiger AN, Hayes AL, et al. Treatment of obstructive sleep apnea alters cancer-associated transcriptional signatures in circulating leukocytes. Sleep. 2014; 37: 709–14, 14A-14T. [DOI] [PMC free article] [PubMed] [Google Scholar]